國立臺灣大學生命科學院生化科學研究所 博士論文
Graduate Institute of Biochemical Sciences College of Life Science
National Taiwan University Doctoral Dissertation
醣胜肽抗生素生合成酵素蛋白晶體結構及反應機制為基礎的 新化學結構生物活性分子設計
Structure- and mechanism-based enzyme design for biologically active new chemical entities on glycopeptide antibiotics
劉祐禎 Yu-Chen Liu
指導教授:李宗璘 博士 蔡明道 博士 Advisor: Tsung-Lin Li, Ph.D.
Ming-Daw Tsai, Ph.D.
中華民國 102 年 7 月 July, 2013
ii
Acknowledgement
First, I would like to appreciate my advisor Dr. Tsung-Lin Li for the guidance of my Ph.D. research. He provides unlimited support to my Ph.D. training. Thanks Dr. Li and Dr. Ming-Daw Tsai for providing supervision on my research work. I also thank committee members Dr. Po-Huang Liang, Dr. Che Ma, and Dr. Su-Chang Lin for their efforts on my thesis evaluation as well as many valuable suggestions and comments.
With all these professors’ help I can complete this dissertation.
Second, I would like to acknowledge my lab members Ms. Yi-Shan Li, Mr. Syue-Yi Lyu, Mr. Li-Jen Hsu, Dr. Yu-Hou Chen, Ms. Yu-Ting Huang, Dr. Hsiu-Chien Chan, Ms.
Chuen-Jiuan Huang, Dr. Gan-Hong Chen, Dr. Chia-Cheng Chou, Ms. Xiao-Wei Zou, Mr. Ning-Shian Hsu, Dr. Chin-Yuan Chang, Mr. Hsien-Wei Yeh and Mr. Kuan-Hung Lin for their help on my experiments. Especially thank Yi-Shan for her biochemical experiments, Syue-Yi for his MIC assay, Li-Jen for his animal study, Yu-Hou for his AUC analysis in Dbv29 project. For MppJ project, I especially appreciate Xiao-Wei for her mutagenesis and activity assay, Ning-Shian for his chiral analysis and binding affinity assay. I cannot finish my experimental works by myself without their assistance.
Finally, I would like to thank my parents and my wife Ms. Pei-Ju Wang for their support and accompany. They are always on my side sharing my happiness and sadness.
With their encouragement I can graduate from school within four years. Now I get my Ph.D. degree and I will move on to the next stage as a postdoctoral research fellow. I hope they will be proud of me and that I can share my success with them in the near future.
iii
Section 1. Interception of teicoplanin oxidation intermediates yields new antimicrobial scaffolds.
iv
摘要
細菌抗藥性問題日益嚴重,使得發展新型抗生素更加困難,藉由改變抗生素生 合成路徑而改變抗生素結構,提供我們一個可能克服細菌抗藥性問題的機會。
Dbv29 為參與抗生素 A40926 生合成之六碳糖氧化酶,利用 X 光蛋白質結晶學與 生物化學的方法,我們解析出 Dbv29 的蛋白質結構以及催化作用機制,並且發現 一對酪胺酸活性基團在輔酶蛋白共價結合、酵素活性及維持蛋白質結構上扮演非 常重要的角色。更特別的是,受質在 Dbv29 與 teicoplanin 複合體蛋白質結構中以 反應中間產物呈現,使我們得以進一步利用 Dbv29 合成各種不同化學結構的衍生 物。在抑菌測試中,部分的衍生物對於具有抗藥性的腸球菌(VRE)比抗生素 vancomycin 與 teicoplanin 表現出更好的抑菌效果。因此利用這嶄新的方法我們可 以發展出更多不同型態的抗生素衍生物來解決細菌抗藥性問題。
關鍵字:醣胜肽抗生素、teicoplanin 衍生物、氧化還原酶、還原胺化、抗萬古黴素 腸球菌(VRE)。
v
Abstract
In the search for new efficacious antibiotics, biosynthetic engineering offers attractive opportunities to introduce minor alterations to antibiotic structures that may overcome resistance. Dbv29, a flavin-containing oxidase, catalyzes the four-electron oxidation of a vancomycin-like glycopeptide to yield A40926. Structural and biochemical examination of Dbv29 now provides insights into residues that govern flavinylation and activity, protein conformation and reaction mechanism. In particular, the serendipitous discovery of a reaction intermediate in the crystal structure led us to identify an unexpected opportunity to intercept the normal enzyme mechanism at two different points to create new teicoplanin analogs. Using this method, we synthesized families of antibiotic analogs with amidated and aminated lipid chains, some of which showed marked potency and efficacy against multidrug resistant pathogens. This method offers a new strategy for the development of chemical diversity to combat antibacterial resistance.
Keywords: glycopeptide antibiotics, teicoplanin analogs, oxidoreductase, reductive amination, vancomycin-resistant Enterococcus (VRE).
vi
Table of Contents
Verification letter from the oral examination committee...i
Acknowledgement ... ii
Section 1. Interception of teicoplanin oxidation intermediates yields new antimicrobial scaffolds. ... iii
Chinese abstract ... iv
Abstract ... v
Table of Contents...vi
List of Figures...ix
List of Tables...x
1. Introduction ... 1
1.1 Emerging of resistant pathogens ... 1
1.2 Glycopeptide antibiotics ... 1
1.3 Modifications of glycopeptide antibiotics by biosynthetic enzymes ... 3
1.4 Dbv29 ... 3
2. Materials and Methods ... 6
2.1 Gene cloning and protein purification ... 6
2.2 Crystallization and data collection ... 6
2.3 Structure determination and refinement ... 7
2.4 Enzymatic activity assay... 8
2.5 Mutagenesis ... 8
2.6 Circular dichroism spectroscopy ... 8
2.7 Analytical ultracentrifuge analysis ... 9
2.8 Synthetic conditions for new analogs ... 9
vii
2.9 Compound characterization ... 10
2.10 In vivo study ... 12
3. Results ... 13
3.1 Dbv29 is a FAD-containing dimer ... 13
3.2 Contributions to cofactor- and substrate-binding sites ... 13
3.3 A diol intermediate leads to catalytic redirection ... 15
3.4 Analogs provide alternate antimicrobial protection ... 17
3.5 Dbv29 has a dynamic quaternary structure ... 18
4. Discussion ... 20
References... 48
Section 2. Structure and mechanism of non-heme iron-SAM dependent methyltransferase and its engineering to hydratase. ... xi
Chinese abstract ... xii
Abstract ... xiii
List of Figures...xiv
List of Tables...xv
1. Introduction ... 52
1.1 Mannopeptimycin ... 52
1.2 Methyltransferases and methylation ... 54
1.3 MppJ ... 55
2. Materials and Methods ... 57
2.1 Gene cloning ... 57
2.2 Protein expression and purification ... 57
2.3 Crystallization and data collection ... 58
2.4 Structure determination and refinement ... 58
viii
2.5 Enzymatic reaction ... 59
2.6 Site-directed mutagenesis ... 59
2.7 Analytical ultracentrifugation analysis ... 60
2.8 Isothermal titration calorimetry analysis ... 60
2.9 X-ray absorption spectroscopy ... 60
2.10 Electron paramagnetic resonance spectroscopy ... 61
2.11 Chiral HPLC analysis ... 61
3. Results ... 62
3.1 Protein crystallization and structure determination ... 62
3.2 Overview of the structure ... 63
3.3 Conformational change and SAM/SAH-binding ... 64
