國
立
交
通
大
學
統計學研究所
碩士論文
利用藥物化學特徵和統計學習建構藥物負向事件預測模型
Prediction for Adverse Drug Events by
Chemical
Descriptors and Statistical Learning
研 究 生:吳宜靜
指導教授:盧鴻興 教授
利用藥物化學特徵和統計學習建構藥物負向事件預測模型
Prediction for Adverse Drug Events by
Chemical Descriptors and
Statistical Learning
研究生:吳宜靜
Student:Yi-Jing Wu
指導教授:盧鴻興 博士 Advisor:Dr. Henry Horng-Shing Lu
國 立 交 通 大 學
統 計 學 研 究 所
碩
士
論
文
A Thesis
Submitted to Institute of Statistics
College of Science
National Chiao Tung University
In Partial Fulfillment of the Requirements
For the Degree of
Master
in
Statistics
June 2012
Hsinchu, Taiwan, Republic of China
利用藥物化學特徵和統計學習建構藥物負向事件預測模型
研究生:吳宜靜
指導教授:盧鴻興 教授
國立交通大學統計學研究所
摘
要
當藥物成分進入人體後,產生複雜的擾動效應稱為藥效。藥效可分為主要治療藥效和額外
的效果,而
藥物不良反應事件
是額外效果的一部分。
每個藥物成分都是一種化合物,由其
化學式可以得到化學資訊特徵。基於藥物在人體系統中產生的生物擾動與其化學結構有關
的假設,我們檢視上市藥物的藥物不良反應事件與其化學資訊特徵之間的關聯。在本研究
中,我們使用決策樹方法指認出與 1384 個藥物不良反應事件的相關化學資訊特徵,並設計
一套自動分析流程。針對我們選定的 35 個有興趣的藥物不良反應事件可以得到模型十折交
叉驗證正確率高於 80%,例如: 糖尿病(87.1%),急性腎功能衰竭(91.0%)和腎功能不全
(94.6%)。
Prediction for Adverse Drug Events by Chemical Descriptors
and Statistical Learning
Student:Yi-Jing Wu
Advisor:Dr. Henry Horng-Shing Lu
Institute of Statistics
National Chiao Tung University
Hsinchu, Taiwan
Abstract
In addition to the medicine treatment effect, side effects are complex undesired phenomena due to
the bio-activity of pharmaceutical compound. For each compound, the chemistry informatics can
delineate its intrinsic chemical formula into chemistry informatics features. Based on the
assumption that the chemical structure is critical to the biological perturbation in the human
system, we investigate different adverse drug events with associated chemistry informatics
features of marketed drugs. In this research, we identify 1,384 ADEs with corresponding
associated chemistry informatics features by decision tree. With an automatic analysis workflow,
we can obtain a concordant drug subset with satisfying 10-fold cross-validation accuracy. The
accuracy of selected 35 ADEs in the test experiment is higher than 80%. For example, there are
three ADEs of interest and their accuracy: Diabetes Mellitus (0.871), Renal Failure Acute (0. 910)
and Renal Impairment (0. 946).
誌 謝
兩年碩士生涯雖然短暫但是收穫卻不少。這兩年能夠有所成果,
首先必須感謝我的指導教授─盧鴻興教授,感謝您在論文上、
課業上以及生活上不厭其煩地指導與幫助。其次,感謝口試委
員許文郁教授、王秀瑛教授以及謝文萍教授辛苦審查,並給指
導和建議使論文更加完善。
也必須感謝交大統計所所有的老師、郭姐和劉小姐,除了提供
我們良好的學習環境,讓我在碩士兩年學到了許多知識外,平
日給予的關懷與幫助讓我們能夠無憂無慮地學習。
再來要感謝所有交大統計所 99 級的同學們,因為有你們的一同
努力與分享,在兩年碩士生活中一起歡笑一起哭,讓我這兩年
的學習過程多了更多的調味劑。
最後,我要感謝我的家人,謝謝他們在我學習生涯中無論是遇
到挫折或是難過都給予我百分百的支持,使我能夠在學習過程
中完全無後顧之憂。
吳宜靜 謹誌于
國立交通大學統計學研究所
中華名國一百零一年六月
Contents
1.
Introduction... 1
1.1.
Background ... 1
1.2.
Literature Review ... 5
1.3.
Purpose of Research ... 6
2.
Method ... 7
2.1.
Materials ... 7
2.2.
Data Analysis ... 9
2.3.
Research Design ... 16
3.
Result ... 20
3.1.
Compare the performance by using different types of chemical feature sets. ... 20
3.2.
Select concordant drugs and then analyze the relation between chemical compounds
and ADE under the regular condition ... 23
4.
Discussion and Conclusions ... 31
4.1.
Discussion ... 31
4.2.
Conclusion ... 33
4.3.
Recommendation for Future Research ... 34
Reference: ... 35
List of Figures
Figure 1.1 The drug-metabolizing capability.
... 3
Figure 1.2 The instance about the therapeutic drug targets existing in multiple cell and
tissue types.
... 4
Figure 1.3 The main purpose of our research.
... 6
Figure 2.1 Flow chart for pre-processing work.
... 11
Figure 2.2 Workflow about the automatic clustering analysis.
... 16
Figure 2.3 The instance of concordant drug subset by concordant clustering analysis.
.... 17
Figure 2.4 The structure of clustering generated from the automatic clustering analysis.
18
Figure 2.5 Flow chart of our research.
... 19
Figure 3.1(a) The result of the first question in the first stage.
... 21
Figure 3.1(b) The result of the second question in the first stage.
... 21
Figure 3.1(c) The result of the third question in the first stage.
... 22
Figure 3.2 Comparison of the IDT and SGDT for each ADEs of interest.
... 23
Figure 3.3(a) Using the prediction accuracy to compare the IDT and SGDT for each
ADEs of interest.
... 24
Figure 3.3(b) Using the ratio between prediction accuracy and guess rate to compare the
IDT and SGDT for each ADEs of interest.
... 25
Figure 3.4 This is similarity guided decision tree of diabetes mellitus.
... 26
Figure 3.5 This is similarity guided decision tree of renal failure acute.
... 28
Figure 3.6 This is similarity guided decision tree of renal impairment.
... 30
Figure 4.1 Using a graph to explain why an unselected drug is concordant
to the most similar selected drug.
... 32
List of Tables
Table 2.1 This is an introduction about ASCII files.
... 8
Table 2.2 A comprehensive list of chemical descriptors used in this study.
... 9
Table 2.3(a) 1411 approval drugs’ information.
... 11
Table 2.3(b) List of what information are contained in DRUGyyQq.txt.
... 12
Table 2.3(c) List of what information are contained in REACyyQq.txt.
... 12
Table 3.1 The chemical description of features we used in the diabetes mellitus prediction
model.
... 26
Table 3.2 The chemical description of features we used in the renal failure acute
prediction model.
... 27
Table 3.3 The chemical description of features we used in the renal impairment.
... 28
Table 4.1 Show the number of unselected drugs and selected drugs for the two ADEs of
interest..
... 31
Table 4.2 Show the approval date of the two unselected drugs and two “the most similar”
selected drugs..
... 32
List of Notation and Abbreviations
Abbreviation Terminology
ADE
Adverse Drug Event
FDA
Food and Drug Administration
AERS
Adverse Event Reporting System
HIV
MedDRA
Medical Dictionary for Regulatory
Activities
CDK
Chemistry Development Kit
ISR
CV
Cross-Validation
IDT
Initial Decision Tree
1. Introduction
1.1. Background
Modern medicine provides more sanative treatment due to the improvement in
pharmaceutical equipment. However, this achievement gives rise to some potential
danger in drug usage. An UK national analysis indicates that each year adverse drug
reactions are responsible for 5% hospital admission which cost approximately 0.5 billion
pounds per year. Besides, fatal adverse drug reactions account for approximately 3% of
all death in the general population [1]. The research of drug monitoring reveals that
adverse drug reactions can turn into social cost.
