國立臺灣大學醫學院微生物學研究所 博士論文
Graduate Institute of Microbiology College of Medicine
National Taiwan University Doctoral Dissertation
發展非切割式 CRISPR/Cas9 衍生之基因編輯系統治療 慢性 B 型肝炎
Development of the CRISPR/Cas9-mediated non- cleavage gene-editing strategies for treatment of
chronic hepatitis B
楊于嬋
Yu-Chan Yang 指導教授:楊宏志 博士
Advisor: Hung-Chih Yang, M.D., Ph.D.
誌謝
歷經風雨後,終於迎來美麗的彩虹。這將近九年的博士生涯,走得跌跌撞撞,
過程中的點點滴滴,也只有自己能細細回味與感受。最最感恩的就是家人的支持 與包容,讓我在沒有包袱下順利完成。
為什麼會唸博士班?總是有不少人這麼問,而初衷只因為對於實驗有熱誠罷 了。但,這樣的熱情,其實會在不斷的挫敗下越磨越殆盡。這些年,自己慢慢有 些轉變與突破,雖然不是一大步,但是很為自己的進步感到愉快。博班的過程中,
總使外人不斷的灌雞湯,給予打氣鼓勵,但在自己無法敞開心胸接納之前,這些 其實都是枉然。心境的轉變與對事物的執著,是在踏上這條路的旅程中,有所體 會與修正的。過程中,本來不懂得享受這樣的旅程,是後面才慢慢釋懷懂得去享 受,人生的酸甜苦辣,學習對於很多事情看的淡然些,對於周遭的人更珍惜彼此 的相遇,也更懂得如何跟自己相處。
感謝楊老師耐心的指導與人生路上的分享。剛開始進到實驗室是充滿幹勁的,
只是考驗出現後,自己的偏執,種下念博士班不順遂的因,好在透過不斷學習與 修正,慢慢享受學習接受失敗的過程。過去總覺得跟老師不在同一頻道,也無法 理解老師總是正面的沒有理由的鼓舞,但在潛移默化中,慢慢增加了自己內在光 明面,也越來越願意跟老師互相討論,因此獲益不少,當然除了學術的部分,老 師的待人接物與溝通上,真的是值得學習的好對象。
感謝諸位口試委員,從報告中聽取您們的建議,並在相互討論問答中,看到
我的不足與缺失。謝謝凌老師與陳老師,兩位老師都是 CRISPR 領域的佼佼者,
多次的報告討論中,讓我能用不同的角度或觀點來切入並深入自己的研究題目,
也總是有新的訊息或資源來分享。謝謝葉老師,您提出來的問題真的是犀利且重 要,是值得不斷思考的。謝謝蔡老師,您的邏輯思路與人生分享,真的讓我獲益 不少,也在多次與您的聊天中,更認識自己。
感謝實驗室的大家,研究所不再只是純粹當學生,她其實是社會的小縮影,
彼此間的互動是原動反動的。謝謝品宏,對於動物實驗總是能給予不斷的指導與 托付,在分生實驗上也能適時的給予建議,除此之外,生活上相互關照、包容、
與健康督促,能從這幾年下來彼此間的成長看出。謝謝芳儀,她在實驗室的時間,
總能放心將實驗室的大小事情交付給她。謝謝阿琦,我真心的欣賞你,將來在學
術上應該會是厲害的角色,自從開啟你的High 模式,跟你什麼都可以討論,從
學術到生活,真的很棒。謝謝煜翔,他在實驗上幫了不少忙,除了分生實驗外,
生物資訊方面都因著他的分析才能完成。謝謝學弟妹們對我超大的包容,我是個
情緒大起大落的人,可以High 到整個七樓都聽到我的吵鬧,也可以低落到什麼
人都不理,謝謝大家可以跟我一起High,以及能包容我的暗黑情緒與婆媽,只願
進到R722 的大家都能快樂有進度的在學術上精進。
僅以此論文,感謝一路走來幫助我的大家,祝福大家一切順心如意。
中文摘要
目前的抗病毒藥物無法根治慢性 B 型肝炎(chronic hepatitis B, CHB)的主要障礙,
是因為受感染的肝臟細胞內存在著 B 型肝炎病毒(hepatitis B virus, HBV)的複製 模板共價閉合環狀去氧核糖核酸(covalently closed circular DNA, cccDNA),以潛 在病毒庫持續存在。理論上,要達到完全根治 CHB,需要在不破壞宿主細胞基因 前提下,消滅所有 HBV 的基因,包含了 HBV cccDNA 和鑲嵌入宿主細胞的嵌入 型去氧核醣核酸(integrated DNA)。近年來,很多研究證實 CRISPR/Cas9 基因編 輯系統透過專一性破壞 HBV 基因體,可以作為治療 CHB 的潛力工具。然而,
CRISPR/Cas9 基因編輯系統不只會破壞 cccDNA,還可以標的作用在鑲嵌在我們 染色體上的 HBV 基因體而誘發雙股去氧核醣核酸的斷裂(double strand breaks, DSBs),導致宿主基因體的重新排列或是損傷的產生。為了增加 CRISPR/Cas9 基 因編輯系統使用上的安全性與應用性,我們使用非切割式 CRISPR/Cas9 衍伸之基 因編輯系統,在不造成 DSBs 去活化 HBV cccDNA 和 integrated HBV DNA。首先,
我們應用 SpCas9 取得的單鹼基編輯器(Base editor; BE)造成無義突變(nonsense mutation)去活化 HBV 基因體。透過篩選取得能造成無義突變的 gRNAs。接著使 用已帶有 HBV 基因體的細胞株,將 SpCas9-BE 和 gRNAs 一起送入細胞後,觀察 到 HBV 基因體能有效地被編輯,且 HBV 的聚合酶(polymerase)和表面蛋白都 有效地被抑制,有些 gRNAs 標地的位置有一石二鳥的作用,具有同時抑制 HBV 聚合酶和表面蛋白的功能。此外,我們也成功的在 HBV 細胞感染系統中,透過 SpCas9-BE 作用在游離基因的(episomal)cccDNA 能有效的抑制病毒基因的表現。
目前正在小鼠模式中實驗,運用腺病毒為基礎的 intein 調節分裂 Cas9 單鹼基編 輯 系 統 ( adeno-associated virus (AAV)-based intein-mediated split-Cas9-BE delivery system)編輯 HBV 基因體。除此之外,我們也在發展其他非切割式 CRISPR/Cas9 衍伸之基因編輯系統來抑制 HBV 基因的表現,像是透過表觀遺傳學(epigenetic)
(pegRNA)作用在 HBV 啟動子的部分,抑制病毒蛋白的表現,或是使用前間隔 序列鄰近構形(protospacer adjacent motif; PAM)限制較小的 SpCas9-NG,尋找適 合的 gRNAs 可以作用在不同基因型上的 HBV。最後,希望透過非切割式 CRISPR/Cas9 衍伸之基因編輯系統對於 HBV 基因體造成永久失去活性能成功,
則最終可能可以根治 HBV 的感染。
Keywords: B 型肝炎病毒, 共價閉合環狀去氧核糖核酸, 單鹼基編輯器, 先導編輯, 前間隔序列鄰近構形
Abstract
Covalently closed circular DNA (cccDNA) of hepatitis B virus (HBV) is a major barrier to a cure of chronic hepatitis B (CHB) by current antiviral therapy. In theory, to cure chronic hepatitis B, it is required to eliminate all the replication-competent HBV DNAs without damaging the host genomic DNAs. Recently, it has been shown that the CRISPR/Cas9 system can be utilized for site-specific cleavage of HBV DNA and bears the potential to cure CHB. However, because cccDNA and integrated HBV DNA share almost the same DNA sequences, the CRISPR/Cas system inevitably targets integrated HBV DNA and induces double-strand breaks (DSBs) of host genome, which bear the risk of genomic rearrangement and damage. To enhance the safety and applicability of the CRISPR/Cas9 system in treating CHB, an ideal strategy with CRISPR/Cas9 needs to effectively inactivate HBV genomes without induction of DSBs of host genomes. To achieve this goal, we examined the utility of recently developed CRISPR/Cas-mediated non-cleavage strategies in inactivating HBV genomes, including base editors (BEs) and DNA methylation. We first adopted CRISPR-BE to determine its use in inactivation of HBV genomes. Candidate target sites of the SpCas9-derived base editors (BE) and its variants in HBV genomes were screened for generating nonsense mutations of viral genes with individual guide RNAs (gRNAs). SpCas9-BE with certain gRNAs
expression in cells harboring integrated HBV genomes, but induced very few indels.
Some point mutations introduced by base editing resulted in simultaneous suppression of both polymerase and surface genes. Interestingly, we demonstrated that the episomal cccDNA could be successfully edited by SpCas9-BE for suppression of viral gene expression in an in vitro HBV infection system, although the efficacy still needs to be improved. In addition, we also evaluated the utility of the CRISPR-methyltransferase system to silence HBV genome. DNA methylation is an epigenetic mechanism that regulates gene expression. We took advantage of the CRISPR/Cas9-mediated DNA methyltransferase system to introduce de novo methylation of HBV genomes that may suppress viral gene expression. Finally, we examined whether the recombinant AAV delivery of intein-mediated split-Cas9 could be utilized for HBV genome editing in vivo. In summary, we demonstrated that Cas9-mediated non-cleavage approach,
particularly Cas9-BE system, it can be utilized for permanent inactivation of cccDNA and integrated HBV DNA without DSBs of host genome, providing a potential safe strategy to cure CHB. Nevertheless, there remain several challenging issues that need to be solved before the realization of CRISPR/Cas9-mediated non-cleavage strategies for HBV cure.
