行政院國家科學委員會專題研究計畫 成果報告
含側鏈之共軛分子的液晶及 LED 性質研究
計畫類別: 個別型計畫
計畫編號: NSC91-2113-M-009-016-
執行期間: 91 年 08 月 01 日至 92 年 12 月 31 日
執行單位: 國立交通大學材料科學與工程學系
計畫主持人: 林宏洲
報告類型: 精簡報告
報告附件: 出席國際會議研究心得報告及發表論文
處理方式: 本計畫可公開查詢
中 華 民 國 93 年 3 月 31 日
計畫名稱:含側鏈之共軛分子的液晶及 LED 性質研究
Studies of LC and LED Properties of Conjugated Molecules Containing
Side Groups
計畫編號:NSC91-2113-M-009-016
執行期限:91 年 8 月 1 日至 92 年 12 月 31 日
主持人:林宏洲 交通大學 材料系
一、中文摘要 在此計畫中將設計及合成出數類新穎之雜 環材料及氫鍵液晶材料,並將各類含雜環及氫鍵 結構之單體分子拓展到各類型高分子,包含側鏈 型高分子、主鏈型高分子、及網狀高分子等高分 子結構中,並探討各類雜環及氫鍵單體液晶分子 材料及其高分子材料之LED 光電特性及物性。 延伸液晶材料之研究將開發數類高效率主鏈型 LED 高分子、側鏈型 LED 高分子、及高效率發 光液晶高分子,並包含LED 小分子發光基團之 LED 高分子摻合材料、LED 小分子發光基團之 氫鍵液晶高分子,以做為發光二極體元件。所欲 發展之發光二極體材料與元件目標如下: (1) 高發光效率 LED 高分子材料:合成新穎之 光電高分子材料包括三至五環共軛的有機分子, 藉由改變其共軛長度、環狀結構、及側鏈取代 基,以期發展出高發光效率之高分子或摻合物。 其中末端取代基為pyridine 環者,更可經由酸之 搭配而形成氫鍵錯合物。 (2) 高性能極化光發光二極體:為得極化光發光 二極體,將探討發光液晶高分子經摩擦電極表層 之高分子處理後形成定向排列,以製備具高效率 偏極效果之極化光 (polarized light) 發光二極體 元件。其極化效率將與其化學結構、液晶相種 類、及配向製程等因素有關。 (3) 奈米 LED 材料:將引用前述發展之含側鏈 的 三 個 或 五 個 共 軛 環 狀 發 光 體 為 共 軛 硬 段 (conjugated rigid blocks) 加 上 軟 段 (coil flexible blocks)合成新穎之 rod-coil diblock or triblock copolymers 製備成奈米級之 LED 材料。不同共軛 硬段及軟段之組成將影響其奈米尺度的大小及 morphology 類型的不同,更將絕對影響其形成 LED 發光體之光電特性。 綜合以上各目標,將比較評估主鏈型、側鏈 型、共聚合、及超級複合等分子結構對於提昇高 分子發光二極體材料與元件之發光效率的影響。 關鍵詞:雜環材料、氫鍵液晶材料、網狀高分子、 主鏈型LED 高分子、側鏈型 LED 高分子、發光 液晶高分子、LED 高分子摻合材料、LED 氫鍵液 晶高分子、極化光發光二極體、奈米LED 材料。 AbstractA few kinds of novel heterocyclic
materials and hydrogen-bonded (H-bonded)
liquid crystalline (LC) materials will be
designed and synthesized in this project, and
these monomers containing heterocyclic and
H-bonded structures will be expanded to
various polymers, including side-chain
polymers, main-chain polymers, and
cross-linking polymers. Both LED
(light-emitting diode) and physical properties
of these heterocyclic and H-bonded monomers
and polymers will be studied. Extending the
study of liquid crystalline materials, several
series of high performance main-chain LED
polymers, side-chain LED polymers, and
electroluminescent LC polymers, including
LED polymer blends containing organic LED
molecules, LED H-bonded LC polymers
containing LED moieties will be explored and
made into LED devices. The goal of
developing these LED materials and devices
are as follows:
(1) LED polymers with high EL efficiency:
Synthesis of novel electro-optical molecules
consisting of three to five conjugated rings by
tuning their conjugation lengths, ring structures,
and lateral substituents to develope high
performance LED polymers or blends. If the
end groups of the LED moieties are pyridine
rings, H-bonded complexes can be obtained by
complexation with acid donors.
(2) High performance polarized LED: To
acquire polarized LED, electroluminescent LC
polymers will be aligned by heating to their LC
phases on a rubbing surface. After cooling to
room temperature, the well defined orientation
of the LC polymers will be fabricated into
polarized LED devices. The polarized
efficiency of LED devices will be related to the
chemical structure, LC type, and alignment
process.
(3) Nano-scaled LED materials: Synthesis of
novel rod-coil diblock or triblock copolymers
containing previous molecules consisting of
three to five conjugated rings as conjugated
rigid blocks plus coil flexible blocks to form
nano-scaled LED materials. Different
conjugated rigid blocks and coil flexible blocks
will influence the dimension of the nano-scale
and the morphology, and thus to affect the
electro-optical properties of the LED moieties.
In general, the influence of the molecular
architecture, including main-chain LED
polymers, side-chain LED polymers, LED
copolymers, and supramolecular LED polymers,
on the promotion of the efficiency of the LED
materials and devices will be compared and
evaluated.
