國
立
交
通
大
學
材料科學與工程學系
碩
士
論
文
機能性金/二氧化錫
核/衛星異質組裝結構之氣體感測應用
Functional Au-SnO
2core-satellite heteroassemblies
for gas sensing applications
研 究 生:陳怡真
指導教授:陳軍華 博士
機能性金/二氧化錫
核/衛星異質組裝結構之氣體感測應用
Functional Au-SnO
2core-satellite heteroassemblies
for gas sensing applications
研 究 生:陳怡真 Student:Chen-Yi CHEN
指導教授:陳軍華 博士 Advisor:Chun-Hua CHEN, Ph.D.
國 立 交 通 大 學
材 料 科 學 與 工 程 學 系
碩 士 論 文
A ThesisSubmitted to Department of Materials Science and Engineering College of Engineering
National Chiao Tung University In partial Fulfillment of the Requirements
For the Degree of Master
In
Materials Science and Engineering
September 2011
Hsin-chu, Taiwan, Republic of China
中華民國一百年九月
i
機能性金/二氧化錫
核/衛星異質組裝結構之氣體感測應用
研究生:陳怡真
指導教授:陳軍華 博士
國立交通大學材料科學與工程學系﹙研究所﹚碩士班
摘要
二氧化錫奈米粒子(SnO
2NPs)及其組裝結構已被廣泛地設計合成,藉由
對象氣體在粒子表面吸附(Adsorption)或脫附(Desorption)時所產生之劇烈電
阻變化,可作為氣體感測之用。本研究主要討論機能性金/二氧化錫核/衛星
異質組裝結構(Au-SnO
2CSHA)之化學法製備及以光學為基礎的氣體感測應
用。透過異質結構參數之調控,來達到以光學訊號感測 CO 濃度之目的。結
構製備主要分為兩個步驟:首先,利用直接還原法合成 Au NPs (~13 nm),
其次透過熱分解合成 SnO
2NPs (~2 nm),並在 Au NPs 表面自組裝形成異質
結 構 。 由 高 解 析 穿 透 式 顯 微 鏡 (High resolution transmission electron
microscopy, HRTEM)發現,組裝層之 SnO
2NPs 形貌及尺寸可利用溶液 pH
而後沈積於經氧電漿親水處理之石英基板上,進行紫外光-可見光吸收光譜
分析及 CO 之光學感測實驗。
從大氣及室溫下之吸收光譜得知,隨著 SnO
2NPs 組裝層厚度的增加(從
5 nm 至 9 nm),Au-SnO
2CSHA 的吸收波峰從 520 nm 最高紅位移至 540 nm,
此時 Au-SnO
2CSHA 之膠體溶液顏色由紅變紫。利用米氏理論搭配簡化之
Au-SnO
2核殼模型,進行吸收光譜之模擬計算發現,當緻密 SnO
2殼層厚度
為 7 nm,Au 核球直徑為 13 nm 時,其波峰位於 556 nm,相較於合成所得
SnO
2組裝層厚度 7 nm CSHA 波峰位置大幅紅位移。為了進一步趨近此理論
與實驗厚度上之差異,故進一步利用等效介質理論(EMT),假設 SnO
2組裝
層為 SnO
2NPs 及水所建構之非緻密殼層(SnO
2體積佔有率為 0.5 時),即可
獲得與實驗值相同之波峰位置 540 nm。當 SnO
2在殼層體積佔有率越低,波
峰發生藍移(Blue shift)。此高依存性之關係,可以作為 SnO
2組裝層厚度及
體積佔有率定量之用。
在 Au-SnO
2CSHA 的光學 CO 感測方面,將具 5 nm SnO
2組裝層之
Au-SnO
2CSHA 置於不同濃度的 CO 環境中(5 ppm~10000 ppm),在不同溫
度下進行臨場吸收光譜檢測時發現,隨著 CO 濃度的增加,全測試波長範圍
(200 nm~1100 nm)之吸收度均微幅增加,不同吸收波段對感測的響應略有不
同,其中最大的吸收變化量約出現在 556 nm。利用此波長進行感測特性之
數值分析發現,此 CSHA 在 1000 ppm 以下有較佳之感測靈敏度,最高吸收
iii
度變化率(Absorbance change ratio, ACR)為 0.014。值得注目的是,本研究成
功地將傳統感測器所需操作溫度(>300
oC)降低至室溫。
Functional Au-SnO
2core-satellite heteroassemblies for gas sensing applications
Student:Chen-Yi CHEN Advisor:Chun-Hua CHEN, Ph.D.
Department of Materials Science and Engineering
National Chiao Tung University
ABSTRACT
SnO
2nanoparticles (NPs) and their assemblies have been widely designed,
synthesized and applied for gas sensing by screening the drastic changes of
electrical resistance while absorbing or desorbing targeting gases. In this work,
we mainly focus on the chemical preparation and optic-based gas-sensing
application of functional Au-SnO
2core-satellite heteroassemblies (Au-SnO
2CSHA). The fabrication of such heteroassemblies involving two steps, i.e. the
fabrication of Au cores (~13 nm) by direct reduction method and the following
formation and assembling of SnO
2NPs (~2 nm) on the suspended Au cores, is
v
sensing and optical observation. From high resolution transmission electron
microscopy (HRTEM) images, it is found that the assembled thickness as well
as the morphology of the tiny SnO
2NPs can be precisely controlled by varying
pH value and concentration of Sn precursors. The prepared Au-SnO
2CSHA
were repeatedly washed with de-ionized water and then deposited onto
plasma-treated glass substrates for a series of UV-visible absorption and CO
sensing characterizations at various CO concentrations and substrate
temperatures.
From the UV-visible spectra measured at atmosphere at room temperature,
as the thickness of the SnO
2NPs assemblies increased from 5 nm up to 9 nm,
the absorption peak of the prepared Au-SnO
2CSHA greatly red shifted from 520
nm to 540 nm, where the solution color changed from red to purple. Based on
classical Mie theory and the assumption of a simplified Au-SnO
2core-shell
spherical model, the calculated spectrum peak of a 13 nm Au core coated with a
dense 7 nm SnO
2shell located at 556 nm which is greatly red-shifted compared
with the experimental one with a porous 7 nm SnO
2shell at 540 nm. In order to
approach the real structural parameters for obtaining a peak at 540 nm, we
further introduced the effective medium theory (EMT) for a theoretical
estimation of the Au-SnO
2CSHA with a porous 7 nm SnO
2and found that with
the assumption of a mixture of 50% (volume ratio) SnO
2NPs and 50% water,
the calculated peak is coincident with the experimental one.
The Au-SnO
2CSHA (5 nm SnO
2) was applied for the in-situ investigation
of CO sensing properties under controlled CO concentration from 5 ppm to
10000 ppm at various temperatures from room temperature to 200
oC. It was
found that with the increase of CO concentration, the absorption intensity
accordingly decreased over the full measured range from 200 nm to 1100 nm
and a maximum change was found at around 556 nm which is the characteristic
absorption of Au NPs. By analyzing the CO concentration dependent variation
of the Au absorption intensity, CSHA exhibit a significant CO sensing sensitivity
below 1000 ppm where an excellent absorbance change ratio (ACR) as high as
0.014 can be approached at room temperature.
