國立臺灣大學醫學院生物化學暨分子生物學研究所 碩士論文
Graduate Institute of Biochemistry and Molecular Biology College of Medicine
National Taiwan University Master Thesis
Rutin 與 EGCG 作用於大鼠胰臟 β 細胞之抗糖毒性 機制探討
Protection of Rat Pancreatic β cells Against Glucotoxicity by Rutin and EGCG
蔡佩珊 Pei-Shan Cai
指導教授:林仁混 博士 Advisor: Jen-Kun Lin, Ph.D.
中華民國 97 年 6 月 June 2008
國 立 臺 灣 大 學 生 物 化 學 暨 分 子 生 物 學 研 究 所
碩 士 論 文
Ru tin
與 E G C G
作 用 於 大 鼠 胰 臟 β 細 胞 之 抗 糖 毒 性 機 制 探 討
蔡 佩 珊 撰
97
6
誌謝
研究所的兩年,不僅是對於科學的思考訓練,更重要的是學習處理事情的態 度與精神。首先感謝林仁混老師,這兩年來對於實驗上的指導,每次的會談老師 總是會給予實驗上不同方向的思考,跳脫固有的模式而有不一樣的創新想法,對 於實驗設計與思考構思都有實質的助益。老師對於實驗與科學的熱情和專注,也 是往後我繼續學習與加油的目標,謝謝老師這兩年來的指導與栽培,希望在接下 來的日子,可以持續保有現在的熱情,繼續為科學努力與奮鬥。感謝給予指導的 口試委員,中國醫藥大學生物科技學系系主任鍾景光教授,中山醫學大學生物醫 學學系李宣佑教授,台大醫學院藥理所蕭水銀教授,台北醫學大學醫學檢驗生物 技術學系何元順教授,謝謝各位老師於百忙中撥冗參加口試過程,所給予的精闢 見解與指證,使本論文更加完善,對於論文與實驗思維實為受益良多。
實驗室裡的大家,非常感謝你們這兩年來的包涵和幫助,琇珍學姐,學姐對 於實驗與生活上的建議,往往能在困頓的時刻開啟不一樣的方向;志立學長,對 於實驗內容上提供的見解與器材,幫助實驗可以順利的進行;鳳蘭學姐,謝謝學 姐給予的打氣與加油,在最緊要關頭的時刻可以舒緩點情緒;俊德學長,雖然只 有一次切磋球技的機會,不過除了實驗在球場上也能得到不一樣的共鳴,也是非 常不凡的回憶;雅姿學姐,學姐總是奔波於實驗之中,雖然只有一年相處的時間,
也謝謝學姐於各方面的指導。凱揚同學,實驗室的好夥伴,一起大笑一起嘆氣,
一起瘋狂一起咆嘯,這兩年很感謝有你的陪伴,在逼近爆炸極點的時候,總是還 有你,我們一起在呼喊,然後互道加油,謝謝你給予的包涵與體諒,我們繼續加 油,向夢想的未來走去。佩吟同學,高中的三年與碩班的兩年,我們當了五年的 同班同學了,謝謝施小妹每天每天的體貼和分享生活上的點點滴滴,在實驗壓力 下得到了些許喘息與心靈安慰。晉志,國輝,勁文學弟,謝謝你們對於我在實驗
上的龜毛的體諒,未來還要一起在實驗室內打拼,繼續加油。
這兩年來,家人還有朋友們對於我追求夢想所給予的支持與鼓勵,是你們讓 我可以繼續揮霍,以最清爽的模樣繼續朝著夢想前去。馨頤,Afra,Lynne,Iris,
謝謝你們的體貼與體諒,謝謝你們的鼓勵和所給予我的勇氣,你們的加油讓我不 再懷疑,堅持下去。我最親愛的家人,我的母親張簡玉英,謝謝你的體諒和支持,
謝謝你從小到大的栽培和教誨;親愛的姐姐純真和姐夫順德,給予我的鼓勵與打 氣,姵勻與虹文,雖然妳們依舊調皮搗蛋,但妳們的可愛是家裡最開心的寶貝,
也是紓解壓力最好的良方;源添舅舅與鳳雀舅媽,謝謝你們給予我的支持與鼓勵,
讓我可以繼續去追逐夢想。
生活,讀書,課業,學習,這一切一切都不單單只是個人的作為,感謝成長 過程中,參與這一切的每個大家,謝謝你們相信與支持著我,未來還有很久遠的 路途,而我於此,希望仍繼續保有著熱情與勇氣,往前勇敢的去面對和實現夢想,
藉由你們的鼓勵,我也將會更充實與完整。
Table of contents
中文摘要………...…..………..…1
Abstract……….…………...3
Abbreviations………...……….…………...5
Introduction………...6
Materials and Methods………..……...………...13
Results………..…...…...….18
Discussion……….….………..27
References……….………....……..35
Figures………..……...………47
Appendices…….……….………....……65
List of Figures
Figure 1. Enhancement of Glucose Stimulated Insulin Secretion and preservation of
glucose sensing ability during the high glucose incubation in the action of
Rutin or EGCG………..…...…47
Figure 2. Effects of Rutin or EGCG on the stimulation of IRS2 signaling……...…..…50
Figure 3. Rutin or EGCG Alters PDX-1 Nuclear Translocation and expression…...53
Figure 4. Inhibition of Glucolipotoxicity via AMPK activation…………...…55
Figure 5. The maintenance of cell mass against chronic hyperglycemic
incubation……...………...…………....…..58
1
中文摘要
第二型糖尿病,此後天慢性疾病對於現今人類有著重大的威脅,目前以周邊 組織產生胰島素抗性與胰臟胰島β細胞功能喪失為主要探討之對象。在長時間因 飲食等因素導致肥胖產生下,當體內為因應高代謝負荷量時,胰島β細胞會大量 釋出胰島素促使糖類代謝,過去研究發現在肥胖個體上胰島β細胞因應體內代謝 需求,產生代償性β細胞增生之現象,如此達到應對體內代謝之需求。但在長期 負荷環境下可導致胰島β細胞失去對糖份感受性,最終可促使β細胞走向細胞凋亡 一途,當胰島β細胞失去代償作用,同時合併周邊組織胰島素抗性之情況下,往 往成為糖尿病致死之因素。過往研究中已顯示茶中富含之EGCG對於抗癌與身體 能量之代謝有著顯著之功效。此外,已有研究顯示,蕎麥濃縮萃取物可有效降低 於糖尿病模式動物之血糖濃度。因而針對天然物質中茶多酚物EGCG與蕎麥富含 之類黃酮Rutin,對於胰島β細胞於高糖濃度的環境下,是否能提供實質上保護作 用與其分子機制為何,進行相關研究與探討。
於本實驗研究顯示,EGCG與Rutin皆可以有效增加β細胞對於高糖環境培育 下之胰島素釋放,並對長時間培育於高糖環境下之β細胞維持對糖份之感受性,