Keywords:
Heterocyclic Materials,
Hydrogen-bonded (H-bonded) Liquid
Crystalline Materials, Cross-linking Polymers,
Main-chain LED Polymer, Side-chain LED
Polymer, Photoluminescent LC Polymer, LED
H-bonded LC Polymer, Polarized LED,
Nano-scaled LED Materials.
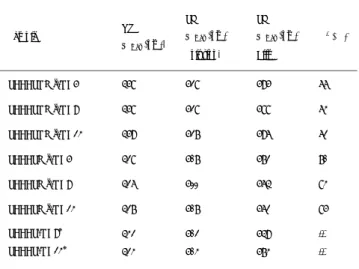
二、計劃緣由與目的 近來使用超級分子複合力製備新穎的液晶 材料已快速發展,含酸之苯甲酸結構最早被發現 具有分子間氫鍵的結構,這類型酸分子本身經由 氫鍵配對而形成雙分子,受分子經氫鍵連接而形 成足夠長度之液晶硬段,因分子間氫鍵作用力促 進了酸分子之液晶性質,但此類為經由液晶配對 而非分子辨識,最早為分子間共價鍵結產生分子 辨識形成氫鍵型液晶是在1989 年由 Kato 等人所 發現的。超級複合分子液晶乃經由不同分子間產 生非共價鍵,例如氫鍵,所形成的分子錯合物。 其液晶性質可輕易地經由混合不同的質子予體 及質子受體來調整,且新的液晶性質可經由超級 複合分子而獲得。許多不同氫鍵及不同組成成份 的氫鍵結構被開發來穩定液晶相。由於超級複合 分子結構的調整及修飾極為簡易,氫鍵液晶材料 具有應用在未來顯示器及光電材料之潛力。但據 我們所知,超級複合分子中雜環部份除了N 原子 以外,亦含有其他異原子,因此大部份的N 原子 僅做為提供產生氫鍵鍵結銜接的位置。氫鍵型液 晶可分為side chain [1]、main-chain [2]、combined [3]及 network [4]四種。在氫鍵液晶小分子部份, 依其組成形態與成份可分類為單氫鍵液晶 [5]、 雙單環單氫鍵組合液晶、具彎曲結構之氫鍵液 晶、異位雙氫鍵液晶和同位雙氫鍵液晶,目前是 針對單氫鍵液晶與雙氫鍵組合液晶來做研究。此 外延續實驗室之前的成果 [6,7],並由文獻中得 知,具有側鏈的五環共軛分子 [8,9],不但具有不 錯的螢光性質 [10-13],也因其共軛硬段部分的 加長,使分子有液晶性質產生,因此藉由改變此 分子側鏈取代基的長度及在分子的兩末端苯環 改成雜環,可藉由與酸形成氫鍵錯合物,而產生 液晶相並調整其範圍,具有側鏈的四環及五環共 軛分子與酸形成之氫鍵錯合物將首先被開發。當 同時具有液晶和螢光行為之分子,將其置入有平 行配向的cell 中,當 cell 中的分子加熱至液晶相 時,若偵測其所發出的光為偏極光,則可成功地 將 液 晶 與 螢 光 兩 者 物 性 相 結 合 。 另 具 有 oxadiazole 硬段之不對稱雜環液晶分子材料亦 將被開發並探討其末端極性取代基對其液晶及 發光性質之影響。 三、結果與討論 第一部分為 1,3,4-oxadiazole 之合成與結 構,這些物質的相轉移溫度如 Table 1 所示, mesophase 的溫度範圍由 3.1 ℃至 62.4℃,由於 NPO-OMe 具有 enantiotropic mesophase,因此 甲基取代的化合物顯示了 nematic 相,然而 10-NPO-Me 有 SmA 相而 10-NPO-OMe 並沒有,這可 由 methoxy 與 oxadiazole 兩官能基的偶極矩的 方向不同來解釋,這效應也可由 n-NPO-Me 的 isotropic 溫度比 n-NPO-OMe 高來推測而得。由 此一系列 PL 光譜的最大波長列於 Table 2 中, 8-NPO-X 衍 生 物 的 放 光 波 長 為 紫 色 , 其 中 8-NPO-Me 最紅位移,這是因末端取代基偶極矩不 同所造成,因為 mesogenic core 拉長了共軛長 度所影響,以 8-NPO-Me 為主所得的其他化合物 相 對 量 子 效 率 中 , 8-NPO-Cl 的 數 值 最 高 而 8-NPO-NO2 最低,這可由 quench 效應來解釋,由 Table 3 中得知 Dpl 於 100 ℃時,其數值介於
1.4~3.4 間,10-NPO-Me 有最高值 3.4。由文獻中 得知 fluorine 比 chlorine 有較大之電負度, chloro 取代的系統較 fluoro 系統產生較大的電 負度,末端基影響了 mesophase 項羽相轉移溫 度,如 Figure 1 所示其熔點與液晶相溫度隨著 末端基的偶極矩增加,除 8-NPO-Me 外,clearing 溫度與結晶溫度也有類似傾向,其他化合物的 mesophase 相範圍如 Figure 2 所示,ㄚ 8-NPO-OMe 由 3.1 ℃增加至 8-NPO-NO2 的 62.4 ℃,由這結 果證明了增加末端基的偶極矩可改善這一系列 化合物的 mesophase 相範圍,熔點與 clearing 溫度也與此一結果相吻合。 第二部分分子結構如 Scheme 2 與 3,由 Table 4.得知 DSC 對照出來的結果,可發現 PBBBrOC4 本身並不具有液晶相,有可能是接在中間苯環 上,側向的烷氧基鏈段太多,構成硬段縱向力的 貢獻不足,使分子不易排列,因而不能形成液 晶。比較三種配成的氫鍵錯合物,由於 Thiophene 雜環酸配成氫鍵錯合物後形成 Trimer, 因此進 入 isotropic 的溫度是三者最高的。