vii
致謝
在交大的時光飛逝,還能記得第一次踏進校園裡充滿心中的悸動,我的碩士求學
生涯即將結束,期間受到許多人的幫助,在此致上我心中滿滿的謝意。 首先,我要感謝指導教授陳軍華老師,因為您總是不厭其煩的指導及注意每一個小 細節,讓我在研究的路上得到成長與進步,其次,要感謝口試委員黃華宗老師以及張文 固老師對本論文的悉心審閱,並提出許多寶貴的意見,使本論文更加地完善;我要感謝 所有熱心的學長姐以及親愛的學弟妹們,相孙學長的經驗就像是資料庫提供很多資訊, 讓我更能掌握實驗的方向感,學長修誠、景荃、宗漢、宗哲以及學姊芳卿、盈婷,無論 在研究還是生活,都給予我很大的幫助,感謝與我一貣奮鬥的同學智仁,除了給予我許 多寶貴的意見外,你也總是為大家帶來歡笑,感謝學弟妹克憲、明修與雅婷,總是在我 需要的時候給予我協助,還一貣分享了許多快樂和憂傷,感謝我的室友書豪,不僅有緣 可以一貣在交大念書,也提供我許多生活上的幫助。最後,我要感謝我的父母親,總是 給我百分百的支持,才使我可以無後顧之憂地完成學業,讓我走自己的路。因為你們, 我才能順利過完碩士求學生涯,希望你們能夠永遠健康快樂,謝謝你們。目錄
中文摘要 ………i ABSTRACT ... iv 致謝.……….vii 目錄………..………...viii 圖目錄……….x 表目錄………..xiv 一、緒論 ………...1 二、理論基礎與文獻回顧 ... 3 2-1 基本理論 ... 32-2 金屬氧化物半導體氣體感測器(MOS gas sensor) ... 6
2-3 MOS 基 CO 感測器 ... 8 2-4 SnO2之 CO 感測特性 ... 11 2-5 光學式氣體感測 ... 14 2-6 研究動機與目的 ... 23 三、實驗方法與步驟 ... 25 3-1 實驗藥品 ... 25 3-2 實驗設備 ... 25 3-3 Au-SnO2 CSHA 之製備法 ... 26 3-4 奈米結構分析 ... 29 3-5 光學式 CO 感測特性 ... 31 四、結果與討論 ... 33 4-1 pH 值對 Au-SnO2 CSHA 結構之影響 ... 33 4-2 前驅物濃度對 Au-SnO2 CSHA 結構之影響 ... 38
ix 4-3 Au-SnO2 CSHA 之光學特性-紫外光可見光吸收光譜分析 ... 42 4-4 光學式氣體感測 ... 46 五、結論 ……….51 參考文獻 ……….52 附錄、紫外光-可見光吸收光譜模擬計算理論 ... 57 A 米氏理論(Mie theory) ... 57 B Drude 修正 ... 59 C 光學核殼模型 ... 60
圖目錄
Fig. 2-1:Schematic band diagrams of an insulator, semi-conductor and conductor. ... 3 Fig. 2-2:Demonstrating the structure of the materials, and the positions of the surface, bulk and particle boundary. ... 5 Fig. 2-3:Schematic diagram of negatively charged chemisorbed oxygen species cause an upward band bending and consequently a depletion layer in the near-surface region. ... 7 Fig. 2-4:The effects of grain size on the response to the carbon monoxide. ... 9 Fig. 2-5:Influence of organic vehicle content in inks without mineral binder on the response of sensors to CO (300 ppm) at 500 oC:(1) 41.3 wt.%, (2) 35.0 wt.%, (3) 23.8 wt.%, (4).17.8 wt.%. ... 10 Fig. 2-6:Demonstrating the response of the sensor to humidity. ... 10 Fig. 2-7:SEM pictures of the SnO2 sensing layer. ... 12
Fig. 2-8:Schematic representation of a porous sensing layer with geometry and energy band. ... 12 Fig. 2-9:(a) TEM micrographs of 1.0 wt% Au/SnO2. (b) Response of Au/SnO2 (solid line) and
SnO2 (dashed line) to various concentrations of CO at different temperatures. ... 13
Fig. 2-10:Illustrations of (a) surface plasmons and (b) localized surface plasmon. ... 15 Fig. 2-11:Experimental (solid line) and Simulated by Drude theory (dash line) absorption spectra of Ag nanoparticles... 16 Fig. 2-12:Optical tunability is demonstrated for nanoshells with a 60 nm silica core radius and gold shells 5, 7, 10, and 20 nm thick. ... 17 Fig. 2-13: Extinction spectra of immobilized monolayers of gold solid colloids. ... 18 Fig. 2-14:(a) Scheme of LSPR–VOC sensor testing system (b) LSPR spectrums of Ag
xi
nanoparticles responding to 0∼100% relative saturated toluene vapor concentrations. (c) spectrum integration area of Ag nanoparticles between
350∼550 nm. ... 19
Fig. 2-15:(a) Scheme of building LSPR sensor array assembly in a flow-cell cube. Real-time response signals tested with (b) m-xylene, insert shows the magnified response curves of NAP-Au-Npat 2000 ppm and (c) n-pentanol (wavelength regions: +, 395–475 nm; −, 480–620 nm; ○, 690–850 nm). ... 20
Fig. 2-16:(a) Schematic presentation of polymer-coated gold island film preparation. Spectral response of the vapor sensor based upon gold island films coated with (b) PS and (c) PSS. ... 21
Fig. 2-17:(a) Schematic of the sol–gel sensor architecture. (b) Effect of H2 in the optical absorption of SiO2-NiO-Au nanocomposite. Absorption spectra of films annealed at 500 oC measured in air and under a 1% v/v H2 mixture at 300 oC operative temperature. ... 22
Fig. 3-1:Process of synthesizing Au NPs by tri-sodium citrate. ... 27
Fig. 3-2:Process of synthesizing Au-SnO2 CSHA. ... 28
Fig. 3-3:Schematic of optical gas sensor architecture. ... 32
Fig. 4-1:(a) TEM and (b) HRTEM images of the Au-SnO2 CSHA prepared with a stannate concentration of 40 mM at pH 10.5. ... 34
Fig. 4-2:(a) TEM and (b) HRTEM images of the Au-SnO2 CSHA prepared with a stannate concentration of 40 mM at pH 8. ... 35
Fig. 4-3:(a) TEM and (b) HRTEM images of the Au-SnO2 CSHA prepared with a stannate concentration of 40 mM at pH 5.8. ... 36
Fig. 4-4:XRD patterns of the Au-SnO2 CSHA prepared with a stannate concentration of 40 mM at pH (a) 10.5, (b) 8, and (c) 5.8. ... 37 Fig. 4-5:(a) TEM and (b) HRTEM images of the Au-SnO2 CSHA prepared with a stannate
concentration of 5.6 mM at pH 10.5. ... 39 Fig. 4-6:(a) TEM and (b) HRTEM images of the Au-SnO2 CSHA prepared with a stannate
concentration of 0.18 mM at pH 10.5. ... 40 Fig. 4-7:XRD patterns of the Au-SnO2 CSHA prepared at pH 10.5 with a stannate
concentration of (a) 40 mM (b) 5.6 mM (c) 0.18 mM... 41 Fig. 4-8:UV-Vis absorption spectra of the Au-SnO2 CSHA prepared at (a) pH 5.8, (b) pH 8,
and (c) pH 10.5. ... 43 Fig. 4-9:UV-Vis absorption spectra of the Au-SnO2 CSHA prepared with a stannate
