A Novel Integrated Framework and
Improved Methodology of Computer-Aided
Drug Design
Calvin Yu-Chian Chen 1,2,3,4,5,6 *
1 College of Pharmaceutical Sciences, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, China
2 Department of Biotechnology, Asia University, Taichung, 41354, Taiwan.
3 China Medical University Beigang Hospital, Yunlin,65152, Taiwan.
4 Laboratory of Computational and Systems Biology, China Medical University, Taichung, 40402, Taiwan.
5 Department of Medical Research, China Medical University Hospital, Taichung, 40447, Taiwan.
6 Computational and Systems Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
* Corresponding author
Tel.: +886-4-2205-2121 ext. 4335
E-mail address: [email protected] 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Abstract
Computer-aided drug design (CADD) is a critical initiating step of drug development, but a single model capable of covering all designing aspects remains to be elucidated. Hence, we developed a drug design modeling framework that integrates multiple approaches, including machine learning based quantitative structure-activity relationship (QSAR) analysis, 3D-QSAR, Bayesian network, pharmacophore modeling, and structure-based docking algorithm. Restrictions for each model were defined for improved individual and overall accuracy. An integration method was applied to join the results from each model to minimize bias and errors. In addition, the integrated model adopts both static and dynamic analysis to validate the intermolecular stabilities of the receptor-ligand conformation. The proposed protocol was applied to identifying HER2 inhibitors from traditional Chinese medicine (TCM) as an example for validating our new protocol. Eight potent leads were identified from six TCM sources. A joint validation system comprised of comparative molecular field analysis, comparative molecular similarity indices analysis, and molecular dynamics simulation further characterized the candidates into three potential binding conformations and validated the binding stability of each protein-ligand complex. The ligand pathway was also performed to predict the ligand “in” and “exit” from the binding site. In summary, we propose a novel systematic CADD methodology for the identification, analysis, and characterization of drug-like candidates.
Keywords: traditional Chinese medicine (TCM); drug design; docking; ligand-based
drug design; pathway; structure-based drug design 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
Introduction
In silico pharmacology is commonly adopted in modern pharmaceutical
industry for drug discovery1-4. Computer aided drug design not only has the advantage of high-throughput analysis, but also greatly reduces the expenses on biochemical laboratory experiments. Many theories have been developed in attempt to best approximate the real molecular interactions and, subsequently, to identify drug-like compounds 5, 6. Predictive models based on the quantitative structure-activity relationship (QSAR) approaches (ligand-based) 7 or the receptor-ligand compatibility (structure-based) have been developed 8. However, a universally applicable method remains to be found.
Standard CADD protocols use either ligand-based drug design (LBDD) or structure-based drug design (SBDD) approaches. For LBDD, a prediction model is built based on the compound features of known drugs, and a new drug can be identified by mapping the properties7. Quantitative structure-activity relationship (QSAR) approaches, such as multiple linear regression (MLR) and supported vector machine (SVM) that build linear and non-linear prediction models respectively 9, 10, were commonly used for LBDD. Although key active features can be identified, specificities and sensitivities of LBDD are limited by predefined molecular scaffolds. As receptor structures are increasingly resolved, SBDD has been developed to simulate novel receptor-ligand interactions, namely molecular dockings. In SBDD, a compound is virtually docked into a predetermined ligand binding site where binding poses and corresponding affinities of the protein-ligand complex are evaluated. Novel drugs, such as aldose reductase inhibitors, adenovirus protease inhibitors, gamma-aminobutyric acid inhibitors, HER2 inhibitors, and other therapeutic agents have been discovered based on structure-based pharmacology 11-13. While SBDD is capable of identifying novel drug scaffolds, results often vary depending on the algorithms, 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26
targets, and descriptors involved in the calculation 14. Nevertheless, these data give valuable background information for drug discovery.
To integrate the advantages of ligand-based and structure-based approaches, our lab constructed the Weighted Score (WS) algorithm in the attempt to develop a robust model for identifying novel compounds of distinct molecular scaffolds. By integrating QSAR algorithm with molecular docking analysis, WS demonstrated significantly higher prediction accuracy than consensus score functions. However, WS also inherited drawbacks from each method due to the restrictions of training models and the limitations of docking algorithms.
It is not surprising that a second CADD process is required for validation. 3D-QSAR protocols, such as comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA)15, are often applied for mapping 3D pharmacophores with each candidate16. Like all QSAR approaches, the 3D-QSAR methods can only validate a restricted range defined by the training data. Alternatively, the binding stability can be monitored through molecular dynamics (MD) simulation, which evaluates the dynamic movements of each atom within a binding conformation 17, 18. Despite its ability to estimate actual binding situations, high resource requirement and long calculation times hinder most SBDD studies from microsecond time-scale evaluations.
Despite the increasing number of algorithms developed, creating robust predictive models as well as discovering novel drug scaffolds remains challenging. Accordingly, we propose an integrated drug discovery system that employs multiple algorithms in a logical order. For a given target of interest, properties of its known ligands are collectively analyzed using machine learning QSARs by multiple linear regression (MLR) and supported vector machine (SVM) 9, 10, pharmacophore model 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
by HypoGen 19, 3D-QSARs by comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) 15, 16, WS, structure-based molecular docking algorithm, and molecular dynamics (MD) simulation. All aforementioned methods have been adopted in our lab for drug discovery analysis 20-27. A normalized ranking system is used to unify the prediction results.
Our integrated protocol comprises of four major steps: preparation, modeling, ranking, and validation. In the preparation step, data collection, ligand database, and protein structure are determined. The prediction modeling step focuses on building multiple drug design models. The ranking step summarizes the results from each in
silico model. A drug candidate or precursor will be further analyzed and characterized
in the validation step. To test the validity of this proposed method, our protocol was applied to identify inhibitors of human epidermal growth factor receptor 2 (HER2) from traditional Chinese medicine (TCM)28-30. HER2 is a member of the epidermal growth factor receptor (EGFR) superfamily, a class of surface receptor tyrosine kinase involved in the signal transduction pathways for cell growth and differentiation 31. Overexpression of HER2/EGFR elevates several downstream signaling pathways, such as the PI3K/Akt pro-survival pathway31. Inappropriate signaling may lead to uncontrolled cell proliferation, enhanced cell motility, and resistance to apoptosis and angiogenesis 32. Elevation of HER2 expression has been reported in multiple cancer studies 33-36. Consequently, HER2-targeting cancer drugs such as Lapatinib, Neratinib, Pertuzumab have been developed 37. However, adverse side-effects ranging from tolerable nausea, diarrhea, and rash, to severe toxic hepatitis are associated with current HER2-targeting drugs 38. Consequently, development of HER2 inhibitors with less side effects is of utmost importance in HER2-based cancer chemotherapy. From a CADD perspective, development of HER2 inhibitors satisfies the requirements of a 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
training data for LBDD as well as having available protein sequences and EGFR models for homology modeling in SBDD, making this drug class unique for evaluating our novel protocol as well as for drug discovery.
Material and Methods
Data and Model Preparations
Compound Database. The comprehensive TCM small molecule database, TCM
Database@Taiwan 39, may be used for western medicine-based and TCM-based drug discovery, respectively. For our purpose of identifying potential HER2 drug leads from TCM, we downloaded all available TCM molecules from TCM Database@Taiwan to conduct HER2 inhibitor screening.
Data Collection. For the identification of potential HER2 inhibitors, molecular
structures and corresponding bioactivities (pIC50) of 401 known HER2 inhibitors were obtained 40-54 to construct the data set for training the LBDD prediction model.
Homology Modeling. The 3D structure of a target protein may be obtained from
RCSB Protein Data Bank (PDB) 55or through homology modeling 56. The HER2 protein sequence (P04626; erbB2_human) used was obtained from Swiss-Prot/TrEMBL. In order to construct a homology model for the HER2 kinase domain, EGFR proteins (PDBID: 2ITY and 2J5E) were used as templates and sequence identities and similarities at the kinase domain (residues 720 – 987) were examined (Figure 1). Homologous crystal structures of the EGFR kinase domain (PDB ID: 1M14, 2GS2, 2GS6, and 2GS7) from RCSB Protein Data Bank 57, 58 were aligned using the MODELER module in Discovery Studio 2.5 59. Five HER2 models were built independently and the HER2 model with the highest Discrete Optimized Protein Energy (DOPE) score, which evaluates conformation stability, was selected and further validated using Ramachandran plot (Figure 2) and Verify-3D (Figure 3). 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
Predictive Modeling
Structure-Based Virtual Screening. All ligands, including those from TCM
Database@Taiwan and the control Lapatinib, were prepared according to specified modeling methodologies. Chemistry at HARvard Molecular Mechanics (CHARMm) force field 60 was applied to both the HER2 homology model and the ligands. Smart minimizer was used as the docking algorithm and calculation of all scoring functions provided by LigandFit module in DS2.5 were selected. The scoring functions included DockScore, LigScore (LigScore1 and LigScore2), piecewise linear potentials (-PLP and -PLP2), Jain, potential of mean force (PMF and -PMF04), LigInternalEnergy (LIE), and Ludi (Ludi 1, Ludi 2, and Ludi 3) 61-65. The first 20 ligands with the highest Dock Score were selected for this study.
Machine Learning QSAR Models. The 298 ligands described in the data
collection section were used for each MLR and SVM model, where 278 ligands were randomly selected for the training set and the remaining 20 for the test set. Molecular property descriptors were calculated from all ligands using Calculate Molecular Properties module in Discovery Studio 2.5 59. Genetic algorithm (GA), namely Genetic Function Approximation (GFA)51, was applied onto the training set and 10 key descriptors were identified. All descriptors were normalized before building machine learning models.
