國立台灣大學生命科學院生化科技學系 碩士論文
Department of Biochemical Science and Technology College of Life Science
National Taiwan University Master Thesis
以轉錄因子 Mxr1 再程序化的策略加強嗜甲醇酵母菌 Pichia pastoris AOX1 啟動子效率
Enhancement of Pichia pastoris AOX1 Promoter Efficiency by Reprogramming the Transcription Factor
Mxr1 張景翔
Ching-Hsiang Chang
指導教授:黃慶璨 博士 Advisor: Ching-Tsan Huang, Ph.D.
中華民國 106 年 7 月
謝誌
時光匆匆、歲月如梭,又到了說掰掰的時候!在Lab106 的時間應該有 4 年 了吧,這段時間說長不長、說短不短,短到讓我覺得自己好像也沒做幾件事就要 走了;但也長到足以讓我從一個不想念碩班的大學生,變成想試試能不能繼續在 研究之路上前進的碩士生。為什麼會有這樣的轉變呢?我想最主要的原因是在這 段時間內,我過得很開心!感謝在這幾年跟我相處的大家!
首先,謝謝我的指導教授-黃慶璨老師!黃教授真的是一個〝老師〞,從老 師那學到很多待人處事的經驗與邏輯,並了解溝通的重要,希望這點我有明顯的 進步!還有也謝謝老師願意相信我、願意給我很的大自由,讓我可以做我想做的 題目、實驗,這樣一個快樂學習的環境對我真的很重要!同時,感謝吳亘承老師,
對我來說吳老師更像是實驗室的大學長,跟我分享了許多寶貴的經驗與新創的想 法,讓我更期待未來研究之路的樣子!以及口試委員:陳浩仁、林晉玄、凌嘉鴻 老師,謝謝老師們在我論文上給予的建議!
再來則是Lab106 的前、今、後人:謝謝宛伶學姐每次畢業典禮都有來看我 們!謝謝莉欣與巧青學姐,教會我很多實驗、給予我很多幫忙!謝謝玉儒學姐、
浩業學長,在醱酵課的過程中,讓我有機會接觸到不一樣的刺激!謝謝俞均與芝 榕學姊,讓實驗室更有趣也更溫馨!謝謝昱伶學姊,處理實驗室的許多事情,carry 菇類組!謝謝同屆的大家,凱琳、孟羲和哲銘,從你們身上我都有學到很多東西、
看到許多我不足之處!謝謝下一屆的學弟妹們,為實驗室注入不一樣的元素,映 希、浩安,酵母菌組就交給你們carry 啦!越來越進入狀況的尚儒、冠陞繼續加 油呀!也感謝其它新進的學弟妹,希望你們也可以在Lab106 快樂學習!
最後,則是我的家人,讓我可以無憂無慮的完成從小到大的學習,支持我每 次選擇要做的事情!
景翔 謹誌 106 年 7 月
摘要
Pichia pastoris 為嗜甲醇酵母菌的一種,是極具潛力的異源蛋白質表達系統,
兼具微生物與真核系統的優勢。搭配甘油培養-甲醇誘導策略,可嚴謹調控 P.
pastoris 的 AOX1 啟動子,並大量生產重組蛋白質。然而,調控過度嚴謹的 AOX1 啟動子,使 P. pastoris 只能侷限在特定的培養條件,必需以有毒、易燃的甲醇作 為唯一碳源時,才可有效率誘導 AOX1 啟動子。本研究希望藉由轉錄因子再程序 化,改變 AOX1 啟動子的調控,以提升 P. pastoris 的生產效率。透過建立轉錄活 化子 Mxr1 (Methanol expression regulator 1)的自我正回饋控制迴路(Positive auto- regulation circuit),降低抑制性碳源對 AOX1 啟動子的干擾,並解決 Mxr1 稀釋效 應所造成轉錄活性下降。以 AOX1 啟動子表現綠色螢光蛋白質基因,並以 AOX2 啟動子表現額外的 Mxr1。在不同種類及濃度的碳源培養下,證實此策略可以提 升 AOX1 啟動子的轉錄活性,並且不會造成細胞生長缺陷。在甘油高於特定濃度 時,AOX1 啟動子仍保留嚴謹的控制。但當甘油降至特定濃度後,Mxr1 正回饋迴 圈的啟動,逐步降低 AOX1 啟動子的嚴謹度,去除殘留抑制性碳源對 AOX1 啟動 子的干擾,進而使 AOX1 啟動子能受甘油受限的誘導。然而,提升額外 Mxr1 之 拷貝數,無法進一步加強 AOX1 啟動子的轉錄效率,顯示 Mxr1 與外源性 AOX1 啟動子的最佳比例仍需更深入目標的探討。最後,AOX1 啟動子提升的轉錄活性,
可能會受限於外泌效率,而影響胞外蛋白質的產量,但未來可以搭配其它策略解 決此問題。總而言之,透過轉錄因子再程序化的策略,可以保有AOX1 啟動子原 有的優勢,加強甲醇誘導的轉錄活性,同時解決AOX1 啟動子調控過於嚴謹的缺 點,使P. pastoris 更具應用性。
Abstract
The methylotrophic yeast Pichia pastrois has been extensively applied in production of recombinant proteins because it combines the advantages of single cell in microbial and post-translational modification in eukaryotic systems. The AOX1 promoter (PAOX1) is the most common promoter used for heterologous protein expression in P. pastoris. A glycerol-methanol-shift induction strategy is applied to achieve high productivity. However, the tightly regulated PAOX1 also led P. pastoris expression to restrictive conditions. To improve the efficiency of protein production, we tried to reprogram the transcriptional regulation of PAOX1 in P. pastoris. The ectopic Mxr1 expressed by the mild AOX2 promoter (PAOX2) did not cause growth defect. The transcriptional efficiency of PAOX1 was enhanced since the limitation of Mxr1 titration effect was broken by extra Mxr1. PAOX1 became more flexible due to the positive feedback of Mxr1 and was regulated by glycerol. With the extra Mxr1 driven by PAOX2, PAOX1 showed better activity without than that with medium replacement. Moreover, glycerol starvation induced GFP production with reprogramming Mxr1 in P. pastoris.
Increasing copy number of ectopic Mxr1 did not enhanced the efficeince of PAOX1. These results showed overexpression of Mxr1 by one copy of PAOX2 might be enough to achieve the maximum activity of PAOX1. Although the improvement of transcriptional efficiency might be limited by secretory ability, these problems could be sloved by combination with other strategies. In conclusion, transcriptional reprogramming of Mxr1 improved the efficiency of P. pastrois under methanol induction and potentially made P. pastrois become methanol-free induction system to eliminate the problems of methanol.
Keywords: Pichia pastoris, methanol, AOX1 promoter, transcription factor, Mxr1
目錄
謝誌... I 摘要... II Abstract ... III 目錄... IV 圖目錄... VII 表目錄... VIII 附圖目錄... IX
第一章前言... 1
一、異源表達系統... 1
二、Pichia pastoris 嗜甲醇酵母菌表現系統 ... 1
1. 具有轉譯後修飾... 2
2. 具外泌異源蛋白質的能力... 2
3. 表現量高、調控嚴謹的甲醇誘導系統... 3
三、AOX1 啟動子的轉錄調控 ... 4
1. MXR1 ... 5
2. PRM1 ... 7
3. MIT1 ... 8
4. NRG1... 9
四、甲醇誘導系統面臨的困境與解決辦法... 11
1. 甲醇的負面影響... 11
2. 過度嚴謹的 AOX1 啟動子 ... 12
五、研究動機... 14
1. 目的... 14
2. 轉錄調控策略... 14
3. 目標... 15
第二章 材料與方法... 18
一、實驗菌株與培養條件... 18
1. 細菌... 18
2. 真菌... 18
二、培養基... 19
三、表現載體建構... 21
1. pPICZ-mEGFP ... 21
5. pAOX2KH-Mxr1 ... 23
四、嗜甲醇酵母菌電穿孔轉形... 28
1. P. pastoris 勝任細胞製備 ... 28
2. 電穿孔轉形... 28
五、嗜甲醇酵母菌染色體DNA 分析 ... 30