concentration of (a) 40 mM, (b) 5.6 mM, and (c) 0.18 mM. ... 43 Fig. 4-10:Simulated absorption spectra of the Au-SnO2 core-shell models with various shell
thicknesses. ... 45 Fig. 4-11:Simulated absorption spectra of the Au-SnO2 core-shell models with different
volume ratios of SnO2 NPs to medium (water). ... 45
Fig. 4-12:Absorption spectra of the Au-SnO2 CSHA with a 7 nm SnO2 shell measured in dry
air and after exposure to 1000 ppm CO at 200 oC. ... 47 Fig. 4-13:The absorption difference between the UV-Vis spectra shown in Fig. 4.12. ... 47 Fig. 4-14:Absorption spectra of the Au-SnO2 CSHA with a 7 nm SnO2 shell measured in dry
air and after exposure to 1000 ppm CO at room temperature. ... 48 Fig. 4-15:The absorption difference between the UV-Vis spectra shown in Fig. 4.14. ... 48 Fig 4-16:Absorption spectra of the Au-SnO2 CSHA with a 5 nm SnO2 shell measured in dry
air and after exposure to 1000 ppm CO at room temperature. ... 49 Fig. 4-17:The absorption difference between the UV-Vis spectra shown in Fig. 4.16. ... 49 Fig. 4-18:Absorbance at 556 nm obtained from the Au-SnO2 CSHA with a 5 nm SnO2 shell
as a function of CO concentration. ... 50 Fig. 4-19:ACR at 556 nm obtained from the Au-SnO2 CSHA with a 5 nm SnO2 shell versus
xiii
Fig. B-1:Drude model of free electron transport action of metal when apply electric field. . 59 Fig. C-1:Core–shell nanosphere geometry:ε1, ε2, and ε3 are the dielectric functions for the
core, shell and embedding regions, respectively, R1 is the core radius and R2 the
total particle radius. ... 60 Fig. D-1:Schematic of core-satellite structure. ... 62
表目錄
Table 1-1:Demonstrating the effects of certain concentrations of carbon monoxide on human health. ... 2 Table 2-1:Sign of resistance change (increase or decrease) in different gas atmospheres. ... 4 Table 2-2:Case studies of CO gas sensing on semiconducting metal oxides. ... 9 Table 4-1:The FWHMs of the SnO2 (111) peaks and the estimated particle sizes of the
Au-SnO2 CSHA prepared at different pH values. ... 37
Table 4-2:The FWHMs of the SnO2 (111) peaks and the estimated particle sizes of the
1
一、 緒論
十八世紀以前,所有的工業都是使用人力、動物以及一些自然的動力,如水力、風 力等,直到蒸汽機的發明才真正解決了人類必須面對的動力問題,然而,現代工業發展 為人類創造巨大財富的同時,卻也給生態環境帶來了嚴重的汙染。工業快速成長及化石 燃料之需求不斷增加,生產過程中所產生的廢棄產物及氣體種類也越來越多,主要是以 煙霧、飄塵等微粒、及一氧化碳(CO)、二氧化硫(SO2)、一氧化氮(NO)和二氧化氮(NO2) 等有毒氣體為主,這些有毒性氣體在對流層中隨著氣流不斷地擴散及飄移,不只汙染工 業區周遭環境,更使得人類健康、生態系統和社會環境受到無形毒害的威脅。 大部分的有毒氣體都是無色、無臭、無味,中毒後的症狀不易被察覺,其中以 CO 議題最為險峻,CO 會直接與血液中血紅蛋白的鐵中心結合,且此結合反應為不可逆, 意味著當血液中運輸氧氣的鐵中心分子優先與 CO 結合後,氧氣不能再被吸收,因此當 人體暴露於高濃度的 CO 環境時可能會導致死亡。許多工業國家的統計顯示,在不當作 業場所中所引貣的 CO 中毒所造成的致死率高達百分之五十以上,美國國家研究所更指 出,在 CO 濃度為 35 ppm 的環境下[1],人體最長的可曝露值僅為八小時(Table 1-1)。根 據我國衛生署統計,民國 95 年共有 301 人死於 CO 中毒。另外,根據臺北榮總毒藥物 諮詢中心的統計資料顯示,CO 中毒經常是集體發生的,尤以冬天為常見,中毒原因是 不當使用熱水器以及蓄意自殺者最多;颱風來襲期間,部分地區停電,有些家庭會以柴 油發電機來發電因應,再加上門窗緊閉,也容易發生 CO 中毒事故。此外,由汽車或引 擎的廢氣所造成的 CO 中毒事件,也屢有所聞;這種情況容易發生在把汽車停泊室內時, 又發動引擎使用空調,進而造成密閉車內或室內人員中毒。有些現代技術的製程,如煉 鐵,亦會產生 CO 副產品,其他諸如鍋爐房、釀酒廠、倉庫、石油煉油廠、造紙廠都是 CO 中毒的高危險作業環境。 CO 中毒可能導致人體中需要大量氧氣的器官(如心臟和大腦)產生永久性損壞,進 而引貣遲發性腦病變、行為退化等症狀;若能以氣體感測器預先偵測 CO 的逸漏,並在第一時間做出正確的危機處理就可以確保人身安全。為了保障居家與工業安全,開發適 用於不同作業場所之高可靠度氣體感測器,成為現代工業安全中相當重要的研究課題。
氣體感測器(Gas sensor)是用來量測氣體的種類、濃度和成分的感測器,由於氣體的 種類很多,性質都不相同,一種感測器無法檢驗所有氣體種類,因此檢測不同氣體之氣 體感測器相繼被開發研究。根據氣體感測器的感測原理與機制,可將其分為:金屬氧化 物 半 導 體 氣 體 感 測 器 (Metal-oxide semiconductor gas sensor) 、 電 化 學 氣 體 感 測 器 (Electrochemical sensor)、觸媒燃燒式氣體感測器(Catalytic combustion gas sensor)。金屬 氧化物半導體氣體感測器因其具備耐熱性、耐蝕性佳、響應速率快、元件製作容易,因 此被廣泛的使用在家庭與工廠環境中,以偵測毒性氣體及燃燒爆炸性氣體。
Table 1-1:Demonstrating the effects of certain concentrations of carbon monoxide on human health. [1]
Carbon monoxide
concentration (ppm) Signs and symptoms
35 Headache and dizziness within 6 to 8 hours of exposure
100 Slight headache in 2 to 3 hours
200 Slight headache within 2 to 3 hours;loss of judgment
400 Frontal headache within 1 to 2 hours
800 Dizziness, nausea and convulsions within 45 minutes, Insensible within 2 hours
1600 Headache, tachycardia, dizziness and nausea within 20 death within 30 minutes
6400 Headache and dizziness in 1 to 2 minutes;convulsions, respiratory arrest, and death in less than 20 minutes
3
二、 理論基礎與文獻回顧
2-1 基本理論
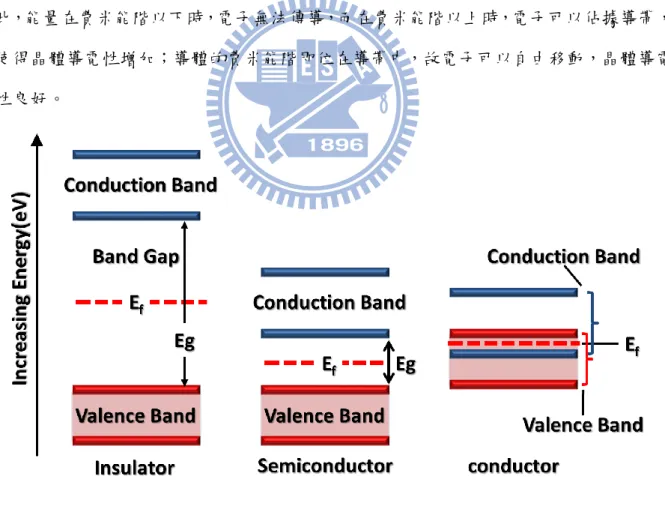
2-1-1 能帶理論(Band theory)
在能帶理論中,晶體裡存在價帶(Valence band )和導帶(Conduction band),費米能階
(Fermi level)定義為在絕對零度下,最高可用電子之能階,Fig. 2-1為三種主要類別材料
的能帶:絕緣體價帶與導帶間的能隙較大(通常為 10 eV 以上),因為激發價帶的電子 到導帶需要大量的能量,所以電子的傳導不會發生;半導體的能隙適中(0.5-5.0 eV),因 此,能量在費米能階以下時,電子無法傳導,而在費米能階以上時,電子可以佔據導帶, 使得晶體導電性增加;導體的費米能階即位在導帶中,故電子可以自由移動,晶體導電 性良好。
2-1-2 能帶理論應用於感測器
氣體與金屬氧化物薄膜的表面相互作用(一般是透過表面吸附氧離子),導致材料 中載子濃度變化,改變了材料的導電度。在 n 型半導體中,多數載子為電子,與還原性 氣體作用導致導電度增加;相反地,一個氧化性氣體消耗感測層的電子,使得導電度下 降。p 型半導體中的多數載子是電洞,氧化性氣體造成電洞的數量增加,使得導電度增 加,而隨著還原性氣體的增加,將負電荷引入該材料,使得電洞濃度降低,電阻增加。 其關係如Table 2-1[1]所示。Table 2-1:Sign of resistance change (increase or decrease) in different gas atmospheres. [1]
Classification Oxidizing gases Reducing gases
n-type Resistance increase Resistance decrease