For MLR, the selected descriptors were used to construct a linear relationship equation in MATLAB (Equation 1):
A AnXn
Y 0 [1]
A0= the Y intercept
An= the coefficients of descriptor Xn Xn= the descriptor 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
For predicting continuous data, a regression version of SVM method, namely supported vector regression (SVR) 66 was used on the training set. Training data in the
form of f(x) = <w,x> + b,wRd,bR, where all property values were scaled to between -1 and 1, were inserted into the Kernel function. The SVR algorithm solves
w by introducing an ε-insensitive loss function (Equation 2):
l
i
y
b
x
w
b
x
w
y
C
w
i i i i i i i i l i i i b w,...,
1
,0
,
,
)
,
s.t.
)
(
2
1
* * 1 * 2 * , ,min
[2]By introducing Lagrange multiplier and kernel k(x,x’) = (x),(x') , the
non-linear transformation of the optimization problem is evaluated by (Equation 3):
l 0 i * i i * i i l 0 i l 0 j * j j i * i i j i l 1 j i, * j j * i i]
[0,
-and
0
)
-(
s.t.
)
-(
y
)
-(
-)
,
(
)
-)(
-(
2
1
-max.
C
x
x
k
[3]The resulting prediction model could be generalized by the Kernel function (Equation 4): C C b x x k x f i l i i i
0 , 0 s.t. ) , ( ) ( ) ( * i 1 i * [4]For MLR and SVM models, a 5-fold cross validation was implemented to validate the training set. Applicability domain (APD) was also used to validate reliability of predictions 67, descriptors of all compounds employed to define the 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
domain of applicability were calculated the matrix distance, based on the Euclidean distances between test compounds and nearest neighbor training compounds. The predictions of test set are reliable when the distance was lower than APD. APD was calculated using Enalos Domain of Applicability Node under KNIME 2.7.1 software 68, 69, with the following equation (Equation 5),
Z d
APD [5] The set of distances lower than the average of Euclidean distances between all pair of training set is formulated, <d> and
was defined as average distance and standard among the set of distances, respectively. Z was set as 0.5 for empirical cutoff value. The prediction model was validated by calculating square correlation coefficients (R2) between predicted and actual pIC50 values of the test set. To estimate the predicting ability of MLR and SVM models, we performed the external validation using 20 compounds in test set as discussed in validation test of Tropsha’s study 70, 71. The validation used the following criteria (Equation 6),15 . 1 ' 85 . 0 15 . 1 85 . 0 1 . 0 ' 7 . 0 2 2 0 2 2 2 0 2 2 k or k R R R or R R R R [6]
Where R is the coefficient of multiple correlation for 20 compounds in test set. 2 2 0 R
and 2 0
'
R are the coefficients of determination with the slopes of k and k' for the
regression lines through the origin for predicted versus observed pIC50 and observed versus predicted pIC50, respectively.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
The constructed MLR and SVM models were applied to predict pIC50 of the candidate compounds resulting from virtual screening.
Weighted Score (WS) Modeling. Weighted Score equation was constructed by
evaluating the weighted contribution of each docking scoring function to the pIC50 prediction. In the WS equation, a weight (correlation) coefficient was assigned to each normalized score based on the docking results, and the sum of the scores was presumed to be in a linear relationship to pIC50 (Equation 7):
b pIC a SF SF W WS n i c i i
50 1 [7] Wi= the weightSFi= the value of scoring function
i= number of scoring function
SFc= the value of scoring function of control pIC50 = –log (IC50)
Coefficients a and b were calculated to predict pIC50 of an unknown compound based on the scores from docking.
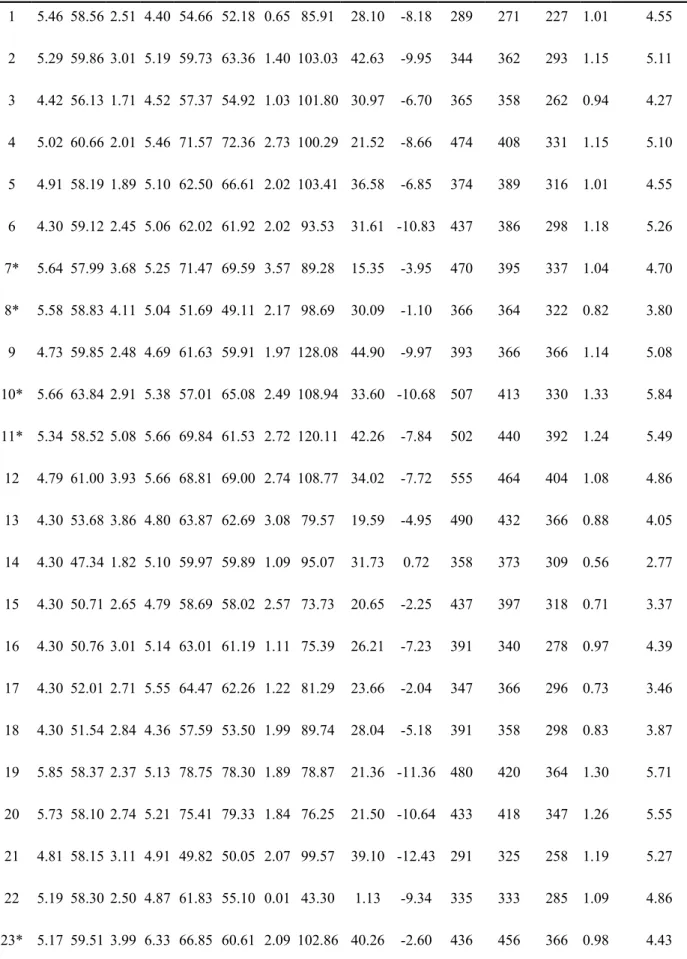
The 36 HER2 inhibitors from Li et al. 47 were used to construct the WS model (Table 1). The training set was obtained by randomly selecting 29 compounds, and the remaining 7 were used as the test set. Scores for each scoring function were generated following the docking procedure previously detailed, normalized, and used to calculate the WS of each ligand. Candidate compounds from virtual screening were subjected to WS model calculations to predict their corresponding pIC50.
Normalized Voting System
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
A normalized voting system is used to summarize the predicted bioactivities based on multiple prediction models. Correlation coefficients of each model are assigned. Prediction results on a given ligand would be normalized based on the prediction ranking. Predictions ranking in the top 20% of each model (indicating a fit to the model) are assigned a score of 1, otherwise 0. The sum of scores of a given compound determines the likeliness of a hit. Candidate compounds are ranked based on the sum of scores and mapped with the docking results. The voting system scoring function is defined as
Mi(x)where Mi(x) is the normalized score of x compound from the ith model.
This method is a simplified approach based evaluation 72 by assuming the independence and the equivalent weight of each model.
In this particular study, the normalized ranking system was used to evaluate pIC50 predictions from MLR, SVM, and WS determined for potential HER2 inhibitors from TCM. Prediction rankings were normalized as described and the sum of scores for each ligand was determined. Ligands with the highest sums were identified as potential inhibitors and subjected to further analyses for further validation.
Validation
QSAR Modeling and Compound Feature Validation. Bayesian network was
constructed using the training and test set data, and descriptors used for building machine learning QSAR models. Descriptor data and pIC50 were discretized into a maximum of three categories to reduce biased data distribution. After data discretization, linear regression analysis for each pIC50 category was applied to the training set. For the ith pIC50 category containing n ligands, y
ij and xijp represent the
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24
pIC50 value and the pth descriptor value in the jth ligand, respectively. The regression
model of the data sets n
j ijp ij ij x x y , 1,..., } 1 { is shown by Equation 8: i i i i X y [8] where in i i i y y y y . . . 2 1 , inp in p i i p i i i x x x x x x X . . . . . . . . . . . . . . . . . . . . . . . . 1 2 21 1 11 ,
and i and i represent regression coefficients and error term in the ith pIC50 category, respectively. The unknown regression coefficienti was estimated using ordinary least squares (Equation 9):
i T i i T i i X X X y 1 ) ( ˆ [9]
The Bayesian network (BN)73 is an acyclic directed graph G that represents a set of discrete random variables V={v1,…,vn} and their conditional probability based on
probabilistic model. In a graph G whose vertices correspond to the random variables
v1,…,vn, each variable vi is only associated to its descended variables. Thus, the joint
probability distribution over random variables V in a BN is given by (Equation 10):
n i i i n P v Pa v v v P( 1,..., ) 1 ( | ( )) [10]where Pa(vi) represents the set of parents of vi in graph G.
To discover the most probable BN from a collection of observed data D={d1,…,dn}
regarding variables V, a common approach is to introduce a scoring function that 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
evaluates each possible graph G by the distribution of the observed data. Banjo (Bayesian Network Inference with Java Objects) is a software for structure learning of static Bayesian networks 74 implemented in Java. This software uses score matrices to assess the possibility for each graph G with the searching method of simulated annealing. The scoring metrics is based on the full Bayesian posterior probability P(G|
D) and incorporates a complexity penalty to avoid data over-fitting. Relationships in
BN structure among the ten descriptors and pIC50 was investigated through the training set using the Banjo package. Observed data are the categories of the ten descriptors and their respective pIC50. The running time of the program was set to one hour. The parameter learning module in the Bayes Net Toolbox (BNT) (https://code.google.com/p/bnt/) package was used to estimate the probability for each node vi conditional upon its parents. For our BN in this study, observed data D is
specified as the categories of the ten descriptors, and the pIC50 category (k) is predicted by the following formula (Equation 11):
) | ( max arg 1P i D k n i [11] where i represents the ith category of pIC50, and n represents the total number of pIC50
categories. The pIC50 value is calculated by Equation 12:
k k X
pIC50 ˆ [12]
The BN algorithm used was written in Matlab codes that integrated both the Banjo
and BNT packages. The marginal probability P(i|D) can also be calculated through
the BNT module. The generated BN was used to validate the bioactivity of the TCM candidates selected based on the voting system.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
3D-QSAR models were constructed using the 36 HER2 inhibitors identified by Li et al. (Li et al., 2010). The conformational template was built by aligning 29 randomly selected inhibitors using the SYBYL 8.0 Atom-Fit module 75. Force field descriptors for CoMFA were calculated using Lennard-Jones potential and Coulombic potential. Steric, electrostatic, hydrophobic and hydrogen bond (H-bond) donors and acceptors fields were calculated for CoMSIA using a Gaussian function for calculating short distance interaction. A grid spacing of 1Å was used throughout the analysis. Partial least squares (PLS) regression was utilized to analyze the 3D-QSAR models. All CoMFA and CoMSIA fields were taken as independent variables.