Naphthalene 酸的結構是比配苯酸者更為剛硬,需要較多的能 量來破壞分子的堆疊,所以進入 isotropic 的溫 度比搭配苯酸者來得高。同樣的情形也發生在 PBBBrOC8 (Table 5.),因接在中間苯環上側向 的烷氧基鏈段太多,雖然大大提升了溶解度,但 分子間間距變大,分子空間變得寬鬆,使分子具 有足夠的空間運動,促使分子無法規則性進行排 列而容易進入 isotropic,因而不具有液晶相。 針對 PBBBrOC4 和 PBBBrOC8 這兩系列的氫鍵錯合 物而言,在冷卻時多呈現類似臘狀物,相轉移的 溫度範圍不易看出,所以可利用 POM 所觀察的溫 度標示。Table 6.中可說明,在 PBBBrOC8 中間 苯環上側向的碳鏈數比 PBBOC8-Br 來得多,造成 分子間距加大,以致分子更容易進入 isotropic 而不易形成液晶相。由此可見,PBBBrOC8 因第三 個苯環上引入了烷氧取代基後,熔點明顯降低約 35℃。PBBPOC4 不具有液晶相,原因是兩末端 pyridine 雜環上氮原子的電負度比環上的碳原 子大,所以在兩末端形成兩各向外拉的偶極炬, 使 兩 鄰 近 分 子 在 dipole-dipole interaction force 的作用下,緊密堆疊在一起,分子因此呈 結晶性而不具液晶相。PBBPOC4 分子在和 C10ONA 配成氫鍵錯合物後,dipole-dipole interaction force 的效應變弱,緊密堆疊的效應降低。另外, 集中在分子共軛環上的電子雲因為 Naphthalene 酸的引入而更能均勻分佈,使分子縱向引力增 強,分子因而不再緊密堆疊而能呈現向列型液晶 相。從 Table 7.所示看出,在加熱到 140℃時, PBBPOC4-ONA 由 結 晶 相 進 入 等 向 性 液 態 (Isotropic);而在冷卻回來到 134℃時,出現絲 帶狀光學條紋(schlieren texture),即分子已 經進入向列相。持續了將近 25℃的液晶溫度範圍 後,再由向列相進入結晶相。相較於 OBA 和 THDA 所配成的氫鍵錯合物,則都沒有顯現液晶性質。 由於 Naphthalene 酸比起苯環具有更佳的硬段延 伸,再加上分子具有兩個末端 pyridine 環,在 配上雙邊氫鍵後,使硬段增強,因而使分子較利 於排列並呈現出液晶向列相的性質。對於 THDA 而言,基於 thiophene 屬於彎曲的結構,破壞了 分子的 dipole,縱然配上氫鍵後,分子也不易排 列,所以並沒有形成液晶。Table 8.所示中知道, 當接在中間苯環上側向的碳鏈數為 8 時,配上 Naphthalene 酸形成氫鍵錯合物後,會和 PBBPOC4 / ONA 氫鍵錯合物一樣具有向列相。但是,可看 出當碳鏈數增為 8 時,由於分子間距變大,液晶 分子的在空間上的運動變得容易,分子的規則性 降低了,使液晶相的穩定度下降,液晶溫度範圍 因而變短,約只有 5℃的溫度範圍而已。從上各 表顯示,本系列化合物多不具有液晶相,原因是 接在中間苯環上側向的烷氧基數目太多,促使分 子縱向力降低而不易排列;再加上隨著碳鏈數的 伸長,分子間距變得更大,使液晶分子的規則性 變差而無法形成液晶。另外,從上各表中也可以 歸納出,碳鏈數的伸長、分子間距拉長是造成熔 點下降的原因。由紫外光-可見光、螢光及量子 產率 (quantum yield)可以得知分子本身基態到 激發態的能差、光色以及發光效率,Table 9.列 出一系列氫鍵受體測得的資料表。其中紫外光-可見光是以 CH2Cl2為溶劑,配置濃度為 10-5~10-6 M 的溶液所測得,這一系列化合物中共軛結構類 似,但是在中間芳香環部分含有推電子基-OR 的 存在,因此除了在 388 nm ~448 nm 有一 π→π* 躍遷的最大吸收峰外,在 325nm∼341nm 處所出 現的波峰可能是 n →π* 躍遷所生成的的吸收 峰,而且隨著有效共軛長度的拉長,發光波長愈 長,對 PL-solution 來探討可發現 PBBPOC4 / PBBPOC8 以及 PBBBrOC4 / PBBBrOC8 兩者間 PL 波 長偏移不大,可見鏈段的長短對 PL-soln 並無多 大的影響,因為分子溶在溶劑 CH2Cl2中配成極稀 溶液 (<10-5 M)後,分子與分子間會彼此完全分 開,不至於有明顯的 π→π interaction 現象 產生。但是,對 PL-film 而言,分子與分子緊密 的堆疊會導致較強的 π-π interaction 而使 excimer 產生。就分子軌域理論而得知,其基態 與激發態之能差會減小,因此會造成放出的螢光 波長變長,所以 PL-film 光譜會有明顯紅位移的 現象產生。由於 PBBPOC4 比 PBBPOC8 更易緊密堆 疊 , π-π interaction 效 應 較 大 , 所 以 其 PL-film 會 較 PBBPOC8 來 得 紅 位 移 。 同 理 , PBBBrOC4 也比 PBBBrOC8 來得紅位移。從 Table. 10 可觀察到,當形成氫鍵錯合物時,幾乎都發生 了紅位移的現象。這是因為 pyridine 雜環上的