1. 轉形株染色體簡易分析... 30
2. 轉形株目標基因拷貝數測定... 30
六、轉形株培養與分析... 32
1. 試管誘導... 32
2. 搖瓶誘導... 32
3. 醱酵槽誘導... 32
七、mRNA 表現量分析 ... 34
八、蛋白質分析... 36
1. 聚丙烯醯胺膠體電泳分析... 36
2. 西方墨點法... 36
3. AOX 活性分析 ... 37
第三章 結果... 40
一、轉形菌建立與分析... 40
1. 胞內型綠色螢光蛋白質生產菌株... 40
2. 外泌型綠色螢光蛋白質生產菌株... 40
3. Mxr1 再程序化之胞內型綠色螢光蛋白質生產菌株 ... 41
3. Mxr1 再程序化之外泌型綠色螢光蛋白質生產菌株 ... 41
二、轉錄因子Mxr1 再程序化對異源蛋白質生產的影響 ... 50
1. 額外表現 Mxr1 加強胞內型綠色螢光蛋白質的生產 ... 50
2. 額外表現 Mxr1 的效果不隨著拷貝數增加而提升 ... 50
3. 額外表現 Mxr1 不會破壞 AOX1 啟動子嚴謹調控的特性 ... 51
4. 額外表現 Mxr1 提升 AOX1 啟動子轉錄效率 ... 51
5. 額外表現 Mxr1 解除 P. pastoris 甲醇誘導需要置換的限制 ... 52
6. 額外表現 Mxr1 賦予 P. pastoris 以碳源受限誘導的能力 ... 52
7. 額外表現 Mxr1 改善 P. pastoris 的甲醇代謝能力 ... 54
8. 異源蛋白質的生產可能受限於外泌效率... 54
第四章 討論... 69
一、額外表現Mxr1 對 AOX1 啟動子轉錄活性的影響 ... 69
二、額外表現Mxr1 對 AOX1 啟動子碳源控制性的影響 ... 70
三、甲醇調控機制之探討... 71
1. Nrg1 與甘油抑制之關係 ... 71
2. Prm1 與氮源之關係 ... 72
四、無甲醇誘導系統發展的可能性... 72
1. 在甘油受限時的 Nrg1 表現量 ... 72
2. Mit1 潛在的轉譯後調控 ... 73
第五章 結論... 74
第六章 未來展望... 75
第七章 參考文獻... 77
圖目錄
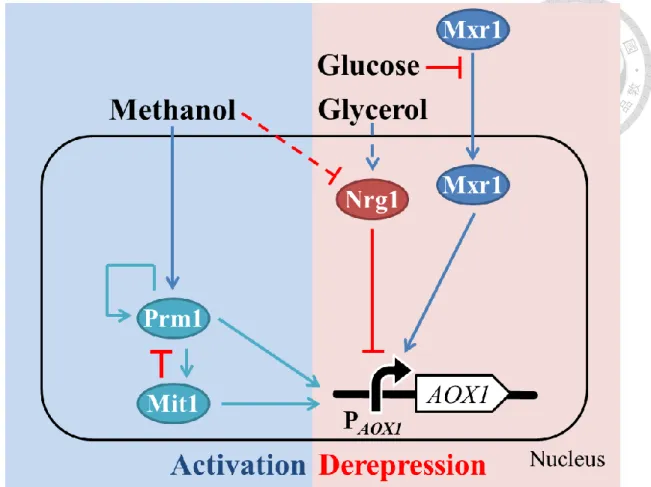
圖一、AOX1 啟動子的轉錄調控示意圖 ... 10
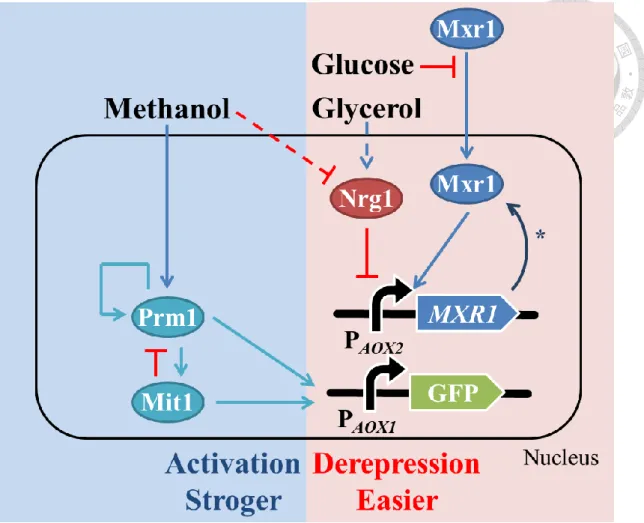
圖二、Mxr1 再程序化菌株轉錄調控示意圖 ... 16
圖三、本論文之研究架構圖... 17
圖四、本研究建構之表現載體圖譜... 24
圖五、以不同培養基篩選pPICZ-mEGFP 轉形株 ... 42
圖六、胞內型mEGFP 生產菌株標準化螢光強度分析 ... 43
圖七、螢光顯微鏡觀察胞內型mEGFP 生產菌株 ... 44
圖八、以不同培養基篩選pPICZα-mEGFP 轉形株... 45
圖九、以不同G418 抗性濃度篩選 Mxr1 再程序化之 E3 轉形株 ... 46
圖十、聚合酶連鎖反應分析Mxr1 再程序化轉形株染色體 DNA ... 47
圖十一、以不同G418 抗性濃度篩選 Mxr1 再程序化之 SE12 轉形株 ... 48
圖十二、以標準方法誘導Mxr1 再程序化轉形株 ... 56
圖十三、不同拷貝數Mxr1 轉形株的標準化螢光強度分析 ... 57
圖十四、外源性MXR1 拷貝數與標準化螢光強度之關係圖 ... 58
圖十五、Mxr1 再程序化轉形株在不同甘油下的濁度與標準化螢光強度 ... 59
圖十六、Mxr1 再程序化轉形株在不同碳源下的標準螢光強度 ... 60
圖十七、Mxr1 再程序化轉形株之 mEGFP 與 MXR1 mRNA 表現量分析 ... 61
圖十八、Mxr1 再程序化轉形株之 NRG1、PRM1、MIT1 mRNA 表現量分析 .... 62
圖十九、Mxr1 再程序化轉形株不置換培養基的標準化螢光強度 ... 63
圖二十、以甘油受限誘導Mxr1 再程序化轉形株 ... 64
圖二十一、Mxr1 再程序化轉形株以醱酵槽進行甘油受限誘導之參數圖 ... 65
圖二十二、Mxr1 再程序化轉形株以醱酵槽進行甲醇誘導之參數圖 ... 66
圖二十三、各菌株在不同碳源下的甲醇代謝能力... 67
圖二十四、分析Mxr1 再程序化轉形株的胞、內外螢光強度 ... 68
表目錄
表一、建構表現載體與定序之引子序列... 25
表二、本研究使用之分生試劑套組... 27
表三、即時定量聚合酶鏈鎖反應之引子序列... 35
表四、蛋白質分析所用試劑之組合成分... 38
表五、本研究建構之轉形株與其外源基因的拷貝數... 49
附圖目錄
附圖一、異源表達系統比較圖... 81 附圖二、嗜甲醇酵母菌甲醇之利用途徑... 82
第一章前言
一、異源表達系統
異源表達(heterologous expression)是透過基因工程,將外源基因送入特定宿 主,並以該宿主的蛋白質合成系統生產重組蛋白質(recombinant protein)。相較於 直接從原物種取得蛋白質,透過異源表達系統生產具備以下幾點優勢:(1)原料取 得容易、(2)生產成本低廉及(3)操作程序簡單。因此異源表達系統已廣泛應用於 學術、農業、工業、生技醫療產業等[1]。而適合用於異源蛋白質表達系統的宿主 也發展出許多不同選擇,從原核的大腸桿菌到真核的酵母菌、植物及哺乳細胞等,
每種系統都有著各自的優缺點,如附圖一。
二、Pichia pastoris 嗜甲醇酵母菌表現系統
嗜甲醇酵母菌(methylotrophic yeast)為一類可利用甲醇為唯一碳源的酵母菌,
此類酵母菌具有特別的甲醇代謝路徑(methanol utilization pathway),並在過氧化 體(peroxisome)進行初步的甲醇代謝,以避免代謝產物的毒性。首先,甲醇先經由 酒精氧化酶(alcohol oxidase, AOX, EC 1.1.3.13)氧化成甲醛,並產生副產物-過氧 化氫,過氧化氫會進一步由過氧化氫酶(catalase)分解成水與氧氣,而甲醛則經由 異化作用(dissimilation)轉換成能量,或是透過同化作用(assimilation)形成生物質 量,如附圖二。甲醇代謝路徑受到碳源的調控,在甲醇為唯一碳源的情況下,相 關基因會大量表現,以避免甲醇、甲醛過量累積而造成細胞死亡。這些表現量高、
調控嚴謹的甲醇代謝基因,加上酵母菌為真核微生物系統的優勢,使嗜甲醇酵母 菌成為廣泛使用的異源蛋白質表現平台,其中常見的菌株有Pichia pastoris、
P. pastoris為本研究中使用的異源表達系統,是目前研究最透徹的嗜甲醇酵 母菌。在1960年代後期,P. pastoris曾被用於生產單細胞蛋白質(single cell protein, SCP),雖然受到石油危機、甲醇價格上漲的影響,而逐建被黃豆取代,但留下大 規模醱酵製程技術,成為未來工業化生產異源蛋白質的基石[5]。1980年,Phillips Petroleum Company和Salk Institute Biotechnology/Industrial Associates, Inc. (SIBIA, La Jolla, CA, USA)合作開發P. pastoris異源蛋白質表現系統,包含菌種、表現載體 及使用流程等[6],至今已有數千種以上的蛋白質成功的以此系統進行表現[7],
其中有一些重組蛋白質藥物已通過美國FDA (Food and Drug Administration)的核 可,抱括:胰島素、抗體和B肝疫苗,顯示P. pastoris在未來蛋白質藥物領域中扮 演重要的角色[8]。
此系統除了具有微生物系統的便宜、快速、產量大等優點[9],還具有以下幾 點優勢:
1. 具有轉譯後修飾
相較於原核大腸桿菌系統,P. pastoris屬於真核生物,能夠對蛋白質進行醣基 化、磷酸化、形成雙硫鍵等轉譯後修飾,更適合用於表現真核生物的異源蛋白質,
而且不像釀酒酵母(Saccharomyces cerevisiae),會造成蛋白質過度醣基化,使其生 產的蛋白質可能導致人體產生過敏反應[10]。因此,同樣為真核微生物表達系統 的P. pastoris,比S. cerevisiae更適合用來生產醫藥用蛋白質[11]。
2. 具外泌異源蛋白質的能力
S. cerevisiae進行有性生殖時,α mating type的交配信息素α-factor,會透過外 泌訊息α-mating factor prepro peptide,進入轉譯後轉移外泌途徑(Post-translational translocation)而送至胞外[12]。因此將異源基因的N端接上來自S. cerevisiae的外泌 訊息,能讓P. pastoris將異源蛋白質分泌至胞外[13],另外因P. pastoris外泌性原生
蛋白質極少的特性,可使上清液大部分皆為異源蛋白質,方便後續純化工作。
3. 表現量高、調控嚴謹的甲醇誘導系統
酒精氧化酶是甲醇代謝的第一個酵素,但由於此酵素效率不佳,菌體需要表 現大量的酵素以代謝甲醇。在 P. pastoris 中,有兩種酒精氧化酶,分別為 Aox1
和 Aox2,這兩種酵素皆受到嚴謹的調控,甲醇能誘導其大量表現,卻會被葡萄
糖、甘油和乙醇抑制。然而,兩種酵素的表現量並不一致,AOX1 啟動子的表現 量為AOX2 啟動子的 10 倍以上,進行甲醇誘導時,AOX1 mRNA 可達細胞內總 mRNA 的 5%,而蛋白質可達總可溶蛋白的 30%[2]。因此,表現量高、調控嚴謹 的AOX1 啟動子十分適合用於生產異源蛋白質。
三、AOX1 啟動子的轉錄調控
AOX1 啟動子是 P. pastoris 中最常被使用的啟動子,根據其受碳源嚴謹調控 的特性,目前也發展出一套誘導模式。然而,科學家對AOX1 啟動子的了解仍停 留在微生物生理等巨觀的角度,詳細的分子調控機制並沒有一個完整的論述。此 外,在不同嗜甲醇酵母菌之間,面對碳源轉換所採取的策略也不太一樣。相較於 P. pastoris 的 AOX1 啟動子,H. polymorpha 的 MOX (methanol oxidase)啟動子能 以更彈性的條件活化,甲醇的誘導效果不會受到甘油的干擾,甚至在甘油受限的 情況下,就可達到甲醇誘導60%的轉錄活性[14]。然而,此碳源適應的差異,並 非因為兩個啟動子序列不同,若是將AOX1 啟動子轉入 H. polymorpha 中,就能 以甘油受限的方式活化 AOX1 啟動子,顯示轉錄因子對甲醇適應的重要性[15]。