3.4 Ppy-binding site and coordination chemistry ... 65
3.5 Reaction mechanism ... 66
3.6 No non-heme oxygnease activity ... 68
3.7 Stereochemistry ... 70
3.7.1 Methylation ... 70
3.7.2 Post-translational modification ... 71
3.7.3 Methoxylation ... 72
4. Discussion ... 73
References... 93
Appendix ... xvi
ix
List of Figures
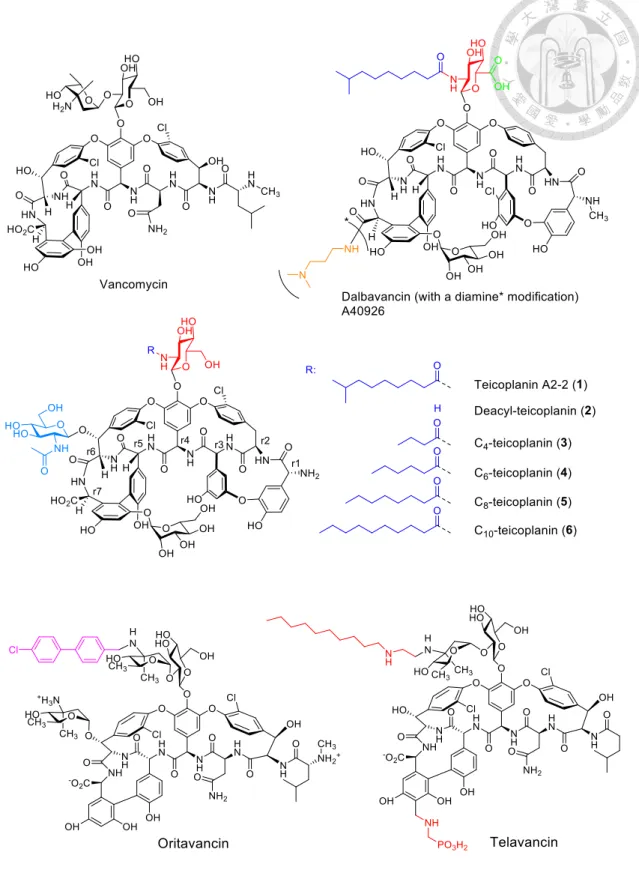
Figure 1. Structures of relevant glycopeptide antibiotics. ... 21 Figure 2. The structure of Dbv29 and its enzymatic mechanism. ... 22 Figure 3. Intermediate trapping strategies to probe the catalytic mechanism. ... 23 Figure 4. Variations in acyl chain length regulate the resultant glycopeptide
analogue. ... 24 Figure 5. Mice infection test for analog 25 in blood bacterial clearance. ... 25 Figure 6. Structural analysis of dimeric and monomeric Dbv29. ... 26 Figure 7. Analytical ultracentrifugation (AUC) analyses for Dbv29 and mutants. 28 Figure 8. Circular dichroism (CD) analyses for Dbv29 and mutants. ... 30 Figure 9. CD analysis of protein stability. ... 31 Figure 10. MS and MSMS spectra of the benzylamine-Tei aminated analog (25). 32 Figure 11. NMR information for de-r4-Tei. NMR spectra include 1H, 13C, HSQC, HMBC, and COSY. ... 35 Figure 12. NMR information for analog 25. NMR spectra include 1H, 13C, HSQC, HMBC, and COSY. ... 38 Figure 13. HRMS data for representative compounds. ... 39
x
List of Tables
Table 1. MICs of vancomycin, teicoplanin and analogs against tested strains. ... 40 Table 2. Data collection and refinement statistics for Dbv29 structures. ... 41 Table 3. Relative enzymatic activities of Dbv29 mutants. ... 42 Table 4. Product yields of oxidized, aminated or amidated teicoplanin analogs synthesized by Dbv29. ... 44 Table 5. NMR assignments for compound 25 as determined from spectra shown in Figures 11 and 12. ... 47
1
1. Introduction
1.1 Emerging of resistant pathogens
The rise of bacterial resistance against long-standing drugs of last resort, such as vancomycin, has created an urgent demand for the development of new antibiotics with enhanced or broadened antimicrobial efficacy1-5. Recent strategy for finding new classes of antibacterial compounds based on targets identified from bacterial genomics has not yet proved successful6. This has led us to consider rationally manipulating potential natural products. Natural products have historically been invaluable as a source of antibacterial drugs, such as penicillins, macrolides and glycopeptides, reflecting their evolutionary origin as ‘weapons’ that bacteria use against each other. Glycopeptide, vancomycin, had long been known as the ‘last resort’ in treating serious infections caused by bacteria resistant to several other antibiotics, such as the ‘super bug’
methicillin-resistant Staphylococcus aureus (MRSA), but even vancomycin resistance has now emerged, further underlining the need for new antibiotics. Thus, compounds with activity against clinically important Gram-positive pathogens, including antibiotic- resistant strains such as MRSA, penicillin resistant Streptococcus peuminiae, and vancomycin resistant Enterococci (VRE) are highly demanded7, 8.
1.2 Glycopeptide antibiotics
Vancomycin (Figure 1) that has outpaced multidrug resistance of methicillin-resistant Staphylococcus aureus (MRSA) for decades is facing mighty backlashes by emerging vancomycin-intermediate S. aureus (VISA) and vancomycin-resistant S. aureus (VRSA) in addition to having lost battle to vancomycin-resistant Enterococci (VRE).
Dalbavancin (Figure 1), a new member of the vancomycin family antibiotics known for active against Staphylococci (e.g. MRSA) and Streptococci while weak against
2
Enterococci (e.g. VRE), was developed based on the drug lead glycopeptide antibiotic A40926 (Figure 1). Two other renowned vancomycin-based derivatives, oritavancin and telavancin, are active against strains resistant to conventional glycopeptides (e.g. VRE) but relatively weak against methicillin-resistant Staphylococcus aureus (MRSA).
Seeking new compounds with enhanced and/or broader antimicrobial spectrum for refractory bacterial infections are never over demanded. A40926 differs from teicoplanin (Tei, 1, Figure 1) mainly in lacking the N-Ac glucosaminyl on residue 6 (r6) and the replacement of the N-acyl glucosaminyl C-6 OH substituent on residue 4 (r4) with a COOH group (Figure 1). These structural features make it an ideal system to exploit the manipulation of genes and enzymes involved in the biosynthetic pathways of both antibiotics to create hybrid structures and new analogs. The close structural similarity of vancomycin and related glycopeptide antibiotics including A40926 and teicoplanin have prompted a variety of studies to determine whether late-stage tailoring enzymes that act on one of the family members will cross-react with other family members to create new hybrid analogs; these strategies have yielded several compounds with altered and improved antimicrobial profiles, demonstrating the utility of this approach. Though many hybrid structures have been developed using these methods or have been identified from natural sources, modifications at the sugars of the residue 4 and 6 are rare.
Three new glycopeptides are currently close to common clinical usage, namely, oritavancin (Targanta Therapeutics Inc) and telavancin (Theravance Inc/Astellas Pharm Inc) which are analogs of vancomycin, and dalbavancin (Pfizer Inc) (Figure 1) which in fact is a A40926 analog. All three compounds have some commonality in structure but with difference in pharmacokinetic and pharmacodynamic properties and in vitro spectrum. In general, the three compounds all have been well tolerated in clinical trials
3
to date but with pros and cons individually. In short, oritavancin and telavancin are active against strains resistant to conventional glycopeptides (e.g. VRE) but relatively weak against strains such as methicillin-resistant Staphylococcus aureus (MRSA), while dalbavancin features active against Staphylococci (e.g. MRSA) and Streptococci but weak against Enterococci (e.g. VRE) or Staphylococci harboring the vanA gene cluster.