p-type Resistance decrease Resistance increase
一個 p 型感測器的反應模型可由 Eq. 2-1 和 Eq. 2-2 表示[2],在材料表面吸附氧原子 造成原子的離子化並產生電洞(V˙ ),電洞和離子可以和還原性氣體反應,如 CO,使其 形成 CO2,電荷載子濃度的不同(此為電洞)表現在感測器電阻值的變化: 1 2𝑂2 ⇄ 𝑂 −+ 𝑉˙ 𝑉˙+ 𝑂−+ CO → 𝐶𝑂 2 在 Naisbitt 等人描述的反應模型中,感測器之微結構只有很細微的影響,且此模型 只適用於一個有限的範圍之內。此模型並假定在金屬氧化物表面上的吸收物為 O2 -,由 Eq. 2-3 可知,這對感測反應是非常不利的,其中電阻的改變與氣體濃度及靈敏度成正比 (靈敏度 A 為在特定溫度時材料的常數),其中 R 是曝露在待測氣體下的電阻和基準電 阻 R0(Eq. 2-4)[3]: 1 2𝑂2 ⇄ 𝑂 2−+ 2𝑉˙ 𝑅 𝑅0 = 1 + A[𝐶𝑂] (Eq. 2-1) (Eq. 2-2) (Eq. 2-3) (Eq. 2-4)
5
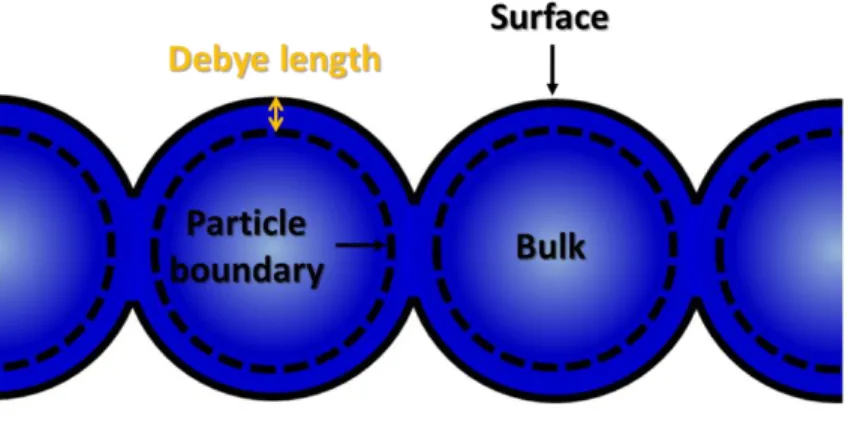
相反地,Naisbitt 等人所建議的 Eq. 2-1 可能性較大,但其為非線性反應,推測一定 還有其他因素影響。我們假設只有氣體可以停留在反應表面,且反應表面只對目標氣體 發生反應,材料可以被分成三個區域(Fig. 2-2):(i)表面(Surface),(ii)塊材(Bulk,較難接 近目標氣體),(iii)顆粒邊界(Boundary)。
Fig. 2-2:Demonstrating the structure of the materials, and the positions of the surface, bulk and particle boundary. [2]
表面和顆粒邊界之間的距離,為可發生電荷分離的距離,稱德拜長度(Debye length),反 應公式為 Eq. 2-5: 𝐺𝑇 = 𝛾𝑃𝐵(1 + 𝐴[𝐶𝑂]) + 1 [(1 𝛾B)+( 1 𝛾𝑆(1+𝐴[𝐶𝑂]))] 其中 GT為響應(GT = RT / RT,0),RT為感測器的總電阻,RT,0是在乾燥空氣中表面的基 準電阻,其中𝛾𝑥 = 𝑅𝑥,0∕ 𝑅𝑇,0(x 可能為以下幾種表示,PB 表示顆粒邊界,B 表示塊材, S 表示表面)。[4] 響應時間與材料的晶粒尺寸和顆粒邊界大小直接相關,n 型和 p 型半導體的響應模 式不同。在形成基準電阻時,氧被吸附到表面並且從材料提取電子,這個過程將決定基 準電阻 R0,p 型的電阻率相對於塊材是減少,而相對於 n 型則為增加。Fig. 2-2 模型中 三個區域電阻的相關貢獻不同;對於非常小尺寸的晶粒,晶粒可以被認為完全不包含塊 材區域(故整個晶粒被認為對表面積有貢獻),在這種情況下,Eq. 2-4 是一個適當的響 應模型。如果晶粒非常大,整個晶粒對電阻或導電度的貢獻會相對很小,其表面電阻可 視為定電阻,此模式普遍適用於 p 和 n 型感測器。 (Eq. 2-5)
2-2 金屬氧化物半導體氣體感測器(MOS gas sensor)
1962 年就已經發現金屬氧化物表面對氣體吸附或脫附會改變物質的導電性,在 ZnO 薄膜中,對表面氣體的靈敏度(Sensitivity)可達十億分之一(ppb),金屬氧化物半導體感測 器具有極大比表面積以作為吸附目標,可使分析物盡可能吸附於表面,進而產生更顯著 的響應,且提高對低濃度氣體的靈敏度。[5]氧化物半導體氣體感測器之感測原理是半導 體氧化物表面導電度會因為氣體吸附而發生顯著改變,當氧分子吸附在 n 型氧化物半導體表面時,如 ZnO 和 TiO2等,表面的電子轉移給氧分子,形成陰離子(O-),以化學吸附
(Chemisorption)型態存在於氧化物表面上,在氧化物表面區域形成載子空乏層(Depletion
layer),進而降低了 n 型氧化物半導體的導電性。當環境中的還原氣體,如 CO 或 H2,
與吸附在氧化物半導體表面的氧離子發生反應,表層電荷密度會因此改變,導致表面空
間電荷(Space charge)區寬度發生改變,而造成導電度變化,例如氧離子吸附在 SnO2(n
型氧化物半導體)上,使得表面形成較寬的空乏區,造成電阻升高,如Fig. 2-3 [6],這類 感測器用來監測空氣中不同的低濃度氣體,操作溫度在 300 o C−500 oC 之間,長久以來, SnO2為主要的氧化物半導體感測材料,其次為 Zn,W,Ti 等氧化物。由於氣體感測器 的導電度變化量是由表面吸附反應所決定,因此感測材料表面積越大,感測靈敏度則越 高,奈米結構材料具有相當大的表面積-體積比,很適合需要大表面積的氣體感測應用, 因此成為近年來被廣泛研究的目標。尤其重要者,當感測材料的組成結構體(如晶粒)尺 寸小於傳導電子的德拜長度時,氣體感測靈敏度與響應速率都可大幅提高,氣體吸附所 引致的感測材料導電度變化是因為材料表面空乏區變化,一般金屬氧化物的空間電荷層 寬度(L)可延伸到數十奈米,如果能將感測材料的結構體尺寸縮小到 2L 以下,氣體吸附 所引致的表面導電度變化將會相當顯著,一般奈米結構氧化物半導體材料即符合如此的 尺度要求,一旦曝露於受測氣體的氛圍中,氣體分子可以立即與氧化物半導體表面發生 吸附反應,所以奈米結構氧化物半導體是一種理想的氣體感測材料。
7
Fig. 2-3:Schematic diagram of negatively charged chemisorbed oxygen species cause an upward band bending and consequently a depletion layer in the near-surface region. [6]
2-3 MOS 基 CO 感測器
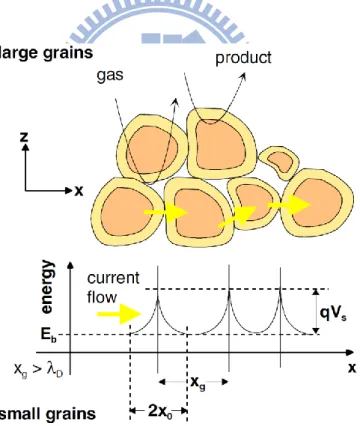
Table 2-2 [7]中列舉具有潛力之 CO 感測材料,其中檢測 CO 最好的金屬氧化物為 SnO2、ZnO 等。 Barbi 等人[14]開發了以 SnO2為基礎之感測器,顯示出對濃度 10 ppm 以上 CO 氣體 的響應,其理想的工作溫度為 250 o C,在 20 ppm 和在 100 ppm 的響應(R0/ R)分別為 2.2 和 4.1,響應隨著濃度穩定增加。Pt 的摻雜(Doping)使得結構改變,一般而言,和純 SnO2 相比,Pt 摻雜使得感測靈敏度下降,響應時間也變慢。此外,該作者亦顯示了控 制晶粒尺寸之重要性(Fig. 2-4)。在晶粒尺寸為 40 Å 時,對氣體有最大的響應,這是因 為整個晶粒可作為反應表面。Riviere 等人[15]採用網版印刷(Screen printing)製造不同前驅物所組成之 SnO2膜,該
材料表現出強烈的響應,在溫度 500 o
C,300 ppm CO 時導電率增加為原來的十倍(Fig.
2-5),不過,值得注意的是,SnO2薄膜在 500 oC 的空氣中保持一個小時後才導入 CO,
有助於維持導電率之安定。
Tischner 於 2008 年[16]利用噴霧熱分解法(Spray pyrolysis)製備之 SnO2薄膜(50-100
nm),在 250-400 oC 溫度區間有極佳之感測表現,可檢測 CO 濃度為 5 ppm,是一個正 常家庭中有可能會暴露的濃度,此薄膜的響應和溫度有關:在 250 o C 時,薄膜在 0-40 ppm 表現出良好的響應;在 400 o C 時,增加了其響應範圍為 0 至 100 ppm。另外還發現,薄 膜對於濕度的改變極為敏感(Fig. 2-6),可應用於預期濕度變化量。作者認為由於晶界效 應,越薄的薄膜可能越敏感。
就目前而言,檢測 CO 最好的金屬氧化物半導體材料是 SnO2,其他的如 CeO2和 TiO2,
雖然已進行研究,但是因為 SnO2有穩定的化學與機械性質,且在低濃度 CO 環境中,
9
Table 2-2:Case studies of CO gas sensing on semiconducting metal oxides. [7]
Material Temperature (oC) Detection limit Reference
SnO2 RT-700 1000 ppm [8] 25-400 200-3000 ppm [9] 200 1% [10] ZnO 700 200 ppm [11] 350 250 ppm [12] 250 60% [13]
Fig. 2-5:Influence of organic vehicle content in inks without mineral binder on the response of sensors to CO (300 ppm) at 500 oC:(1) 41.3 wt.%, (2) 35.0 wt.%, (3) 23.8 wt.%, (4).17.8 wt.%. [15]
11
2-4 SnO
2之 CO 感測特性
SnO2是一種寬能隙 n 型半導體,其導電特性與內部的氧空位相關,被廣泛應用於 檢測有毒氣體,其優點如成本低、靈敏度高,非常有吸引力,而缺點如缺乏選擇性和水 氣的干擾效應強烈,所以阻礙測量設備的使用。SnO2感測器一般在空氣中的工作溫度介 於 200 和 400 o C,在此溫度下材料可傳導電子,表面也對環境中的氣體產生化學吸附, 感測材料的導電率決定於表面的反應,即電極間半導體材料的電荷轉移和傳輸機制,例 如普遍認為氧氣離子吸附(Ionosorption)的效果為 O2-或 O-附著在材料表面建立一個負電 荷的表面,增加表面電阻,還原性氣體如 CO 和表面的氧離子反應,將電子釋放回感測 材料,此轉導(Transduction)步驟,即實際的電荷轉移以減少感測電阻。在 SnO2表面氧 的離子種類已用光譜技術被證明:在低溫下(<200 o C),SnO2表面上的氧為分子性吸附,即吸附中性氧,O2(abs),或是吸附帶有電荷的氧,O2(abs)-;然而在較高溫度下,為氧的解
離吸附,即 O(abs)或 O(abs) -。 在較低溫度: CO + 𝑂2− → 𝐶𝑂 3− 𝐶𝑂3−+ 𝐶𝑂 → 2𝐶𝑂 2+ 𝑒− 在較高溫度: CO + 𝑂− → 𝐶𝑂 2− 𝐶𝑂2− → 𝐶𝑂 2 + 𝑒− 此機制預測在低溫下兩個 CO 分子的一個電子被釋放(Eq. 2-6 及 Eq. 2-7),而在較高溫度 下,每 CO 分子的一個電子被釋放(Eq. 2-8 及 Eq. 2-9),因此,感測器的響應在較高溫度
較強。在 Bârsan 和 Weimar 的研究中[17],感測層由單一尺寸的晶粒組成(Fig. 2-7),自
由電荷載子(SnO2的電子)必須克服晶粒的表面障礙才能夠傳導(Fig. 2-8),這取決於感
(Eq. 2-6)
(Eq. 2-7)
(Eq. 2-8)
測層的型態,即使化學性質完全相同的表面,在感測 CO 氣體時,感測器的電阻可能會 因為感測層的孔隙度(Porosity)而非常不同。
Fig. 2-7:SEM pictures of the SnO2 sensing layer. [17]
Fig. 2-8:Schematic representation of a porous sensing layer with geometry and energy band.