氮原子有一孤對電子,和質子予體的-COOH 基形 成氫鍵後,會有類似 charge transfer 的行為發 生,而在氮原子帶正電荷。此正電荷對整個分子 的效應有如一個拉電子基,因此當所用的酸越 強,越容易形成氫鍵錯合物。當此拉電子效應越 強,分子 LUMO 越低,energy band gap 就變得越 窄,所以酸度最強的 THDA 其紅位移現象的發生 最為明顯,偏移了約 70nm,具有應用在調變光色 的應用潛能。PBBBrOC8 比 PBBOC8-Br 來得紅位 移,這說明了 PBBBrOC8 因為在第三個苯本環上 多了一對推電子基-OC8,以致共軛分子 HOMO 上 昇,energy band gap 減少而產生紅位移的現象。 本系列化合物僅能測得 irreversible 氧化電 位,經由計算得知 HOMO 能階位於 5.30 eV ~ 5.82 eV;而利用 λmax onset去推算,可得其 LUMO 能階,
約位於 2.89 eV ~3.58 eV,如 Table 11 所示。 PBBPOC4-ONA 最大的 polarization ratio (I// /I┴)
最大比值是 1.43,而且 I//>I┴。
第三部份分子結構如 Scheme 4,所合成之氫
鍵受體以及氫鍵予體[41]
(如圖 3-4)個別的相變 化 列 於 Table 12 中 。 Table 13-17 分 別 為 PBBBPOMe-OC8 PBBBPOMe-OC12 、 PBBBPMe-OC8 、 PBBBPMe-OC12 其氫鍵錯合物整理所得之數據,以 上為在 2,4 環上取代基為 Methoxy Group 的系 列。經 DSC 和偏光顯微鏡分析發現其不易有液晶 相產生(PBBBPOMe-OC8 系列中僅具 5℃的液晶相 範圍)。推測可能原因為中間苯環上有 OC8 和 O12 長碳鏈段,使的液晶分子不易堆疊排列外,再加 上 Methoxy Group 本身也會影響液晶分子排列, 所以即使形成氫鍵錯合物,其液晶性質還是不明 顯。綜合這幾組實驗結果可以發現,以 Methyl Group 取代的化合物在形成氫鍵錯合物後,其液 晶性質明顯比 Methoxy Group 系列為佳。原因可 能為 Methyl Group 的分子基團較小,對液晶排 列影響不大,所以易於形成向列相液晶;另外, 隨著中間苯環上取代基的鏈段增加,其向列相的 液晶範圍也會隨著縮小,其形成氫鍵錯合物之相 轉移溫度如表 Table 17 所示。Table 18 列出一 系列氫鍵受體測得的資料表。其中紫外光-可見 光吸收光譜是以二氯甲烷為溶劑,配置濃度為 10-5 ~10-6 M 的溶液所測得,這兩種系列化合物由 於共軛結構類似,因此大部分最大吸收峰 425 nm ~460 nm,為較長共軛鏈的π→π* 躍遷,而且隨 著 2,4 環取代基推電子強度的增加,會有紅移 的現象 (λmax(abs): Methoxy Group 系列 > Methyl
Group 系列)。並且經由分析紫外光-可見光吸收 光譜起始吸收(UV λonset)可以算出各分子的譜帶 間隙(Eg)的大小。Methyl 系列約為 2.53 eV, Methoxy 系列則為 2.38 eV。PL-Solution 的光譜 可以發現隨著 2,4 環取代基隨著的不同,λmax(PL) 位置也不同,而且 λmax(PL) 波長位置為 Methoxy
Group 系列 > Methyl Group 系列>無取代基系 列 , 這 是 因 為 Methoxy Group 推 電 子能 力 較 Methyl Group 強,所以比較紅位移,無取代基的 樣品波長為最短。而中間取代基碳鏈長度則不對 PL 光譜有影響,所以整體光譜位置落在 511 nm(Methyl Group 系列)和 527 nm(Methoxy Group 系列)左右。PL-Film 趨勢同於 PL- Solution, 同樣是 Methoxy 系列比較紅位移。但由於分子與 分 子 緊 密 的 堆 疊 , π - π Interaction 導 致 Excimer 的產生,所以 PL-film 光譜會具更紅位 移的現象。由於 PBBBPMe-OC4 和 PBBBPOMe-OC4 兩者中間取代基碳鏈較短,分子較易堆疊,所以 在 PL-film 其紅位移程度都較同系列分子多。取 少許以配好的氫鍵錯合物溶於二氯甲烷中配成 溶液後,滴在玻璃片上,使其自然揮發乾,形成 薄膜,而測得 PL-film。而 Blend-Film 是取合成 出化合物與 PVK 依 4:3 (Sample:PVK)比率混合 溶解後,滴於玻璃片上自然揮發乾。其結果列於 Table 19。質子受體與質子予體形成氫鍵錯合物 後,會有三種因素導致其螢光性質改變[41] :(1) 氫鍵形成所引起的紅位移:吡啶雜環上的氮原子 有一孤對電子,在形成氫鍵錯合物後,孤對電子 和質子予體上的酸形成氫鍵,氮原子上會形成一 個正電荷,而正電荷對整個分子的影響有如一個 拉電子基,因此當所用的酸越強越容易形成氫鍵 錯合物,質子受體分子上原有從烷氧基到吡啶環 之電荷。在氮原子帶正電荷後,分子的 LUMO 軌 域能量也隨之降低,Optical Energy Gap 縮小, 所以有螢光紅位移的發生,而且隨著著氫鍵受體 2,4 環上推電子基團增加,其 HOMO 能階也會提 升,進而產生更紅位移現象(Methoxy Group 系列 搭配酸後,其紅位移最明顯)。(2)立體障礙導致 的藍位移:部分氫鍵受體形成的 PL-Film 其光譜 最大放射鋒位置與其氫鍵錯合物做比較發現氫 鍵錯合物反而產生藍位移。