Molecular Dynamics (MD) Simulation. The Simulation package in Discovery
Studio 2.5 was applied for MD simulation 59. The simulation system was established to mimic the cytoplasmic environment with a solvation shell of TIP3P water at 0.90% NaCl concentration. Protein-ligand complexes selected from molecular docking were subjected to energy minimization, heating, equilibration, and dynamic cascade production. The minimization protocol consisted of 500 steps of deepest descent followed by 500 steps of conjugated gradient. The input complex was heated for 50 ps from 50K to 310K without constraint and equilibrated for 150 ps at 310K. Production step was conducted for 30ns at 310K with constant temperature dynamics method. Temperature coupling decay was set to 0.4ps. SHAKE algorithm was applied to restrain bonds involving hydrogen. The Analyze Trajectory module was used to analyze trajectories of total energy, root mean square deviations (RMSD) of the ligands and ligand-protein complexes, and H-bond formation and distances during MD. LigandPath 76 was used to estimate possible pathways of each ligand within the complexes. Possible ligand pathways were identified by mapping protein free space at every conformation during the initial 5 ns and final 5 ns. Pathways identified during 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
the initial and final 5 ns were termed entry and exit pathways, respectively. Minimum clearance was set at 4 Å and a surface probe of 6 Å was used.
Results/Discussion
A schematic representation of our proposed method is summarized in Figure 4.
Modeling
Structure-based Virtual Screening. Virtual screening was applied to evaluate
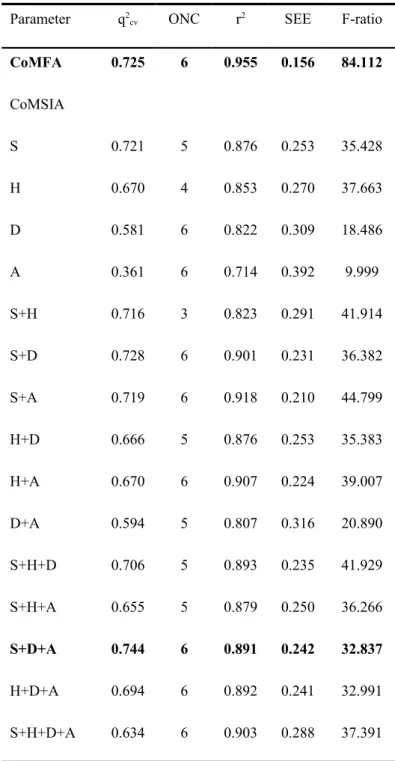
binding affinity of each ligand in the TCM Database@Taiwan with the HER2 binding site. The top 20 ligands ranked by Dock Score are listed in Table 2. The screening outcome identified 13 TCM compounds with DockScores higher than Lapatinib, suggesting higher binding affinity to the HER2 binding site than the commercial drug. However, since binding affinity is not a definite indicator of bioactivity, all 16 ligands with slightly lower Dock Scores than Lapatinib (DockScore = 67.330) were subsequently mapped to ligand-based models.
Machine Learning QSAR Models. The 10 key molecular property descriptors identified used
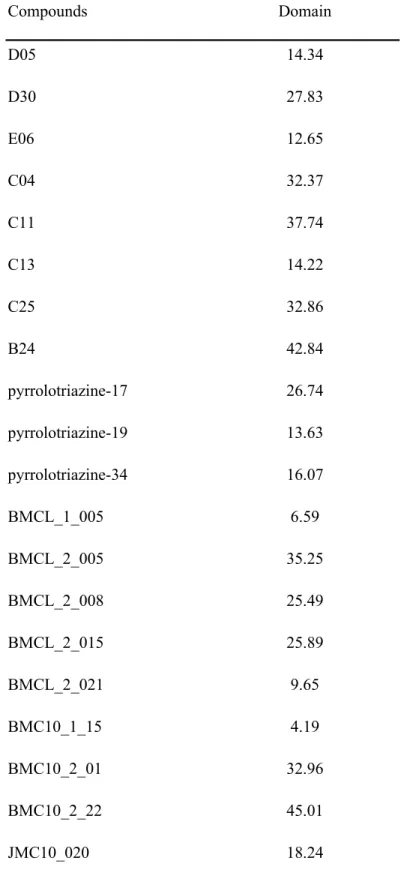
to construct QSAR models were Num_Rings6, Num_Atom Classes, Num_H_Acceptors, Num_H_Donors_Lipinski, Molecular_Fractional Polar SASA, Dipole_Y, Jurs_DPSA_1, Jurs_DPSA_3, Jurs_FPSA_2, and PMI_X. The linear relationship model built by MLR algorithm enclosed most training data points within the 95% confidence interval (Figure 5A). Test set predictions were also distributed closely along the 95% confidence interval. The values of applicability domain and validation compounds were shown in Table 3. The predictions of test set were considered reliable as all domains of validation compounds are lower than APD (225.88). The results of external validation for MLR and SVM models indicated significant predicting ability from the following statistics,
For MLR model 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24
1 994 . 0 ' , 1 . 0 005 . 0 ' 1 001 . 1 , 108 . 0 7 . 0 805 . 0 2 2 0 2 2 2 0 2 2 k with R R R k with R R R R For SVM model 1 970 . 0 ' , 1 . 0 002 . 0 ' 1 026 . 1 , 1 . 0 030 . 0 7 . 0 810 . 0 2 2 0 2 2 2 0 2 2 k with R R R k with R R R R
The prediction distribution and the R2 value of 0.8963 suggested that the MLR model was
sufficient to predict inhibition activity. However, MLR model over-predicted several inhibitory activities due to the less descriptive nature of the linear model. For the SVM regression model, almost all training set data points were distributed within 95% confidence interval except for a few outliers (Figure 5B). This also applied to most ligands in the test set. The SVM regression model had high correlation and low ε-insensitive loss, making the SVM regression a sensitive model for predicting inhibitor binding activity. Predicted pIC50 of the 16 TCM candidates using the generated MLR and
SVM models are summarized in Table 4.
Weighted Score Modeling. A total of 12 docking algorithms were applied to
evaluate ligand-protein interactions based on the 36 ligands listed in Table 1. Table 5 lists calculated scores of each ligand. Most DockScore values ranged around or above 50, implying strong binding affinity of the protein-ligand complex. Correlation analysis between observed pIC50 and each evaluation score indicated that LIE and DockScore had higher correlation values than the other scores (Figure 6). By integrating all docking algorithm scores into the WS algorithm, the final WS prediction model demonstrated a higher prediction correlation of 0.4086 than individual docking algorithm predictions (Figure 5C). This suggested that WS could improve the prediction modeling system by integrating the scoring functions into a 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
linear correlation function. Corresponding WS scores of each selected TCM candidate are listed in Table 4.
Normalized Voting System
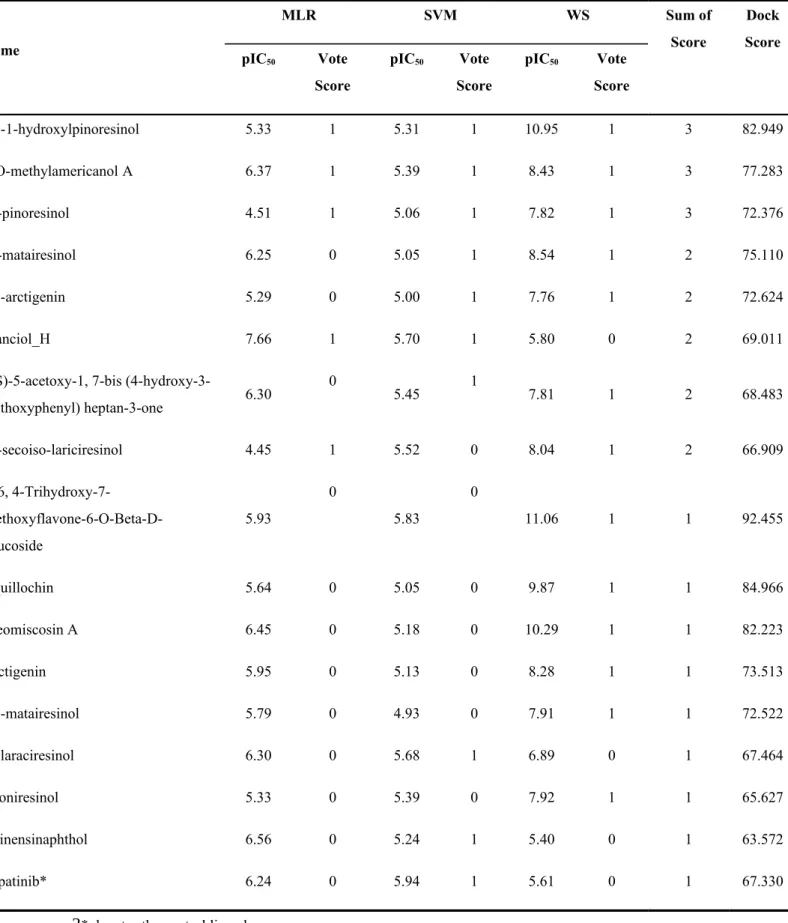
The 16 TCM ligands with Dock Scores exceeding 60 were mapped to the generated MLR, SVM, and WS models. Ligands with predicted pIC50 values ranking in the top 20% of each model were assigned a voting score of 1, 0 if otherwise. A ligand with a high sum of score and DockScore implied a stable binding conformation with HER2. As shown in Table 4, eight TCM ligands had sum of scores higher than 1 in addition to Dock Scores similar if not higher than Lapatinib. Lapatinib, the control ligand, received only one vote, suggesting that the novel voting system may be able to identify more potent drug candidates from natural sources than previously used methods.
