國 立 交 通 大 學
生物科技研究所
碩士論文
溶藻弧菌胺醯組胺酸雙胜肽酶之生化特性分析
及其功能性胺基酸之研究
Characterization of Functional Residues for Catalysis and
Kinetics of Aminoacylhistidine Dipeptidase from Vibrio
alginolyticus
研究生: 陳怡親
指導教授: 吳東昆 博士
Characterization of Functional Residues for Catalysis and Kinetics of
Aminoacylhistidine Dipeptidase from Vibrio alginolyticus
研究生:陳怡親 Student: Yi-Chin Chen
指導教授:吳東昆 博士 Advisor: Dr. Tung-Kung Wu
國 立 交 通 大 學
生物科技研究所
碩士論文
A Thesis
Submitted to Department of Biological Science and Technology
College of Science
National Chiao Tung University
in partial Fulfillment of the Requirements
for the Degree of
Master
in
Biological Science and Technology
July, 2007
Hsinchu, Taiwan, Republic of China
溶藻弧菌胺醯組胺酸雙胜肽酶之生化特性分析及其功能性胺基酸
之研究
研究生:陳怡親 指導教授:吳東昆 博士
國立交通大學 生物科技研究所碩士班摘要
胺醯組胺酸雙胜肽酶(PepD, EC 3.4.13.3)為胜肽酶家族M20 中的一員。胜肽酶家族M20 中的酵素皆屬於金屬雙胜肽酶,而經由研究發現可被應用於抗菌、癌症的臨床治療及神 經傳導物質的調控等方面。過去對於細菌中胺醯組胺酸雙胜肽酶的研究很少,只針對其 序列和部分生化特性進行探討,並無其生理角色或活性區胺基酸相關之研究。本論文將 溶藻弧菌pepD基因殖入pET-28a(+)質體中,表現出N端帶有His-tag之重組蛋白,並利用 Ni-NTA親和層析管柱純化之。純化出的蛋白質對於特定的Xaa-His雙胜肽( 例如: L-carnosine)具有水解的能力,但無水解三胜肽之活性。經酵素動力學研究,溶藻弧菌PepD蛋白對雙胜肽L-carnosine之Km與kcat值分別為0.36 mM與 8.6 min-1。經由序列分析
預測溶藻弧菌PepD蛋白上胺基酸位置His80、Asp82、Asp119、Glu149、Glu150、Asp173 及His461 為活性區胺基酸。其中將所預測影響金屬鍵結之胺基酸Asp119 以及扮演催化 角色之胺基酸Glu149 分別進行飽和定點突變,發現大部分之突變蛋白皆失去或降低原 有之活性。此外,以同屬M20 胜肽酶家族之PepV蛋白結晶結構為模板做出溶藻弧菌PepD 蛋白之同源模擬,顯示出相同之活性區胺基酸。因此,根據本論文實驗結果將首次提出 胺醯組胺酸雙胜肽酶活性區胺基酸之分佈情形與其可能扮演之功能。
Characterization of Functional Residues for Catalysis and Kinetics of
Aminoacylhistidine Dipeptidase from Vibrio alginolyticus
Student : Yi-Chin Chen Advisor : Dr. Tung-Kung Wu
Institute of Biological Science and Technology National Chiao Tung University
Abstract
Proteins of the aminoacylase-1/metallopeptidase 20 (Acyl/M20) family were characterized to contain a zinc-binding domain at their active site. Aminoacylhistidine dipeptidase (PepD, EC 3.4.13.3) is a member of peptidase family M20 which catalyzes the cleavage and release of N-terminal amino acid, usually are neutral or hydrophobic residue, from Xaa-His peptide or polypeptide. We have cloned a PepD gene, which shared high sequence identity with PepD from various Vibrio spp. and 63% from Escherichia coli and
Salmonella typhimurium, from Vibrio alginolyticus. V. alginolyticus PepD was expressed and
purified by Ni-NTA column. The kinetics values including kcat (8.6 min-1), kcat/Km (0.398
mM-1s-1) of bacterial PepD were first identified. Sequence analysis revealed that Asp82 and Glu149 were probable active site residues and that Asp119, Glu150, Asp173, and His461 were probable metal ion binding residues of V. alginolyticus PepD. Site-directed mutations of D119 (putative metal ion binding site residue) and E149 (putative active site residue) residues of PepD exhibited activity decreasing or losing, as compared with wild-type PepD. The homology model of V. alginolyticus PepD, based on that of L. delbrueckii PepV structure, exhibited similar active site pocket as predicted. The functional role of these residues on enzyme catalysis and kinetics will be discussed in the thesis.
謝誌
(Acknowledgement)
在過去兩年的研究生活中,有苦有甘並且充滿刺激。不管在實驗上或是生活上,都 接觸到許多新鮮的人、事、物。在這過程中,我對生物科技這個領域有了更深入的了解, 也習得許多專業知識及實驗操作技巧,在待人接物方面也學習到很多道理。回顧這兩 年,檢視自己,相信在專業領域的拓展及心靈上的成長都日益成熟。而首先要感謝的就 是指導教授吳東昆 博士,給予我加入實驗室學習的機會,不僅在專業的知識背景、實 驗技術及實驗設計觀念上給予指導,在生活上及待人接物方面也給予很多幫助,教導我 對事情所應有的積極態度及不畏懼失敗挫折的毅力並且給予適時的鼓勵及肯定,在最後 也花費了很多的心力協助論文的修改。感謝口試委員張大慈 教授與楊裕雄 教授於百忙 之中抽空審閱及修改我的論文初稿,並對實驗設計觀念、操作技巧、實驗過程、結果與 討論提供諸多寶貴意見,使本論文能更加完善。 感謝實驗室的大學長程翔學長,總是不斷的鼓勵我,讓我擁有更多對實驗的熱情而 不會因為失敗而喪志,在最後分子模擬實驗上也給予了我很多的幫助;感謝行動百科全 書裕國學長,把我從一個什麼都不會的小毛頭教導成到現在可以獨立操作實驗,不管是 在實驗的設計上或是技術上的指導,都給予我相當多的幫助。並且教導我積極汲取新的 知識技術的態度,感謝過去兩年來的教導和照顧;感謝庭翊學長,總是不厭其煩的替我 解答大大小小的實驗問題,並且教導我學會獨立解決問題的能力,不管在實驗上或是生 活事物上認真負責的態度,一直都是我努力學習的典範;感謝善解人意的媛婷學姊,在 生活上對我的照顧及關心,在我低潮時給予我很大的支持及鼓勵;感謝晉豪學長,在 Circular Dichroism 和 HPLC 分析實驗上的教導和幫忙,遇到困難時總是積極的協助我 找出可能的問題所在,陪我渡過只有儀器操作聲的難熬夜晚;感謝晉源學長,在我剛進 入實驗室時幫助我很快的適應實驗室生活,在辛苦的純化及養晶的實驗上付出相當多的 心力及幫助;感謝文鴻學長,在日常生活上的照料,並且很有耐心的替我解答一些實驗 上奇奇怪怪的問題,總是在我需要幫忙時給予協助。另外感謝我最好的戰友大景,文宣 和皓宇,謝謝你們不論在實驗或生活上的幫忙及照顧,總是默默的聽我抱怨並且給予我鼓勵及支持,那些無數個一起打拼的夜晚還有講不完的蠢事都會是最好的回憶。感謝學 弟妹文祥、良瑋和采婷,在我實驗期間的協助,生活上的關心及照顧,還有最後這段時 間給予我的支持與鼓勵。感謝新成員禕婷、聖慈、天昶和亦諄在口試的時候細心負責的 幫忙準備餐點和設備。感謝所有關心與指導過我的人,陪伴著我渡過這兩年的人,我很 開心我是身為Wu lab 的其中一員,這兩年的碩士生涯將會是我人生旅程中不可抹滅的 一段精采又美麗回憶!在此分享我的成果及對所有的人致上最深的謝意和祝福。
Keywords
Vibrio alginolyticus, aminoacylhistidine dipeptidase, PepD, metallopeptidase, L-carnosine,
Abbreviations
APS Ammonium persulfate
AUC Analytical Ultracentrifugation
bp base pair
BSA bovine serum albumin
CD Circular dichroism
CDC Centers for Disease Control and Prevention
CO2 carbon dioxide
dH2O distilled water
ddH2O double distilled water
DMF dimethylformamide
DMSO dimethyl sulfoxide
DNA deoxyribonucleic acid
dNTP deoxynucleoside triphosphate
EDTA ethylenediamine-tetraacetic acid
ELISA enzyme-linked immunosorbent assay
FBS fetal bovine serum
GABA-His γ-Amino-butyryl-histidine dipeptide
HCl hydrogen chloride
HRP horseradish peroxidase
H2SO4 sulferic acid
ICP-MS inductively coupled plasma-mass spectrometry IPTG isopropyl-1-thio-β-D-galactopyranoside
kb kilobase(s) kDa kiloDalton(s)
KPi potassium phosphate LB Luria-Bertani
mAb monoclonal antibody
NaCl sodium chloride
NaN3 sodium azide
NaOH sodium hydroxide
NC nitrocellulose
NCBI National Center for Biotechnology Information
NC-IUBMB Nomenclature Committee of the International Union of Biochemistry and Molecular Biology
NEM N-ethylmaleimide
OPA o-phthaldialdehyde
pAb polyclonal antibody
PBS phosphate buffered saline
PCR polymerase chain reaction
PEG polyethylene glycol
RT room temperature
SDS-PAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis
SEM Scanning Electron Microscopy
spp. species
TCA trichloroacetic acid
TEMED N,N,N’,N’-tetramethylethylenediamine
TMB 3,3’,5,5’-tetramethylbenzidine Tris base Tris(hydroxymethyl)aminomethane
TSB tryptic soy broth
Table of Contents
Abstract (Chinese) ... I
Abstract (English)...II
Acknowledgement... III
Keywords... V
Abbreviations...VI
Table of Contents ... VIII
List of Figures ...XI
List of Tables ... XIII
Chapter 1 Introduction ...1