1.3 Modifications of glycopeptide antibiotics by biosynthetic enzymes
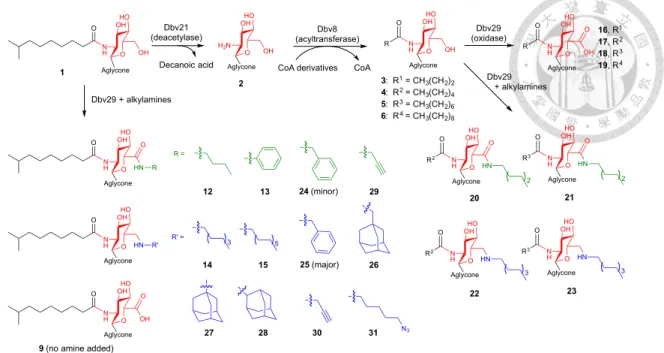
Previous reports have shown that even modest structural modifications of vancomycin can overcome resistance. We have elucidated the biological functions for enzymes Dbv8, 9, 21, and 29. They together were characterized to be responsible for the synthesis of the N-acyl glucosamine moiety of A40926 in the filamentous Actinomycete Nonomuraea sp. ATCC39727. Given the “diversity-oriented synthesis”
and “synthesis on privileged compound” as new concepts in the field, we are keen to build up a useful platform using native and engineered Dbv8, 9, 21, and 29 as biocatalysts. New analogs by modifications of teicoplanin will be dissimilar to any reported compound (they are almost vancomycin and A40926 derivatives). New analogs demonstrated superior in anti-VRE activity will be expected to be at least not less than oritavancin and/or telavancin to the given strains tested and they are very likely to be more effective to some oritavancin- and telavancin-insensitive strains. As a consequence, diversification of the “privileged compound” teicoplanin may help conquer bacterial resistance and ease the strong demand for new antibiotics.
1.4 Dbv29
Though many hybrid structures have been developed using these methods or have been identified from natural sources, modifications at the C6 position are rare. We have been interested in Dbv29, a hexose oxidase that catalyzes the last step in A40926 biosynthesis in the filamentous Actinomycete Nonomuraea sp. ATCC397279, 10. Our
4
previous efforts demonstrated that this flavoenzyme performs a four-electron oxidation to convert the glucosaminyl C6 alcohol found on the ‘top’ of the peptide scaffold to a carboxylic acid. Dbv29 is unusual among enzymes known to perform oxidations at the C6 site, both in the coenzyme used (for example, NAD is used in the UDP-glucose dehydrogenase family) and in lacking a typical cysteine residue in the active site11-13. Our previous studies showed that Dbv29 activity is dependent on the presence of an acyl group on the C2 amine of the same carbohydrate residue9, 10, 14, 15
, though the exact nature of this acyl group proved somewhat flexible, as Dbv29 could oxidize both teicoplanin—containing an extended acyl chain—and analogs of A40926 with an acetyl or acyl group attached (and lacking a distant N-acetyl glucosamine substituent).
Bioinformatics analysis pointed to two tyrosine residues, Tyr165 and Tyr473, as potentially important for the reaction mechanism16, but single phenylalanine mutants did not significantly affect activity in an overnight incubation experiment. Mutagenesis of His91 and Cys151 did provide strong support that the flavin cofactor, characterized at that time as flavin mononucleotide (FMN), was covalently bound to the enzyme through these residues; unexpectedly, the double mutant of these two enzymes retained some level of activity, unlike other flavoenzymes that covalently link to their cofactor9,
16-19
. Thus, the function of this enzyme remained enigmatic.
In this study, we sought to both understand the mechanism of this unusual enzyme and—given our prior demonstration of acyl chain promiscuity—explore whether it could be employed to generate non-natural glycopeptide analogs. We report crystallographic, biophysical and biochemical characterization of Dbv29 that demonstrates the critical role of Tyr165 and Tyr473 in the enzyme reaction.
Additionally, through the serendipitous discovery of a reaction intermediate in the crystal structure, we design a new synthetic strategy to rationally intercept the reaction
5
mechanism. This approach allows access to new classes of products that would be extremely difficult to obtain by other means and that offer improved and complementary profiles for antimicrobial drug discovery efforts.
6
2. Materials and Methods
2.1 Gene cloning and protein purification
The dbv29 gene was amplified from genomic DNA by PCR. The products were each ligated according to the manufacturers' instruction (Novagene) into the pET-28a(+) expression vector which provides an N-terminal His6-tagged protein. E. coli cells were grown at 37°C in 1 L of LB medium until an A600 of about 0.7 was reached. Protein expression was induced with 0.2 mM IPTG at 16°C, and, after 12 h, the cells were harvested by centrifugation, resuspended in 10 mM imidazole-HCl buffer, pH 7.8 (binding buffer), and ruptured using a microfluidizer. The cell-free extract prepared by centrifugation was applied to an Ni2+- resin column, which was then washed successively with binding buffer and wash buffer before eluting the target protein with m imida ole- Cl. Gel filtration was performed using an kta FPLC system equipped with an S-200 Superdex column (Amersham Bioscience) under isocratic conditions (20 mM Tris, pH 7.6, 100 mM KCl). The buffer was exchanged for 50 mM HEPES buffer, pH 7.2 using Millipore centrifugal filters. Protein concentrations were estimated using the Bradford assay. The purified protein was confirmed by SDS-PAGE, Western blotting, and electrospray mass spectrometry (ESI-MS).
2.2 Crystallization and data collection
Dbv29 was crystallized by using hanging drop vapor diffusion method at 20 °C. 10 mg ml−1 Dbv29 in 20 mM TRIS (pH 8.0, 100 mM NaCl) was mixed with the same volume of reservoir solution. Hexagonal crystals can be obtained in two crystallization conditions. One contains 50 mM calcium chloride, 100 mM BIS-TRIS (pH 6.5), 30%
(v/v) PEGMME 550; the other contains 200 mM imidazole malate (pH 5.5), 33% (v/v) PEG 600. Crystals were obtained after one-week incubation. A different crystal shape was achieved in 200 mM di-ammonium hydrogen citrate (pH 5.0), 17% (w/v) PEG
7
3350, C10 teicoplanin (5 mM; C10 teicoplanin was purified from commercially available teicoplanin mixtures by HPLC; the purified C10 teicoplanin was salt-free, so it required 10% (v/v) DMSO to dissolve), whereby a crystal was obtained after a five- month incubation. Data sets were collected on an ADSC Quantum-210 CCD using the synchrotron radiation -ray source tuned at a wavelength of . and an operating temperature of 100 K at beamline 13C1 of the National Synchrotron Radiation Research Center in Taiwan. Data were indexed and scaled with the HKL2000 package20. The hexagonal crystal belongs to P6122 space group with unit cell dimensions a b 66. and c . . ecause of the large cell dimensions, the maximum resolution that can be reached is . . The crystal obtained from the second condition belongs to P21 space group with unit cell dimensions a = 61. , b 0.8 , c = 124.9 and β 8.4° and was diffracted to 1.93 resolution. he contents of both asymmetric units were estimated from the Matthews coefficient21. The data suggest that a value of . 3 Da-1 with 40.8% solvent corresponds to two molecules per asymmetric unit in the P6122 crystal, and a value of 2.44 3 Da−1 with 49.7% solvent content indicates four molecules per asymmetric unit in the P21 crystal.
2.3 Structure determination and refinement
The molecular replacement method was used to obtain phase information, and MolRep22 was used to find the phase solution. nitial phases were obtained at resolution using the coordinates of AknOx (PDB ID: 2IPI) as the search model. Phase extension yielded electron density maps into which a polypeptide model was built with the program XtalView23. The model was further refined with CNS and REFMAC24, 25. Water molecules were added with a water-pick routine in the CNS program. The final model has an factor of 6. for all reflections between and . resolution and an Rfree of 20.5% under 5% randomly distributed reflections. In the Ramachandran
8
plot, 99.5% of all residues are in the allowed region. Figures were generated using PyMOL (http://www.pymol.org). Detailed refinement statistics are given in Table 2.
