國立宜蘭大學環境工程學系 碩士論文
Department of Environmental Engineering National Ilan University
Master Thesis
合成胺基磁性吸附劑應用於吸附水中銅離子
Application of Magnetic Particles Modified with Amine Groups to Adsorb Copper ions in Aqueous Solution
指導教授:邱求三博士 Chiou, Chyow-San, Ph. D.
研究生:錢柏勳
英文姓名:Chien, Poh-Sun
誌謝 誌謝 誌謝 誌謝
當初進來的時候是以專題生的身分進入實驗室,如今離開實驗室卻是以碩士生的身 分離開,最感謝的當然是在屏東的家人對我的支持,讓我沒有後顧之憂好好的完成我的 學業。再來要感謝的就是從專題老師變成我的指導教授的邱求三教授,四年的光陰,對 老師的感激不是一兩句話能表達出來,最想對老師說的就是:感謝老師這四年來的栽 培,不僅是學業上的指導還有做人處事的態度,能跟到老師您真是我的福氣啦!還有感 謝化工系的雅芬老師,情意相挺讓我們能自由的使用系上的儀器來分析我的實驗樣品;
也要感謝我的口試委員:陳華偉與郭俞麟老師對於論文上的建議與指導,使我的論文能 夠更加的完整。
接著要感謝的是實驗室的學弟妹們:接在我後面考上這裡,年資比我還深的世瑋,
在實驗上遇到瓶頸時總是能一起討論,想辦法來解決問題,遇到外敵時一定會同一陣 線,共同抵抗外侮;專題生的 RURU(艾儒)與喵(欣怡),在暑假 2 個月瘋狂做實驗的時 候,陪我走過實驗從零到一的日子,多虧有她們才能讓我的實驗順利完成。雖然實驗室 的人員不多,但是實驗室的成員卻都能相互鼓勵吐槽,相處融洽像個大家庭一樣,讓我 再次慶幸能夠進來這個實驗室。
感謝研究所的同學們,特別是同在五樓的親朋好友,在埋首各自的實驗、辛苦打拼 之餘仍不忘相約上頂摟吹吹風,呼吸一下新鮮空氣相互鼓勵,能夠來到這樣一個氣氛融 洽、充滿歡笑的班級,讓我的研究生活不會枯燥沉悶且充滿快樂的回憶,多謝大家並致 上我誠摯的祝福。
最後要感謝在實驗及生活上幫助過我的人,陪我走過兩年的歲月,在此,衷心的獻
摘要 摘要 摘要 摘要
磁性顆粒作為吸附劑可以克服水溶液中微小顆粒的回收問題,磁性顆粒(Fe3O4)先 以水玻璃對其表面做 SiO2 層包覆,以保護磁性顆粒使其避免在水溶液中因水解而遭受 破壞,由於胺基對銅離子有很大的錯合常數,故將以 SiO2 包覆完的 Fe3O4 再利用 N-[3-(Trimethoxysilyl)propyl]-ethylenediamine (TPED)作表面胺基修飾,再利用此胺基磁 性吸附劑對水溶液中的銅離子進行吸附實驗,實驗中分別對水溶液 pH 值、銅離子濃度、
溶液離子強度及吸附劑添加量等參數進行探討,並進行等溫吸附實驗(25℃)、動力吸附 及脫附實驗。經由 pH 値影響實驗發現,最佳吸附條件的 pH 値為 5.5±0.1;氯離子對吸 附行為的干擾在氯離子濃度為 500 mg L-1最為顯著。將等溫吸附實驗數據代入分析後發 覺其符合 Langmuir 吸附模式分析,並估算其最大吸附量及自由能分別為 11.237 mg g-1 與-18.819 kJ mol-1。由動力吸附實驗求得吸附的活化能為 26.915 kJ mol-1。實驗後的胺基 磁性吸附劑經外加磁場回收,以酸性溶液脫附所吸附的重金屬後,進行重複吸附實驗,
發現其對銅離子的吸附能力稍有衰減,以重複使用三次為佳。
關鍵詞 關鍵詞 關鍵詞
關鍵詞::::磁性顆粒、SiO2、胺基、吸附、銅離子
Abstract
A magnetic adsorbent permits easy recovery of the adsorbent from the treated water by magnetic force, without the need for further downstream treatment processes. In this research, a magnetic adsorbent with amine groups has been developed by covering SiO2 film on the surface of Fe3O4 and the followed amino-functionalization using N-[3-(Trimethoxysilyl)propyl]-ethylenediamine (TPED) via covalent bonding with SiO2. The synthesized magnetic amine-adsorbents were applying to adsorbe copper ions in aqueous solution in the batch system, and the maximum adsorption was occurred at pH 5.5 ± 0.1. The adsorption equilibrium data fit Langmuir isotherm equation well with a maximum adsorption capacity of 11.237 mg g-1. Langmuir adsorption equilibrium constant and the Gibbs free energy were 0.0956 L mg-1 and -18.819 kJ mol-1, respectively, at 298 K. A pseudo-second-order model could best describe adsorption kinetics and the derived activation energy was 26.915 kJ mol-1. The adsorbed copper ions on the magnetic amine-adsorbent could be desorbed by using acidic solution, and the recycled adsorbent could be reused up to three times.
Keyword: magnetic adsorbent, SiO2, amine, adsorbe, copper ion.
目錄 目錄 目錄 目錄
摘要 ...I
Abstract ... II 目錄 ... III 圖目錄 ...VII 表目錄 ...IX
第一章 緒論 ... 1
1.1 研究背景...1
1.1.1 磁性顆粒簡介 ... 1
1.1.2 磁性顆粒合成 ... 1
1.1.3 磁性載體技術 ... 2
1.1.4 磁性載體技術應用 ... 4
1.1.5 含銅廢水 ... 4
1.2 研究目標...4
第二章 文獻回顧... 7
2.1 磁性鐵氧化物...7
2.1.1 氧化鐵基本性質 ... 7
2.1.2 磁性載體 Fe3O4... 10
2.1.3 磁學特性 ... 11
2.2 SiO
2/Fe
3O
4顆粒 ...192.2.1 膠體粒子之生成機制 ... 19
2.2.1.1 均質成核(homogeneous nucleation)與表面成長(surface growth) .... 19
2.2.1.2 膠體穩定與凝聚機制 ... 19
2.2.2 SiO2特性介紹...23
2.2.3 SiO2包覆 Fe3O4... 29
2.3 粒子表面修飾...33
2.3.1 粒子的表面修飾 ... 33
2.3.2 胺官能基修飾 SiO2/Fe3O4顆粒 ...37
2.3.3 合成胺基文獻整理 ... 37
2.4 銅的介紹與吸附理論...40
2.4.1 銅 ... 40
2.4.2 吸附理論 ... 41
2.4.2.1 等溫吸附理論 ... 41
2.4.2.2 熱力學研究 ... 43
2.4.2.3 吸附動力學 ... 43
2.4..2.4 競爭吸附 ... 45
2.4.2.5 影響吸附因子 ... 45
第三章 材料與方法... 47
3.1 實驗藥品與儀器...47
3.1.1 實驗藥品 ... 47
3.1.2 儀器 ... 48
3.2 特性分析...49
3.2.6 BET 比表面積分析... 51
3.2.7 磁性分析(Vibrating Sample Magnetometer, VSM)... 53
3.3 合成方法...53
3.3.1 SiO2包覆 Fe3O4... 53
3.3.2 胺基修飾 SiO2/Fe3O4... 54
3.4 吸附實驗...54
3.4.1 等溫吸附實驗 ... 54
3.4.2 動力吸附實驗 ... 55
3.4.3 離子強度實驗 ... 55