近年來,陸續有研究探討AOX1 啟動子的調控機制,啟動子活化過程大致上 可以分為兩個階段,第一個階段為去抑制化(derepression),當環境中的抑制性碳 源降低到一定濃度之下,AOX1 啟動子的轉錄活性約可達到甲醇誘導的 2%;第 二個階段則是甲醇誘導活化(activation),轉錄活性可高達受葡萄糖抑制時的 1000 多倍[16]。在這兩個調控階段,已知有 4 個轉錄因子參與其中,並且研究較為透 徹,分別為3 個活化子(activator) Mxr1、Mit1、Prm1 與 1 個抑制子(repressor) Nrg1,
如圖一。
1. MXR1
2006 年,Lin-Cereghino 等人發現 MXR1 (PAS_chr4_0487)對於甲醇代謝的重 要性,MXR1 突變會影響 P. pastoris 甲醇代謝路徑與過氧化體形成的相關基因表 現,進而使P. pastoris 無法生存於碳源為甲醇的情況。轉錄因子 Mxr1 的序列與 S. cerevisiae 的轉錄因子 Adr1 相似,其中又以 N 端區域相同度最高(70% identity)。
Adr1 是 S. cerevisiae 代謝乙醇、甘油與油酸很重要的轉錄因子,Mxr1 與 Adr1 的 相似性意味兩者可能是同源基因,都負責調控碳源代謝相關基因[17]。
Mxr1 由 1156 個胺基酸組成,分子量約為 130 kDa,N 端包含兩個 DNA 結 合鋅指 (Type I C2H2 DNA-binding zinc finger),透過辨認核心序列 5' CCYC 3',
結合在AOX1 啟動子的 6 個不同位置。因此,若是剔除這 6 個結合位,同樣會導 致AOX1 啟動子的轉錄活性下降[18]。此外,若是在 P. pastoris 轉入過多外源性 AOX1 啟動子,可能導致 Mxr1 稀釋效應。染色體上 Mxr1 結合位會因為外源性 基因數量提升而增加,使Mxr1 的相對濃度被稀釋,進而可能導致 Mxr1 無法結 合在所有的結合位,除了使異源蛋白質表現量下降外,甚至會影響到甲醇代謝 [19]。而 Mxr1 的轉錄活化區域主要位於第 246 到第 280 號胺基酸之間,此區段 由許多疏水性與酸性胺基酸組成,是轉錄活化區域常見的特色。綜合以上兩點,
Mxr1 的 N 端 400 個胺基酸(包含 DNA 結合能力與轉錄活化能力),即可具有 Mxr1 全長的功能,至於剩下區域功能尚未明瞭[20]。
不同於其它嗜甲醇酵母菌,P. pastoris 的 MXR1 是一個持續性表現的基因,
並不會因為碳源不同而有所差異。但是當環境中的碳源缺乏葡萄糖時,Mxr1 會 由細胞質移動到細胞核中,此現象指出葡萄糖對AOX1 啟動子的抑制可能是透過 影響Mxr1 在細胞中的位置[17, 21]。而乙醇的抑制則可能是透過磷酸化 Mxr1 的 第215 號胺基酸 Serine,雖然磷酸化的 Mxr1 並不會改變在細胞中的位置,也不 會喪失與AOX1 啟動子結合的能力,但磷酸化的區域(胺基酸 212 到 225)會被 14-
存在於細胞核中,但是否仍保有結合AOX1 啟動子的能力,或是否有特殊的轉譯 後調控,至今仍無詳細的探討。
在AOX1 啟動子啟動過程中,雖然可分為去抑制化與甲醇活化的兩個階段,
但是這兩者之間並仍沒有一個明顯的界線。Mxr1 可能在這兩階段都扮演一定的 角色,不過 Mxr1 目前主要被認為參與去抑制化的過程[22]。MXR1 突變而導致 AOX1 啟動子轉錄活性的喪失,無法以過量表現 Mit1 或 Prm1 修復,顯示 Mxr1 的功能與其他兩個轉錄活化子並不重疊[23]。Mxr1 除了影響甲醇代謝,同時還影 響油酸、乙酸的代謝[17, 24],以胺基酸為碳源的能力也受到 Mxr1 的調控[21],
甚至P. pastoris 在利用抑制性碳源甘油、乙醇時的生長速度,都會因為 MXR1 突 變而降低[17],因此 Mxr1 可能是一個全面性的碳源調控子。此外,Mxr1 面對不 同碳源時的反應,並不是透過耗時的轉錄活化、改變胞內濃度,而是以快速的轉 譯後調控(細胞內位置差異、磷酸化)影響下游基因的表現,因此 Mxr1 或許能幫 助 P. pastoris 在碳源(尤其是葡萄糖)缺乏時快速的去抑制化啟動子,並進一步協 同其它的轉錄因子,以適應多樣化的碳源。
2. PRM1
CbTrm1 最早於嗜甲醇酵母菌 Candia boidinii 中發現,已知對甲醇代謝扮演 重 要 的 角 色 , 並 透 過 序 列 比 較 發 現 與 P. pastoris 的 同 源 基 因 PpTrm1 (PAS_chr4_0203)的胺基酸序列有 58%的相同度[22]。PpTrm1 在後續的研究稱為 Prm1,此蛋白質由 989 個胺基酸組成,分子量約為 111 kDa,為 Zn(II)2Cys6類型 之轉錄因子。PRM1 突變的性狀主要在甲醇代謝基因的缺乏,雖然可能會因為缺 乏酒精氧化酶與二羥基丙酮合成酶 (dihydroxyacetone synthase, Das),而使過氧化 體體積不正常,但 PRM1 突變並不會影響過氧化體的增殖(proliferation)。過氧化 體除了用於代謝甲醇,對油酸代謝也十分重要,因此相較於全面性的碳源調控者 Mxr1,Prm1 可能更專注於甲醇的調控[23]。
Prm1 對甲醇代謝影響與環境中氮源有關。相較於營養培養基,PRM1 突變 株在基礎培養基(minimal medium)中的甲醇代謝缺陷較嚴重,但此現象可能與 Prm1 在細胞中的位置無關,Prm1 會持續存在於細胞核中,並不會因為環境中的 碳、氮不同而改變[22]。並由於 P. pastoris 轉錄因子的研究大多是在基礎培養基 中進行,氮源對 Prm1 功能的影響至今仍不太清楚。
在基礎培養基中,Prm1 的表現量會受到甲醇的誘導,但活化倍率並不明顯。
因此,Prm1 調控 AOX1 啟動子可能是在碳源不同時,對啟動子有不同親和力而 造成。在 AOX1 啟動子上有 2 個 Prm1 結合位,兩個位置與 Prm1 的結合力會在 甲醇為碳源時顯著上升,但確切的機制仍不清楚。除了調控甲醇代謝相關基因外,
Prm1 也會結合自身啟動子,產生的正回饋控制(positive auto-regulation)對於 Prm1 的表現十分重要[23]。
3. MIT1
2016 年,MIT1 (PAS_chr3_0836)被證明參與 P. pastoris 甲醇調控,在 H.
polymorpha 中存在同源基因 MPP1,不過兩者的相似度並不高。Mit1 為 Zn(II)2Cys6類型之轉錄因子,由888 個胺基酸組成,分子量約為 98 kDa,比 Mpp1 多出幾段冗餘的序列,若是剔除這些序列,可使 P. pastoris AOX1 啟動子 不受甘油抑制,顯示Mit1 與 Mpp1 可能有不同的轉譯後調控機制,進而導致 H.
polymorpha 與 P. pastoris 碳源適應的差異[23]。
MIT1 與 PRM1 有許多相似點,同樣主要參與在甲醇調控。MIT1 突變會影 響甲醇代謝基因的表現,但不直接影響過氧化體的增殖。Mit1 在細胞內位置並 不會因碳源而改變,但對AOX1 啟動子上 3 個位置的結合力則是在甲醇時最 高。然而,Mit1 與 Prm1 的表現量並不太一樣,在基礎培養基中,Mit1 的表現 量會受到甲醇強烈的誘導,活化倍率可達700 倍。對 AOX1 啟動子的重要性也 不盡相同,缺乏Mit1 的影響比 Prm1 明顯,剔除 AOX1 啟動子上 Mit1 結合位的 影響也相對嚴重。此外,過量表現 Mit1 可以回補缺乏 Prm1 所造成的問題,反 之無法,這些現象顯示 Mit1 與 Prm1 功能的關聯性與層級性[23]。
進一步發現,Mit1 與 Prm1 會相互調控。Prm1 可透過結合 MIT1 啟動子,
促進Mit1 的生成。但 Mit1 卻會抑制 Prm1 的表現,不過由於尚未發現 Mit1 與 PRM1 啟動子結合位,仍無法確定詳細的因果關係。最後,雖已清楚 Mxr1、
Prm1、Mit1 在甲醇調控中扮演重要的角色,但由於這 3 個轉錄因子並不具直接 的交互作用[23],且甲醇感應路徑(methanol sensing pathway)也不清楚,因此 P.
pastoris 如何藉由這三個轉錄因子傳遞甲醇存在的訊號還需要更多的研究。
4. NRG1
Mxr1 在甘油為碳源時會進入細胞核中,但 AOX1 啟動子仍受到抑制,顯示 還有其他機制參與在甘油抑制中。2015 年,NRG1 (PAS_chr3_1242)被證明在甘 油抑制扮演重要的角色,NRG1 突變株可在甘油為碳源時,表現甲醇代謝相關 的基因。Nrg1 由 209 個胺基酸組成,分子量為 23 kDa。不同於 Mxr1,Nrg1 會 持續在細胞核中,不會因為碳源而改變[25]。Nrg1 的表現量會受碳源調控,甘 油轉換至甲醇會使表現量下降為原本的1/3[26]。在 AOX1 啟動子上有 5 個 Nrg1 的結合位,其中2 個與 Mxr1 結合位重疊,雖然並不清楚兩個轉錄因子對結合 位的親和力,但此現象指出Nrg1 可與 Mxr1 競爭結合位,導致 AOX1 啟動子受 甘油抑制,但當環境中的碳源轉換成甲醇時,表現量下降的Nrg1 可使此抑制現 象消失[25]。
圖一、AOX1 啟動子的轉錄調控示意圖
AOX1 啟動子的啟動大致上可分為去抑制化與甲醇誘導活化,有 3 個活化子 Mxr1、Mit1、Prm1 與 1 個抑制子 Nrg1 參與在這兩個階段。
Figure 1. The schematic diagram of transcription regulation of AOX1 promoter The activation of PAOX1 was separated into two stages, including derepression and activation. Transcription factors involved in PAOX1 regulation were found, including Mxr1, Prm1, and Mit1 as activators, as well as a repressor, Nrg1.