1.1 Vibrio species...1 1.2 Vibrio alginolyticus ...3 1.3 Metallopeptidase ...5 1.4 Acyl/M20 family ...6 1.5 Aminoacylhistidine Dipeptidase ...7 1.6 Carnosine ...8 1.7 Carnosinase ...101.8 Other Carnosine-Hydrolyzing Enzymes...11
1.9 Peptidase V ...12
Chapter 2 Methods ...16
2.1 Expression of Vibrio alginolyticus pepD gene in E.coli...16
2.2 Purification of expressed Vibrio alginolyticus PepD ...16
2.3 Protein concentration determination ...17
2.4 SDS-PAGE and Native-PAGE analysis...17
2.5 Western blot analysis ...19
2.6 Enzymatic activity assay of Vibrio alginolyticus PepD...19
2.7 Substrate specificity of Vibrio alginolyticus PepD...20
2.8 Enzyme kinetics...20
2.9 Site-directed mutagenesis on Vibrio alginolyticus pepD...21
2.10 Circular dichroism (CD) spectroscopy...22
2.11 Crystallization ...23
2.12 Analytical Sedimentation velocity Ultracentrifugation ...23
Chapter 3 Results ...24
3.1 Expression and Purification of Vibrio alginolyticus PepD...24
3.2 Enzymatic activity assay of Vibrio alginolyticus PepD...25
3.3 Substrate specificity of Vibrio alginolyticus PepD...25
3.4 Enzyme kinetics of Vibrio alginolyticus PepD...26
3.5 Site-directed mutagenesis analysis of Vibrio alginolyticus pepD ...28
3.6 Enzyme kinetics of the mutant PepD ...34
3.7 The secondary structure of Vibrio alginolyticus PepD...36
3.8. Structure features of Vibrio alginolyticus PepD...38
Chapter 4 Discussion and Conclusion ...43
Chapter 5 Future Work ...50
Chapter 6 Reference...52
Appendix 1 ...59
Appendix 2 ...60
List of Figures
Fig. 1. Reaction result in liberated L-histidine from L-carnosine hydrolyzation ...8
Fig. 2. Common histidine-containing dipeptides present in mammals ...10
Fig. 3. The overall structure of PepV and the stereo view of catalytic and zinc binding residues of PepV. ...13
Fig. 4. Schematic of the active site of PepV...14
Fig. 5. SDS-PAGE and Western blot analysis of purified PepD. ...24
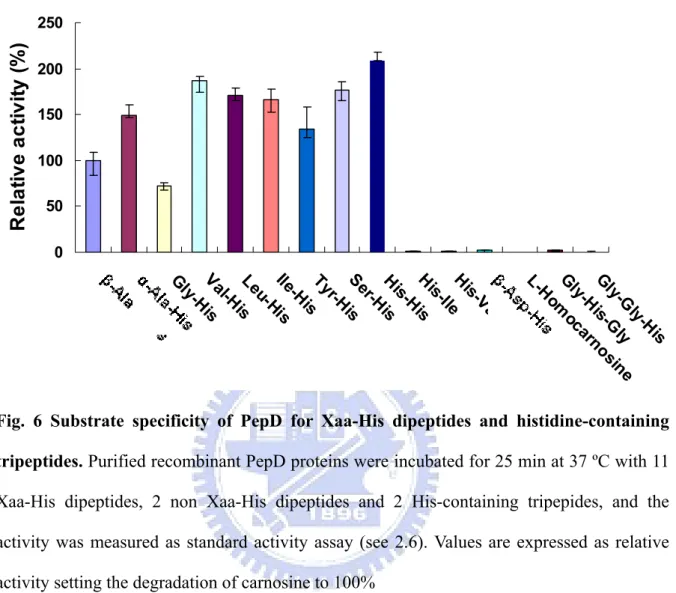
Fig. 6. Substrate specificity of PepD for Xaa-His dipeptides and histidine-containing tripeptides. ...26
Fig. 7. Enzyme kinetic of V. alginolyticus PepD...27
Fig. 8. Multiple sequence alignment with PepD and PepV...29
Fig. 9. SDS-PAGE of purified wild-type and mutant proteins of D119...30
Fig. 10. SDS-PAGE of purified wild-type and mutant proteins of E149...30
Fig. 11. Nativ-PAGE analysis of purified PepD wild-type and mutant proteins...31
Fig. 12 Western blotting analysis of purified PepD wild-type and mutant proteins...32
Fig. 13. Analytical ultracentrifugation determination of PepD protein. ...32
Fig. 14. Analytical ultracentrifugation determination of denatured PepD protein ...33
Fig. 15. Enzymatic activities of wild-type and mutant PepD on L-carnosine. ...34
Fig. 16. Enzyme kinetics of wild-type and mutant PepD...35
Fig. 17. The CD spectra of V. alginolyticus PepD wild-type and mutant proteins ...37
Fig. 18. Three-dimensional ribbon of crystal structure of PepV and the generated PepD model based on PepV. ...38
Fig. 19. Stereo view of PepD superimposed with the active site residues of PepV. ...39
Fig. 20. Local view of PepD superimposed with the active site residues of PepV and CPG2...40
Fig. 22. The V. alginolyticus PepD crystals were grown at 20 in two condition℃ ...42 Fig. 23. Stereo view of orientation relationship between zinc ion and the putative metal
binding residues Asp119 ...45 Fig. 24. Proposed catalytic mechanism for the hydrolysis of N-terminal amino acid
residues...46 Fig. 25. Stereo view of orientation relationship between catalytic water and the putative
List of Tables
Table 1. Association of Vibrio spp. with different clinical syndromes. ...2 Table 2. Differentiation of V. parahemolyticus and V. alginolyticus. ...4 Table 3. Solutions and volumes for preparing SDS-PAGE and Native-PAGE separating
gel and stacking gel. ...18 Table 4. The fluent gradient program ...21 Table 5. Reaction conditions and cycling parameters for PCR mutagenesis reaction ...22 Table 6. Kinetic Parameters for hydrolysis of L-carnosine at 37ºCand pH 6.8 of
wild-type and mutant V.alginolyticus PepD ...36 Table 7. The secondary structure content of wild-type and mutant V. alginolyticus PepD
Chapter 1 Introduction
1.1 Vibrio species
Members of the genus Vibrio are defined as gram-negative bacteria possessing a straight or curved rod shape. They are motile organisms, using a single polar flagellum to travel. Most of Vibrio spp. are halophilic bacterial and a few species are nonhalophilic, depending on their sodium chloride requirements. Vibrio bacteria are commonly found in marine or estuarine environments and most species are sensitive to acid pH. They are heterotrophic organisms, obtaining nutrients from their mutualistic, parasitic, or pathogenic relationships with other organisms and can undergo both respiratory and fermentative metabolism.
In the last four decades, researches on taxonomic, environmental, virulent, and medical aspects of Vibrio species have expanded greatly. Vibrio spp. are considered as pathogens in aquacultured species. Vibriosis is a systemic bacterial infection of estuarine fishes and marine lives. On the basis of phenotypic data, the major species causing vibriosis in shrimp are V.
alginolyticus, V. anguillarum, V. harveyi, and V. parahaemolyticus.1 The V. anguillarum, V. damsela, and V. carchariae are major vibriosis-causing species in fish. It resulted in high
mortality and severe economic loss for shrimp aquaculture in all producing countries.
Vibrio species can infect human by taking seafood or when the wound contacted with the
brine. Vibriosis is caused by taking seafood contaminated with Vibrio parahemolyticus or
Vibrio vulnificus. These bacteria damage the inner wall of the intestine and get into the
bloodstream, which causes diarrhea and related symptoms. Another major disease caused by
Vibrio species is cholera. Cholera is a disease of the small intestine, unlike other enteric
illnesses. V. cholerae infect the small intestine, they get through the mucus layer and adhere to the mucosal cells where they release enterotoxins. In recent years, there is a low occurrence of V. cholerae by improving sewage and water treatment.