13
Bahrami 等人[18]報告了當存在 CO、CH3和 C3H8的環境中,Au NPs 在以 SnO2為基
礎的感測器中對靈敏度和選擇性的影響,1% Au/SnO2 粉末以共沉法(Co-precipitation
method)製備,清洗後在 150 oC 烘乾,而後在 300 oC 下煅燒 3 小時,Au NPs 大多 3 至 5 nm,分佈在 SnO2表面,Fig. 2-9(a),SnO2和 Au/SnO2的 BET 表面積測定分別為 210 和
110 m2/g,分別對應到 4 和 7.5 nm 的球形鬆散顆粒,研究在 40-1000 ppm 的 CO 的環境 中,溫度區間 170‒300 o
C,Au/SnO2和 SnO2傳感器的響應。和 SnO2感測器相比,Au/SnO2
感測器對 CO 具有更高的響應,Au/SnO2和 SnO2分別在 250 oC 和 275 oC 表現出最大靈
敏度,由於 Au 的促進作用,Au/SnO2感測器對 CO 的最高響應發生在相對較低的溫度,
Fig. 2-9(b)。在 Au/SnO2感測器的感測器對 CO 的感測中,體積小且高度分散的 Au NPs
提高了靈敏度和選擇性。
Fig. 2-9:(a) TEM micrographs of 1.0 wt% Au/SnO2. (b) Response of Au/SnO2 (solid line) and SnO2 (dashed line) to various concentrations of CO at different temperatures. [18]
2-5 光學式氣體感測
表面電漿子的研究已有百年歷史,在二十世紀初,就已經在金屬光柵的反射光譜中 觀察到表面電漿子相關的光學現象。電漿子(Plasmon)是一種貴金屬表面自由電子隨著電 磁波而產生的集體式電偶極振盪現象,以反抗外在電磁場的穿透,如果將這種集體振盪 行為量子化,我們稱為表面電漿共振(Surface plasmon resonance),存在外加電場將會造 成金屬自由電子相對於固定離子核的位移,對塊材電漿子(Bulk plasmon)來說,這些振盪 會發生在電漿振動頻率(Plasma frequency),並具有能量[19]: Ep = ℏ√𝑚ℰ𝑛ℯ2 0 其中ε0為自由空間的介電常數,n 為電子密度,e 是電子電荷,m 是電子質量。 在金屬表面,電漿子的形式是表面電漿子極化(SPPs),也簡稱為表面電漿子(Surface
plasmon),Fig. 2-10(a) [20],是存於金屬與介電質介面上的表面電磁波。表面電漿子可
以光學方式被激發,當入射光通過金屬表面的光柵或缺陷時,可以被耦合到駐波或表面 電漿子模式,因為它是由入射帄行波所激發的表面電漿子振盪電場,以表面波(Surface wave)的形式在金屬與介電物質形成的介面上傳播,所以高角度入射的光(即與表面幾 近帄行的波相量κ)可以最有效地達到耦合現象。
考慮一個遠比入射光波長λ還小的圓球形金屬奈米粒子,因為奈米粒子非常小,金 屬粒子的集膚效應(Skin depth effect)可以被忽略,當它受到入射光的照射後,奈米粒子 上的自由電子會受到入射電場的影響而發生集體振盪的現象,我們稱之為奈米粒子的電 漿子現象(Particle plasmon),由於這樣的振盪行為發生在非常小的奈米粒子上,也稱之 為局域化表面電漿子(Localized surface plasmon, LSP),如Fig. 2-10(b)。局域化表面電漿 子會造成兩個重要的影響,首先,在金屬表面附近,形成高度增強的近場(Highly enhanced near-field),電場附近粒子的表面都被大幅增強,這種增強在表面會達到最大值,接下來 迅速地隨著距離的增加而下降,這表面增強的特性已被利用在各種表面光譜的量測上 [21],例如表面增強拉曼光譜(Surface-enhanced spectroscopy, SERS);其次,貴金屬奈米
15 粒子發生在可見光波長的消光,在電漿子共振頻率有一個最大值,消光波峰取決於周圍 介質的折射率。 表面電漿子在金屬的表面傳播,離開金屬表面,其電場大小依指數函數迅速衰減, 因此表面電漿子被約束在金屬表面,同時也增強金屬表面的電場,使表面電漿子對金屬 表面的特性格外敏感,也因此可利用來作為化學、生物之感測應用。
Fig. 2-10:Illustrations of (a) surface plasmons and (b) localized surface plasmon. [20] (a)
在色散關係上,局域化表面電漿子表現不若表面電漿子,其頻率與波向量之間的關 係,並非一條曲線,而是呈現一模糊區域,所以局域化表面電漿子的激發會直接表現在 其吸收或散射光譜。Kondow [22]將 Ag NPs 吸收峰,歸因於粒子電漿子共振,當入射光 頻率符合粒子電漿子共振頻率時,會因吸收入射光能量而激發產生粒子電漿子,形成吸 收峰,如Fig. 2-11。 金屬奈米球受到入射光之電磁場作用下,金屬球上導電電子團受到電場作用,以球 體為中心發生電偶極振盪現象並產生極化(Polarizability)。靜電極化 α 可表示如下: α= 4π𝑅3 𝜀 − 𝜀𝑚 𝜀 + 2𝜀𝑚 上式為假設一金屬球半徑為 R,介電常數為 ε,處在一個介電常數 εm的環境條件下所得 之結果。 影響材料介電常數ε 的因素有很多,其中最重要的為材料成分、尺寸以及形狀,此 即電漿子共振效應所需考量之重要參數。此外,金屬由於自由電子存在而擁有電漿子共 振,其中 Au、Ag 以及 Cu 因具備散射截面積(Scattering cross section)之優勢[23],成 為電漿子效應研究中最為重要的參考指標成分。
Fig. 2-11:Experimental (solid line) and Simulated by Drude theory (dash line) absorption spectra of Ag nanoparticles. [22]
17
對於核殼(Core-shell)奈米粒子,形狀對於電漿子共振關係則是反應在核心半徑與殼
層厚度之比例上,Halas [24]率先提出奈米殼(Nanoshell)名詞,並利用固定 SiO2核心半徑
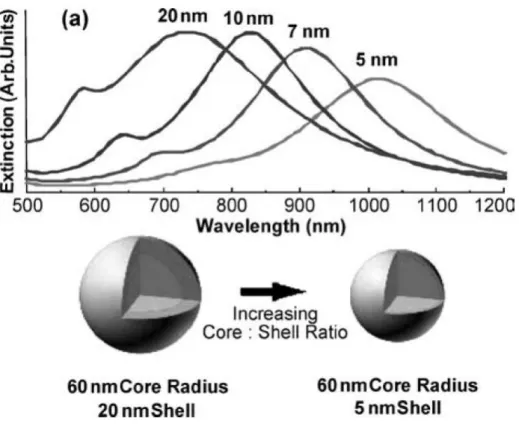
(120 nm),僅改變 Au 殼層厚度(5 nm~20 nm),來了解核殼型奈米粒子之電漿子共振之粒
子形狀依存關係,隨著 Au 殼層厚度的增加,電漿子共振波長會出現藍位移現象,如Fig.
2-12。以此理論觀點為依據,本論文以成功合成 Au NPs 及 Au-SnO2核/衛星異質組裝結
構(CSHA)為目標。
Fig. 2-12:Optical tunability is demonstrated for nanoshells with a 60 nm silica core radius and gold shells 5, 7, 10, and 20 nm thick. [24]
表面電漿子共振是最近大家相當感興趣的奈米尺度現象,貴金屬奈米結構之合成在 過去十年有了重大的進步,由於金屬奈米粒子的特徵吸收峰會受到介質環境的影響而產 生位移,例如Fig. 2-13 [25]為 50 nm 之 Au NPs 分散在不同的溶液中,隨著溶液介電常 數增加,Au NPs 之特徵吸收峰產生愈明顯的紅移現象。此外,表面電漿共振氣體感測 器量測快速、無須標記,透過奈米粒子表面電漿共振之光譜變化,可以檢測分子間相互 作用,此技術已被應用在生物及化學感測器。Cheng 等人[26]已經證明表面電漿子感測 可以感測揮發性有機化合物,如甲苯,在研究報告中,透過粒子消光光譜之峰值波長和 總積分面積,針對吸附在 Au NPs、Ag NPs 和 Au 殼層上的化合物進行分析,此反應快 速並可逆,如Fig. 2-14所示;後來,使用堆疊基板系統,設計針對不同有機物的三種自 組裝單分子膜(SAMS),如Fig. 2-15 [27]所示,發現九種不同氣體與相互微反應模式,檢 測濃度的極限可下降至 16 ppm。由 Karakouz 等人[28]發展的表面電漿共振氣體感測器 結構為熱蒸鍍之 Au 島狀薄膜上披覆聚苯乙烯磺酸(PSS)和聚苯乙烯(PS),如Fig. 2-16所 示,氣體(氯仿、水蒸氣等)造成這些聚合物的膨脹或收縮,進而影響折射率並導致表面 電漿共振波峰改變。Gaspera 等人[29]在 2011 年回顧他們對於光學氣體感測器的研究, 其合成多孔性金屬氧化物奈米材料與 Au NPs 的複合薄膜,孔隙提供了氣體分子一個達 到奈米粒子表面的路徑來進行化學反應,Au NPs 的導入提高了對氣體的感測,而且 Au NPs 的表面電漿子共振峰可以被利用來作為選擇性光學氣體感測器,如Fig. 2-17所示。
19
Fig. 2-14:(a) Scheme of LSPR–VOC sensor testing system (b) LSPR spectrums of Ag nanoparticles responding to 0∼100% relative saturated toluene vapor concentrations. (c) Spectrum integration area of Ag nanoparticles between 350∼550 nm. [26]
(a)
(b)
Fig. 2-15:(a) Scheme of building LSPR sensor array assembly in a flow-cell cube. Real-time response signals tested with (b) m-xylene, insert shows the magnified response curves of NAP-Au-Npat 2000 ppm and (c) n-pentanol (wavelength regions: +, 395–475 nm; −, 480–620 nm; ○, 690–850 nm). [27]
(a)
(b)
21
Fig. 2-16:(a) Schematic presentation of polymer-coated gold island film preparation. Spectral response of the vapor sensor based upon gold island films coated with (b) PS and (c) PSS. [28]