Table of Contents
中文摘要...I
Abstract………... II Table of contents………III Introduction
1.1 HBV epidemiology and public health………...1
1.2 Virion structure and genome of HBV………2
1.3 HBV life cycle………..4
1.4 HBV DNA integration………..5
1.5 Integrated HBV DNA and cccDNA are two major obstacles for eradicating HBV from CHB patients………7
1.6 Genome-editing tools………...8
1.7 CRISPR/Cas9 system………...9
1.8 Cas9-mediated Base editors………11
1.9 CRISPR/Cas9 system in inactivation of HBV genomes……….13
1.10 The potential and challenge for curing CHB using the CRISPR/Cas9 system…..14
Specific Aims………...16
Material and Methods 2.1 Plasmids………..17
2.3 Transfection of cell lines……….19
2.4 Lentiviral production and transduction………...19
2.5 Transduction with lentiviruses………20
2.6 Preparation and infection of HBV………...21
2.7 Sanger and MiSeq sequencing of base edited genomic DNA and cccDNA……….22
2.8 Immunoblotting assay……….23
2.9 ELISA of HBsAg………23
2.10 Quantitative real-time PCR (qRT-PCR)………...24
2.11 HBV DNA extraction and Southern blotting……….24
2.12 DNA-library preparation and MiSeq sequencing………..25
2.13 Off-target assessment………26
2.14 Immunofluorescence Assay………..27
2.15 DNA bisulfited and pyrosequencing assay for DNA methylation analysis……...28
2.16 Statistics………29
Results 3.1 Application of Cas9-BE for treatment of CHB 3.1.1 To design and screen HBV-specific gRNAs for inducing nonsense mutations by spCas9 base editors………...30
3.1.2 Introducing nonsense mutations into integrated HBV genomes by base editing suppresses the expression of HBsAg and polymerase………...31 3.1.3 To induce dual suppression of HBsAg and polymerase by HBV base-editing specific gRNAs of HBV genome………..34 3.1.4 To validate the dual suppression phenomenon by specific point mutations of HBV genome……….35 3.1.5 Suppression of HBV protein expression through base editing HBV cccDNA by spCas9 base editor………37
3.1.6 Using the intein-mediated split Cas9-BE system via rAAV to base-editing HBV genomes………....38 3.2 Application of Cas9-mediated DNA methylation for CHB treatment
3.2.1 Suppressing the expression of HBV genome by CRISPR/Cas9-mediated de novo methylation………...39 Discussion
4.1 Summary of this study………42 4.2 Approaches to improve the base editing in HBV genomes………..42 4.3 The advantages of Cas9-BE for inactivation of HBV genome………46 4.4 Kill two birds with one stone: dual suppression of two genes by base editing in the
4.5 Silencing the HBV promoters by Cas9-DNA methyltransferase or prime editing..49 4.6 Obstacles and advancement of CRISRP-Cas9 technologies………...50 4.7 Conclusion and perspectives………...53 References………...54 Figures
Figure 1. Schematic illustration of the HBV life cycle………..62 Figure 2. Current and future HBV virological targets for treatment and cure of CHB...63 Figure 3. Cytosine base editing……….64 Figure 4. Screening gRNAs for SpCas9-mediated base editing in HBV-HEK293T cells………...…66 Figure 5. Screening gRNAs for SpCas9 variants-mediated base editing in HBV- HEK293T cells……….68 Figure 6. Sanger sequencing of base-edited sites in the polymerase and surface genes of HBV genome………69 Figure 7. The base editing of specific HBV loci to effect viral gene expression of HepG2.2.15 cells………..70 Figure 8. The off-target analysis for the top three genomic loci of two gRNAs gP9 and gS8 by BE4-mediated SpCas9 in HepG2.2.15 cells………..72
Figure 10. Validation of the effect of base-edited missense mutations on the expression of HBV surface and polymerase proteins in Huh7………76 Figure 11. Base editing of HBV cccDNA……….…79 Figure 12. Efficiency of delivery of BE3 and HBV infection in HepG2-NTCP cells………...83 Figure 13. The intein-mediated split–Cas9………...84 Figure 14. The methylation levels in HBV genome………..85 Tables
Table1. The base-editing efficiency of SpCas9-BE with individual gRNAs……….…88 Table 2. The conserveness of gRNAs indifferent HBV genotypes………....89 Table 3. The base-editing efficiency of SpCas9-BE variant VQR, VRER, and EQR with individual gRNAs……….90 Table 4. Plasmid list and function……….92 Table 5. Primer and probe list………...…93 Appendix
Figure S1. Mechanisms of Cas9-mediated prime editing………..97
1. Introduction
1.1 HBV epidemiology and public health
According to the WHO’s estimation, approximately 250 million individuals live with
chronic hepatitis B virus (HBV) infection, and most of them reside in the Western Pacific
Region and the African Region. Based on the genome sequences, HBV is divided into eight
genotypes (A-H) with distinct geographical distribution.1 Chronic hepatitis B (CHB) often
leads to adverse clinical outcomes, including cirrhosis, hepatic failure, and hepatocellular
carcinoma (HCC).2
The nature history of CHB patients via mother-to-child (or vertical) transmission often
consists of four phases, including immune tolerance, immune clearance, inactive carrier, and
immune escape.3,4 Vertical transmission is associated with a higher risk of persistence chronic
HBV infection.5 The initial immune clearance phase is characterized with positive hepatitis B
e antigen (HBeAg), and high serum HBV DNA, but with normal alanine aminotransferase
(ALT). Many patients enter the immune clearance phase and develop HBeAg-positive hepatitis
(elevated ALT levels), which increases the risk of liver fibrosis, cirrhosis, and HCC. The
majority of patients will eventually undergo HBeAg seroconversion with suppression of HBV
DNA and normalized ALT, marking the transition to an inactive phase. The inactive replication
phase may persist indefinitely. However, occasionally reactivation of HBV with hepatitis flare
can occur spontaneously or be triggered by immunosuppressive therapy, and it is termed
HBeAg-negative hepatitis.6 Current guidelines suggest patients with HBeAg-positive or
HBeAg-negative hepatitis to receive antiviral therapy to prevent the long-term detrimental
outcomes.
The underlying mechanisms that maintain the immune tolerance phase remain unclear.
Nevertheless, viral proteins, namely, HBeAg and HBsAg have been demonstrated to suppress
inflammatory cytokines and interferon-stimulated gene (ISG) transcription, which diminishes
the function of Kupffer cells and monocytes. In addition, excessive HBsAg may promote virus-
specific T cell anergy and deletion.7-10 Therefore, reduction of HBsAg levels may be able to
restore HBV-specific immune responses.
1.2 Virion structure and genome of HBV
HBV belongs to Hepadnaviridae family, and contains a 3.2-kb relaxed circular DNA
(rcDNA) genome. The virion, called Dane particle, consists of lipid membrane and icosahedral
nucleocapsid. The nucleocapsid is made up by core proteins and protein-linked partially
double-strand DNA (pdsDNA). The endoplasmic reticulum (ER)-derived membrane is made
up by the viral envelop proteins, large (L), middle (M), and small (S) at the ratio of around
1:1:4, and surrounds the nucleocapsid. In addition to infectious viral particles, HBV also
produces noninfectious subviral particles (SVP), which contain envelope proteins, but no viral
capsid nor genomes inside.11,12
HBV contains four partially overlapping open reading frames (ORFs) encoding
precore/core (preC/C), polymerase, envelope, and X proteins, and generates 7 proteins. Four
majors viral RNAs, preC/pregenomic, preS1, preS2/S, and X, with 3.5, 2.4, 2.1, and 0.7 kb in
length, respectively, they are transcribed from four promoters and two enhancers. All the four
transcripts terminate at the same 3’ polyadenylation signal, but start with variable 5’ start sites.
The precore protein is translated from preC mRNA and further processed to generate
secreted HBeAg, which may play immunoregulatory functions. The pregenomic mRNA
(pgRNA) is translated to core and polymerase proteins, which are involved in encapsidation of
pgRNA and reverse transcription. The preS1 transcripts encode the large (L) surface protein,
and the preS2/S transcripts encode the middle (M) and small (S) surface proteins. These three
envelope proteins share the same C-terminal. The remaining 0.7kb transcripts encode the
multifunctional X protein, which interfere with transcription, cell signaling, protein
degradation, and apoptosis in the host.13
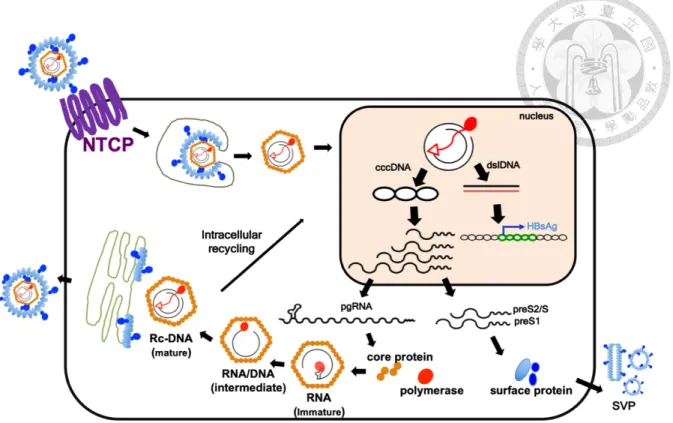
1.3 HBV life cycle
HBV enter hepatocytes by binding the pre-S1 domain of L envelop protein to sodium
taurocholate cotransporting polypeptide (NTCP) receptor and heparan sulfate proteoglycans,
and subsequently internalize into the host cells through endocytosis.14,15 The nucleocapsid is
then released into the cytoplasm, and viral rcDNA is translocated into the nucleus following
viral uncoating. In the nucleus, the rcDNA is converted into cccDNA probably via host factors,
although the detailed mechanisms remain largely unknown. The cccDNA is associated with
histone proteins, non-histone proteins, and probably some viral proteins to form a mini-
chromosome structure and plays a key role as the transcriptional template for pgRNA and
subgenomic RNAs. The pgRNA serves as a template for translating polymerase and core
proteins. Then the polymerase binds to pgRNA to trigger encapsidation, which initiates (-)-
stand DNA synthesis. As DNA synthesis progresses, most of the pgRNA is degraded by
polymerase protein’s RNase H activity and the remaining non-degraded part of pgRNA acts as
a primer for (+)-strand DNA synthesis. The mature nucleocapsid can be either enveloped with
surface proteins in ER and budding out into extracellular space or enter the intracellular
recycling to amplify cccDNA pools (Figure 1).
