୯ҥᆵεᏢғڮࣽᏢଣނᏢࣴز܌
ᅺγፕЎ
Institute of Zoology, College of Life Science
National Taiwan University Master Thesis
чᆵለ܄ࢨύ༾ғނဂޑᕴᡏ୷Ӣᡏϩ
!
Metagenomic Analysis of Microbial Communities from an Acidic Hot Spring in Northern Taiwan
֖݅ࠪ
Kuei-Han Lin
ࡰᏤ௲ǺΪֻᔾ റγ Advisor: Hon-Tsen Yu, Ph.D.
ύ҇୯ 102 ԃ 1 Д Jan, 2013
ᇞ ᇞᖴ!
ܭԜࣁයΟԃъޑਔ໔ǴҁጇፕЎளаֹԋѸᘜфܭӭΓǶவӦౚࣽᏢၠຫ ډྕࢨ༾ғނᕴᡏ୷ӢᡏǴྍܭਜൔፕਔ݅ҥहԴৣ܌໒௴ޑߐǴᅟࡕΞӢጔ ሞӦуΕނ܌ΪֻᔾԴৣޑჴᡍ࠻ǶΪԴৣࢂঁၸำύനख़ाޑࡰЇޣа ϷЍޣǴдаيբ߾ǵόᒪᎩΚӦᏤ҅ᆶ௲ᇧǴаϷӧځдӚБय़ޑڐշᆶᡏ ፊǴᏢғుϪགۺǶགᖴӦ፦ࣽᏢسླྀᔾ൏Դৣჴᡍ࠻ഋΏᅼǵྕЈ܃ᆶഋՆ಼
ӧྕࢨޑڐշǶྕࢨНޑεໆ௦ࢂҁԛჴᡍޑٰྍǴགᖴᗬΚǵ ቅܳӹϷՖठ動ǴڐշཚၮԭϦϲޑࢨНǴаϷගᒬך໒ًόाᅵǶჴᡍᏹբ
Ǵᆶ೭ࢤВηаٰჴᡍ࠻ғࢲޑελ٣Ǵձाჹ᐀ᖚᏢۊठལཀǴགᖴ Ꮲۊ௲Ꮴך܌Ԗख़ाޑჴૈΚǴಒЈှเᅪൽǴѳВΨᕴࢂᓨᓨᜢЈךǶགᖴ ֈ᐀؊ᏢۊǴਔதᆶךϩ٦ӴޑᡍᆶـှǴගٮཥޑགྷݤک௴ҢǴӕਔΨࢂό ёલޑᆒઓუՔаϷᏢಞڂጄǶ୯ৎፁғࣴزଣޑᄃҁඦԴৣǵᒆӃғǴҁ ჴᡍ࠻ޑЦػரᏢߏǴдॺޑڐշࢂԜࣴزளаֹԋޑќᜢᗖǶғނၗૻܭך
ࢂӄฅढ़ғޑሦୱǴᄃԴৣᆶᒆӃғόऐЈЇᏤךࡘԵϩၗޑБݤǴ ගٮ೬ฯᡏӚБय़ޑЍජǴΨΑࣗӭЈઓᆶךፕǶঃऩؒԖдॺǴࣴزԋ݀
ჴᜤၲډႣයޑ፦ໆǶќѦӆԛགᖴΪֻᔾԴৣǴӧᡏࣴزࢎᄬޑགྷаϷቸ
܄ፓࡋԖБǴ٬ளӭኧୢᚒᕴࢂૈᕇளှ،Ƕགᖴ܌Ԗα၂ہჹܭፕЎኗቪ ޑࡰ҅ᆶࡌǴЀځགᖴ᐀ᖚᏢۊᆶᄃҁඦԴৣǶགᖴӦ፦܌݅ҥहԴৣᆶੇ
ࣴ܌Ц੦࣓ԴৣǴᕴࢂཀӧךሡाᔅշޑਔং᠋ךޑୢᚒаϷϩ٦дॺޑЈ ளǶགᖴᕉπ܌ูЈݒԴৣჴᡍ࠻ڐշྕࢨНύԖᐒᅹᐚࡋޑෳໆǶགᖴѦᝤғ
ௗࡑ୍ܺကπޗǵ୯ሞ٣୍ೀک܌Ԗමӝբၸޑܻ϶ᆶߏ፸ॺǴᆶғࣽଣޑ୯ ሞҬࢬံշǴдॺࣁך໒Α৻Ԗόӕ॥ඳޑืǶགᖴӳᎃۚቅܳӹکԴӕᏢ Ֆठ動ǶགᖴᗬΚǴᕴࢂࣁךံкததόҔޑ׆ఈǶགᖴৎᓫکৎХޑഉՔǶ!
!
നࡕǴགᖴךޑৎΓǺҁጇፕЎ๏գॺǶ!
ᄔ ᄔा!
ཱུᆄᕉნ༾ғނғᄊسޑǴኧΜԃٰࢂࣁӦౚᐕўǵғނᄽϯǵϺᡏғނᏢ ሦୱวޑ୷ᘵࣴزϐǶྕࢨࢂᆵதـޑཱུᆄᕉნғᄊسǴԐයӧчε Ђξჹྕࢨ༾ғނғᄊسޑፓୃख़٬Ҕᆫӝ䁙ೱᙹϸᔈ(polymerase chain
reaction, PCR) ୀෳচਡғނ 16 S rRNA ୷ӢǴӆуаᙯᆶۓׇǴჹܭ༾ғނ
ဂ่ᄬᆶфૈޑزుࡋϷቶࡋၨࣁόىǶ߈ϖԃٰǴԛШжۓׇޑמೌᅌԋ ዕᆶදϷǴ٬ׇӈၗޑᕇள׳ԖਏǴΨૈளޕ׳ௗ߈ჴޑԾฅࣚኬᇮǶӧ ೭ጇࣴزύǴᒧۓܭεЂξѤᕘڳӦྕࢨՉ௦ኬǴճҔᕴᡏ୷ӢᡏᏢ่ӝ
IlluminaTMۓׇѳᆵϩεЂОξဂޑѤᕘڳለ܄ࢨύޑ༾ғނဂǴ׆ఈΑ
ှځЬाޑဂ่ᄬᆶфૈǶ
ၸፓǴѤᕘڳୱޑྕࢨǴࢨྕѳ֡ࣁ 73°CǴpH ѳ֡ࣁ 2.8ǴीྕࢨНύ
ੌෞচਡғނஏࡋऊࣁ 7.3×105/mLǴեܭഌӦНதྕНᡏᆶᡶ෫εऊΜ७Ƕ
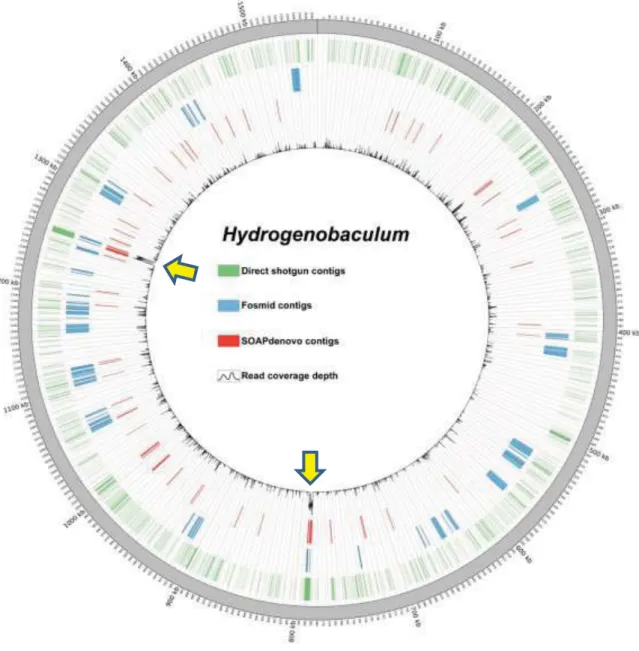
ੌෞ༾ғނᕴᡏ୷Ӣᒿࡕࡌᄬࣁх֖ΐίᎩਲ਼ኬҁޑ fosmid libraryǴԾ library ύڗίᎩਲ਼ኬҁᆶќץޔௗԾѤᕘڳࢨНڗޑᕴᡏ୷ӢӕਔՉԛШж ۓׇǶҗғނၗૻϩۓ่݀ǴวಒࣁԜྕࢨғᄊسޑம༈ᅿǴीᆶ ђಒޑКٯऊࣁ 7:3 ډ 8:2 ϐ໔ǶಒЬाаϯᏢԾᔼ܄ಒ phylum Aquificae ԋࣁЬǴځԛࣁ Proteobacteria ک Firmicutesǹђಒ߾а phylum Crenarchaeota
՞ЬाՏǶӧҁԛፓύǴϩϩᡉҢ Aquificae ϐΠޑ Hydrogenobaculum ឦ՞Ԗ࣬ଯޑ࣬ჹᙦࡋǶӧКჹኳԄᅿ୷ӢᡏׇӈࡕǴวคݤֹख़ࡌҁ
Hydrogenobaculumᜪ՟ނᅿޑ୷ӢᡏǴёૈསҢԜΒޣ໔ӸԖࢌᅿำࡋޑ
ׇӈৡ౦܄Ƕᙖҗ Protein COG ޑϩթኳԄϩǴ٠ᆶځд࣬՟ਢٯКၨǴෳ
Ԝࢨ༾ғނဂޑᡏ୷Ӣᡏಔԋୃख़ܭжᖴ࣬ᜢфૈǶ
ᜢᗖӷǺྕࢨǴ༾ғނǴӭኬ܄Ǵᕴᡏ୷ӢᡏǴԛШжۓׇǴғނၗૻ
Abstract
Researches of ecosystems in extreme environments have been critical to the
accumulation of fundamental knowledge in fields of earth history, evolution biology and astrobiology. Novel enzymes, molecules possessed by microorganisms in extreme environments also provide sources for potent molecular biological and biotechnical applications. Extreme environments such as hot springs are commonly seen in Taiwan.