Structures and origins of the 8 identified TCM candidates ranked by the voting system in conjunction with Dock Score are shown in Figure 7.These compounds originated from a variety of sources traditionally used against sepsis, inflammation, toxicity, and skin ailments (Li & Luo, 2003). Although correlation between these symptoms and cancer were not evident, these medical conditions are common among cancer patients 77-79. In addition, Arctium lappa has been reported as an alternative treatment for breast cancer though the therapeutic mechanisms remain to be elucidated 80. These findings suggest the potential therapeutic value of these TCM for cancer treatment and provide support for our proposed voting system.
Validation
Visual structure analysis. Structural similarities were observed in the TCM
candidates selected from our analysis (Figure 7). Generally speaking, candidates other than 9-O-methylamericanol_A (Figure 7B) and shanciol_H (Figure 7F) shared similar structural topologies with a phenol group on each end and two or more polar oxygen 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26
atoms in the central bridge regions. A more detailed review indicated that (+)-1-hydroxylpinoresinol (Figure 7A) and (-)-pinoresinol (Figure 7C) shared similar structures; similarity was also found between (-)-matairesinol (Figure 7D) and (+)-arctigenin (Figure 7E). Intriguingly, candidates from different plant origins also showed high structural resemblance (Figure 7C, 7D, 7E, 7H). Such results indicate that our proposed voting system was able to identify candidates with common structures that might be key features for HER2 inhibitors.
QSAR Validation.
Figure 8 shows correlation of observed and predicted bioactivities using the generated BNT model. An R2 value of 0.495 indicated that the model was acceptable. Predicted activities of the TCM candidates by BNT are listed in Table 6. BNT results are consistent with MLR and SVM results, further verifying the bioactivities of the TCM candidates.
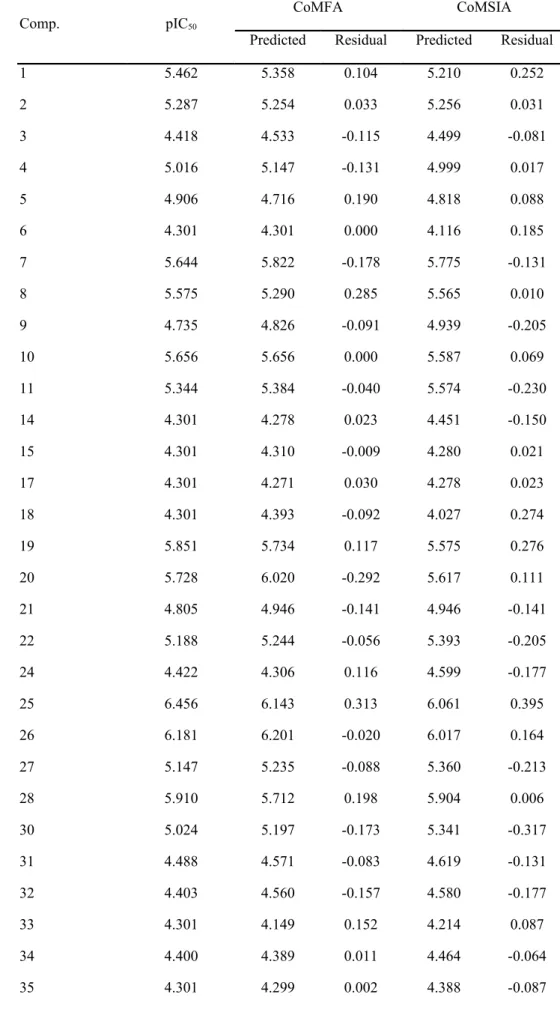
CoMFA and CoMSIA were applied for systematic validation of the structure-activity relationship of each ligand. PLS analysis for CoMFA and CoMSIA models are shown in Table 7 with the selected models shown in bold. The CoMFA model was predominated by the steric field since electrostatic interactions were negligible. Contribution from multiple fields, including steric (S), hydrophobic (H), hydrogen bond donor (D), and hydrogen bond acceptor (A) were evaluated for the CoMSIA model. Among all 3D-QSAR models, the highest cross validation correlation coefficient was found in the CoMSIA S+D+A model. This suggests that in addition to steric interactions, H-bond donor/acceptor interactions were also major contributors to the CoMSIA model. Cross and non-cross validation of the CoMFA (q2cv = 0.725, r2=0.955) and CoMSIA (q2cv = 0.744, r2=0.891) models implied that both models were 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24
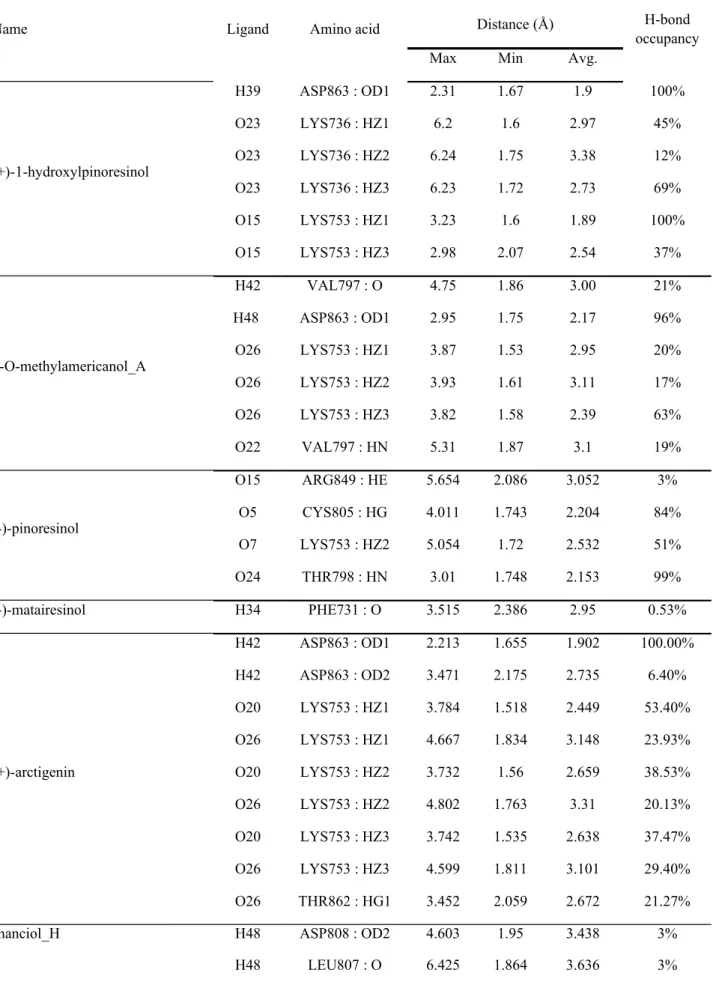
accurate (Table 7). Both models were further validated by the accuracy of predicted pIC50 with actual observed pIC50 of the training set (Table 8).
Validation of the selected TCM candidates were conducted by mapping ligand docking conformations to the identified steric and H-bond donor/acceptor fields in the CoMSIA model (Figure 9). All TCM candidates and Lapatinib demonstrated higher occupancy at the steric bulk region, which was in close approximation to the center of the binding site cavity. The docking conformations also showed consistent H-bond formations at K753. Furthermore, some candidates formed additional H-bonds with D863. These binding conformations matched the H-bond donor/acceptor features identified in the CoMSIA model. Similar binding conformations were observed in candidates sharing structural similarities such as pinoresinol (Figure 9C) and (-)-secoiso-lariciresinol (Figure 9H), and (-)-matairesinol (Figure 9D) and (+)-arctigenin (Figure 9E). This implies two distinct conformations that potentially favor HER2-ligand binding. In general, all TCM candidates conformed to the 3D-QSAR model with the exception of shanciol_H (Figure 9F) and (5S)-5-acetoxy-1, 7-bis (4-hydroxy-3-methoxyphenyl) heptan-3-one (Figure 9G), which along with Lapatinib, contoured partially due to either slight disposition in the binding conformation (Figure 9F) or protrusion of ligand structure outside the predicted feature space (Figure 9G, 9I). These observations were in agreement with ligand ranking from the voting system.
Molecular Dynamics (MD) Simulation. In addition to ligand structure and feature
validation, binding affinities as well as receptor-ligand complex stabilities are also key determinants of a drug-like compound. Based on Figure 9, all TCM candidates formed H-bonds with residue K753. In addition, (+)-1-hydroxylpinoresinol (Figure 9A), (-)-matairesinol (Figure 9D), (+)-arctigenin (Figure 9E), and (-)-secoiso-lariciresinol (Figure 9H) formed additional bonds with residue D863. These H-1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
bonds may contribute to ligand binding affinity as well as binding conformation stability. Root mean square deviation (RMSD) analysis on MD trajectories of receptor-ligand complexes showed that all complexes stabilized after 14ns of MD simulation (Figure 10). Total energy trajectories of each ligand suggest that stable conformation were reached after 14ns simulation (Figure 11). Closer evaluation of each complex RMSD trajectory showed that the top five TCM candidates formed stable binding conformations throughout the simulation. This observation corresponded to the 3D-QSAR validation analysis of the top five candidates. Transient increases in complex RMSDs were observed for the remaining three TCM candidates. These complex changes may be brought on by ligand alterations as suggested by corresponding changes observed in the ligand RMSD trajectories. Despite intermediate changes during the process, all compounds demonstrated stable binding conformations at the end of MD simulation.