Since 1988, the Centers for Disease Control and Prevention (CDC) has maintained a database of reported Vibrio isolations and infections from human. There are at least twelve pathogenic Vibrio species recognized to cause human illness (Table 1).2 Three species of most medical significance are V. cholerae, V. vulnificus, and V. parahaemolyticus. V.
alginolyticus, V. fluvialis, V. furnissii, V. hollisae, and V. metschnikovii, these species are
associated with gastroenteritis.3 V. cincinnatiensis, V. damsela, and V. carchariae are not associated with gastroenteritis, but are pathogenic to human on rare occasions.
Table 1. Association of Vibrio spp. with different clinical syndromes. Clinical Syndromea
Species Gastroenteritis Wound
Infection Septicemia V. alginolyticus +b ++ V. cholerae O1 ++ V. cholerae non-O1 ++ + + V. cincinnatiensis V. damsela ++ V. fluvialis ++ (+) (+) V. furnissii ++ V. hollisae ++ (+) (+) V. metschnikovii (+) V. mimicus ++ (+) (+) V. parahaemolyticus ++ + (+) V. vulnificus + ++ ++
a Data from Levine and Griffin,1993.
b ++ = common presentation, + = less common presentation, and (+) = rare presentation.
Gastroenteritis, wound infection, and septicemia are the major clinical symptoms of
Vibrio infections. The most common clinical symptom is self-limited gastroenteritis. It often
involves in stomachache, abdominal pain, diarrhea, nausea, vomiting and fever, with non-inflammatory infection of the upper small bowel, or inflammatory infections of the colon. Wounds usually occurred at the fingers, foots and palms, with rapidly progressing
swelling, hemorrhagic and severe pain after Vibrio infection for 3 to 24 hours. In patients with medical conditions such as cirrhosis or malignancies, the wound infection may progress very rapidly, with formation of hemorrhagic bullae and extensive soft tissue necrosis. Septicemia is a serious, life-threatening infection that gets worse very quickly and can rapidly lead to septic shock and death. It can be arised from infections throughout the body, including infections in the lungs, abdomen, and urinary tract. Patients of septicemia frequently develop fever, shaking chills, generalized myalgia, edema, and severe pain in the lower extremities. For these reasons, the infection of Vibrio is becoming an important public health problem to human.
1.2 Vibrio alginolyticus
Vibrio. alginolyticus was first recognized and named Oceanomonas alginolytica by
Miyamoto et al. in 1961.2 It was renamed V. alginolyticus by Sakazaki in 1968.3 V.
alginolyticus is one of the 12 recognised marine Vibrio species that have been identified as
being pathogenic for humans and marine animals. It was found with worldwide distribution in marine and estuarine waters, especially in bathing areas4 and had been isolated from coastal water, sediments, and seafood taken from the temperate and tropical areas.5 The
bacterium is able to multiply in salty waters at elevated water temperatures. It is usually isolated in the spring and summer from marine sources. However, the isolation is dependent on water temperature and it is possible to assume that V. alginolyticus may be isolated at any water temperatures greater than 10℃.6 Studies by Baross and Liston showed that the minimum growth temperature for V. alginolyticus is 8℃.7
V. alginolyticus and V. parahaemolyticus were isolated from similar types of marine
are very similar on biochemical properties. V. alginolyticus can be differentiated from V.
parahaemolyticus on the basis of sucrose fermentation, the Voges-Proskauer reaction,
sodium chloride tolerance, and swarming on blood agar (Table 2).6
Table 2. Differentiation of V. parahemolyticus and V. alginolyticus
Characteristic V. parahaemolyticus V. alginolyticus
Sucrose fermentation - +
Vogs-Proskauer - +
Growth in broth with 8% NaCl + +
Growth in broth with 10% NaCl - +
Swarming on blood agar - +
V.alginolyticus might infect fish with the biofilm formation on the intestine, causing fish
mortalities and important economic losing.3, 8 V. alginolyticus is also an important pathogen
to human, first recognized as human pathogen in 1973.9 In recent years, several studies have reported the clinical infection caused by V. alginolyticus.10-12 Most human infections caused by V. alginolyticus were on account of consuming the raw or undercooked seafood obtained from fish, shellfish, shrimps, or squid.13 The major clinical syndromes V. alginolyticus causes are gastroenteritis, wound infections, and septicemia.14 Other clinical syndromes reported in association with V alginolyticus infection include ear infection, chronic diarrhea in a patient with AIDS, conjunctivitis, and post-traumatic intracranial infection.15-17 Vibriosis caused by pathogenic V. alginolyticus is also a common problem in the intensive culture of grouper with a gastroenteritis syndrome (swollen intestine containing yellow fluid).18 Therefore, Vibrio
alginolyticus is not only an important pathogen to marine animals including fish, shellfish
1.3 Metallopeptidase
The peptidase required metal ion for its catalytic activity was named metallopeptidase which could be divided into two board types depending on the number of the required metal ions. Metallopeptidases are the most diverse of the catalytic types of proteases with 15 clans and more than 30 families identified to date.19, 20 In these identified families, there are seventeen endopeptidases, twelve exopeptidases and one metallopeptidases held both functions. Most of metallopeptidases require only one metal ion, but in some families they require two metal ions that act together or so called “co-catalytically”. All known co-catalytic metallopeptidases are exopeptidases which include aminopeptidases, carboxypeptidases, dipeptidases, and tripeptidases, whereas metallopeptidases with only one catalytic metal ion might be exopeptidases or endopeptidases. In these enzymes, the nucleophilic attack on a peptide bond is mediated by a water molecule, which is also observed in aspartic and glutamic peptidases.21 The water molecule was activated by a divalent metal cation, usually zinc but sometimes cobalt, manganese, nickel, or copper.
Several metallopeptidases containing co-catalytic metallo-active sites are key players in carcinogenesis, tissue repair, neurological processes, protein maturation, hormone-level regulation, cell-cycle control and protein-degradation processes.22, 23 They are widely regarded as promising targets for drug discovery, but the detailed mechanistic information is still lack to hamper the therapeutic development. The importance of understanding their mechanism of action is underscored by their central role in several disease states including stroke, diabetes, cancer, HIV, bacterial infections, and neuropsychiatric disorders associated with the dysregulation of glutamatergic neurotransmission, such as schizophrenia, seizure disorders, and amyotrophic lateral sclerosis (ALS). For these reasons, several co-catalytic metallopeptidases have become the target for therapeutic drug designs.24
The majority of metallopeptidases contain two zinc ions at their active site and the residues involved in zinc binding site have been identified by X-ray crystallography. These residues usually are His, Glu, Asp, or Lys, and at least one another residue is required for catalysis, which might play an electrophilic role. With in the identified metallopeptidase familes, an HEXXH motif that form part of the metal-binding site was observed in thirteen metallopeptidases according to the crystallographic studies.19 These zinc metallopeptidases plays roles in metabolic and signaling pathways throughout all kingdoms of life and some are regarded as potential pharmaceutical targets.24
1.4 Acyl/M20 Family
According to the MEROPS database (http://merops.sanger.ac.uk), each peptidase is assigned to a “family” on the basis of statistically significant similarities in amino acid sequence, whereas families that are thought to be homologous are grouped together in a “Clan”. Metallopeptidases were classified into 15 clans including MA, MC, MD, ME, MF, MG, MH, MJ, MK, MM, MN, MO, MP, MQ, and M-. The peptidases in clan MH contains a variety of co-catalytic zinc-dependent peptidases that binding two zinc atoms via five amino acids per monomer, which is held by five amino acid ligands20 and are inhibited by the general metal chelator ethylenediamine-tetraacetic acid (EDTA). In recent reports, it was considered that members of MH and MF clan of dizinc peptidases compared with MC clan of monozinc peptidases displayed three different catalytic zinc centers that have evolved in a similar structural scaffold.25
The peptidase clan MH is further classified into four families: M18, M20, M28, and M42. Enzymes of peptidase family M20 were characterized as water associated with two zinc ions which were bound by five residues in the order His/Asp, Asp, Glu, Glu/Asp, and His at the active site. In general, the general active site residues arrangement of metal-binding residues
with the addition of two catalytic residues (bold) would be His/Asp, Asp, Asp, Glu, Glu, Glu/Asp, His. The Asp residue between two catalytic residues was considered to bind both metal ions. The essential histidine and carboxyl residues in the metal binding sites of all enzymes of this family, were found to be completely conserved.