四、甲醇誘導系統面臨的困境與解決辦法
在過去P. pastoris 的醱酵策略主要是先以葡萄糖、甘油培養菌體至高細胞密 度,再轉換碳源為甲醇誘導目標蛋白質生產。雖然此方法搭配AOX1 啟動子有著 表現量高、嚴謹調控的優勢,然而過度嚴謹的調控與其誘導物甲醇同時也會造成 很多的問題。
1. 甲醇的負面影響
甲醇是一種有毒且易燃的物質,因此在工業化生產需要注意安全,避免甲醇 的滲漏、殘留甚至是造成火災。對酵母菌而言,除了甲醇本身的毒性外,其代謝 產物-甲醛與過氧化氫同樣會對細胞造成傷害[27]。因此在甲醇誘導階段,細胞 會因過多的毒性物質而死亡,菌體破裂進一步導致胞內蛋白酶釋出,使目標蛋白 質降解[28]。此外,代謝甲醇是一個耗氧、產熱的反應,因此在工業化生產階段,
過高的氧氣需求、維持溫度額外的能量花費,都會增加P. pastoris 生產異源蛋白 質的難度與成本[29]。
在過去的研究中,試圖透過幾種方式解決代謝甲醇而產生的問題。首先是透 過改變 P. pastoris 的甲醇利用率,進而降低對甲醇的需求。藉由修改 P. pastoris 的AOX 基因,以建構不同甲醇利用率的菌株:野生型的 P. pastoris 擁有兩種 AOX 基因,為甲醇利用率快的Mut+ (methanol utilaiztion plus);或是透過去除 AOX1、
保留表現量較低的 AOX2,建構出甲醇利用率較慢的 MutS (methanol utilization slow)菌株;而 Mut- (methanol utilaiztion minus)菌株則是同時去除兩個 AOX 基因,
而無法以甲醇為單一碳源[5]。儘管 Mut+菌株甲醇利用率較好,理論上異源蛋白 質的產量比較高[30],但過高的甲醇需求不利於工業化生產,因此 MutS菌株的低 甲醇利用率反而可降低甲醇代謝的負面影響[29]。
偵測甲醇濃度,藉此維持甲醇在適當的添加速率[32]。然而當培養體積上升時,
混勻所需時間增加,會使各種偵測器的反應速度變慢,因此過於複雜的饋料策略 並不一定適用於工業化的生產。也有許多研究提出混和添加甲醇與其他碳源的饋 料策略,透過適量添加其他碳源,包含抑制性較弱的甘油或是不具抑制性的山梨 糖醇,補足降低甲醇添加而缺少的碳源,藉此雖可減輕代謝甲醇的負面影響,但 也導致AOX 啟動子轉錄活性下降,或是使成本提升[16, 33, 34]。
2. 過度嚴謹的 AOX1 啟動子
醱酵策略的改進與 Muts 菌株的改良除了遇到上述問題外,過於嚴謹 AOX1 啟動子是一個關鍵。混合碳源饋料的瓶頸便是AOX1 啟動子太過於限制,必須要 以甲醇為唯一碳源才可有效的生產。甚至在以葡萄糖、甘油培養菌體後,些微抑 制性碳源殘留於培養基,都會影響甲醇誘導效果[5]。除了降低異源蛋白質的產量,
還可能導致菌體需要更長的時間以表現甲醇代謝基因,因此若是在碳源轉換的階 段快速添甲醇,會導致菌體在完全適應甲醇之前,就因甲醇濃度累積過高而死亡,
此問題在甲醇代謝速率較慢的 MutS菌株更需嚴謹的處理[35]。抑制性碳源殘留 的問題雖然可透過置換培養基解決[36],但僅限於實驗室的規模,在工業化生產 階段,並沒有辦法有效地置換反應槽中的培養基。
隨著合成生物學的發展、甲醇調控機制的發現,陸續開始有研究試圖調整 AOX1 啟動子的嚴謹性,透過去除葡萄糖運輸蛋白質 Hxt1 或是葡萄糖感應蛋白 Gss1,使 P. pastoris 無法偵測到環境中的葡萄糖,使 AOX 啟動子可以在有葡萄 糖的情況下活化[37, 38]。同樣地,透過去除甘油激酶 Gut1 或是二羥丙酮激酶 Dak 也可以消除對應碳源的抑制性,顯示磷酸化的碳源代謝形式對於碳源的偵測十分 重要[39]。雖然這些方法可以初步調整 AOX1 啟動子的調控性,但是活化效果仍 不如甲醇誘導。此外,缺乏這些基因也會影響碳源的代謝,而導致菌體生長缺陷。
除了調整碳源偵測路徑外,直接改變AOX1 啟動子相關轉錄因子也可以達到
類似的效果。2017 年,Wang 等人透過去除轉錄抑制子 Mig1、Mig2 以及 Nrg1,
並同時過量表現Mit1,使 P. pastoris 可以在甘油為碳源時啟動 AOX1 啟動子,其
活化效果可以達到甲醇誘導的70%,雖然此方法會降低碳源調控的嚴謹性,不過
仍可以透過高濃度的葡萄糖關閉AOX1 啟動子。此團隊進一步根據改造菌株的特 性,設計出以葡萄糖-甘油二階段培養策略生產重組蛋白質,產量可以達到野生
型菌株以傳統甘油-甲醇培養的50%,同時由於不需要使用甲醇,可以有效的降
低耗氧、產熱的問題[8]。雖然在 Wang 等人的研究中並沒有詳細的探討改造菌株 的生長速度,但在過去與轉錄因子的相關研究中,去除AOX1 啟動子的轉錄抑制 子,會降低P. pastoris 在甘油、葡萄糖中的生長速度,顯示此策略還有提升的空 間[25]。
五、研究動機
Pichia pastoris 為嗜甲醇酵母菌的一種,是極具潛力的異源蛋白質表達系統,
兼具微生物與真核系統的優勢。搭配甘油培養-甲醇誘導策略,可嚴謹調控 P.
pastoris 的 AOX1 啟動子,並大量生產重組蛋白質。然而,調控過度嚴謹的 AOX1 啟動子,使 P. pastoris 只能侷限在特定的培養條件,必需以有毒、易燃的甲醇作 為唯一碳源時,才可有效率誘導 AOX1 啟動子。隨著合成生物學的發展,研究人 員不需侷限在 P. pastoris 原有的特性,透過破壞 P. pastoris 偵測、代謝抑制性碳 源的機制,可去除碳源對 AOX1 啟動子的抑制性。藉由去除轉錄抑制子與過量表 現轉錄活化子,也可以改變 AOX1 啟動子的特性。雖然這些的方法可讓 P. pastoris 以更彈性的條件生產異源蛋白質,但全面性的改變 P. pastoris 的碳源調控機制,
可能使 AOX1 啟動子完全喪失其原本的優勢,甚至造成 P. pastoris 的生長缺陷。
綜合以上問題,顯示 P. pastoris 還有進一步提升的空間。
1. 目的
本研究希望藉由不同的方式改變 P. pastoris 的碳源調控機制,不採取剔除或 持續過量表現基因的策略,而是透過特殊的基因迴路,幫助 P. pastoris 在抑制狀 態與誘導狀態之間的轉換。讓 P. pastoris 在抑制狀態時,可以保有原本的可調控 性,並避免造成生長的缺陷;而在誘導狀態時,降低抑制性碳源對 AOX1 啟動子 的干擾,甚至是進一步加強 AOX1 啟動子的表現量。
2. 轉錄調控策略
轉錄因子 Mxr1 為本研究主要切入點,相較於其他轉錄活化子,Mxr1 主要 參與在 AOX1 啟動子的去抑制化,是相對上游的調控者。在不同嗜甲醇酵母菌中,
碳源對 Mxr1 的影響也不盡相同,此差異或許是菌種間碳源適應不同的原因之一 [40]。因此透過調整 Mxr1 的表現量,應有助於改善 AOX1 啟動子調控過於嚴謹
的問題。此外,增加轉錄活化子 Mxr1 的表現量,同時可解決提升外源基因拷貝 數所造成的 Mxr1 稀釋效應,以增加 AOX1 啟動子的轉錄活性。額外的 Mxr1 不 會以持續性表現的 GAP 啟動子或是甲醇誘導的 AOX1 啟動子,以保有 P. pastoris 受碳源調控的特性,並避免過多的 Mxr1 累積而使 P. pastoris 死亡。透過表現量 較弱的 AOX2 啟動子生產 Mxr1,建立 Mxr1 的自我正回饋控制迴路(positive auto- regulation curcuit),Mxr1 的表現量必須當抑制性碳源降低至一定程度或是環境中 有甲醇時,才會藉由正回饋迴路逐漸上升,進而降低 AOX1 啟動子的嚴謹性,並 提升 AOX1 啟動子的轉錄活性,如圖二。
3. 目標
為了驗證此策略的可行性,以內生型或外泌型綠色螢光蛋白質作為 P.