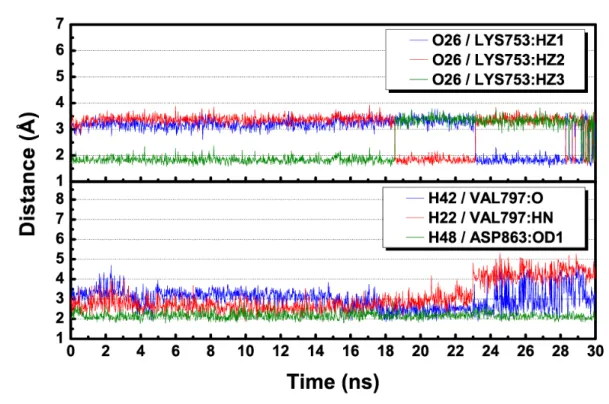
The stability of each receptor-TCM candidate complex was further validated through the molecular interaction analysis. H-bond occupancies, which are critical for molecular stability (Desiraju, 2005), are listed in Table 9. Notably, the top three candidates, (+)-1-hydroxylpinoresinol (Figure 12), 9-O-methylamericanol_A (Figure 13), and (-)-pinoresinol (Figure 14) had at least two H-bonds with high occupancies and stabilities. Comparatively, the other candidates had more fluctuating H-bonds (Figure 15–19). Nevertheless, the intermolecular forces were sufficient in maintaining the overall binding stability. Intriguingly, structurally similar candidates (-)-matairesinol and (+)-arctigenin had distinctively different H-bonds and H-bond occupancies (Table 9). This suggested a second intermolecular force that contributes significantly to ligand stability. As suggested by 3D-QSAR validation, steric interaction may be an important secondary force contributing to protein-ligand 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
stability. According to the 2D snapshots of each TCM compound at the end of MD simulation, residues K753 and D863 appeared to be the key residues for binding stability (Figure 20). Interaction between polar residues and electronegative ligand atoms further contributed to binding affinities. The presence of van der Waals (vdW) interactions appeared to stabilize the binding conformations by “locking” the steric bulk regions of each candidate within a restricted space (Figure 20). The importance of the two residue are further supported by the dihedral torsion angle changes measured during MD (Figure 21). Locations where H-bonds are formed with K753 and D863 have relatively small torsion fluctuations, suggesting the formation of stable H-bonds which help anchor the ligands within the binding site. Interaction between polar residues and electronegative ligand atoms further contributed to binding affinities. The presence of van der Waals (vdW) interactions appeared to stabilize the binding conformations by “locking” the steric bulk regions of each candidate within a restricted space (Figure 20). Based on the positions of H-bonds and vdW interaction regions, three potential binding conformations could be identified. In the first pose, the H-bond acceptors interacted with the key residues K753 and D863. The phenyl rings on each end of the ligand were stabilized through vdW interactions (Figure 20A, 20B, 20H). The second binding conformation favored vdW interactions as the major interaction forces (Figure 20C, 20D). The third pose formed H-bonds with K753 and D863 through the oxygen atoms on the phenyl ring at one end of the ligand (Figure 20E, 20F, 20G). The third binding conformation was comparatively linear, with the non H-bond anchored regions stabilized by vdW forces.
The final question to be asked was whether or not the ligands could access and exit from the binding site. Possible pathways of entry and exit for each ligand are shown in Figure 22. According to our simulation results by LigandPath, all ligands could access and exit the binding site under dynamic conditions. Most ligands, with 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26
the exception of had similar entry and exit pathways With the exception of (-)-pinoresinol (Figure 22C) and shanciol_H (Figure 22F). In the case of (-)-(-)-pinoresinol and shanciol_H, an exit pathway distinct from the original entry sites were observed. Nevertheless, our results imply that the top candidates selected through the voting system were not only predicted to have bioactivity and binding stability, but could indeed gain access to and from the binding site.
Conclusion
In summary, we present a sophisticated CADD method that can systematically identify, analyze, and characterize potential drug candidates and the interaction features from a given ligand database and protein of interest. Identification of HER2 inhibitors from TCM using the proposed method was used to validate its applicability. By summarizing analysis results from virtual screening, machine learning-based QSARs and WS using the voting score system, eight TCM candidates were identified with higher sum of scores and Dock Scores than Lapatinib. The identified candidates shared similar structures and molecular properties. Intriguingly, the TCM origins of some candidates have been reported as alternative cancer treatments, suggesting the reliability of the screening and voting system described in this report. All eight candidates were validated using 3D-QSAR model and displayed features that favor H-bonds as well as steric interactions. The analysis identified key H-bond formation regions around K753 and D863, which suggested an important feature in drug design. The MD simulation further classified three distinctive binding poses based on the interplay between H-bonds and vdW interactions. Using this method, we identified eight TCM candidates that possess inhibitory properties against HER2 activities as well as characterized three possible binding conformations for future drug design and analysis. By integrating parallel analysis of LBDD and SBDD properties, the voting system suggested a possible improvement to current drug screening processes.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26
Acknowledgements
The research was supported by grants from the National Science Council of Taiwan (NSC101-2325-B-039-001), Asia University (100-asia-56, asia100-cmu-2, 101-asia-59), China Medical University (DMR-101-094), and China Medical University Hospital (DMR-102-001, DMR-102-003, DMR-102-051). This study is also supported in part by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH102-TD-B-111-004) and Taiwan Department of Health Cancer Research Center of Excellence (DOH102-TD-C-111-005). We are grateful to Asia University for computer time and facilities
1 2 3 4 5 6 7 8 9
References
[1] Gleeson, M. P.; Hersey, A.; Hannongbua, S. In-silico ADME models: A general
assessment of their utility in drug discovery applications. Curr. Top. Med. Chem.,
2011, 11 (4), 358-381.
[2] Gozalbes, R.; Carbajo, R. J.; Pineda-Lucena, A. Contributions of computational
chemistry and biophysical techniques to fragment-based drug discovery. Curr. Med.
Chem., 2010, 17 (17), 1769-1794.
[3] Mah, J. T.; Low, E. S.; Lee, E. In silico SNP analysis and bioinformatics tools: A
review of the state of the art to aid drug discovery. Drug Discov. Today, 2011, 16
(17-18), 800-809.
[4] Villoutreix, B. O.; Eudes, R.; Miteva, M. A. Structure-based virtual ligand
screening: recent success stories. Comb. Chem. High Throughput Screening, 2009, 12
(10), 1000-1016.
[5] Marrero-Ponce, Y.; Casanola-Martin, G. M.; Khan, M. T.; Torrens, F.; Rescigno,
A.; Abad, C. Ligand-based computer-aided discovery of tyrosinase inhibitors.
Applications of the TOMOCOMD-CARDD method to the elucidation of new
compounds. Curr. Pharm. Des., 2010, 16 (24), 2601-2624.
[6] Zhang, S. Computer-aided drug discovery and development. Methods Mol. Biol.,
2011, 716, 23-38. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
[7] Bacilieri, M.; Moro, S. Ligand-based drug design methodologies in drug
discovery process: An overview. Curr. Drug Disc. Technol., 2006, 3 (3), 155-165.
[8] Kalyaanamoorthy, S.; Chen, Y. P. Structure-based drug design to augment hit
discovery. Drug Discov. Today, 2011, 16 (17-18), 831-839.
[9] Burbidge, R.; Trotter, M.; Buxton, B.; Holden, S. Drug design by machine
learning: support vector machines for pharmaceutical data analysis. Comput. Chem.,
2001, 26 (1), 5-14.
[10] Livingstone, D. J.; Salt, D. W. Judging the significance of multiple linear
regression models. J. Med. Chem., 2005, 48 (3), 661-663.
[11] Iwata, Y.; Naito, S.; Itai, A.; Miyamoto, S. Protein structure-based de novo
design and synthesis of aldose reductase inhibitors. Drug Des. Discov., 2001, 17 (4),
349-359.
[12] Pang, Y. P.; Xu, K.; Kollmeyer, T. M.; Perola, E.; McGrath, W. J.; Green, D. T.;
Mangel, W. F. Discovery of a new inhibitor lead of adenovirus proteinase: steps
toward selective, irreversible inhibitors of cysteine proteinases. FEBS Lett., 2001, 502
(3), 93-97.
[13] Yi, P. L.; Tsai, C. H.; Chen, Y. C.; Chang, F. C. Gamma-Aminobutyric Acid
(GABA) receptor mediates suanzaorentang, a traditional Chinese herb remedy,
-induced sleep alteration. J. Biomed. Sci., 2007, 14 (2), 285-297. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
[14] Cummings, M. D.; DesJarlais, R. L.; Gibbs, A. C.; Mohan, V.; Jaeger, E. P.
Comparison of automated docking programs as virtual screening tools. J. Med.
Chem., 2005, 48 (4), 962-976.
[15] Kubinyi, H. Chemogenomics in drug discovery. Ernst. Schering Res. Found
Workshop, 2006, (58), 1-19.
[16] Akamatsu, M. Current state and perspectives of 3D-QSAR. Curr. Top. Med.
Chem., 2002, 2 (12), 1381-1394.
[17] Huang, N.; Kalyanaraman, C.; Bernacki, K.; Jacobson, M. P. Molecular
mechanics methods for predicting protein-ligand binding. PCCP, 2006, 8 (44),
5166-5177.
[18] Rognan, D. Molecular dynamics simulations: A tool for drug design. Perspect.
Drug Discovery Des., 1998, 9-11 (0), 181-209.
[19] Li, H.; Sutter, J.; Hoffman, R., In Pharmacophore Perception, Development, and
Use in Drug Design, Güner, O. F., Ed. International University Line, La Jolla, CA:
2000; pp 171–189.