(a)
(c) (b)
Fig. 2-17:(a) Schematic of the sol–gel sensor architecture. (b) Effect of H2 in the optical absorption of SiO2-NiO-Au nanocomposite. Absorption spectra of films annealed at 500 oC measured in air and under a 1% v/v H2 mixture at 300 oC operative temperature. [29]
(a)
23
2
-6 研究動機與目的
科技發展歷程顯示出人類認識的世界正逐步從巨觀走到微觀,再從微觀走向原子的 世界。微觀是指建構在微米基礎上的物質世界,而現在正好是處於微觀與基本粒子世界 的中間階段―奈米階段,而奈米技術(Nanotechnology)被認為是最有潛力成為下一個影響 人類的革命性技術,足以改變我們生存的世界。當粒子的尺度小於0.1 μm,就會被歸類 為奈米材料的範疇,由於奈米化後位於表面之原子數明顯增多,表面原子配位不飽和度 高且表面張力大,因此,這些表面原子具有較高的活性;此外,奈米材料表面原子的熱 力學(Thermodynamics)穩定性較差,很容易與其他原子結合以降低其表面張力,呈現出 非常活潑的化學性質,以及十分活躍的擴散效應[30]。本研究乃針對現行的金屬氧化物 氣體感測器之感測材料進行結構設計,藉由奈米技術的導入,期能提高感測器對有毒氣 體的選擇性與靈敏度,並在較低的操作溫度下維持其感測訊號的強度。過去二十年來,奈米金屬氧化物半導體如 SnO2、ZnO、TiO2、NiO 等均已被證明具
有感測氣體分子應用之潛力[31]。如前所述,感測特性的增強主要是由化學反應性和比 表面積來決定[32],而此兩者均與材料結構直接相關,藉由合成方法的設計改良與控制, 來形成具有高選擇性和高靈敏度特性之奈米結構。 其中,添加金屬奈米粒子為一有效之改良策略。金屬奈米粒子具有催化能力,因此 可以大幅提高感測過程中分析氣體和氧化物之間的化學反應性。另外,金屬奈米粒子亦 能增加金屬氧化物之導電性,有利於提高量測之方便性。更重要的是,金屬奈米粒子在 可見光範圍內顯示電漿子共振峰,如 Au 或 Ag,故此類奈米複合材料就可以用來作為選 擇性光學氣體感測器,圍繞表面電漿子共振帶的介電常數變化將隨著氣體種類的不同而 有所不同,這會導致在不同波長下光學特性多樣化的變異,如此就可以提高對感測氣體 的選擇性,此一特點是傳統電阻式感測器所無法實現。 目前已有文獻報導指出[33,34],當 SnO2摻雜入貴金屬 M{M=金(Au)、銀(Ag)、鉑(Pt)、 鈀(Pd)等}形成複合材料或核衛星結構時,其氣體感測性質可獲得改善,但是在 300-400 o C 的操作溫度下,會使得核衛星的金屬奈米粒子產生熔融團聚,使之結構維持困難。為
改善上述之團聚現象,Yu[35]提出以 Au 奈米粒子為核、SnO2奈米粒子為殼之結構,利 用 SnO2殼層去干擾奈米金屬核在高溫下可能產生的聚集反應,且金屬核也可以幫助二 氧化錫奈米粒子之形成並抑制其在高溫下之晶粒成長。結果顯示:其可將最佳操作溫度 降至 200 o C 並維持其效果。然而,對於一般作業環境或居家環境而言,若能將氣體感測 器之最佳操作溫度再更進一步降至室溫,則除了提高其商業價值外,在市場上的實用性 與普及性亦會隨之擴大,進而提昇整體社會與工業安全。
本研究所設計的 Au core- SnO2 satellite 奈米異質複合材料為一利用光學特性變化來
感測氣體之濃度。主要是利用觀察貴金屬(Au, Ag)奈米粒子位於可見光區表面電漿子共 振峰之光學變化,來達到量測 CO 濃度之目的。其主要量測原理就是依靠金屬奈米粒子 表面電漿子共振峰對周圍界質介電係數極度敏感之特性,故當 CO 吸附或是脫附時,如 前所述, SnO2 NPs 將產生顯著之電阻變化,故將直接對位於核心之 Au NPs 產生影響, 導致表面電漿子共振峰值及位置產生變化。相較於傳統電阻式金屬氧化物半導體感測器 需要在 300 o C 以上進行操作,光學氣體感測器將具有室溫可操作且環保之特性,可大幅 提昇氣體感測器在不同作業環境之應用與人員安全。
25
三、 實驗方法與步驟
3-1 實驗藥品
1. 錫酸鈉(Sodium TinOxide Trihydrate):Na2SnO3,98%,Alfa Aesar。
2. 四氯金酸(Hydrogen Tetrachloroaurate (III), hydrate):HAuCl4,99.99%,Alfa Aesar。
3. 檸檬酸三鈉(Tri-sodium Citrate, Dihydrate):HOC(COONa)(CH2COONa)2,J.T. Baker。
4. 氫氧化納(Sodium Hydroxide):NaOH,98%,Mallinckrodt Baker Inc.。 5. 乙醇(Ethanol):C2H6O,95%,友和貿易有限公司。 6. 丙酮(Acetone):C3H6O,99%,友和貿易有限公司。
3-2 實驗設備
儀器名稱 廠牌及型號 加熱器 CORNING, PC-420D 低溫高速離心機 HITACHI, CF15RX II 超音波共振機 DELTA, DC200H 烘箱 DENGYNG, DO45桌上型 PH 計 EUTECH, Cyber Scan PH510
紫外光-可見光光譜儀 THERMO SCIENTIFIC, Evolution 300
X光繞射儀 BRUKER, D2
熱場發射掃描式電子顯微鏡 JEOL, JSM-6500F
冷場發射掃描式電子顯微鏡 JEOL, JSM-6700F
場發射高分辨穿透式電子顯微鏡 Philips Technai G2 Dispersive X-ray Spectrometer (EDS)
3-3 Au-SnO
2CSHA 之製備法
本實驗中 Au-SnO2 CSHA 之合成分為兩階段[36]:第一階段利用檸檬酸鈉還原法製備 Au 奈米粒子膠體溶液。步驟如下:將 HAuCl4水溶液(500 ml, 0.4 mM)中速攪拌並加熱到 沸騰,同時將 tri-sodium citrate 水溶液(25ml, 34mM)加熱至沸騰,混和快速攪拌,持溫 30 min 後冷卻至室溫,即可得 Au 膠體溶液,此溶液是由檸檬酸離子維持穩定。第二階 段則是將 Na2SnO3水溶液(2 ml, 40 mM)加入前述 Au 膠體溶液中,攪拌 30 min,使溶液 溫度上升至 60 o C,持溫 1 hr 形成 Au-SnO2 CSHA,此膠體溶液可維持穩定約 1 個月。 製程溫度、pH 值和錫酸鈉離子濃度均會影響此 Au-SnO2複合奈米粒子殼層的均質程度。 在溶液中也包含了獨立存在之 SnO2 NPs,可以透過離心清洗降低佔有率,卻也會使得 Au-SnO2 CSHA 產生團聚。我們嘗試調整錫酸鈉濃度和 Au 膠體溶液之 pH 值來控制殼層 厚度及孔隙度,還有提升 Au-SnO2 CSHA 的產率。27
3-3-1 Au NPs 之製備法
(1) 配製 500 ml 的 0.4 mM HAuCl4水溶液
(2) 配製 25 ml 的 34 mM tri-sodium citrate 水溶液
(3) 分別將 HAuCl4水溶液和 tri-sodium citrate 水溶液加熱至沸騰
(4) 將 tri-sodium citrate 水溶液加入 HAuCl4水溶液中:以磁石攪拌子快速攪拌
(5) 在 100 oC 下持溫 30 min
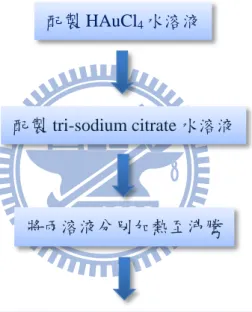
Fig. 3-1:Process of synthesizing Au NPs by tri-sodium citrate.
配製 HAuCl4水溶液
配製 tri-sodium citrate 水溶液
將兩溶液分別加熱至沸騰
將 tri-sodium citrate 水溶液加入 HAuCl4水溶液
在 100 o
以 NaOH 溶液調整 Au 奈米粒子溶液之 pH 值 將 Na2SO3水溶液加入 Au 奈米粒子溶液 製備 Na2SnO3水溶液 攪拌 30 min 加熱至 60 o C,並持溫 1hr
3-3-2 Au-SnO
2CSHA 之製備法
(1) 配製 0.1 M NaOH 水溶液 (2) 取 10 ml 的 0.4 mM Au 奈米粒子溶液 (3) 將步驟(2)之 Au 奈米粒子溶液以步驟(1)之 NaOH 水溶液調整 pH 值:分次少 量加入以調整至 pH 10.5 (4) 配製 2 ml 的 Na2SnO3水溶液 (5) 將步驟(4)之 Na2SnO3水溶液加入已調整好 pH 值之 Au 奈米溶液 (6) 以磁石攪拌子將溶液中速攪拌 30 min (7) 使溶液在水浴環境中溫度緩緩上升至 60 oC (8) 在 60 oC 下持溫 1 hr (9) 將(8)所得之溶液離心,離心參數為轉速 15000 r.p.m., 溫度 5 oC,時間 30 min。 離心之後將上層溶液取出廢棄,並將所得之奈米粒子散佈於酒精中清洗,再次 離心,以確保奈米粒子表面之潔淨度。29
3-4 奈米結構分析
3-4-1 場發射穿透式電子顯微鏡(FE-TEM)
穿透式電子顯微鏡(TEM)為近代分析奈米材料/奈米結構最有力的分析工具之一,與 SEM不同的是,TEM的高能電子束可穿透厚度約100 nm以下的薄樣品,藉由高能穿透電 子與樣品內部原子的交互作用,產生不同程度的散射。經由偵測器的訊號收集與後續的 訊號處理,即可得到材料的形貌圖或繞射點圖譜,可用於分析材料之形貌、尺寸分布、 生長方向、結晶性等等性質。 試片製作: 將欲觀測之奈米材料分散於水、酒精、丙酮等溶液中,溶液濃度控制在 20~50 ppm 為佳,接著使用 200 mesh 之鍍碳銅網浸入含有奈米材料之溶液中,等待幾秒鐘後取出, 將鍍碳銅網放入 40 o C 烘箱中乾燥 1 天,待其完全乾燥後即可進行 TEM 之觀察。3-4-2 X光繞射分析(XRD)