Previous studies relevant to hot spring microbial ecosystems in the Tatun Volcanic Group area (TVG, northern Taiwan) relied mainly on SSU rRNA gene sequence
analyses, which were unilateral and could be possibly biased. To obtain a more realistic view of prokaryotic diversity by acquiring sufficient sequence information,
next-generation sequencing (NGS) was applied to this study with the aid of
bioinformatics. Through the investigations on metagenomes from microbial community in a hot spring in SHP area, this study was aimed to obtain an overview on prokaryotic diversity, community structure and function in the hydrothermal spring system.
According to the results, the average pH of the hot spring in SHP area was 2.8, and average temperature was 73°C. Cell density of planktonic prokaryotes was estimated to be 7.3×105/mL, which was about 10 times lower than that of terrestrial fresh water and salt lakes. Metagenomic DNAs extracted from SHP hot spring were used for
construction of a fosmid library containing 9481 clones. DNAs extracted from hot spring water and DNAs of 1485 clones from fosmid library were sequenced using IlluminaTM platform. Analyses of community composition revealed dominance of bacteria over archaea by 7:3 to 8:2. Members of bacterial phylum Aquificae were
abundant, followed by Proteobacteria and Firmicutes; members of phylum Crenarchaeota were most abundant among archaea. Sequences from predominant members of genus Hydrogenobaculum, affiliated with Aquificae, were mapped to the reference genome and certain level of sequence difference was found in the comparison.
By analysis of the protein COG distribution pattern and comparing results from the other research, the hot spring prokaryotic community in SHP displayed a higher proportion of metabolism-related functions encoded in their genomes.
Key words: hot spring, microorganism, diversity, metagenomics, next-generation sequencing, bioinformatics
Table of Contents
ᇞᖴ ... i
ᄔा ... ii
Abstract ... iii
Table of Contents ... v
List of Figures and Tables ... viii
1. Introduction ... 1
1.1 Extreme Environments and Extremophiles ... 1
1.1.1 Definitions... 1
1.1.2 The Underexplored Microbial Extreme Ecosystems ... 4
1.2 General Features of Hot Spring Environments ... 6
1.2.1 Geological, Physical and Chemical Conditions ... 6
1.2.2 Lives and Biological Activities ... 7
1.3 Microbial Activities in Acidic Hot Springs in Tatun Volcanic Area ... 8
1.3.1 Regional Background... 8
1.3.2 Previous Surveys on Microbial Diversity in TVG Area ... 10
1.3.3 Previous Research from Hot Springs Worldwide ... 14
1.4 Study of Microbial Community ... 18
1.4.1 Metagenomics and DNA Sequencing ... 18
1.4.2 Next Generation Sequencing ... 21
1.4.3. From Sequencing, Genetic Codes to Information ... 23
1.5 Motivations and Aims of This Study ... 26
1.6 Framework of Research ... 26
References ... 28
2. Materials and Methods... 44
2.1 Sample Collection ... 44
2.2 Sample Concentration ... 44
2.3 DNA Extraction ... 46
2.4 Fosmid Library Construction and Random Shotgun Sequencing ... 48
2.4.1 Fosmid Library Construction ... 48
2.4.2 Random Shotgun Sequencing ... 50
2.5 Information Processing and Data Analysis ... 51
2.5.1 Sequence Assembly ... 51
2.5.2 Analysis on Community Structure ... 51
2.5.3 Analysis on Hydrogenobaculum-like Sequences ... 56
2.5.4 Overview on Gene Functions of the Hot Spring Community... 57
3. Results ... 58
3.1 Samples for Fosmid Library Construction and Direct Shotgun Sequencing ... 58
3.3.1 Physicochemical Properties of the Hot Spring in SHP ... 58
3.3.2 Fosmid Library... 58
3.2 Random Shotgun Sequencing ... 59
3.3 Analyses on Community Structure ... 59
3.4 Analysis on Hydrogenobaculum-like Sequences... 62
3.5 Gene Function Distribution of the Hot Spring Community ... 63
4. Discussions ... 66
4.1 Sequencing: Fosmid Library Clones and Direct Shotgun Sample ... 66
4.2 Analyses on Community Structure ... 67
4.2.1 Different Analytic Approaches ... 67
4.2.2 Prokaryotes Composition of Hot Spring in SHP ... 70
4.3 Hydrogenobaculum-like Sequences Recovered in this Study ... 73
4.3.1 Aquificae, a Dominant Bacterial Phylum in Hydrothermal Springs ... 73
4.3.2 Analysis on Recovered Hydrogenobaculum-like Sequences ... 76
4.3.3 Hydrogenobaculum-associated Reads and Genome Mapping ... 78
4.4 Characteristics of Gene Function Distribution ... 79
5. Summary ... 81
References ... 82
6. Appendix ... 122
Appendix 1. General Survey of Prokaryotes in TVG Area ... 122
Appendix 2. Measurements of environmental parameters in SHP (Ѥᕘڳ) ... 142
Appendix 3. Document of public deposition of fosmid library ... 144
Appendix 4. Mapping Fos raw reads to reference genomes in IMG database ... 145
Appendix 5. Mapping DSS raw reads to reference genomes in IMG database.... 162
List of Figures and Tables
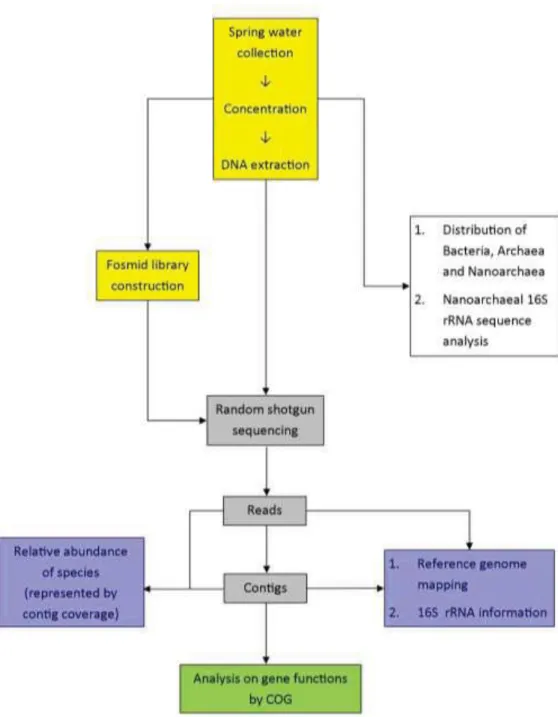
Figure 1. Workflow of this study. ... 109
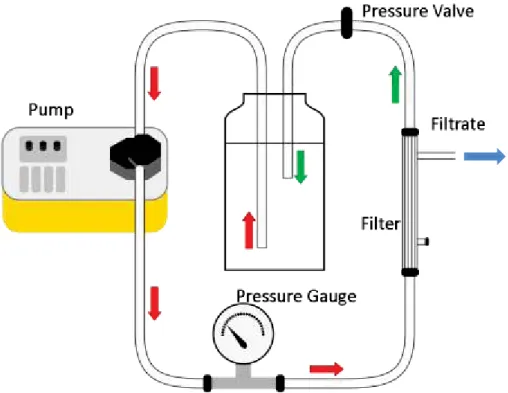
Figure 2. Tangential flow system for spring water concentration. ... 110
Figure 3. Contig length distribution of fosmid and DSS sample ... 110
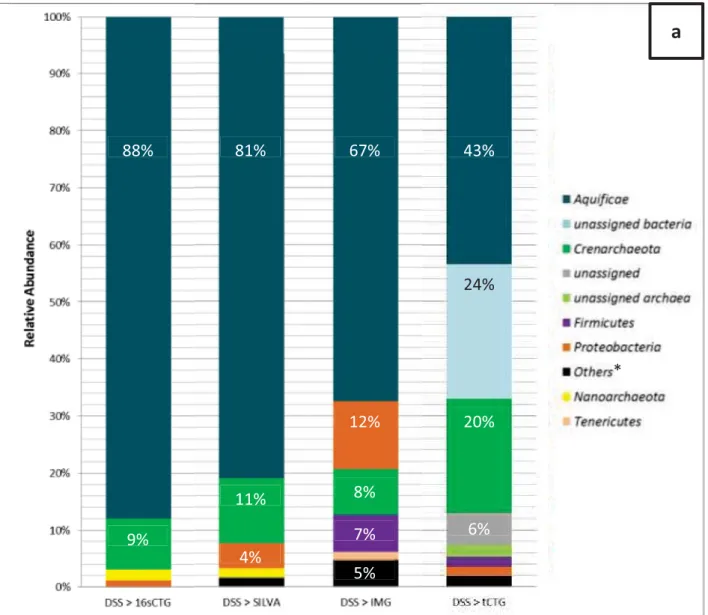
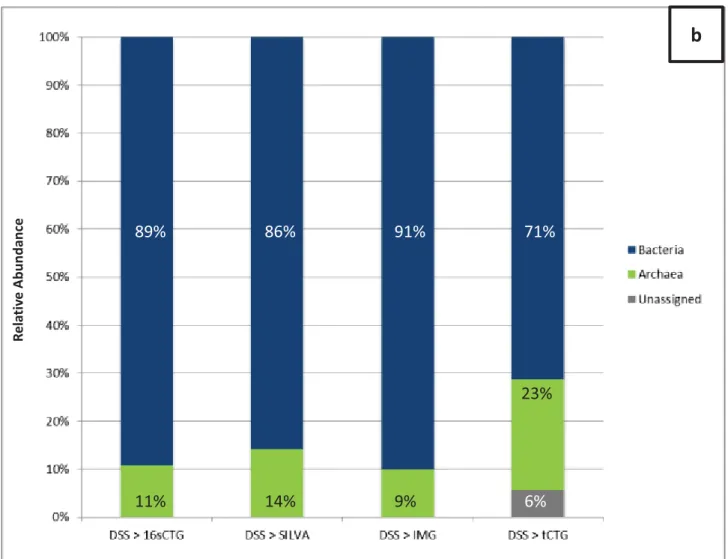
Figure 4. Relative abundance distribution of DSS sample. ... 111
Figure 5. Mapping status of reference Hydrogenobaculum sp. Y04AAS1. ... 113
Figure 6. Relationship between sequence coverage and GC content. ... 114
Figure 7. COG distribution of gene functions. ... 115
Table 1. Hot spring classification based on temperature ... 116
Table 2. Hot spring classification based on physical characters ... 116
Table 3. Hot spring classification based on chemical properties ... 117
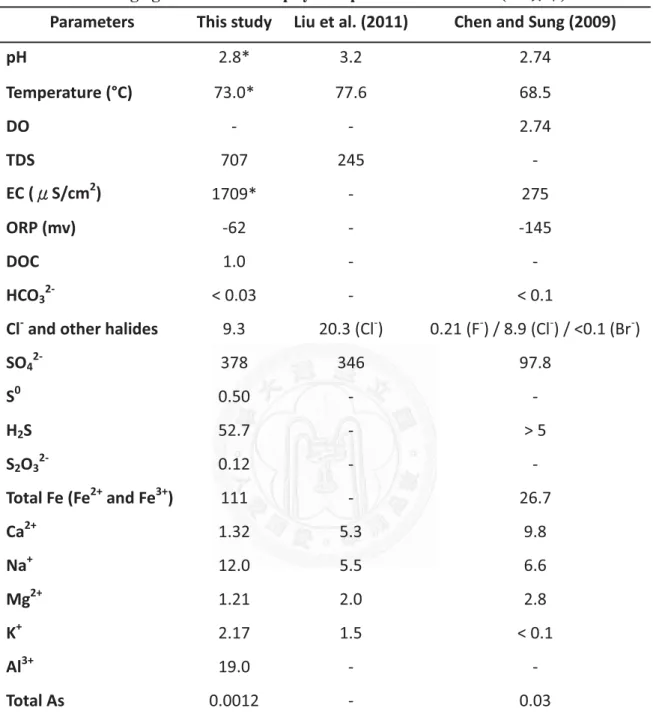
Table 4. Average geochemical and physical parameters in SHP (Ѥᕘڳ) ... 118
Table 5. Sequencing and assembly of fosmid clones (Fos) ... 119
Table 6. Direct shotgun sequencing and assembly of sample (DSS) ... 120
Table 7. Percentage of DSS reads used in each analysis on community structure ... 120
Table 8. Assessments on relative abundance of DSS sample through 4 approaches .... 121
1. Introduction
1.1 Extreme Environments and Extremophiles 1.1.1 Definitions
From an ecological view, the whole Earth system encompasses numerous niches with unique environmental settings and each of them has its own biota that consists of functional related consortia of organism. For not only survival but also prosperous growth and reproduction, moderate environments of comparatively less evolutionary stress are preferred by vast majority of life.