2.4 Enzymatic activity assay
Dbv29 activity was determined by LC-MS. The assay mix containing en yme ( μg) and the corresponding substrate (1 mM) in buffer (50 mM HEPES pH 7.2, 100 mM aCl, m D ) (total volume μl) was incubated for 2 h at 37 °C. Each reaction mixture was then centrifuged at 16,000 g for 5 min (Heraeus Biofuge Pico) and filtered on an ultracentrifugal filter unit (5 kDa cut-off membrane, Millipore). The filtrate was directly subjected to HPLC-ESI/Q-Tof (Waters HPLC 2695 interfaced with an ESI source coupled to a Micromass micro Q-Tof mass spectrometer) or HPLC-ESI-LTQ (Agilent 1200 Series interfaced with an ESI source coupled to a Thermo-Finnigan LTQ XL ion trap spectrometer), using a gradient of 0–60% acetonitrile in 0.1% TFA in water over 30 min. Online LC-MS spectra were recorded by MassLynx (Waters) or Xcalibur (Thermo Fisher Scientific, Inc.).
2.5 Mutagenesis
Site-directed mutagenesis was carried out by using QuickChange (Stratagene), and the wild-type Dbv29 was used as the template for single mutation. For double mutation, the single or double mutants, respectively, served as templates. All mutations were confirmed by DNA sequencing. The pET/His plasmid was used for protein expression.
Mutant enzymes were purified with the same protocol as recombinant wild-type Dbv29.
2.6 Circular dichroism spectroscopy
For structural analysis, far UV circular dichroism spectra of wild-type Dbv29 and mutants were recorded between 240 nm and 190 nm on a Jasco circular dichroism spectropolarimeter at 25 °C. Proteins were present at 1 mg ml−1 in a standard buffer
9
solution. Each spectrum represents an average of three scans, wherein the buffer background is subtracted. For stability analysis, proteins of wild-type Dbv29 and Y165F/Y473F mutant were dissolved in a standard buffer solution at 1 mg ml−1 for thermal melting experiments. Circular dichroism measurement was carried out with a Jasco J-815 spectropolarimeter in a temperature range of 20–100 °C at 222 nm wavelength. Melting temperatures, Tm, were determined by using two-state approximation.
2.7 Analytical ultracentrifuge analysis
The sedimentation velocity experiments were performed with a Beckman-Coulter XL-I analytical ultracentrifuge. Samples and buffers were loaded into 12-mm standard double-sector Epon charcoal-filled centerpieces and mounted in an An-60 Ti rotor. We introduced μl of a mg ml−1 sample into the cell. Sedimentation velocity experiments were performed at rotor speed of 40,000 r.p.m. at 20 °C. The signals of samples were monitored at 280 nm and collected every 3 min for 6 h. The raw data of experiments were calculated using SedFit software26. The density and viscosity of buffer were calculated using Sednterp software (http://
www.jphilo.mailway.com/default.htm). All samples were visually checked for clarity after ultracentrifugation, and no indication of precipitation was observed.
2.8 Synthetic conditions for new analogs
Teicoplanin (a mixture of five analogs) was purchased from AAPIN Chemicals Ltd (UK). The mixture was subjected to HPLC purification to obtain C10-Teicoplanin with purity >95%. 5-Azide pentylamine (99%) was obtained from Dr. Chung-Yi Wu (Academia Sinica) as a gift. All other chemicals were obtained from Sigma/Aldrich Chemical Co. (St. Louis, MO, USA) without further purification unless otherwise stated.
Teicoplanin analogs (2-6) were enzymatically synthesized as described previously9, 10, 14,
10
15. In brief, compound 2 was prepared by adding Dbv21 or Orf2* (10 g) into a typical buffer solution (50 mM HEPES, pH 7.2, 100 mM NaCl, 1 mM DTT) containing Tei (1, 1 mM) for 5 h at 37°C; compounds 3-6 were prepared by adding Dbv8 (10 g) into the same buffer solution but containing compound 2 and butanoyl-, hexanoyl-, octanoyl-, or decanoyl-CoA for compounds 3-6, respectively, at 25°C overnight. For the final reaction, Teicoplanin analogs (9, 10, 11, 25 and 32, which showed relatively higher yields from initial tests) were enzymatically synthesized using the optimized conditions described below. Enzyme stability (and, indirectly, the extent of exposure of the aldehyde for functionalization) was tested in an array of organic solvents (MeCN, MeOH, EtOH, DHF, DMF, DCM, DMSO etc.); DMSO turned out to be the most appropriate solvent as Dbv29 is highly stable in up to 90% DMSO. 50% DMSO, however, is included in the current protocol as it gives relatively higher yields. Other conditions, such as the amounts of alkylamine and reductant27, were also optimized one at a time against the fixed concentration of Tei (0.5 mM) for a better yield (below). In general, 10-fold of alkylamine and the reducing agent versus Tei was determined to be most appropriate for the current protocol, which is summarized as follows: Tei (0.5 mM), alkylamine (5 mM; carbon number >6), and Na(CN)BH3 (5 mM) in 50% DMSO buffer solution at 37°C overnight incubation.
2.9 Compound characterization
All compounds were characterized by MS and HPLC. For select compounds, NMR analyses were performed on a Bruker Avance 600 spectrometer equipped with CryoProbeTM, with tetramethylsilane (TMS) as an internal standard. Compounds were dissolved in deuterated dimethyl sulfoxide (DMSO-d6) if not otherwise stated, and spectra were recorded at room temperature. NMR peaks of reference compound (de-r4-
11
Tei) and compound 25 are listed below (also see Table 5); original spectra can be found in Figures 11 and 12.
Reference compound (de-r4-Tei). HRMS ES(-): 780.1677 [M-2H]-2, calc. for C72H68O28N8Cl2 780.1760 [M-2H]-2 (Figure 13). 1H-NMR (600 MHz, DMSO-d6) (Figure 11): 9.68 (m, 2H), 9.27 (m, 1H), 8.65 (m, 1H), 8.53 (s, 1H), 8.42 (s, 1H), 7.92 (m, 2H), 7.80 (m, 1H), 7.26 (m, 3H), 7.18 (m, 2H), 7.10 (m, 4H), 6.97 (m, 1H), 6.75 (m, 3H), 6.46 (m, 2H), 6.23 (m, 1H), 5.27 (m, 2H), 5.13 (m, 2H), 4.42 (m, 2H), 4.34 (m, 1H), 4.28 (d, J 11.5, 1H), 4.12 (m, 1H), 3.06 (m, 2H), 2.95 (m, 1H), 1.88 (m, 2H), 1.26 (m, 1H). 13C NMR (600 MHz, DMSO-d6): 172.79, 172.72, 169.87, 169.63, 169.32, 168.59, 168.44, 167.36, 167.27, 167.19, 158.76, 158.67, 158.56, 158.49, 158.29, 158.08, 157.87, 157.46, 157.38, 155.52, 155.09, 150.27, 150.23, 149.06, 148.56, 148.49, 147.63, 143.51, 135.58, 134.67, 134.41, 128.26, 128.19, 127.13, 126.20, 125.66, 125.25, 123.43, 120.71, 120.19, 120.08, 118.29, 116.30, 105.66, 99.26, 99.13, 97.52, 96.65, 76.91, 76.81, 73.80, 73.66, 73.00, 72.55, 70.59, 70.00, 69.92, 69.81, 65.99, 65.92, 63.11, 61.13, 60.75, 59.25, 56.12, 56.07, 55.86, 55.79, 55.17, 54.59, 23.21, 23.12. An expanded version of HSQC, HMBC and 1H COSY spectra of the reference compound is shown in Figure 11.