The market drugs are required to show indications on the package or in the
instructions leaflet. If the pharmacists want drugs to be approved for sale, they must
report clinical results to the Food and Drug Administration (FDA). The clinical trial
should control some of the confounding variable effect, such as, age, gender, or changes
in hormone level. Subjects of the clinical trial are randomly assigned into two groups in
double blind procedure. Subjects in the experiment group are given the drug, and the
others in control group are receiving placebo. The main purpose of the clinical trial is to
confirm the principal treatment effect and the secondary purpose is to find possible
adverse reactions.
Side effect is a usual term for the negative consequence of adverse drug reaction.
And the medical record about side effect or adverse drug reaction is a report of adverse
drug event (ADE). Since 2004, the FDA set up a Pharmacovigilance system to collect the
negative consequence of market drug reports from the health professionals and patients.
All the information about adverse drug events is recorded in adverse effect reporting
system (AERS).
Besides, FDA is asked to do some analysis related to adverse drug effects and
provides these results to the public. If FDA find some adverse drug effects are not listed
on the package or in the instructions leaflet, FDA will ask pharmaceutical company to
provide inspection reports, and may have three possible procedures:
i.
The pharmaceutical company should list “new” adverse effects on the package
or in the instructions leaflet.
ii.
The pharmaceutical company should indicate the eligibility of patients on the
package or in the instructions leaflet.
iii.
In the worst case, the drugs will be removed from the shelves.
We notice the importance to investigate adverse drug events. In consequence, we
search the literatures about the hidden risk of approval drugs and start the following
research in this thesis for the drug safety issue.
“The occurrence of drug side effect” is one of the most popular topics in the field of
modern medicine. Nowadays, experts and scholars have proposed the possible causes of
adverse drug effects into four categories [2]:
(1)
Adverse events related to the therapeutic effects of drugs:
The first category of adverse drug effects occurs when the therapeutic effects have
other additional negative consequences. Generally, these drug side effects occur as the
concentration of medication is higher than required for beneficial drug effects. The drug
metabolism variation is related to several enzyme activities. The required dose of drug
might be different among people due to the individual genomic factor.
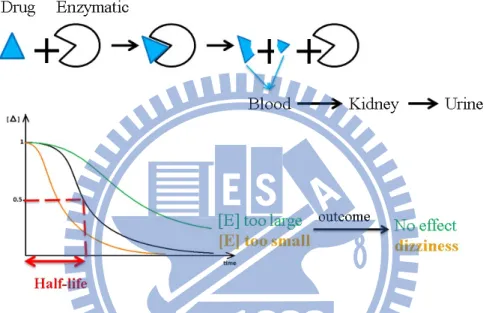
In general case, drugs are catalyzed by some enzymes in the liver. The drug effects
in human system will become vanished as the time passed. After enzymatic reaction, the
catalyzed drug waste will be transported to the kidney and then excreted into urine.
However, the enzyme activity is diverse among people. If the enzyme activity is higher;
that is, the rate of metabolism increases, then the time of drugs functioning in vivo is
shorter than average. Therefore, it can result in less treatment effect. However, if the
enzyme activity is lower; that is, the rate of metabolism decreases, then the time of drugs
functioning in vivo is longer than average. Thus, it results in higher treatment effect and
adverse drug effect (Figure 1.1).
For example, anticoagulant (Warfarin) is to inhibit vitamin K epoxide reductase
(VKOR). However, if the concentration of medication in vivo is higher than that required
for beneficial drug effect, this will result in over-inhibited clotting, which may lead to
hemorrhagic stroke.
Figure 1.1: This figure shows that the drug-metabolizing capability. The black curve
denotes the general case, the orange curve denotes the lower enzyme
activity, and the green curve denotes the higher enzyme activity.
(2)
The therapeutic drug targets existing in multiple cell and tissue
types:
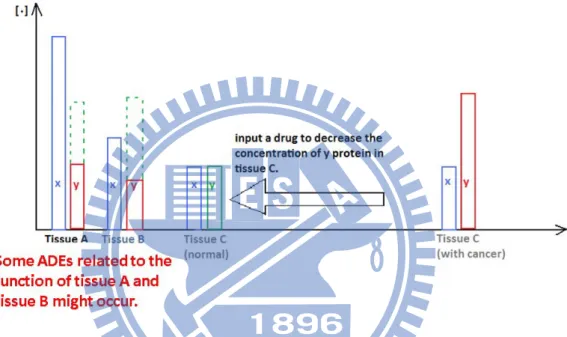
The second category of adverse drug events occurs when a drug’s target (a specific
protein) serves multiple functions in different tissues of the body.
For illustration, given three different tissues, A, B, and C, all of them contain two
proteins x and y and their corresponding expression level in normal case. If the
expression of protein y in tissue C is higher, tissue C becomes cancerous. And the design
a chemical compound, which can hydrolyze protein y or block its bio-activity, aims to
drug inhibits protein y in tissue C, as well as in tissue A and tissue B. Thus, it causes the
function of protein y in tissue A and tissue B becomes less than normal, which we
consider as a possible scenario of the cause of adverse drug effect (Figure 1.2).
In real case, Morphine is designed to achieve analgesia by binding with µ-opioid
receptors in the brain. However, it may affect the same type of opiate receptors in the
intestines, which inhibits peristalsis.
Figure 1.2: Initially, we want to regulate tissue C by inputting an anti-cancer drug. The
function of the anti-cancer drug inhibits the protein y in tissue C. It as well as
inhibits protein y in tissue A and tissue B at the same time. Thus, ADEs will
be resulted.
(3)
Adverse drug events mediated by off-targets of a drug:
The third category of adverse drug events occurs when one drug interferes with
other unexpected proteins or pathways. This category is different from the second
category. The former considers that one drug affects single protein among more than one
tissue. The protein or pathway that the drug intends to function is called “drug-target”,
and the drug affecting unexpected proteins or pathway is called “off-target”. It should be
mentioned that the off-target will be another scenario of side effects.
For example, Delavirdine, HIV reverse transcriptase inhibitor, not only inhibits the
reverse transcriptase of HIV, but also interacts with histamine H4 receptor, which will
result in severe rash.
(4)
Mixture of several drug side effects:
The fourth category is a mixture of the adverse drug events in above three categories.
For example, Domperidone is designed to promote gastrointestinal motility, but also
inhibits the human Ether-à-go-go-Related Gene K+ channels (HERG K+ channels),
which will result in cardiac arrhythmias.
1.2. Literature Review
Hammann et.al has reported a set of ADE prediction models with high prediction
accuracy in chemistry and pharmaceutical field [3]. This research has been published in a
journal of nature group (Clinical Pharmacology & Therapeutics, with impact factor
6.961*).
In this article, they state the relation between the drug chemical features and the
ADE as the perturbed “human body functioning mechanism”.
In the study, they used decision tree to establish the association between drugs’
chemical features and ADEs. They analyzed four kinds of ADEs, allergy, central nervous
system (CNS), hepatotoxicity and nephrotoxicity. The importance of drug chemical
descriptors can be revealed, because the proposed the four ADE prediction models
achieved high prediction accuracy rate, 89.74%, 90.22%, 88.68%, and 78.94%,
respectively.
However, in the four ADE models, only few market drugs are considered. The
number of market drugs of each ADE is 164, 286, 334, and 338 respectively. In addition,
they do not clearly explain how to select the “drug subset” from the initial drug set (507
drugs) for each ADE by using the reporting frequency threshold.