其原因應為形成氫鍵 錯合物後,質子予體的導入使整個分子的體積變 的更為龐大,立體障礙的情形變的比較嚴重,使 原本堆積緊密的質子受體在形成氫鍵錯合物之 後變的不易堆疊,所以螢光體彼此之間的 π-π Interaction 效應降低,因此相較於 Film 之 PL 光譜有藍位移的發生;不過若與 Blend Film 比 較則可發現,形成氫鍵錯合物後,其 PL-Film 還 是都會產生紅位移現象。(3)酸性程度造成不同 紅位移現象:三種質子予體相比,OBA 與 ONA 的 pka 值差不多,而 THDA 的 pka 值最小,所以其酸 性最高,是較佳的質子予體,所以形成的氫鍵較 強,因而 THDA 所導致的紅位移最明顯。註:OBA: pka = 4.21;ONA:pka = 4.17。PBBBPMe-OC8-ONA 分子冷卻至 90 ℃時,有最大的 Polarization Ratio (I(//)/ I(⊥)),比值為 2.13,由 CV 圖分析
Irreversible 的氧化電位,所以利用紫外光/可 見 光 光 譜 中 長 波 長 的 λonset 來 決 定 光 學 能 帶
(Optical Band Gap,Eg = 1240/λonset),再利用
Ea = I p - Eg,即可求出還原電位。比較其結果如
Table 20 顯示,PBBBPOMe-OC12 跟 PBBBPMe-OC12 兩者 LUMO 差距不大,但在 HOMO 的改變上, PBBBPOMe-OC12 提 升 較 多 , 這 應 是 分 子 上 面 Methoxy Group 推電子能力較 Methyl Group 顯著 所導致。
四
、計畫成果自評 本計畫完成開發數系列含 oxadiazole 硬段 之不對稱雜環液晶分子材料,與含四環及五環共 軛結構的螢光材料,其中有部份結構具有優異的 液晶性質,將有機會製備成高亮度之發光元件。 五、參考文獻
1. Kawakami, T.; Kato, T. Macromolecules, 1998,
31, 4475.
2. Aoki, K.; Nakagawa, M.; Ichimura, K. J. Am.
Chem. Soc., 2000, 122, 10997.
3. Kato, T.; Ihata, O.; Ujiie, S.; Tokita, M.; Watanabe, J. Macromolecules, 1998, 31, 3551. 4. Kihara, H.; Kato, T.; Uryu, T.; Frechet, J. M. J.
Chem. Mater., 1996, 8, 961.
5. Kihara, H.; Kato, T.; Uryu, T.; Ujiie, S . J. M. J.
Macromolecules, 1996, 29, 8734.
6. Ko, C. W.; Lin, H. C. Thin Solid Films, 2000, 363, 298.
7. Lin, H. C.; Sheu, H. Y.; Chang, C. L.; Tsai, C.
J. Mater. Chem., 2001, 11, 2958.
8. Gill, R. E.; Hilberer, A.; Hadziioannou, G.
Synth. Met., 1997, 84, 637.
9. Gill, R. E.; Meetsma, A.; Hadziioannou, G. Adv.
Mater., 1996, 8, 212.
10. Hadziioannou, G.; Gill, R. E.; Van Hutten, P. F.
Chem. Mater., 1996, 8, 1341.
11. Brouwer, H. J.; Krasnikov, V. V.; Hadziioannou, G. Chem. Phy., 1998, 227, 65.
12. Barashkov, N. N.; Guerrero, D. J.; Ferraris, J. P.
Synth. Met., 1995, 75, 153.
13. Gebhardt, V.; Bacher, A.; Thelakkat, M.; Haarer, D.; Synth. Met., 1997, 90, 123.
HO O OH H2n+!CnO O NHNH O X O Cl X Br(CH2)nH NH2NH2.H2O POCl3 MeOH X N N O H2n+1CnO n= 6, 8, and 10
X = Me, OMe, Cl, F, CN, and NO2
MeOH/H2SO4 K2CO3/Acetonitriale 1 2a n = 6 2b n = 8 2c n = 10 3a n = 6 3b n = 8 3c n = 10 4a-X n = 6
4b-X n = 8 X = Me, OMe, Cl, F, CN, and NO2
4c-X n = 10 where n = 6, 8, and 10;
and X = Me, OMe, F, Cl, CN, and NO2
n-NPO-X HO O O H2n+!CnO O O H2n+!CnO O NHNH2
Scheme 1. The synthetic route of
1,3,4-oxadiazole-based materials.
Table 1. Phase behavior of n-NPO-X
derivatives
a.