Compound 25. HRMS ES(+): 984.3033 [M+2H]2+, calc. for C95H104O32N10Cl2 984.3098 [M+2H]2+ (Figure 13). 1H-NMR (600 MHz, DMSO-d6) (Figure 12): 9.67 (m, 2H), 8.61 (m, 1H), 8.40 (m, 1H), 7.93 (m, 2H), 7.77 (m, 2H), 7.18 (m, 14H), 6.71 (m, 3H), 6.48 (m, 2H), 6.23 (m, 1H), 5.24 (m, 7H), 4.96 (m, 1H), 4.62 (m, 1H), 4.39 (m, 5H), 4.08 (m, 1H), 3.70 (m, 2H), 3.00 (m, 4H), 2.14 (m, 1H), 1.99 (m, 1H), 1.86 (m, 3H), 1.48 (m, 2H), 1.37 (m, 1H), 1.15 (m, 12H), 0.84 (d, J 6.5, 6H). 13C NMR (600 MHz, DMSO-d6):
172.87, 172.79, 172.41, 172.36, 169.94, 169.81, 169.39, 169.24, 168.88, 168.76, 168.35, 167.28, 158.54, 158.41, 158.21, 158.00, 157.80, 157.67, 157.48, 157.41, 155.59, 148.27,
12
135.73, 135.62, 134.68, 131.88, 131.82, 130.21, 130.09, 129.96, 129.88, 129.20, 129.12, 128.86, 128.80, 128.38, 125.02, 120.79, 120.74, 120.20, 118.28, 116.29, 114.62, 109.20, 98.91, 96.62, 76.95, 76.83, 73.80, 73.73, 72.85, 72.80, 72.71, 72.56, 72.44, 72.20, 70.76, 70.68, 70.09, 69.81, 66.02, 63.13, 61.30, 61.20, 61.08, 60.76, 56.58, 56.38, 56.19, 55.77, 54.57, 51.74, 51.00, 50.16, 35.98, 31.37, 29.23, 29.10, 29.02, 28.97, 28.92, 28.87, 28.80, 27.47, 27.40, 26.76, 26.71, 26.67, 25.11, 24.97, 23.25, 23.17, 22.65, 22.18,14.09. An expanded version of HSQC, HMBC and 1H COSY spectra of the compound 25 is shown in Figure 12.
2.10 In vivo study
ICR female mice were purchased from the National Laboratory Animal Breeding and Research Center, Taipei, Taiwan. Mice with average body weight of 27 to 30 g were subjected to infection via intravenous (i.v.) injection with 1.3 × 105 cfu/mouse of E.
faecalis (ATCC 51559) at day 0. For treatment study, mice were randomized into four groups at the start of the experiment and administered 10 mg/kg of either vancomycin, teicoplanin, benzylamine-teicoplanin (25) or saline (control) by i.v. twice a day for three days (from day 1 to day 3, a total of 6 doses). Mice were subjected to anesthesia; whole blood was then sampled from orbital sinus on day 1, day 2, and day 3. The whole blood underwent serial dilutions with PBS, which were plated on Brain Heart infusion agar (BHI agar; Difico, Detroit, MI, USA) for enumeration of cfu (colony formation unit).
The graphs and statistical analyses were performed using SigmaPlot® and SigmaStat® . Differences were considered significant if the P value was < 0.05.
13
3. Results
3.1 Dbv29 is a FAD-containing dimer
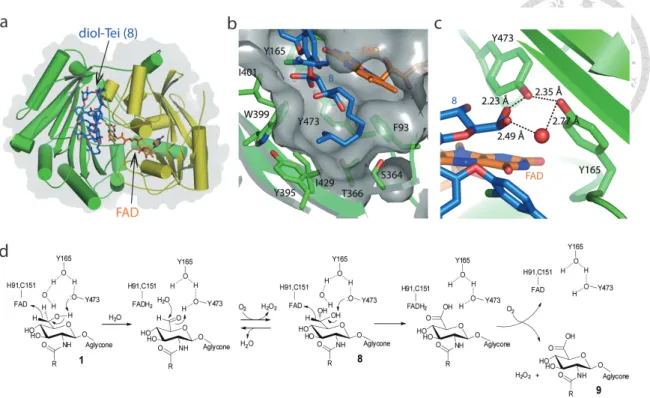
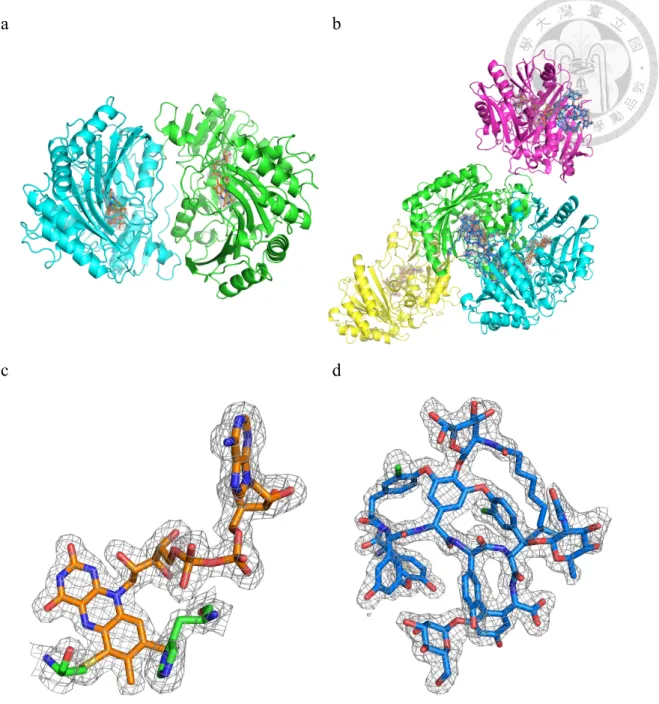
To begin our investigations, we solved crystal structures of Dbv29 free and in complex with substrate. The free structure superimposes very well with the complexed form (r.m.s. deviation of . for C), suggesting that binding of ligand does not induce substantial conformational transitions. As shown for the bound enzyme (Figure 2a), the structure consists of two domains: the F domain, which binds the flavin cofactor, and the S domain, which recognizes substrates, confirming that Dbv29 belongs to the flavin-dependent p-cresol methylhydroxylase (PCMH) superfamily19. The crystal structure also offers clear evidence that the flavin cofactor is flavin adenine dinucleotide (FAD) and not FMN. We suspect that the previous suggestion of an FMN- containing enzyme was likely because of hydrolysis of FAD in solution. The major difference between the two structures is that apo Dbv29 appears to form a dimer on the basis of a large subunit-subunit interface ( , 6 2 per monomer). However, in the complex, an asymmetric unit contains four molecules but does not demonstrate substantial protein-protein interaction, suggesting that the bound form exists as a monomer. This is in contrast to three other members of the PCMH family, glucooligosaccharide oxidase (GOOX)17, aclacinomycin oxidoreductase (AknOx)16 and berberine bridge enzyme (BBE)28, which were solved recently in monomeric forms.
3.2 Contributions to cofactor- and substrate-binding sites
As discussed above, our prior studies had highlighted unusual roles for four residues in controlling flavin binding and reactivity. The crystal structure now reveals a possible explanation for these functions (Figure 2). Though the isoalloxazine ring of flavin is covalently linked to the side chains of His91 and Cys151, it is closely connected to Tyr165 and Tyr473 via the substrate and a bridging water (Figure 2c). We revisited our
14
enzyme activity studies to obtain improved semi-kinetic data and observed that, as previously reported, single H91A and C151A mutants, as well as the H91A/C151A double mutant sequence, retained low levels of activity (11%, 23% and 5% activities, respectively, relative to wild type; Table 3). The mutated enzymes also retained their characteristic yellow color, in stark contrast to the corresponding mutants of GOOX, AknOx and BBE, which lost their color and activities. In contrast to our earlier work, however, the improved kinetic analysis demonstrated that Y165F and Y473F mutations did substantially affect enzyme activity, reducing function to 70% or 23% of the wild- type function, respectively; the Y165F/Y473F double mutant further led to loss of the yellow color and total loss of catalytic activity. Notably, the Y473E mutation similarly abolished activity; we suspect the lower pKa of this side chain or the alternate conformation adopted by the nonplanar ligand might explain this observation. We additionally examined the impact of mutations at the nearby positions Arg360, Ser364 and Thr366, as well as six other tyrosine residues to search for other contributing interactions. However, aside from the Y135F mutant protein, which could not be expressed, none of these mutations disrupted flavin binding (Table 3).