1.3. Purpose of Research
Although a majority of patients can reach the most of efficacy after taking medicine,
still some patients will experience some particular side effects or adverse events.
Furthermore, some side effects will result in serious injury in patients, such as
hepatotoxicity and rhabdomyolysis which lead to acute cardiac death. In our study, we
want to predict more ADEs based on Adverse Event Reporting System (AERS) and more
market drugs. Our goals of the present study are to expand the chemical descriptors
database, and to develop an automatic concordant analysis to obtain an explainable drug
subset in the feature space. In this research, we aim to determine the chemical, physical,
and structural properties of compounds that are associated with certain ADE.
The impact of this research is profound, for patients’ safety related to prescription
medicine is an important issue in public health. In this study, we propose a new approach
for investigate more adverse drug events of market drugs based on the current chemistry
informatics. The new approach can help researchers and clinicians to have an estimated
ADE propensity for a new designed drug. (Figure 1.3)
2. Method
2.1. Materials
Adverse drug report
AERS is a computerized information database [4]. It is a monitoring program
designed to support the safety of drugs on the shelf. When we download the AERS
database, we can get two kinds of formats, one is SGML and the other is ASCII. ASCII
data files contain seven different types of folders and each folder contains the relative
information of adverse drug events. For example, the “REAC” folder contains several
data files (notated: REACyyQq.txt), and each file contains the adverse events of each
drug. Table 2.1 lists each folder in detail. We use the ASCII data files which are collected
from the first quarter of 2004 to the fourth quarter of 2010 to get the number of reports
for each drug and each ADE.
In order to get the ADE list, we download the pt.asc file from the Medical
Dictionary for Regulatory Activities (MedDRA). MedDRA is a medical dictionary and is
used to classify the information about adverse event associated with the use of
biopharmaceuticals and other medical products. Because the drugs’ names which
recorded in the AERS database are not always generic names, we need a comprehensive
list of drugs’ names. Therefore, we find the corresponded generic name, brand names,
and synonyms of each approval drug in the DrugBank database. The DrugBank database
combines the complete information about drug target and detailed drug [5].
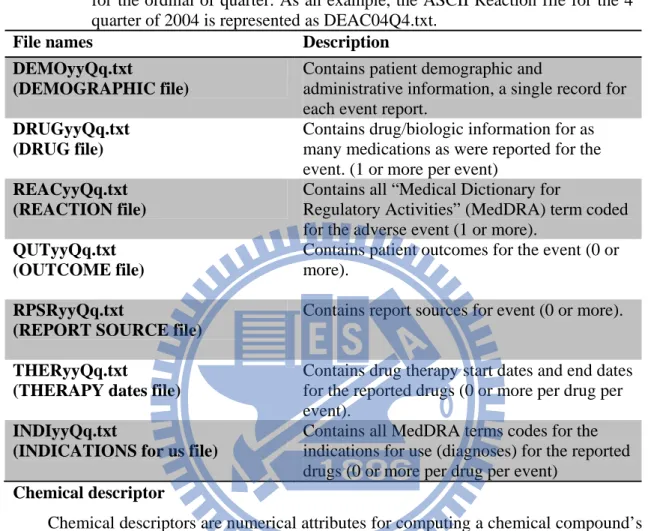
Table 2.1: In the ASCII format, file names have the format <file-descriptor>yyQq, where
<file-descriptor> is a 4-letter abbreviation of the data source, “yy” is a 2-digit
identifier for the year, “Q” stands for the quarter, and “q” is a 1-digit identifier
for the ordinal of quarter. As an example, the ASCII Reaction file for the 4
thquarter of 2004 is represented as DEAC04Q4.txt.
File names
Description
DEMOyyQq.txt
(DEMOGRAPHIC file)
Contains patient demographic and
administrative information, a single record for
each event report.
DRUGyyQq.txt
(DRUG file)
Contains drug/biologic information for as
many medications as were reported for the
event. (1 or more per event)
REACyyQq.txt
(REACTION file)
Contains all “Medical Dictionary for
Regulatory Activities” (MedDRA) term coded
for the adverse event (1 or more).
QUTyyQq.txt
(OUTCOME file)
Contains patient outcomes for the event (0 or
more).
RPSRyyQq.txt
(REPORT SOURCE file)
Contains report sources for event (0 or more).
THERyyQq.txt
(THERAPY dates file)
Contains drug therapy start dates and end dates
for the reported drugs (0 or more per drug per
event).
INDIyyQq.txt
(INDICATIONS for us file)
Contains all MedDRA terms codes for the
indications for use (diagnoses) for the reported
drugs (0 or more per drug per event)
Chemical descriptor
Chemical descriptors are numerical attributes for computing a chemical compound’s
structure. These include elemental analysis, charge analysis, geometry, partitioning
coefficient and other characteristics. Some of chemical descriptors include large
information about measures of molecular connectivity, connectivity indexes.
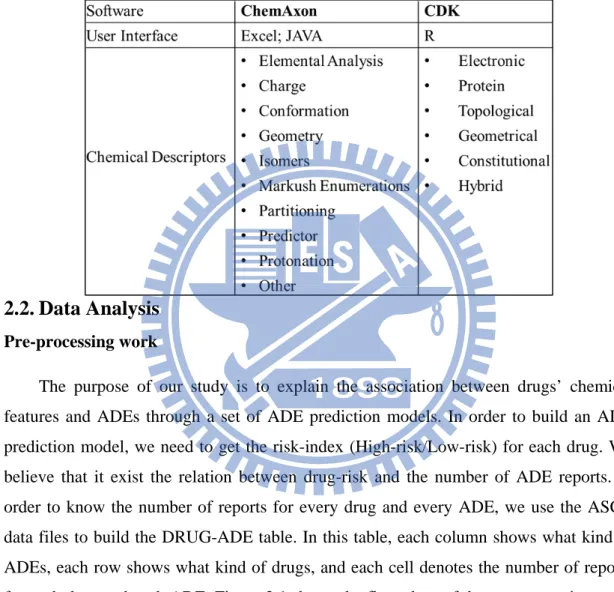
There are two sources to get chemical descriptors, one is ChemAxon, and the other
is Chemistry Development Kit (CDK). ChemAxon is a providing chemical software
development platform for characterizing chemical structures and substructures [6].
Additional chemical descriptors are calculated by using the open-source cheminformatics
package, CDK
[7]. This package allows users to access functionality by a Java
framework for cheminformatics including load molecules, evaluate fingerprints, and
calculate molecular descriptors, etc.
A list of chemical descriptors used in our study is given in Table 2.2. It is
worth
noting that for some chemical descriptors they do not have only one
chemical feature. A
comprehensive list of chemical features used in this study is given in Appendix A.
Table 2.2: This is a comprehensive list of chemical descriptors we used in this study.
There are two sources of chemical descriptors, one is ChemAxon and the
other is CDK.
2.2. Data Analysis
Pre-processing work
The purpose of our study is to explain the association between drugs’ chemical
features and ADEs through a set of ADE prediction models. In order to build an ADE
prediction model, we need to get the risk-index (High-risk/Low-risk) for each drug. We
believe that it exist the relation between drug-risk and the number of ADE reports. In
order to know the number of reports for every drug and every ADE, we use the ASCII
data files to build the DRUG-ADE table. In this table, each column shows what kind of
ADEs, each row shows what kind of drugs, and each cell denotes the number of reports
for each drug and each ADE. Figure 2.1 shows the flow chart of the pre-processing work
and Table 2.3 introduces these files used in the work. The following are the procedure of
the pre-processing work:
(1)
Define the columns and rows of DRUG-ADE table:
We work for the union of the adverse reaction names in the REACyyQq.txt and
the adverse reaction names in pt.asc file. The union becomes the columns of
DRUG-ADE table. Then, we use the Drug_ATC.txt data file to get the generic
name of each drug. The drugs’ generic names become the rows of DRUG-ADE
table.