[20] Chang, T. T.; Sun, M. F.; Chen, H. Y.; Tsai, F. J.; Fisher, M.; Lin, J. G.; Chen,
C. Y. C. Screening from the World's largest TCM database against H1N1 virus. J.
Biomol. Struct. Dyn., 2011, 28 (5), 773-786.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
[21] Sun, M. F.; Chen, H. Y.; Tsai, F. J.; Liu, S. H.; Chen, C. Y.; Chen, C. Y. C.
Search for novel remedies to augment radiation resistance of inhabitants of
Fukushima and Chernobyl disasters. J. Biomol. Struct. Dyn., 2011, 29 (2), 325-338.
[22] Lin, C. H.; Chang, T. T.; Sun, M. F.; Chen, H. Y.; Tsai, F. J.; Chang, K. L.;
Fisher, M.; Chen, C. Y. C. Potent inhibitor design against H1N1 swine influenza:
Structure-based and molecular dynamics analysis for M2 inhibitors from traditional
Chinese medicine database. J. Biomol. Struct. Dyn., 2011, 28 (4), 471-482.
[23] Sun, M. F.; Chang, T. T.; Huang, H. J.; Lee, K. J.; Yu, H. W.; Chen, H. Y.; Tsai,
F. J.; Chen, C. Y. C. Key features for designing phosphodiesterase-5 inhibitors. J.
Biomol. Struct. Dyn., 2010, 28 (3), 309-321.
[24] Sun, M. F.; Chang, T. T.; Chang, K. W.; Huang, H. J.; Chen, H. Y.; Tsai, F. J.;
Lin, J. G.; Chen, C. Y. C. Blocking the DNA repair system by traditional Chinese
medicine? J. Biomol. Struct. Dyn., 2011, 28 (6), 895-906.
[25] Chang, S. S.; Huang, H. J.; Chen, C. Y. C. Two birds with one stone? Possible
dual-targeting H1N1 inhibitors from traditional Chinese medicine. PLoS Comp. Biol.,
2011, 7 (12), e1002315.
[26] Yang, S. C.; Chang, S. S.; Chen, H. Y.; Chen, C. Y. C. Identification of potent
EGFR inhibitors from TCM Database@Taiwan. PLoS Comp. Biol., 2011, 7 (10),
e1002189. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
[27] Chen, K. C.; Sun, M. F.; Yang, S. C.; Chang, S. S.; Chen, H. Y.; Tsai, F. J.;
Chen, C. Y. C. Investigation into potent inflammation inhibitors from traditional
Chinese medicine. Chem. Biol. Drug Des., 2011, 78 (4), 679-688.
[28] Kuo, J. H.; Chu, Y. L.; Yang, J. S.; Lin, J. P.; Lai, K. C.; Kuo, H. M.; Hsia, T.
C.; Chung, J. G. Cantharidin induces apoptosis in human bladder cancer TSGH 8301
cells through mitochondria-dependent signal pathways. Int. J. Oncol., 2010, 37 (5),
1243-1250.
[29] Lin, K. T.; Lien, J. C.; Chung, C. H.; Kuo, S. C.; Huang, T. F. A novel
compound, NP-184, inhibits the vascular endothelial growth factor induced
angiogenesis. Eur. J. Pharmacol., 2010, 630 (1-3), 53-60.
[30] Wu, P. P.; Liu, K. C.; Huang, W. W.; Ma, C. Y.; Lin, H.; Yang, J. S.; Chung, J.
G. Triptolide induces apoptosis in human adrenal cancer NCI-H295 cells through a
mitochondrial-dependent pathway. Oncol. Rep., 2011, 25 (2), 551-557.
[31] Le, X. F.; Lammayot, A.; Gold, D.; Lu, Y.; Mao, W.; Chang, T.; Patel, A.; Mills,
G. B.; Bast, R. C., Jr. Genes affecting the cell cycle, growth, maintenance, and drug
sensitivity are preferentially regulated by anti-HER2 antibody through
phosphatidylinositol 3-kinase-AKT signaling. J. Biol. Chem., 2005, 280 (3),
2092-2104.
[32] Yarden, Y. Biology of HER2 and its importance in breast cancer. Oncology,
2001, 61 Suppl 2, 1-13. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
[33] Engel, R. H.; Kaklamani, V. G. HER2-positive breast cancer: current and future
treatment strategies. Drugs, 2007, 67 (9), 1329-1341.
[34] Sakai, K.; Mori, S.; Kawamoto, T.; Taniguchi, S.; Kobori, O.; Morioka, Y.;
Kuroki, T.; Kano, K. Expression of epidermal growth factor receptors on normal
human gastric epithelia and gastric carcinomas. J. Natl. Cancer Inst., 1986, 77 (5),
1047-1052.
[35] Scher, H. I. HER2 in prostate cancer--a viable target or innocent bystander? J.
Natl. Cancer Inst., 2000, 92 (23), 1866-1868.
[36] Signoretti, S.; Montironi, R.; Manola, J.; Altimari, A.; Tam, C.; Bubley, G.;
Balk, S.; Thomas, G.; Kaplan, I.; Hlatky, L.; Hahnfeldt, P.; Kantoff, P.; Loda, M.
Her-2-neu expression and progression toward androgen independence in human prostate
cancer. J. Natl. Cancer Inst., 2000, 92 (23), 1918-1925.
[37] Gajria, D.; Chandarlapaty, S. HER2-amplified breast cancer: mechanisms of
trastuzumab resistance and novel targeted therapies. Expert Rev. Anticancer Ther,
2011, 11 (2), 263-275.
[38] Frankel, C.; Palmieri, F. M. Lapatinib side-effect management. Clin. J. Oncol.
Nurs., 2010, 14 (2), 223-233.
[39] Chen, C. Y. C. TCM Database@Taiwan: The World's largest traditional Chinese
medicine database for drug screening in silico. PLoS ONE, 2011, 6 (1), e15939. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
[40] Cai, X.; Zhai, H. X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C. J.; Bao, R.;
Qian, C. Discovery of
7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanam ide (CUDc-101) as a potent multi-acting HDAC, EGFR, and
HER2 inhibitor for the treatment of cancer. J. Med. Chem., 2010, 53 (5), 2000-2009.
[41] Cheng, Z. Y.; Li, W. J.; He, F.; Zhou, J. M.; Zhu, X. F. Synthesis and biological
evaluation of 4-aryl-5-cyano-2H-1,2,3-triazoles as inhibitor of HER2 tyrosine kinase.
Biorg. Med. Chem., 2007, 15 (3), 1533-1538.
[42] Chiosis, G.; Lucas, B.; Shtil, A.; Huezo, H.; Rosen, N. Development of a
purine-scaffold novel class of Hsp90 binders that inhibit the proliferation of cancer cells and
induce the degradation of Her2 tyrosine kinase. Bioorg. Med. Chem., 2002, 10 (11),
3555-3564.
[43] Fink, B. E.; Vite, G. D.; Mastalerz, H.; Kadow, J. F.; Kim, S. H.; Leavitt, K. J.;
Du, K.; Crews, D.; Mitt, T.; Wong, T. W.; Hunt, J. T.; Vyas, D. M.; Tokarski, J. S.
New dual inhibitors of EGFR and HER2 protein tyrosine kinases. Bioorg. Med.
Chem. Lett., 2005, 15 (21), 4774-4779.
[44] Hadden, M. K.; Hill, S. A.; Davenport, J.; Matts, R. L.; Blagg, B. S. Synthesis
and evaluation of Hsp90 inhibitors that contain the 1,4-naphthoquinone scaffold.
Biorg. Med. Chem., 2009, 17 (2), 634-640.
[45] Huang, K. H.; Veal, J. M.; Fadden, R. P.; Rice, J. W.; Eaves, J.; Strachan, J. P.;
Barabasz, A. F.; Foley, B. E.; Barta, T. E.; Ma, W.; Silinski, M. A.; Hu, M.; Partridge, 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
J. M.; Scott, A.; DuBois, L. G.; Freed, T.; Steed, P. M.; Ommen, A. J.; Smith, E. D.;
Hughes, P. F.; Woodward, A. R.; Hanson, G. J.; McCall, W. S.; Markworth, C. J.;
Hinkley, L.; Jenks, M.; Geng, L.; Lewis, M.; Otto, J.; Pronk, B.; Verleysen, K.; Hall,
S. E. Discovery of novel 2-aminobenzamide inhibitors of heat shock protein 90 as
potent, selective and orally active antitumor agents. J. Med. Chem., 2009, 52 (14),
4288-4305.
[46] Kuduk, S. D.; Zheng, F. F.; Sepp-Lorenzino, L.; Rosen, N.; Danishefsky, S. J.
Synthesis and evaluation of geldanamycin-estradiol hybrids Bioorg. Med. Chem.
Lett., 1999, 9 (9), 1233-1238
[47] Li, H. Q.; Yan, T.; Yang, Y.; Shi, L.; Zhou, C. F.; Zhu, H. L. Synthesis and
structure-activity relationships of N-benzyl-N-(X-2-hydroxybenzyl)-N '-phenylureas
and thioureas as antitumor agents. Biorg. Med. Chem., 2010, 18 (1), 305-313.
[48] Lippa, B.; Kauffman, G. S.; Arcari, J.; Kwan, T.; Chen, J. S.; Hungerford, W.;
Bhattacharya, S.; Zhao, X. M.; Williams, C.; Xiao, J.; Pustilnik, L.; Su, C. Y.; Moyer,
J. D.; Ma, L.; Campbell, M.; Steyn, S. The discovery of highly selective erbB2 (Her2)
inhibitors for the treatment of cancer. Bioorg. Med. Chem. Lett., 2007, 17 (11),
3081-3086.
[49] Lv, P. C.; Zhou, C. F.; Chen, J.; Liu, P. G.; Wang, K. R.; Mao, W. J.; Li, H. Q.;
Yang, Y.; Xiong, J.; Zhu, H. L. Design, synthesis and biological evaluation of 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
thiazolidinone derivatives as potential EGFR and HER-2 kinase inhibitors. Biorg.