1.4 HBV DNA integration
Integration of HBV DNA into host genomes occurs through double-strand (ds)DNA
break which is a common event occurring upon HBV infection.16,17 However, neither the
timing nor mechanisms of HBV integration into host genomes is well understood. Unlike
retrovirus, HBV integration is not a requisite for viral life cycle because integrated HBV DNA
does not serve as a template for productive viral replication, and hence represents a replicative
dead-end for the virus. However, the structure of integrated HBV DNA affects all viral ORFs
except the HBsAg ORF and the integrated HBV DNA which has recently been proved to be a
crucial source for the continuous secretion of HBsAg. Excessive amount of secreted HBsAg
likely has the immunosuppressive effect and act as a ‘decoy’ for antibody responses in order
to allow HBV to escape from host immunological control.18
Analysis of integrated HBV genomes in primary liver tissues including tumor and non-
tumor tissues discovered genome rearrangements and/or deletions within the HBV genome.19-
21 So far, it remains unclear whether these rearrangements occur at pre-integration, or post-
integration, or a combination of both. Integration of HBV DNA into host genome can cause
chromosomal instability, gene mutations, and truncated protein production, that can lead to
selective advantage for HBV-induced liver cancer.22 Recently, next-generation sequencing
(NGS) technology has been used to analyze the preferential HBV integration sites of the entire
human genome in the liver cancer. While integration occurs randomly across the host genome,
several genes, including TERT, MLL4, and CCNE1 were identified as HBV preferential
integration sites and are related with carcinogenesis.23,24 The most frequent mutation (around
60%) is at the promoter of TERT gene, which is associated with telomere length maintenance
and cell mortality.24-26 MLL4 encodes histone methyltransferase, associated with chromatin
structure and gene transcription. CCNE1 is a regulatory subunit of cell cycle.27,28 The NGS
technology not only identifies the hot spots of HBV integration sites, but also characterizes the
flanking sequences of host genomes. Taken together, the random HBV integration causes
mutagenesis of host genomes, and leads to carcinogenesis.
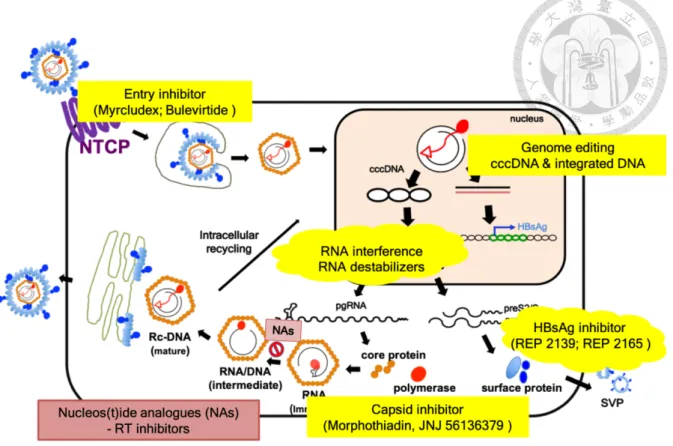
1.5 Integrated HBV DNA and cccDNA are two major obstacles for eradicating HBV from
CHB patients
Recently, there has been emerging enthusiasm for functional cure of HBV, which is
defined as loss of HBsAg and HBV DNA in serum with or without seroconversion to hepatitis
B surface antibody (anti-HBs). Although current antiviral therapies, including reverse
transcriptase inhibitor, nucleos(t)ide analogues (NAs) or less commonly interferon (IFN)
therapy, they have dramatically improved the outcomes of CHB patients, but most of them
have experienced a rebound viremia after discontinuation of nucleos(t)ide analogues (NAs).29
The major obstacle for eradicating HBV infection by NAs is the persistent covalently closed
circular (ccc) DNA, which is the episomal form of virally replicative template.30,31 Curative
strategies for CHB need to either eliminate all the infected hepatocytes or purge all the
replication-competent cccDNAs. This is considered as a complete cure.12,32,33 So far, there are
a lot of drug developments based on different steps of HBV life cycles, but curing HBV remains
extremely challenging because still no drugs can specifically target and destroy cccDNA
(Figure 2). However, disruption of cccDNA alone may not necessarily result in HBsAg loss.
As mentioned above, it is reasonably assumed that functional HBV cure cannot be achieved
without targeting integrated HBV genomes.
In order to evaluate the reduced efficacy and the number of cccDNA after therapeutic
strategies, the adequate and accurate methodologies to measure cccDNA are required.30,34
There are two major ways: (I) Southern blotting, which is still a gold standard for detection of
cccDNA and rcDNA. However, it is neither sensitive nor quantitative.35,36 (II) PCR-based
method, which utilizes cccDNA-specific primers, but it still has a chance to amplify rcDNA if
rcDNA is much more abundant than cccDNA.37-39 In addition, T5 exonucleases or Plasmid-
safe DNase in preparation HBV DNA samples is able to reduce contamination of rcDNA, but
no evidence can prove its entire elimination of contamination entirely.40 Besides, over-
digestion by T5 exonuclease may also affect the accuracy of the measurement. Therefore, more
appropriate methods for cccDNA detection have to be further developed.
1.6 Genome-editing tools
The advance of genome-editing tools has provided a novel approach to treat viral
infections by cutting and destroying viral genomes in a sequence-specific manner. There are
several gene-editing tools including meganucleases (MNs), zinc-finger nucleases (ZFNs),
transcription activator-like effector nucleases (TALENs), and recently developed cluster
regularly interspaced short palindromic repeat (CRISPR)-associated nuclease Cas9, which has
led to revolutionary progress in this field.41 All of these programmable nucleases produce site-
specific DNA double-strand breaks (DSBs), which may trigger error-prone nonhomologous
end-joining (NHEJ) that results in frameshift mutations. The resultant gene deletion or
insertions (indels) or nonfunctional truncated protein production can inactivate the target DNA
genome. Previous studies utilized ZFNs and TALENs for gene disruption and knockdown in
HBV DNA genomes in in vitro cell cultures and in in vivo mouse model.42-44 MNs, ZFNs, and
TALENs achieve site-specific manner through protein-DNA interaction that is difficult to
design and time-consuming; CRISPR-Cas9 is through the RNA-guided system which is
convenient to use and easy to design. Therefore, CRISPR-Cas9 system provides a flexible way
to specifically target the desired HBV DNA sequences.