(2)
The reported code-ISR for each quarter and each year:
ISR denotes the number uniquely representing an adverse event report and is
the primary link field between data files. We work for the union of the ISR in
DRUGyyQq.txt data file and the ISR in the REACyyQq.txt data file. The file
name of output file is called ISRyyQq.txt.
(3)
Get used drugs’ codes of each ISR for each drug’s role in event:
There are four types of drug’s reported role in event, primary suspect drug (PS),
secondary suspect drug (SS), concomitant (C), and interacting (I). For each
type of drug’s reported role, we use DRUGyyQq.txt and the rows of
DRUG-ADE table to list the code of used drug for each ISR. (The details of
each output data are: ISR $i$j$k…, where i, j, and k denotes the i
th, j
th, and k
throws in the Drug-ADE table; “$” is a delimiter.)
(4)
Get the codes of adverse events for each ISR, where the code represents which
column adverse events is in the DRUG-ADE table:
For each ISR in the REACyyQq.txt, we can get the corresponded adverse
reactions, and then we use the columns of DRUG-ADE table to get which
column matches the adverse reaction. The output file is called
“Out_REACyyQq.txt” which records the code of adverse reactions for each
ISR.
(5)
Get the number of reports for each drug and each ADE:
Use these output files in step (2), (3), and (4) to compute the number of adverse
reports for each drug and each ADE
Figure2.1: This is a flow chart about pre-processing work. The finally output file is the
DRUG-ADE table. Each cell of the table denotes the number of reports per
drug per ADE.
Table 2.3(a): This data file contains 1,411 approval drugs’ information. We can
Table 2.3(b): The DRUGyyQq.TXT contains drug/biologic information about
medications reported for the event (1 or more per event).
Table 2.3(c): The REACyyQq.TXT contains all MedDRA term coded for the adverse
event (1 or more).
Risk-index
After the pre-processing work, we want to give a risk-index to each drug and each
ADE by the DRUG-ADE table. In order to determine the risk-index for each drug and
each ADE, we define some criteria:
If the number of reports is zero, then we define that the risk-index is low risk.
Otherwise, we compare the average ADE reports with a cut value. If the average ADE
repots is larger than the cut value, we define the ADE as high risk. Otherwise, the ADE is
not deterministic.
0.
.
0
For deciding which cut value is appropriate, we try different quantile, such as 0.05,
0.1, 0.15, etc. And then, we choose a cut-value which makes the ratio between the
number of high risk and low risk closed to 1.
Classification rule- decision tree [8] [9]
Generally, a decision tree is built by a root at the top and several leaves at the
bottom. Both the root and the leaves are called “node”, but there is only one special node
which is called “root”. We start a decision tree at the root node, and for each node, there
is a test applied to determine what the next node will be. This process is repeated until all
the data points arrive at a terminal node (leaf). The terminal node gives what group the
data point belongs to. The data points that end up at the same leaf of the tree are
classified as one group. It should be mentioned that there are a lot of ways to grow a tree,
but this does not mean the types of all of leaves in each tree are different. Therefore, it
needs some trim after growing up one tree.
Suppose that Y is an indicator variable and there is a single feature X. First, we
choose a split point
that partitions the feature X into two disjoint
sets
∞
, and
,
∞
:
Let
be the proportion of observation in
such as
:
∑
,
∈
∑
∈
,
,
1, 2
0, 1.
The impurity of the split
is defined to be
,
This particular measure of impurity is known as the Gini index. We choose the split
point
to minimize the impurity. We decide split points that bring about the smallest
impurity among features. This process is continued until some stopping criterion is met.
Use 10-fold cross validation to estimate accuracy rate [9]
The basic idea of cross-validation is to partition randomly the observed data into two
groups: the training set and the testing set. The training set is used to produce an
estimated classifier
, and the testing set to obtain an estimate
A
of the accuracy rate of
.
The accuracy rate is defined by
1
1
∈
,
.
The algorithm of 10-fold cross-validation is listed as following:
(1)
The data are randomly partitioned into 10 subsets of approximately equal sizes.
(2)
For
1,2, … , 10, do the following steps:
(2.1) Take subset k as the testing set and take the others as the training set.
(2.2) Use the training set to compute the classifier
.
(2.3) Use
to predict the data in subset k. Let
denote the observed
accuracy rate.
(3)
The average accuracy rate,
, would be
∑
Find concordant drug subset determined by ADE risk-index and chemical features
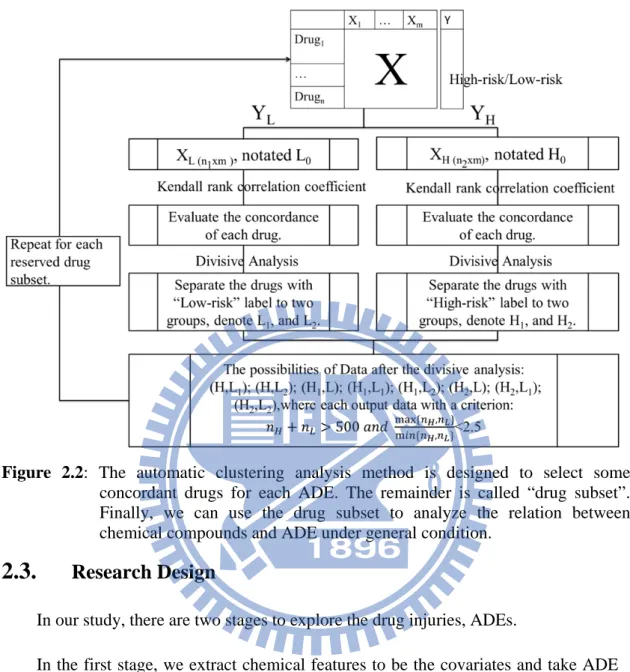
We build an automatic work flow to find concordant drug subset for each ADE. The
drug subset contains two types of ADE risk-index (high-risk or low-risk). For drugs in
each type, they are more similar in drugs’ chemical feature set.
The workflow is described as the following, and Figure 2.2 shows the flow chart of
it:
(1)
Given an ADE, we separate the drug set into two groups (Notation:
,
),
one is collected low-risk drugs ( ), and the other group is collected high-risk
drugs ( ).
(2)
Use Kendall rank correlation coefficient to evaluate the concordance between
each two drugs on feature space.
―
For
group with
high-risk drugs, we build an
Kendall
table notated as
TABLE
:
TABLE
―
For
group with
high-risk drugs, we build an
Kendall
table notated as
TABLE
.
(3)
Use divisive analysis to divide items into two clusters.
― For
group, we take the
1
TABLE
as a distance matrix. Then,
we divide
items into two clusters (Notation:
and ).
― For
group, we take the
1
TABLE
as a distance matrix. Then,
we divide
items into two clusters (Notation:
and ).
(4)
We consider these eight possibilities of drug subset:
,
,
,
,
,
,
,
,
,
,
,
,
,
,
,
(5)
If the drug subset,
,
,
,
0, 1, 2, satisfy the number of drugs lager
than 500 and
#
,#
#
,#
2.5,
then the drug subset
,
is the possibility of concordant drug subset. We
take this drug subset as
,
and repeat the steps from (1) to (4).
Figure 2.2: The automatic clustering analysis method is designed to select some
concordant drugs for each ADE. The remainder is called “drug subset”.
Finally, we can use the drug subset to analyze the relation between
chemical compounds and ADE under general condition.
2.3.
Research Design
In our study, there are two stages to explore the drug injuries, ADEs.