Med. Chem., 2010, 18 (1), 314-319.
[50] A. Maddry, J. A.; Kussner, C.; W. Truss, J. W.; Niwas, S.; White, E. L.; Kwong,
C. D. Inhibition of the Her2 tyrosine kinase and characterization of a hydrophobic site
near the nucleotide binding domain. Bioorg. Med. Chem. Lett., 1997, 7 (16),
2109-2114.
[51] Rogers, D.; Hopfinger, A. J. Application of genetic function approximation to
quantitative structure-activity relationships and quantitative structure-property
relationships J. Chem. Inf. Comput. Sci., 1994, 34 (4), 854-866
[52] Sun, L.; Cui, J.; Liang, C.; Zhou, Y.; Nematalla, A.; Wang, X.; Chen, H.; Tang,
C.; Wei, J. Rational design of
4,5-disubstituted-5,7-dihydro-pyrrolo[2,3-d]pyrimidin-6-ones as a novel class of inhibitors of epidermal growth factor receptor (EGF-R) and
Her2(p185(erbB)) tyrosine kinases. Bioorg. Med. Chem. Lett., 2002, 12 (16),
2153-2157.
[53] Xu, G.; Abad, M. C.; Connolly, P. J.; Neeper, M. P.; Struble, G. T.; Springer, B.
A.; Emanuel, S. L.; Pandey, N.; Gruninger, R. H.; Adams, M.; Moreno-Mazza, S.;
Fuentes-Pesquera, A. R.; Middleton, S. A.
4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors. Bioorg.
Med. Chem. Lett., 2008, 18 (16), 4615-4619.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
[54] Xu, G.; Searle, L. L.; Hughes, T. V.; Beck, A. K.; Connolly, P. J.; Abad, M. C.;
Neeper, M. P.; Struble, G. T.; Springer, B. A.; Emanuel, S. L.; Gruninger, R. H.;
Pandey, N.; Adams, M.; Moreno-Mazza, S.; Fuentes-Pesquera, A. R.; Middleton, S.
A.; Greenberger, L. M. Discovery of novel
4-amino-6-arylaminopyrimidine-5-carbaldehyde oximes as dual inhibitors of EGFR and ErbB-2 protein tyrosine kinases.
Bioorg. Med. Chem. Lett., 2008, 18 (12), 3495-3499.
[55] Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.;
Shindyalov, I. N.; Bourne, P. E. The Protein Data Bank. Nucleic Acids Res., 2000, 28
(1), 235-242.
[56] Baker, D.; Sali, A. Protein structure prediction and structural genomics. Science,
2001, 294 (5540), 93-96.
[57] Stamos, J.; Sliwkowski, M. X.; Eigenbrot, C. Structure of the epidermal growth
factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline
inhibitor. J. Biol. Chem., 2002, 277 (48), 46265-46272.
[58] Zhang, X.; Gureasko, J.; Shen, K.; Cole, P. A.; Kuriyan, J. An allosteric
mechanism for activation of the kinase domain of epidermal growth factor receptor.
Cell, 2006, 125 (6), 1137-1149.
[59] Accelerys Software Inc. Discovery Studio Modeling Environment, 2.5; Accelerys
Software Inc.: San Diego, 2009. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
[60] Brooks, B. R.; Brooks, C. L.; Mackerell, A. D.; Nilsson, L.; Petrella, R. J.; Roux,
B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; Caflisch, A.; Caves, L.; Cui, Q.;
Dinner, A. R.; Feig, M.; Fischer, S.; Gao, J.; Hodoscek, M.; Im, W.; Kuczera, K.;
Lazaridis, T.; Ma, J.; Ovchinnikov, V.; Paci, E.; Pastor, R. W.; Post, C. B.; Pu, J. Z.;
Schaefer, M.; Tidor, B.; Venable, R. M.; Woodcock, H. L.; Wu, X.; Yang, W.; York,
D. M.; Karplus, M. CHARMM: The biomolecular simulation program. J. Comput.
Chem., 2009, 30 (10), 1545-1614.
[61] Bohm, H. J. The development of a simple empirical scoring function to estimate
the binding constant for a protein-ligand complex of known three-dimensional
structure. J. Comput. Aided Mol. Des., 1994, 8 (3), 243-256.
[62] Gehlhaar, D. K.; Verkhivker, G. M.; Rejto, P. A.; Sherman, C. J.; Fogel, D. B.;
Fogel, L. J.; Freer, S. T. Molecular recognition of the inhibitor AG-1343 by HIV-1
protease: conformationally flexible docking by evolutionary programming. Chem.
Biol., 1995, 2 (5), 317-324.
[63] Jain, A. N. Scoring noncovalent protein-ligand interactions: a continuous
differentiable function tuned to compute binding affinities. J. Comput. Aided Mol.
Des., 1996, 10 (5), 427-440.
[64] Krammer, A.; Kirchhoff, P. D.; Jiang, X.; Venkatachalam, C. M.; Waldman, M.
LigScore: a novel scoring function for predicting binding affinities. J. Mol. Graph.
Model., 2005, 23 (5), 395-407. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
[65] Muegge, I.; Martin, Y. C. A general and fast scoring function for protein-ligand
interactions: a simplified potential approach. J. Med. Chem., 1999, 42 (5), 791-804.
[66] Fan, R. E.; Chen, P. H.; Lin, C. J. Working set selection using second order
information for training support vector machines. J. Mach. Learn. Res., 2005, 6,
1889-1918.
[67] Zhang, S.; Golbraikh, A.; Oloff, S.; Kohn, H.; Tropsha, A. A novel automated
lazy learning QSAR (ALL-QSAR) approach: method development, applications, and
virtual screening of chemical databases using validated ALL-QSAR models. J. Chem
Inf. Model., 2006, 46 (5), 1984-1995.
[68] Melagraki, G.; Afantitis, A.; Sarimveis, H.; Igglessi-Markopoulou, O.;
Koutentis, P. A.; Kollias, G. In silico exploration for identifying structure-activity
relationship of MEK inhibition and oral bioavailability for isothiazole derivatives.
Chem. Biol. Drug Des., 2010, 76 (5), 397-406.
[69] Afantitis, A.; Melagraki, G.; Koutentis, P. A.; Sarimveis, H.; Kollias, G.
Ligand-based virtual screening procedure for the prediction and the identification of novel
beta-amyloid aggregation inhibitors using Kohonen maps and Counterpropagation
Artificial Neural Networks. Eur. J. Med. Chem., 2011, 46 (2), 497-508.
[70] Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model., 2002, 20 (4),
269-276. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
[71] Zhang, S. X.; Golbraikh, A.; Oloff, S.; Kohn, H.; Tropsha, A. A novel automated
lazy learning QSAR (ALL-QSAR) approach: Method development, applications, and
virtual screening of chemical databases using validated ALL-QSAR models. J. Chem
Inf. Model., 2006, 46 (5), 1984-1995.
[72] Bonissone, P. P.; Xue, F.; Subbu, R. Fast meta-models for local fusion of
multiple predictive models. Appl. Soft. Comput., 2011, 11 (2), 1529-1539.
[73] Friedman, N.; Linial, M.; Nachman, I.; Pe'er, D. Using Bayesian networks to
analyze expression data. J. Comput. Biol., 2000, 7 (3-4), 601-620.
[74] Yu, J.; Smith, V. A.; Wang, P. P.; Hartemink, A. J.; Jarvis, E. D. Advances to
Bayesian network inference for generating causal networks from observational
biological data. Bioinformatics, 2004, 20 (18), 3594-3603.
[75] Tripos Associates SYBYL Molecular Modeling Software, 8.0; Tripos Associates:
St. Louis, MO, 2007.
[76] Lin, T. L.; Song, G. Efficient mapping of ligand migration channel networks in
dynamic proteins. Proteins, 2011, 79 (8), 2475-2490.
[77] Coussens, L. M.; Werb, Z. Inflammation and cancer. Nature, 2002, 420 (6917),
860-867.
[78] De Conno, F.; Ventafridda, V.; Saita, L. Skin problems in advanced and terminal
cancer patients. J. Pain Symptom Manage., 1991, 6 (4), 247-256. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
[79] Williams, M. D.; Braun, L. A.; Cooper, L. M.; Johnston, J.; Weiss, R. V.; Qualy,
R. L.; Linde-Zwirble, W. Hospitalized cancer patients with severe sepsis: analysis of
incidence, mortality, and associated costs of care. Crit. Care, 2004, 8 (5), R291-R298.
[80] Zick, S. M.; Sen, A.; Feng, Y.; Green, J.; Olatunde, S.; Boon, H. Trial of Essiac
to ascertain its effect in women with breast cancer (TEA-BC). J. Altern. Complement.