並有效減緩β細胞長期培育於高糖環境下所導致之細胞衰亡之發生,有助於細胞 生長與存活。進一步探討其分子機制,Rutin與EGCG可有效促進其PDX-1轉錄因 子進入細胞核,並促進其活化。PDX-1為對於胰島發育與β細胞維持正常功能之 重要因子,有效活化PDX-1可幫助β細胞生長、胰島素生成與第二型葡萄糖運輸 蛋白(Glucose transporter 2)等蛋白質表現,同時發現EGCG於長時間的作用下,更 可有效增加PDX-1蛋白質表現量。胰島素接受器受質IRS-2在過去研究顯示,對 於β細胞生長、維持糖份感受性和胰島素生成等相關生理功能皆扮演重要角色,
而β細胞長期處於高糖環境下會降低IRS-2表現而影響細胞正常功能,在同時給予 細胞Rutin與合併於高糖的環境下,發現對於胰島素接受器受質(IRS-2)蛋白質表
2
現量有顯著增加的效果,對於其活性的增加也藉由Rutin與EGCG之給予有著顯著 的效果,並有效影響至下游訊號,包括Akt與FoxO1活性之表現。過去研究顯示β 細胞在長期高糖環境下會促進細胞內脂肪堆積,造成對細胞的損傷與破壞,直接 影響其正常功能運作。EGCG及Rutin於高糖之環境下對於β細胞皆能快速促進活 化細胞內之AMPK(AMP-activated protein kinase)此激酶活性,並能有效抑制脂肪 酸 合 成 酶 FAS(Fatty acid synthase) 之 生 成 , 同 時 也 抑 制 乙 醯 輔 酶 A 羧 化 酶 ACC(Acetyl-CoA carboxylase)活性之上升與脂質合成相關之轉錄因子SREBP1的 表現,並有效減緩細胞於高糖環境下細胞內脂肪之囤積。Rutin與EGCG有效幫助 胰島β細胞於調控醣類與脂質類代謝,此外,也可藉由影響細胞週期相關之蛋白 質表現幫助維持細胞活性與增生,如Cyclin D1與p21,同時也作用於BAX與Bcl-2 等蛋白質表現,幫助抑制高糖下導致之細胞凋亡現象。
胰島β細胞於長期高糖環境下,促使其逐漸失去正常生理功能與反應,而 EGCG與Rutin兩種物質則可幫助β細胞面對於高糖所誘導產生之毒性,延續其細 胞活性與存活能力,可望有效延緩糖尿病致病過程之發展。
3
Abstract
Pancreatic β cell is a fundamental element for the development of diabetes.
Chronic hyperglycemia is associated with insulin insufficiency and peripheral insulin resistance, in which β cells have to meet overloaded metabolic demands, but
gradually will cause the deteriorating cell function, even leading to irreversible damage, cell death. The decompensation of pancreatic β cell followed by adaptation
of increased demand represents the onset of diabetic progression. Therefore, how to maintain the intact cellular function under long term glucose induced toxicity could be strategies for detaining the progression of diabetes.
Rutin and EGCG, natural occurring compounds, have been abundantly found in buckwheat and tea separately, which have been shown the potential of anti-diabetes and anti-obesity in past studies. The actions of Rutin and EGCG on pancreatic β cell are discussed in this study, manifesting the underlying molecular mechanism in regulating the cellular glucose and lipid metabolism. Rutin and EGCG preserved the glucose sensing and glucose-stimulated insulin secretion ability under high glucose incubation. IRS2 signaling was enhanced in the actions of Rutin and EGCG, facilitating the delivery to downstream signals Akt, FoxO1, and PDX-1, which have been implicated as crucial factors in pancreatic β cell growth and function. AMPK is
4
considered as a fuel sensor that enables to response the cellular energy expenditure, and also exerts numerous regulations in metabolism. AMPK was activated in the treatment and effectively suppressed the cellular lipogenesis via inhibition of FAS expression, inactivation of ACC, and manipulation of SREBP1 maturation. Cyclin D1, p21, Bcl-2, and BAX expression levels are also affected in the treatment of Rutin and EGCG, which enhance the cell viability to deal with chronic exposure of elevated
glucose. Long term action of glucose caused multiple abnormalities in metabolism, however, Rutin and EGCG presented comprehensive protection on pancreatic β cell
against glucotoxicity, and exhibited the potential to be candidates for anti-diabetes.