In the first stage, we extract chemical features to be the covariates and take ADE
record to be the drugs’ risk-index as the response variable. C4.5 Decision tree is used to
obtain the mapping function from chemical feature space onto the binary risk-index
space. Then, we use 10-fold cross-validation to estimate the prediction model accuracy.
The resulted decision tree is the build ADE prediction model with associated chemical
features. The ADE prediction models on the first stage are called Initial Decision Trees
(IDTs). It serves as feature selection for each ADE. We use “rpart” package of
statistical software R to obtain the IDTs for each ADE. Finally we compare the
prediction accuracy among three chemical feature sets for each ADE. The aim of the
first stage is to compare the performance of different chemical feature sets and obtain
the associated chemical features for each ADE.
The performance of IDT implies that there exists an instance subset that is
relatively more separable in the associated feature space. We call this instance subset as
concordant drug subset. It is illustrated in Figure 2.3.
Figure 2.3: This figure illustrates the concordant drug subset is red-surrounded. The
triangle and circle drugs are shown for binary ADE risk index.
In the second stage, we develop an automatic analysis work flow to obtain
concordant drug subset on each ADE of interest. After divisive clustering, we can get
several drug subsets which are candidate concordant drug subsets of a particular ADE.
Finally, we take the drug subset as the data set and use 10-fold CV and decision tree to
build a set of candidate models for the fixed ADE. Among all of candidate models, we
choose the optimal model with the highest prediction accuracy. For instance, we have
29 drug subsets as illustrated in Figure 2.3. The 29 drug subset is obtained by divisive
clustering analysis. Each node stands for a drug set with binary risk index. The high risk
the low risk drugs. There can be 8 children of each node at most due to the combination
illustrated in the first level in Figure 2.3. Among 29 drug subset, each has a candidate
model and each model represents a decision tree with its own 10-fold CV accuracy. The
model with the highest 10-fold CV accuracy is the final ADE prediction model. The
optimal prediction models of ADEs are called “Similarity Guided Decision Tree”
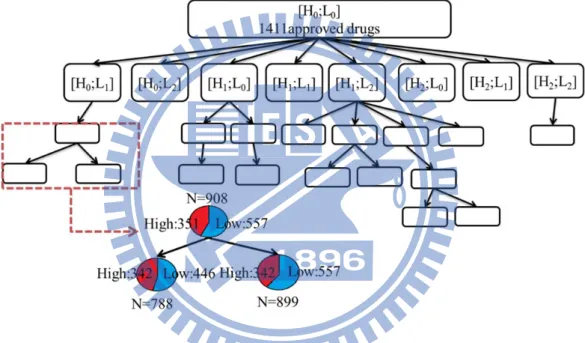
(SGDT). Figure 2.4 gives an overall view of our research.
Figure 2.4: For an ADE, we have an initial drug set and use automatic clustering analysis
to get numerous drug subsets which contain concordant drugs. Put the initial
drug set into automatic clustering analysis, then we can get several drug
subsets which satisfy the condition we set. Each drug subset could be another
initial drug set in the next level, and iteratively execute the automatic
clustering analysis. The iteration stops until none of output file satisfies the
criteria we set.
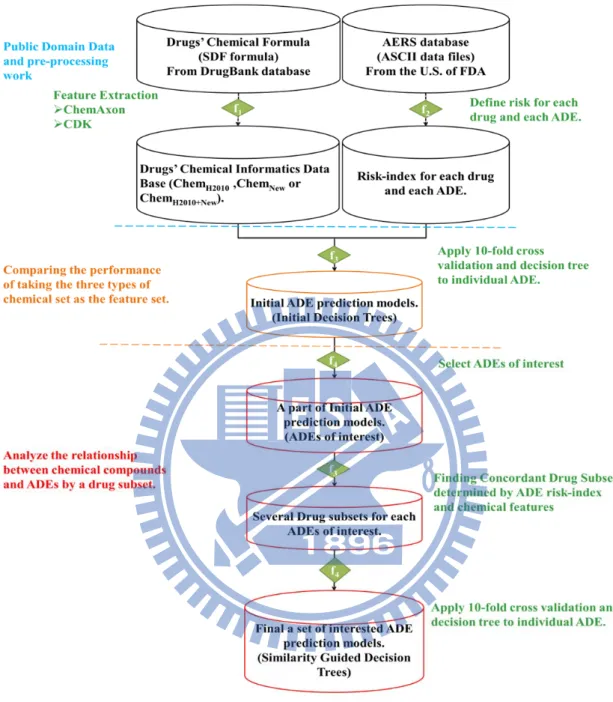
Figure 2.5: The flow chart of our research. The first step is material collection and
pre-processing work. The pre-processing work is to get the risk-index for
each drug and each ADE. The second step is to compare the performance
of ADE prediction models which generate by taking three types of
chemical sets as the feature set, such as, Chem
H2010, Chem
New, and
Chem
H2010+New. The most important of our research is the part which
locates below the orange dashed line. This step is to analyze the relation
between chemical compounds and ADEs of interest under some regular
3. Results
The most important of our research is that we build an automatic concordant
analytic method. We use automatic clustering analysis, 10-fold cross-validation and
decision tree to analyze the relation between chemical compounds and ADEs under some
regular condition. We define the prediction accuracy and the ratio of
to
evaluate the performance of ADE prediction.
3.1. Compare the performance by using different types of chemical
feature sets.
There are three issues to compare the performance of prediction model by using
three different types of chemical feature sets,
,
, and
:
Q 1.
Can we find some ADEs (
∗′
s), satisfying X
→
∗
, with a
good prediction function?
Q 2.
Given those
∗′
s, can we verify if the performance of X
→
∗
is better than that of X
→
∗
?
Q 3.
If we get the negative answer in Q 2, can we verify if the performance of
X
→
∗
is better than that of
X
→
∗
?
In the first question, we define some criterion to judge the performance of ADE
prediction models. Let the upper bound of 95% C.I. of
be the threshold. If
of an ADE model is larger than the threshold, then we consider this
ADE model as an acceptable prediction model. We find 642 out of 1,483 ADEs that has
the prediction accuracy higher than the threshold, shown in Figure 3.1(a). Considering
the second question, we can find 121 ADEs whose prediction accuracy of using
Chem
as the feature set is higher than using Chem
, shown in Figure 3.1(b). For
answering the third question, we use 841 ADEs which have negative response in the
second question. Then, we test whether the prediction accuracy of using Chem
as the feature set is higher than using Chem
. The result shows that there are only 44
ADEs which indicate that the prediction accuracy of using Chem
is better
than using Chem
, shown in Figure 3.1(c).
Figure 3.1(a): Each red point denotes that the ADE model’s
is higher than the
threshold. And each blue point denotes that the ADE model’s
is not higher than the threshold.
0.50 0.55 0.60 0.65 0.70 0 .45 0. 5 0 0. 5 5 0. 60 0. 65 0 .70
(a) Question1:Cut Value:0.9712839
Random Guess
P
re
di
ct
ion
A
c
c
ur
ac
y
w
it
h
Ch
em
.(
H
20
10
)
Figure 3.1(b): In the scatter plot, the red points (121 points) denote that the performance
of using the
feature set to build an ADE model is better than
using the
feature set. The blue points denoted that the
performance of using the
feature set to build model is not
better than using
.
Figure 3.1(c): Among 841 ADEs have a negative answer in Q 2; there are only 44 ADEs
(red points) which satisfy that the performance of using
is better than using
.