K K1 I K N I K I N K I N SmA K K1 N K2 I K N I K N I K N I K SmA I K K1 SmA I K SmE SmA I K K1 I SmA K SmA I K K1 N I 6-NPO-CN K K1 SmA 8-NPO-CN I K K1 SmA 10-NPO-CN I K SmA 6-NPO-NO2 I SmE SmX SmA 8-NPO-NO2 I K SmE SmA I K SmA I SmE SmE SmE 6-NPO-Me 112.5 (29.6) 114.8 (-9.5) 122.0 (16.7 ) 106.7 (-0.3) 86.1 (-24.0) 8-NPO-Me 121.7 (29.5) 104.6 (-0.4 ) 96.1 (-26.0) 10-NPO-Me 106.7 (-0.6) 92.9 (-0.6) 85.3 (-25.9) 121.0 (32.9) 6-NPO-OMe 91.7 (0.4) 99.2 (24.1) 112.5 (0.1) 107.5 (-14.2) 119.5 (18.5 ) 111.0 (-0.3) 58.1 (-21.7) 8-NPO-OMe 109.4 (31.6) 114.6 (0.2) 113.1 (-0.4) 79.5 (-26.6) 10-NPO-OMe 110.1 (30.9) 113.2 (0.2) 111.6 (-1.4) 76.5(-27.4) 6-NPO-Cl 153.7 156.0 153.8 (-4.8) 130.3 (-31.5) 8-NPO-Cl 105.1 (4.3) 143.1 (35.0) 159.9 (6.1) 157.3 (-6.2) 122.4 (-30.0) 100.9 (-4.3) 10-NPO-Cl 88.0 (1.2) 131.6 (26.6) 157.5 (-5.6) 118.0 (-25.8) 85.2 (-1.0) 6-NPO-F 117.7 (1.9) 139.3 (32.1) 128.2 (-3.6) 109.7 (-24.5) 99.5 (-1.3) 8-NPO-F 100.0 (3.6) 127.0 (28.3) 132.4 (-4.5) 105.9 (-26.7) 92.7 (-3.2) 134.9 (4.4) 160.0 (5.5) 130.0 (-1.2) 162.7 (30.0) 167.1 (0.1) 165.5 (-0.3) 118.4 (-30.4) 65.5 (9.4) 143.3 (26.5) 167.1 (1.8) 165.0 (-1.9) 126.4 (-25.8) 42.0 (-8.6) 89.7 (13.9) 132.2 (26.5) 172.5 (3.0) 170.5 (-2.9) 118.9 (-26.3) 64.9 (-29.4) 88.2 (-7.1) 165.4 (35.4) 190.4 (3.1) 187.9 (-3.2) 139.5 (-10.4) 99.9 (-1.5) 91.9 (14.1) 143.7 (10.2) 202.6 (4.4) 91.0 (19.7) 141.5 (11.2) 203.9 (5.3) 201.6 (-5.3) 137.3 (-11.1) 50.0 (-14.9) 10-NPO-NO2 10-NPO-F 80.5 (3.7) 115.9 (19.5) 131.8 (-3.8) 96.4 (-19.0) 75.6 (-3.4) 134.2 (3.7) 200.3 (-4.4) 139.1 (-10.1)Table 2. Absorption and photolumines- cence
spectra of 8-NPO derivatives.
8-NPO-Me 8-NPO -OMe 8-NPO -F 8-NPO -Cl 8-NPO -CN 8-NPO -NO2 Absorption peaks, nm 323 325 323 329 338 346 Emission peak (PL), nm 374 377 375 383 417 390 Rel. quantum yield 1 1.05 0.95 1.3 1.05 0.05
Table 3. PL dichroic ratio (D
PL) and order
parameter of n-NPO-X in various conditions.
10-NP O-Me 6-NPO-O Me 10-NP O-F 10-NP O-Cl 10-NP O-CN 10-NPO -NO2 Maximum PL dichroic ratioa, (DPL= I∥/ I⊥) 3.4 (100 ℃) 1.8 (85℃) 1.9 (110 ℃) 1.4 (150 ℃) 1.7 (130 ℃) 1.6 (150℃) Order parameterb 0.44 0.21 0.23 0.11 0.19 0.17
80 100 120 140 160 180 200 220 8-NPO-X NO2 CN Cl F OMe X=Me te mpe rat ur e ( oC) A B C D
Figure 1. The effect of polar substituents on
the transition temperatures (dipole increases
from left to right). (A) clearing point,
(B)melting point, (C) liquid crystallization
temperature (on cooling), (D)crystallization
temperature.
0 10 20 30 40 50 60 70 NO2 CN Cl F OMe X=Me m esophase rang e ( o C) 8-NPO-X heating coolingFigure 2. The effect of polar substituents on
the mesophase range during heating and
cooling cycles. (dipole increases from left to
right).
N N (P) - PBBPCH3 N N OCH3 OCH3 H3CO H3CO (Q) - PBBPOCH3 N Br OC4H9 OC4H9 C4H9O C4H9O (R) - PBBBrOC4 N N OC4H9 OC4H9 C4H9O C4H9O (S) - PBBPOC4 N Br OC8H17 OC8H17 C8H17O C8H17O (T) - PBBBrOC8 N N OC8H17 OC8H17 C8H17O C8H17O (U) - PBBPOC8Scheme 2. Proton acceptor.