5
Abbreviations
ACC, acetyl-CoA carboxylase
AMPK, AMP-activated protein kinase
CaMMK, calmodulin-dependent protein kinase kinase Cdk, Cyclin-dependent kinase
EGCG, (-)-epigallocatechin-3-gallate FAS, fatty acid synthase
FoxO1, Forkhead-O transcription factor 1, FKHR GK, glucokinase
GLUT2, glucose transporter 2
GSIS, glucose-stimulated insulin secretion
HMG-CoA reductase, 3-hydroxy-3-methylglutaryl-coenzyme A reductase IRS2, insulin receptor substrate protein 2
MCD, malonyl-CoA decarboxylase
MODY, maturity-onset diabetes of the young PDX-1, pancreas-duodenum homeobox-1 PEPCK, phosphoenolpyruvate carboxylkinase
SREBP1, sterol-regulatory-element-binding transcription factor 1 TZD, thiazolidinedione
6
Introduction
Diabetes mellitus is the most common metabolic disorder that causes about 5%
of all death globally each year (1). The etiology of diabetes could be genetics or environmental influences (2), including MODY (maturity-onset diabetes of the young) (3), deregulated glucose sensing or insufficient insulin secretion (pancreatic β cell dysfunction), autoimmune-mediated diabetes (type 1), or insulin resistance in the peripheral tissue. Type 2 diabetes comprises 90% of diabetic individuals and its epidemic expansion associated with the diet behavior and the problem of obesity (4, 5). Two major characteristics of type 2 diabetes are pancreatic β cell dysfunction and peripheral insulin resistance (6), which both could arise from imbalance energy metabolism, however, which one is the cause or the consequence of type 2 diabetes that still needs to be resolved. To discard the controversy, pancreatic β cell indeed
play a critical role in the pathogenesis of type 2 diabetes (7-9). The failure of pancreatic β cell after adaptation resulted from high metabolic demand (6), that leads
to decompensation and accelerates the progression of diabetes accompanied with the
higher mortality (10, 11).
The chronic stress of hyperglycemia forced pancreatic β cell to compensate for
the continuously increased metabolic load and eventually resulted in dysregulation of
7
glucose and lipid homeostasis. Insulin is secreted from the islet cells with glucose stimulation to maintain the normal blood glucose levels; however, β cells contribute
to insufficient secretion of insulin to meet the chronic hyperglycemia. Impaired glucose-stimulated insulin secretion (GSIS) is the first event of pancreatic β cell dysfunction and the defect could accompany by deterioration of mitochondrial ATP production (12). Pancreatic β cell act as a glucose sensor and the metabolism-secretion coupling mechanism is triggered by glucose transportation, glycolysis, ATP production, depolarization of mitochondrial membrane, calcium ion influx, and the final step of the exocytosis of insulin. The Elevation of ATP/ADP ratio generated from the glucose flux is a crucial factor to regulate glucose-stimulated insulin secretion (13), thus the blockade of oxidative mitochondrial metabolism would directed to inhibit the insulin secretory machinery.
Glucose or carbohydrate is regard as fuel for life; nevertheless, prolonged exposure of high glucose condition would lead body to a pathological circumstance called glucotoxicity. Impairment of glucose-stimulated insulin secretion (GSIS),
gradually diminished the insulin/IRS signaling and cell failure are results from the glucose induced toxicity to pancreatic β cell. The mechanism of glucotoxicity
involved in several aspects of energy homeostasis, and one of which is the deterioration of IRS2 signaling. IRS2, insulin receptor substrate protein 2, regulates β
8
cell growth, development, function and survival. Furthermore, IRS2 acts as a critical role in the maintenance of peripheral insulin sensitivity, and central leptin sensitivity (14, 15). Disruption of IRS2 would cause the spontaneous apoptosis of pancreatic β cell and the mice lacking Irs2 display similarities to diabetic individuals, whereas the upregulation of IRS2 might prevent the onset of diabetes in old and obese mice and
delay islet destruction (16).
Pancreatic β cell initially compensates for the peripheral insulin resistance and
hyperglycemia by expanding the cell mass and insulin secretory capacity; however, the chronic stress of increased metabolic demand could compromise IRS2 function or diminish its downstream signal cascade. It is considered that to stimulate the IRS2
signaling might be a potential therapeutic strategy for diabetes (15), and effectively activate its downstream signal could preserve the integrity of pancreatic β cell. IRS2
mediates the signaling through Akt phosphorylation to inactivate FoxO1 (Forkhead-O transcription factor 1, FKHR) via Ser phosphorylation, and which increases PDX-1 (pancreas-duodenum homeobox-1) expression (17). The role of Akt in β cell is considered as an essential factor that regulates cell proliferation, survival and insulin secretion(18), moreover negatively regulates the downstream factor FoxO1 (19).