pastoris 生產的目標蛋白質,並加以編輯轉錄因子的表現量。本研究之架構圖如 圖三,具體預達成事項為:
(1) 建構綠色螢光蛋白質生產菌株 (2) 建構轉錄再程序化之載體
(3) 以不同培養程序檢驗此策略的成效 (a) AOX 啟動子的轉錄活性
分析綠色螢光強度與異源基因mRNA 表現量 (b) AOX 啟動子的可調控性
不同濃度、種類的碳源或不同誘導策略對AOX 啟動子的影響 (c)對 P. pastoris 原生性基因的影響
分析培養過程的濁度、Aox 酵素活性與各轉錄因子的 mRNA 表現量
圖二、Mxr1 再程序化菌株轉錄調控示意圖
以AOX2 啟動子建立外源性 Mxr1 之自我正回饋調控,如星號所示。
Figure 2. The schematic diagram of transcription regulation in the cell with reprogramming of Mxr1
Asterisk represenst the positive auto-regulation of extra Mxrt regulated by PAOX2
圖三、本論文之研究架構圖
Figure 3. The schematic structure of this study
第二章 材料與方法
一、實驗菌株與培養條件
1. 細菌
本研究用於建構及保存質體之宿主細胞為大腸桿菌 Escherichia coli EPI300 (Epicentre Technologies Corp, USA)。以 Luria-Bertani 液態培養基於 37oC、轉速 250 rpm 震盪培養,或另添加 1.5%洋菜膠固態培養於 37 oC。
2. 真菌
本研究使用的重組蛋白質表達菌株為嗜甲醇酵母菌 Pichia pastoris KM71H (MutS, aox1::ARG4, arg4),購買自 Invitrogen (Carlsbad, CA, USA)。以 YPD 液態 培養於30oC、轉速 250 rpm 震盪培養,或另添加 1.5% 洋菜膠固態培養於 30oC。
二、培養基
Luria-Bertani broth (LB):2.5 g LB (Acumedia)溶於 100 mL 蒸餾水,121 oC、20 分鐘高溫高壓滅菌後備用。
Luria-Bertani agar plate (LA):配置 LB 時,另加入 1.5% Bacto agar (Acumedia),
滅菌後降溫至50 oC 時倒於 9 cm petri dish,待凝固後保存於 4 oC。
Low salt LB broth (LSLB):2 g Lennox L Broth (Alpha Bioscience)溶於 100 mL 蒸 餾水,121 oC、20 分鐘高溫高壓滅菌後備用。
Low salt LB agar plate (LSLA):配置 LSLB 時,另加入 1.5% Bacto agar (Acumedia),
滅菌後降溫至50 oC 時倒於 9 cm petri dish,待凝固後保存於 4 oC。
10x dextrose (20%, w/v):20 g 葡萄糖(dextrose, D-Glucose Anhydrous, Biotech)溶 於100 mL 蒸餾水,121 oC、20 分鐘高溫高壓滅菌後備用。
YPD broth:10 g YE (yeast extract, Bio Basic)及 20 g peptone-A (from meat (Bio Basic))溶於 900 mL 蒸餾水,滅菌冷卻後加入 100 mL 10x dextrose。
YPD agar plate:配置 YPD 時,另加入 1.5% Bacto agar (Acumedia),滅菌後降溫 至50 oC 時倒於 9 cm petri dish,待凝固後保存於 4 oC。
Zeocin stock solution (100 mg/mL (Invitrogen, R250-01)):避光保存於-20oC G418 stock solution (250 mg/mL):2.5 g G418 sulfate 溶於 10 mL 蒸餾水,以 0.22 μm 濾膜過濾滅菌後保存於 4oC。
Kanamycin stock solution (50 mg/mL):500 mg Kanamycin sulfate (USP)溶於 10 mL 蒸餾水,以 0.22 μm 濾膜過濾滅菌後保存於 4oC。
Ampicillin stock solution (100 mg/mL):1 g Ampicillin (Sigma)溶於 10 mL 蒸餾水,
以0.22 μm 濾膜過濾滅菌後保存於 4oC。
10x 甘油 (10, 20, 40%, w/v):10、20 或 40 g 甘油 (Glycerol Anhydrous, J.T.Baker)
(Amresco)及 2.30 g K2HPO4以蒸餾水配置成100 mL 溶液,調整 pH=6.0±0.1,121
oC、20 分鐘高溫高壓滅菌後備用。
500x biotin stock solution (0.02%, w/v):20 mg D-biotin (Sigma)溶於 100 mL 蒸餾 水,以0.22 μm 濾膜過濾滅菌後保存於 4oC。
甲醇 (100%, v/v):甲醇(CH3OH, J.T. Baker)以 0.22 μm 濾膜過濾滅菌後保存於 4oC。
10x YNB (13.4%, w/v):3.4 g yeast nitrogen base w/o amino acid and ammonium sulfate (YNB, Bio Basic)及 10 g (NH4)2SO4(Amresco)溶於 100 mL 蒸餾水,以 0.22 μm 濾膜過濾滅菌,4oC 避光保存。
BMDY 培養基:10 g YE (yeast extract, Biotech)及 20 g peptone-A (from meat (Biotech))溶於 700 mL 蒸餾水,滅菌後加入 100 mL 10x YNB、100 mL 10x 磷酸 鉀緩衝液、2 mL 500x biotin、100 mL 10x dextrose。
BMGY 培養基:10 g YE (yeast extract, Biotech)及 20 g peptone-A (from meat (Biotech))溶於 700 mL 蒸餾水,滅菌後加入 100 mL 10x YNB、100 mL 10x 磷酸 鉀緩衝液、2 mL 500x biotin、100 mL 10x 對應濃度之甘油。
BMNY 培養基:10 g YE (yeast extract, Bio Basic)及 20 g peptone-A (from meat (Bio Basic))溶於 800 mL 蒸餾水,滅菌後加入 100 mL 10x YNB、100 mL 10x 磷酸鉀緩 衝液、2 mL 500x biotin。
BMMY 培養基:使用前於 BMNY 培養基添加 100%甲醇至適當甲醇濃度。
BMGMY 培養基:使用前於 BMGY 培養基添加 100%甲醇至適當甲醇濃度。
FBS 醱酵槽培養基:0.93 g CaSO4、18.2 g K2SO4、14.9 g MgSO4•7H2O、4.13 g KOH、40 mL 甘油、26.7 mL 磷酸(85%)加水定量至 1 L。
PTM1 (Pichia trace metal 1):6 g CuSO4•5H2O、0.08 g NaI、3 g MnSO4•H2O、
0.2 g Na2MoO4•2H2O、0.02 g H3BO3、0.5 g CoCl2、20 g ZnCl2、65 g FeSO4•7H2O、
0.2 g biotin、5 mL H2SO4加水定量至1 L。
三、表現載體建構
本研究所使用的表現載體pPICZA、pPICZαA、pPIC3.5K,購買自 Invitrogen (Carlsbad, CA, USA),為嗜甲醇酵母菌 P. pastoris 常使用的載體,載體具有酒精 氧化酶啟動子 (Alcohol oxidase promoter, AOX),能藉由甲醇誘導後,驅動下游異 源基因表現。pPICZA、pPICZαA 之篩選標誌為 Zeocin。pPIC3.5K 帶有 HIS4 基 因可以回補histidine 缺陷株,並具有篩選標誌 G418 (Geneticin)或 Kanamycin。
使用之引子序列可參考表一,合成委託明欣(Mission Biotech)或基隆米克斯 (Genomics)。將建構完成之載體轉形進大腸桿菌勝任細胞中,菌液塗至含有對 應抗生素(100 μg/mL Ampicillin、100 μg/mL Zeocin、50 μg/mL Kanamycin)的篩 選培養基;待轉形株長出後,進行單一菌落聚合酶鏈鎖反應挑選轉形株,先以 牙籤挑取單一菌落點至二次水中,加熱95 oC、10 分鐘破菌後做為模板,以專 一性引子進行聚合酶鏈鎖反應,並以電泳確認轉形株是否帶有目標片段,將含 有正確片段之菌落抽取質體,送定序以確認序列正確 (Genomic),建構過程使 用之分生試劑套組可參考表二。
1. pPICZ-mEGFP
利用引子F-PmlI-mEGFP、R-mEGFP-stop-SalI 於前人質體中擴增出 mEGFP 片段,以限制酶 PmlI 與 SalI 接入以相同限制酶處理之線狀載體 pPICZA,完成 pPICZ-mEGFP,如圖四(a),以 5'AOX1 定序確認,進行 P. pastoris 轉形時,以 SaclI 切成線狀。
2. pPICZα-mEGFP
利用引子F-EcoRI-mEGFP、R-mEGFP-stop-SalI 於前人質體中擴增出 mEGFP 片段,以限制酶 EcoRI 與 SalI 接入以相同限制酶處理之線狀載體 pPICZαA,完 成pPICZα-mEGFP,如圖四(b),以 5'AOX1 定序確認,進行 P. pastoris 轉形時,
以SaclI 切成線狀。
3. pPIC3.5KH
利用引子F-AvrII-6xHis、R-partialHIS4-XbaI 於質體 pPIC3.5K 中擴增出 5'端 帶有6x His tag 的 AOX1 終止子(terminator),以限制酶 AvrII 與 XbaI 接入以相同 限制酶處理之線狀載體 pPIC3.5K,完成 pPIC3.5KH,如圖四(c),以 5'AOX1 定 序確認。
4. pAOX2KH
利用引子F-AatII-AOX2、R-AOX2-BamHI 於質體 P. pastrois 的染色體 DNA 中擴增出AOX2 啟動子,由於引子對起始 Tm 值較低,採用 Touch up PCR 技術,
先以annealing 溫度 47oC 反應 11 個周期後,再以 58oC 反應 20 個周期。擴增完 的片段以taq polymerase 於末端加 A,接入 yT&A vector,以 M13F、M13R 定序 確認。接著以限制酶AatII 與 BamHI 處裡,並接入以相同限制酶處理之線狀載體 pPIC3.5KH,完成 pAOX2KH,如圖四(d) 進行 P. pastoris 轉形時,以 SalI 切成線 狀。
5. pAOX2KH-Mxr1