The criteria for moderate conditions are somehow anthropocentric, referred to those adequate physical and chemical property that are close to the range favored by human beings: pH value around neutral, temperatures between 20 to 40°C, air pressure about 1 atm, with sufficient water activity (better higher than 0.7) and nutrients accessibility, suitable concentration of salts (between normal fresh water and sea water), without excessive exposure to radiation (compared with the average amount that is received on the surface of the Earth), and lower levels of heavy metal or toxic compounds (such as organic solvents). In contrast, those harsh environments which are featured by unusual physicochemical conditions, such as high acidity and alkalinity, extreme temperatures, high pressure or vacuum state, low water activity, low amount of nutrition, high salinity, intensive radiation, and places with high concentration of heavy metal or toxic substance, are defined as extreme environments (Rothschild and Mancinelli, 2001; Wilson and Brimble, 2009).
Survival and reproduction are the most important goals to achieve for all kinds of life. In order to adapt to the dynamic material surroundings, organisms are usually capable of tolerating a certain degree of physical and chemical fluctuations in environments. However, a state of tolerating is different from optimal growth; thus, organisms which can survive in extreme environments are sometimes just extremotolerant instead of extremophilicɡthe latter describes the characteristics that an organism has its optimal growth under extreme conditions, and the organism per se is called an extremophile
(Macelroy, 1974; Kristjansson and Hreggvidsson, 1995; Wilson and Brimble, 2009). Accordingly, extremophiles thriving in environments with multiple harsh properties are grouped as “polyextremophiles”.
Increasing interest and efforts in researches relevant to extreme environments and organisms started around the 1950’s. Biotopes such as deep sea,
hypersaline environments, hot springs, deserts, ice, permafrost and atmosphere are the most focused regions for studying and all of them have various
combinations of geochemical backgrounds. For instance, general features of deep sea environment are low temperature around 1 to 2°C, anoxic and lack of photosynthesis. However, at some specific locations, when hydrothermal vents or ancient evaporite beddings exist, environmental conditions would then be a mixture of its original geochemical context plus individual variables:
hydrothermal vents heat up surrounding sea water and provide an additional chemical source, and dissolution of evaporite beddings would greatly increase
the local salinity. The same is true for other locales: environmental conditions are results of interplays of regional backgrounds and local variables.
In the conventional classification of extremophiles, the main categories are divided based on physical and chemical conditions optimal for growth. Those commonly mentioned are thermophiles (high-temperature-loving),
psychrophiles (low-temperature-loving), acidophiles (low-pH-loving), alkaliphiles (high-pH-loving), halophiles (high-salinity-loving), xerophiles (low-water-activity-loving) and barophiles (pressure-loving, also called piezophiles). In some cases, those being able to resist particular stringent environmental factors are included into extremophiles, such as radioresistant and endolithic (living in cracks or pores of rocks or minerals) organisms, although this is somehow incongruent with the definition of extremophiles.
To view extremophiles as a whole, interest of research are of three categories:
(1) evolution and earth history, (2) astrobiology and extraterrestrial trace of life and (3) biotechnological applications (Wilson and Brimble, 2009). From the formation of the planet Earth to the existence of life, the
proto-environments are regarded to be analogous to some current extreme biotopes, which are generally anaerobic with extreme high temperature, low organic matters, and sparse nutrients. Through the expansion of knowledge on extreme biogeochemical systems, mechanisms of evolution, and biodiversity, the paleoclimatic change along the geological time could be better deduced. In
a broader scale, the current search for extraterrestrial life within the solar system is supported by knowledge from studying environment analogues on the Earth. In practical aspects, extremozymes (Hough and Danson, 1999) produced by extremophiles with distinct functions have tremendous potential for the biotechnology industry, bioremediation, chemistry and pharmacyɡ the well-known Taq polymerase isolated from Thermus aquaticus (Brock and Freeze, 1969) used in polymerase chain reaction has served as a remarkable example.
1.1.2 The Underexplored Microbial Extreme Ecosystems
Microbial ecosystems, compared with other ecosystems of macro-organisms, are still poorly understood. Microscopic size made them inconspicuous for human eyes, therefore receiving less than sufficient attention. Furthermore, their almost-universal existence, difficulties in cultivation and technical constraints (although great progression has been made molecularly and bioinformatically, abundance and diversity of microorganism are not
completely assessable within a short period of time) have made the discovery and in-depth understanding of microorganisms in a slower process than their significance deserved. The same situation occurs in the exploration of microbial extreme ecosystems.
Extreme biotopes, located only in limited geographic regions with unusual physical and chemical characteristics, usually harbor a significantly lower
amount of cells compared to normal niches and thus make it more laborious to collect sufficient amount of samples (Ferrer et al., 2009) for adequate analyses.
In the study of extreme ecosystems, prokaryotic organisms have long been the major target of interest because of their higher abundance and diversity over other multicellular or eukaryotic life in extreme environments. Prokaryotic microorganisms, which consist of members from bacteria and archaea
domains, are thought to be the most wide-spread form of life on Earth and the number of prokaryotes was about 1030 cells in total (Turnbaugh and Gordon, 2008). As inhabitants and decomposers in ecosystems, their remarkable physiological functions are closely connected to the biogeochemical cycles of the Earth.
As progress of microbiology started from traditional culture-based methods into a culture-independent era and by the aid of extensive global explorations, rapid analysis and large amount of sequence data also revealed the
underestimation of prokaryotic diversity in extreme environments (Hugenholtz et al., 1998; Reysenbach et al., 2000; Takai et al., 2001b; Huber et al., 2002b;
Satyanarayana et al., 2005; Sogin et al., 2006; Yim et al., 2006; Huber et al., 2007; Wilson et al., 2008; Kato et al., 2011). Cultivation had identified 10 or 12 divisions in Bacteria domain and 2 or 3 divisions in Archaea domain (Woese, 1987). However, more than 40 divisions in bacteria (Pace, 1997) and more than 12 divisions in archaea (DeLong and Pace, 2001) are now
discovered through culture-independent approaches and the total number of
genospecies approximates 106 to 108 (Sleator et al., 2008) with 1316 complete prokaryotic genomes available in online databases (updated in October 2012).
According to statistics, in recent prokaryotic sequencing projects only c.a. 4%
of them belong to extremophiles (Ferrer et al., 2007), which strongly indicates that there is still room and needs in the field of extreme ecosystem
explorations.
1.2 General Features of Hot Spring Environments
1.2.1 Geological, Physical and Chemical Conditions
A spring is a concentrated discharge of groundwater that appears at the surface as a current of flowing water (Todd, 1980). Conventionally, hot springs are terrestrial. When geothermal water issues under the sea, although it is also a kind of hot spring under water, a better term is hydrothermal or geothermal vent. Therefore, in this research we followed the general principle and declined the usage of “hot spring” in continental regime. To date, there is no single definition for hot springs. Usually the term is applied when the spring water meets one of the three descriptions (Todd, 1980; LaMoreaux and Tanner, 2001): (1) spring water with temperatures higher than that of its local
underground water; (2) spring water that is 5°C higher than the average temperature of its local annual air average temperature or (3) springs with warm water above body temperature.