0.50 0.55 0.60 0.65 0.70 0. 45 0. 50 0. 5 5 0. 60 0. 65 0 .70
(b) Question2
Prediction Accuracy with Chem(H2010)
P
red
ic
ti
on A
c
c
u
ra
c
y
w
ith C
h
em(
N
e
w
)
0.50 0.55 0.60 0.65 0.70 0. 50 0. 55 0. 60 0. 65 0. 70(c) Question3
Prediction Accuracy with Chem(H2010)
Pr
edi
c
ti
on
Ac
c
ur
ac
y
w
it
h C
he
m
(H
20
10+N
ew
)
After comparing the performance of using different types chemical feature sets, we
find that no matter which feature set we take to build ADE models, the prediction
accuracy of each ADE is not higher than 0.8. The accuracy rate will decrease rapidly if
we consider multiple ADEs. For illustration, suppose we are interested in a new drug is
high-risk or low-risk drug corresponding to 3 specific ADEs and of each ADEs, the
prediction rate is 0.8. Then the total prediction accuracy would be reduced to 0.8
0.512. Therefore, we need to increase the prediction accuracy of ADE models as higher
as possible.
3.2. Select concordant drugs and then analyze the relation between
chemical compounds and ADE under some regular condition
We build an automatic concordant analysis to select concordant drugs. Then, we
take the remaining drugs as data set to build prediction models by decision tree. Finally,
we use 10-fold cross-validation to estimate the prediction accuracy of each ADE model.
3.2.1. ADEs of interest filtering.
In order to check the feasibility of this automatic concordant analysis, we need to
construct testing data. There are a lot of ADEs in our study (1,483 ADEs), so we select
some ADEs to be the testing data. In this thesis, we filter out the ADE of interest by
considering the 10-fold CV accuracy to the guess rate:
# # ,#
.
We use the ADEs’ prediction accuracy as the response variable and the guess rate as the
covariate to build a simple linear regression, where the prediction accuracy is generated
by taking
Chem
as the feature set and the prediction model is called “Initial
Decision Tree” (IDT). If the accuracy of an ADE model is higher than the upper bound of
95% prediction interval of accuracy, then the ADE is one of the “ADEs of interest”.
Among 1,384 ADEs, we obtain 35 ADEs which satisfy that the accuracy is higher than
the upper bound.
Now we use these ADEs of interest to analyze the relationship between drugs’
chemical compounds and ADEs under some regular condition. These prediction models
are called “Similarity Guided Decision Tree” (SGDT). In Figure 3.2 (a), we compare the
prediction accuracy between IDT (blue bars) and SGDT (red bars) for each “ADEs of
interest”, where the drug subset contains the remaining drugs after doing automatic
clustering analysis. Obviously, the prediction accuracy of SGDT is higher than IDT for
each ADEs of interest.
What we are most interested in is the ratio between prediction accuracy and guess
rate, for the guess rate has a significant effect on the prediction accuracy. The high
random guess shows that the majority of drugs belong to one kind of drug-risk level
(high-risk or low-risk). As a result, we just guess all of drugs are belong to this drug-risk
level, and then the prediction accuracy rate is naturally high. In order to avoid this
situation, we need to consider the ratio,
. The higher ratio illustrates
that the prediction model is more reliable. In Figure 3.2(b), we compare the ratio,
between IDT and SGDT for each ADEs of interest. Since the red bar
(SGDT) is higher than the blue bar (IDT) of each interest ADEs. Consequently, we
conclude that the performance of using an appropriate drug subset is better than using a
fixed drug set for these ADEs of interest.
Figure 3.2(a): In this bar plot, each red bar denotes that the performance of an ADE
prediction model after doing the automatic clustering analysis, and each
blue bar denotes the performance of ADE model by using initial drug set.
Figure 3.2(b): For each ADEs of interest, the ratio,
by using a drug
subset (red bar) is higher than using a fixed drug set (blue bar).
In summary, the results of interested ADEs’ prediction models show that 24 ADEs
have the prediction accuracy greater than 80%. However, if we consider the ratio
between prediction accuracy and guess rate, we can find 33 ADEs of interest have
1.3 among 35 ADEs of interest.
Among these 35 ADEs of interest, there are three more serious ADEs: Diabetes
Mellitus (PT
5768), Renal Failure Acute (PT
15877) and Renal Impairment (PT
15894).
(1)
DIABETES MELLITUS (PT
5768):
Feature Set:
Drug Size: 1,411 approval drugs
By automatic clustering analysis (the second stage), the prediction accuracy is
listed as following:
IDT
0.0085
524
538
1062
0.493
0.527
SGDT
489
244
733
0.667
0.871
Figure 3.3: The SGDT of the ADE-DIABETES MELLITUS for 489 high-risk drugs and
244 low-risk drugs (
733), with a prediction accuracy of 0.871.
Table 3.1: The chemical description of features we used in the ADE model.
Feature Code Feature Name
Parameter/
Discriptive Statistic
X1_0_113_2
JCStericEffectIndex
Atom=false
X1_0_75_1
JCMinimalProjectionArea
None
X1_0_77_1
JCMinZ
None
X2_0_41_1
JAR_hmoelectrophiliclocaliz
ationenergy_Localization
energy L(+)
IQR
X2_0_81_1
JAR_sterichindrance
None
X2_0_90_1
JAR_wienerpolarity
None
X3_0_2_1
SDF_ALOGPS_LOGS
None
Table 3.1 (Continue)
(2)
RENAL FAILURE ACUTE (PT
15877)
Feature Set:
Drug Size: 1,411 approval drugs
By divisive analysis (the second stage), the prediction accuracy is listed as
following:
Cut
Value High Low N
Guess
Rate
Accuracy
IDT
0.027
539
438
977
0.552
0.531
SGDT
508
231
739
0.687
0.910
Feature Code Descriptor
Definition
X1_0_113_2
JCStericEffectIndex
Steric effect index
X1_0_75_1
JCMinimalProjectionArea Calculates the minimal projection area
X1_0_77_1
JCMinZ
Returns the minimum z coordinate
of the bounding box.
X2_0_41_1
JAR_hmoelectrophilicloc-alizationenergy
HMO Electrophilic localization energy L(+).
X2_0_81_1
JAR_sterichindrance
Steric hindrance
X2_0_90_1
JAR_wienerpolarity
Wiener polarity
X3_0_2_1
SDF_ALOGPS_LOGS
"The extent to which a compound will
dissolve in water. The log of solubility is
generally inversely related to molecular
weight."- U.S. Environmental Protection
Agency, 2009
Figure 3.4: The SGDT of the ADE- RENAL FAILURE ACUTE for 508 high-risk drugs
and 231 low-risk drugs (
739), with a prediction accuracy of 0.910.
Table 3.2 (Continue)
(3)
RENAL IMPAIRMENT (PT
15894)
Feature Set:
Drug Size: 1,411 approval drugs
By divisive analysis (the second stage), the prediction accuracy is listed as
following:
Cut Value
High
Low
N
Guess Rate
Accuracy
IDT
0.006
561
548
1109
0.506
0.526
SGDT
247
257
504
0.510
0.946
Feature Code
Descriptor
Definition
X2_0_21_29
JAR_composition
Elemental composition calculation (w/w%).
X2_0_39_9
JAR_hmoelectro-ndensity
HMO Electron density.
X2_0_44_1
JAR_hmolocalizat-ionenergy
HMO Electrophilic localization energy L(+).
X2_0_52_6
JAR_localization-energy
Localization energy L(+)/L(-).
X2_0_57_1
JAR_msacc
Hydrogen bond acceptor average
multiplicity over microspecies by pH.
X2_1_8_1
JAR_aromatic-bondcount
Aromatic bond count.
X4_0_29_7
RCDK_KierHall-SmartsDescriptor
A fragment count descriptor
that uses e-state fragments
X4_1_20_6
RCDK_CPSA-Descriptor
Calculates 29 Charged Partial Surface Area
(CPSA) descriptors. One of them is FNSA.3.
FNSA.3=the ratio between charge weighted partial
negative surface area and total molecular surface area.