FoxO1, a transcription activator of pdx1 gene, shuttles between the cytoplasm and the nucleus switched by phosphorylation, and phosphorylated FoxO1 leads to
9
sequestration in cytoplasm and facilitate the PDX-1 expression. Previous studies
indicated that FoxO1 as a repressor of PDX-1 expression (20) and constitutive nuclear expression would prevent β cell hyperplasia and accelerates the onset of diabetes,
furthermore, it has been suggested that FoxO1 nuclear exclusion could be a initiator of pancreatic β cell proliferation during the compensation to insulin resistance (21).
The insulin is exclusively expressed in pancreatic β cell and plays a critical role of controlling the whole body metabolic harmony. PDX-1 is a homeodomain protein that can bind to the proximal region of the insulin gene promoter, and involved in the pancreas development(22). The mutation of PDX-1 have been shown to the onset of
one form of diabetes (MODY4) (23), in contrast to upregulate the PDX-1 expression could restore pancreatic β cell function in Irs2 knockout mice (24). The glucotoxicity
could diminish the binding activity of PDX-1 mediated by the oxidative stress, in the end, to impair the insulin gene expression. Moreover, disorder of lipid metabolism or
called lipotoxicity contributed from chronic metabolic stress would also cause pancreatic β cell dysfunction and cell failure (25), and inhibit the PDX-1 nuclear
translocation (26).
Obesity is one well-known risk factor for the development of metabolic syndrome. Abnormalities in lipid metabolism have been considered as contributing factors for type 2 diabetes, which could influence the peripheral tissue insulin
10
sensitivity and pancreatic insulin secretory capacity (27). In addition to disorder of
lipid metabolism, previous studies have indicated that hyperglycemia and hyperlipidemia could synergize in causing toxicity in pancreatic β cell called
glucolipotoxicity (28). Pancreatic β cell would lose the adaptation ability under chronic excessive glucose or fatty acid level exposure, which alter the expression level of key transcription factors and enzymes related to metabolic network. Multiple metabolic abnormalities is dysregulation of the AMP-activated protein kinase (AMPK) signaling cascade, which is a heterotrimetic protein and involved in glucose and lipid metabolism (29). AMPK acts as a fuel sensor and could be regulated by multiple mechanisms, which could be activated by upstream kinases LKB1(STK11) or CaMMK (calmodulin-dependent protein kinase kinase) via Thr172 phosphorylation (30).
Long-term actions of glucose on pancreatic β cell lipid metabolism were possibly mediated by suppressing or decreasing the AMPK activity (27). AMPK regulates the expression or activity of the lipid metabolism related proteins, such as Acetyl-CoA Carboxylase (ACC), 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase), malonyl-CoA decarboxylase (MCD), fatty acid synthase (FAS), moreover, AMPK could inhibit the generation of sterol-regulatory-element-binding transcription factor 1 (SREBP1) (31, 32). Furthermore, numerous studies indicated that the
11
accumulation of lipid droplets could lead to proceed the dysfunction of pancreatic β
cell (28, 33-35). The effect of chronically elevated glucose not only interferes with the glucose metabolism, but the lipid metabolism. In other words, disturbance of the
metabolic harmony eventually leads to a comprehensive effect.
Chronic exposure to high glucose condition would induce β cells damage by
failing to compensation, and disorder of cellular metabolism caused from the ablation of molecular mechanism. In addition to cell dysfunction, cell mass would
dramatically reduce after the decompensation and in the end, the onset of diabetic progression. Pancreatic β cell has exclusive role in metabolism, however, diabetes or
other metabolic syndromes could cause the deleterious effect resulted from disorder of energy metabolism. How to preserve the β cell function or delay the onset of
progressive diabetes might be a good starting point for diabetes therapeutic strategy.
Tea (Camellia sinensis), one of the global most popular beverages, was regarded as potential agent with anti-diabetes and anti-obesity activity (36). Previous studies have shown that tea polyphenols could activate AMPK to attenuate hepatic lipid accumulation (37), and the activation of IRS1 in hepatoma cells (38). Except for the effect of tea, buckwheat (Fagopyrum esculentum) concentrate has been shown the ability to reduce blood glucose concentration in diabetic model mice (39, 40), thus in this current study, two natural occurring compounds EGCG (Epigallocatechin gallate)
12
and Rutin are used, which have been abundantly found in green tea and buckwheat separately, to examine the effect in pancreatic β cell. The investigation of the action of EGCG and Rutin in pancreatic β cell, which to reveal the underlying molecular
mechanism of the protection from glucotoxicity.
13
Materials and Methods
Materials
The pure compound (-)-epigallocatechin-3-gallate (EGCG), Rutin, D-glucose, sodium pyruvate, HEPES, and ycciuv3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide were obtained from Sigma-Aldrich (St. Louis, MO, USA). The anti-PDX1, anti-Foxa2, anti-SREBP1, anti-pTyr and anti-LKB1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-pAkt (Ser473) and anti-Bax were obtained from Cell signaling Technology, Inc. (Beverly, MA, USA). The anti-pAMPK (Thr172), anti-AMPK, anti-IRS2 and anti-p21 were from Upstate Biotechnology (Lake Placid, NY, USA). The anti-FAS and anti-Cyclin D1 were from BD Bioscience (Franklin Lakes, NJ, USA). The anti-pFoxO1 (Ser256) and anti-FoxO1 were obtained from Abcam Inc. (Cambridge, MA, USA). The anti-pACC (Ser79) was purchased from Transduction Laboratory (Lexington, KY, USA).