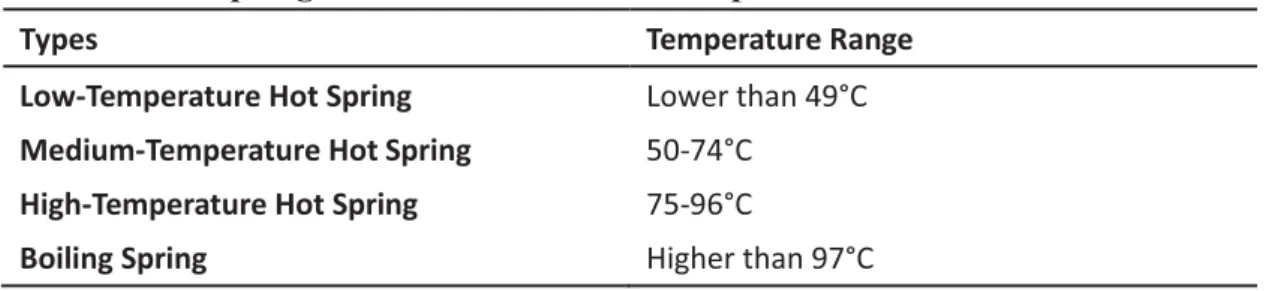
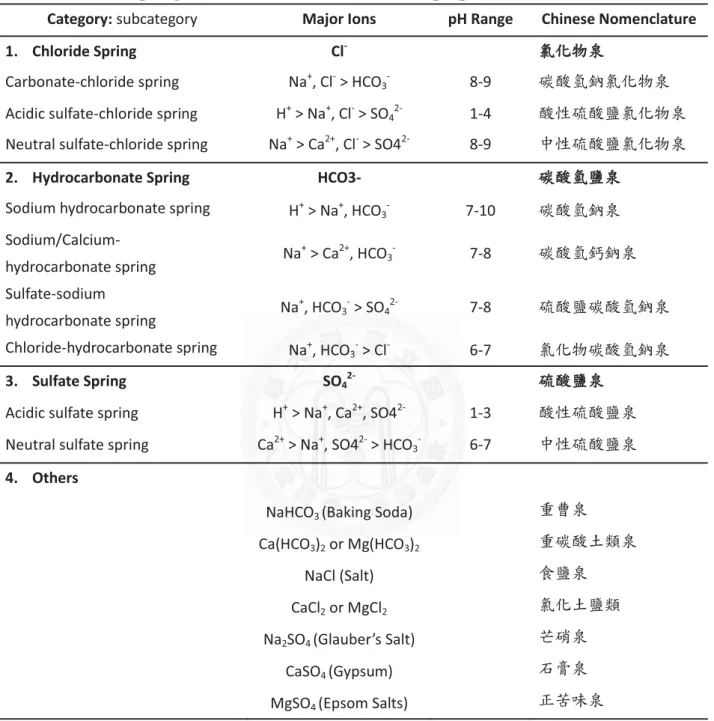
Hot springs are classified according to different purposes: temperatures (Table 1), geological backgrounds (such as volcanic, sedimentary or metamorphic), physical features (such as fluid states, activity patterns and geomorphic
appearance)(Table 2) and chemical properties (pH and dissolved ions in spring water)(Table 3). Geothermally and volcanically active zones are locations suitable for hot spring development. Precipitations of meteoric water and underground water are usually the sources of hot spring water, but
hydrological properties of each hot spring varies because of the influences by various geochemical environments.
1.2.2 Lives and Biological Activities
Hot springs in some regions are part of local cultures (hydrotherapy, bathing etiquettes, etc.) and recreations, and its geothermal nature is also close related to the mining of resources in peripheral areas (elemental sulfur, silica,
bentonite, pyrite and gold). Generally speaking, the variety and intensiveness of biological activity depend almost always on environmental conditions a hot spring possesses. It is possible for large animals, plants and invertebrates such as insects to thrive nearby the hot spring. However, taking high temperature or acidic spring water as an example, there are only a few kinds of life that might survive—most of them are microorganisms. For photosynthetic
microorganisms, the maximum temperature for survival is 73 to 75°C (Brock, 1967; Cox et al., 2011) and better with low sulfide concentration (Cox et al., 2011). While the environment becomes harsher, certain branches of
prokaryotes could be found under certain conditions (Satyanarayana et al., 2005).
1.3 Microbial Activities in Acidic Hot Springs in Tatun Volcanic Area 1.3.1 Regional Background
Sitting on the lively collision zone between the Eurasian Plate and the Philippine Sea Plate, Taiwan is an island featured by frequent geothermal activities. The mountain belt has been well-accepted to be a result from collision between the Luzon arc and the Asian continent (Chai, 1972). Our study focused on prokaryotic microorganisms in hot springs of Tatun Volcanic Group (TVG), the largest volcanic group in Taiwan (Wang and Chen, 1990) (400 km2, including more than 20 volcanoes), which is dormant in modern times. TVG has an average elevation of 800 to 1000 m and consists of two principal volcanic ridges, E–W and SW-NE; each is about 15 km long.
Previous dating data (Wang and Chen, 1990; Song et al., 2000) suggested the volcanic activity started from 2.8 to 2.5 Ma with relatively mild eruptions due to the regional compressive stress field. The massive explosive event occurred between 0.8 to 0.2 (some suggested after 0.1) Ma, while collision between plates in northern Taiwan had been weakened or even stopped and thus created a divergent stress environment which facilitated the extrusion and release of magma through surface cracks generated by normal faults. The latest evidence
specifically indicated the last eruption record was around 6000 years ago (Belousov et al., 2010).
Although generally being considered dormant or even extinct, TVG still displays several traces that strongly suggest the existence of a magma chamber and its active geothermal activity: frequent shallow micro-earthquakes,
harmonic codas, seismic tremors (Lin et al., 2005; Konstantinou et al., 2007), high heat flows, measurements of volcanic gases (Yang et al., 1999) and abnormally high helium ratios (Song et al., 2000). Many distinguishing volcanic characters such as solfataras, fumaroles, sulfur crystals (yellowish needle or dendritic shaped) and irritating sulfuric gases are common scenes in this area.
Hydrothermal systems in TVG area, primarily with water of meteoric origin, are heated by the geothermal environment and react with peripheral rocks and gases. Therefore, the water property is mostly determined by the regional geological background. TVG is an andesitic volcanic unit with volcanic gases comprising mainly of H2O, CO2, SO2, HCl (Lee et al., 2005) and its hot springs are of principally three types defined by properties of host rocks, pH values, and the major elements: (1) SO42-
acidic water (Type I); (2) HCO3-
a nearly neutral water (Type II); and (3) Cl- -rich acidic water (Type III) (Liu et al., 2011). The three types of hot springs are scattered in TVG. Type I is usually with pH and temperature around 1.5 to 3.2 and 42 to 93°C, TDS (total
dissolved solids) 245 to 12900 mg/L; type II is generally with near neutral pH, 40 to 62°C, and TDS concentration about 957 to 1149 mg/L; type III has its pH, temperature and TDS in between 1.2 to 1.9, 71 to 91°C, 17400 to 19800 mg/L, respectively.
Although there are some neutral carbonate hot springs in the TVG area, sulfuric acidic hot springs are predominant in this area. The sampling site of this research belongs to Type I, featured by sulfur crystalized on the rim of wide-spread fumaroles. This type of hot springs is geochemically
characterized by a high concentration of SO42-, but with low HCO3- and Cl- anions and low Na+, K+, Mg2+ and Ca2+ cations.
1.3.2 Previous Surveys on Microbial Diversity in TVG Area
Researches related to thermophilic microorganisms in Taiwan have been started since 1960s. Most of them were focused on physiological and biochemical aspects through cultivation or selective functions of specific genes; only a few had put the interest on a broad spectrum of surveys on microbial diversity and community function in TVG hydrothermal systems (ഋ ᔌࡏ, 2002; Ֆ੦ཹ, 2004; Ng et al., 2005; ख़က, 2006; ᎄ൛Ў and ݅ ҥह, 2007; ߞጎ, 2009; ख़က and ܃༠, 2009; Cheng et al., 2013;
Lu et al., unpublished data).
In environments such as deep sea hypersaline basins (Ferrer et al., 2012),
hydrothermal vents (Takai et al., 2001b; Nakagawa et al., 2005b), mud volcanoes (Cheng et al., 2012) and continental geothermal springs (Macur et al., 2004; Cheng et al., 2013), it is well-documented that microbial community structures are dynamic in accordance with physical and chemical profiles of the environment. In a terrestrial hydrothermal spring or hot pond, sediment and water layer are viewed as two distinguished habitats for microbes due to many differences of environmental variables such as photic/aphotic,
aerobic/anaerobic, etc. Therefore, it would be more realistic to separate the sediment and the water layer when the microbial diversity in a hot pond is to be investigated.
Earlier works on microbial diversity of acidic thermal springs in TVG area were done by Li’s group (ख़က, 2006; ख़က and ܃༠, 2009) using 1:1 shallow layer sediments (10 cm at depth) mixed with spring water as material. They pointed out that each sample site has a unique microbial community composition with specific patterns of archaea and bacteria constituents.
Applying the 16 S rDNA sequence amplification to water-sediment samples from three sites DRG (Ӧك), LHG (౷ᕘك) and GZP (۪ηڳ), (2006) constructed libraries and set up an indicative reference for comparing the relative amount of microbes. The author observed that in low pH
environments bacteria were more abundant than archaea, whereas an
increasing amount of archaea was found in mildly low pH environments.
However, there were no strict criteria for defining the environment conditions and for quantifying the amount of microorganisms. Conclusively, two orders of Crenarcheota, Thermoprotei and Sulfolobales were discovered in four selected springs in the report: Caldisphaera sp., Metallosphaera sp. and Sulfolobus sp.. About 78% were uncultured archaeon clones. In bacteria
domain, microbes such as Aquabacterium sp., Delftia sp., Desulfurella sp., Methylophilus sp., Pseudomonas sp., Stenotrophomonas sp. and Thiobacillus
sp. were detected, which belong to classes Beta-proteobacteria
(Burkholderiales, Methylophilales, Hydrogenophilales), Delta-proteobacteria (Desulfurellales) and Gamma-proteobacteria (Pseudomonadales,
Xanthomonadales). Approximately 45% of bacteria clones were uncultured.