1.7 CRISPR/Cas9 system
The CRISPR/Cas system is an adaptive immune system of prokaryotes to provide
protections from invading genetic elements. Two classes of CRISPR/Cas system are
discovered. Class 1 is composed by multiprotein CRISPR RNA (crRNA)-effector complexes,
and Class 2 has a single effector protein, such as Cas9. The prototype of Cas9 protein is derived
from Streptococcus pyogenes. Cas9 has the ability to recognize a short DNA sequence, 5’-
NGG-3’, called the protospacer adjacent motif (PAM), and is complexed with guide RNAs
(gRNAs) to form an active DNA endonuclease. The trans-activating crRNA (tracrRNA)-
crRNA duplex or an artificial loop sequence that is generated by linking tracrRNA and crRNA
together, called gRNA, guides the Cas9 protein to scan a target DNA genome for the PAM
sequence to the specific DNA sites. Cas9 uses its HNH domain to cleave the DNA strands that
is complementary to the 20-nucleotide sequence of the crRNA, and uses the RuvC domain to
cleave the other DNA strand opposite the complementary strand. The DSBs are induced by
these two enzymatic sites, which are often repaired by the non-homologous end-joining (NHEJ)
pathway. The NHEJ pathway frequently leads to nucleotide insertions or deletions (indels) and
thus disrupts the open reading frames (ORFs) of genes. Therefore, the risk of CRISPR/Cas9
system is caused by DSBs which causes unexpected gene rearrangement and large gene
deletions.45
To expand its utility for genome editing, Cas9 is further engineered to modify or
inactivate its endonuclease activity. Alanine substitution of key residues in RuvC domain
(D10A) produces nick in complementary domain, while H840A mutation of HNH domain
nicks in the non-targeting strand. Both D10A/H840A mutations generate catalytically
inactivate or “dead” Cas9 (dCas9). According to different gene-editing demands, Cas9 protein
is engineered to fuse with other proteins or domain insertion for different applications, such as
P65 protein for gene activation, KRAB protein for gene inhibition, and DNMT3s for epigenetic
regulation.46
1.8 Cas9-mediated Base editors
Cas9 has been also fused with cytidine deaminase or adenine deaminase for developing
base editors (BEs). The goal of BEs is to specifically correct point mutations related to disease,
without generating random indels caused by DSBs. The ABEs contain hypothetical
deoxyadenosin deaminase with catalytically impaired Cas9 that mediate conversion of A-G/T-
C in genomic DNA.47 The CBEs utilizes a catalytically impaired Cas9 endonuclease (dCas9)
tethered with APOBEC deaminase at the N’-terminal. It has been shown to generate precise C-
T/G-A conversion without DSBs at specific genome loci (Figure 3a). Both ABEs and CBEs
have the ability to convert one base pair to another directly and irreversibly but only CBEs
enables to create stop codon. Therefore, CBEs is an ideal tool for introducing premature stop
codons in a gene to suppress its expression. To enhance the C-T/G-A conversion, the dCas9-
deaminase construct is fused with a uracil glycosylase inhibitor (UGI) that suppresses uracil
excision following deamination to prevent the reversion of the U:G pair to a C:G pair. In order
to improve the editing efficiency, restoration of H840 in dCas9 to D10A Cas9 nickase (nCas9),
resulting in nicking of the non-edited DNA strand. All the above modifications helped to
develop the third-generation BE (BE3). Therefore, BE3 is designed with the combination of
APOBEC1, nCas9, and UGI that elevate the editing efficiency in mammalian cells. The fourth-
generation BE4 increases the efficiency of C-T/G-A conversion, while halving the frequency
of undesired base changes compared to BE3. BE4Gam is generated by fusing BE4 to DSB-
binding protein Gam from bacteriophage Mu to further reduce indel formation (Figure 3b). In
addition, the efficacy of base editing can be significantly improved by optimizing codon usage
(BERA), and further enhanced by including nuclear targeting motifs at the N-terminus of BE
enzymes (FNLS-BEs).48,49 Theoretically, base-editing the target nucleotides without DSBs of
DNA should reduce the risk of genome rearrangement and carcinogenesis.
1.9 CRISPR/Cas9 system in inactivation of HBV genomes
There have been several studies targeting HBV genomes with CRISPR/Cas9 system.
Previously, we demonstrated that the CRIPSR/Cas9 system could be used to disrupt the HBV
genome both in vitro and in vivo.50 Several other studies also showed that the CRISPR/Cas9
system could efficiently destruct HBV genomes including cccDNA and integrated HBV
sequences.51 However, cleavage of integrated HBV DNA by the CRISPR/Cas9 system will
lead to DSBs of host genome that induce complex genome rearrangement. In addition, a few
studies pointed out that CRISPR/Cas9 system is subjected to the off-target DNA cleavage that
could be noted using the highly sensitive deep sequencing analysis. These studies have
demonstrated that the CRISPR/Cas9 system is a useful tool in destroying HBV genomes, and
bear the therapeutic potential for curing CHB. Nevertheless, the limitations of the
CRISPR/Cas9 system cannot be neglected, either.
1.10 The potential and challenge for curing CHB using the CRISPR/Cas9 system
Although the CRISPR/Cas9 system provides a strong evidence to specifically destroy
HBV genomes, there are still some significant issues that needs to be addressed before its
translation to clinical HBV treatments. First, the potential off-target effects from the
CRISPR/Cas9 system has to be eliminated. Several strategies have been developed to reduced
off-target effects. For example, Fu et al. have demonstrated that the paired gRNAs and nCas9s
can improve the target specificity to reduce undesired indels at off-target sites without reducing
the on-target efficiency.52 Another variant Cas9 (fCas9) was generated by fusing catalytic
inactivated Cas9 and FoxI nuclease. The specificity at the on-target sites of fCas9 system was
higher and the off-target sites were reduced than the original Cas9, but the fCas9 system
remains less efficient. 53 Therefore, the off-target effects of Cas9 with HBV-specific gRNAs
still need to be cautiously examined.
Another issue is the delivery efficiency of the CRISPR/Cas9 system to the liver. In
CHB patients, cccDNA copies of each hepatocyte may not be evenly distributed. To eradicate
HBV, it has to effectively deliver Cas9-gRNAs into all infected hepatocytes. Adeno-associated
viral vectors (AAVs) is commonly used in gene therapy because of the low immunogen, non-
integrated nature, and high infection rate. Thus, the AAV vector is a proper choice for
CRISPR/Cas9 delivery. However, the larger size of Cas9 gene (~4.2 kb) challenges the
packaging capacity of AAV (~4.5kb). This packaging obstacle can be solved by using a smaller
Cas9 orthologs, such as Staphylococcus aureus Cas9 (SaCas9), which is >1kb shorter or using
the split-intein Cas9 system. 54 Alternatively, non-viral vectors to deliver messenger RNAs
(mRNAs) or proteins of Cas9 into the hepatocytes can be used, but there remains a room to
improve the efficacy.55
Furthermore, cleavage of integrated HBV DNA in host chromosomes are a serious
safety concern of CRISPR/Cas9 application to achieve complete cure of CHBs. Integration of
HBV genomes into host genomes is the by-products during virus replication. Since the HBV
integrated genomes have the same sequences with cccDNAs, the cleavage of cccDNA by
CRISPR/Cas9 system is accompanied by cutting the integrated HBV genomes. Although
disruption of integration HBV caused inactivation of HBV protein production especially
decreased HBsAg expression, the risk for DSBs of host genome that may damage or yield
truncated gene products leading to carcinogenesis cannot be ignored.
Specific Aims
In order to inactivate HBV genomes without making DSBs, my work aimed to examine
the utility of CRISPR/Cas9-mediated non-cleavage genome tools including base editing and
de novo DNA methylation in inactivation of HBV genome for the cure of CHB. We would first
use the CRISPR/Cas9-derived BEs in introducing nonsense mutations to cccDNAs and
integrated HBV genomes. We would also examine whether CRISPR/Cas9-derived DNA
methyltransferase could silence HBV genome by DNA methylation. In addition, we would use
AAV-split-inteins platform for base editing HBV genome in vivo.
2. Materials and Methods
2.1 Plasmids
The human codon-optimized base editing vectors pLenti-FNLS-P2A-Puro (BE3) and pLenti-
BE4Gam-P2A-Puro (BE4), U6-gRNA were obtained from Addgene (Watertown, MA, USA).
The variant VQR (D1135V, R1335Q and T1337R), VRER (D1135V, G1218R, R1335E and
T1337R), and EQR (D1135E, R1335Q and T1337R) were generated by site-directed
mutagenesis of the pLenti-BE4Gam-P2A-Puro backbone based on previous report.56 The DNA
methylation vector phCas9.D3A.puro and phCas9.dD3A.puro were obtained from Addgene
(Watertown, MA, USA). The pLenti-D3A.puro, pLenti-dD3A.puro, pLenti-D3A.D3L.puro,
and pLenti-dD3A.D3L.puro were generated through Gibson assembly. The pLenti-U6-gRNA-
BSD was generated through Gibson assembly, by combing the U6-BsmBI-sgRNA scaffold
and the blasticidin-resistant gene which was derived from the pLVX.AcGFP.N1 (catalog#
632154, Clontech, Mountain View, CA, USA) backbone. The resultant gRNAs were
subsequently cloned into the plasmid pLenti-U6-gRNA-BSD. The non-vector helper plasmids
for lentiviral production, p8.91 and pMD.G were obtained from RNAi core of Academia Sinica,
Taiwan. BE-designer-CRISPR RGEN tools were used to identify the 20-bp protospacer
sequences of gRNAs targeting HBV core, polymerase, surface, and X
(http://www.rgenome.net/be-designer/). 1.3×HBV-WT was derived from the pCMV-HBV
backbone using Gibson assembly. 1.3×HBV-W156X-S and 1.3×HBV-W414X-P were
generated by site-directed mutagenesis of the 1.3×HBV-WT backbone. Summary all plasmids
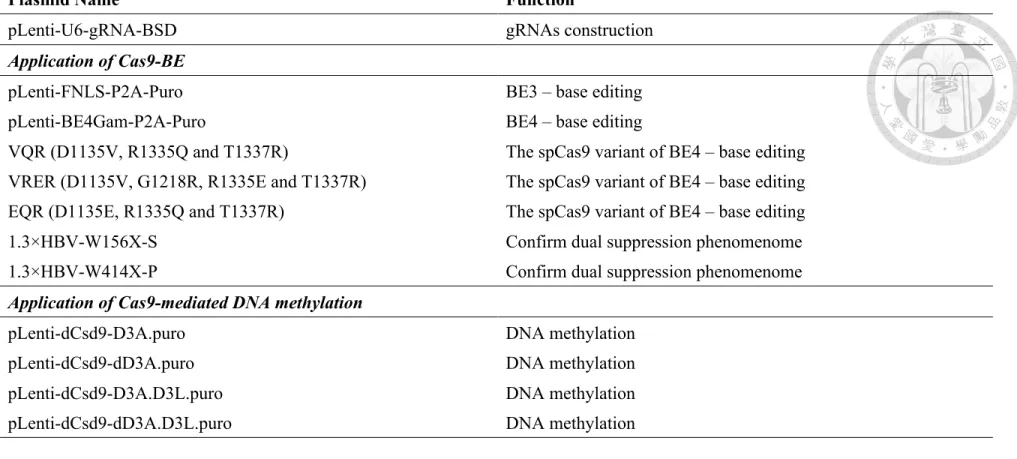
and it function in Table 4.