Cell culture
RIN-m5F rat insulinoma pancreatic β cells were obtained from the NHRI cell bank (National Health Research Institutes, Taiwan) and maintained in RPMI 1640 containing 11 mM glucose supplemented with 10 mM HEPES, 1 mM sodium
14
pyruvate, 10% (v/v) fetal bovine serum, 100 units/ml penicillin and 100 μg/ml
streptomycin (HyClone, Logan, UT, USA) in a humidified 5% CO2 incubator at 37
℃.
Immunoprecipitation and Western blotting
Cells were incubated in media containing 11 mM glucose followed by 2 h starvation and treated with 33 mM glucose contained media (defined as high glucose stimulation state) with or without EGCG or Rutin for indicated duration of time. Cells were lysed with buffer (10% glycerol, 1% Triton X-100, 0.1% SDS, 10 mM NaF, 50 mM Tris-HCl, pH 8.0, 5 mM EDTA, 150 mM NaCl, 0.5 mM phenylmethysulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 0.5 mM dithiothreitol), and the lysates were centrifuged at 12,000 rpm for 30 min, and then to collect the supernatants
as whole cell extracts. For western blotting, equal amounts of total cellular protein (20 μg) were resolved by SDS-PAGE transferred onto polyvinylidene difluoride
membranes (Millipore), and probed with primary antibody followed by secondary antibody conjugated with horseradish peroxidase. The immunocomplexes were visualized with enhanced chemiluminescence system (Perkin Elmer life sciences, Boston, MA, USA). For immunoprecipitation, equal amount of cell lysates were precipitated with anti-IRS2 antibodies, and then immobilized on protein G-Sepharose
15
beads followed by gently rocking overnight at 4 ℃. To collect the immunocomplexes by centrifugation at 12,000 rpm for 30 min at 4 ℃, and then washed by ice-cold PBS, incubated at 100℃ for 10 min with 20 μl electrophoresis buffer, and the supernatant was analyzed by western blotting.
ATP detection assay and insulin secretion detection
The cells were incubated in the media contained with maintained glucose condition (11 mM) or high glucose condition (33 mM) for 48 h, and then to stimulate the cells by replacing the media contained with high glucose condition with or without Rutin or EGCG. After 2 h stimulation, the cell media were obatined to quantification by rat insulin ELISA kit (Mercodia Inc., Uppsala, Sweden), and the ATP contents were quantificated by luminescence ATP detection assay system (Perkin Elmer life sciences, Boston, MA, USA).
Immunohistochemistry
Cells were incubated in high glucose condition with or without Rutin or EGCG for 24 h. Cells were washed with PBS, fixed with 10% formalin for 30 min on ice, washed twice with PBS, and blocked with 1% BSA/PBS for 1 h at room temperature, and then incubated with anti-PDX1 antibodies (100X) at room temperature for 1 h.
16
The secondary antibody conjugated with FITC (200X, Santa Cruz biotechnology) was used for 1 h incubation at room temperature. The nuclei were stained by DAPI for 1 h and all images were acquired from fluorescence microscope (Carl Zeiss Inc., AXIO Image A1).
Cell cycle Analysis and Flow cytometry
Cells were incubated in 25 mM glucose contained media with or without Rutin or EGCG after 2 h starvation and replaced the media every 24 h. For indicated duration of time, cells were trypsinzed and fixed in PBS with 70% ethanol. After fixation and discard of ethanol, Cells were resuspended in PBS, then treated with 2%
Triton X-100 and RNase A incubated at 37 ℃ for 30 min and exposed to propidium iodide (PI). Cell cycle analysis was performed by BD FACSCalibur system (BD Biosciences, Mountain View, CA) and the fraction of cell cycle was calculated by Acquisition software of BD CellQuest (BD Biosciences, Mountain View, CA).
Oil Red O staining
To measure the cellular lipid droplet accumulation, cells were incubated in maintained glucose condition or high glucose condition with or without Rutin or EGCG after 2 h starvation. These cells were incubated for 5 days and replaced the
17
media every 24 h. After the incubation, cells were fixed by 10% formalin for 60 min followed by washing with iced PBS. Dehydration with 60% isopropanol and stained with Oil Red O solution (3 mg/ml in 60% isopropanol) for 15min at room temperature.
After staining, cells were washed with water and then added 100% isopropanol to elute the bound dye for spectrophotometry at 510 nm detection.
MTT assay
Cells were incubated in 33 mM glucose contained media with or without Rutin or EGCG after 2 h starvation. The cell mass was examined by the MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) assay at the indicated duration of time. The MTT working solution (2 mg/ml in PBS) was exposed to each cultured well and incubated for 2-4 h at 37 ℃. The MTT-formazan crystals were dissolved in 1ml of DMSO and the absorbance of 550 nm were performed by spectrophotometer.