由於Mxr1 序列上帶有一個 SalI 切位,會影響後續 P. pastoris 的轉形,因此 以Overlap PCR 技術,在不影響氨基酸序列的情況下,將 SalI 切位去除。利用引 子 對 F-BamHI-Mxr1 、 R-SalImutant-Mxr1 與 引 子 對 F-SalImutant-Mxr1 、 R- MXR1P-AvrII,分別擴增出 Mxr1 5'端與 3'端序列,將 PCR 產物純化,並稀釋到 適當濃度。混合兩種PCR 產物做為模板,搭配引子 F-BamHI-Mxr1、R-MXR1P- AvrII 擴增出完整 Mxr1 片段,以限制酶 BamHI 與 AvrII 接入以相同限制酶處理之 線狀載體pAOX2KH,完成 pAOX2KH-Mxr1,如圖四(e),以 5'AOX2、3'AOX1、
F-SalImutant-Mxr1、R-SalImutant-Mxr1、Mxr1p-interal 定序確認,進行 P. pastoris 轉形時,以SalI 切成線狀。
圖四、本研究建構之表現載體圖譜 Figure 4. The maps of the plasmid (a) pPICZ-mEGFP,
(b) pPICZα-mEGFP, (c) pPIC3.5KH, (d) pAOX2KH, (e) pAOX2KH-Mxr1
(a) (b)
(c) (d)
(e)
表一、建構表現載體與定序之引子序列
Table 1. The primer used for plasmids construction and sequencing
名稱 序列(5'端到 3'端)
用於建構表現載體的引子
F-PmlI-mEGFP CACGTGATGGTGAGCAAGGGC F-EcoRI-mEGFP CTAGAATTCGTGAGCAAGGGCG R-mEGFP-stop-SalI ACCGTCGACTTACTTGTACAGCTCG
F-AvrII-6xHis
CCCTAGGCATCATCATCATCATCATTGATCAA GAGGATGTC
R-partialHIS4-XbaI CATCTAGATGCTCACCGCAATGCTG F-AatII-AOX2 GCGGACGTCTTTTTTTCAGACC R-AOX2-BamHI GCGGGATCCTTTTTCTCAGTTG F-BamHI-mEGFP GTGGGATCCATGGTGAGCAAGG R-mEGFP-AvrII TGGCCTAGGCTTGTACAGCTCG F-BamHI-Mxr1 GCGGGATCCATGAGCAATCTACC
R-SalImutant-Mxr1 CCCAGTTCTTAGTGGACTCATTCTCATC F-SalImutant-Mxr1 GATGAGAATGAGTCCACTAAGAACTGGG R-MXR1P-AvrII GCGCCTAGGGACACCACCATCTA
用於定序的引子
M13F CAGTATCGACAAAGGACACACT M13R GTTTTCCCAGTCACGAC
5' AOX1 GACTGGTTCCAATTGACAAGC 3' AOX1 GCAAATGGCATTCTGACATCC
Mxr1p-interal GGTGTGCCCACTCCAACTCTTC
表二、本研究使用之分生試劑套組
Table 2. The cloning reagents or kits used in this study
實驗 分生試劑套組
質體抽取 HiYieldTM Plasmid Kit Mini (ARROWTEC)
DNA 聚合酶鏈鎖反(PCR)
Taq DNA polymerase (ARROWTEC) Pfu DNA polymerase (ARROWTEC)
Pushion DNA polymerase (New England Biolabs) Q5 DNA polymerase (New England Biolabs)
PCR 產物純化 HiYieldTM Gel/PCR Fragment Exrtraction Kit (ARROWTEC)
酵素結切與接合
AatII (New England Biolabs) AvrII (New England Biolabs) BamHI-HF (New England Biolabs) BstBI (New England Biolabs) EcroI-HF (New England Biolabs) PmlI (New England Biolabs) SacI-HF (New England Biolabs) SalI-HF (New England Biolabs) StuI (New England Biolabs)
YEA T4 DNA ligase (Yeastern biotech)
四、嗜甲醇酵母菌電穿孔轉形
1. P. pastoris 勝任細胞製備
將單一菌落之P. pastoris 以 3 mL YPD,於 30 oC、轉速 250 rpm 活化 20 小 時,此時OD600約15;取 1 mL 菌液加至 100 mL YPD 中使 OD600介於0.1~0.2 之 間,於30 oC、轉速 250 rpm 活化 7 小時,此時 OD600約2;以 3000 g、4 oC 離心 5 分鐘,倒去上清液後加入 50 mL Pre-treat buffer (100 mM LiOAC, 10mM DTT, 0.6 M sorbitol, 10 mM Tris-HCl, pH7.5)於 30 oC 靜置 30 分鐘。以 1500 g、4 oC 離心 10 分鐘後,以2 mL 1 M sorbitol 清洗三次,最後以 500 μL 1 M sorbitol 回溶菌體,
每管分裝80μL 後放置冰上備用。
2. 電穿孔轉形
將表現載體以對應限制酶切成線狀後,純化並測量其濃度,取總共 1 μg 線 狀質體並與P. pastoris 勝任細胞加至 0.2 mm Gap cuvette (No.620 BTX, San Diego, USA)中,冰浴 10 分鐘,以 Electro Cell Manipulator ECM630 Electroporation System (BTX, San Diego, CA, USA)進行電穿孔轉形,條件為 1.5 kV、25 μF、200 Ω,電 穿孔完畢以1 mL 1M sorbitol 稀釋菌體,並於 30 oC 下靜置 60 分鐘,接著根據不 同的質體有對應不同的步驟:
(1) pPICZ-mEGFP 與 pPICZα-mEGFP
取250 μL 菌液塗至 YPDSZ 篩選培養基上(1% Yeast extract, 2% Peptone, 2%
Dextrose, 1 M Sorbitol, 1.5% Agar, 100 μg/mL Zeocin),於 30oC 靜置培養 2~3 天。
(2) pAOX2KH、pAOX2KH-Mxr1
取250 μL 菌液塗至 RDB(Z)上(1.34% YNB, 4 x 10-5% Biotin, 2% Dextrose, 2%
Agar, 1 M Sorbitol),Zeocin 添加與否根據 P. pastoris 勝任細胞是否有帶有 Zeocin 抗性基因。於30oC 靜置培養 1~2 天。
五、嗜甲醇酵母菌染色體DNA 分析
1. 轉形株染色體簡易分析
以牙籤挑取合適大小轉形株之單一菌落,點至 breaking buffer (Epicentre Technologies Corp, USA)中,以 65 oC、30 分鐘,95 oC、10 分鐘破菌後做為模板,
以專一性引子進行聚合酶鏈鎖反應,並以電泳確認轉形株是否帶有目標基因。
2. 轉形株目標基因拷貝數測定
酵母菌以3 mL YPD、30 oC、轉速 250 rpm 培養 20 小時,抽取其染色體 DNA (NautiaZ Bacteria/Fungi DNA Extraction Mini Kit, Nautia Gene),並測定濃度。
以即時定量聚合酶鏈鎖反應(real-time PCR)測定目標基因之拷貝數,此技術
是透過螢光染劑反應出每個 PCR 循環後生成產物之總量,並當某一循環中螢光
強度達到閾值時,此時的循環數便稱為Ct (threshold cycle),Ct 值的高低會與啟
始模板濃度成反比,測定出各樣品之 Ct 值後,進一步可以透過絕對定量法、相
對定量法,相對-絕對定量法測定未知樣品的濃度。
本研究使用相對-絕對定量法,每次實驗會以目標基因與參考基因都為一個
拷貝數的對照組菌株,作出兩基因不同濃度對 Ct 值的標準曲線,再以此分別計
算出實驗組菌株的目標基因與參考基因的濃度,兩者的比值即為目標基因的拷貝 數。計算公式如下:
Copy number
Target gene= Copy quantity 𝑇𝑎𝑟𝑔𝑒𝑡 𝑔𝑒𝑛𝑒
Copy quantity 𝑅𝑒𝑓𝑒𝑟𝑒𝑛𝑐𝑒 𝑔𝑒𝑛𝑒
使用參考基因為P. pasotris 的 1 拷貝基因 MET2[41, 42],測定的目標基因為 AOX1 啟動子、AOX2 啟動子與 MXR1,對應的引子如表三。測得之目標基因拷 貝數減去內生性的1 拷貝後,即為外源基因之拷貝數。
PCR 反應配方為 5 L genomic DNA、各 1 L 順反引子 (4 M),10 L 2xIQ2 SYBR Green FAST qPCR System Master Mix-HIGH ROX,補水至 20 L。採用 ABI 即時定量儀器進行螢光量的測定,PCR 擴增條件為 95oC, 3 min 進行 1 循環,
95oC, 5 sec,65oC, 30 sec 共進行 40 循環。
六、轉形株培養與分析
1. 試管誘導
將帶有目標基因之轉形株單一菌落接至 3 mL YPD 中,於 30oC、轉速 250 rpm 活化 16 小時,以 3000 g 離心 10 分鐘,用無菌水懸浮菌體並測定 OD600,取 1.5 × 108菌體數量(1 OD600 = 5 × 107 CFU/mL),3000 g 離心 10 分鐘,以不同碳 源X 之 3 mL BMXY 回溶菌體至 OD600等於1,於 30oC、轉速 250 rpm 培養 10 小時後取樣分析。
2. 搖瓶誘導
將帶有目標基因之轉形株單一菌落接至 3 mL YPD 中,於 30 oC、轉速 250 rpm 活化 20 小時,此時 OD600約15,取適量菌液加至 100 mL BMGY 中使 OD600
等於 0.15,於 30 oC、轉速 250 rpm 培養 24 小時,以 3000 g 離心 10 分鐘,將 BMGY 去除,重新以 20 mL 不同碳源之 BMXY 懸浮菌體。每 24 小時加入甲醇 至正確的終濃度,每日取樣測定濁度、螢光強度(SpetraMax M2e Microplate Reader, Molecular device),各轉形株的標準化螢光強度為扣除野生型的背景螢光度後,
除以各轉形株的濁度。同時樣品以3000 g 離心 10 分鐘、保留適量菌體於-80 oC,
待之後分析胞內蛋白質、mRNA 表現量。
3. 醱酵槽誘導
將帶有目標基因之轉形株單一菌落接至 3 mL YPD 中,於 30 oC、轉速 250 rpm 活化 20 小時,此時 OD600約15,取適量菌液加至 100 mL BMGY 中使 OD600