2.2 Cell lines and culture
HEK293T-C, -Pol, and -S cells were generated by transduction of HEK293T cells with
lentiviral vector containing harboring part of the entire ORFs of core, polymerase, and surface
genes, respectively, from genotype D HBV. HEK293T, HEK293T-C, -Pol, -S, HepG2.2.15,
and HepAD38 were all maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco,
Waltham, MA, USA) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and
100 μg/mL streptomycin (Gibco, Waltham, MA, USA) at 37°C and 5% CO2. Additionally, 1.5
ug/ml puromycin was added to HEK293T-C, Pol, and -S cells, 400 μg/mL G418 was added to
HepG2.2.15, 400 μg/mL G418 and tetracycline were added to HepAD38. HepG2-NTCP-C4
cells were maintained in DMEM/F12, GlutaMAXTM (Gibco, Waltham, MA, USA) containing
10% FBS, 100 U/ml penicillin, 100 μg/mL streptomycin (Gibco, Waltham, MA, USA), 5
μg/mL human insulin (ProSpec, Ness-Ziona, Israel), 10mM HEPES (Gibco, Waltham, MA,
USA) and 1 mg/mL G418.
2.3 Transfection of cell lines
DNA transfection was performed using LipofectamineTM 3000 (Thermo Fisher Scientific,
Waltham, MA, USA) according to the manufacture’s protocol with some modification. For
transfection-based editing experiments in HEK293T, HEK293T-C, HEK293T-P, and
HEK293T-S, cells were seeded 70-80% confluence and cotransfected by the expression vectors
containing the base editor (pLenti-FNLS-P2A-Puro; BE3) and the sgRNA at the ratio of 4:1.
For the experiments comparing the viral expression of 1.3×-HBV-WT and the derived HBV
with site-directed mutagenesis, Huh7 cells were transfected by indicated plasmids (1.3×HBV-
WT, W414X-P, or W156X-S) and were then harvested at 3 days or 5 days post transfection.
Subsequently, genomic DNAs was extracted by blood and tissue kit (Qiagen, Hilden, Germany)
and subjected to Sanger sequencing, or Hirt’s DNA was extracted for Southern blotting assay.
2.4 Lentiviral production and transduction
For the production of lentivirus of base editors pLenti-FNLS-P2A-Puro (BE3), pLenti-
BE4Gam-P2A-Puro (BE4), pLenti-D3A.puro, pLenti-dD3A.puro, pLenti-D3A.D3L.puro, and
pLenti-dD3A.D3L.puro, HEK293T were seeded in 10-cm dishes containing 5 μg/mL poly-D-
lysin (Sigma, St. Louis, MO, USA). Cells were seeded one day before transfection, and the
next day, cells at 95% confluence were transfected with a prepared mix in Opti-MEM (Gibco,
Waltham, MA, USA) containing 6 μg of lentiviral backbone, 4 μg of p8.91 and 2 μg of pMD.G.
The media of HEK293T were replaced with Opti-MEM containing 5% FBS, and the culture
media were collected after 48 h and 72 h. The supernatant was filtered with a 0.4 μm filter
(Millipore, Billerica, MA, USA), and subsequently ultracentrifuged with 20% sucrose cushion
at the bottom of the tube and incubated at 26,000 r.p.m. (4°C) for 2 h. The precipitated viral
pellet was resuspended in Opti-MEM overnight, and then stored at -80°C. For the production
of the lentivirus of sgRNAs, HEK293T cells were seeded in a six-well plate containing 5
μg/mL poly-D-lysin (Sigma, St. Louis, MO, USA) one day before transfection. Next day, the
cells at 95% confluence were transfected with a prepared mix in Opti-MEM (Gibco, Waltham,
MA, USA) containing 1.5 μg of lentiviral backbone, 1 μg of p8.91 and 0.5 μg of pMD.G. The
following procedures for collection, purification and storage of lentiviruses were the same as
described above.
2.5 Transduction with lentiviruses
For transduction of HepG2.2.15 and HepG2-NTCP-C4 cells, 5×105 individual cells were
seeded in a twelve-well plate. After 24 h, cells were transduced with viral supernatants in the
presence of polybrene (8 μg/μl) and the plates were centrifuged for 1 h at 1250 g, 32°C. Three
days after transduction, the cells were treated with puromycin (2.5 μg/mL) and blasticidin S (5
μg/mL) for 7 days of selection. The transduced cells were trypsinized and reseeded at the same
number, and subsequently transduced by the same lentivirus again as the above procedures.
For the BEs lentivirus transduction into HepG2.2.15 cells, the supernatants were collected at 3
and 5 days post twice pLenti-BE4Gam-P2A-Puro (BE4) transduction, and the cell lysates were
collected at 5 days post transduction. For the HepG2-NTCP-C4 transduction, the cells were
transduced two times of pLenti-FNLS-P2A-Puro (BE3) and gRNAs. For the methylation
experiments, HepG2.2.15 cells were transduced one time and the cell lysates were collected at
14 days post transduction and selection.
2.6 Preparation and infection of HBV
Infectious HBV was produced from HepAD38 cells as previously described.57 The supernatant
was harvested and concentrated by 20% sucrose cushion. For the HBV infection experiment,
HepG2-NTCP-C4 cells were seeded in a 12-well plate and transduced by pLenti-FNLS-P2A-
Puro (BE3) and gRNA lentiviruses. After repeated lentiviral transduction for 2 times, HBV
was infected at 5,000 genome equivalent (GE)/cell. All infections were performed as
previously described.15,58 In addition, HBV was infected into HepG2-NTCP-C4 cells in a 12-
well plate at 50,000 GE/cell for cccDNA detection by Southern blot. Briefly, the cells were
mixed with HBV in the presence of 8% PEG8000 and 5% DMSO at 37°C for 16 h in
suspension. To suppress the formation of newly synthesized RC-DNA, infected HepG2-
NTCP-C4 cells were treated with 20 μM 3TC (Lamivudine, sigma, St. Louis, MO, USA) from
3 days after infection.
2.7 Sanger and MiSeq sequencing of base edited genomic DNA and cccDNA
Genomic DNAs of harvested cells were extracted using the DNase Blood & Tissue Kit (Qiagen,
Hilden, Germany) according to the manufacturer’s instructions. The genomic regions of
interest were amplified by PCR with the site-specific primers (Table 5) and PfuUltra II Fusion
HS DNA Polymerase (Agilent Technologies, Santa Clara, CA, USA) according to the
manufacturer’s protocol. The PCR products were purified by the Illustra GFX PCR DNA and
Gel band Purification Kits (GE Healthcare, Chicago, IL, USA), and were subjected to Sanger
sequencing.
To remove linear and RC-form HBV DNAs for Sanger and Miseq sequencing of cccDNA,
genomic DNAs were extracted and digested with T5 exonuclease (New England Biolabs,
Ipswich, MA, USA) in the reaction mixture of 50 μl containing 500 ng DNA, 5 μl 10× reaction
buffer and 1 μl T5 Exo at 37°C for 1 h and afterwards 11 mM EDTA was added to stop the
reaction.
2.8 Immunoblotting assay
Cells were washed with phosphate-buffered saline (PBS) and lysed with RIPA buffer (50 mM
Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1%
SDS, protease inhibitor cocktail (Roche, Basel, Switzerlan)). Whole cell extracts were
subjected to 12%-sodium dodecyl sulfate polyacrylamide gel electrophoresis (12% SDS-
PAGE), followed by Western blot analysis using primary antibodies (anti-"-actin (Merck,
Kenilworth, NJ, USA), anti-HBs (Ad/Ay) antibody (ab9193; Abcam, Cambridge, UK)),
secondary antibody and detected by Millipore Immobilon Western Chemiluminescent HRP
substrate (Millipore, Burlington, MA, USA).
2.9 ELISA of HBsAg
Elecsys® HBsAg II (Roche Diagnostics, Basel, Switzerlan) was used for HBsAg qualitative
determination in the culture supernatant of G2.2.15. Samples with a signal/cutoff ratio (S/CO)
of ≥ 1 are considered positive, and the values are considered as a semiquantitative level of
HBsAg. The quantitative levels of HBsAg in the culture supernatant of HepG2-NTCP-C4 was
measured using ARCHITECT HBsAg kit (Abbott, Chicago, IL, USA). The calibration range
recommended by the manufacturer was from 0 to 250 IU/mL. The positivity criteria of HBsAg
was ≥ 0.05 IU/mL.
2.10 Quantitative real-time PCR (qRT-PCR)
The viral DNAs were purified from the supernatant of G2.2.15 using the DNase Blood &
Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The PCR
reaction was performed in a total volume of 10 μl which contain 4 μl of DNA template, 0.25
μM of each primer, 0.1 μM probe, and 5 μl TagMan Mastrmix. The program is 2 min at 50°C,
10 min at 95°C and 40 cycles of 95°C for 15 sec and 60°C for 1 min. The probe and primers
sequence are listed in Table 5.