Statistical Analysis
All results were expressed as means ± S.D. The significance of difference between experimental groups was performed with Student’s t-tests. p <0.05 was
considered significant. (*, p <0.05; **, p< 0.01; ***, p< 0.001)
18
Results
Enhancement of glucose stimulated insulin secretion and preservation of the glucose sensing ability during the high glucose incubation in the actions of Rutin or EGCG
The cellular ATP content is responded to the glucose stimulation, and acts as a critical factor to regulate the insulin secretory machinery. Pancreatic β cells were
stimulated by 33 mM glucose for 2 h incubation, and this high glucose condition made cells to go through glycolysis to produce ATP. In Fig. 1A, under the high glucose stimulation, cells treated with Rutin or EGCG both effectively elevated the
contents of ATP, and that indicated that these two nature occurring compounds might be agents to enhance the efficiency of ATP production in pancreatic β cells. The
Correspondences of the insulin secretion also be observed in Fig. 1C, which shown that Rutin and EGCG both increased the insulin to release from cells under high glucose stimulation.
Diabetic individuals might loss the response of glucose stimulation from chronic glucose induced β cell dysfunction, thus to examine whether Rutin or EGCG could still stimulate the insulin secretory machinery of cells that have been long time incubated in high glucose that might be vital for manipulating the β cell function.
19
Cells were incubated in 33mM glucose contained media for 48 h, and then to detected the cellular ATP content and the released insulin followed by another 2 h high glucose stimulation. In Fig. 1B, Rutin effectively elevated the ATP content, thus EGCG only
elevated the ATP content by treating with high dose (10 μM), but not showed significant in low dose treatment (0.1 μM, p=0.224). The insulin secretion of control
cells were dramatically suppressed after 48 h high glucose incubation, however, the diminished insulin secretory machinery could be rescue by Rutin or EGCG treatment
(Fig. 1D). The actions of Rutin and EGCG might be potent effects to preserve the glucose sensing ability in pancreatic β cells to meet the chronic metabolic demand.
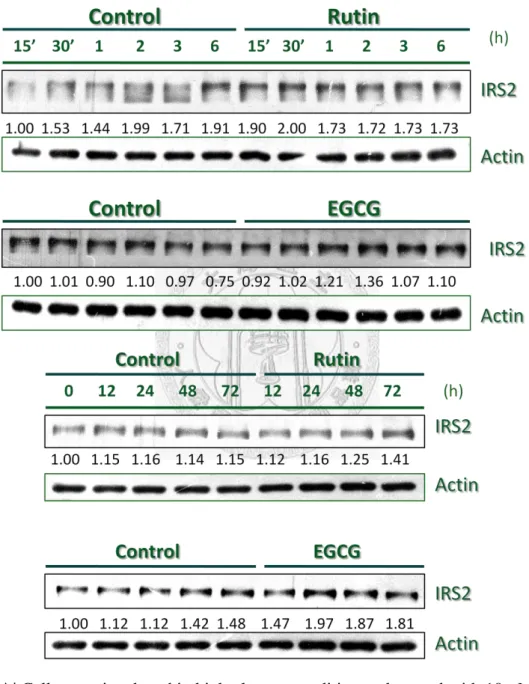
Effects of Rutin or EGCG on the stimulation of IRS2 signaling
The IRS2 signaling in pancreatic β cell regulates the cell survival and maintains the cell function. Pancreatic β cells were stimulated by 33 mM glucose and IRS2 gradually decreased the expression level, nevertheless, cells treated with Rutin or
EGCG reversed the effect of high glucose (Fig. 2A). Disruption of IRS2 in mice model has been found that the β cell failure through apoptosis mechanism, and
developed to similarities in diabetes, however, Rutin and EGCG both increase the IRS2 protein level against the high glucose incubation.
To determine the action of Rutin and EGCG on IRS2 signaling, the
20
immunoprecipitation was performed to detect Tyr phosphorylation level of IRS2.
When insulin recognized insulin receptor of cell membrane, this bound action would stimulate tyrosine autophosphorylation in the β subunit of receptor, which recruited
cellular substrate IRS proteins for Tyr phosphorylation. In Fig. 2B and Fig. 2C, cells were incubated in high glucose condition, cells treated with Rutin or EGCG both elevated the Tyr phosphorylation level after 2 h, furthermore, the effects were also
observed after 24 h high glucose incubation. The activation of IRS2 signal would initiate the downstream signal which involved in pancreatic β cell function and
proliferation. In Fig. 2D, EGCG rapidly enhanced the FoxO1 phosphorylation level compared with control, and combined Fig.2 C and Fig. 2D results which revealed that EGCG via IRS2 activation to increase the FoxO1 nuclear exclusion, however, phosphorylated form Akt was slightly increased from EGCG effect. In this result, the phosphorylation of FoxO1 was induced by high glucose but gradually diminished the phosphorylation after long term high glucose incubation, which could be found in control result; nevertheless, EGCG consistently maintained the phosphorylation rate to make FoxO1 to exclude from nucleus, and that would lead to increase the expression of downstream signal PDX-1.
In Fig. 2E, Rutin enhanced the phosphorylation level of FoxO1 between 6-24 h incubation, comparing with the effect of EGCG, which found that Rutin had less
21
power to increase and maintain the phosphorylation level of FoxO1. To combined Fig.
2B and Fig. 2E results, which revealed that Rutin could rapidly stimulate the IRS2 signal after 2 h incubation, and then enhance the phosphorylation level of FoxO1 in 6h incubation but the effect would diminish after 24 h incubation, even the phosphorylated form Akt was increased in 12 h incubation. In general, these two natural products both could enhance the IRS2/Akt/FoxO1 signaling under high glucose condition, even EGCG has much powerful effect than the stimulation of Rutin.