Figure 3.5: The SGDT of the ADE- Renal Impairment for 247 high-risk drugs and 257
low-risk drugs (N=504), with a prediction accuracy of 0.946.
Table 3.3: The description of features we used in the ADE model.
Feature Code
Feature Name
Parameter/
Discriptive Statistic
Descriptor
Definition
X4_0_19_9
RCDK_VP.0
None
RCDK_ChiPathDescriptor
Evaluates chi path
4. Discussion and Conclusions
4.1. Discussion
In our research, we find that the size of drug subset is smaller for each ADEs after
conducting automatic concordant analysis. Therefore, we do some check-up on these
“unselected drugs”.
Given an ADE, we can examine the Kendall τ rank correlation coefficient
between “unselected drugs” and “selected drugs” in the chemical feature space. For each
“unselected drug”, we choose a most similar “selected drug” which has the maximum τ
value. Then, we compare the corresponding risk-indices.
If the risk-indices are different between some unselected drug and some most similar
selected drug, the unselected drug is called “mislabel” drug. Otherwise, if the drug-risk of
some unselected drug and some most similar selected drug are the same, the unselected
drug is called “concordant” drug. For example, we can get results for Renal Failure Acute
and Diabetes Mellitus as the following table:
Table 4.1 Show the number of unselected drugs and selected drugs for the two
ADEs of interest.
We list out the possible speculations for mislabel and concordant as the following:
(1)
Mislabel:
Mislabel drugs can be separated into two cases:
(1.1) The unselected drug is a low-risk drug, and the most similar selected
drug is a high-risk drug. There are several possible reasons of this case:
(1.1.1) The drug is used for particular patients. The patient population
(1.1.2.1) AERS does not cover all adverse event occurrences
globally.
(1.1.2.2) The newly marketing drugs documented in shorter history
tend to have less observation.
(1.2) The other case is that the unselected drug is a high-risk drug, and the
most similar selected drug is a low-risk drug. The possible reason of this
case is that the drug may be a concomitant drug, such as, second suspect
drug, concomitant drug, and interacting drug. This kind of drugs tends
to have higher reports.
For example, in Renal Failure Acute prediction model, the approval history duration
of the two low-risk drugs (DB01193, DB00488) are at least less than ten years comparing
to the most similar selected drugs (DB00190, DB01168). Therefore, the observed ADE
report frequency may lead us to underestimate their risk propensity.
Table4.2 Show the approval date of the two unselected drugs and two “the most
similar” selected drugs.
(2)
Concordant
Given an ADE risk-index (high or low), some unselected drugs are actually
relatively closer to the selected drug group. For instance, drug A, drug B and
other four drugs are the same ADE risk-indexed. However, drug A and drug B
are determined as unselected by our automatic analysis method. As shown in
Figure 4.1, drug B are actually closer to the four selected drugs.
Unselected Drug Approval date Risk-index
Most similar
selected drug
Approval date
Risk Index
DB01193
1999/12/30
Low-risk
DB00190
Approved Prior to
jan 1, 1982
High-risk
DB00488
1990/12/26
Low-risk
DB01168
Approved Prior to
Figure 4.1: (a) The first division separates drug A from the other five drugs. (b) The
second division separates drug B from the other four drugs. (c) The
remained four drugs are determined as selected drugs. Drug A is relatively
far from the four selected drugs in chemical feature space, while drug B is
relatively closer to the four selected drugs.
4.2. Conclusion
In our research, we identify 1,384 ADEs with corresponding associated chemistry
informatics features by decision tree. With an automatic analysis work flow, we can
obtain a concordant drug subset with satisfying 10-fold cross-validation accuracy. The
test experiment about selected 35 ADEs of interest results in accuracy higher than 80%.
Three observations from the results are worth emphasizing:
(1)
After conducting the automatic analytic method, these drugs give a significant
drug-risk level. This leads to significant results of decision tree.
(2)
Compared with the research of Hammann et al., we consider more ADEs,
chemical features and approval drugs to build a set of ADE prediction models.
(3)
There are 35 ADE models with the prediction accuracy higher than 0.8.
The practical benefits of our experimental results are of great interest both for the
R&D in medicine industry and in public health. We can apply the trained model to
forecast the new designed drugs (potential compounds) for its ADE. In addition, we can
recognize the probability of some ADE occurrence when some chemical compound exits.
Our models can not only guide future clinical trials, but also run monitoring and test
during such trials. Moreover, they allow pharmacists for more advanced and efficient
drug design.
4.3. Recommendation for Future Research
Our analysis is focused on the relation between chemical compounds and ADEs. In
fact, the process of drug-related injuries resulted in some adverse reaction is still a “black
box”. After years of study, many experts believe that the types of elements which result
in drug-related injuries are not only chemical compounds, but also target gene. Some
scholars have proposed some other view about the causes of adverse reactions (Berger,
2011). In their approach, they believe that drug side effect is mediated by complex
cellular networks. In response to a drug, the variations in cellular networks can expose
silent phenotypes. This type of adverse drug event is caused by inheritance. Namely, if
we only use chemistry features, we cannot thoroughly explain this type of adverse drug
events.
In the future studies, to break the limitation of ADEs’ study, we can consider other
factors which may result in drug-related injuries, such as, drugs’ target gene and ADME
mechanism. Thus, it may be of interest for future research that we combine the systems
biology and drugs' chemistry to build ADE prediction model. The ADE models not only
have higher prediction accuracy but also contain more "selected drugs". That means the
"concordant drug" are really dissimilar to a "selected drug" on the "new" feature space
(combine chemistry and system biology), where the two drugs have the same risk-index.
Reference:
[1] Ritter, J. M. (2008). Minimising Harm: Human Variation and Adverse Drug
Reactions (ADRs). British Journal of Clinical Pharmacology.
[2] Berger, S. I., and Iyengar R. (2011). Role of systems pharmacology in understanding
drug adverse events. WIREs Systems Biology and Medicine.
[3] Hammann, F., and Gutmann, H. (2010). Prediction of Adverse Drug Reactions
Using Decision Tree Modeling. Clinical Pharmacology & Therapeutics, 88 (1), 52–
59.
[4] U.S. Food and Drug Administration. (2012). Adverse Event Reporting System
(AERS). Retrieved July 01, 2011, from http://www.fda.gov/Drugs/default.htm
[5] Departments of Computing Science & Biological Sciences, University of Alberta.
(2012). DrugBank. Retrieved July 5, 2011, from http://www.drugbank.ca/
[6] Marvin was used for drawing, displaying and characterizing chemical structures,
substructures and reactions, Marvin 5.9.3 (version number), 201n (insert year of
version release), ChemAxon (http://www.chemaxon.com)
[7] Guha, R. (2007). 'Chemical Informatics Functionality in R'. Journal of Statistical
Software 6(18)
[8] Michael J. A. (1997). Data Mining Techniques. New York: WILEY
[9] Wasserman, L. (2004). All of statistics a concise course in statistical inference. New
York: Springer.
[10] Keiser M. J., and Setola V. (2009). Predicting new molecular targets for known
drugs. Nature, 462:175–181.
[11] Campillos M., and Kuhn M. (2008). Drug target identification using side-effect
similarity. Science, 321:263–266.
[12] Pouliot, Y., and Chiang, A.P. (2011). Predicting Adverse Drug Reactions Using
Publicly Available PubChem BioAssay Data. Nature, 90(1):90-99.