Peptidase family M20 could be further devided into 4 subfamilies, M20A, M20B, M20C, and M20D, which their active site residues were different among subfamilies. The type protein of peptidase family M20C is aminoacylhistidine dipeptidase which could hydrolyze Xaa-His dipeptides. Several available crystal structures of M20 family enzymes, including PepV, CPG2, showed a dizinc-binding domain. Enzymes of the Acyl/M20 family have shown potential for different applications. In biocatalysis, the high stereoselectivity of Acy1 allows the preparation of L-amino acids from racemic mixtures of N-acyl-L-amino acids.26 Succinyldiaminopimelate desuccinylase is considered as a potential anti-bacterial target, and CPG2 is considered as a therapeutic agent in ADEPT for cancer treatment.27
1.5 Aminoacylhistidine Dipeptidase
The pepD gene exists extensively among the prokaryotes and eukaryotes. Some studies have suggested that expression of pepD negatively affected biofilm formation which was considered for infection, causing fish mortalities and important economic loss. Therefore, PepD could be a promising target to control bacterial biofilm formation and infection.28 In E.
coli, pepD encodes a 52 kDa protein and is active as a homodimer with molecular mass of
100 kDa. The board substrate specificity of PepD was activated by Co2+ and Zn2+ and deactivated by metal chelators.29, 30 The first direct proof of aminoacylhistidine dipeptidase (EC 3.4.13.3, also Xaa-His dipeptidase, X-His dipeptidase, carnosinase, and PepD) activity for hydrolysis of an unusual dipeptide L-carnosine (β-Ala-L-His) in bacteria was obtained with Pseudomonas aeruginosa in 1974.31 In the following years, this carnosine-hydrolyzing
enzyme is discovered from a number of bacterial species,32 but only PepD from Escherichia
coli have been characterized genetically and biochemically.30 Aminoacyl-histidine
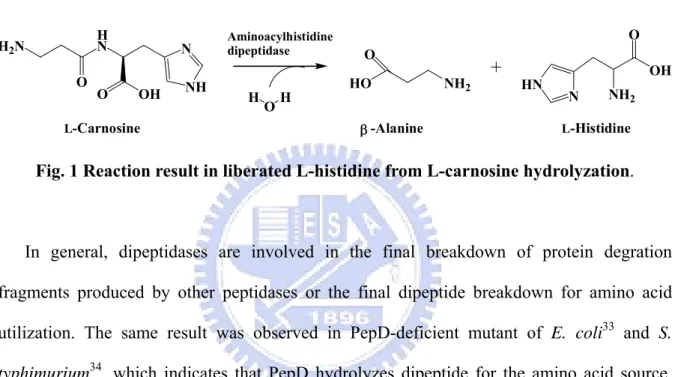
dipeptidases are zinc-containing metallopeptidase, whose catalytic reaction involved the release of an N-terminal amino acid, usually neutral or hydrophobic, from a polypeptide. PepD could hydrolyze Xaa-His dipeptides even include an unusual dipeptide carnosine (β-Ala-L-His) toβ-Alanine and L-Histidine(Fig. 1).19
L-Carnosine N NH O OH H N H2N O OH H Aminoacylhistidine dipeptidase HO NH2 O -Alanine L-Histidine N HN NH2 OH O β
Fig. 1 Reaction result in liberated L-histidine from L-carnosine hydrolyzation.
In general, dipeptidases are involved in the final breakdown of protein degration fragments produced by other peptidases or the final dipeptide breakdown for amino acid utilization. The same result was observed in PepD-deficient mutant of E. coli33 and S.
typhimurium34, which indicates that PepD hydrolyzes dipeptide for the amino acid source.
However, the biological impact of PepD still remains unclear.
1.6 Carnosine
The enzymes with L-carnosine hydrolyzing activity were also observed in mammals and named as carnosinase in general. The unique substrate of carnosinase, L-carnosine (β-alanyl-L-histidine), was first isolated in 1990 from meat extracts and subsequently found to be widely distributed in excitable central and peripheral vertebrate tissues. It is abundant in skeletal muscles of most vertebrates and constantly present in cardiac muscle and brain in millimolar concentrations (1.7–2.5 mM in whole brain,0.15–0.25 mM in medulla oblongata and ≥ 10 mM in olfactory bulb), but not in several other organs, such as kidney, liver, and
lung.35 The concentration of this dipeptide is regulated by two enzymes, carnosine synthase and carnosinase, both of which are present in brain.
The complete role of these dipeptides is still unknown, even though their function has been studied intensively in recent years. Available studies indicated that carnosine has a range of antioxidant or cytoprotective properties,36 to act as a cytosolic buffer,37 an antioxidant,38 and an antiglycation agent.39 The unique dipeptide has been proposed that could act as a natural scavenger of dangerous reactive aldehydes from the degradative oxidative pathway of endogenous molecules such as sugars, polyunsaturated fatty acids (PUFAs) and proteins. In particular, it has been recently demonstrated that carnosine is a potent and selective scavenger of α,β-unsaturated aldehydes, typical by-products of membrane lipids peroxidation and considered second messengers of the oxidative stress, and inhibits aldehyde-induced protein-protein and DNA-protein cross-linking in neurodegenerative disorders such as Alzheimer's disease, in cardiovascular ischemic damage, in inflammatory diseases.40
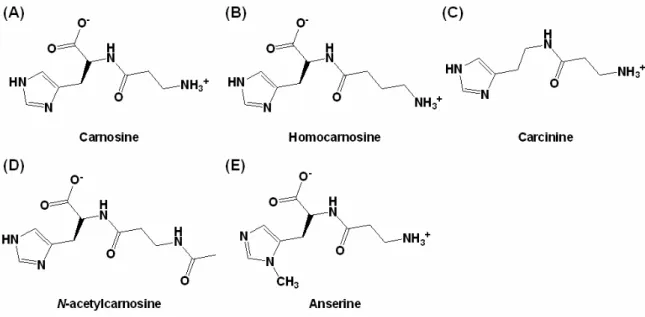
Moreover, carnosine represents the archetype of a series of histidine-containing dipeptides in mammals, such as homocarnosine (γ-aminobutyric acid-L-histidine), carcinine,
N-acetylcarnosine, and anserine (Fig. 2). Homocarnosine was suggested to be a precursor for
the neurotransmitter GABA. Being controlled by one or several carnosinases, it acts as a GABA reservoir and may mediate the antiseizure effects of GABAergic therapies.41
Fig. 2 Common histidine-containing dipeptides present in mammals.
1.7 Carnosinase
Carnosine is synthesized by carnosine synthase (EC 6.3.2.11) from β-alanine and histidine in many tissues and degraded by intra- or extracellular dipeptidases, also named carnosinases, all belonging to the large family of metalloproteases. In mammals, at least two types of carnosinases with differentproperties. The first enzyme is serum carnosinase (CN1, EC 3.4.13.20)42 and the second one is known as a cytosolic form (also named tissue carnosinase, CN2, EC 3.4.13.18). Serum carnosinase (CN1)was identified as a homodimeric dipeptidase with a narrow substrate specificity for Xaa-His dipeptides including L-carnosine. The nature of metal ion in serum carnosinase remains unknown and could be activiated by Cd2+ and citrate ions.43 It could also hydrolyze homocarnosine and anserine, andthese activities were not inhibited by bestatin, a compound known to specifically inhibit various amino- and dipeptidases.
Serum carnosinase was assumed to be involved in some important pathological conditions. Decreased concentrations of serum carnosinase have been observed in patients
with Parkinson’s disease, multiple sclerosis, or after a cerebrovascular accident.44 It was also suggested that monitoring of serum carnosinase might be useful to predict the clinical symptom of patients with acute stroke.45 Deficiency of human carnosinase has been associated with neurological deficits including intermittent seizures and mental retardation.46,
47 Study on serum carnosinase was also approached by computational analysis that suggested
a therapeutic usefulness of either inhibiting by L-carnosine analogues (e.g., in diabetes) or activating the enzyme by the rational design of citrate-like, non-toxic allosteric modulators (e.g., in homocarnosinosis).48
Tissue carnosinase (CN2) was first isolated from porcine kidney by Hanson and Smith in 194949 and subsequently found widely distributed in tissues of rodents and higher mammals. It acted as an ubiquitous nonspecific dipeptidase rather than a selective carnosinase with broad substrate specificity butcould not hydrolyze homocarnosine or anserin.42 This enzyme requiresMn2+ ions for its activity and is strongly inhibited by lowconcentrations of bestatin.
1.8 Other Carnosine-Hydrolyzing Enzymes
There are also some other proteins reported to have the dipeptidase activity on L-carnosine. BapA from Pseudomonas sp. proposed as β-Ala-Xaa dipeptidase (EC 3.4.13.-) was found to hydrolyze peptide bonds of β-alanyl dipeptides (β-Ala-Xaa).50 Pep581 from
Prevotella species (Prevotella spp.) shared similar sequence identity of 47% with E. coli
PepD and has a calculated molecular weight of 53.2 kDa. Pep581 hydrolyzed both dipeptides and single amino acid from the N-terminus of tri- and oligopeptides, which is different from PepD enzymes.51 Anserinase (Xaa-methyl-His dipeptidase, EC 3.4.13.5) mainly catalyzing the hydrolysis of N-acetylhistidine in all poikilothermic vertebrates is also active on carnosine.