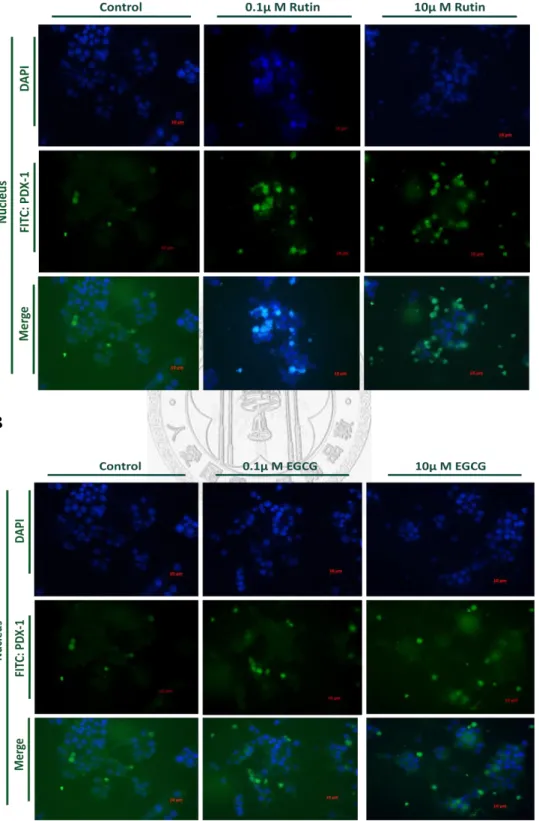
Rutin or EGCG Alters PDX-1 Nuclear Translocation and expression
Previous studies have been shown that pancreatic β cell under glucotoxicity condition would suppress the transcription factor PDX-1 activity, which was an essential element for insulin expression level and cell survival. Cells were incubated in high glucose condition, and treated with Rutin or EGCG. In Fig. 3A and Fig. 3B, Rutin and EGCG both effectively enhanced the nuclear translocation activity of PDX-1 after 24 h high glucose incubation. PDX-1 has a crucial role in the
determination and differentiation of pancreas, and its transactivation confers the expression of pancreatic β cell specific genes, included insulin, GLUT2 (glucose
transporter 2),GK (glucokinase), islet amyloid polypeptide, and another critical
22
pancreatic transcription factor Nkx 6.1 (22, 41). Rutin and EGCG could promote the PDX-1 nuclear translocation, which mentioned that Rutin and EGCG might be potential agents for glucose sensing and cell survival.
In Fig. 3C, cells were incubated in high glucose condition, and the effect of EGCG elevated the PDX-1 expression level under long term incubation; however, Rutin had no effect on the PDX-1 expression. To combined Fig. 3A and Fig. 3C results, which revealed that Rutin could promote PDX-1 translocation to nuclei, thus the expression level was not influenced. EGCG could enhance the ability of PDX-1 translocation against high glucose incubation; moreover, it also increased the expression level of PDX-1 (Fig. 3B and Fig. 3C).
Inhibition of Glucolipotoxicity via AMPK activation
AMPK act as a cellular energy sensor and dysregulation of its signaling network is considered as a key event in causing multiple abnormalities of metabolism (42).
Cells were incubated in high glucose condition, and treated with Rutin or EGCG. In Fig. 4A and Fig. 4B, the activity of AMPK was rapidly increased by the treatment of Rutin or EGCG via Thr phosphorylation, furthermore, these effects could prolong to 72 h. The reciprocal effect was observed in FAS (Fatty acid synthase) expression, in which the enhancement of AMPK activity by Rutin or EGCG might lead to suppress
23
the cellular lipogenesis.
The Rutin or EGCG effects of AMPK activity were mediated by upstream signal LKB1 (Fig. 4C and Fig. 4D), following the activated AMPK to downstream signals, which suppress the FAS expression level, in addition, it also negatively regulated the activity of ACC (Acetyl-CoA Carboxylase) through Ser phosphorylation. Pancreatic β Cells elevated the cellular lipogenesis activity under high glucose incubation;
however, Rutin and EGCG both effectively suppress the mechanism via AMPK
signaling network. Long term actions of glucose could disturb the lipid metabolism of β cells (43), nevertheless, Rutin and EGCG might be potential agents for reversing the
glucose induced dysregulation of lipogenesis.
The intracellular triglyceride content was correlated to insulin resistance in liver and muscle (44-46), thus the ectopic lipid accumulation in pancreatic β cell might be correlated to cellular dysfunction, even lead to apoptosis (34, 35, 47-49). The cellular lipid accumulation was performed by Oil red o staining, in which cells were incubated in long term high glucose condition for 5 days (Fig. 4E). The treatment of Rutin or EGCG both dramatically suppress the lipid droplets accumulation under high glucose incubation, however, the control cells increased about 43.3% of the cellular fat content to compared with the maintenance glucose condition control. In the Fig. 4 results, both of Rutin and EGCG could provoke the activity of AMPK in pancreatic β
24
cells under high glucose incubation, and suppress the lipogenesis, eventually effectively decrease the lipid accumulation to delay the toxicity of chronic metabolic stress.