Med., 2006, 12 (10), 971-980. 1 2 3 4 5 6 7
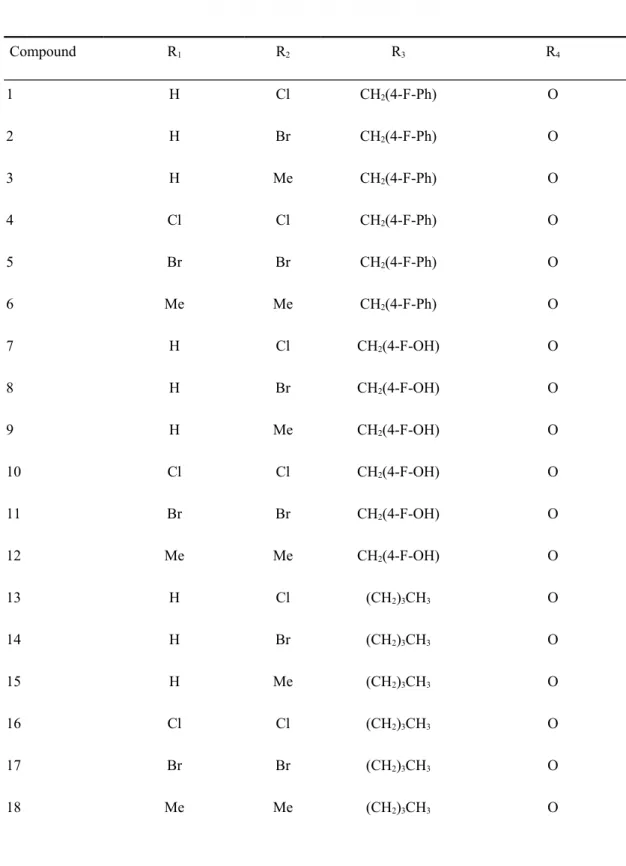
Table 1. Scaffold of thirty-six compounds* used for training the WS model. Compound R1 R2 R3 R4 1 H Cl CH2(4-F-Ph) O 2 H Br CH2(4-F-Ph) O 3 H Me CH2(4-F-Ph) O 4 Cl Cl CH2(4-F-Ph) O 5 Br Br CH2(4-F-Ph) O 6 Me Me CH2(4-F-Ph) O 7 H Cl CH2(4-F-OH) O 8 H Br CH2(4-F-OH) O 9 H Me CH2(4-F-OH) O 10 Cl Cl CH2(4-F-OH) O 11 Br Br CH2(4-F-OH) O 12 Me Me CH2(4-F-OH) O 13 H Cl (CH2)3CH3 O 14 H Br (CH2)3CH3 O 15 H Me (CH2)3CH3 O 16 Cl Cl (CH2)3CH3 O 17 Br Br (CH2)3CH3 O 18 Me Me (CH2)3CH3 O 1
19 H Cl CH2(4-F-Ph) S 20 H Br CH2(4-F-Ph) S 21 H Me CH2(4-F-Ph) S 22 Cl Cl CH2(4-F-Ph) S 23 Br Br CH2(4-F-Ph) S 24 Me Me CH2(4-F-Ph) S 25 H Cl CH2(4-F-OH) S 26 H Br CH2(4-F-OH) S 27 H Me CH2(4-F-OH) S 28 Cl Cl CH2(4-F-OH) S 29 Br Br CH2(4-F-OH) S 30 Me Me CH2(4-F-OH) S 31 H Cl (CH2)3CH3 S 32 H Br (CH2)3CH3 S 33 H Me (CH2)3CH3 S 34 Cl Cl (CH2)3CH3 S 35 Br Br (CH2)3CH3 S 36 Me Me (CH2)3CH3 S
* Compounds adopted from 47
1
Table 2. Top 20 TCM ligands obtained from virtual screening. Ligands are ranked by DockScore. Name DockScore 5, 6, 4-Trihydroxy-7- Methoxyflavone-6-O-Beta-D-Glucoside 92.455 Aquillochin 84.966 (+)-1-Hydroxylpinoresinol 82.949 Cleomiscosin_A 82.223 9-O-Methylamericanol_A 77.283 (-)-Matairesinol 75.110 Arctigenin 73.513 (+)-Arctigenin 72.624 (+)-Matairesinol 72.522 (-)-Pinoresinol 72.376 Shanciol_H 69.011
(5S)-5-Acetoxy-1, 7-bis (4-hydroxy-3-methoxyphenyl) heptan-3-one 68.483
Isolaraciresinol 67.464 (-)-Secoiso-lariciresinol 66.909 Lyoniresinol 65.627 Chinensinaphthol 63.572 Sanjidin_B 56.578 Bletilol_C 55.579 Shanciol_G 54.696 (-)-Hydnocarpin 53.334 Lapatinib* 67.330
* denotes the control ligand 1
2
Table 3 Applicability domain of test set descriptors for SVM and MLR models (APD = 225.88) Compounds Domain D05 14.34 D30 27.83 E06 12.65 C04 32.37 C11 37.74 C13 14.22 C25 32.86 B24 42.84 pyrrolotriazine-17 26.74 pyrrolotriazine-19 13.63 pyrrolotriazine-34 16.07 BMCL_1_005 6.59 BMCL_2_005 35.25 BMCL_2_008 25.49 BMCL_2_015 25.89 BMCL_2_021 9.65 BMC10_1_15 4.19 BMC10_2_01 32.96 BMC10_2_22 45.01 JMC10_020 18.24 1
Table 4. Results of the top 16 TCM candidates based on the voting system. Prediction values ranking in the top 20% of each model was assigned a voting score of 1; otherwise 0.
Name MLR SVM WS Sum of Score Dock Score pIC50 Vote Score pIC50 Vote Score pIC50 Vote Score (+)-1-hydroxylpinoresinol 5.33 1 5.31 1 10.95 1 3 82.949 9-O-methylamericanol A 6.37 1 5.39 1 8.43 1 3 77.283 (-)-pinoresinol 4.51 1 5.06 1 7.82 1 3 72.376 (-)-matairesinol 6.25 0 5.05 1 8.54 1 2 75.110 (+)-arctigenin 5.29 0 5.00 1 7.76 1 2 72.624 shanciol_H 7.66 1 5.70 1 5.80 0 2 69.011
(5S)-5-acetoxy-1, 7-bis
(4-hydroxy-3-methoxyphenyl) heptan-3-one 6.30 0 5.45 1 7.81 1 2 68.483 (-)-secoiso-lariciresinol 4.45 1 5.52 0 8.04 1 2 66.909 5, 6, 4-Trihydroxy-7- Methoxyflavone-6-O-Beta-D-Glucoside 5.93 0 5.83 0 11.06 1 1 92.455 Aquillochin 5.64 0 5.05 0 9.87 1 1 84.966 Cleomiscosin A 6.45 0 5.18 0 10.29 1 1 82.223 Arctigenin 5.95 0 5.13 0 8.28 1 1 73.513 (+)-matairesinol 5.79 0 4.93 0 7.91 1 1 72.522 Isolaraciresinol 6.30 0 5.68 1 6.89 0 1 67.464 Lyoniresinol 5.33 0 5.39 0 7.92 1 1 65.627 Chinensinaphthol 6.56 0 5.24 1 5.40 0 1 63.572 Lapatinib* 6.24 0 5.94 1 5.61 0 1 67.330
* denotes the control ligand 1
2
Table 5. Docking evaluation of each ligand used for training WS model.
Name pIC50 DS LS-1 LS-2 -PLP1 -PLP2 Jain -PMF -PMF04 LIE Ludi_1 Ludi_2 Ludi_3 WS Predicted pIC50
1 5.46 58.56 2.51 4.40 54.66 52.18 0.65 85.91 28.10 -8.18 289 271 227 1.01 4.55 2 5.29 59.86 3.01 5.19 59.73 63.36 1.40 103.03 42.63 -9.95 344 362 293 1.15 5.11 3 4.42 56.13 1.71 4.52 57.37 54.92 1.03 101.80 30.97 -6.70 365 358 262 0.94 4.27 4 5.02 60.66 2.01 5.46 71.57 72.36 2.73 100.29 21.52 -8.66 474 408 331 1.15 5.10 5 4.91 58.19 1.89 5.10 62.50 66.61 2.02 103.41 36.58 -6.85 374 389 316 1.01 4.55 6 4.30 59.12 2.45 5.06 62.02 61.92 2.02 93.53 31.61 -10.83 437 386 298 1.18 5.26 7* 5.64 57.99 3.68 5.25 71.47 69.59 3.57 89.28 15.35 -3.95 470 395 337 1.04 4.70 8* 5.58 58.83 4.11 5.04 51.69 49.11 2.17 98.69 30.09 -1.10 366 364 322 0.82 3.80 9 4.73 59.85 2.48 4.69 61.63 59.91 1.97 128.08 44.90 -9.97 393 366 366 1.14 5.08 10* 5.66 63.84 2.91 5.38 57.01 65.08 2.49 108.94 33.60 -10.68 507 413 330 1.33 5.84 11* 5.34 58.52 5.08 5.66 69.84 61.53 2.72 120.11 42.26 -7.84 502 440 392 1.24 5.49 12 4.79 61.00 3.93 5.66 68.81 69.00 2.74 108.77 34.02 -7.72 555 464 404 1.08 4.86 13 4.30 53.68 3.86 4.80 63.87 62.69 3.08 79.57 19.59 -4.95 490 432 366 0.88 4.05 14 4.30 47.34 1.82 5.10 59.97 59.89 1.09 95.07 31.73 0.72 358 373 309 0.56 2.77 15 4.30 50.71 2.65 4.79 58.69 58.02 2.57 73.73 20.65 -2.25 437 397 318 0.71 3.37 16 4.30 50.76 3.01 5.14 63.01 61.19 1.11 75.39 26.21 -7.23 391 340 278 0.97 4.39 17 4.30 52.01 2.71 5.55 64.47 62.26 1.22 81.29 23.66 -2.04 347 366 296 0.73 3.46 18 4.30 51.54 2.84 4.36 57.59 53.50 1.99 89.74 28.04 -5.18 391 358 298 0.83 3.87 19 5.85 58.37 2.37 5.13 78.75 78.30 1.89 78.87 21.36 -11.36 480 420 364 1.30 5.71 20 5.73 58.10 2.74 5.21 75.41 79.33 1.84 76.25 21.50 -10.64 433 418 347 1.26 5.55 21 4.81 58.15 3.11 4.91 49.82 50.05 2.07 99.57 39.10 -12.43 291 325 258 1.19 5.27 22 5.19 58.30 2.50 4.87 61.83 55.10 0.01 43.30 1.13 -9.34 335 333 285 1.09 4.86 23* 5.17 59.51 3.99 6.33 66.85 60.61 2.09 102.86 40.26 -2.60 436 456 366 0.98 4.43 1