2.11 HBV DNA extraction and Southern blotting
HBV DNA was extracted by the modified Hirt method as previously described.50 Infected
HepG2-NTCP-C4 was lysed in Hirt’s buffer (0.7% SDS, 10 mM Tris-HCl, pH 8.0, and 10 mM
EDTA, pH 8.0). The lysates were treated with 5 M NaCl and incubated at 4°C overnight and
then centrifuged at 10,000 r.p.m. for 30 minutes at 4°C. For extraction of the DNA, the
supernatant was treated by saturated phenol twice and phenol:chloroform (1:1) once. DNA was
precipitated with twice the volume of 100% ethanol at room temperature overnight and
subsequently precipitated by 10,000 r.p.m. centrifugation at 4°C for 30 minutes. 30 μg Hirt
DNA were analyzed by the modified Southern blot method as previously described.50
2.12 DNA-library preparation and MiSeq sequencing
Thermo Scientific Phusion High-Fidelity DNA polymerases kit was used according to the
manufacturer’s recommendations (Illumina, San Diego, CA, USA) for DNA-library
preparation. Adaptor-ligated DNA was indexed and enriched through limited-cycle PCR. The
DNA library was quantified with NanoDrop and through real-time PCR. The DNA-library was
loaded on an Illumina MiSeq instrument according to the manufacturer’s instructions (Illumina,
San Diego, CA, USA), and sequenced with 600 cycles by the Medical Microbiota Center of
the First Core Laboratory, National Taiwan University College of Medicine.
The quality of raw reads were evaluated by FastQC. Base editing efficiency and indel rates of
each sample were calculated using a Python script. Briefly, the sequence of gRNA target
regions in each read was identified by splitting the reads by 10-bp flanking sequences with
exact matches on both sides of each target regions. Indels were calculated as the number of
reads with target regions which contain insertions or deletions divided by the total read number.
Base editing efficiency was measured by counting the number of A, T, C, G bases at each
position on target sequences and then the numbers were divided by the number of total reads.
The ratios of induced premature stop codons in each samples were determined by dividing the
number of reads containing induced premature stop codons with the number of total reads.
2.13 Off-target assessment
To characterize the specificity of CRISPR-mediated base editor, we analyzed the off-target
effects of base editing for two of the most effective gRNAs gP9 and gS8. We used the online
software CRISPOR (http://crispor.org) to find the top three predicted off-target sites for each
gRNA. This software dose not only scores gRNA specificity but also gives a lot of information
including off-targets prediction and primer designs according to the CRISPOR Manual. DNA
samples were the same as those used in Figure 7 and the off-target sequences were amplified
by PCR with indicated primers listed in Table 5. The procedures for DNA-library preparation
and MiSeq sequencing are the same as those described in the main article.
2.14 Immunofluorescence Assay
To evaluate HBV infection efficiency, cells were seeded on the glass coverslip were fixed with
4% paraformaldehyde (PFA) in phosphate-buffered salin (PBS) for 20 minutes at room
temperature (RT) and then washed with PBS twice. The cells were permeabilized with 0.1%
Triton-X-100 for 5 minutes at RT and blocked with 1% BSA in PBS for 30 minutes. The cells
were incubated with anti-HBc (Dako, Santa Clara, CA, USA; 1:1500) and anti-FLAG antibody
(Sigma, St. Louis, MO, USA; 1:100) in a humidified chamber at 4oC overnight. After washing
with PBS, the cells were stained with rhodamine-conjugated anti-mouse IgG (Jackson
ImmunoResearch, West Grove, PA, USA) and fluorescein isothiocyanate (FITC)-conjugated
anti-rabbit IgG antibodies (Jackson ImmunoResearch, West Grove, PA, USA) for 1.5
hour. DNA was stained with Hoechst 33258 at 37 oC and the cells were covered with mounting
medium (catalog number #H-1000; Vector, Burlingame, CA, USA). The staining patterns were
observed by confocal microscopy (LSM 780; Zeiss, Oberkochen, Germany) in the Imaging
Core at the First Core Lab of National Taiwan University College of Medicine.
2.15 DNA bisulfited and pyrosequencing assay for DNA methylation analysis
Sodium bisulfite modification of genomic DNA was conducted with EZ DNA Methylation-
GoldTM kit (Zymo Research, Irvine, CA, USA) according to the manufacture’s protocols. PCR
was performed with PyroMark PCR kit (Qiagen, Hilden, Germany) according to the
manufacture’s protocols. Each 25 μl of PCR mixture was prepared as follow: 2 μl of bisulfite
modified DNA, 12.5 μl 2x pyroMark PCR Master Mix, 2.5 μl 10x CoralLoad concentrate, 5 μl
5x Q-solution, 1 μl RNas-free water, and 2 μl primers. PCR was performed according to the
optimized cycling protocol by PyroMark PCR kit as follows: initial 15 min of PCR activation
step followed by 45 amplification cycles at 94℃ for 30 min, 56℃ for 30 min and 72℃ for 30
min. The program was ended with a final elongation at 72℃ for 10 min. PCR products were
stored at -20℃. Then, PCR product was analyzed by 1.5% TAE agarose gel to check whether
a single product of the expected size was obtained before Pyrosequencing.
PCR products were mixed with Streptavidin-Resin beads (2 μl per sample; Qiagen, Hilden,
Germany) and shaked for 15 min. DNA was denatured at 95℃ to produce ssDNA templates
for Pyrosequencing assay. Finally, the ssDNA was released and combined with the sequencing
primer, which was extended during the Pyrosequencing reaction to provide the sequence of the
template DNA. Pyrosequencing was detected by PyroMark Q24 and the data was produced in
the form of pyrograms.59 The primer sequences and information are listed in Table 5.
2.16 Statistics
An unpaired, two-sided Student’s t test was used to compare the difference between two
independent groups.
3. Results
3.1 Application of Cas9-BE for treatment of CHB
3.1.1 To design and screen HBV-specific gRNAs for inducing nonsense mutations by SpCas9 base editors
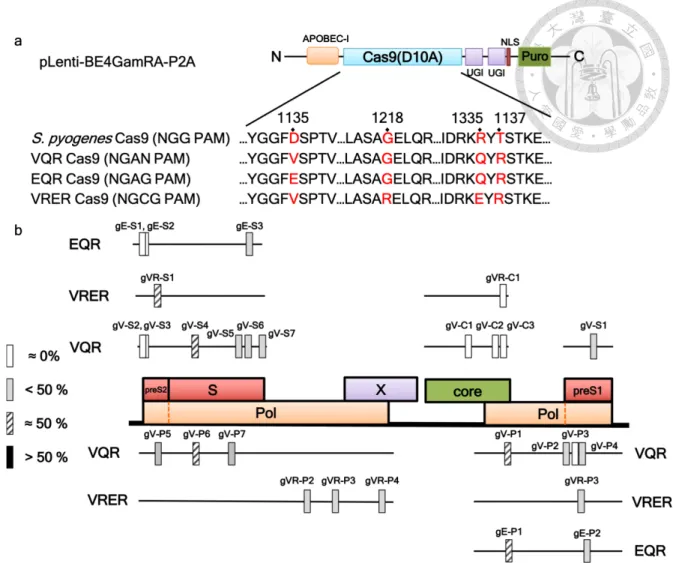
To construct the HBV-specific gRNAs that are suitable for SpCas9 base editor (SpCas9-
BE), we first searched for the candidate protospacer sequences across the four ORFs of HBV
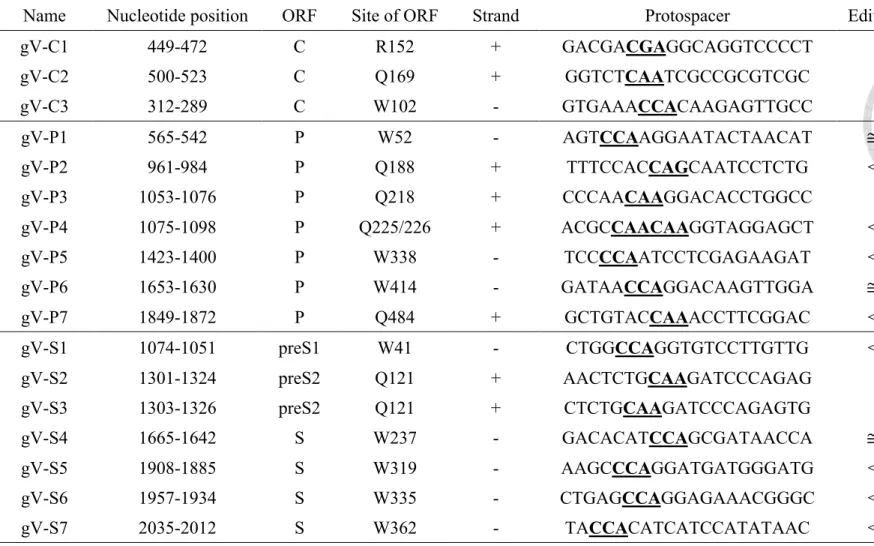
genome by using the website software BE designer.11 We identified 23 candidate target
sequences, including 3 in core, 9 in polymerase, 9 in surface, and 2 in X ORFs (Figure 4a and
Table 1). We constructed HBV-specific gRNAs and then co-transfected with codon-optimized
FNLS-P2A-Puro, hereafter named BE3 for the sake of simplicity, into integrated HBV
genome-containing HEK293T cells (HBV-HEK293T cells), including HEK293T-core,
HEK293T-pol, and HEK293T-surface cells. The efficiency of C-T/G-A conversion in these
gRNA targeting sites were evaluated by Sanger sequencing (Figure 4b). The ratio of C-T/G-
A conversion at each target site was roughly categorized as approximate 0%, less than 50%,
approximate 50%, and greater than 50%. The information and base-editing efficiency of each
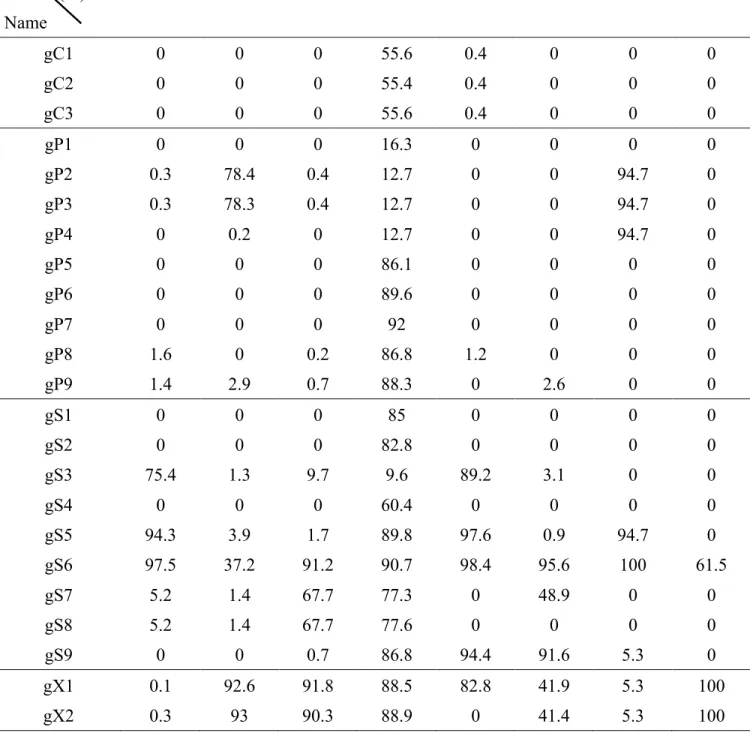
gRNA is summarized in Table 1. Overall, among all the screened HBV-specific gRNAs, we
discovered fourteen gRNAs targeting the polymerase and surface ORFs with base-editing
efficiency of approximate or greater than 50%, whereas the gRNAs targeting core and X ORFs
has less optimal base editing efficiency (approximate 0% or less than 50%).