3.4.4 競爭實驗 ... 55
3.4.5 脫附實驗 ... 56
3.4.6 重複實驗 ... 56
第四章 結果與討論... 57
4.1 合成觸媒之討論...57
4.1.1 FT-IR 分析... 57
4.1.2 SEM 分析 ... 62
4.1.3 XRD、BET 及磁性分析 ... 64
4.1.4 UV-VIS 分析 ... 67
4.2 吸附實驗討論...69
4.2.1 pH 値影響 ... 69
4.2.2 空白吸附實驗 ... 73
4.2.3 等溫吸附實驗 ... 74
4.2.4 動力吸附實驗 ... 77
4.2.5 離子強度與競爭吸附實驗 ... 82
4.2.6 脫附實驗 ... 87
4.2.7 重複實驗 ... 90
第五章 結論與建議... 91
5.1 結論...91
5.2 建議...93
參考文獻 ... 94
圖目錄 圖目錄 圖目錄 圖目錄
圖 2-1 尖晶石結構的鐵氧化物示意圖(邱意為,2005) ...8
圖 2-2 Fe2+和 Fe3+在 Fe3O4中旋轉磁力矩組態示意圖(邱意為,2005) ...13
圖 2-3 經過磁壁之磁力矩自旋示意圖(Cullity, 1972) ...16
圖 2-4 磁力矩與磁壁隨磁場強度變化示意圖(Cullity, 1972) ...16
圖 2-5 材料磁化狀態與外加磁場之磁滯曲線關係圖(Cullity, 1972) ...17
圖 2-6 磁區結構與材料粒子大小關係圖(陳育裕,1998) ...17
圖 2-7 保磁力(HC)與顆粒大小關係圖(Cullity, 1972) ... 18
圖 2-8 各磁性物質的粒徑對保磁力之關係圖(Cullity, 1972) ...18
圖 2-9 兩奈米粒子間之總位能(VT)與距離(dm)關係圖(陳彥至,2004) ... 21
圖 2-10 DLVO 總位能曲線圖(黃柏源,2004) ...21
圖 2-11 電雙層之示意圖(黃柏源,2004) ...23
圖 2-12 膠體粒子間穩定之最終型態(陳彥至,2004) ...24
圖 2-13 酸鹼值對矽膠體粒子聚合反應影響之示意圖(Iler, 1979)...27
圖 2-14 鹽類存在對聚合時間的影響示意圖(Iler, 1979)...27
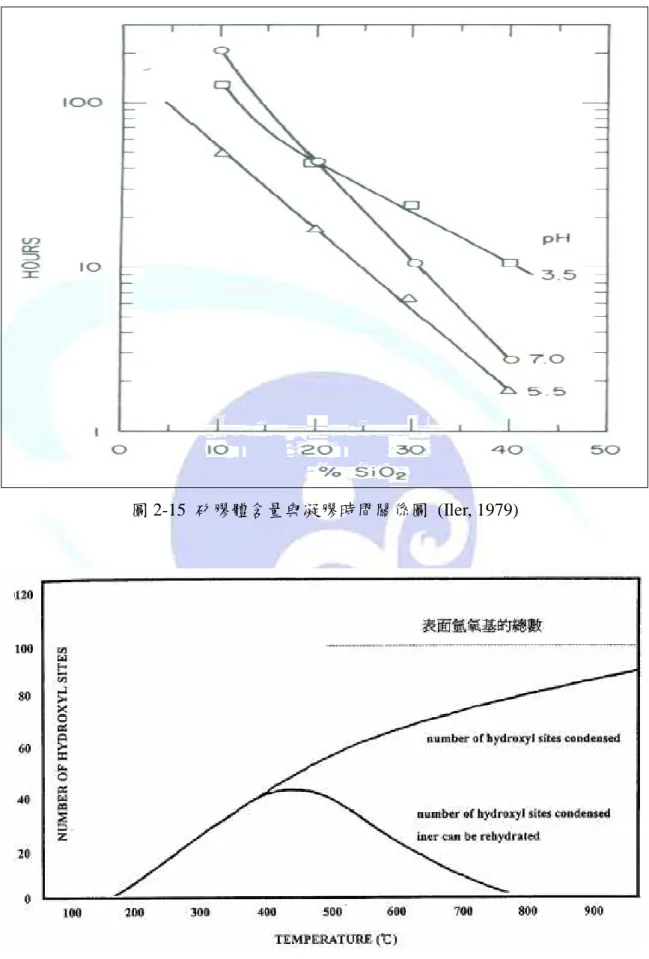
圖 2-15 矽膠體含量與凝膠時間關係圖 (Iler, 1979)...28
圖 2-16 溫度對矽膠體表面氫氧基的影響(Leyden, 1985) ...28
圖 2-17 生物分子修飾粒子表面圖(Niemeyer, 2001)...34
圖 2-18 無機物質修飾粒子表面圖(Caruso, 2001)...36
圖 2-19 TPED 與 SiO2表面反應機制(Liu et al., 2004)...37
圖 2-18 銅的形態與 pH 值變化示意圖(Kumar et al., 2007) ...40
圖 4-1 不同觸媒之 FT-IR 圖 ...59
圖 4-2 不同包覆方法包覆 Fe3O4之 FT-IR 圖 ...60
圖 4-3 TPED 之 FT-IR 圖譜 ...61
圖 4-4 胺基磁性吸附劑( NH2 / SiO2 / Fe3O4 )結構示 ... 61
圖 4-5 Fe3O4放大 1000 倍之 SEM 圖 ...62
圖 4-6 SiO2/Fe3O4放大 6000 倍之 SEM 圖...63
圖 4-7 NH2/SiO2/Fe3O4放大 1000 倍之 SEM 圖 ...63
圖 4-8 不同觸媒(Fe3O4、SiO2/Fe3O4、NH2/SiO2/Fe3O4)之 XRD 圖譜 ... 65
圖 4-9 不同觸媒(Fe3O4、SiO2/Fe3O4、NH2/SiO2/Fe3O4)之磁滯曲線圖 ... 66
圖 4-10 胺基磁性吸附劑與 4-nitrobenzaldehyde 反應圖(Campo et al., 2005) ...68
圖 4-11 Fe3O4、SiO2/Fe3O4以及胺基磁性吸附劑 UV-VIS 比較圖 ... 68
圖 4-12 單純 Fe3O4、SiO2/Fe3O4與胺基磁性吸附劑之零電點測試圖 ...70
圖 4-13 不同 pH 值吸附銅離子比較圖...72
圖 4-14 Fe3O4、SiO2/Fe3O4與胺基磁性吸附劑在不同 pH 值時對銅離子移除效率圖..73
圖 4-15 Fe3O4、SiO2/Fe3O4及胺基磁性吸附劑吸附銅離子比較圖 ...74
圖 4-16 胺基磁性吸附劑吸附銅之 Langmuir 等溫吸附圖 ...76
圖 4-17 胺基磁性吸附劑吸附銅之 Freundlich 等溫吸附圖...76
圖 4-18 不同溫度下胺基磁性吸附劑吸附銅之 (a) 時間動態圖 (b) 假二階動力吸附 模式 ...79
圖 4-19 不同溫度動力反應速率常數對 1 / T 作圖 ...80
圖 4-20 不同濃度下胺基磁性吸附劑吸附銅之 (a) 時間動態圖 (b) 假二階動力吸附 模式 ...81
圖 4-21 NaCl 濃度影響胺基磁性吸附劑吸附銅離子效果圖 ...83
表目錄 表目錄 表目錄 表目錄
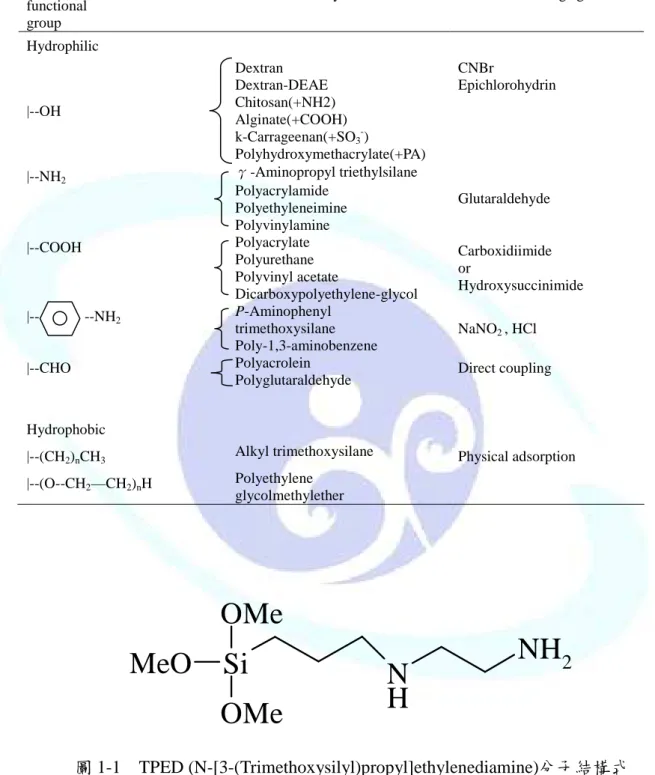
表 1-1 常用修飾材料表面之高分子(Pieters, 1992) ...3
表 2-1 鐵氧化物種類...9
表 2-2 氧化鐵物理性質(Cornell and Schwertmann, 2003) ...9
表 2-3 Fe2+和 Fe3+在 Fe3O4單位晶胞中磁力矩之分佈(邱意為,2005) ...13
表 2-4 SiO2包覆 Fe3O4作法整理 ...31
表 2-5 粒子表面常見生物分子修飾系統(Niemeyer, 2001)...35
表 2-6 合成胺基文獻整理...38
表 4-1 不同觸媒(Fe3O4、SiO2/Fe3O4、NH2/SiO2/Fe3O4)的 BET 分析數據 ... 65
表 4-2 不同 pH 時銅離子之理論濃度...72
表 4-3 胺基磁性吸附劑等溫吸附實驗數據...75
表 4-4 胺基磁性吸附劑等溫吸附之 Langmuir 模式與 Freundlich 模式比較...75
表 4-5 在不同溫度、濃度處理下,由假一階及假二階模式所求得胺基磁性吸附劑吸 附銅的反應速率常數(k2)、相關係數(r2)及活化能 ... 82
表 4-6 胺基磁性吸附劑對不同陽離子濃度配比的吸附量...85
第一章 第一章 第一章
第一章 緒論 緒論 緒論 緒論
1.1 研究背景 研究背景 研究背景 研究背景 1.1.1 磁性顆粒簡介 磁性顆粒簡介 磁性顆粒簡介 磁性顆粒簡介
磁性流體由美國太空總署(NASA)的 Papell 於 1960 年代中期所發明,其原本主要 構想為將火箭燃料與磁性流體相混合,於無重力狀態下,利用磁性流體所具有的磁性與 流變性質,控制火箭液態燃料在管內之移動方向;由於其特殊的流變性質與應用性,引 起許多科學研究者高度的興趣。磁性流體是由強磁性粒子、溶媒以及界面活性劑所組 成,磁性流體與普通流體最主要的區別為其流體中含有磁性粒子,故磁性粒子為磁性流 體之重要組成物質。磁性流體內含的強磁性微粒子,其主要可分為鐵氧體氧化物與金屬 膠體粒子兩類,鐵氧體氧化物的構造屬尖晶石(spinel)結構,包括 Fe3O4、MnFe2O4、NiFe2O4
或 MnZn、NiZn 系鐵氧磁體,不過目前大多以 Fe3O4為主,磁性流體最初主要是應用於 太空開發產業上,隨著後繼研究者發現磁性流體在磁場作用下具有許多特殊物理性質,
而被廣泛地應用於如能源、醫療等新用途上,如將抗體接枝在磁性流體表面,利用其去 分離溶血性病人的血液等。(周子琪,2004;王炳傑,2004)。
磁性分離技術為利用物質的磁性不同,於磁場作用下而達到分離的目的,理想的磁 性載體應具有超順磁性(當無外加磁場時物質無殘磁性,此性質可避免奈米級磁性聚合 物顆粒之間,因磁性互相吸引團聚而造成使用過程中的分散困難。)、良好的機械強度 及化學穩定度,若能具備製作便利及成本低廉等優點,將更具應用潛力。
解法及火花侵蝕法等(鄭景軒,2003 ),其中化學共沉法具備成本較為低廉,利於工業上 大量生產等優點,故於實際應用上較為可行,此法由日本東北大學下飯坂達(1996)所提 出,其將鹼性溶液加入含三價鐵的 FeCl3及含有二價鐵的 FeCl2混合水溶液中,以生成 細微粒的 Fe3O4磁粒,其化學反應如方程式(1)所示:
O H O Fe OH
Fe
Fe3+ +2 2++8 − → 3 4 +4 2 (1)