1.9 Peptidase V
Lactobacillus delbrueckii pepV is 1413 nucleotides in length and consists of 470 amino
acid residues, encodes a protein with predicted molecular mass of 52 kDa. Lactobacilli are organisms with multiple amino acid auxotrophies making them critically dependent on their proteolytic abilities to efficiently degrade milk protein casein and used as starting materials in their dairy fermentations. Deletion of the dipeptidase pepV gene from Lactobacilli resulted in significantly decreased growth rates but did not reduce the final cell density.52
The Peptidase V (PepV) from Lactobacillus delbrueckii ssp. lactis DSM 7290 was originally identified as a carnosinase, cleaving L-carnosine as a source of histidine, in the E.
coli mutant strain UK197 (pepD, hisG).53 It has also been characterized as a relatively
unspecific dipeptidase cleaving a variety of dipeptides, especially those with the unusual β-alanyl residue in the N-terminus, and removing the N-terminal amino acid from a few distinct tripeptides.53 Moreoner, PepV is related not only to peptidases but also to acetylornithine deacetylase (ArgE, EC 3.5.1.16) and succinyldiaminopimelate desuccinylase (DapE, EC 3.5.1.16), and has been described as a member of the aminoacylase-1 family recently.54 These enzymes share the characteristics of hydrolyzing amide bonds in a zinc- (or cobalt-) dependent manner. Therefore, PepV is recognized as a metallopeptidase in M20A subfamily from MH clan. It could be fully inhibited with metal chelating agent 1, 10-phenanthroline or EDTA.
PepV is the first crystallized dinuclear dipeptidase with carnosine-hydrolyzing enzymatic activity in M20 family. The 3D structure of PepV protein consists of two distinc domains, named the lid (lower) domain and the catalytic (upper) domain. The upper domain contains the polypeptides from Met1 to Gly185 and from Ser388 to Glu468, whereas the lid domain comprises the residues from Glu186 to Gly387. In the crystal structure of PepV, two zinc
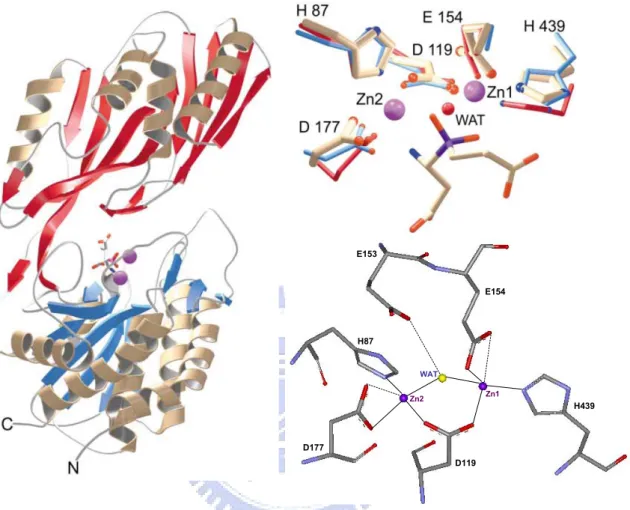
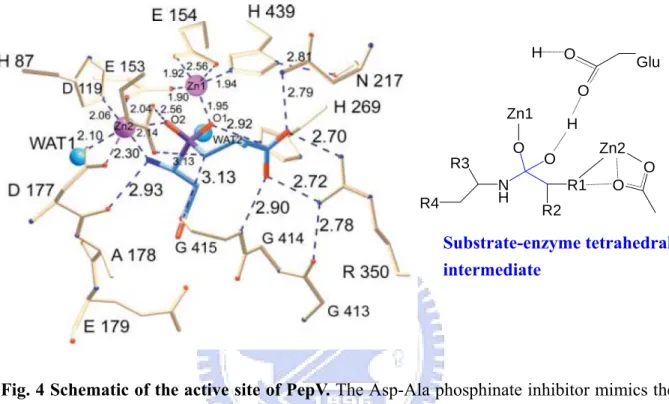
ions is associated in one monomer protein.55 Zinc ions are located in the catalytic domain and held by five residues including His87, Asp119, Glu154, Asp177, and His439 (Fig. 3).55
D119 E153 E154 H439 H87 D177 WAT Zn2 Zn1 D119 E153 E154 H439 H87 D177 WAT Zn2 Zn1 D119 E153 E154 H439 H87 D177 WAT Zn2 Zn1 D119 E153 E154 H439 H87 D177 WAT Zn2 Zn1
Fig. 3 The Overall structure of PepV and the stereo view of catalytic and zinc binding residues of PepV. The inhibitor of PepV (beige) superimposed with the zinc binding residues
of AAP (blue) and CPG2 (red). Residues are numbered according to the PepV sequence. The catalytic water molecule of CPG2 is depicted in red (WAT).
These two zinc ions, as described by Jozic et al.,55 were considered to play two different roles for hydrolyzing substrates: for stabilization of the substrate-enzyme tetrahedral intermediate as well as for activation of the catalytic water molecule (Fig. 4). Zinc 1 which associated with the imidazole group of H439, carboxylate oxygen of E154 and D119 primarily appeared to facilitate substrate binding via a “oxyanion binding hole” with H269
resulting in polarize the scissile carbonyl group and therefore to promote the nucleophilic attack by the catalytic water molecule. Zinc 2 was coordinated by the carboxylate oxygen of H87, D177 and the bridging D119. It seemed primarily to activate the catalytic water molecule and to promote binding and hydrolysis.
R4 R3 N H O O R2 R1 O O O O H Zn1 H Glu Zn2 Substrate-enzyme tetrahedral intermediate
Fig. 4 Schematic of the active site of PepV. The Asp-Ala phosphinate inhibitor mimics the
dipeptide substrate is shown in blue. The bridging catalytic water attacks the carbonyl carbon of the scissile peptide bond to form a sp3-orbital substrate-enzyme tetrahedral intermediate.
The PepV catalytic domain has similar folding relative to that previously found dinuclear carboxyl exo-peptidases AAP,56 SGPA57 and CPG2.58 There is a cis-peptide bond between the bridging Asp119 and the adjacent residue Asp120 in PepV which was also observed in these enzymes. In both di-zinc and mono-zinc carboxypeptidases of M20 family, this cis-conformation generally occurred in those regions of a structure intimately associated with catalysis and seems to be necessary to force the important zinc bridging carboxylate into the correct geometry.
1.10 Research Goal
V. alginolyticus, an important pathogen to human and fish, might infect fish with the biofilm formation on the intestine. Some studies have suggested that expression of pepD negatively affects biofilm formation, it could be a promising target to control bacterial biofilm formation and infection. Aminoacyl-histidine dipeptidases (pepD) which exists extensively among prokaryotes and eukaryotes belong to metallopeptidase 20 (M20) family from metallopeptidase H (MH) clan (MEROPS: The peptidase database). It could hydrolyzes Xaa-His dipeptides including an unusual dipeptide carnosine (β-Ala-L-His), which is benefited to organisms at several physiological aspects. Both the biological importance and function of aminoacylhistidine dipeptidase and L-carnosine are less known. Enzymes of peptidase M20 family have showed the potential for different applications which can act as anti-bacterial target, therapeutic agent for cancer treatment and possibly play roles in aging and neurodegenerative or psychiatric. Since the latent importance of bacterial aminoacylhistidine dipeptidase in biological aspect, additional with the studies on it is indispensable either in the genetic or biochemical aspect from E. coli and S. typhimurium. We perform a study on aminoacylhistidine dipeptidase from V. alginolyticus through gene expression, protein purification and biochemical properties characterization. Due to neither investigations on its functional residues nor crystal structure are reported, a study of the functional residues of V. alginolyticus PepD through site-directed mutagenesis analysis are performed.
Chapter 2 Methods
2.1 Expression of Vibrio alginolyticus pepD gene in E. coli
We used pET-28a(+) as the expression vector. The constructed plasmid pET-28a(+)-pepD was transformed into E. coli BL21(DE3)pLysS competent cell by heatshock method and spread the cell on LBkan agarose plate and incubated at 37 ºC for 12 to 16 hrs. Picked up the
clonies harboring pET-28a(+)-pepD and cultured in 3 ml LBkan medium for several hours
then transferred into 300 mL LBkan medium. Added 150 μL 1 M IPTG (final concentration
was 0.5 mM) when the OD600 approach 0.5 ~ 0.6, then incubated at 37 ºC for another 6 hrs
for induction of the expression of pepD. The pET-28a(+) plasmid was also transformed into
E. coli BL21(DE3)pLysS competent cell and following the same experimental procedure as a
control.
2.2 Purification of expressed Vibrio alginolyticus PepD
After 6 hrs incubation at 37 ºC with rotary shaking, the cell was collected by centrifugation at 6,500rpm for 30 min at 4 ºC. The bacterial pallet was resuspended with 20 ml 20 mM Tris-HCl, 0.5 M NaCl, pH 6.8 buffer (buffer A). The resuspended cells were disrupted by sonication method using a sonicator with pulsing on 2 sec and pulsing off 1 sec for the toatal sonication time of 3 min at 30% energy. The whole experimental procedure of sonication was operated on ice. Repeat the sonication steps at least 3 times. After sonication, the cell lysate was centrifuged at 9,500rpm for 30 min at 4 ºC to remove the cell debris and intact cell. The supernatant was collected for further purification.