C10H21O
COOH COOH
C10H21O
(A) - OBA (B) - ONA
(C) - THDA
S COOH HOOC
4-Decyloxy-benzoic acid 6-Decyloxy-naphthalene-2-carboxylic acid
Thiophene-2,5-dicarboxylic acid
Scheme 3. Proton donor
Table 4. The phase transition temperature of
donor and acceptor.
Compound Phase Behavior - ℃(J/g)
PBBBrOC4 K 86.4(24.1) I I 60.4(-23.4) K H C PBBPOC4 K 217.7(30.8) I I 206.5(-33.4) K H C PBBBrOC8 K 61.0(20.9) I I 44.2* K H C PBBPOC8 K 154.9(28.2) I I 145.2(-28.9) K H C
Table 5. The phase transition temperature of
H-bonding complexes.
Compound Phase Behavior-℃(J/g)
PBBBrOC4 PBBBrOC4 -OBA PBBBrOC4 -ONA PBBBrOC4 -THDA K 86.4 (24.1) I I 60.4(-23.4) K K 84.1(36.6) I I 38.6(-18.3) K K 104.9(3.3) I I 82.2(-3.8) K K 157.9(38.8) I I 131.5(-22.3) K H C H C H C H C
Table 6. The phase transition temperature of
PBBBrOC8 and H-bonding complexes.
Compound Phase Behavior-℃(J/g)
PBBBrOC8 PBBBrOC8 -OBA PBBBrOC8 -ONA PBBBrOC8 -THDA K 61.0(20.9) I I 44.2*K K 71.73(4.3) I I 51.6(-2.7) K K 102.7(5.7) I I 74.8(-4.0) K I 200* K K 181* I H C H C H C H C
Table 7. The phase transition temperature of
PBBBrOC4 and H-bonding complexes.
Compound Phase Behavior-℃(J/g)
PBBPOC4 PBBPOC4 -OBA PBBPOC4 -ONA PBBPOC4 -THDA K 217.7(30.8) I I 206.5(-33.4) K K 89.3(3.3) K’ 97.3(4.3) K’’131.8(41.0) I I 114.3(-35.2) K’’ 90.8(-5.1) K’ 68.9(-3.8) K K 139.6(42.8) I I 134* N 100* K K 137.5(1.4) K’ 186.5(18.5) I I 164.4(-16.6) K H C H C H C H C
Table 8. The phase transition temperature of
PBBPOC4 and H-bonding complexes.
Compound Phase Behavior-℃(J/g)
PBBPOC8 PBBPOC8 -OBA PBBPOC8 -ONA PBBPOC8 -THDA K 154.9(28.2) I I 145.2(-28.9) K K 110.5(42.1) I I 96.1(-43.9) K 125* I I 100* N 95*K K 175.6(2.1) I I 143.2(-2.1) K H C H C H C H C
Table 9. The λmax of H-bonding acceptor
for optical properties.
Pure sample UV(nm) PL(nm)-soln PL(nm)-film Φ(%)
PBBPMe 394 456, 476 505 41 PBBPOMe 330, 388 454, 470 507 35 PBBBrOC4 325, 412 474 532 62 PBBPOC4 341, 448 499 559 67 PBBBrOC8 326, 412 475 510 59 PBBPOC8 341, 441 500 552 70
Table 10. The λmax of H-bonding complexes
of photoluminescent properties.
Pure OBA ONA THDA
PBBBrOC4 532 559 573 600
PBBPOC4 559 596 590 -
PBBBrOC8 510 518 523 585
PBBPOC8 552 556 585 -
Table 11. The data of CV and band gap.
Pure sample λonset(nm) Epc(mV) IP(eV)-
HOMO Ea(eV)- LUMO Eg(eV) PBBBrOC4 515 819 5.30 2.89 2.41 PBBPOC4 570 1286 5.76 3.58 2.18 PBBBrOC8 525 933 5.41 3.05 2.36 PBBPOC8 547 1347 5.82 3.55 2.27
Scheme
4.Table 12. The phase transition temperature of
acceptors and their H-bonding complexes.
Sample Phase Behavior
℃ (J/g) PBBBPOMe-OC4 PBBBPOMe-OC8 PBBBPOMe-OC12 PBBBPMe-OC4 PBBBPMe-OC8 PBBBPMe-OC12 PBBBP-OC8# PBBBP-OC12# OBA ONA THDA K 287* I I 258* K K 141.8(-6.4) K’ 180.0(50.8) I I 113.6 (-38.7) K K 97.4(-14.5) K’ 120.0(13.7) I I 92* K K 233.1 (38.1) I I 212 (-38.8) K K 164.2 (32.4) I I 146.7 (-31.4) K K 149.6 (34.3) I I 132.3 (-34.0) K K 128.5(10.1) K’ 230.2(52.6) I I 220.6(-54.8) K’ 120.4(-11.0) K K 68.3(8.1) K’ 218.7(63.7) I I 194.9(-64.9) K’ 63.7(-8.8) K K 85.1(8.9) Sx 96.0(30.3) Sc 123.7(4.5) N 142.4(7.7) I I 139.7(-7.7) N 120.5(-4.3) Sc 90.2(-31.1) Sx 69.4(-21.5) K K 104.8(0.7) K’ 136.3(39.4) Sc 140.5(2.9) N 175.9(7.7) I I 173.3(-4.7) N 138.4(-3.6) Sc120.3(-40.3) K’ 89.7(-1.7) K K 300* I H C H C H C H C H C H C H C H C H C H C H
Table 13. The phase transition temperature of
PBBBPOMe-OC8 and their H-bonding
complexes.