由方程式(1)可得知,理論上 Fe3+ / Fe2+的比值為 1/2 時,溶液中有足量的鹼便可完 全反應生成 Fe3O4,但於實際進行方程式(1)於製造 Fe3O4時,水溶液中之溶氧及空氣中 之氧氣,將可能改變 Fe3+/ Fe2+的比值,進而影響 Fe3O4的產率及性質,故有許多研究 者[5,6]嚐試探討反應溶液之 Fe3+ / Fe2+比值、pH 值、鹼液濃度及鹼液種類等因素對產物 Fe3O4的影響,目前已大致能經由適當的控制反應條件,而得到較小的 Fe3O4顆粒(奈米 等級)及較大的飽和磁化量。
1.1.3 磁性載體技術 磁性載體技術 磁性載體技術 磁性載體技術
磁性載體技術的發展已有多年歷史,最早運用在分離廢水中的有機物質,原理主要 是利用具有磁性的材料與目標物相互結合,再透過外部磁場的操控達到分離與純化的目 的。磁性材料經過物理或化學適當的表面修飾後,可成為具有選擇性或非選擇性的結合 巨分子、生物分子、細胞、膠體粒子等物質的吸附劑;天然或合成的高分子是常使用在 修飾磁性材料表面的物質,使材料表面具有適當的官能基,便可參與特定的表面反應,
所以磁性材料表面的官能基是磁性載體技術應用能否成功的關鍵因素(麥守義,2005),
一般常見具有官能基之天然或合成的高分子如表 1-1 所示。以本研究為例,為了保護磁 性顆粒(Fe3O4)在酸性條件下不會崩解,便在磁性顆粒外層包覆一層二氧化矽(SiO2)以避 免發生崩解情況,且因二氧化矽外層含有 OH 鍵,方便再修飾上具有特定官能基的高分 子 , 如 本 研 究 所 合 成 具 有 胺 基 的 磁 性 吸 附 劑 便 以 表 面 有 SiO2 層 的 磁 性 顆 粒 與 N-[3-(Trimethoxysilyl)propyl]ethylenediamine (TPED,其結構示如圖 1-1)反應而得。
表 1-1 常用修飾材料表面之高分子(Pieters, 1992)
Polymer functional group
Polymer Derivatizing agent
Hydrophilic
|--OH
Dextran Dextran-DEAE Chitosan(+NH2) Alginate(+COOH) k-Carrageenan(+SO3-)
Polyhydroxymethacrylate(+PA)
CNBr
Epichlorohydrin
|--NH2 γ-Aminopropyl triethylsilane Polyacrylamide
Polyethyleneimine Polyvinylamine
Glutaraldehyde
|--COOH Polyacrylate
Polyurethane Polyvinyl acetate
Dicarboxypolyethylene-glycol
Carboxidiimide or
Hydroxysuccinimide
|-- --NH2 P-Aminophenyl trimethoxysilane Poly-1,3-aminobenzene
NaNO2 , HCl
|--CHO Polyacrolein
Polyglutaraldehyde Direct coupling
Hydrophobic
|--(CH2)nCH3 Alkyl trimethoxysilane Physical adsorption
|--(O--CH2—CH2)nH Polyethylene glycolmethylether
Si N
H
NH 2 OMe
OMe
MeO
1.1.4 磁性載體技術應用 磁性載體技術應用 磁性載體技術應用 磁性載體技術應用
磁性載體技術應用發展之初,主要是用於廢水的處理以及生化分離方面;到了 70 年代中期,磁性載體發展的技術漸漸受到重視,廣泛使用在蛋白質與酵素的固定化、藥 物傳送、免疫分析與生醫感測上。進入到 90 年代,奈米級的磁性載體也逐漸被合成出 來,除了一開始的應用方面,另外在癌症的治療與核磁共振顯像等方面,也展現了不錯 的成效,顯示磁性奈米載體之應用具有相當大的發展潛力。(麥守義,2005)
1.1.5 含銅廢水 含銅廢水 含銅廢水 含銅廢水
由於工業之發展及高科技產業的發達,來自於印刷電路板、電鍍或金屬表面處理及 電池等工業製造程序中所排放之重金屬汙染物已造成一定程度之環境問題,其中又以重 金屬銅佔多數,因此,去除重金屬銅便成為日益重要之課題。
傳統之含銅廢水的處理方法主要為利用調整 pH 值的方式,使銅離子在鹼性環境下 形成氫氧化物沉澱,以去除廢水中所存在之銅離子。一般較常作為調整劑的物質包含氫 氧化物、石灰、碳酸鹽及硫化物等,但是此種方法會產生出大量之含水重金屬污泥,需 要另外的設備與土地空間來對所產生的污泥進行處理,反而需要額外的成本來做後續處 理。再者,由於處理過程須添加大量的藥劑,使得經處理後之放流水中的鹽類及總固體 物濃度增高,往往造成大量的放流水無法回收再利用,導致水資源的浪費。(李嘉宜,
2005)
1.2 研究 研究 研究 研究目 目 目 目標 標 標 標
本研究的主要目的是在於研究一有效且便於回收的新型吸附劑,利用傳統的吸附方 法來去除廢水中之重金屬銅,而吸附完後的吸附劑之需稍加磁場便能輕易的回收再利 用,不但不會造成污泥排放濃度的問題,且放流水也可簡易的回收再利用。
吸附法是一種常見的廢水處理方法,其原理為透過吸附劑表面的分子與吸附質分子 間產生的交互作用力,如靜電作用力、凡得瓦爾力、氫鍵、共價鍵...等,來達到污染物 的分離與廢水純化的目的。目前商業上有使用的吸附劑,如:活性碳(active carbon)、鋁 凝膠(alumina)、矽凝膠(silica gel)、矽藻土(diatomaceous earth)、酸性白土(acid clay)、骨
碳(bone charcoal)以及矽酸鋁分子篩(aluminum silicate molecular sieve)等,粒徑大都落在 微米級或次微米級,而本研究所合成的胺基修飾磁性顆粒粒徑大小將位在微米的範圍。
一般廢水吸附程序中的吸附劑若其顆粒越小,將可增加吸附劑的表面積而提升吸附 劑對吸附質的吸附效率,但後續的吸附劑回收程序將造成很大的困擾,應用具有磁性的 吸附劑將為可行的解決方法,因為只需利用外加的磁場,便可使固液兩相快速分離,達 到吸附劑的回收與再使用的目的。
本研究所合成之新型吸附劑(胺基磁性吸附劑)為表面經過二氧化矽包覆後磁性顆 粒,再於二氧化矽表面以胺基來進行修飾,因吸附劑表面帶有共價鍵結的胺基(-NH2),
與銅離子具有強烈錯合的能力,且因以磁性顆粒為載體,所以合成出的吸附劑亦帶有磁 性,反應完後的吸附劑只需利用外加磁場便可快速回收,相較於一般吸附劑需沉澱、過 濾或離心等的回收方式較為簡便且快速。因此本研究是基於有效與技術之可行性,解決 廢水中重金屬污染及反應後簡易回收再利用之構想。
本論文之研究內容分為新型吸附劑之合成及吸附兩種方式,探討合成之吸附劑與銅 離子的去除效果,其研究內容包括:
一 一 一
一、、、、胺基磁性吸附劑胺基磁性吸附劑胺基磁性吸附劑胺基磁性吸附劑之合成之合成之合成:之合成:: :
1. 利用不同來源(矽酸鈉、四乙氧基矽烷)之二氧化矽包覆磁性顆粒,探討最佳操 作步驟及選定最佳之包覆來源。
2. 藉由各種儀器(SEM、XRD、磁力曲線、BET、FT-IR、UV-Vis)的分析,與空 白觸媒的比較(單純 Fe3O4、SiO2/Fe3O4),探討胺基是否有成功修飾上經過二氧 化矽包覆的磁性顆粒表面。
3. 以恆溫震盪反應器進行摻雜不同離子之競爭實驗,了解胺基磁性吸附劑對銅離 子之選擇性。
4. 以不同濃度的硝酸來清洗反應過後的胺基磁性吸附劑,探討不同濃度的酸對吸 附劑的脫附效果;再以恆溫震盪反應器進行重複實驗,探討胺基磁性吸附劑的 重複利用性。
第二章 第二章 第二章
第二章 文獻回顧 文獻回顧 文獻回顧 文獻回顧
2.1 磁性鐵氧化物 磁性鐵氧化物 磁性鐵氧化物 磁性鐵氧化物 2.1.1 氧化鐵 氧化鐵 氧化鐵 氧化鐵基本性質 基本性質 基本性質 基本性質
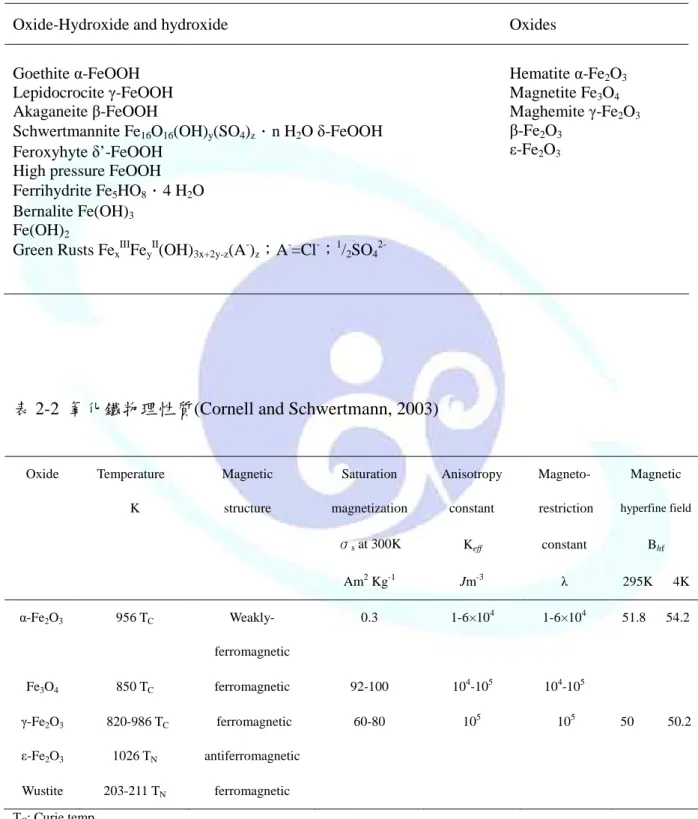
氧化鐵的種類大致上可分為:氧-氫氧化合物(Oxide-Hydroxide)與氧化物(Oxide)兩 大類,如表 2-1,氧-氫氧化合物為氧化鐵的結構中帶有氫氧根存在,而氧化物則為鐵離 子單純與氧鍵結而成。表 2-2 為介紹常見氧化鐵之物理性質,由表中可看出各鐵氧化物 的飽和磁化量(Am2 Kg-1)等性質(Cornell and Schwertmann, 2003)。
以應用方面分類,可將鐵氧化物分為兩類:軟磁(Soft ferrite)與硬磁(Hard ferrite)。
一般來說,軟磁材料特性容易磁化,也容易退磁,大部分的軟磁材料極容易被磁化,外 加約 10 或 20 Oe (oersted)磁場,即可達到飽和磁化量(Saturation magnetization),當外加 磁場消失時,感應生成之磁化便消失,材料其本身並無保磁力(Coercivity,Hc),如 Fe3O4。 而硬磁一般均具有高飽和磁化量與高保磁力值之特性,通常硬磁具有的保磁力値(Hc)都 大於 50,000 A(安培)/m 以上,大部份硬磁材料都堅硬但易碎,且具有永久的磁性,如 γ-Fe2O3。