In order to expand candidate gRNAs, we also performed the screening for candidate
gRNAs using spCas9-BE variants including VQR, VRER, and EQR, which recognize altered
PAMs, NGAN, NGCG, and NGAG, respectively (Figure 5a).56 The results are summarized in
Figure 5b and Table 3, showing that the overall editing efficiency of SpCas9-BE variants was
lower than that of the natural SpCas9-BE.
3.1.2 Introducing nonsense mutations into integrated HBV genomes by base editing suppresses the expression of HBsAg and polymerase
As mentioned above, the integrated HBV genomes can continuous produce HBsAg
expression. We thus examined whether the selected gRNAs combined with BE4Gam-P2A-
Puro were able to suppress the expression of viral genes from integrated HBV genomes,
particularly HBsAg, in HepG2.2.15, which harbors integrated replication-competent dimeric
HBV genomes.60 BE4Gam-P2A-Puro, hereafter named BE4, contains codon-optimized Cas9
nickase (nCas9) and nuclear targeting motifs and Gam at the N-terminus, and thus has higher
base-editing efficacy and reduced undesired base changes and indel formation compared to
BE3.49 We chose a number of gRNAs targeting surface ORFs with high base-editing efficiency,
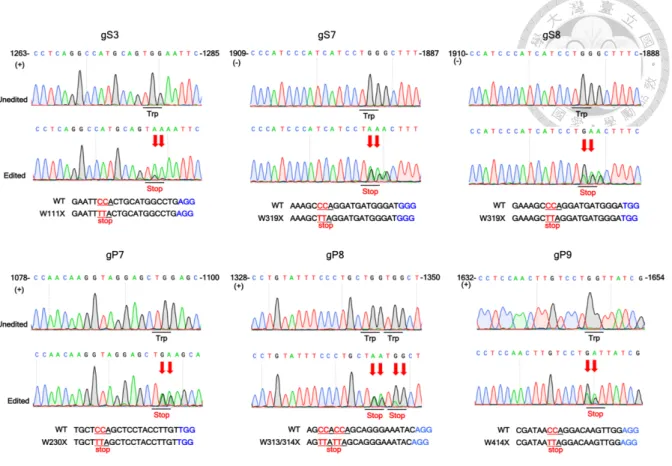
including gS3 (preS2), gS7 (S), and gS8 (S). Following lentiviral transduction of HepG2.2.15
cells with gRNAs and BE4 twice, the supernatants and lysates of HepG2.2.15 cells were
collected to examine the expression of HBsAg by the semiquantitative ELISA and
immunoblotting, respectively. The genomic DNA was subsequently extracted from transduced
cells and subjected to Sanger sequencing and NGS. The results show the ratios of successful
C-T/G-A conversion at target sites of gS3, gS7, and gS8 were 50%, 50% and
approximate 50%, respectively (Figure 6). Additionally, HBsAg secretion was observed in the
supernatants of HepG2.2.15 cells treated with gS7 and gS8 which target S gene significantly
decreased, whereas cells treated with gS3, which targets preS2 gene, it did not show significant
HBsAg reduction (Figure 7a). The expression of HBsAg was also confirmed by
immunoblotting analysis, which shows a pattern of HBsAg reduction similar to the ELISA
results. Only HepG2.2.15 cells treated by gS7 and gS8, but not those treated by gS3, exhibited
significantly decreased expression of small surface protein (Figure 7b).
Furthermore, we examined the effect of base-editing inactivation on HBV polymerase
by utilizing gRNAs with high efficiency, gP7, gP8, and gP9, which target protospacer
sequences that are conserved in 88% of genotype D HBV strains. The extent of sequence
conservation for the protospacer sequence and PAM of each gRNA is summarized in Table 2.
We generated lentiviruses of gRNAs and BE4 and co-transduced into HepG2.2.15 with them
twice in an interval of 14 days. Genomic DNA was extracted for Sanger sequencing and NGS.
The results show that the C-T/G-A conversion rates at all three sites are approximate 50%
(Figure 6). The HBV DNA level of the supernatants in HepG2.2.15 cells treated by gP7, gP8
and gP9 decreased by more than 60%, indicating effective inactivation of HBV replication
(Figure 7c). In addition, the effective base editing was further confirmed by NGS, which
showed that the C-T/G-A conversion rates at the target sites of gP7, gP8, gP9, gS3, gS7, and
gS8 were around 35% to 80% (Figure 7d). We also analyzed the frequency of Cas9-BE-
induced on-target indels by NGS. We used measurement of indels to evaluated the risk of DSB.
Our results showed that base editing with all the above 6 gRNAs (gP7, gP8, gP9, gS3, gS7,
and gS8) resulted in low on-target indels (0.5%~5%) (Figure 7e). We further compared the
frequencies of indels caused by wild-type Cas9 and Cas9-BE at the target sites of gP9, gS7,
and gS8; and found that wild-type Cas9 indeed induced significantly much higher levels of
indels (> 70%) than Cas9-BE (Figure 7e). In addition to the on-target specificity of Cas9-BE,
we also analyzed the off-target effects of base editing for two of the most effective gRNAs
(gP9 and gS8). We chose top three predicted off-target sites for each gRNA. The mutagenesis
rates of all the off-target sites measured by NGS were very low (Figure 8).
3.1.3 To induce dual suppression of HBsAg and polymerase by HBV base-editing specific gRNAs of HBV genome
HBV genome is compactly organized and arranged into four ORFs with substantial
overlapping regions. Therefore, we were interested to determine whether the nucleotide change
in the ORF of polymerase would introduce missense mutation and influence the expression of
surface protein and vice versa. Actually, the three gRNAs gP7, gP8 and gP9 not only generated
nonsense mutations in the polymerase gene, but also resulted in G50L (preS1), G25/26N
(preS2), and G71N (S) missense mutations in the surface ORFs. We found that the HBsAg
levels of the supernatants from HepG2.2.15 treated by gP8 and gP9 decreased significantly
using the semiquantitative HBsAg ELISA assay (Figure 9a). The decline of HBsAg was no
significant in the supernatant of gP7 treated cells but more profound in gP9 treated cells.
Consistently, immunoblotting analysis of cell lysates also showed that a similar pattern of the
reduced surface antigen expression (Figure 9b). Likewise, the three gRNAs gS3, gS7 and gS8
not only caused nonsense mutations in the surface gene, but also led to E292L (gS3) and G500S
(gS7 and S8) missense mutations in the polymerase gene. Interestingly, the HBV DNA levels
in the supernatants of HepG2.2.15 cells treated by gS3, gS7, and gS8 also decreased
significantly (Figure 9c). Taken together, we demonstrate that the expression of both
polymerase and surface genes can be significantly reduced by simultaneous introduction of
missense mutation into polymerase gene and nonsense mutation into surface gene, respectively,
or vice versa using gRNAs targeting the overlapping regions of these two genes.
3.1.4 To validate the dual suppression phenomenon by specific point mutations of HBV genome
To further confirm the effective dual suppression of polymerase and surface gene
expression by the particular gRNAs, including gP9, gS7, and gS8, we intentionally introduced
these nonsense mutations into HBV genomes by site-directed mutagenesis including W156X
in surface (W156X-S) and W414X in polymerase (W414X-P), which corresponding with the
base-editing sites of gS7/gS8 (same base-editing site) and gP9 (Figure 10a). Because the
surface and polymerase genes of HBV genome are extensively overlapped, W156X-S nonsense
mutation also introduces G500S in polymerase, and W414X-P causes G71N in S gene as well.