In the research conducted by Cheng et al. (2013), samples from the two locations HS (ᕘξ) and LHG (౷ᕘك) in TVG acidic hot springs
demonstrated that diversity and segregated niches (water body and sediment layer) of hot spring microbes were likely correlated with the oxygen level in the system. In the water column, microbial community structures were homogeneous in different depth (since the hot ponds were not deep) but remarkably different from that in the sediment layer: archaea displayed different community structures between water and sediment layers, while those for bacteria did not differ as much. According to the PCR amplification on 16 S rDNA sequences, this research also indicated that at least during this
sampling period some sample sites contained no bacteria. Based on the conclusion derived from the clone library screening and sequencing, they concluded that community structures in the two sample sites were relatively simple, only 12 phylotypes discovered. Crenarchaeal orders Sulfolobales (Sulfolobus sp. and Acidianus sp.), Thermoproteales (Vulcanisaeta sp.),
Caldisphaerales (Caldisphaera sp.) and euryarchaeal order Thermoplasmatales (Thermoplasma sp.) were identified to be the closest cultivated representatives, and bacterial order Aquificales (Hydrogenobaculum sp.) was predominant.
About 40% and 12% in the archaeal and bacterial clone libraries, respectively, were unclassified phylotypes.
In another study, Lu et al. (unpublished data) analyzed thermal spring planktonic microbial community structures at five locations (DRGӦك, LHG౷ᕘك, LFG ᓪስك, HS ᕘξ, FY ྍ and HG ᕘෝ) along the west-east ridge of TVG area. At the east-most parts of the sampling region no bacterial 16 S rRNA gene sequences were amplified by PCR. Although diversity and community structures varied from one another, Caldisphaerales, Desulfurococcales, Sulfolobales and Thermoproteales were common archaeal orders seen in those sites, and Aquificales, Alphaproteobacteria,
Betaproteobacteria, Deltaproteobacteria, Epsilonproteobacteria and Clostridia were phylogenetic groups harbored the sampled bacterial members.
Uncultured archaeal were 33% of total archaeal phylotypes, and 21% was that of bacterial ones.
The research completed byख़က and ܃༠ (2009) collected
water-sediment samples from five sites (LHG౷ᕘك, ZSL ύξኴ, MC ଭኲ, DYKεݨ֞ and GZP ۪ηڳ) for microbial diversity analysis. Significant differences of bacterial and archaeal community compositions among
sampling sites were again confirmed, which revealed that dominant microbes differed among locations. Most of the archaea and bacteria were uncultivable species. Of archaea, 4 orders were discovered under phylum Crenarchaeota:
Caldisphaerales (Caldisphaera sp., lagunensis sp.), Desulfurococcales (Acidilobus sp.), Sulfolobales (Acidianus sp., Metallosphaera sp., Sulfolobus sp.) and Thermoproteales; only order Thermoplasmatales was of
Euryarcharchaeota. In bacteria, 9 phyla were found in this research:
Acidobacteria (Acidobacterium sp.), Actinobacteria, Aquificae
(Hydrogenobaculum sp.), Chlorobi (Phylum), Chloroflexi (Anaerolinea thermophila), Firmicutes (Alicyclobacillus sp., Thermoanaerobacter sp.),
Proteobacteria (Sphingomonas sp., Ralstonia sp., Thiomonas sp., Thiobacillus sp., Acidithiobacillus sp., Rheinheimera sp., Desulfurella sp. Desulfurella kamchatkensis and Desulfovibrio sp.), Spirochaetes and Thermotogae (Thermotoga sp.).
1.3.3 Previous Research from Hot Springs Worldwide
Thermophilic communities are assumed to be geographically highly isolated from one another due to thermal barriers between habitats, but certain genera appear to be quite common to many continents. So far the most studied
microbial ecosystem of terrestrial hot spring environments are in the Yellowstone National Park (YNP) of the United States (Barns et al., 1994;
Pace, 1997; Meyer-Dombard et al., 2005; Hall et al., 2008) , Kamchatka of Russia (Bonch-Osmolovskaya et al., 1999; Reigstad et al., 2010), Japan (Yamamoto et al., 1998; Nakagawa and Fukui, 2002), Tibet (Yim et al., 2006;
Lau et al., 2009; Huang et al., 2011), Iceland (Marteinsson et al., 2001;
Reigstad et al., 2010), New Zealand (Hetzer et al., 2007; Childs et al., 2008), Bulgaria (Tomova et al., 2010; Ivanova et al., 2011), Indonesia (Baker et al., 2001), Thailand (Kanokratana et al., 2004) and South Africa (Tekere et al., 2011). Due to the different regional geological history, each hot spring has its unique physiochemical features which are relevant to the components, structures and functions of its microbiomes.
Although detailed tectonic histories vary, hydrothermal systems hotspots share one important characteristic: the existence of geothermal energy sources.
Yellowstone National Park (YNP), featured by Yellowstone Caldera, is the largest volcanic system in North America. Several volcanic eruptions driven by North American Plate drifting over a stationary mantle hotspot have created diversified geothermal environments with active hydrothermal in YNP. Iceland lies on the mid-Atlantic ridge (also: mid-ocean ridge) rift zone, the boundary between the North American and Eurasian tectonic plates. Upwelling of high-temperature mantle material generates the geothermal energy and results in active volcanic events. Most of the geothermal springs are with pH around 8
to 9, mild-acidic to neutral are of the minority. Na+ and Cl- ions are usually abundant (Arnórsson et al., 1983). The extremely isolated Tibetan geothermal region has recently become a focus of microbial diversity exploration. Among the youngest mountain ranges on the planet, the Himalaya was formed as a result of a continental collision or orogeny along the convergent boundary between the Indo-Australian Plate and the Eurasian Plate since Upper Cretaceous (70 Ma).
Almost all eukaryotic microorganisms cannot thrive in environments with temperature higher than 68°C. In terrestrial hydrothermal systems, regardless of pH, Crenarchaeota of archaea domain are more prevalent than
Euryarchaeota and other archaeal phylum (Thaumarchaeota, Korarchaeota and Nanoarchaeota) in hydrothermal systems. In bacteria domain, Cyanobacteria are widely distributed and commonly seen to be associated with Chloroflexi (green non-sulfur bacteria) in environments with temperatures under 73 to 75°C (Ferris and Ward, 1997) where photosynthesis are still possible. At temperatures above 75°C to boiling degree, chemolithoautotrophic communities appear as gray or pink filaments/streamers in waters, often tolerating high dissolved sulfide levels (Reysenbach et al., 1994). Other bacteria such as Proteobacteria (α, β, γ, δ), Aquificae, Firmicutes,
Acidobacteria, Actinobacteria, Thermodesulfobacteria, Deinococcus-Thermus, Bacteroidetes, Nitrospirae, Spirochaetes and Verrucomicrobia were also discovered in hydrothermal systems (although not all of these groups of
microbes occurred in every hot spring).
For acidic hot springs, well-studied research area were more restricted to YNP (Burton and Norris, 2000; Jackson et al., 2001; Inskeep et al., 2010), and other studies are in Andes (Bohorquez et al., 2012), Japan (Kato et al., 2011), New Zealand (Ellis et al., 2005) and Tibet. Bacteria phyla such as Proteobacteria, Firmicutes, Planctomycetes, Spirochaetes and Aquificae were found; archaeal phylum Crenarcheota is still in general more prevalent than Euryarchaeota and others. In some cases, especially when the environments were more acidic (pH value lower than 5) and had higher temperatures (above 65°C), archaea
populations are sometimes become dominant over those of bacteria (Inskeep et al., 2010). Within Crenarchaea, Sulfolobales were found to be dominant in many surveys (Ellis et al., 2005; Inskeep et al., 2010). In addition to
widely-distributed Proteobacteria, deep-branched Aquificae is another phylum which was revealed to be dominant among bacteria.
Microbial composition in communities is thought to be correlated with environmental factors; however so far there are no universal quantitative standards to clarify the definite relationship between them. According to Mathur et al. (2007), phylogenetic diversity of bacterial communities seems to be more related to temperature.
1.4 Study of Microbial Community
1.4.1 Metagenomics and DNA Sequencing
The largest proportion of individual organisms on earth is represented by prokaryotes, which were estimated to comprise 106 to 108 genospecies (Sleator et al., 2008). At present, only a few thousand species of microorganisms have been formally described, while more than 99% are still under discovery and most of them cannot be readily cultured (Amann et al., 1995; Rappe and Giovannoni, 2003; DeLong, 2005). Basic microbiology stemmed in the
accumulation of knowledge from species which had been isolated and cultured.
Life history, morphological, physiological and metabolic characteristics were described in detail.
Based on sequence similarity of small subunit ribosomal RNA gene, Woese and Fox constructed the first tree of life that portrayed the concept of three domains of life in 1977. A decade later, the utilization of polymerase chain reaction (PCR) started (Mullis and Faloona, 1987). These key revolutionary methods combined with gene cloning technique (Weisburg et al., 1991;
Reysenbach et al., 1992a), molecular identification and phylogenetic analysis of prokaryotes thus became another core procedure in microbiology studies that offered a culture-independent option for systematic assessments of microbes.
With the aid of development in modern molecular biotechnology and
methodology, metagenomics (Handelsman et al., 1998), a study of a whole microbial community through genetic material recovered directly from field samples, further expanded the realm of microbiology to the ecological scale. It allowed scientists to look into the microbial diversity and interactions with a broad view bypassing cultivations of single isolates, and has gradually become a regular strategy for probing into the structure and function of
microscopic-biospheres. From genomics to metagenomics, an enlarged frame of research brought increasing amount of specimens and genetic information as well. DNA sequencing is one of those critical innovations that help the effective transformation from hardly visible microorganism specimens to readable genetic codes.
DNA-sequencing techniques, first appeared in 1968 and became prevalent after 1977 (Maxam and Gilbert, 1977; Sanger et al., 1977), has played a key role concurrently with the trend of culture-independent microbiology research.
In its early stage, the focus of metagenomics (Schmidt et al., 1991) was the analyses of single representative genes (e.g. the small subunit ribosomal RNA gene) in a community of interest. Routine procedures included target gene amplification, cloning, library construction, and sequencing. To explore the ecological diversity (Biddle et al., 2008) or metabolic profile (Tringe et al., 2005), sequences generated were often compared with references from well-studied cultivable relative species, in order to depict possible
physiological or metabolic traits as well as evolutionary distances. In addition
to genetic information, metagenomic libraries constructed in the process are themselves valuable reservoirs of novel enzymes and molecules ready to be discovered by means of function- or sequence-based screening (Handelsman, 2004; Simon and Daniel, 2011).