就結晶結構來分類,可將鐵氧化物分成三大類:
(1) 尖晶石結構(Spinel structure):具有高導磁率、磁束密度,並擁有陶鐵磁性
(Ferrimagnetism),主要為複合 Zn、(Mn+Zn)、(Ni+Zn)、Li 之鐵氧化物。
(2) 石榴石結構(Garnet structure):具反鐵磁性,主要應用於通訊與雷射,如釔鐵氧化物。
三價的陽離子,則此結構即稱為正尖晶石結構(Normal spinel structure),分子通式:
(M2+)(Fe32+)O4;若 A 全為二價陽離子,B 則是二價與三價離子各佔一半,此結構便為反 尖晶石結構(Inverse spinel),分子通式:(Fe3+)(M2+Fe3+)O4。其中陽離子的分布將會對飽 和磁化量具有相當大的影響(邱意為,2005)。
A:四方體心 B:八面體心
圖 2-1 尖晶石結構的鐵氧化物示意圖(邱意為,2005)
表 2-1 鐵氧化物種類(Cornell and Schwertmann, 2003)
Oxide-Hydroxide and hydroxide Oxides
Goethite α-FeOOH Lepidocrocite γ-FeOOH Akaganeite β-FeOOH
Schwertmannite Fe16O16(OH)y(SO4)z.n H2O δ-FeOOH Feroxyhyte δ’-FeOOH
High pressure FeOOH Ferrihydrite Fe5HO8.4 H2O Bernalite Fe(OH)3
Fe(OH)2
Green Rusts Fex IIIFey
II(OH)3x+2y-z(A-)z;A-=Cl-;1/2SO4 2-
Hematite α-Fe2O3
Magnetite Fe3O4
Maghemite γ-Fe2O3
β-Fe2O3
ε-Fe2O3
表 2-2 氧化鐵物理性質(Cornell and Schwertmann, 2003)
Oxide Temperature Magnetic Saturation Anisotropy Magneto- Magnetic
K structure magnetization constant restriction hyperfine field
σs at 300K Keff constant Bhf
Am2 Kg-1 Jm-3 λ 295K 4K
α-Fe2O3 956 TC Weakly- 0.3 1-6×104 1-6×104 51.8 54.2
ferromagnetic
2.1.2 磁性載體 磁性載體 磁性載體 磁性載體 Fe
3O
4磁性顆粒的基本要求,不只要具有較強的磁性,也需要表面攜帶能與親和配位基共 價鍵合的有機活性官能基團,較常用的磁性顆粒有金屬合金 (Fe、Co、Ni)、氧化鐵 (γ-Fe2O3、Fe3O4)、鐵氧體 (Co Fe2O4、Ba Fe12O19)、氧化鉻 (CrO2)和氮化鐵 (Fe4N) 等,
而 Fe3O4 (Magnetite)為最常應用的磁性顆粒,主要原因為容易於水溶液中沉澱製備,若
欲獲得較大粒徑(> 100 nm) Fe3O4顆粒,只需使亞鐵鹽溶液在鹼性條件下氧化沉澱即可;
若利用亞鐵鹽溶液與高鐵鹽溶液按一定比例在鹼性條件下共沉所合成之 Fe3O4則為奈米 級的顆粒(< 10 nm),且具有超順磁性。
Fe3O4之化學式為(Fe2+)(O2-)2-(Fe3+)2(O2-)2,鐵離子同時以+2 和+3 價狀態存在,莫 耳比為 1:2。每個 Fe2+和 Fe3+離子存在淨旋轉磁力矩,分別為 4 和 5 個 Bohr 磁子,而其 中的 O2-離子為磁中性。在鐵離子間因具有反平行旋轉偶合的相互作用,使其特性與反 鐵磁性相似,但是,因為旋轉力矩的不完全抵銷,導致產生淨陶鐵磁力矩。
Fe3O4具有反尖晶石的結構,以對稱而言屬於立方體,類似於尖晶石結構,可將其 視為 O2-離子的密集堆積面所堆積形成。因為具有反尖晶石結構,Fe3+離子有一半在八面 體位置,另外一半在四面體的位置,而 Fe2+離子則均勻的分布於八面體位置。
鐵離子旋轉力矩之排列如圖 2-2 和表 2-3。由圖 2-2 可知,所有位於八面體位置的 Fe3+離子,其旋轉力矩會平行排列於四面體者;但是旋轉力矩的方向卻直接相反於 Fe3+
離子排列在四面體位置者,主要歸因於相鄰鐵離子之反平行耦合。由此可知,所有 Fe3+
離子的旋轉力矩會相互抵銷,對固體的磁化不帶有影響。而所有 Fe2+離子其力矩因為都 以相同的方向排列,總力矩反應出淨磁化作用,由表 2-3 可得知,表中箭號方向即代表 力矩方向。所以,陶鐵磁性固體的飽和磁化量可由每個 Fe2+離子的淨旋轉磁力矩與 Fe2+
離子個數的乘積計算而得知。
Fe3O4經過空氣氧化後會形成三氧化二鐵(Fe2O3),而 Fe2O3的三種型態如表 2-2 所 示,其中又以 γ-Fe2O3最為常見,主要原因為其晶格的排列與 Fe3O4最相似,只要經過
空氣的氧化就會變此型態。γ-Fe2O3 也屬於陶鐵磁性物質材料,但是其具有自生磁化 (Spontaneous magnetization)、磁滯(Hysteresis)及飽和(Saturation)等磁現象,因其本身具 自生磁化之特性,會導致顆粒互相吸引而團聚,使顆粒表面修飾不利進行(官月平,2000)。
2.1.3 磁學特性 磁學特性 磁學特性 磁學特性
2.1.3.1 磁性材料分類磁性材料分類磁性材料分類磁性材料分類
物質在磁場中可被磁化的稱為磁性物質,磁性(magnetism)可分為好幾種,各具有 不同的磁性結構,磁性物質每單位體積(1m3)所含之磁矩稱作磁化強度(magnetization intensity, M),假設磁場為 H,則兩者關係:
M = χ H (2.1) χ:磁化率(magnetic susceptibity), henry/m
χ與μo(導磁率)之單位相同,因此可用μo為單位來量度χ。此量度的磁化率稱作相對
磁化率(relative magnetic susceptibity, χr),可依χr的各種變化形式來解釋物質的磁性結 構,以此觀點可將磁性分為下面五類:
(1) 反磁性(diamgnetism)
反磁性呈現的磁化與外加磁場方向相反,磁化率為負,χr之値約在 10-5~10-6, 為一種弱磁性,而導磁率略小於 1,如果物質中有磁原子,則磁原子所顯現的順磁 性會很輕易的掩蓋反磁性。典型的反磁性材料有 Cu、Zn、Si 和具 NaCl 晶型結構之 無機材料與大部分有機分子等。
(2) 順磁性(paramagnetism)
與鹽類、部分金屬...等。
(3) 鐵磁性(ferromagnetism)
鐵磁性材料有強烈的交互作用在原子與相鄰原子之磁陀間,導致磁陀彼此之間 以同方向排列,而具有較強的自生磁化(spontaneous magnetization)性(磁化的產生不 是靠外加磁場作用),但鐵磁性物質之磁化強度會有飽和現象,也就是在一特定溫度 下其磁化強度到達某定値時,將不再隨磁場強度增強而有增大的趨勢。溫度對鐵磁 性物質的影響很大,當溫度上升時,磁陀受到熱激發(thermal agitated)而使排列被擾 亂,使磁性隨溫度上升而減弱,當溫度超過各物質之居里溫度(Curie temp., Tc),材 料磁性會由鐵磁性轉為順磁性,其磁化率之倒數與絕對溫度呈直線關係,此關係稱 為 Curie-Weiss 定律(方程式 2.1)。典型的鐵磁性材料有鐵、鈷、鎳等。
θ
= − T
k C ( 2.2 )
其中 C 為居里常數,T 與θ為溫度 (4) 反鐵磁性(antiferromagnetism)
反鐵磁性也屬於弱磁性,與順磁性一樣,具有小而正的磁化率,溫度對磁化率 有明顯的影響,在χ-T 曲線上,當溫度低於 Neel 溫度時,磁陀形成逆平行排列,使 正向與反向磁陀彼此相互抵消,磁陀間的作用力隨外界溫度的上升而減弱,使得外 加磁場對此種物質磁化能力隨之增加,因此磁化率隨溫度增高而增大,與順磁相反;
但是當溫度超過 Neel 溫度時,磁陀呈現雜亂的排列,磁化率隨溫度增高而減少,磁 性會由反鐵磁性轉變為順磁性。典型的材料有鉻和錳。
(5) 陶鐵磁性(ferrimagnetism)
陶鐵磁性是由 Neel 所提出,此類的材料在宏觀上類似鐵磁性的行為,磁離子佔 有兩種的格子位置(A 與 B),由於 A 和 B 位置的磁陀間具有強烈的負交互作用,假 設 A 位置的磁陀指向正向,則 B 位置的磁陀即指向負向,因為 A 和 B 位置的磁離 子數目及離子的磁陀大小皆不相同,故產生淨磁矩,即為自生磁化。當陶鐵磁體在 溫度升高時,磁矩排列會受到熱擾亂,自生磁化便會減少,達到居里溫度時,磁矩
的排列會完全雜亂,自生磁化也隨之消失。典型的陶鐵磁體有一結構通式 MO.