The supernatant was purified by affinity chromatography with Ni-NTA column. One mL Ni-NTA resin was packed in 20 mL plastic column and pre-equilibrated with 10 mL buffer A containing 20 mM imidazole (10 bed volumn). The supernatant was load into the column then washed with 10 ml buffer A contained 20 mM imidazole (5 bed volumes). Five bed
volumes of buffer A contained 80 mM, 150 mM, 300 mM, and 500 mM imidazole were used sequentially to elute the expressed pepD protein. At last, washing the Ni-NTA column with buffer A containing 1 M imidazole. The eluted fractions were collected for SDS-PAGE analysis and enzymatic activity assay. By SDS-PAGE analysis, the high purity eluted fractions were collected and dialyzed with 2 L 50 mM Tris-HCl pH 6.8 buffer for 2 hrs and following 3 L for 8 hrs. After enzymatic activity analysis, the purified proteins were stored at -80 ºC before ready for following experiments. PepD could be stored at -80 ºC without losing of activity for six months.
2.3 Protein concentration determination
The protein concentrations of purified proteins were measured by BCA Protein Assay Reagents. Each well of the F96 MicroWellTM plate were added 20 μL sample and mixed with 200 μL BCATM Working Reagents (BCATM Reagent A:BCATM Reagent B = 50:1). The reaction were incubated at 37 ºC for 30 min in dark. The absorbances of samples were measured at 562 nm on Multiskan Ascent Microplate Reader. A 2 mg/mL bovine serum albumin (BSA) stock and successive dilutions (1.5, 1.0, 0.75, 0.5, 0.25, 0.125, 0.025 mg/mL) followed in the same procedure as described above were served as standards.
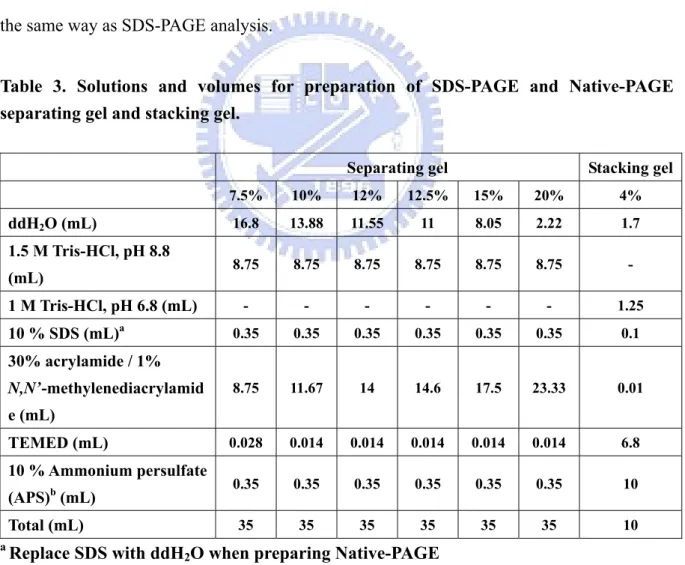
2.4 SDS-PAGE and Native-PAGE analysis
After expression and purification, gel electrophoresis was used to check the expression level, protein purity, and determination of the molecular weight. The samples were electrophoresed on a 12.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Table 3). Each 10 μL sample was mixed with 2 μL 5X SDS-PAGE sample buffer and incubated at 95 ºC for 5 min to denature proteins. The electrophoresis was performed with 1X SDS-PAGE running buffer at 90 Volt for 30 min following 120 Volt for 1.5 hrs. The SDS-PAGE was stained with stain buffer containing Coomassie Brilliant blue
R-250 for 30 min and destained with destain buffer I (methanol/acetic acid/water = 4:1:5, v/v/v) for 20 min and following destain buffer II (methanol/acetic acid/water = 1.2:0.05:8.75) overnight.
Native-PAGE was performed to check the native form of PepD. The purified and dialyzed proteins fractions were electrophoresed on a 7.5% Native-PAGE (Table. 3). The experimental steps were similar to SDS-PAGE analysis besides the gel containing no SDS and without denaturing treatment. Each 10 μL sample was mixed with 2 μL 5X Native-PAGE sample buffer and was performed immediately with iced 1X Native-PAGE running buffer at 90 Volt for 3 hrs with 4 ºC circulating water bath. The proteins were stained and destained in the same way as SDS-PAGE analysis.
Table 3. Solutions and volumes for preparation of SDS-PAGE and Native-PAGE separating gel and stacking gel.
Separating gel Stacking gel
7.5% 10% 12% 12.5% 15% 20% 4% ddH2O (mL) 16.8 13.88 11.55 11 8.05 2.22 1.7 1.5 M Tris-HCl, pH 8.8 (mL) 8.75 8.75 8.75 8.75 8.75 8.75 - 1 M Tris-HCl, pH 6.8 (mL) - - - 1.25 10 % SDS (mL)a 0.35 0.35 0.35 0.35 0.35 0.35 0.1 30% acrylamide / 1% N,N’-methylenediacrylamid e (mL) 8.75 11.67 14 14.6 17.5 23.33 0.01 TEMED (mL) 0.028 0.014 0.014 0.014 0.014 0.014 6.8 10 % Ammonium persulfate (APS)b (mL) 0.35 0.35 0.35 0.35 0.35 0.35 10 Total (mL) 35 35 35 35 35 35 10 a Replace SDS with ddH
2O when preparing Native-PAGE bRecommended to prepare freshly and mix at last
2.5 Western blotting analysis
After gel electrophoresis, the resultant Native-PAGE and a nitrocellulose (NC) membrane were soaked instantly in the transfer buffer. Following transferred the protein immediately to an NC membrane with a blotting apparatus at 90 Volt for 1 hr, the NC membrane was blocked with blocking buffer for 1 hr at RT. The membrane was then washed 3 times with 1X PBS buffer and incubated with the primary anti-PepD monoclonal antibody (mAb) at 1:1,000 dilutions with 1X PBS buffer for 1 hr at RT with gentle shaking, followed by washed 5 times with 1X PBST buffer to remove the unbound primary antibodies. The washed membrane was further incubated with the goat anti-mouse IgG conjugated HRP at 1:5,000 dilutions with 1X PBS buffer for 1 hr at RT with gentle shaking. Finally, the membrane was washed with 1X PBST buffer for 5 times. The immunoreactive bands were visualized with a chemiluminescence reagent and the autoradiography film.
2.6 Enzymatic activity assay of Vibrio alginolyticus PepD
The PepD activity was determined according to Teufel et al.42 on the basis of measurement of histidine by using of the o-phthalaldehyde (OPA) reagent. The substrate L-carnosine (β-Ala-L-His) would be hydrolyzed to β-Alanine and L-Histidine. The fluorescence of the derivative of histidine with OPA was detected at λEx: 355 nm and λEm: 460 nm.
L-Carnosine N NH O OH H N H2N O OH H PepD HO NH2 O -Alanine L-Histidine N HN NH2 OH O β N HN N N H N
OPA (Schiff base)
NH2 OH O L-Histidine H O H O OH O
There were 20 μL purified enzyme (0.5 mg/mL) and 80 μL 50 mM Tris-HCl pH 6.8 buffer reacted with 0.5 mM L-carnosine for 20 min. Liberated histidine was derivatived by adding 100 μL OPA reagent and incubated at 37 ºC in darkness for 5 min. The reaction containing only buffer with L-histidine and L-carnosine reacted with OPA were served as positive and negative control, respectively. All reactions were carried out in triplicate. Fluorescence of the histidine derivatived with OPA was measured by Fluoroskan Ascent FL. (λEx: 355 nm and λEm: 460 nm).
2.7 Substrate specificty of Vibrio alginolyticus PepD
To investigate the substrate specificity of PepD, various Xaa-His dipeptides, including β-Ala-L-His (L-carnosine), α-Ala-L-His, Gly-His, Val-His, Leu-His, Ile-His, Tyr-His, Ser-His, His-His, β-Asp-L-His, and γ-Amino-butyryl-His (GABA-His, homocarnosine) and two histidine-containing tripeptides, Gly-Gly-His and Gly-His-Gly, were used. The activity on L-carnosine was defined as 100%. The enzymatic activity analysis method and reaction condition were as described on 2.6.
2.8 Enzyme Kinetics
For determination of Vmax, Km, and kcat of V. alginolyticus PepD and compared the hydrolysis efficiency with the wild-type and mutant PepD, the method described by Csámpai
et al.59 was modified to use by using High Performance Liquid Chromatography (HPLC)
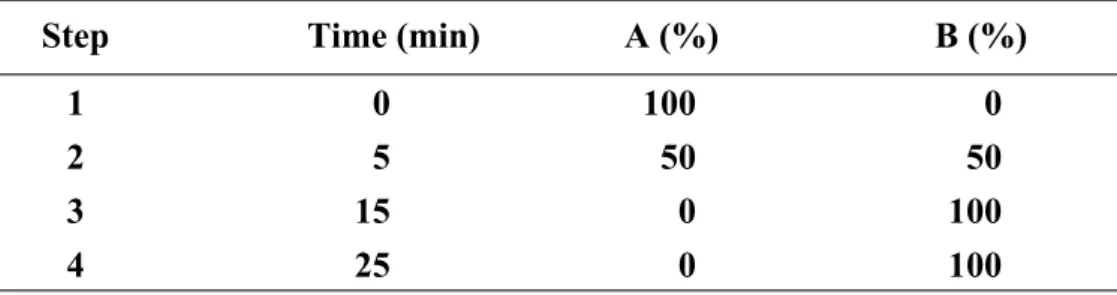
with Fluorescence Detector (FLD). The system, which consists of Agilent 1100 Series Quaternary pump, Autosampler, Fluorescence Detector and Inertsil ODS-3 (7 μm, 7.6 mm×250 mm) column, was used. The eluent system consisted of two components: eluent A was 0.05 M sodium acetate of pH 7.2, while eluent B was prepared from 0.1 M sodium acetate–acetonitrile–methanol (46:44:10, v/v/v) (titrated with glacial acetic acid or 1 M sodium hydroxide to pH 7.2). The gradient program was as described on Table 4. The fluent
flow-rate was 0.8 mL/min at 30 ºC.