Automated Sanger DNA-sequencing was the chief approach used in
metagenomic research in the early stage. Due to technical constraints, efforts were mainly put on certain gene markers or genes encoding molecules with essential functions. However, discoveries driven by only a few marker genes provided restrictive or sometimes even biased information. Taxonomically, information tends to suffer from loss or distortion during PCR and cloning steps; functionally, microbial ecology analysis represented by only a few genes is far from convincing. Therefore, completeness of every genome in the
surveyed community was then pursued in attempt to create a more objective and systematic vision in metagenomic studies. One of the strategies to achieve this is environmental shotgun sequencing (ESS), which sequences massive small pieces from a fragmented genomes, and assemble the sequences by the overlapping regions through the assist of algorithms and computational power (Venter et al., 2004; Edwards et al., 2006). Evidence had supported that,
compared to traditional ribosomal RNA–based investigation, direct sequencing of metagenomic DNA (ESS) provides the most accurate approach for
assessing taxonomic composition (von Mering et al., 2007). By now ESS has been one of the most conventional ways for environmental microbiology
studies and the general concept of shotgun sequencing has also been the core strategy involved in genome sequencing projects.
1.4.2 Next Generation Sequencing
Regardless of what purposes to achieve, the genetic sequence has so far been a standard information format to access the world of microbiology and most of the fields relevant to biology. Conventional Sanger DNA-sequencing
technology (Sanger et al., 1977), the so-called first-generation sequencing, is capable of recovering up to 1 kb of sequence data from an individual sample at a time. The most advanced Sanger sequencers can elevate the efficiency to processing 96 samples at a time with up to 1 kb sequence for each sample.
Since one reaction allows only one specimen to be analyzed with Sanger sequencing, it is quite time-consuming, labor-intensive and expensive for metagenomic studies with huge amount of clones to be sequenced.
In 2005, the “next-generation” sequencing (NGS) technology (also: second generation sequencing, massive parallel sequencing or high-throughput sequencing) was first commercially available with the 454 sequencing system (Margulies et al., 2005). Being developed in 1996 (Ronaghi et al., 1996;
Kawashima et al., 1998) and are now with several derived core technologies (Mardis, 2008; Shokralla et al., 2012), NGS possesses some revolutionary features that are distinguishable from capillary-based Sanger sequencing. First is the throughput of data, which is at least an order of magnitude higher than
Sanger method, measured by base pairs per day of run time. Second, the cost of NGS by base pair is at least an order of magnitude lower. Third, the reads from NGS are generally shorter than those generated by capillary sequencing.
Last, an environmental sample can be recovered and analyzed directly through NGS and bypass the cloning procedure as well as the PCR amplification.
Suppose that one human genome is to be sequenced by the two approaches.
By utilizing ABI 3700 (Mitchelson, 2001) of Sanger capillary sequencing technology (could read up to 1 million base per day), eight years of non-stop processing is needed to complete the task. In contrast, only 2 hours are required if an Illumina HiSeq 2000 (Ajay et al., 2011) of NGS technology is applied (which is capable of producing 50 Gbp per day).
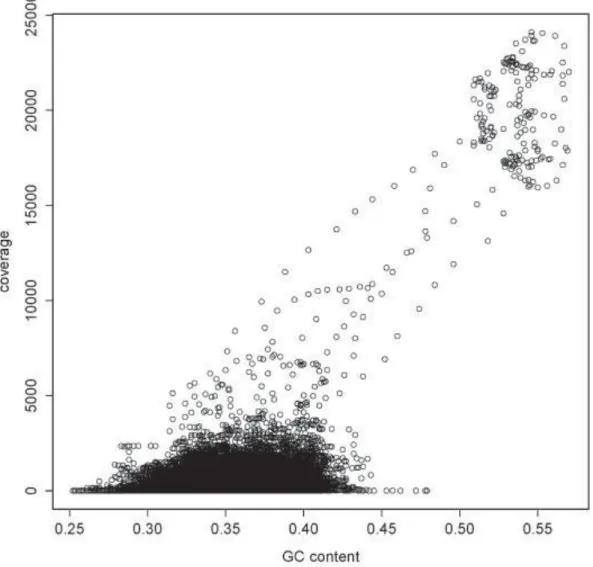
Bypassing the cloning step in NGS sequencing makes it possible to obtain result in an objective way: DNA fragments are usually not cloned into host with equal possibility, some may lost during processing, some may subject to selective bias such as GC content (Temperton et al., 2009). However, for NGS samples which have no library archives, genes are discovered only as a series of sequences, their actual physiological functions or relative gene locations are so far impossible to be re-accessed from picking up clones that harbor the desired inserts. NGS has advantages over capillary-based method mainly in its affordable price for high-throughput data production, but reduction in average read length has always been a drawback even advances in bioinformatics are gradually narrowing the gap between immense data and proper solutions for
analysis and interpretation.
Since 2005, researches incorporating NGS technique have increased dramatically. Unlike first generation sequencing, NGS constitutes various strategies that rely on complex interplay of enzymology, chemistry, high-resolution optics, hardware and software engineering. By now,
454-prosequencing GS FLX + system (Roche) and HiSeq system (Illumina) are the main players in high throughput sequencing despite the fact that there are some other competitors and new comers.
1.4.3. From Sequencing, Genetic Codes to Information
From capillary-based sequencing to the era of NGS massive parallel sequencing, output data has dramatically expanded in size but meanwhile shortened in the read length. Careful considerations must be taken when evaluating if a sample is suitable to be analyzed through the shotgun
sequencing approach, since the output sequences from all members within the community will be fragmented and mixed. To choose a community with less structural complexity (such as one dominant species and low diversity) can effectively reduce the difficulty during assembly, especially when shotgun sequencing is to be carried out by an NGS sequencer. Supporting evidences came from several metagenomic studies on extreme or oligotrophic
environments (although were sequenced through capillary-based method) such as acidophilic biofilm (Tyson et al., 2004), acid mine drainage (Edwards et al.,
2000) and the Sargasso Sea (Venter et al., 2004), which were successfully profiling the community structures or even recovered whole genomes of some dominant species (Baker and Banfield, 2003).
Sufficient quantity and high quality of extracted DNA is critical to sequencing.
In general, for pyrosequencing or cloning, some micrograms of genomic DNA are required. Although library construction is not necessary for other NGS systems such as Illumina, at least 20 μg would be suggested. For low-biomass samples derived from environments with harsh conditions, usually it would be the major challenge to obtain enough metagenomic material for starting up (Ferrer et al., 2009).
Once the sequencing stage has been finished, reads are base-called from signals generated by sequencer, screened to remove vectors (if vectors were used) and barcodes and then undergo a quality check to trim off bases or reads with poor-quality. The next step is to assemble reads according to overlapping regions into longer fragments termed contigs. Assembly is a process which consumes enormous computing power and also a step which might introduce artifacts to the final output (Kunin et al., 2008). The finished length of contig can vary widely from around 100 bp to more than 100 kb, depending on microbial community composition and sample quality.
After contigs are generated, the diversity, gene functions and metabolic
pathways of the sample are three basic directions for further analyses.
Databases such as SILVA (Pruesse et al., 2007), Greengenes (DeSantis et al., 2006), or Ribosomal Database Project II (RDP II) (Cole et al., 2003) offer useful resources for rRNA gene-based classification of microorganisms so that sequences containing taxonomic information or sequences derived directly from 16 S rRNA gene amplicon can be analyzed readily.
For gene function annotation and metabolic pathway investigation, the Gene Ontology (GO) database (Ashburner et al., 2000), Clusters of Orthologous Groups (COG) database (Tatusov et al., 2001), Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa et al., 2008), Pfam (Finn et al., 2010), NCBI (Sayers et al., 2009), SEED (Overbeek et al., 2005) are
commonly used databases for metagenomic data sets generated by NGS. To compare data on hand with other known species, reference genomes deposited in database like IMG (Joint Genome Institute) can be downloaded to a local server and mapped with reads or contigs.
After the sequence information was annotated, taxonomic relative abundance, main metabolic functions and unique pathways are usually integrated with environmental factors and cross-compared with other studies. Public domain depositions of all available data are also essential for environmental
metagenomic research; the accumulation of results from surveys facilitates later cross-comparison and also contributes to the discovery of the whole
picture of microbial biosphere on Earth.
1.5 Motivations and Aims of This Study
Hot springs, one of extreme environments that had been proven to be a reservoir of valuable knowledge to the past (earth history), present (microbial ecology) and future (bioprospecting and astrobiology), are worth being further explored and understood. In the past few decades, studies of this field in Taiwan have accumulated a certain amount of knowledge that provided a rough image of local hot spring ecosystems. However, more thorough and systematic researches on prokaryotic diversity are needed. In this study, the high-throughput sequencing technique was applied to obtain sufficient sequence data that facilitated a more comprehensive analysis on prokaryotic community structure in an acidic hot spring ecosystem.
1.6 Framework of Research
Samples of hot spring water were collected, concentrated and from which metagenomic DNAs were extracted for fosmid library construction and direct shotgun sequencing. Results of sequencing were analyzed for assessments of prokaryotic community composition, genome mapping and community gene function distribution. Prokaryotic community structure analysis and gene functions were the main topics of this study (blue and green boxes of Figure 1).
Prior to the sample collection, a general survey on the bacteria, archaea and nanoarchaea distribution of six candidate hydrothermal springs in TVG area was done (white box of Figure 1 and Appendix 1).
References
Ajay, S. S., Parker, S. C. J., Abaan, H. O., Fajardo, K. V. F. and Margulies, E. H.
(2011). "Accurate and comprehensive sequencing of personal genomes."
Genome Research 21(9): 1498-1505.
Amann, R. I., Ludwig, W. and Schleifer, K. H. (1995). "Phylogenetic Identification and in-Situ Detection of Individual Microbial-Cells without Cultivation."