等於0.15,於 30 oC、轉速 250 rpm 培養 24 小時,使 OD600約為16~20,作為種 菌接種至醱酵槽中。本實驗使用5 L 醱酵槽(FS-01-A05, Major Science),槽內含 有4%甘油為碳源的 2 L FBS 培養基,並添加 8.7 mL 的 PTM1,培養溫度為 30
oC,並以 10%氨水或 2 N 硫酸調整至 pH = 5,通氣量為 2 vvm,攪拌速率為 800 rpm。培養階段分為甘油批次培養(Batch)、甘油饋料培養(Fed-batch)與甲醇誘導。
階段轉換主要藉由溶氧值(DO)的變化判斷,當碳源用盡時,菌體耗氧速度降低而 使溶氧值上升,表示可以轉換至下一個階段。饋料使用的碳源中含有12 mL/L 的 PTM1。
七、mRNA 表現量分析
以NautiaZ Bacteria/Fungi RNA Mini Kit (Nautia Gene)抽取菌體之總 mRNA,
測定濃度後,稀釋至125 ng/μL。8 μL (1000 ng)樣品加入 1 μL DNase (Promega)與 1 μL10x Reaction Buffrer 於 37 oC 反應 30 分鐘,去除樣品中殘留的染色體 DNA,
接著加入1 μL Stop Solution 於 65 oC 反應 10 分鐘終止反應。
此樣品進一步以ARROW-Script Reverse Transcriptase III (ARROWTEC)反轉 錄成cDNA,11 μL 的樣品加入 0.2 μL Radom Hexamer (1 μg/μL, ARROWTEC)與 1.8 μL 之 DEPC 水,於 70 oC 反應 10 分鐘後置於冰上冷卻,接著加入 4 μL 5x First-Strand Buffer、1 μL dNTP mix (10 mM)、1 μL RNase inhibitor (40 unit/μL)、1 μL ARROW M-MLV Reverse Transcriptase III (200 unit/μL),於 25 oC 反應 5 分鐘 後以 50oC 反應 60 分鐘,最後以 70 oC 反應 15 分鐘終止反應,並加入 80 μL ddH2O 稀釋致理論cDNA 濃度 10 ng/μL。
以即時定量聚合酶鏈鎖反應(Real-time PCR)測定目標基因 mRNA 表現量,
使用相對定量法(∆∆C 法),以 18s rRNA 作為內控制基因(Internal control),各樣 品的mRNA 表現量以參考樣品的倍率呈現。PCR 反應配方為 5 L cDNA、各 1
L 順反引子(4 M),10 L 2xIQ2 SYBR Green FAST qPCR System Master Mix- HIGH ROX,補水至 20 L。採用 ABI 即時定量儀器進行螢光量的測定,PCR 擴 增條件為95oC, 3 min 進行 1 循環,95oC, 5 sec,65oC, 30 sec 共進行 40 循環。使 用到的引子如表三。
表三、即時定量聚合酶鏈鎖反應之引子序列 Table 3. The primer used for real-time PCR
名稱 序列(5'端到 3'端)
MET2-qF CGTTCTCGCAACTCTTTCGAAGAA MET2-qR CAATGGCATCAGTTATGACGGAAG AOX1p-qF TTCTCACACATAAGTGCCAAACG AOX1p-qR AAAAGTGGGTGTTGAGGAGAAGAG AOX2p-qF CACCCAGCCCTCTTCATCAA
AOX2p-qR TCGCCCTTGGATGGAAAA 18s rRNA-qF GAGGATTGACAGGATGAGAGC 18s rRNA-qR CAAGGTCTCGTTCGTTATCGC mEGFP-qF GGCACAAGCTGGAGTACAACT mEGFP-qR ATGTTGTGGCGGATCTTGAAG MXR1-qF CTGATGCTATGAATGCCAAGGA MXR1-qR CTGAATCGTTATTACGACCGGAAT PRM1-qF TGGCGGCAAGAAGAACGTATA PRM1-qR TGGTTGCTGTGCCTTTGCTA MIT1-qF GTGGCACGCTGGAAAGCTAAT MIT1-qR ATAGGCCAGCCTGTTGCACA NRG1-qF CCCAACACAGCCATCAGAAA NRG1-qR GTGAATACGTGTGTGACGAGC
八、蛋白質分析
1. 聚丙烯醯胺膠體電泳分析
將誘導後的蛋白質產物上清液以4x loading dye 在 99 oC 加熱 10 分鐘,菌體 則是取2.5 × 108以50 L 4x loading dye 懸浮,在 99 oC 加熱 30 分鐘,放室溫備 用。配置上層5% SDS 聚丙烯醯胺焦集膠體,和下層 12% SDS 聚丙烯醯胺分離 膠體,架設於直立式電泳槽 (HoeferTM Dual Gel Caster; Amersham Biosciences, USA),並在內外槽加入適量 Tris-glycine SDS running buffer。將備好的蛋白質,
注入上層焦集膠體齒梳槽內,先以通電70 伏特,使蛋白質在上下層膠體間聚集
形成一直線,提高至120 伏特後,約 90 分鐘即可取出膠片以 Coomassie brilliant blue R-250 染色。上述使用試劑之配方如表四。
2. 西方墨點法
將誘導後的蛋白質產物上清液以4x loading dye 在 99 oC 加熱 10 分鐘,菌體 則是取2.5 × 108以50 L 4x loading dye 懸浮,在 99 oC 加熱 30 分鐘,放室溫備 用。配置上層5% SDS 聚丙烯醯胺焦集膠體,和下層 12% SDS 聚丙烯醯胺分離 膠體,架設於直立式電泳槽 (HoeferTM Dual Gel Caster; Amersham Biosciences, USA),並在內外槽加入適量 Tris-glycine SDS running buffer。將備好的蛋白質,
注入上層焦集膠體齒梳槽內,先以通電70 伏特,使蛋白質在上下層膠體間聚集
形成一直線,提高至120 伏特後,約 90 分鐘即可取出膠片浸泡於 Transfer buffer 中5 分鐘,即可進行轉印。將 0.45 μm PVDF 轉印膜 (PerkinElmer, USA)浸泡於 100%甲醇 10 分鐘,同時也將濾紙浸泡於 Transfer buffer。以濕式電泳轉印槽(Bio- rad)進行轉印,由負極至正極依序放置海綿、濾紙兩片、電泳膠片、PVDF 轉印 膜、濾紙兩片、海綿,於400 v、150 mA 條件下轉印 2 小時。轉印完成,將 PVDF
轉印膜浸泡於gelatin-NET 震盪 60 分鐘。再置換為以 gelatin-NET 稀釋 5000 倍之 rabbit anti-His 一次抗體於 4 oC 靜置過夜。以 PBST buffer 震盪清洗三次,每次 10 分鐘。再置換為以gelatin-NET 稀釋 5000 倍之 goat anti-rabbit IgG HRP conjugate 二次抗體於室溫震盪 60 分鐘,加上 WESTERN LIGHTINGTM Plus-ECL 基質溶 液,並以UVP 偵測冷光訊號,上述使用試劑之配方如表四。
3. AOX 活性分析
取5 × 107 CFU 菌體分析,離心去除上清液後,以 100 L AOX assay reaction mixture 懸浮,於 30 oC 反應 30 到 60 分鐘後,比較顏色深淺,紅色越深表示 AOX 活性越強,上述使用試劑之配方如表四。
表四、蛋白質分析所用試劑之組合成分 Table 4. The reagents used for protein analysis
試劑名稱 成分
5% SDS 聚丙烯醯胺焦集膠體 5% Acrylamide (29:1) 0.125 M Tris-HCl (pH 6.8)
0.1% SDS (Sodium dodecyl sulfate) 0.1% APS (Ammonium persulfate) 0.1% TEMED (Tetramethylethylenediamine) 12% SDS 聚丙烯醯胺分離膠體 12% Acrylamide (29:1)
0.375 M Tris-HCl (pH 8.8)
0.1% SDS (Sodium dodecyl sulfate) 0.1% APS (Ammonium persulfate) 0.04% TEMED (Tetramethylethylenediamine) 4x Protein loading dye (pH 6.8) 8% SDS
200 mM Tris-HCl (pH=6.8) 0.4% Bromophenol blue 20% Glycerol
400 mM 2-ME (2-Mercaptoethanol) Tris-glycine SDS running buffer
(pH 8.4)
25 mM Tris 250 mM Glycine 0.01% SDS
Transfer buffer Tris-glycine SDS running buffer 10% (v/v) methanol
AOX assay reaction mixture 100 mM K-phosphate buffer (pH 7.5) 0.05% chromogen o-dianisidine
0.25% cetyltrimethylammonium bromide (CTAB)
1% (v/v) methanol
3 U/ml horseradish peroxidase
第三章 結果
一、轉形菌建立與分析
1. 胞內型綠色螢光蛋白質生產菌株
將表現載體pPICZ-mEGFP 以 SacI 切成線狀後,以電穿孔轉形送入 P. pastoris KM71 勝任細胞,並將菌液塗至 YPDSZ 篩選培養基,待 2~3 天菌落長成後,接 著利用不同濃度 zeocin 的 YPDZ 篩選抗性較高的轉形株,如圖五(a)。進一步將 可耐受1000 μg/mL zeocin 之轉形株,培養於 2000 μg/mL zeocin YPDZ 培養基上 與MD 培養基,挑選出 3 個抗性最強的菌株,並命名為 E1、E2、E3,如圖五(b),
並確定轉形株仍為Histidine 缺陷株,如圖五(c)。
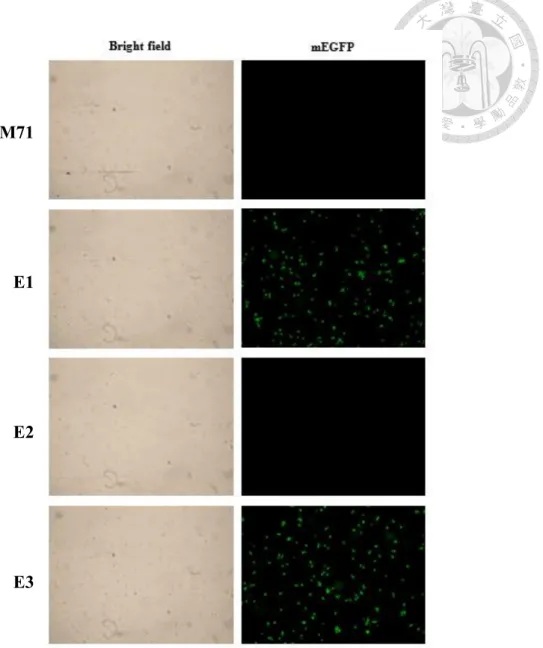
以搖瓶誘導上述 3 個轉形株與野生形控制組 5 天,每天添加 0.5%甲醇並取 樣分析,測定其標準化螢光強度,E1 與 E3 的螢光強度會隨著誘導天數逐漸上
升,而野生形控制組與 E2 的螢光不會,如圖六。樣品同時以螢光顯微鏡觀察,
細胞螢光亮度與直接測定其螢光強度相符,如圖七。
能成功表現出綠色螢光蛋白質的菌株E1 和 E3,進一步以 real-time PCR 測 定轉形株之外源基因 mEGFP 拷貝數,其中又以 E3 帶有較高的拷貝數,約有 9
個外源基因,結果如表五。後續實驗會以E3 作為母株,進一步編輯其轉錄因子
的表現量。
2. 外泌型綠色螢光蛋白質生產菌株
將表現載體 pPICZα-mEGFP 以 SacI 切成線狀後,以電穿孔轉形送入 P.