We observed that W156X-S nonsense mutation led to dramatic HBsAg reduction in the
supernatant and cytoplasm to below the detection limit, and G71N missense mutation (W414X-
P) also caused significant decrease of HBsAg by transfection of Huh7 cells with the WT or
mutant HBV-expression plasmid (Figure 10b,c). In addition, for the mutations in polymerase
gene, both W414X-P nonsense mutation and G500S missense mutation (W156X-S) resulted
in significant reduction of the viral DNA in the supernatant at 5 days post-transfection (Figure
10d). We further performed Southern blot to measure the intracellular replicative intermediates
of transfected cells, and found that the two mutant HBV genomes with polymerase mutations
W414X-P and G500S (W156X-S) caused significant reduction of RC-DNA to beyond the
detection limit (Figure 10e). Taken together, our results confirm that the nucleotide changes
at these two particular loci of HBV genome can result in profound dual suppression of the
polymerase and surface gene expression.
3.1.5 Suppression of HBV protein expression through base editing HBV cccDNA by SpCas9 base editor
Since cccDNA is the replicative template of HBV, we further determined whether the
SpCas9 base editor could indeed generate nonsense mutations in cccDNA, which is an
extrachromosomal DNA. We first showed that following HBV infection of HepG2-NTCP-C4
cells in vitro, HBV RC-DNA and cccDNA, it could be detected by Southern blotting (Figure
11a). The identity of cccDNA was further validated by the appearance of 3.2 kb DNA after
linearization of RC-DNA and cccDNA with EcoRI digestion. Additionally, T5 exonuclease
can degrade ssDNA and linear and nicked dsDNA, and leave supercoiled dsDNA. Therefore,
it is significant enhanced the purity of cccDNA isolation after T5 exonuclease treatment
(Figure 11a). To prove C-T/G-A conversion in cccDNA, we then conducted an experiment by
initially repeated transduction of HepG2-NTCP-C4 cells with gRNAs/SpCas9-BE followed by
HBV infection. Our previou results showed that BE3 exhibited higher base-editing efficacy on
cccDNA than BE4 (Figure 11b,c), so we have chosen BE3 for further experiments. We
isolated cccDNA from HBV-infected HepG2-NTCP-C4 by T5 exonuclease treatment, and
analyzed C-T/G-A conversion in cccDNA by Sanger sequencing and NGS (Figure 11d,g). The
results showed that gP9 and gS8 target sites were effectively edited by SpCas9 base editors at
the efficiency close to 50% estimated by Sanger sequencing (Figure 11d). The results of NGS
analysis showed 25% to 35% of base editing that is consistent with the results of Sanger
sequencing (Figure 11g). In addition, BE3 also induced far fewer undesired on-target indels
(< 0.5%) (Figure 11h). Finally, we demonstrated that the secreted HBsAg levels were
significantly decreased in HBV-infected cells treated by gP9 and gS8; and viral DNAs were
also significantly reduced in cells treated by all these three gRNAs (Figure 11e,f). We also
demonstrated the HBV infection efficacy in Cas9 transduced HepG2-NTCP-C4 cells by
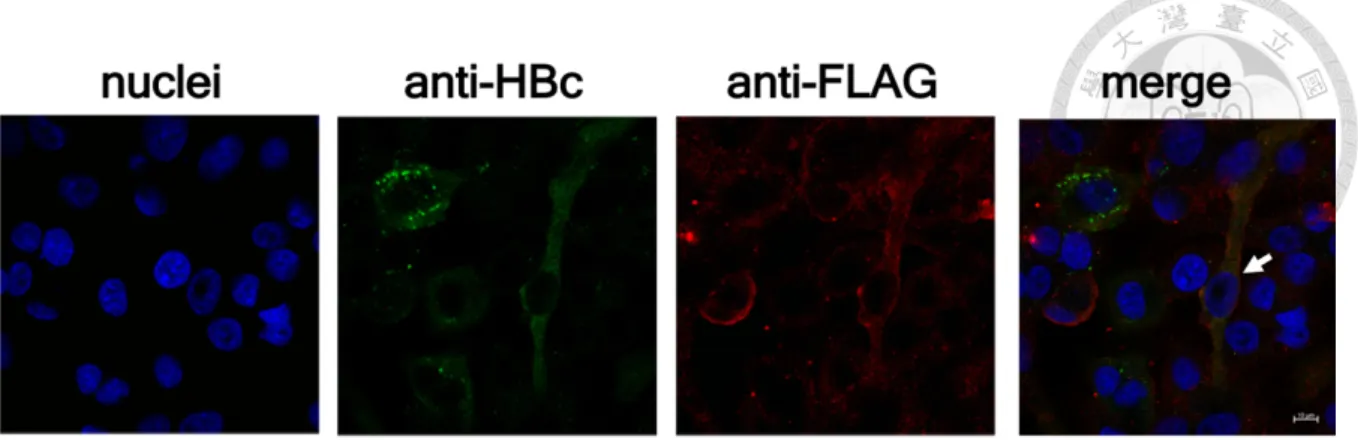
immunofluorescence assay (IFA). We showed that the efficiency of HBV infection and
delivery of Cas9 was around 22.1% and 15.4%, respectively, and only 4.8% cells were double-
positive. Consistently, the results of NGS analysis that around 21.7% of HBV infected cells
were positive for Cas9-FLAG (Figure 12). Collectively, our results prove that cccDNA could
be base edited to reduce the expression of viral proteins.
3.1.6 Using the intein-mediated split Cas9-BE system via rAAV to base-editing HBV genomes
The large size of Cas9-BE has hampered its application for gene therapy because of the
challenging and inefficient in vivo delivery by common viral vectors, like rAAV. We decide to
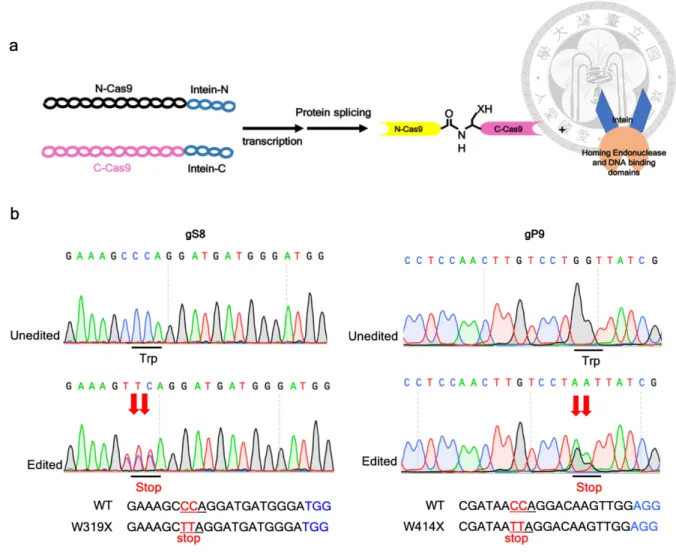
take advantage of the intein-mediated split Cas9 system which is a protein splicing tool.54 The
full Cas9 protein split into two dysfunctional fragments and fused with split inteins; when the
intein-mediated trans-splicing occurs and the full Cas9 protein is reconstituted (Figure 13a).
The split Cas9-BE system was also created and cloned to a pair of rAAVs by the RNAi core at
Academia Sinica. We co-transfected with the plasmids of AAV-split-intine-Cas9 into
integrated HBV genome-containing HEK293T cells (HBV-293T-pol and HEK293T-surface
cells) and the efficiency of C-T/G-A conversion in gS8 and gP9 targeting sites were evaluated
by Sanger sequencing. The ratio of C-T/G-A conversion at each target site was approximate
50% (Figure 13b). We will further exam the base-editing efficiency of HBV genomes by
delivery of the intein-mediated split Cas9-BE via the pair of rAAVs in vivo.
3.2 Application of Cas9-mediated DNA methylation for CHB treatment
3.2.1 Suppressing the expression of HBV genome by CRISPR/Cas9-mediated de novo methylation
DNA methylation is involved in many regulatory activities of the genome including
silencing of gene expression and maintenance of gene integrity and stability. The methylation
from distinct CpG sites of HBV genome is shown to be related with the HCC.61 In addition,
the methylation profile of integrated HBV genome is associated with the flanking human
sequences.62 Previous study analyzed the post-transcriptional modifications (PTMs) of HBV
cccDNA chromatin which regulates the HBV transcription.63 However, the biological
functions of CpG sites of HBV genome are still obscure. We used dCas9 harbored the catalytic
domain of DNA methyltransferase (DNMT) to catalyze de novo methylation of HBV genomes.
As previous study, the DNA methylation region introduced by dCas9-DNMT is ~200 bp
upstream and downstream of the PAM sites.64 There are two to three CpG islands distributed
in HBV genomes across different HBV genotypes. The three CpG islands in genotype D are
mapped in Figure 14a.65 We identified 19 HBV-specific gRNAs (4 gRNAs in CpG I, 11
gRNAs in CpG II, and 4 gRNAs in CpG III) and co-transduced dCas9-D3A/D3A.D3L and
gRNAs into HepG2.2.15. The methylation of dCas9-D3A.D3L is higher than dCas9-D3A and
the methylation levels were distinct at different CG sites (Figure 14b,c). Therefore, 6 CG sites
in CpG I and 2 CG sites could be methylated so far (Figure 14d,e). Currently, we have not yet
determined the methylation levels of CpG island II, which hosts the HBV core and X promoter
region. We will analyze whether de novo methylation of HBV genome can silence HBV
replication.