Fe2O3,M 為二價的金屬離子,如 Mn、Ni、Fe、Co、Mg...等。
圖 2-2 Fe2+和 Fe3+在 Fe3O4中旋轉磁力矩組態示意圖(邱意為,2005)
表 2-3 Fe2+和 Fe3+在 Fe3O4單位晶胞中磁力矩之分佈(邱意為,2005)
八面體格子位置 四面體格子位置 淨磁力矩
2.1.3.2 磁滯曲線與磁區磁滯曲線與磁區磁滯曲線與磁區磁滯曲線與磁區
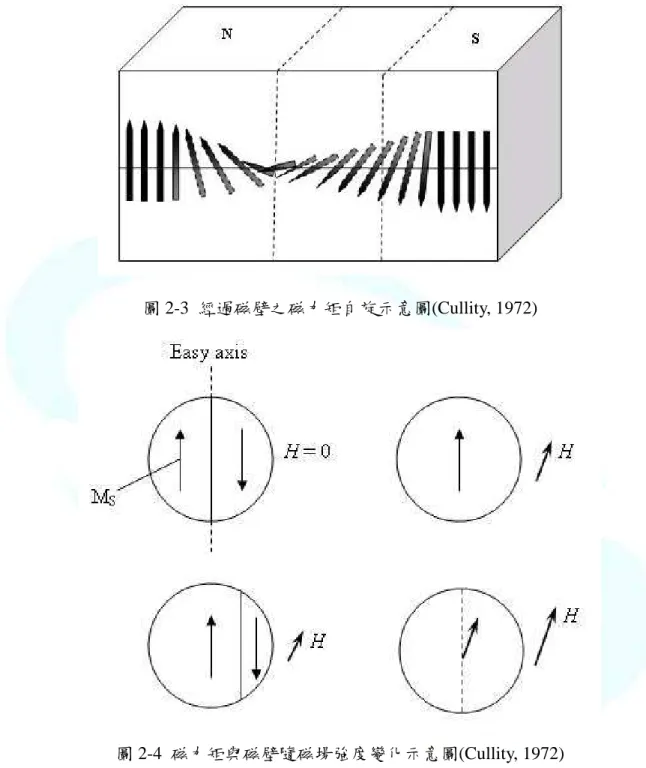
在一般具有鐵磁性體的材料內部之原子磁矩耦合,導致電子自旋方向平行排列成一 小區域,此小區域稱為磁區(magnetic domain)。當沒有外部磁場時,物質全體的磁矩為 零,磁區之間的交界稱為磁壁(domain wall),如圖 2-3 所示。如對鐵磁性體施加一外部 磁場,磁區之間的磁壁會有一磁矩做連續性的方向改變,起先是由於磁壁的移動,最後 因為磁區的旋轉使得磁區增大,如圖 2-4 所示,全體磁力矩便在磁場方向磁化。
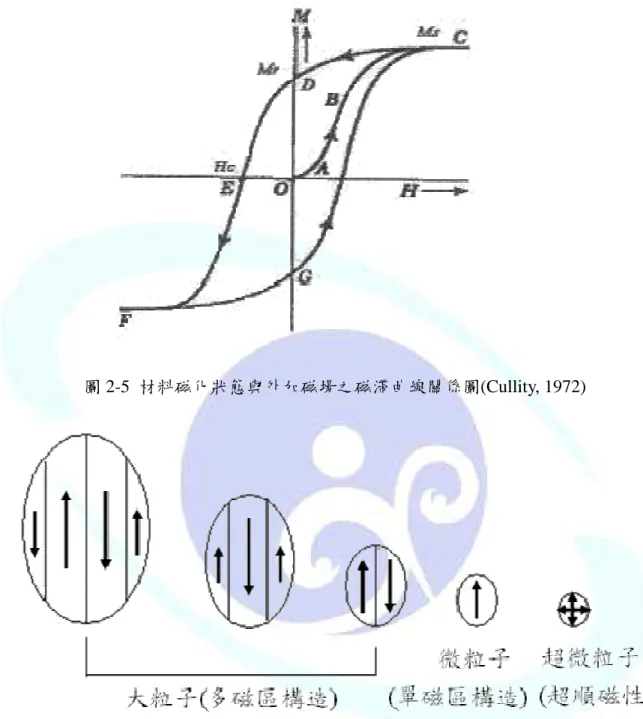
材料磁化的狀態與外加磁場的關係可用磁滯曲線來描述,如圖 2-5。當磁場開始施 加時,磁區便開始偏轉,材料磁化的強度由去磁狀態開始(H=M=0),隨著磁場強度增加 而沿著曲線 OABC 變化,最終達到飽和磁化(saturation magnetization,MS)。在圖中 OA 的範圍內,磁化的過程是可逆的,也就是說,當磁場消失後,磁化便會回復為零。曲線 OA 的斜率稱為初導磁率(initial susceptibility),若超出此範圍,則磁化便不再是可逆的。
當磁化達到飽和磁化狀態(MS)時,因為全體磁力矩已隨磁場方向磁化,如圖 2-4 所示,
因此即使再提高外加磁場強度亦無法使磁化強度增加。
若將磁場從飽和磁化狀態下開始遞減,則磁化 M 會隨著曲線 CD 逐漸減少,當 H 為 0 時 , 磁 化 會到達 一 有 限 值 Mr( 線 段 OD) , 此 值 便 稱 為 殘 留 磁 化 量 (remanent magnetization)。假設磁場在反方向持續地增加,磁化會繼續減少,最後降到零,此時的 磁場強度(HC)稱作保磁力(coercivity)。曲線 DE 的部份,通常稱作去磁曲線。當反向磁 場持續增加,導致磁化也在反方向增加,最終會達到負飽和磁化。若磁場再轉換成正向,
則磁化曲線會沿著 FGC 變化,形成一個封閉的迴圈(CDEFGC),此封閉迴圈稱為磁滯線 (hysteresis loop),如圖 2-5 所示。
基本上,可以由保磁力的大小來區別軟磁與硬磁的材料,根據定義來說明,保磁力 大於 200 Oe 為硬磁性材料;小於 20 Oe 為軟磁性材料;介於兩者之間則為伴媵磁性材 料(楊明勳,2004)。
2.1.3.3 磁性與粒徑之關聯磁性與粒徑之關聯磁性與粒徑之關聯磁性與粒徑之關聯
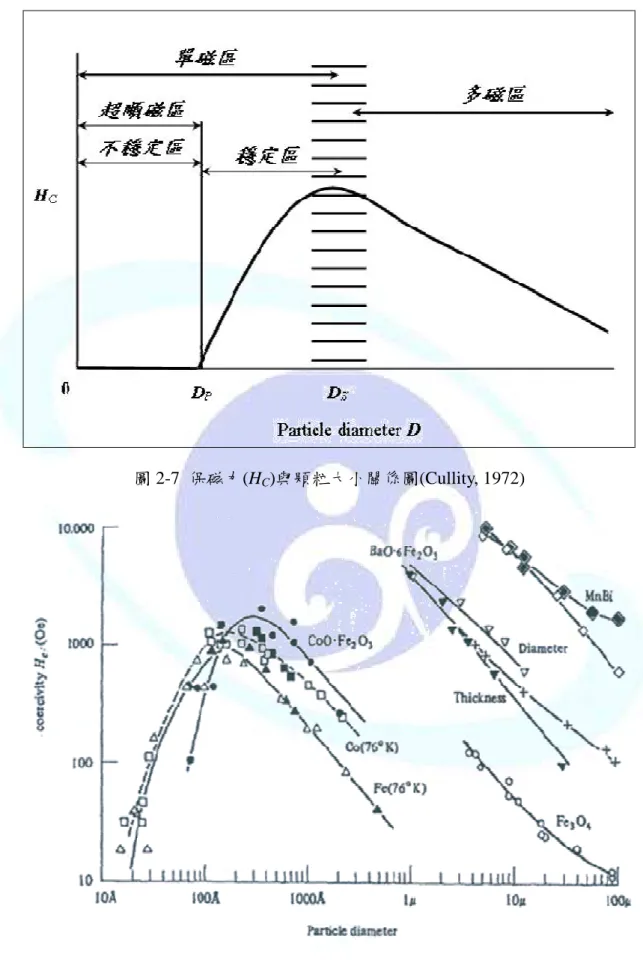
磁性材料的磁區結構與材料粒子的大小有關,如圖 2-6 所示,可分為多磁區、單磁 區,及比單一磁區更小的超順磁區。此三個區域與保磁力的關係顯示於圖 2-7。
(1) 多磁區(multidomain)
當磁性材料粒徑大於單磁區粒徑的臨界值 DS時,粒子的內部會形成多個磁區,此 區域的磁化是由於磁壁的位移而發生,粒徑越小時保磁力越大,其關係式如下:
D a b HC= +
(2.3) 其中 a、b 為常數,D 為粒徑。
(2) 單磁區(single domain)
當磁性材料粒徑小於單磁區粒徑的臨界值 DS時,一個粒子只會有一個磁區,此種 磁化的結構為旋轉磁化,當粒徑接近臨界值時,會得到最大的保磁力,若粒徑小於臨界 值,保磁力會受到熱效應影響而降低,關係式如下:
2
3
D c d HC= −
(2.4) 其中 c、d 為常數,D 為粒徑(介於 DS和 DP之間)。
(3) 超順磁區(superparamagnetic domain)
當磁性材料粒徑小於 DP時,熱效應的影響會導致材料消磁,造成保磁力為零,這 種材料在較弱的磁場中即可呈現磁飽和狀態,且不會有磁滯的現象產生。
不同磁性材料的單磁區臨界半徑 DS與超順磁區的臨界半徑 DP都有所不同,圖 2-8 為各磁性物質的粒徑對保磁力之關係圖。一般來說,飽和磁化量只與材料的種類有關,
與粒徑無關(麥守義,2005)。
圖 2-3 經過磁壁之磁力矩自旋示意圖(Cullity, 1972)
圖 2-4 磁力矩與磁壁隨磁場強度變化示意圖(Cullity, 1972)
圖 2-5 材料磁化狀態與外加磁場之磁滯曲線關係圖(Cullity, 1972)
圖 2-6 磁區結構與材料粒子大小關係圖(陳育裕,1998)
圖 2-7 保磁力(HC)與顆粒大小關係圖(Cullity, 1972)
圖 2-8 各磁性物質的粒徑對保磁力之關係圖(Cullity, 1972)
2.2 SiO
2/Fe
3O
4顆粒 顆粒 顆粒 顆粒 2.2.1 膠體粒子之生成機制 膠體粒子之生成機制 膠體粒子之生成機制 膠體粒子之生成機制
2.2.1.1 均質成核均質成核均質成核均質成核(homogeneous nucleation)與表面成長與表面成長與表面成長(surface growth) 與表面成長
當物質在改變溫度、壓力、時間等環境條件時,可能從原來所存在的相轉變為另一 相,產生相的變化,而由氣體或液體變成固體的過程即是成核理論。溶液中顆粒之成核 與成長,與反應時溶質於溶液中的過飽和度有明顯的關係,廣義的定義過飽和度乃為溶 質於過飽和溶液中與平衡狀態時的濃度差。因為過飽和度會影響成核與成長,所以,過 飽和度的大小將會決定成核與成長的時間。要使溶質產生過飽和狀態有物理及化學方 法,物理方面有冷卻、蒸發、鹽析等方法,而經由化學反應亦可造成飽和度的增加。除 了過飽和度會造成影響外,分子間的作用力也會影響到成核與成長。