Microbiological Reviews 59(1): 143-169.
Arnórsson, S., Gunnlaugsson, E. and Svavarsson, H. (1983). "The chemistry of geothermal waters in Iceland. II. Mineral equilibria and independent variables controlling water compositions." Geochimica Et Cosmochimica Acta 47(3):
547-566.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M., Sherlock, G. and Consortium, G. O. (2000).
"Gene Ontology: tool for the unification of biology." Nature Genetics 25(1):
25-29.
Baker, B. J. and Banfield, J. F. (2003). "Microbial communities in acid mine drainage." Fems Microbiology Ecology 44(2): 139-152.
Baker, G. C., Gaffar, S., Cowan, D. A. and Suharto, A. R. (2001). "Bacterial
community analysis of Indonesian hot springs." Fems Microbiology Letters 200(1): 103-109.
Barns, S. M., Fundyga, R. E., Jeffries, M. W. and Pace, N. R. (1994). "Remarkable Archaeal Diversity Detected in a Yellowstone-National-Park Hot-Spring Environment." Proceedings of the National Academy of Sciences of the
United States of America 91(5): 1609-1613.
Belousov, A., Belousova, M., Chen, C. H. and Zellmer, G. F. (2010). "Deposits, character and timing of recent eruptions and gravitational collapses in Tatun Volcanic Group, Northern Taiwan: Hazard-related issues." Journal of
Volcanology and Geothermal Research 191(3-4): 205-221.
Biddle, J. F., Fitz-Gibbon, S., Schuster, S. C., Brenchley, J. E. and House, C. H.
(2008). "Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment." Proceedings of the National
Academy of Sciences of the United States of America 105(30): 10583-10588.
Bohorquez, L. C., Delgado-Serrano, L., Lopez, G., Osorio-Forero, C., Klepac-Ceraj, V., Kolter, R., Junca, H., Baena, S. and Zambrano, M. M. (2012). "In-depth Characterization via Complementing Culture-Independent Approaches of the Microbial Community in an Acidic Hot Spring of the Colombian Andes."
Microbial Ecology 63(1): 103-115.
Bonch-Osmolovskaya, E. A., Miroshnichenko, M. L., Slobodkin, A. I., Sokolova, T.
G., Karpov, G. A., Kostrikina, N. A., Zavarzina, D. G., Prokof'eva, M. I., Rusanov, I. I. and Pimenov, N. V. (1999). "Biodiversity of Anaerobic lithotrophic prokaryotes in terrestrial hot springs of Kamchatka."
Microbiology 68(3): 343-351.
Brock, T. D. (1967). "Life at High Temperatures - Evolutionary Ecological and Biochemical Significance of Organisms Living in Hot Springs Is Discussed."
Science 158(3804): 1012-&.
Brock, T. D. and Freeze, H. (1969). "Thermus Aquaticus Gen N and Sp N a
Nonsporulating Extreme Thermophile." Journal of Bacteriology 98(1): 289-&.
Burton, N. P. and Norris, P. R. (2000). "Microbiology of acidic, geothermal springs of
Montserrat: environmental rDNA analysis." Extremophiles 4(5): 315-320.
Chai, B. H. T. (1972). "Structure and Tectonic Evolution of Taiwan." American Journal of Science 272(5): 389-&.
Cheng, T.-W., Chang, Y.-H., Tang, S.-L., Tseng, C.-H., Chiang, P.-W., Chang, K.-T., Sun, C.-H., Chen, Y.-G., Kuo, H.-C., Wang, C.-H., Chu, P.-H., Song, S.-R., Wang, P.-L. and Lin, L.-H. (2012). "Metabolic stratification driven by surface and subsurface interactions in a terrestrial mud volcano." Isme Journal.
Cheng, T. W., Wang, P. L., Chen, K. C., Wu, J. J., Song, S. R. and Lin, L. H. (2013).
"Segregated planktonic and bottom-dwelling archaeal communities in
high-temperature acidic, sulfuric ponds of the Tatung Volcano Group, northern Taiwan." Terrestrial, Atmospheric and Oceanic Sciences (in press).
Childs, A. M., Mountain, B. W., O'toole, R. and Stott, M. B. (2008). "Relating Microbial Community and Physicochemical Parameters of a Hot Spring:
Champagne Pool, Wai-o-tapu, New Zealand." Geomicrobiology Journal 25(7-8): 441-453.
Cole, J. R., Chai, B., Marsh, T. L., Farris, R. J., Wang, Q., Kulam, S. A., Chandra, S., Mcgarrell, D. M., Schmidt, T. M., Garrity, G. M. and Tiedje, J. M. (2003).
"The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy." Nucleic Acids Research 31(1): 442-443.
Cox, A., Shock, E. L. and Havig, J. R. (2011). "The transition to microbial photosynthesis in hot spring ecosystems." Chemical Geology 280(3-4):
344-351.
Delong, E. E. (2005). "Microbial community genomics in the ocean." Nature Reviews Microbiology 3(6): 459-469.
Delong, E. E. and Pace, N. R. (2001). "Environmental diversity of Bacteria and Archaea." Systematic Biology 50(4): 470-478.
Desantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., Huber, T., Dalevi, D., Hu, P. and Andersen, G. L. (2006). "Greengenes, a
chimera-checked 16S rRNA gene database and workbench compatible with ARB." Applied and Environmental Microbiology 72(7): 5069-5072.
Edwards, K. J., Bond, P. L., Gihring, T. M. and Banfield, J. F. (2000). "An archaeal iron-oxidizing extreme acidophile important in acid mine drainage." Science 287(5459): 1796-1799.
Edwards, R. A., Rodriguez-Brito, B., Wegley, L., Haynes, M., Breitbart, M., Peterson, D. M., Saar, M. O., Alexander, S., Alexander, E. C. and Rohwer, F. (2006).
"Using pyrosequencing to shed light on deep mine microbial ecology." Bmc Genomics 7.
Ellis, D. G., Bizzoco, R. L. W., Maezato, Y., Baggett, J. N. and Kelley, S. T. (2005).
"Microscopic examination of acidic hot springs of Waiotapu, North Island, New Zealand." New Zealand Journal of Marine and Freshwater Research 39(5): 1001-1011.
Ferrer, M., Beloqui, A., Timmis, K. N. and Golyshin, P. N. (2009). "Metagenomics for Mining New Genetic Resources of Microbial Communities." Journal of
Molecular Microbiology and Biotechnology 16(1-2): 109-123.
Ferrer, M., Golyshina, O., Beloqui, A. and Golyshin, P. N. (2007). "Mining enzymes from extreme environments." Current Opinion in Microbiology 10(3):
207-214.
Ferrer, M., Werner, J., Chernikova, T. N., Bargiela, R., Fernandez, L., La Cono, V., Waldmann, J., Teeling, H., Golyshina, O. V., Glockner, F. O., Yakimov, M. M.,
Golyshin, P. N. and Consortium, M. S. (2012). "Unveiling microbial life in the new deep-sea hypersaline Lake Thetis. Part II: a metagenomic study."
Environmental Microbiology 14(1): 268-281.
Ferris, M. J. and Ward, D. M. (1997). "Seasonal distributions of dominant 16S rRNA-defined populations in a hot spring microbial mat examined by denaturing gradient gel electrophoresis." Applied and Environmental Microbiology 63(4): 1375-1381.
Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., Gavin, O. L., Gunasekaran, P., Ceric, G., Forslund, K., Holm, L., Sonnhammer, E. L. L., Eddy, S. R. and Bateman, A. (2010). "The Pfam protein families database."
Nucleic Acids Research 38: D211-D222.
Hall, J. R., Mitchell, K. R., Jackson-Weaver, O., Kooser, A. S., Cron, B. R., Crossey, L. J. and Takacs-Vesbach, C. D. (2008). "Molecular characterization of the diversity and distribution of a thermal spring microbial community by using rRNA and metabolic genes." Applied and Environmental Microbiology 74(15):
4910-4922.
Handelsman, J. (2004). "Metagenomics: Application of genomics to uncultured microorganisms." Microbiology and Molecular Biology Reviews 68(4):
669-+.
Handelsman, J., Rondon, M. R., Brady, S. F., Clardy, J. and Goodman, R. M. (1998).
"Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products." Chemistry & Biology 5(10): R245-R249.
Hetzer, A., Morgan, H. W., Mcdonald, I. R. and Daughney, C. J. (2007). "Microbial life in champagne pool, a geothermal spring in Waiotapu, New Zealand."
Extremophiles 11(4): 605-614.
Hough, D. W. and Danson, M. J. (1999). "Extremozymes." Current Opinion in Chemical Biology 3(1): 39-46.
Huang, Q. Y., Dong, C. Z., Dong, R. M., Jiang, H. C., Wang, S., Wang, G. H., Fang, B., Ding, X. X., Niu, L., Li, X., Zhang, C. L. and Dong, H. L. (2011).
"Archaeal and bacterial diversity in hot springs on the Tibetan Plateau, China."
Extremophiles 15(5): 549-563.
Huber, H., Hohn, M. J., Rachel, R., Fuchs, T., Wimmer, V. C. and Stetter, K. O. (2002).
"A new phylum of Archaea represented by a nanosized hyperthermophilic symbiont." Nature 417(6884): 63-67.
Huber, J. A., Mark Welch, D., Morrison, H. G., Huse, S. M., Neal, P. R., Butterfield, D. A. and Sogin, M. L. (2007). "Microbial population structures in the deep marine biosphere." Science 318(5847): 97-100.
Hugenholtz, P., Pitulle, C., Hershberger, K. L. and Pace, N. R. (1998). "Novel division level bacterial diversity in a Yellowstone hot spring." Journal of Bacteriology 180(2): 366-376.
Inskeep, W. P., Rusch, D. B., Jay, Z. J., Herrgard, M. J., Kozubal, M. A., Richardson, T. H., Macur, R. E., Hamamura, N., Jennings, R. D., Fouke, B. W.,
Reysenbach, A. L., Roberto, F., Young, M., Schwartz, A., Boyd, E. S., Badger, J. H., Mathur, E. J., Ortmann, A. C., Bateson, M., Geesey, G. and Frazier, M.