The substrate-binding site in the S domain is somewhat hidden by a long loop (residues 351–362) that crosses over the binding pocket. The electron density for the aglycone shows some discontinuity, whereas that for the N-acyl amino sugar moiety is apparent and clear. The sugar ring anchors against the si face of the isoalloxazine ring of FAD and exposes the C5 carbon close to the N5 nitrogen of FAD, while the C4 hydroxyl group forms a hydrogen bond with Asn427. The N-acyl group extends from the sugar ring into a hydrophobic lipid cavity (Figure 2b). We tested the importance of packing interactions in this lipid cavity by comparing the enzymatic turnover of two substrates with short (C4, 3) and long (C10, 6) alkane chains; the longer chain substrate
15
was preferred by approximately ten-fold, indicating contributions from but not a strict requirement for hydrophobic packing.
Beyond Tyr165 and Tyr473, which contact the C6 hydroxyl group, most residues that line the ligand-binding site are located in different loop regions. Mutational analysis of some of these residues showed that Ser364, Thr366, Tyr370 and Tyr470—though they did not impact flavin binding—did significantly alter enzyme function, with resultant activities of 4–21% of wild type. Additionally, Trp399 and Ile401 proved to be critical for function, as the mutated enzymes displayed either no activity or no more than 10%
of wild-type function (Table 3).
3.3 A diol intermediate leads to catalytic redirection
Although the structure of the complex was solved by cocrystallization with teicoplanin, the electron density is best fitted with neither teicoplanin (the substrate) nor oxo-teicoplanin (the product). Instead, the appearance of extra out-of-plane electron density on the C6 terminus of the amino sugar was best explained by the presence of two OH groups in sp3 configuration or a water-coordinated diol (Figure 2c). We had previously suggested a basic series of reactions needed to convert the C6 alcohol to a carboxylate in which a diol intermediate, or aldehyde equivalent, is used as the transformed substrate for the second half of the oxidation. The structural information obtained here now allows us to build on our prior analysis to propose a specific model for Dbv29 function (Figure 2d): in the first oxidation step, the C6-OH is deprotonated by the hydroxyl group of Tyr473, which in turn is activated by the hydroxyl group of Tyr165. These residues together drive the pro-R hydride transfer from the C6 carbon to the N5 of the isoalloxazine ring and yield the aldehyde intermediate corresponding to the diol found in the crystal structure. In the second oxidation, a second hydride transfer to FAD is likely facilitated by a proton relay network involving the tyrosine pair, the
16
substrate diol and the diol-coordinated water molecule. In each two-electron oxidation reaction, the reduction of FADH2 back to FAD converts one molecule of molecular oxygen to hydrogen peroxide.
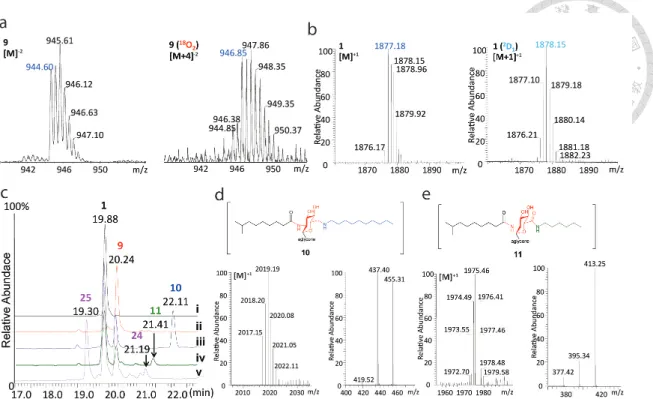
Beyond the mere presence of this unexpected chemical group on the protein substrate, we also discovered that this moiety appeared to be directly exposed to solvent. To test this hypothesis, we conducted two sets of reactions using isotopically labeled reactants.
When the teicoplanin and Dbv29 were mixed in 18O-labeled water, a dominant mass of M+4 instead of M+2 was found, indicating both oxygens of the diol intermediate were labeled with 18O (Figure 3a). The simplest explanation for this result is that the aldehyde is accessible to water and that the aldehyde and diol are in equilibrium. In addition, when we reduced teicoplanin with NaBD4 in the presence and absence of Dbv29, MS analyses of the product recovered from the reaction with protein showed a mass increased by 1 mass unit (Figure 3b). Again, the simplest explanation for this result is that the NaBD4 was able to access the oxidized aldehyde in the reactive site and reduce it to a deuterated alcohol species.
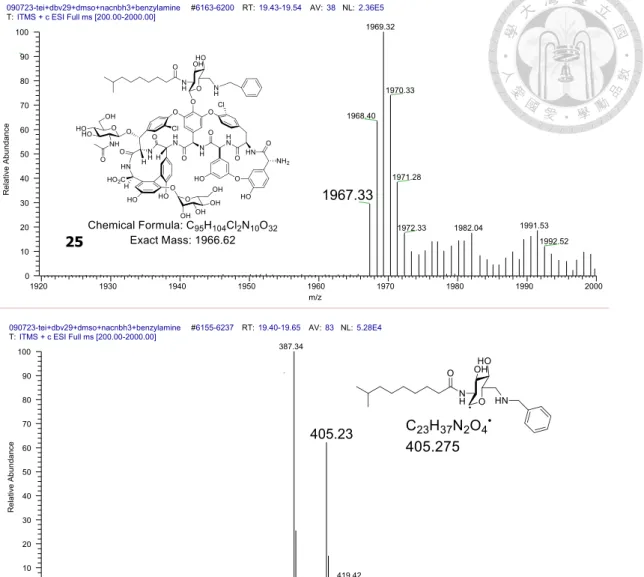
The accessibility of the aldehyde further led us to explore the possibility of trapping and modifying this intermediate as a way to produce new antibiotic analogs. First, we tested the reductive amination of an aldehyde with an amine27. Enzymatic reactions were carried out in the presence of teicoplanin, Dbv29, sodium cyanoborohydride (Na(CN)BH3) and either n-decylamine or n-hexylamine. Remarkably, the LC-MS traces (Figure 3c) indicated that although reaction with n-decylamine resulted in the expected formation of n-decylaminated teicoplanin (10), the reaction with n-hexylamine led to n- hexylamidated teicoplanin (11), both of which were confirmed by MS (Figure 3d,e).
These results can be explained by differential insertion of the amine into the active site.
Thus, the hexylamine would react with the aldehyde intermediate, and the resulting
17
gem-hydroxyl-alkylamine could remain in the active site to undergo the second oxidation reaction. In contrast, decylamine appears to be too bulky to enter the active site and so instead reacts with the aldehyde functional group to form an imine that remains exposed to solution or is even released into solution, where it is subsequently reduced. Thus, the reactive intermediates can be trapped at varying stages during the catalytic course.
To take advantage of this reactivity, we constructed a chemoenzymatic strategy to create additional analogs with variations in both the length and the structure of the lipid chains at both C2- and C6-positions of teicoplanin and oxo-teicoplanin (Figure 4). Our protocol used Dbv21, an enzyme catalyzing deacetylation of the GlcNac moiety10, 14, and Dbv815, an enzyme catalyzing the acylation of the glucosamine moiety, in combination with Dbv29 and various coenzyme A derivatives. When mixed in the presence of organic solvents (for example, 50% DMSO) and selective reducing agents (for example, 5 mM sodium cyanoborohydride), we were able to obtain a series of new analogs 12–31. The enzymes were quite tolerant to new functional groups, as demonstrated by the inclusion of amantidine and related compounds as well as alkyne and azide handles that can be further manipulated through ‘click’ chemistry for applications such as identification of a second mode of action for broader spectrum analogs29, 30.