XRD 分析為應用相當廣泛的分析技術之一,由於每種結晶材料皆有其特殊的晶面 間距,而 X 光只有在入射角度與材料晶面間距滿足布拉格繞射定律(Bragg Law),且繞 射光強度經過材料的結構因子(Structure factor)計算後不為零時,才會因產生建設性干涉, 在 X 光繞射圖譜上顯示出特定的峰值,因此透過分析所得的 XRD 圖譜,可得到待測物 之成份、晶粒大小、結晶性、結晶方向、相比例等等資訊,且其分析為非破壞性分析, 對具有結晶性之物質的鑑定特別有利。 各種結晶物質的 XRD 繞射峰位置與強度可由 JCPDS 資料庫查詢而得,而經由 Scherrer equation 計算圖譜峰值的半高寬,則可得知該材料之晶粒尺寸,為 XRD 分析相 當常用的工具,Scherrer equation 如 Eq. 3-1 所示。L = 0.91𝜆 𝐵 ∙ cos 𝜃
L:帄均粒徑大小(Å ) B:繞射峰半高寬(rad) θ:布拉格繞射角(degree) λ:X-ray 波長(Å ) 分析條件: 操作電壓:30 kV 操作電流:10 mA 掃描模式:Theta-two theta 掃描速率:0.04 °/sec。
3-4-3 紫外光-可見光吸收光譜(UV-vis absorption spectra)
紫外光-可見光吸收光譜常用於金屬奈米粒子之鑑定,製備完成之金屬奈米粒子可 直接散佈於溶液中進行檢測,也可分散於基板上形成薄膜再進行檢測。其原理為利用金 屬的表面電漿子共振效應,當金屬奈米粒子受到入射光照射時,其表面電荷與入射光之 電磁場交互作用而形成表面電漿子共振,不同的材料有不同的共振情形,利用紫外光-可見光吸收光譜可以很容易地觀察到。因為不同的材料、成份比例、粒徑大小、形貌皆 會對其表面電漿子共振效應造成影響,因此除了成份鑑定之外,也有許多人利用紫外光 -可見光吸收光譜來探討奈米材料的尺寸、核殼結構奈米粒子的成份比例、厚度等性質。 試片製作: 將待測之材料分散於水、酒精、丙酮等溶液中,或塗佈於石英玻璃上即可。第一次 量測時需準備另外兩組用於承載待測樣品之溶液或基板,量測建立基準線,以做為扣除 背景值之用,背景值扣除完畢後再將所要量測之樣品放入儀器中進行量測即可。 分析條件: Photometric Mode:Absorbance Band Width:1.0 nm Data interval:0.5 nm
31
3-5 光學式 CO 感測特性
光學氣體感測是利用消光光譜(Extinction spectra)收集峰值的變化以監測氣體濃度, 波長範圍為 200-1100 nm,試片基材為尺寸 2 cm×2 cm×1 mm 石英玻璃,薄膜試片被夾 在加熱器上,此加熱器固定在一個訂製的不銹鋼氣體槽中,由兩片 9.5 cm×7.5 cm×1 mm 石英玻璃夾住氣體槽的兩邊,以供光入射試片,供氣體進出之通道用內徑 4 nm 的血清 圔覆蓋,此氣體槽安裝在紫外光可見光光譜儀的樣品槽位置,如 Fig. 3-3。操作溫度由 常溫至 300 o C,溫度是利用加熱器上的感溫棒回饋系統控制,不同濃度的 CO 氣體由乾 燥空氣稀釋,並利用氣密式玻璃注射筒注入氣體槽中,放氣時將兩個通道同時打開,並 利用小幫浦將氣體槽內之氣體抽出、乾淨氣體進入。 試片製作: 石英基材在去離子水、酒精和丙酮中置於超音波震盪機中各震盪 15 min,取出後在 40 oC 的烘箱中乾燥,利用反應式離子蝕刻機(RIE)將石英基材做氧電漿處理 10 min,以 增加基材表面的親水性,氧電漿處理具有時效性,所以需要立即將樣品滴在基材上。已 合成之 Au-SnO2 CSHA 膠體溶液需經過三次的離心清洗,以維持粒子表面乾淨,並且可 分散在酒精溶液中,將已離心清洗之 Au-SnO2 CSHA 分散至 1 ml 酒精中,取 0.03 ml 分 散溶液滴在基板上,使其在空氣中自然乾燥,形成巨觀均勻的薄膜。 量測步驟: 第一次量測時需在參考槽夾上三片 1 mm 厚度的石英玻璃當作背景值,在氣體槽中 夾上乾淨表面的石英玻璃,並以石英玻璃夾住氣體槽的兩邊,而後將氣體槽至於樣品槽 位置,注意石英玻璃必須位於光路的焦點上,量測建立基準線,以做為扣除背景值之用, 背景值扣除完畢後再將欲量測之薄膜試片夾上氣體槽中進行量測,作為乾燥空氣中的參 考光譜,將不同濃度之 CO 氣體注入氣體槽中,濃度由低到高改變,每隔 5 min 量測一 次,每個濃度量測 0.5 hr,放氣時每隔 5 min 測一次,直到量測結果與未注入氣體前的 結果相同。33
四、 結果與討論
4-1 pH 值對 Au-SnO
2CSHA 結構之影響
Fig. 4-1為溶液 pH 10.5 合成之 Au-SnO2 CSHA,此結構是由尺寸約 1 nm 之 SnO2 NPs
緊密地堆積在直徑約 13 nm 球狀 Au NPs 表面上,形成厚度約為 7 nm 的殼層。Fig. 4-2
為溶液 pH 8 合成之 Au-SnO2 CSHA,由尺寸約 2 nm 之 SnO2 NPs 堆積成約 9 nm 較鬆散
的殼層。而在溶液 pH 5.8 合成之 Au-SnO2 CSHA,如 Fig. 4-3,是形成一大片 SnO2 NPs
團聚,Au NPs 則散布其中,稱不上為 CSHA。當溶液 pH 值增加,小尺寸之 SnO2 NPs
較均質且緊密地沉積在 AuNPs 表面,殼層之厚度亦隨之增加。
Fig. 4-4顯示了在不同 pH 環境中 Au-SnO2 CSHA 之 XRD 圖譜,將所得之 XRD 圖
譜與 JCPDS 比對,其繞射峰分別為 SnO2之(110)、(101)、(211)結晶面及 Au 之(111)、(200)
結晶面,而 SnO2 (JCPDS 41-1445)之繞射峰可對應至 Rutile 結構,此外,寬廣之繞射峰
顯示合成所得之 SnO2 NPs 尺寸極小,利用(110)繞射峰半高寬(FWHM),配合 Scherrer
公式可估算 SnO2 NPs 帄均晶粒尺寸,其與 pH 值之關係如table 4-1所示。結果發現在
Fig. 4-1:(a) TEM and (b) HRTEM images of the Au-SnO2 CSHA prepared with a stannate concentration of 40 mM at pH 10.5.
(a)
35
Fig. 4-2:(a) TEM and (b) HRTEM images of the Au-SnO2 CSHA prepared with a stannate concentration of 40 mM at pH 8.
(a)
Fig. 4-3:(a) TEM and (b) HRTEM images of the Au-SnO2 CSHA prepared with a stannate concentration of 40 mM at pH 5.8.
(a)
37
20 30 40 50 60
(111)
(200)
Two theta (deg.)
Int
ens
it
y
(a.
u.
)
(c) pH 5.8
(b) pH 8
(a) pH 10.5
(101)
(211)
(110)
Fig. 4-4:XRD patterns of the Au-SnO2 CSHA prepared with a stannate concentration of 40 mM at pH (a) 10.5, (b) 8, and (c) 5.8.
Table 4-1:The FWHMs of the SnO2 (111) peaks and the estimated particle sizes of the Au-SnO2 CSHA prepared at different pH values.
pH value FWHM of SnO2 (110) (deg.) Estimated grain size (nm)
10.5 7.4 1.1 8 4.1 2.0 5.8 7.5 1.1 JCPDS 41-1445 SnO2 JCPDS 04-0784 Au
4-2 前驅物濃度對 Au-SnO
2CSHA 結構之影響
Fig. 4-1為錫酸鈉溶液濃度 40 mM 所合成之分散性良好的 Au-SnO2 CSHA,其殼層
厚度約 5 nm。但是除了 Au-SnO2 CSHA 外,尚有許多單獨存在之 SnO2 NPs,換言之,
Au-SnO2 CSHA 的產率較低。當錫酸鈉之濃度降低至 5.6 mM,則 Au-SnO2 CSHA 的產
率大幅提升,如Fig. 4-5所示,SnO2殼層約 5 nm 且厚度較不均勻。但當錫酸鈉濃度降
低至 0.18 mM 時,如Fig. 4-6所示,SnO2前驅物不足以在 Au NPs 表面形成殼層,只形
成 Au NPs 間之橋狀連結。由上述觀察,在錫酸鈉濃度為 5.6 mM 時可得產率最高、殼 層最薄之 Au-SnO2 CSHA。
Fig. 4-7為不同錫酸鈉濃度下合成 Au-SnO2 CSHA 之 XRD 圖譜,清楚呈現出 Au NPs
繞射峰(JCPDS 04-0784)。隨著錫酸鈉濃度增加,SnO2之繞射峰的相對強度亦隨之增加。
利用 SnO2 (111)之 FWHM 推估晶粒尺寸,如table 4-2所示,發現隨著錫酸鈉濃度增加,
39
Fig. 4-5:(a) TEM and (b) HRTEM images of the Au-SnO2 CSHA prepared with a stannate concentration of 5.6 mM at pH 10.5.
(a)
Fig. 4-6:(a) TEM and (b) HRTEM images of the Au-SnO2 CSHA prepared with a stannate concentration of 0.18 mM at pH 10.5.
(a)
41
20 30 40 50 60
Two theta (deg.)
(111)
(200)
(a) Au@SnO2 40 mM (b) Au@SnO2 5.6 mM(101)
Int
ens
ity
(a.
u.
)
(c) Au@SnO2 0.18 mM(110)
(211)
Fig. 4-7:XRD patterns of the Au-SnO2 CSHA prepared at pH 10.5 with a stannate concentration of (a) 40 mM (b) 5.6 mM (c) 0.18 mM.
Table 4-2:The FWHMs of the SnO2 (111) peaks and the estimated particle sizes of the Au-SnO2 CSHA prepared at different stannate concentrations.
Ion conc. (mM)
FWHM of SnO2 (110)
(deg.)