Sample Phase Behavior ℃ (J/g) PBBBPOMe-OC8 PBBBPOMe-OC8 -OBA PBBBPOMe-OC8 -ONA PBBBPOMe-OC8 -THDA K 141.8(-6.4) K’ 180.0(50.8) I I 113.6 (-38.7) K K 122.5(39.8) I I 100.4(-19.4) N 95.7(-18.3) K K 131.1(16.6) K’ 150.9 (48.9) I I 130 (-1.7) N 125.7(-45.5) K’ 74.3(-17.4) K K 280* I I 230* K H C H C H C H C
Table 14. The phase transition temperature of
PBBBPOMe-OC12 and their H-bonding
complexes.
Sample Phase Behavior ℃ (J/g) PBBBPOMe-OC12 PBBBPOMe-OC12 -OBA PBBBPOMe-OC12 -ONA PBBBPOMe-OC12 -THDA K 97.4(-14.5) K’ 120.0(13.7) I I 92* K K 96.0(0.9) K’ 132.0 (46.9) I I 108.6(-47.3) K’ 87.7(-1.0) K K 149.8(49.1) I I 123.8(-44.8) K K 255* I I 215* K H C H C H C H C
Table 15. The phase transition temperature of
PBBBPMe-OC8 and their H-bonding
complexes.
Sample Phase Behavior ℃ (J/g) PBBBPMe-OC8 PBBBPMe-OC8 -OBA PBBBPMe-OC8 -ONA PBBBPMe-OC8 -THDA K 164.2 (32.4) I I 146.7 (-31.4) K K 127 (50) N 141.2(1.37) I I 139.1 (-1.5) N 89.8 (-36.1) K K 102.9 (4.1) K’ 141.7 (40.4) N 164.6(1.8) I I 162.4 (-1.6) N 78.5 (-35.0) K K 271.5* I I 239* K H C H C H C H C
Table 16. The phase transition temperature of
PBBBPMe-OC12 and their H-bonding
complexes.
Sample Phase Behavior ℃ (J/g) PBBBPMe-OC12 PBBBPMe-OC12-OB A PBBBPMe-OC12 -ONA PBBBPMe-OC12-TH DA K 149.6 (34.3) I I 132.3 (-34.0) K K 74.5 (-20.5) K’ 110.9(49.6) N 124.2(1.4) I I 121.9(-1.7) N 88.1 (-21.9) K’ 70.1(-10.1) K K 105.9(9.4) K’ 115.7 (27.9) N 144.7(1.4) I I 142.5(-1.5) N 81.2 (-20.7) K’ 75.7(-9.2) K K 230* I I 192* K H C H C H C H C
Table 17. The phase transition temperature of
PBBBP-OR H-bonding complexes.
Sample Phase Behavior ℃ (J/g) PBBBP-OC8 -OBA PBBBP-OC8 -ONA PBBBP-OC12 -OBA PBBBP-OC12 -ONA K 148.6 (52.3) N 191.3(1.0) I I 187.4(-0.8) N 136.7 (-56.3) K K 128.9 (-11.0) K’ 141.9(48.7) N 215.7(0.8) I I 213.0* N 118 (-38.2) K K 135.3(43.8) N 175.6(1.4) I I 166.0(-1.6) N 116.3 (-53.9) K K 87.8 (2.5) K’ 128.0(37.1) N 201* I I 200.0* N 107.1(-107.1) K’ 76.7(-75.7) I H C H C H C H C HOOC OC10H21 HOOC OC10H21 S COOH HOOC (OBA) (ONA) (THDA) 6-Decyloxy-naphthalene -2-carboxylic acid 4-Decyloxy-benzoic acid Thiophene-2,5-dicarboxylic acid CH3 H3C N H3C CH3 N OR RO R=C4H9 (PBBBPMe-OC4) R=C8H17 (PBBBPMe-OC8) R=C12H25 (PBBBPMe-OC12) OCH3 H3CO N H3CO OCH3 N OR RO R=C4H9 (PBBBPOMe-OC4) R=C8H17 (PBBBPOMe-OC8) R=C12H25 (PBBBPOMe-OC12)
Table 18. The λmax of H-bonding complexes
for UV-Vis, photoluminescence and quantum
yield properties.
Sample UV λmax (nm)* PL λmax (nm)# Solution* PL λmax (nm) # Film Φ+ (%) PBBBPOMe-OC4 458 528 595 66 PBBBPOMe-OC8 458 528 588 63 PBBBPOMe-OC12 459 527 596 62 PBBBPMe-OC4 428 507 572 70 PBBBPMe-OC8 426 511 564 83 PBBBPMe-OC12 427 507 562 85 PBBBP-OC8※ PBBBP-OC12※ 432 423 502 503 549 573 -- --Table 19. The λmax of H-bonding complexes
for photoluminescence spectra.
λmax (nm) Pure Blend OBA ONA THDA
PBBBPOMe-OC8 588 578 611 638 668 PBBBPOMe-OC12 599 581 642 659 677 PBBBPMe-OC8 564 557 562 565 630 PBBBPMe-OC12 562 553 569 577 633 PBBBP-OC8# 549 -- 555 551 -- PBBBP-OC12# 573 -- 571 568 --
Table 20. The data of CV and band gap.
Sample λonset (nm) Eg (Ev) Ip (eV) HOMO Ea (eV) LUMO PBBBPOMe-OC12 520 2.38 5.27 2.89 PBBBPMe-OC12 491 2.52 5.48 2.86