Table 4. The fluent gradient program
Step Time (min) A (%) B (%) 1 0 100 0 2 5 50 50 3 15 0 100 4 25 0 100
Different concentrations of L-carnosine (1, 0.5, 0.25, 0.1, 0.05, 0.025, 0.01, 0.005, and 0.0025 mM) were added as substrates to initiate enzymatic reactions. After 20 min incubation at 37 ºC, the samples were mixed with OPA reagent for 5 min incubation at 37 ºC then injected by autosampler. Fluorescence of the histidine with derivatived OPA was measured by FLD (λExc: 355 nm and λEm: 460 nm). Various concentration of L-histidine solution (0.05,
0.025, 0.01, 0.005, 0.0025, 0.001, 0.0005, 0.00025, and 0.0001 mM) derivatived with OPA reagent were detected as method described above to serve as standards.
2.9 Site-directed mutagenesis on Vibrio alginolyticus pepD
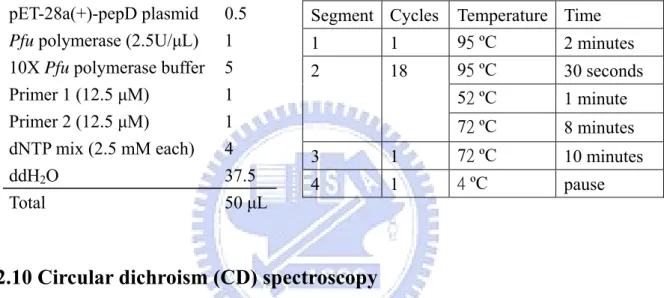
Site-directed mutagenesis was performed by using the QuickChange site-directed mutagenesis kit to create the mutants. Mutagenic primers were designed and pET-28a(+)-pepD plasmid (wild-type) was used as the template: the PCR reaction was carried out by using the nonstrand-displacing action of pfuTurbo DNA polymerase to extend and incorporate the mutagenic primers (Appendix 1.), and resulting in the nicked circular strands. The PCR mutagenesis reaction was performed in the 96-well GeneAmp® PCR System 9700 Thermal Cycler as recommended by the manufacturer of PfuUltraTM High-Fidelity DNA polymerase. Each reaction added 100 ng of wild-type plasmid, 5 μL 10X
Pfu polymerase buffer, 4 μL 2.5 mM dNTP mix, 1 μL of each 12.5 μM primer, 1 μL (2.5 U) Pfu polymerase and ddH2O to the final volume of 50 μL (Table 5). The PCR products with
wild-type and mutant plasmids were incubated with DpnI for 4 hrs at 37 ºC to selectively digest the methylated, non-mutated parental wild-type plasmids. After DpnⅠdigestion, the mutant plasmid was transformed into E. coli. XL1-Blue competent cells, with selection for kanamycin resistance. After the successful mutagenesis confirmed by restriction enzymes and DNA sequencing of plasmid, the desired mutant plasmids were transformed into E. coli BL21( DE3 ) pLysS competent cells for expression of the mutant pepD proteins.
Table 5. Reaction conditions and cycling parameters for the PCR mutagenesis reaction
Segment Cycles Temperature Time
1 1 95 ºC 2 minutes 95 ºC 30 seconds 52 ºC 1 minute 2 18 72 ºC 8 minutes 3 1 72 ºC 10 minutes 4 1 4 ºC pause pET-28a(+)-pepD plasmid 0.5
Pfu polymerase (2.5U/μL) 1
10X Pfu polymerase buffer 5 Primer 1 (12.5 μM) 1 Primer 2 (12.5 μM) 1 dNTP mix (2.5 mM each) 4
ddH2O 37.5
Total 50 μL
2.10 Circular dichroism (CD) spectroscopy
The secondary structure of the wild-type and the mutant pepD proteins were confirmed by monitoring CD spectra. The protein sample concentration was 0.2 mg/mL in 50 mM Tris-HCl, pH 6.8 buffer. The CD spectra were recorded every 1 nm between 200 to 300 nm wavelength used a quartz cuvette of 1 mm path-length in a Jasco J-715 spectropolarimeter, Only 50 mM Tris-HCl, pH 6.8 buffer was as the control. The results were scanned 4 times and averaged. Converted the data into mean residue ellipticity (MRE) by using the equation : [θ]MRE = (MRW × θobs/c × d).60 θobs is the observed ellipticity (in millidegrees) at the
respective wavelength, MRW is the mean residue of the enzyme (MRW = M/n, M = 53548.8 g/mole, n = 490 amino acid residues), d is the cuvette path-length in cm, and c is the protein concentration in mg/mL.60
2.11 Crystallization
Purified recombinant PepD was produced as previously described on 2.2. The purified enzyme was concentrated to 10 mg/mL and dialysed to against Tris-HCl buffer with 20 mM HEPES buffer by Centricon YM30. Using the hanging drop technique, one small droplet of the sample mixed with crystallization reagent was dropped on a
siliconized glass cover slide, and the cover slide would invert to over the reservoir in vapor equilibration with the reagent. In this experiment, hanging drops were formed by mixing 1 μL enzyme solution with 1 μL of crystallization reagent at 20 ºC with the reservoir solution.
2.12 Analytical Sedimentation Velocity Ultracentrifugation
Sedimentation velocity is an analytical ultracentrifugation (AUC) method that measures the molecular moved rate for providing both the molecular mass and the shape of molecules.61 This technique can distinguish the native state of the protein in either a monomer, dimmer, or even tetramer form. The data were evaluated according to the g*(s) method developed by Walter Stafford.62 Since the g*(s) analysis yields both the sedimentation coefficient s from the peak of the curve, the apparent molecular weight can also be determined. Depending on the application and optical system, the protein concentration ranging 0.1 mg/mL to 0.5 mg/mL was used and the sample volume was about 500 μL. Sample was equilibrated with 20 mM Tris-HCl pH 6.8 buffer and this equilibrated buffer was used as another reference control into the reference sector. The sedimentation velocity analysis was performed at National Tsing Hua University.
Chapter 3 Results
3.1 Expression and Purification of Vibrio alginolyticus PepD
Vibrio alginolyticus PepD was sucessfully expressed in E. coli BL21(DE3)pLysS and
purified by Ni-NTA column chromatography. The Ni-NTA resin-bound PepD would be eluted with high purity by 20 mM Tris-HCl, 0.5 M NaCl, pH 6.8 buffer containing 150 mM imidazole. The pure elutent fraction was collected and dialyzed with 20 mM Tris-HCl, pH 6.8 buffer at 4 ºC to remove the salts. The dialyzed protein on SDS-PAGE revealed a single band with molecular mass of approximately 55 kDa (Fig. 5), quite close to the calculated molecular mass, 53.6 kDa, of Vibrio alginolyticus PepD. The purified Vibrio alginolyticus PepD was also confirmed by Western blotting with an anti-PepD mAb (Fig. 5).
M 1 2 3 4
ig. 5 SDS-PAGE and Western blot analysis of purified PepD
f E.coli BL21(DE3)pLysS
The concentration of purified PepD was determined by BCA Protein Assay Reagent and in total 5 mg pure PepD from
97 30 66 45 (kDa) F
(a) Lane M:LMW protein marker;Lane 1:cell crude extracts o
carrying pET-28a(+); Lane 2:cell crude extracts of E.coli BL21(DE3)pLysS carrying pET-28a(+)-pepD; Lane 3:purified PepD from Ni-NTA column; Lane 4:western blotting analysis of purified PepD with anti-PepD mAbs
300 mL E. coli cells could be abtained. The purified PepD was further characterized with the activity assay as described in 2.6.
3.2 Enzymatic activity assay
As described in 2.6, the purified wild-type PepD was subjected to the activity assay with L-c
.3 Substrate Specificity of Vibrio alginolyticus PepD
ase with broad substrate spe
arnosine as a substrate, which would be hydrolyzed to β-alanine and L- histidine. According to Teufel et al.42 on the basis of measurement of histidine by use of
o-phthalaldehyde (OPA) reagent. The histidine-OPA derivative was detected at λEx: 355nm
and λEm: 460nm. The purified PepD was confirmed as a member of carnosine-hydrolyzing
enzymes capable of catalyzing the hydrolysis of L-carnosine to β-alanine and L-histidine which further producing the detectable fluorescence derivative of L-histidine while reacting with OPA reagent in 50 mM Tris-HCl, pH 6.8 buffer.