3.4 Analogs provide alternate antimicrobial protection
Having gained access to new chemical space with our modified teicoplanin analogs, we wanted to examine whether we had similarly reached new biological space;
accordingly, we determined the minimum inhibition concentrations (MICs) of several of the compounds against collections of vancomycin-resistant Enterococcus (VRE) strains (Table 1). We observed that inhibition is proportional to the length of acyl side chains in
18
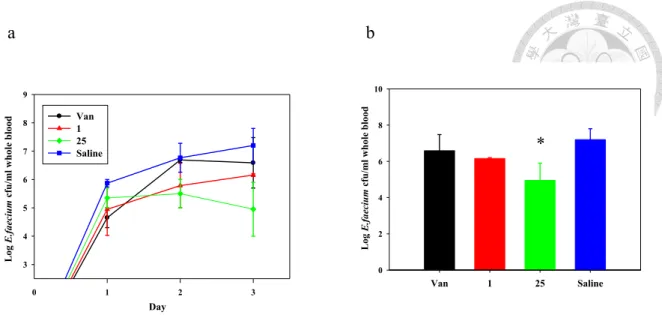
compounds 2–7, with the carboxylate (9) showing no change in activity as compared to teicoplanin. The bi-lipid analogs 10 and 11, however, showed significantly enhanced bactericidal activities against all five tested strains of Enterococcus fecalis when compared to vancomycin and teicoplanin. Notably, the different functional groups in compounds 10 and 11 yielded opposite outcomes for the different strains: the shorter- chain analog 11 is more effective against antibiotic-sensitive strains (for example, ATCC 33186), whereas the longer-chain analog 10 is more effective against drug- resistant strains (for example, ATCC 51559 and 700221). Strikingly, analog 25, tailored with benzylamine (Figure 10), is equally effective against sensitive and resistant strains with dose potency one to two orders of magnitudes lower than vancomycin and teicoplanin and thus may complement compounds such as the A40926 derivative dalbavancin, which is active against Staphylococci (for example, MRSA) and Streptococci but weak against VRE31-34. As a first proof of principle for in vivo efficacy, we tested analog 25 against mice infected with VRE (ATCC 51559) via intravenous injection and found that this molecule was more effective than either vancomycin or teicoplanin at reducing blood bacterial counts (Figure 5).
3.5 Dbv29 has a dynamic quaternary structure
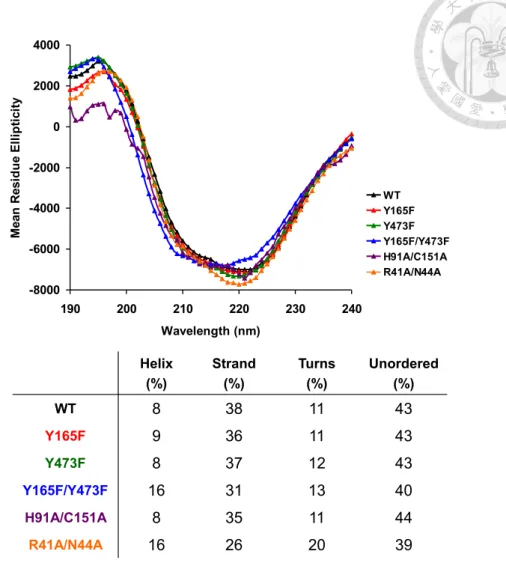
We remained intrigued by the different quaternary forms of the protein observed in the apo and bound forms (Figure 6). To investigate this matter further, we biophysically characterized several constructs, including the wild-type protein, the double mutant that lacks covalent linkages to flavin (H91A/C151A), the nonfunctional double mutant (Y165F/Y473F) and an additional construct with alanine mutations at each of two residues located at the dimeric interface (R41A/N44A). The latter two constructs did not form dimers as monitored by gel filtration analysis or analytical ultracentrifuge experiments and showed some minor deviations from wild type by circular dichroism
19
(Figures 7, 8). Our earlier functional test had demonstrated that the Y165F/Y473F double mutant lacked catalytic activity, but we were also surprised to observe that the enzyme structure was significantly destabilized, as indicated by a decrease in melting temperature of almost 35 °C (Figure 9). Additionally, though Arg41 and Asn44 were not anticipated to participate in the enzyme mechanism directly, the catalytic efficiency of the double mutant is substantially reduced (59% of wild-type activity; Table 3).
Although the mechanistic importance of this is not fully understood, we speculate that the protein quaternary structure dynamics may be correlated to subtle structure changes when a ligand is loaded on or off the enzyme.
20
4. Discussion
The development of new chemical entities for antibacterial applications remains a pressing and challenging goal. Our chemoenzymatic strategy toward this end was made possible by elucidating the reaction mechanism of Dbv29, coupled with the identification of several unusual features of this catalytic process. In particular, we took advantage of a promiscuous lipid cavity as well as a solvent-exposed reaction intermediate to create a small library of new compounds, several of which show promising results against a panel of bacterial strains. Because we focused our attention on an unusual site of modification—C6—we anticipate that the inevitable resistance profiles of these molecules should differ from other glycopeptides for which modifications occur at a dissimilar position, thus extending the effective lifetime of both classes of compounds and suggesting combination treatment as an attractive therapeutic strategy. Toward this end, our combined synthetic protocol provides a cost-effective and environment-friendly method that presages scalable production of these and other next generation drugs.
21
Figure 1. Structures of relevant glycopeptide antibiotics.
22
Figure 2. The structure of Dbv29 and its enzymatic mechanism.
(a) The ligand-bound structure of Dbv29, in which the ‘F’ domain is colored in yellow and the ‘S’ domain is colored in green. (b) View of the lipid tunnel in Dbv29 and substrate binding residues. (c) View of the Tei-binding site, wherein the residues Tyr- 473 and -165 interact with the water-coordinated diol intermediate. (d) The proposed catalytic course.
23
Figure 3. Intermediate trapping strategies to probe the catalytic mechanism.
(a) Mass spectra of Dbv29 enzymatic assays carried out in a H216O or H218O buffer solution. (b) Mass spectra of the substrate with NaBD4 in the absence (left) and presence (right) of Dbv29. (c) HPLC traces demonstrating the formation of n- decylaminated Tei (10) and n-hexylamidated Tei (11); the reactions were conducted in the presence of Na(CN)BH3, Dbv29, and decylamine (trace iii) or hexylamine (trace iv);
controls contained either no Dbv29 (trace i) or no Na(CN)BH3 (trace ii). (d) MS and MS/MS spectra of n-decylaminated Tei (10). An m/z of 455 represents the mixed alkyl- acyl glucosaminyl moiety. (e) MS and MS/MS spectra of bis-acyl oxo-Tei (11). An m/z of 413 represents the bis-acyl aminoglucuronyl moiety.
24
Figure 4. Variations in acyl chain length regulate the resultant glycopeptide analogue.
Reaction with large alkylamines yield amine products, smaller amines yield amide products, and the absence of an amine yields the carboxylic acid. Black, green and blue colors denote variations in carbon length at C2, amidations at C6 and aminations at C6, respectively. See Table 4 for full descriptions of acyl groups used and product yields.