Estimated grain size (nm)
40 8.792 0.928 5.6 12.208 0.668 0.18 14.229 0.573 JCPDS 41-1445 SnO2 JCPDS 04-0784 Au
4-3 Au-SnO
2CSHA 之光學特性-紫外光可見光吸收光譜分析
將在不同 pH 值環境下合成之膠體溶液進行紫外光-可見光吸收光譜分析,如Fig. 4-8
所示,圖中隨著 pH 值從 5.8 增加至 8 及 10.5,波峰位置由 531 nm、534 nm 移至 540 nm。
Fig. 4-9顯示 13 nm Au NPs 膠體溶液和不同 Na2SnO3濃度合成之 Au-SnO2 CSHA 其紫外
光-可見光吸收光譜。隨著濃度的增加,波峰位置從 520 nm (pure Au NPs) 分別移動到 535、538 及 540 nm,溶液顏色由紅變紫。我們可以利用 Au NPs 核之表面電漿子共振波 峰的位移去判斷 SnO2 殼層沉積狀態。Oldfield 等人[36-38]提出一個近似式用來估算在 Au NPs 表面塗覆 SnO2 NPs 之表面電漿子共振波峰: 𝜆2 = 𝜆𝑝2[12.2 + 2𝑛2𝐻2𝑂+ 2𝑔(𝑛𝑆𝑛𝑂2 2 − 𝑛𝐻22𝑂) ∕ 3] (Eq. 4.1) 其中 g 是殼層所占粒子之體積分率(Volume fraction),n 是材料在測定波長的折射率 (Refractive index),λP是金屬的塊材電漿共振吸收波長。SnO2的折射率(nSnO2=2.0)大 於水的折射率(nH2O=1.33),故當 Au NPs 被 SnO2 NPs 披覆時,會發生的強烈的紅移現
43 200 300 400 500 600 700 800 A bso rb an ce Wavelength (nm) (a) pH 5.8 (b) pH 8 (c) pH 10.5
Fig. 4-8:UV-Vis absorption spectra of the Au-SnO2 CSHA prepared at (a) pH 5.8, (b) pH 8, and (c) pH 10.5. 200 300 400 500 600 700 800 Wavelength (nm) (a) Au colloid 0.4 mM (b) Au@SnO2 40 mM (c) Au@SnO2 5.6 mM (d) Au@SnO2 0.18 mM
stannate ion concentration(M)
0 0.04 A b so rb a n ce
Fig. 4-9:UV-Vis absorption spectra of the Au-SnO2 CSHA prepared with a stannate concentration of (a) 40 mM, (b) 5.6 mM, and (c) 0.18 mM.
pH value
在此將實驗數據利用上述理論進行模擬計算,由 TEM 影像得知所合成之 Au 核心 直徑約為 13 nm,SnO2殼層厚度為 5-9 nm,而水介電常數固定為 1.33,模擬過程中一次 只變動一個參數,其餘參數皆保持不變。Fig. 4-10為 SnO2殼層厚度分別為 1 nm、3 nm、 5 nm、7 nm 以及 10 nm 之 Au-SnO2核殼粒子模型計算所得之 UV-Vis 光譜。結果發現, 當厚度為 1 nm 時,波峰位置與純 Au NPs 相同,為 520 nm,故此厚度對 Au NPs 之表面 電漿子現象造成之影響不大。當厚度為 3 nm 時,波峰開始產生紅移現象,當厚度至 10 nm, 波峰逐漸向長波長方向移動,殼層厚度越厚,所造成之紅移現象越明顯。將此模擬光譜
與實驗數據比較,由 TEM 觀察得厚度為 7 nm 的 Au-SnO2 CSHA 其波峰位置在 540 nm,
與模擬光譜上之殼核厚度 3 nm 曲線之波峰位置吻合,此結果可能與樣品核殼粒子是由 SnO2 NPs 堆積而成之非連續殼層,而計算模擬假設為緻密殼層,故可得此樣品之等效緻 密厚度為 3 nm。 進一步利用等效介質理論(EMT),建立殼層為 SnO2 NPs 與介質混合之殼層,固定 殼層為 7 nm,搭配核殼奈米粒子之模擬計算可得等效介質理論吸收光譜,如Fig. 4-11 所示,當 SnO2所占體積比例為 1 時,波峰位置在 575 nm。比例降低為 0.5 時,波峰位 置剛好在 540 nm,與樣品之波峰相符。當比例為 0.1 時,波峰位置在 520 nm,幾乎對 Au NPs 之表面電漿效應不造成影響,當殼層的介電常數變小,SnO2所占體積比例越低, 表面電漿共振波峰發生強烈的藍移(Blue shift),這一點在以表面電漿共振波峰為基礎的 檢測技術上很重要。
45 200 300 400 500 600 700 800 10 1 shell thickness (nm) A bso rb an ce Wavelength (nm) t=1 t=3 t=5 t=7 t=10
Fig. 4-10:Simulated absorption spectra of the Au-SnO2 core-shell models with various shell thicknesses. 200 300 400 500 600 700 800 A bso rb an ce Wavelength (nm) f = 1 f = 0.5 f = 0.1
Volumn ratio (f) of shell
Fig. 4-11:Simulated absorption spectra of the Au-SnO2 core-shell models with different volume ratios of SnO2 NPs to medium (water).
4-4 光學式氣體感測
在本研究中,將討論不同 SnO2殼層厚度對 CO 感測特性之影響。曝露在 CO 中的
Au-SnO2 CSHA,其氣體濃度變化可透過可見光波段光吸收度的變化得知,這種變化是
由於 CO 與奈米粒子表面產生物理吸附或化學吸附的交互作用而造成,吸收度變化率 (Absorbance change ration, ACR)被定義為(ABSCO – ABSair)/ ABSair [39],在空氣中
的吸收度(ABSair)被用來做為數據的正規化,以除去任何可能出現的差異,例如儀器校
正差異。
Fig. 4-12和Fig. 4-14分別為 7 nm SnO2 殼層之 Au-SnO2 CSHA,在 200 oC 和常溫
下,於空氣和 1000 ppm CO 中之吸收光譜。而Fig. 4-13和Fig.4-15則分別為空氣和 1000 ppm CO 二光譜之差異。量測 544 nm 吸收峰時發現,整個波長範圍內吸收度為增加,吸 收度變化率在 200 o
C 時為 0.021,而常溫下則是 0.019。由此推知,核/衛星結構設計能
在常溫下維持可辨視之光學訊號變化率,且與高溫之變化率差異甚微。Fig. 4-16為 5 nm
SnO2 殼層 Au-SnO2 CSHA 於空氣和 1000 ppm CO 中之吸收光譜,根據Fig. 4-17,最大
的吸收變化量皆出現在 556 nm 周圍,樣品薄膜之吸收度變化率為 0.014,此吸收度的增 加可歸因為在感測一氧化碳氣體時複合粒子中的電子密度增加[40]。 為了評估 Au-SnO2 CSHA 對 CO 的靈敏度,將其置於不同 CO 濃度的環境中進行臨 場動態檢測,最低測試濃度為 5 ppm,如Fig. 4-18所示,可發現在 1000 ppm 以下吸收 度的變化非常劇烈,Fig. 4-19為 ACR 和 CO 濃度對數關係圖,發現非常接近線性關係, 由此可見,在較低 CO 濃度具有較高之 ACR。 有兩種機制可以用來解釋 Au NPs 對氧化物檢測還原性氣體之影響[41]:第一種 是 Au 促進了催化活性的增加;第二種則為 Au NPs 對電漿子吸收變化的增強,它與催 化沒有直接關係,表面電漿子共振頻率強烈地被介質的物理性質影響,如介電常數。本 研究設計合成之 Au-SnO2 CSHA,因為 Au 的催化性質,成功地將 CO 氣體感測的操作 溫度降至室溫,SnO2與 CO 氣體分子作用而改變其介電性質而影響了 Au 奈米粒子的電 漿吸收峰,使得 CO 氣體在常溫之可見光環境中可被檢測。
47 300 400 500 600 700 800 0.02 0.03 0.04 0.05 0.06 0.07 0.08 0.09 0.10 0.11 0.12 Abs orbanc e Wavelength (nm) air 1000 ppm CO
Fig. 4-12:Absorption spectra of the Au-SnO2 CSHA with a 7 nm SnO2 shell measured in dry air and after exposure to 1000 ppm CO at 200 oC.
400 500 600 700 800 0.0000 0.0005 0.0010 0.0015 0.0020 0.0025 ABS(C O)-ABS(air) Wavelength (nm)
400 500 600 700 800 0.02 0.03 0.04 0.05 0.06 0.07 0.08 0.09 0.10 0.11 0.12 Abs orbanc e Wavelength (nm) air 1000ppm CO
Fig. 4-14:Absorption spectra of the Au-SnO2 CSHA with a 7 nm SnO2 shell measured in dry air and after exposure to 1000 ppm CO at room temperature.
400 500 600 700 800 0.0000 0.0005 0.0010 0.0015 0.0020 ABS(C O)-ABS(air) Wavelength(nm)
49 400 500 600 700 800 0.00 0.01 0.02 0.03 0.04 0.05 0.06 0.07 0.08 0.09 0.10 0.11 Abs orbanc e Wavelength(nm) air 1000ppm CO
Fig 4-16:Absorption spectra of the Au-SnO2 CSHA with a 5 nm SnO2 shell measured in dry air and after exposure to 1000 ppm CO at room temperature.
400 500 600 700 800 0.0000 0.0005 0.0010 0.0015 0.0020 ABS(C O)-ABS(air) Wavelength (nm)
0 2000 4000 6000 8000 10000 0.0995 0.1000 0.1005 0.1010 A b so rb a n ce CO concentration(ppm)
Fig. 4-18:Absorbance at 556 nm obtained from the Au-SnO2 CSHA with a 5 nm SnO2 shell as a function of CO concentration. 0.1 1 10 100 1000 10000 0.000 0.005 0.010 0.015 A CR CO concentration(nm)
Fig. 4-19:ACR at 556 nm obtained from the Au-SnO2 CSHA with a 5 nm SnO2 shell versus CO concentration in logarithmic scale.

![Fig. 2-4:The effects of grain size on the response to the carbon monoxide. [14]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8543635.187820/25.892.119.805.243.486/fig-effects-grain-size-response-carbon-monoxide.webp)

![Fig. 2-10:Illustrations of (a) surface plasmons and (b) localized surface plasmon. [20]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8543635.187820/31.892.169.838.368.1008/fig-illustrations-surface-plasmons-b-localized-surface-plasmon.webp)
![Fig. 2-13: Extinction spectra of immobilized monolayers of gold solid colloids. [25]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8543635.187820/34.892.328.644.800.1110/fig-extinction-spectra-immobilized-monolayers-gold-solid-colloids.webp)