(2010). "Metagenomes from High-Temperature Chemotrophic Systems Reveal Geochemical Controls on Microbial Community Structure and Function." Plos One 5(3).
Ivanova, I., Atanassov, I., Lyutskanova, D., Stoilova-Disheva, M., Dimitrova, D., Tomova, I., Derekova, A., Radeva, G., Buchvarova, V. and Kambourova, M.
(2011). "High Archaea diversity in Varvara hot spring, Bulgaria." Journal of
Basic Microbiology 51(2): 163-172.
Jackson, C. R., Langner, H. W., Donahoe-Christiansen, J., Inskeep, W. P. and Mcdermott, T. R. (2001). "Molecular analysis of microbial community structure in an arsenite-oxidizing acidic thermal spring." Environmental Microbiology 3(8): 532-542.
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., Katayama, T., Kawashima, S., Okuda, S., Tokimatsu, T. and Yamanishi, Y. (2008). "KEGG for linking genomes to life and the environment." Nucleic Acids Research 36:
D480-D484.
Kanokratana, P., Chanapan, S., Pootanakit, K. and Eurwilaichitr, L. (2004). "Diversity and abundance of Bacteria and Archaea in the Bor Khlueng hot spring in Thailand." Journal of Basic Microbiology 44(6): 430-444.
Kato, S., Itoh, T. and Yamagishi, A. (2011). "Archaeal diversity in a terrestrial acidic spring field revealed by a novel PCR primer targeting archaeal 16S rRNA genes." Fems Microbiology Letters 319(1): 34-43.
Kawashima, E., Farinelli, L. and Mayer, P. (1998). Method of nucleic acid amplification.
Konstantinou, K. I., Lin, C. H. and Liang, W. T. (2007). "Seismicity characteristics of a potentially active Quaternary volcano: The Tatun Volcano Group, northern Taiwan." Journal of Volcanology and Geothermal Research 160(3-4): 300-318.
Kristjansson, J. K. and Hreggvidsson, G. O. (1995). "Ecology and Habitats of Extremophiles." World Journal of Microbiology & Biotechnology 11(1):
17-25.
Kunin, V., Copeland, A., Lapidus, A., Mavromatis, K. and Hugenholtz, P. (2008). "A Bioinformatician's Guide to Metagenomics." Microbiology and Molecular
Biology Reviews 72(4): 557-578.
Lamoreaux, P. E. and Tanner, J. T. (2001). Springs and Bottled Waters of the World:
Ancient History, Source, Occurrence, Quality and Use Springer.
Lau, M. C. Y., Aitchison, J. C. and Pointing, S. B. (2009). "Bacterial community composition in thermophilic microbial mats from five hot springs in central Tibet." Extremophiles 13(1): 139-149.
Lee, H. F., Yang, T. F., Lan, T. F., Song, S. R. and Tsao, S. (2005). "Fumarolic gas composition of the Tatun Volcano Group, northern Taiwan." Terrestrial Atmospheric and Oceanic Sciences 16(4): 843-864.
Lin, C. H., Konstantinou, K. I., Liang, W. T., Pu, H. C., Lin, Y. M., You, S. H. and Huang, Y. P. (2005). "Preliminary analysis of volcanoseismic signals recorded at the Tatun Volcano Group, northern Taiwan." Geophysical Research Letters 32(10).
Liu, C. M., Song, S. R., Chen, Y. L. and Tsao, S. (2011). "Characteristics and Origins of Hot Springs in the Tatun Volcano Group in Northern Taiwan." Terrestrial Atmospheric and Oceanic Sciences 22(5): 475-489.
Lu, H. P., Lin, Y. K., Lin, L. H., Wang, P. L. and Yu, H. T. (unpublished data). The microbial community structure in acidic hot springs.
Macelroy, R. D. (1974). "Some Comments on Evolution of Extremophiles."
Biosystems 6(1): 74-75.
Macur, R. E., Langner, H. W., Kocar, B. D. and Inskeep, W. P. (2004). "Linking geochemical processes with microbial community analysis: successional dynamics in an arsenic-rich, acid-sulphate-chloride geothermal spring."
Geobiology 2(3): 163-177.
Mardis, E. R. (2008). "Next-generation DNA sequencing methods." Annual Review
of Genomics and Human Genetics 9: 387-402.
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., Berka, J., Braverman, M. S., Chen, Y. J., Chen, Z. T., Dewell, S. B., Du, L., Fierro, J. M., Gomes, X. V., Godwin, B. C., He, W., Helgesen, S., Ho, C. H., Irzyk, G. P., Jando, S. C., Alenquer, M. L. I., Jarvie, T. P., Jirage, K. B., Kim, J.
B., Knight, J. R., Lanza, J. R., Leamon, J. H., Lefkowitz, S. M., Lei, M., Li, J., Lohman, K. L., Lu, H., Makhijani, V. B., Mcdade, K. E., Mckenna, M. P., Myers, E. W., Nickerson, E., Nobile, J. R., Plant, R., Puc, B. P., Ronan, M. T., Roth, G. T., Sarkis, G. J., Simons, J. F., Simpson, J. W., Srinivasan, M., Tartaro, K. R., Tomasz, A., Vogt, K. A., Volkmer, G. A., Wang, S. H., Wang, Y., Weiner, M. P., Yu, P. G., Begley, R. F. and Rothberg, J. M. (2005). "Genome
sequencing in microfabricated high-density picolitre reactors." Nature 437(7057): 376-380.
Marteinsson, V. T., Hauksdottir, S., Hobel, C. F. V., Kristmannsdottir, H.,
Hreggvidsson, G. O. and Kristjansson, J. K. (2001). "Phylogenetic diversity analysis of subterranean hot springs in Iceland." Applied and Environmental Microbiology 67(9): 4242-4248.
Mathur, J., Bizzoco, R. W., Ellis, D. G., Lipson, D. A., Poole, A. W., Levine, R. and Kelley, S. T. (2007). "Effects of abiotic factors on the phylogenetic diversity of bacterial communities in acidic thermal springs." Applied and Environmental Microbiology 73(8): 2612-2623.
Maxam, A. M. and Gilbert, W. (1977). "A new method for sequencing DNA."
Proceedings of the National Academy of Sciences 74(2): 560-564.
Metzker, M. L. (2010). "Applications of Next-Generation Sequencing Sequencing Technologies - the Next Generation." Nature Reviews Genetics 11(1): 31-46.
Meyer-Dombard, D. R., Shock, E. L. and Amend, J. P. (2005). "Archaeal and bacterial communities in geochemically diverse hot springs of Yellowstone National Park, USA." Geobiology 3(3): 211-227.
Mitchelson, K. (2001). Overview: The Application of Capillary Electrophoresis for DNA Polymorphism Analysis. Capillary Electrophoresis of Nucleic Acids. K.
Mitchelson and J. Cheng, Humana Press. 162: 3-26.
Mullis, K. B. and Faloona, F. A. (1987). "Specific Synthesis of DNA Invitro Via a Polymerase-Catalyzed Chain-Reaction." Methods in Enzymology 155:
335-350.
Nakagawa, S., Takai, K., Inagaki, F., Chiba, H., Ishibashi, J., Kataoka, S., Hirayama, H., Nunoura, T., Horikoshi, K. and Sako, Y. (2005). "Variability in microbial community and venting chemistry in a sediment-hosted backarc hydrothermal system: Impacts of subseafloor phase-separation." Fems Microbiology
Ecology 54(1): 141-155.
Nakagawa, T. and Fukui, M. (2002). "Phylogenetic characterization of microbial mats and streamers from a Japanese alkaline hot spring with a thermal gradient."
Journal of General and Applied Microbiology 48(4): 211-222.
Ng, C. C., Chang, C. C. and Shyu, Y. T. (2005). "Archaeal community revealed by 16s rRNA and fluorescence in situ hybridization in a sulphuric hydrothermal hot spring, northern Taiwan." World Journal of Microbiology & Biotechnology 21(6-7): 933-939.
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H.-Y., Cohoon, M., De Crécy-Lagard, V., Diaz, N., Disz, T., Edwards, R., Fonstein, M., Frank, E.
D., Gerdes, S., Glass, E. M., Goesmann, A., Hanson, A., Iwata-Reuyl, D., Jensen, R., Jamshidi, N., Krause, L., Kubal, M., Larsen, N., Linke, B.,
Mchardy, A. C., Meyer, F., Neuweger, H., Olsen, G., Olson, R., Osterman, A., Portnoy, V., Pusch, G. D., Rodionov, D. A., Rückert, C., Steiner, J., Stevens, R., Thiele, I., Vassieva, O., Ye, Y., Zagnitko, O. and Vonstein, V. (2005). "The Subsystems Approach to Genome Annotation and its Use in the Project to Annotate 1000 Genomes." Nucleic Acids Research 33(17): 5691-5702.
Pace, N. R. (1997). "A molecular view of microbial diversity and the biosphere."
Science 276(5313): 734-740.
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W. G., Peplies, J. and Glockner, F. O. (2007). "SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB."
Nucleic Acids Research 35(21): 7188-7196.
Rappe, M. S. and Giovannoni, S. J. (2003). "The uncultured microbial majority."
Annual Review of Microbiology 57: 369-394.
Reigstad, L. J., Jorgensen, S. L. and Schleper, C. (2010). "Diversity and abundance of Korarchaeota in terrestrial hot springs of Iceland and Kamchatka." Isme Journal 4(3): 346-356.
Reysenbach, A. L., Ehringer, H. and Hershberger, K. (2000). "Microbial diversity at 83 degrees C in Calcite Springs, Yellowstone National Park: another
environment where the Aquificales and "Korarchaeota" coexist."
Extremophiles 4(1): 61-67.
Reysenbach, A. L., Giver, L. J., Wickham, G. S. and Pace, N. R. (1992). "Differential Amplification of Ribosomal-Rna Genes by Polymerase Chain-Reaction."
Applied and Environmental Microbiology 58(10): 3417-3418.
Reysenbach, A. L., Wickham, G. S. and Pace, N. R. (1994). "Phylogenetic Analysis of the Hyperthermophilic Pink Filament Community in Octopus Spring,