當過飽和度達到一定值的時候,若溶液中沒有晶種存在時,會發生成核現象,此現 象稱作均質成核,若過飽和度較小,且溶液中有晶種存在時,此時均質成核所需要的活 化能會大於表面成長之所需,所以表面成長較為有利,晶種會藉此過程慢慢成長,此過 程便稱作表面成長。此兩種程序主要由過飽合度控制,為一種平行競爭性的程序,有時 成核與成長亦會同時發生,端視顆粒在成長的程序上變數的控制而定(黃柏源,2004)。
2.2.1.2 膠體穩定與凝聚機制膠體穩定與凝聚機制膠體穩定與凝聚機制膠體穩定與凝聚機制
一般來說,膠體為一系統中具有一物質,其均勻的分散在另一個物質中,此兩物質 可能為固態、液態或氣態。膠體大小約在 10-9 m(nm)至 10-6 m(µm)之間,此範圍內之顆 粒具有較高的比表面積與較大的吸附能力,膠體的特性決定於膠體與分散介質間的界面
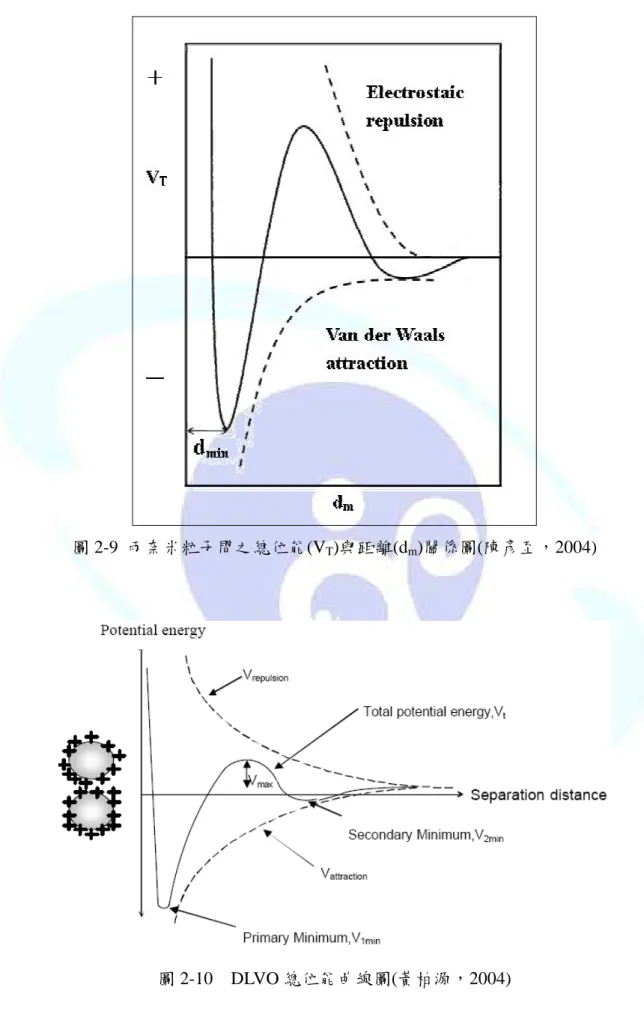
演相當重要的角色。為了讓膠體顆粒容易凝聚,就必須減少顆粒之間排斥的作用與增加 顆 粒 間 的 碰 撞 頻 率 。 膠 體 系 統 之 穩 定 理 論 稱 為 DLVO (Darjaguin-Landan-Verwey-Overbeek)理論,由 Darjaguin 與 Landan 於 1941 年及 Verwey 與 Overbeek 於 1948 年分別提出,此理論認為帶電粒子的膠體同時存在有兩種不同的作用 力,一是靜電斥力,一是凡得瓦爾吸引力,當排斥力大於吸引力,膠體處於穩定的狀態;
反之,則膠體粒子便會互相吸引而產生凝聚(圖 2-9) (陳彥至,2004 )。圖 2-10 為 DLVO 理論中膠體顆粒之排斥能及吸引能結合而成的總位能曲線(黃柏源,2004)。
圖 2-9 兩奈米粒子間之總位能(VT)與距離(dm)關係圖(陳彥至,2004)
因為布朗運動的理論,懸浮液中的膠體顆粒會不停的運動,當顆粒間相互接近時,
擴散層中的反離子(counter ion)便會開始作用,使顆粒相互排斥而無法接近,此時便需要 克服顆粒間的排斥力使顆粒進行凝聚反應,通常用來破壞膠體系統穩定的方法有:
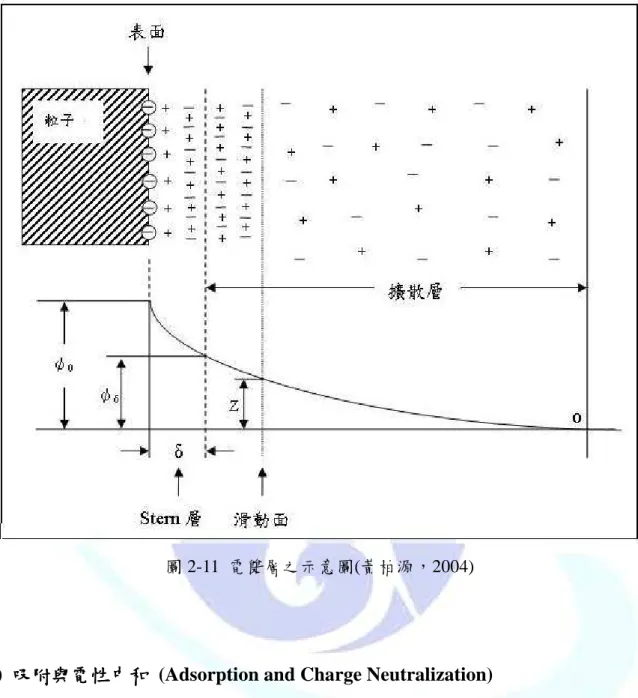
(1)電雙層之壓縮電雙層之壓縮電雙層之壓縮( 電雙層之壓縮 Double Layer Compression ))))
粒子在溶液中,表面離子團會離子化,使膠體粒子皆帶正電或負電。為了要使電荷 平均,粒子表面會於溶液中吸引微量電性相反之離子,而在粒子表面形成一電位差,被 吸引之離子就會在粒子的表面形成電雙層(electrical bouble layer),如圖 2-11。內層稱為 Stern layer,此層為粒子表面帶相反電荷之離子層;外層為 diffuse layer,粒子表面電荷 依然吸引相反電荷靠近粒子,但是被吸引的粒子做自由熱運動,若定義距離粒子無限遠 之電位為零時,粒子表面電位則為ψ0,於 Stern layer 外層(厚度 δ)的電位為ψδ,另外,
膠體化學中需考慮滑動面之存在。於滑動面以內媒體會隨著粒子做共同移動,滑動面以 外則否,滑動面位置接近 Stern layer 的外層或位於擴散層間,在滑動面上的電位定義為 介達電位(Zeta potential,ζ),因為ψ0 及ψδ沒有辦法直接測量,所以介達電位即表示為 粒子表面之電位,其絕對值愈大,表示越不容易發生凝集現象。加入含有較高離子之電 解質於水中時,由於擴散層中含有較高濃度的陽離子,為了保持電中性,擴散層之體積 就必須隨之減小,進而壓縮了電雙層的厚度,導致顆粒間的排斥作用下降,而促使接觸 之機會大增。(黃柏源,2004)
圖 2-11 電雙層之示意圖(黃柏源,2004)
(2) 吸附與電性中和吸附與電性中和吸附與電性中和吸附與電性中和 (Adsorption and Charge Neutralization)
於膠體懸浮液中加入某些離子,能使其吸附於膠體粒子的表面,若添加之離子帶有 與膠體表面相反之電荷,則使表面電位減少,即破壞膠體系統之穩定,造成凝聚現象。
coagulation)模型。第一個模型為假設有一臨界矽酸濃度存在(遠高於飽和濃度),當系統 濃度高於此數值時便會有粒子產生;假設有核的存在時,當系統矽酸濃度高於飽和濃度 時,矽膠粒子便會成長。由此可知,在 TEOS (Tetraethyl Orthosilicate)水解初期,矽酸濃 度高於臨界矽酸濃度時,成核現象即產生足夠之核種,後來因矽酸濃度低於臨界矽酸濃 度,便無新核種的產生,而只有粒子成長的現象發生。
另外,在 DLVO 理論中有提到,欲穩定膠體粒子,則粒子間的排斥力須大於凡得 瓦爾力,可知帶電粒子的穩定性與粒子大小成正比。因為小粒子較不穩定,所以傾向聚 集而生成大粒子,當大粒子生成後,新生成的核便會與其聚集生成最終的穩定膠體粒子 系統,如圖 2-12(陳彥至,2004),兩個膠體粒子間因最外層帶有正電荷而互相排斥,形 成穩定的膠體粒子。
圖 2-12 膠體粒子間穩定之最終型態(陳彥至,2004)
為了保持矽膠體溶膠的型態,可藉由改變溶膠之 pH 值,使二氧化矽表面帶負電荷或加 入一種帶有正電荷,稱為安定對離子( Stabilizing counter ion )之鹽類,使粒子間不會相互 碰撞便不會凝結。
矽膠體之性質會因為環境條件的改變而有所不同,例如:酸鹼值對矽膠體粒子聚合 反應之影響、鹽類存在與膠體濃度對矽膠體安定性的影響、溫度對矽膠體表面氫氧基的 影響等(黃柏源,2004)。
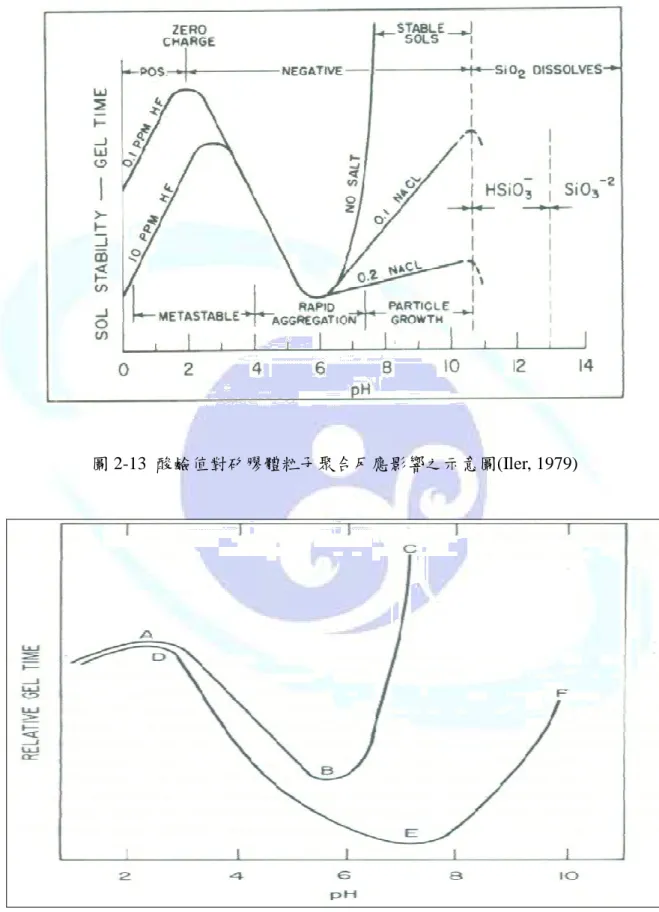
(1) 酸鹼值對矽膠體粒子聚合反應之影響酸鹼值對矽膠體粒子聚合反應之影響酸鹼值對矽膠體粒子聚合反應之影響酸鹼值對矽膠體粒子聚合反應之影響
在不同酸鹼值的條件下,二氧化矽表面的電荷亦有不同的變化,對聚合反應的影響 也不同,如圖 2-13 所示,茲針對此圖說明如下:
(a) pH<2:此範圍之矽酸體粒子的表面帶正電荷,但帶電量不多,為親電子性反應,因
此在[H+]的催化下,還是可以緩慢的進行聚合,聚合時間(Gel time)與聚合速率成正比,
而聚合速率與[H+]的濃度成正比,反應機制如下:
O H Si
O H OH
Si − +
3→= + 2
2=
+ ++
+
+ − =→= − − = +
= Si HO Si Si O Si H
(b) pH=2:此時粒子不帶任何電荷,因為矽膠體之零電點(Zero Point Charge)在 pH = 2,
膠體粒子呈安定狀態,因為在零電點時,膠體粒子只能靠本身的擴散運動進行聚合反 應,所以聚合速率非常緩慢。
(c) 2<pH≦6:此範圍之矽酸體粒子表面帶負電荷,為親核性反應,聚合速率則與[OH-]
的濃度成正比,而聚合的時間會隨著 pH 值上升而減少,其反應機制如下列方程式所示:
O H O Si OH
OH
Si − + →= − +
2=
− + −−
−
+ − =→= − − = +
−
= Si O HO Si Si O Si OH
以緩慢的進行聚合。但是在加入鹽類後,鹽類會幫助膠體達到電中性,加速聚合作用。
圖 2-14 為鹽類存在對聚合時間的影響示意圖,曲線 A-C 為沒有鹽類存在,而曲線 D-F 為有鹽類存在。由圖可知,當 pH 值<2 時,由於膠體帶電量極小,其 ABC 線與 DEF 線相當接近。當 pH 值>2 時,膠體粒子表面逐漸吸附OH−而帶電荷。故 DEF 線慢慢 拉開 ABC 線,表示加入鹽類後可促使膠體聚合。pH 值>6 後,粒子表面吸附大量的OH− 增加帶電量,可看出添加鹽類後的效果更明顯。
而當膠體粒子的濃度增加時,膠體粒子間相互碰撞的機會變多,更容易形成聚合,
圖 2-15 為濃度對矽膠體安定性之影響。
(3) 溫度對矽膠體表面氫氧基的影響溫度對矽膠體表面氫氧基的影響溫度對矽膠體表面氫氧基的影響溫度對矽膠體表面氫氧基的影響
當矽膠體粒子在溫度<400℃時,表面存在的氫氧基(=Si-OH)會彼此縮合脫水而形 成=Si-O-Si=的鍵結,但如果再讓粒子接觸水分一段時間後,即可完全恢復原來=Si-OH 的狀態,圖 2-16 為溫度對矽膠體表面氫氧基的影響,但是當溫度>400℃時,開始會有 部分的鍵結無法再回復,即使長時間放入水中,無法全部回復原來的狀態,當達到 800
℃以上時則完全無法回復。
圖 2-13 酸鹼值對矽膠體粒子聚合反應影響之示意圖(Iler, 1979)
圖 2-15 矽膠體含量與凝膠時間關係圖 (Iler, 1979)
圖 2-16 溫度對矽膠體表面氫氧基的影響(Leyden, 1985)
2.2.3 SiO
2包覆 包覆 包覆 包覆 Fe
3O
41994 年:Philipse 等人以鹽酸當催化劑,tetramethylammonium hydroxide (TMA) 為 穩定劑,利用離子穩定法得到 Fe3O4 的磁流體,並以此為初始物,加入以 Na2O(SiO2)3-5 製備,經酸性離子交換配製出的矽酸溶液,將反應的 pH 值控制在 10,即可得到初步包 覆二氧化矽的磁流體,再來將產物加入純乙醇中,以 TEOS 當作二氧化矽的前驅物,反 應 24 小時,即可得到表面包覆完整的 Fe3O4 奈米粒子。
1998 年:(1) Correaduarte 等人也以 TMA 當作離子穩定劑製備 Fe3O4 磁流體,但與 Philipse 等人不同的是,Correaduarte 只以矽酸鈉溶液來進行包覆,包覆經過稀釋的磁流 體(4 mL 稀釋至 100 mL),反應 pH 控制在 10.5,反應時間 2 小時即可得到產物。(2) Liu 等人先以 TEOS 來包覆 Fe3O4,得到初步包覆的粒子後,為了使包覆較為完整,再以矽 酸鈉進行第二次的包覆。
2001 年:Santra 等人利用三種不同的界面活性劑—Brij-97、Triton X100、Igepal CO-520 形成的逆微胞來製備 Fe3O4,再添加 TOES 於溶液中來包覆。
2002 年:Lu 等人利用異丙醇( 20 mL )與水( 4 mL )的混合溶液作為反應系統,使用 Ferro TEC 公司所販賣的 EMG304 磁流體為初始物,添加不同濃度的 TEOS 來控制包覆 的厚度。
2003 年:Gao 等人利用 cetryltrimethylammonium bromide (CATB)作為界面活性劑,
甲苯為溶劑,形成逆微胞,先於逆微胞內以氨水催化 Fe(II)與 Fe(III)溶液生成 Fe3O4,再 添 加 TEOS 於 反 應 系 統 中 包 覆 Fe3O4, 反 應 後 以 高 溫 除 去 CTAB , 再 以 3-aminopropyltriethoxysilane 改變產物之表面後,將酵素接於產物中。
2005 年:邱以氫氧化鈉當作催化劑,利用化學共沉法製備 Fe3O4,再配製 TEOS、
水及異丙醇(莫耳比為 1:0.05:0.05)的混合溶液作為反應系統,加入已烘乾之 Fe3O4為 初始物,以氨水催化後反應三小時即可完成包覆。
表 2-4 為 SiO2包覆 Fe3O4的文獻整理表。
表 2-4 SiO2包覆 Fe3O4作法整理
作者 方法簡介 出處
Philipse, A. et al. (1) 以鹽酸當催化劑,tetramethylammonium hydroxide (TMA) 為穩定劑,利用離子穩 定法得到 Fe3O4 的磁流體。
(2) 以第一步得到之產物當作初始物,加入以 Na2O(SiO2)3-5製備,經酸性離子交換配製 出的矽酸溶液,將反應的 pH 值控制在 10,即可得到初步包覆二氧化矽的磁流 體。
(3) 將產物加入純乙醇中,以 TEOS 當作二氧 化矽的前驅物,反應 24 小時,即可得到 表面包覆完整的 Fe3O4 奈米粒子。
Langmuir 10, 92 (1994)
Correaduarte A. M. et al.