3
The PepD from E. coli has been identified as a dipeptid
cificity.30 The substrate specificity of PepD from Vibrio alginolyticus was determined with eleven Xaa-His dipeptides, two non Xaa-His dipeptides, and two His-containing tripepides, and compared with the data from E. coli. The experimental method was as described in 2.7. The enzyme activity on L-carnosine (β-Ala-L-His), the known substrate of aminoacylhistidine dipeptidase (PepD), was defined as 100% (Fig. 6). The highest enzyme activity was observed from the hydrolysis of His-His, which was about two times higher than that of the L-carnosine. Moreover, the hydrolysis of α-Ala-L-His, which only differs in the orientation of the alanine also showed 1.5 times higher activity than L-carnosine hydrolysis. The relative dipeptidase activities with the other Xaa-His dipeptides substrates including Val-His, Leu-His, Tyr-His, Ile-His and Ser-His were also superior to that of carnosine degradation, and the enzyme could also hydrolyze Gly-His with good activity. The enzyme showed no apparent activity toward β-Asp-L-His and γ-Amino-butyryl-His (GABA-His, homocarnosine). In addition, the non-Xaa-His dipeptides including His-Ile, His-Val, as well as tripeptides containing histidine in the central or C-terminal position were not degraded,
indicating that V. alginolyticus PepD is a dipeptidase in activity. 0 50 100 150 200 250 Gly-Hi s Va l-HisLeu-H is Ile-Hi s Ty r-H is Se r-H is His -His Hi s-IleHis-V al L-Hom oc arn os in e Gly -H is-G ly Gly-G ly -His
R
el
at
ive act
ivi
ty (
%
)
Fig. 6 Substrate specificity of PepD for Xaa-His dipeptides and histidine-containing
.4 Enzyme Kinetics of Vibrio alginolyticus PepD
-carnosine was performed as desc
tripeptides. Purified recombinant PepD proteins were incubated for 25 min at 37 ºC with 11
Xaa-His dipeptides, 2 non Xaa-His dipeptides and 2 His-containing tripepides, and the activity was measured as standard activity assay (see 2.6). Values are expressed as relative activity setting the degradation of carnosine to 100%
3
The enzyme kinetics of V. alginolyticus PepD for L
ribed in 2.8. The Vmax and Km values of V. alginolyticus PepD (2 μg, 0.186 μM) for L-carnosine calculated from the respective Lineweaver-Burk plot were 1.6 μM/min and 0.36 mM, respectively (Fig. 7). Therefore, the turnover number (kcat, kcat = Vmax/[E]T) of V.
alginolyticus PepD for L-carnosine in 50 mM Tris-HCl, pH 6.8 at 37 ºC was 8.6 min-1 and the
catalytic efficiency (kcat/Km) was 0.398 mM-1s-1. The determined Km value of PepD was 5.64 mM from E. coli30 and 0.25 mM from S. typhimurium34. The other kinetics values including
kcat, kcat/Km, and specific activity of PepD, however, were first identified.
(a)
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 0 0.5 1 1 [L-carnosine] (mM) V0 (u M /m in ) .5(b)
y = 0.2149x + 0.6028 R2 = 0.9865 0 5 10 15 20 25 0 20 40 60 80 100 120 1/[S] (1/mM) 1/ V0 ( m in /u M )Fig. 7 Enzyme Kinetics of V. alginolyticus PepD (a) Michaelis-Menten plot for PepD
catalyzed the hydrolysis of L-carnosine in 50 mM Tris-HCl, pH 6.8 at 37 ºC. (b) Lineweaver-Burk plot calculated from the respective Michaelis-Menten plot.
3.5 Site-directed mutagenesis analysis of Vibrio alginolyticus pepD
is analysis was
was notable that the conserved residue Asp119 of PepD was equal to the adjacent resi
In order to identify the putative active site of PepD, site-directed mutagenes
performed to investigate the essential amino acids. Recent studies reported that the active site residues of enzymes in M20 family were almost conserved.55, 58 Pep V, the first dinuclear dipeptidase with carnosine-hydrolyzing enzymatic activity protein in M20 family has been crystallized in 2002. PepD showed 20.9% identity and 34.3% similarity with PepV based on the sequence alignment employing. Sequence alignment between PepD and PepV also revealed the proposed active site. Surprisingly, the active site residues of PepV were almost conserved in PepD. These residues, including His80, Asp119, Glu150, Asp173, and His461 were expected for the metal binding, whereas Asp82 and Glu149 were expected for the catalysis (Fig. 8).
It
due Asp 120 of PepV that is the adjacent residue next to its metal binding residue Asp119. It is common that aspartic acid residue exhibited the metal binding role extensively at the active site of the enzymes in M20 family. The PepV peptide group between the bridging Asp119 and the adjacent residue Asp120 exhibited a cis-conformation to affect the binding of the metal, whereas the Asp 119 from PepD might be considered to associate with two zinc ions simultaneously. The cis-conformation is thought to be necessary to force the important zinc bridging carboxylate into the correct geometry as described in 1.7. Therefore, the residue Asp119 of PepD may play a more important role on metal binding hence involve in the enzymatic activity. In the study Asp119 and another proposed catalytic residue Glu149 were initially investigated by site-directed mutagenesis.
Fig. 8 Multiple sequence alignment with PepD and PepV
are marked in black and gray,
p119,Glu150, Asp173, His461
The desired mutants were generated by QuickChange site-directed mutagenesis kit as described in 2.9 and the mutant plasm
Identical and conserved amino acids between the sequences
respectively. Dashed lines indicate the gaps introduced for better alignment.
▼ Proposed active site residues : Asp82 ,Glu149
* Proposed metal ion binding residues : His80, As
ids were transformed into E. coli. BL21(DE3)pLysS to express the mutant proteins. Following the same purification experimental procedure of V.
alginolyticus wild-type PepD, the mutant PepD proteins were carried out with 20 mM
Tris-HCl pH 6.8 buffer containing 150 mM imidazole by Ni-NTA column chromtography. The purified wild-type and mutant PepD proteins showed the same molecular weight about 55 kDa on SDS-PAGE (Fig. 9~10.).
-type and mutant proteins of D119.
ild type; Lane 2:PepD D119E mutant; Lane 19L mutant; Lane 5:PepD D119I mutant; Lane 19F mutant; Lane 8:PepD D119A; Lane 9:PepD Lane 11:PepD D119C mutant; Lane 12:PepD
Fig. 10 SDS-PAGE of E149.
Lane M:LMW protein marker; Lane 1:PepD wild type (WT); Lane 2:PepD E149A mutant; Lane 3:PepD E149D mutant; Lane 4:PepD E149G mutant; Lane 5:PepD E149S mutant; Lane 6:PepD E149Q mutant; Lane 7:PepD E149H mutant; Lane 8:PepD E149R mutant
Fig. 9 SDS-PAGE (12%) of purified wild
Lane M:LMW protein marker; Lane 1:PepD w 3:PepD D119M mutant; Lane 4:PepD D1 6:PepD D119 R mutant; Lane 7:PepD D1 D119S mutant; Lane 10:PepD D119T mutant; D119P mutant; Lane 13:PepD D119N mutant
(12%) of purified wild-type and mutant proteins
M 1 2 3 4 5 6 7 M 8 9 10 11 12 13 30 97 66 45 M 1 2 3 4 5 6 7 8 97 66 45 30
In E. coli, PepD was indicated as a homodimer in native sate.29 To observe the native form of PepD . alginolyticus, the wild-type and mutant PepD derivatives were analyzed
with 7.5% Native-PAGE analysis (Fig. 11) A major band with molecular mass near 66 kDa of the protein marker was observed. Interestingly, several weak bands were also examined on the Native-PAGE of the wild-type and mutant PepD. Therefore, it was proposed that PepD in
V. alginolyticus might exist in many forms in the native state. The major band with molecular
weight about 66 kDa might be the monomer that PepD tended to form in its native state. The r form. To
estern blotting (Fig. 13). However, the calculated molecular weight from sedimentation coef
in V
minor weak band with molecular weight near 140 kDa might be the homodime
ensure the native form of PepD in V. alginolyticus, the Westeewrn blotting, that examining a clear band on the film was carried out using anti-PepD mAb (Fig. 12). Besides, Analytical Ultracentrifugation (AUC) was also used to confirm the result of Native-PAGE analysis and W
ficient (s) indicated that PepD preferred to form homodimer in its native state. The calculated molecular weight of denatured PepD protein was as a control comparing to the molecular weight of wild-type PepD protein (Fig. 14).
M 1 2 3 4 5 6
Fig. 11 Nativ-PAGE (10%) analysis of purified PepD wild-type and mutant proteins
Lane M:HMW Native protein marker; Lane1: PepD wild-type(20 μL); Lane 2:PepD D119E mutant (20 μL); Lane 3:PepD E149D mutant (20 μL); Lane 4:PepD wild-type (40 μL); Lane
66
5:PepD D119E mutant (40 μL); Lane 6:PepD E149D mutant (40 μL)
232 140