pastoris KM71 勝任細胞,並將菌液塗至 YPDSZ 篩選培養基,待 2~3 天菌落長成 後,接著挑選轉形株培養於幾種不同培養基上,(1) MD 培養基確定轉形株仍為
Histidine 缺陷株,(2) MMH 培養基誘導轉形株產生綠色螢光蛋白質,並以藍光燈 觀察螢光表現,(3)不同濃度 zeocin 的 YPDZ 篩選出抗性較高的轉形株,根據上 述分析方法,挑選出12 個轉形株,依序命名為 SE1~12,如圖八。
進一步以 real-time PCR 測定轉形株 SE5、SE6、SE9、SE12,外源基因 α- mEGFP 的拷貝數以 SE12 最高,結果如表五。後續實驗會以 SE12 作為母株,進 一步編輯其轉錄因子的表現量。
3. Mxr1 再程序化之胞內型綠色螢光蛋白質生產菌株
將表現載體pAOX2KH-Mxr1 與控制組載體 pAOX2KH 以 SalI 切成線狀後,
以電穿孔轉形送入胞內型綠色螢光蛋白質生產菌株 E3,並將菌液塗至 RDBZ,
待 2~3 天菌落長成後,接著利用不同濃度 G418 的 YPDG 篩選轉形株,實驗組 (pAOX2KH-Mxr1)挑選出 10 個轉形株,依序命名為 E3M1~10,而控制組挑出 1 個轉形株,命名為E3A1,如圖九。經由 PCR 分析後,確定轉形株 E3A1、E3M1~6、
E3M10 中皆都帶有正確的外源基因,如圖十,同樣以 real-time PCR 測定轉形株 之外源性Mxr1 拷貝數,結果如表五。
3. Mxr1 再程序化之外泌型綠色螢光蛋白質生產菌株
將表現載體pAOX2KH-Mxr1 與控制組載體 pAOX2KH 以 SalI 切成線狀後,
以電穿孔轉形送入外泌型綠色螢光蛋白質生產菌株SE12,並將菌液塗至 RDBZ,
待 2~3 天菌落長成後,接著利用不同濃度 G418 的 YPDG 篩選轉形株,實驗組 (pAOX2KH-Mxr1)挑選出 2 個轉形株,依序命名為 SE12M1 與 SE12M2,而控制 組(pAOX2KH)挑出 1 個轉形株,命名為 SE12A1,圖十一。以 real-time PCR 測定
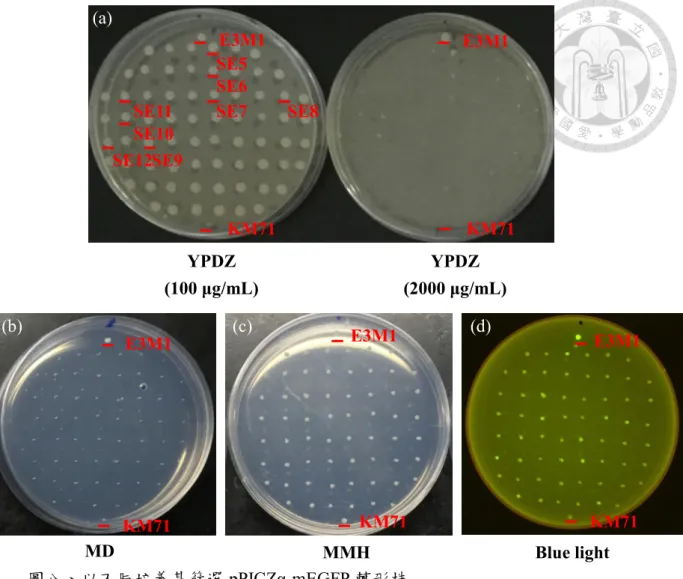
圖五、以不同培養基篩選pPICZ-mEGFP 轉形株
(a)含有 100、500、1000 μg/mL Zeocin 之 YPD 培養基。(b)含有 2000 μg/mL Zeocin 之YPD 培養基。(c) MD 培養基。KM71 (His4-)與 KM71H (His4+)為不具 Zeocin 抗性之菌株。1、2、3 為挑選出之轉形株。
Figure 5. Screening of pPICZ-mEGFP transformants by different plates
(a) YPDZ plate (100、500、1000 μg/mL Zeocin). (b) YPDZ plate (2000 μg/mL Zeocin).
(c) MD plate. KM71 (His4-) and KM71H (His4+) were not resistant to zeocin. The strain 1, 2, and 3 were picked for further experiments.
圖六、胞內型mEGFP 生產菌株標準化螢光強度分析
以搖瓶誘導挑選出3 株轉形株,每天添加 0.5%甲醇並誘導 5 天,其中 KM71 為 沒有螢光的控制組。
Figure 6. The normaolized fluorescent intensity of intracellular mEGFP
Fluorescent intensity of transformants cultured in flask with 0.5% methanol. KM71 was control group and did not express fluorescent intensity.
圖七、螢光顯微鏡觀察胞內型mEGFP 生產菌株
藍光激發 (450-490 nm),曝光 1/4 秒下,400 倍放大觀察甲醇誘導第 5 天之轉形 株。左圖為對應之明視野結果。
Figure 7. Result of transformants observed by fluorescent microscopy
After five days induction, the cells at 400x magnification were excited by blue light (450-490 nm) and the images were captured under 1/4 seconds.
E1
E2
E3 KM71
圖八、以不同培養基篩選pPICZα-mEGFP 轉形株
(a)含有 100 和 2000 μg/mL Zeocin 之 YPD 培養基。(b) MD 培養基。(c)MMH 培 養基。(d)藍光激發下的 MMH 培養基。KM71 (His4-)為不具 Zeocin 抗性與螢光之 負控制組。E3M1 (His4+)為具 Zeocin 抗性與螢光之正控制組。SE1~12 為挑選出 之轉形株。
Figure 8. Screening of pPICZα-mEGFP transformants by different plates
(a) YPDZ plate (100、2000 μg/mL Zeocin). (b) MD plate. (c) MMH plate. (d)MMH plate under blue light. KM71 (His4-) was negative control which was not resistant to zeocin and did not express mEGFP. E3M1 (His4+) was positive control which resistant
MD MMH
E3M1 E3M1
KM71 KM71
Blue light E3M1
KM71 SE5
SE6
SE7 SE8 SE10
SE11
E3M1
SE9 SE12
KM71 KM71
YPDZ (100 μg/mL)
E3M1
YPDZ (2000 μg/mL) (a)
(b) (c) (d)
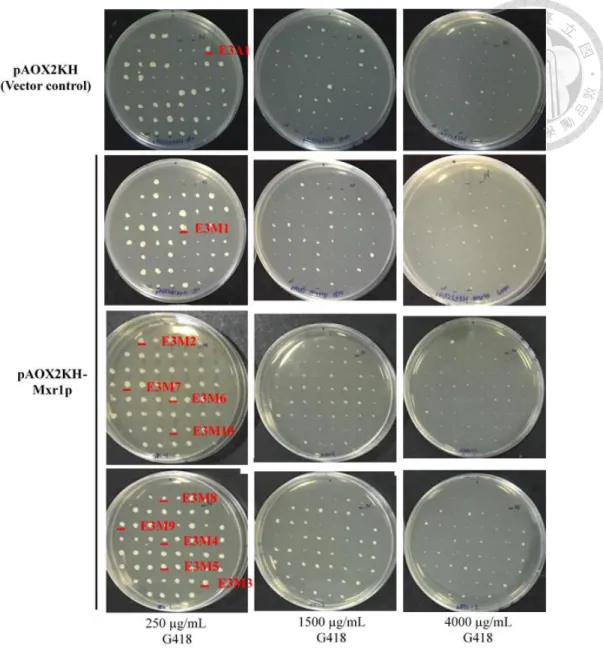
圖九、以不同G418 抗性濃度篩選 Mxr1 再程序化之 E3 轉形株 含有250、1500、4000 μg/mL G418 之 YPD 培養基。
Figure 9. G418 tolerance plate assay
YPDG plate (250, 1500, 4000 μg/mL G418).
圖十、聚合酶連鎖反應分析Mxr1 再程序化轉形株染色體 DNA
轉形株染色體DNA 以引子對 F-EcoRI-mEGFP 與 R-mEGFP-stop-SalI 偵測外源性 mEGFP,或以引子對 F-SalImutant-Mxr1 與 3' AOX1 偵測外源性 Mxr1,確認轉 形株帶有正確的遺傳訊息。P 代表添加質體為模板股之正控制組;N 代表添加水 為模板之負控制組;W 代表野生型 P. pastoris 之染色體為模板之負控制組。
Figure 10. Colony PCR of transformants
The mEGFP fragment was amplified by F-EcoRI-mEGFP/R-mEGFP-stop-SalI primer pairs and the ectopic Mxr1 fragment was amplified by F-SalImutant-Mxr1/3' AOX1 primer pairs. Lane P: plasmid, positive control; lane N: ddH2O, negative control; lane W: wild type, negative control.
圖十一、以不同G418 抗性濃度篩選 Mxr1 再程序化之 SE12 轉形株 含有250、1500、4000 μg/mL G418 之 YPD 培養基。
Figure 11. G418 tolerance plate assay
YPDG plate (250, 1500, 4000 μg/mL G418).
pAOX2
pAOX2-Mxr1p
YPDG (250 μg/mL)
YPDG (1500 μg/mL)
YPDG (4000 μg/mL)