[13] Huang, L.C., and Wu, X. (2011). Predicting adverse side effects of drugs. BMC
Genomics
No. Feature Name Chemcial Descriptors #ofNA Source v5.1Code 1 JCChainAtomCount JCChainAtomCount 0 ChemAxonByExcel 1_1_27_1 2 JCCarboRingCount JCCarboRingCount 0 ChemAxonByExcel 1_1_25_1 3 JClogP JClogP 0 ChemAxonByExcel 1_1_68_1 4 JCFusedAromaticRingCount JCFusedAromaticRingCount 0 ChemAxonByExcel 1_1_50_1 5 JCFusedRingCount JCFusedRingCount 0 ChemAxonByExcel 1_0_51_1 6 JClogD(9) JClogD 0 ChemAxonByExcel 1_1_67_5 7 JClogD(5) JClogD 0 ChemAxonByExcel 1_1_67_3 8 JCChainBondCount JCChainBondCount 0 ChemAxonByExcel 1_1_28_1 9 JCIsoelectricPoint JCIsoelectricPoint 0 ChemAxonByExcel 1_0_60_1 10 JCSmallestRingSize JCSmallestRingSize 0 ChemAxonByExcel 1_1_111_1 11 JCBalabanIndex JCBalabanIndex 0 ChemAxonByExcel 1_1_19_1 12 JClogD(11) JClogD 0 ChemAxonByExcel 1_1_67_2 13 JCDonorCount JCDonorCount 0 ChemAxonByExcel 1_1_41_1 14 JCResonantCount JCResonantCount 2 ChemAxonByExcel 1_1_100_1 15 JCAcceptorSiteCount JCAcceptorSiteCount 0 ChemAxonByExcel 1_1_3_1 16 JCAromaticAtomCount JCAromaticAtomCount 0 ChemAxonByExcel 1_1_12_1 17 JCBasicpKa(strongness=1) JCBasicpKa 87 ChemAxonByExcel 1_1_20_1 18 JCBasicpKa(strongness=2) JCBasicpKa 292 ChemAxonByExcel 1_1_20_2 19 JCAcidicpKa(strongness=1) JCAcidicpKa 306 ChemAxonByExcel 1_1_4_1 20 JCAcidicpKa(strongness=2) JCAcidicpKa 732 ChemAxonByExcel 1_1_4_2 21 JCRingCount JCRingCount 0 ChemAxonByExcel 1_1_104_1 22 JCAliphaticBondCount JCAliphaticBondCount 0 ChemAxonByExcel 1_1_7_1 23 JClogD(7.4) JClogD 0 ChemAxonByExcel 1_1_67_4 24 JCDonorSiteCount JCDonorSiteCount 0 ChemAxonByExcel 1_1_42_1 25 JCPSA(PH=12) JCPSA 0 ChemAxonByExcel 1_0_83_1 26 JCQ_N JCQ_N 0 ChemAxonByExcel 1_1_93_1 27 JCQ_O JCQ_O 0 ChemAxonByExcel 1_1_95_1 28 JCQ_C JCQ_C 0 ChemAxonByExcel 1_1_87_1 29 JCQ_halogen JCQ_halogen 0 ChemAxonByExcel 1_1_90_1 30 JCQ_azole JCQ_azole 0 ChemAxonByExcel 1_1_85_1 31 JCQ_phenol JCQ_phenol 0 ChemAxonByExcel 1_1_96_1 32 JCQ_ketone JCQ_ketone 0 ChemAxonByExcel 1_1_91_1 33 JCQ_amine JCQ_amine 0 ChemAxonByExcel 1_1_84_1 34 JCQ_COOH JCQ_COOH 0 ChemAxonByExcel 1_1_88_1 35 JCQ_benzene JCQ_benzene 0 ChemAxonByExcel 1_1_86_1 36 JCQ_ester JCQ_ester 0 ChemAxonByExcel 1_1_89_1 37 JCQ_nitro JCQ_nitro 0 ChemAxonByExcel 1_1_94_1 38 JCQ_methyl JCQ_methyl 0 ChemAxonByExcel 1_1_92_1 39 JCQ_S JCQ_S 0 ChemAxonByExcel 1_0_97_1 40 JCAcceptorCount JCAcceptorCount 0 ChemAxonByExcel 1_0_2_1 41 JClogD(0) JClogD 4 ChemAxonByExcel 1_1_67_1 42 JCMolecularPolarizability JCMolecularPolarizability 4 ChemAxonByExcel 1_0_78_1 43 JCAliphaticAtomCount JCAliphaticAtomCount 0 ChemAxonByExcel 1_1_6_1 44 JCBondCount JCBondCount 0 ChemAxonByExcel 1_1_23_1 45 JCFusedAliphaticRingCount JCFusedAliphaticRingCount 0 ChemAxonByExcel 1_1_49_1 46 JCHeteroRingCount JCHeteroRingCount 0 ChemAxonByExcel 1_1_58_1 47 JCHyperWienerIndex JCHyperWienerIndex 0 ChemAxonByExcel 1_1_59_1 48 JCRandicIndex JCRandicIndex 0 ChemAxonByExcel 1_1_98_1 49 JCAcceptor(PH=0) JCAcceptor 0 ChemAxonByExcel 1_0_1_1 50 JCAcceptor(PH=1) JCAcceptor 4 ChemAxonByExcel 1_0_1_2 51 JCAcceptor(PH=2) JCAcceptor 9 ChemAxonByExcel 1_0_1_8 52 JCAcceptor(PH=3) JCAcceptor 10 ChemAxonByExcel 1_0_1_9 53 JCAcceptor(PH=4) JCAcceptor 10 ChemAxonByExcel 1_0_1_10 54 JCAcceptor(PH=5) JCAcceptor 11 ChemAxonByExcel 1_0_1_11 55 JCAcceptor(PH=6) JCAcceptor 13 ChemAxonByExcel 1_0_1_12 56 JCAcceptor(PH=7) JCAcceptor 13 ChemAxonByExcel 1_0_1_13 57 JCAcceptor(PH=8) JCAcceptor 15 ChemAxonByExcel 1_0_1_14 58 JCAcceptor(PH=9) JCAcceptor 18 ChemAxonByExcel 1_0_1_15 59 JCAcceptor(PH=10) JCAcceptor 23 ChemAxonByExcel 1_0_1_3 60 JCAcceptor(PH=11) JCAcceptor 27 ChemAxonByExcel 1_0_1_4 61 JCAcceptor(PH=12) JCAcceptor 33 ChemAxonByExcel 1_0_1_5 62 JCAcceptor(PH=13) JCAcceptor 39 ChemAxonByExcel 1_0_1_6 63 JCAcceptor(PH=14) JCAcceptor 48 ChemAxonByExcel 1_0_1_7 64 JCAcidicpKaLargeModel(strongness=1) JCAcidicpKaLargeModel 303 ChemAxonByExcel 1_0_5_1 65 JCAcidicpKaLargeModel(strongness=2) JCAcidicpKaLargeModel 723 ChemAxonByExcel 1_0_5_2 66 JCAcidicpKaLargeModel(strongness=3) JCAcidicpKaLargeModel 1006 ChemAxonByExcel 1_0_5_3 67 JCAcidicpKaLargeModel(strongness=4) JCAcidicpKaLargeModel 1150 ChemAxonByExcel 1_0_5_4 68 JCAcidicpKaLargeModel(strongness=5) JCAcidicpKaLargeModel 1209 ChemAxonByExcel 1_0_5_5 69 JCAcidicpKaLargeModel(strongness=6) JCAcidicpKaLargeModel 1222 ChemAxonByExcel 1_0_5_6 70 JCAcidicpKaLargeModel(strongness=7) JCAcidicpKaLargeModel 1224 ChemAxonByExcel 1_0_5_7 71 JCAcidicpKaLargeModel(strongness=8) JCAcidicpKaLargeModel 1226 ChemAxonByExcel 1_0_5_8 72 JCAliphaticRingCount JCAliphaticRingCount 0 ChemAxonByExcel 1_0_8_1