O OH OHHO H2N Dbv21 (deacetylase)
O OH OHHO NH R Dbv8 O
(acyltransferase)
O OH OHHO NH R Dbv29 O
(oxidase) O
Decanoic acid Aglycone CoA derivatives Aglycone Aglycone
Aglycone O OH OHHO NH
3: R1 = CH3(CH2)2 4: R2 = CH3(CH2)4 5: R3 = CH3(CH2)6 6: R4 = CH3(CH2)8
16, R1 17, R2 18, R3 19, R4
O HN OHHO NH R2
O O
Aglycone
O HN OHHO NH R3
O O
Aglycone
20 21
O HN OHHO NH R2
O
Aglycone
O HN OHHO NH R3
O
Aglycone
22 23
1
2
15 O HN
OHHO NH
O
Aglycone
12
14 R
24 (minor)
25 (major) 26
27 28 30 31 N3
CoA
29
9 (no amine added)
3 3
2 2 Dbv29 + alkylamines
Dbv29 + alkylamines
R =
13
O HN OHHO NH Aglycone
R' R' =
3 5
O OH OHHO NH
O
Aglycone O
O O
O
25 a b
Figure 5. Mice infection test for analog 25 in blood bacterial clearance.
(a) The plot shows the whole blood bacterial counts of mice infected by E. faecalis (ATCC 51559) and treated with vancomycin (Van, in black) or teicoplanin (1, in red) or analog 25 (in green) or saline (control, in blue) for three days. The bacterial counts for the control are steadily increased over time, while the counts for groups treated with drugs are all suppressed but in various extents. Teicoplanin and analog 25 are more prominent than vancomycin in terms of overall drug efficacy. (b) The bar chart shows the bacterial counts at day 3 (end of treatment). In general, these three drugs all show positive effects against the infection, while only analog 25 displays significant. The asterisk (*) indicates significant difference (P < 0.05); data were expressed as mean ± SD for a group of three mice)
0 1 2 3
3 4 5 6 7 8 9
Van 1 25 Saline
Log E.faecium cfu/ml whole blood
Day
Van 1 25 Saline
0 2 4 6 8 10
Log E.faecium cfu/ml whole blood
*
26 a b
c d
Figure 6. Structural analysis of dimeric and monomeric Dbv29.
(a) The dimer of Dbv29 in a P6122 space group (two polypeptide chains in an asymmetric unit); FAD cofactors are represented as stick models. (b) Monomer of Dbv29 in complex with a water-coordinated-diol intermediate in a P21 space group (an asymmetric unit consists of four polypeptide chains in which there are no typical protein-protein interactions with one another); the FAD cofactors and the Tei intermediate are represented as stick models in orange and blue, respectively. (c) The 2Fo-Fc electron density map of FAD contoured at 1.0 σ. (d) The 2Fo-Fc electron density map of the diol intermediate contoured at 1.0 σ.
27
WT Dimer s=5.97 Mcal=103.9 (kDa) fr=1.33
Y165F Dimer s=5.92
Mcal=109.7 (kDa) fr=1.44
Y473F Dimer s=6.00
Mcal=98.1 (kDa) fr=1.00
Y165F/Y473F Monomer s=3.44, 4.12
Mcal=68.9, 64.1 (kDa)
fr=1.66
28
Figure 7. Analytical ultracentrifugation (AUC) analyses for Dbv29 and mutants.
(a-n) The raw experimental data were analyzed by Sedfit (http://www.analyticalultracentrifugation.com/default.htm) and the plots were generated by MATLAB (MathWork, Inc.). The spectra in left panels (a, c, e, g, i, k, m) are the calculated c(s, fr) distributions that are shown as two-dimensional distribution with grid lines representing the s and fr grid in the thermograph. Below this c(s, fr) surface a contour plot of the distribution is projected into the s-fr plane, where the magnitude of c(s, fr) is indicated by contour lines at constant c(s, fr) in equidistant intervals of c.
Contour plots in the right panels (b, d, f, h, j, l, n) are transformed from the calculated 0
H91A/C151A Dimer s=6.28
Mcal=126.2 (kDa) fr=1.44
10
WT + Tei (1) Monomer s=4.17
Mcal=79.7 (kDa)
R41A/N44A
Mix of monomer and dimer s=3.9, 4.4
Mcal=67.7, 58.8 (kDa) s=6.4
Mcal=95.7 (kDa)
m n
fr=1.22
29
c(s, fr) distribution and are shown as c(s, M) distribution. The dotted lines (colored in blue) indicate lines of fr (frictional ratio). The signal of the c(s, M) distribution is indicated by the color temperature. The insert grayscale bars in the right panels indicate the residuals bitmap of each fit. (a, b) Dbv29 WT (dimer, s=5.97 (red dashed line), fr=1.33 (blue dashed line)); (c, d) Dbv29 Y165F (dimer, s=5.92, fr=1.44); (e, f) Dbv29 Y473F (dimer, s=6, fr=1.00); (g, h) Dbv29 Y165F/Y473F (monomer, s=3.44, fr=1.66);
(i, j) Dbv29 H91A/C151A (dimer, s=6.28, fr=1.44); (k, l) Dbv29 WT in the presence of 10 mM Tei (1) (monomer, s=4.17, fr=1.44); (m, n) Dbv29 R41A/N44A (monomer, s=3.9, fr=1.22). The wild-type enzyme and the two single mutants (Y165F and Y473F) (a-f) remain associated as dimers as opposed to the double mutant that dissociates into monomers (g, h). The mutant also has a larger frictional ratio (fr), suggesting its molecular shape has been altered considerably.
30
Figure 8. Circular dichroism (CD) analyses for Dbv29 and mutants.
CD spectra of WT and mutants (Y165F, Y473F, Y165F/Y473F, H91A/C151A and R41A/N44A), in which Y165F/Y473F and R41A/N44A double-mutations are somewhat deviated from others as shown in secondary structure distributions.
-8000 -6000 -4000 -2000 0 2000 4000
190 200 210 220 230 240
Wavelength (nm)
Mean Residue Ellipticity
WT Y165F Y473F Y165F/Y473F H91A/C151A R41A/N44A
Helix (%)
Strand (%)
Turns (%)
Unordered (%)
WT 8 38 11 43
Y165F 9 36 11 43
Y473F 8 37 12 43
Y165F/Y473F 16 31 13 40
H91A/C151A 8 35 11 44
R41A/N44A 16 26 20 39
31 a
b
Figure 9. CD analysis of protein stability.
(a) The denaturation temperature (Tm) for WT was determined by CD to be 84.7°C, meaning Dbv29 is unusually stable. (b) The denaturation temperature (Tm) for the Y165F/Y473F mutant was determined to be 50.4°C; the protein was significantly destabilized by lacking the two oxygen atoms on the tyrosine pair. Therefore, residues Y165 and Y473 have considerable influences on protein stability. (ΔTm = 34.3°C)
32
Figure 10. MS and MSMS spectra of the benzylamine-Tei aminated analog (25).
090723-tei+dbv29+dmso+nacnbh3+benzylamine #6163-6200 RT:19.43-19.54 AV:38 NL:2.36E5 T:ITMS + c ESI Full ms [200.00-2000.00]
1920 1930 1940 1950 1960 1970 1980 1990 2000
m/z 0
10 20 30 40 50 60 70 80 90 100
Relative Abundance
1969.32
1970.33 1968.40
1971.28
1967.33
1991.53 1982.04
1972.33
1992.52
25
090723-tei+dbv29+dmso+nacnbh3+benzylamine #6155-6237 RT:19.40-19.65 AV:83 NL:5.28E4 T:ITMS + c ESI Full ms [200.00-2000.00]
260 280 300 320 340 360 380 400 420 440 460 480 500 520 540
m/z 0
10 20 30 40 50 60 70 80 90 100
Relative Abundance
387.34
405.23
419.42 369.38
33
10 9 8 7 6 5 4 3 2 1 ppm
1H NMR spectrum for compound de-r4-Tei
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 0 ppm 13C NMR spectrum for compound de-r4-Tei
34
ppm
9 8 7 6 5 4 3 2 1 ppm
160 140 120 100 80 60 40 20 0 1H-13C HSQC spectrum for compound de-r4-Tei
ppm
11 10 9 8 7 6 5 4 3 2 1 ppm
180 160 140 120 100 80 60 40 20 1H-13C HMBC spectrum for compound de-r4-Tei