(1) 以 TMA 當作離子穩定劑製備 Fe3O4 磁流 體。
(2) 以矽酸鈉溶液來進行包覆經過稀釋的磁 流體( 4 mL 稀釋至 100 mL ),反應 pH 控 制在 10.5,反應時間 2 小時即可得到產 物。
Langmuir 14, 6430 (1998)
Liu, Q. et al. (1)0.9g Fe3O4加入 40ml 的純酒精混合,以超 音波震盪 30min,接著加入 14ml 的 TEOS 溶液(0.2ml TEOS+13.8ml 純酒精),6ml 的 氨水(30%)逐滴加入當作催化劑,以磁石攪 拌,反應 5hr 確保 TEOS 完全水解,反應完 後以酒精沖洗,置入 110 度的真空烘箱乾 燥後以乾燥箱保存。
(2) 以第 1 步得到的產物當作初始物,加入 90ml 的水置於 500ml 的三頸瓶中,以 0.1N 的 NaOH 調整 pH 值至 9.5±0.1 且以 90±3 度加熱,加入 10ml 的矽酸鈉水溶液,此時 pH 值會上昇,以 0.1M 的 H2SO4調整 pH 值為 9.5±0.2,以磁石攪拌,反應 1hr。反 應完後以蒸餾水沖洗三次,置於真空烘箱 乾燥後以乾燥箱保存。
Chem. Mater. 10, 3936 (1998)
(1) 利用三種不同的界面活性劑—Brij-97、
Gao, X. et al. (1) 利 用 cetryltrimethylammonium bromide (CATB)作為界面活性劑,甲苯為溶劑,
形成逆微胞。
(2) 於逆微胞內以氨水催化 Fe(II)與 Fe(III)溶 液生成 Fe3O4。
(3) 添加 TEOS 於反應系統中包覆 Fe3O4,反 應 後 以 高 溫 除 去 CTAB , 再 以 3-aminopropyltriethoxysilane 改變產物之 表面後,將酵素接於產物中。
Chem. Commun. 2998 (2003)
Yang, H. H. et al. (1) 參照 2001 年 Swadshmukul 的研究製程,
利 用 Triton X-100 與 環 己 烷 (cyclohexan) 所 形 成 的 逆 微 胞 製 備 Fe3O4。
(2) 再以 teyramethoxysilane (TMOS)當作前驅 物,氨水作為催化劑,將 SiO2 包覆於 Fe3O4 表面。
Anal. Chem. 76, 1316 (2004)
陳彥至 (1) 以氨水當催化劑,利用共沉法製備出油酸 水 基 磁 流 體 與 硝 酸 水 基 磁 流 體 作 為 Fe3O4 之來源。
(2) 探討不同參數對 TEOS 包覆 Fe3O4 之影 響。
清華大學碩士論文(2004)
邱意為 (1) 以氫氧化鈉當作催化劑,利用化學共沉法
製備 Fe3O4。
(2) 配製 TEOS、水及異丙醇( 莫耳比為 1:
0.05:0.05 )的混合溶液作為反應系統,
加入已烘乾之 Fe3O4 為初始物。
(3) 以氨水催化後反應三小時即可完成包覆。
暨南大學碩士論文(2005)
2.3 粒子 粒子 粒子 粒子表面修飾 表面修飾 表面修飾 表面修飾 2.3.1 粒子的表面修飾 粒子的表面修飾 粒子的表面修飾 粒子的表面修飾
藉由對粒子以物理或化學方法改質粒子表面的結構和狀態,可達到以下所列之目 的:(1) 改變或改善粒子的分散性;(2) 使粒子產生新的物理、化學、機械性能及新的 性質;(3) 改善粒子與其他物質之間的相容性。對粒子表面修飾的修飾方法有很多,新 的表面修飾技術也持續地發展中(麥守義,2005),以下就目前已發展的方法敘述如下:
(一) 以有機物質修飾粒子表面
以有機物質來修飾奈米粒子,可將粒子的性質改質成親水性或是親油性,或者與有 機物質達到很好的相容性的目的,改質的方法可分為共價鍵結或直接吸附在粒子的
表面上(廖敏宏,2002)。
(二) 以生物分子修飾粒子表面
由於生物分子具高辨識性,因此以生物分子修飾粒子表面時,在粒子上會產生辨識 的功能,此特性可應用到免疫分析、生化分離和生物標識上,改質的方式是先將一 連接物(linker)連接在粒子上,然後再以生物分子上的官能基(functional group, FG) 接在連接物上,如圖 2-17 所示。圖中編號 1-7 為一般粒子表面上所連接之連接物 (linker)。表 2-5 為粒子表面常見的生物分子修飾系統(Niemeyer, 2001)。
(三) 以無機物質修飾粒子表面
以無機物質修飾粒子表面的方式,主要是先將粒子的表面接上官能基,接著再加入 所欲修飾的無機物質與粒子表面上的官能基進行表面反應而得,如圖 2-18 所示。
圖 2-17 生物分子修飾粒子表面圖(Niemeyer, 2001)
表 2-5 粒子表面常見生物分子修飾系統(Niemeyer, 2001)
Particle Linker FG(Functional group) Biomolecule
Au - HS-Cys immunoglobulins, serum
albumins
Au citrate H2N-Lys proteins
Au streptavidin biotin-(CH2)6- immunoglobulins, serum albumins
Au 3 immunoglobulin, streptavdin
Au streptavidin biotin-(CH2)6- DNA
Au - HS-(CH2)6- DNA
Au - (HS-PO3R2)5- DNA
Au 2 DNA
Au 4 HOOC-Glu proteins
Au 5 HS-Cys proteins
Ag citrate H2N-Lys heme proteins,
immunoglobulins
ZnS HS-Cys gluthathione
CdS HS-Cys peptides
CdS Cd2+, HS-(CH2)2-OH DNA
CdSe/ZnS HS-(CH2)6- DNA
CdSe/CdS/Si O2
6 NHS-biotin streptavidin
CdSe/ZnS HS-(CH2)-COOH H2N-Lys immunoglobulin, transfernn CdSe/ZnS 1 H2N-Lys leucine zipper fusion proteins SnO2, TiO2 HOOC-(CH2)n-NH2 HOOC-Glu proteins
GaAs, InP phosphoramide ε-NH2 HOOC-Glu proteins
圖 2-18 無機物質修飾粒子表面圖(Caruso, 2001)
2.3.2 胺 胺 胺 胺官能 官能 官能 官能基 基 基修飾 基 修飾 修飾SiO 修飾
2/Fe
3O
4顆粒 顆粒 顆粒 顆粒
以 N-[3-(Trimethoxysilyl)propyl]ethylenediamine (TPED)為例,胺基與 SiO2表面的反 應機制如圖 2-19,TPED 的 Si 鍵與 SiO2表面的 OH 鍵產生鍵結,TPED 中的矽氧鍵再與 鄰近的 SiO2表面被帶走 OH 鍵的 Si 鍵結,形成穩定的高分子結構(Liu et al., 2004)。
圖 2-19 TPED 與 SiO2表面反應機制(Liu et al., 2004)
2.3.3 合成胺基文獻整理 合成胺基文獻整理 合成胺基文獻整理 合成胺基文獻整理
不同載體合成胺基的方法大同小異(如表 2-6),主要的差異在於溶劑與當作胺基的 來 源 不 同 , 溶 劑 的 選 擇 有 水 、 乙 醇 及 甲 苯 等 , 胺 基 的 選 擇 來 源 有 aminopropyl triethoxysilane (APTS), N-[3-(trimethoxysilyl)-propylethylene] diamine (TPED), trimethoxysilyl propyl diethylenetriamine (TPDT)等,合成的方法整理於下表 2-6。
表 2-6 合成胺基文獻整理
作者 方法簡介 出處
Campo, A. et al.
0.15 g(SiO2)Fe3O4+APTS(2% v/v)(加水後總體積 15mL),70℃(產量最高之溫度)攪拌 24 小時,清 洗後 95℃真空烘乾。
Journal of Magnetism and Magnetic Materials 293,33–40(2005)
Phan and Jones.
前處理:
(1) 加入 CoFe2O4 1.1 g 於 150 mL 水溶液中(乙 醇:水=1:1)
(2) 在室溫下超音波震盪 30 分鐘
(3)加 15 mL (29℃)NH4OH,通入氬氣,在 60℃混 合攪拌 24 小時
合成:
(1)加水、乙醇、己烷清洗,在室溫下真空乾燥 (2)取經前處理後的產物 1.1 g 於 150 mL 水溶液中 (乙醇:水=1:1)
(3) 在室溫下超音波震盪 30 分鐘
(4) 加 入 1 克 N- 〔 3-(Trimethoxysilyl)propyl 〕 ethylenediamine
(5) 加水、乙醇、己烷清洗,在室溫下真空中乾 燥
Journal of Molecular Catalysis A: Chemical 253, 123–131 (2006)
Blitz, I. P. et al.
1. 4g 的二氧化矽加入含有 20 mmol 有機矽烷的 200 mL 甲苯中迴流反應,反應時間最少 2 小時,
反應後過濾並以甲苯清洗,清洗後產物置於 150℃真空烘箱中乾燥。
2. 0.2g MCM-48 + 5 mL 甲苯 + 0.2g
N-[3-(trimethoxysilyl)propyl]ethylenediamine 在 室溫下通入氬氣混合攪拌 30 小時。
3. 1g SBA-15 + 30 mL 甲苯 + 1g
N-[3-(trimethoxysilyl)propyl]ethylenediamine 在 室溫下通入氬氣混合攪拌 24 小時。
Colloids and Surfaces A:
Physicochem. Eng.
Aspects 307, 83–92 (2007)
Zhang, L. et al.
1g SBA-15 與 5 mL c-aminopropyl triethoxysilane (APTS), N-[3-(trimethoxysilyl)-propylethylene]
diamine (TPED), trimethoxysilyl propyl
diethylenetriamine (TPDT) 分別加入 100 mL 除 水的甲苯,且在通入氮氣下迴流反應至少 12 小 時,反應後完全過濾甲苯,於 70-80℃真空烘乾。
Journal of
Non-Crystalline Solids 353 ,4055–4061(2007)