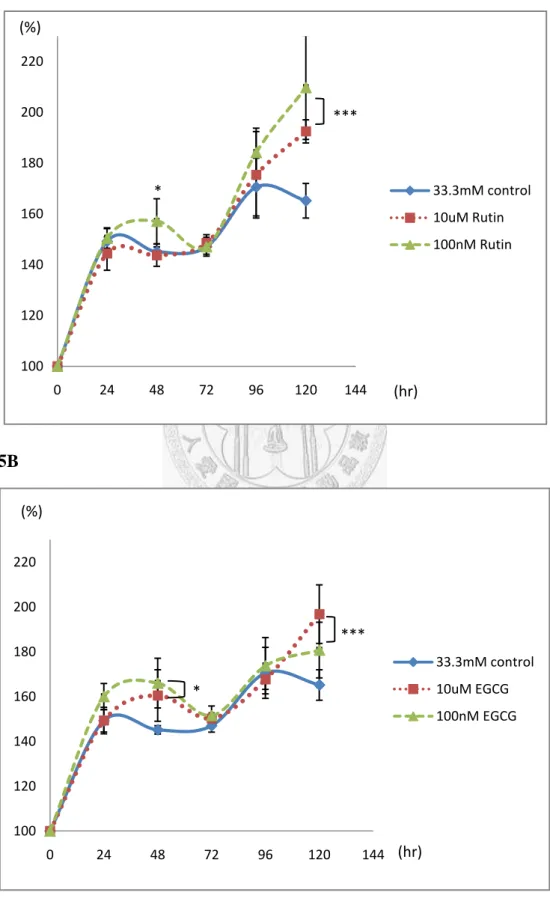
The maintenance of cell mass against chronic hyperglycemic incubation
Hyperglycemic condition could lead to the glucose-induced cell deterioration.
The progression of diabetes accompanied with decompensation of pancreatic β cell,
which revealed that the preservation of cell mass could be a crucial factor for sustaining the chronic metabolic stress, even coupling with insulin resistance (7, 50-52). Cells were incubated in high glucose condition, and treated with Rutin or EGCG. In Fig. 5A and Fig. 5B, the viable cell mass was determined by MTT assay, and the treatment of Rutin or EGCG both effectively increased the cell viability after 120 h high glucose incubation, however, cells without the treatment led to downregulate the viability. To investigate the mechanism of accelerating proliferation or preserving the cell mass, the Cyclin D1 and anti-apoptotic protein Bcl-2 expression levels were increased in the action of Rutin or EGCG (Fig. 5C and Fig. 5D);
furthermore, the pro-apoptotic protein Bax and the Cyclin-dependent kinase (Cdk) inhibitor p21 were successful suppressed by Rutin or EGCG. The effects of Rutin and EGCG in preservation of pancreatic β cell might retain the cell mass against chronic
25
exposure of hyperglycemic condition.
Long term actions of glucose on pancreatic β cell would lead the occurrence of decompensation through apoptosis mechanism. In Fig. 5E and Fig. 5F, cell cycle pattern was analyzed after long term high glucose incubation, in which the subG1 population gradually increased with the incubation time. The effects of Rutin and EGCG delayed the subG1 phase progression after long term incubation, in other words, these natural occurring compounds could delay the process of decompensation.
To determine the underlying mechanism, the western blotting results revealed that Rutin effectively suppressed the p21 expression after long term incubation, furthermore, upregulated Cyclin D1levels (Fig. 5H). In Fig. 5I, Rutin inhibited the
glucose induced maturation of SREBP1, which has been considered as a critical factor related to pancreatic β cell dysfunction (53, 54).
The effect of EGCG to protect pancreatic β cell from glucose induced toxicity, in which EGCG slightly increased the expression level of Cyclin D1, and inhibited the p21 accumulation. Moreover, EGCG consistently suppressed the ACC activity via Ser79 phosphorylation and the expression of SREBP1 (Fig. 5J). In conclusion, the effects of Rutin and EGCG protect pancreatic β cells from glucotoxicity, in which they suppress p21 accumulation and increase Cyclin D1 expression level to driving the cell cycling, and restrain the intracellular lipogenesis from long term induction of
26
glucose.
27
Discussion
The prevalence of diabetes is estimated to be 2.8% in 2000, and predicted that could be 4.4% in 2030. Based on the global survey of diabetes implicated the devastating difference of the prevalence of diabetes between urban and rural populations in developing countries, which revealed that the association of diet alteration, stress, obesity, and decreased physical capacity (55). To resolve this modern disease, several drugs have been developed to treat diabetes; however, the complicated disorder of metabolism has no complete cure yet (56-58). Tea is a common traditional beverage in ordinary life of Asian people, and previous studies have suggested the novel function in anti-diabetes and anti-obesity from its ingredients. EGCG, most abundant tea polyphenol in green tea, has been reported in attenuation of insulin signaling and insulin mimetic effect (38, 59, 60). Furthermore, buckwheat, a natural herb, has been demonstrated the ability to reduce blood glucose (39), and improvement of glucose homeostasis from its ingredient called Rutin.
Natural occurring compounds are easy to obtain and have lower price to chemical drugs, hence, the advantages of natural products might let them to be novel agents for the treatment of diabetes.
Patients with the metabolism syndrome manifest complex abnormalities of
28
glucose and lipid metabolism, and gradually develop to interference of multiple tissues (61-63). Excess energy intake and decreased exercise capacity would lead to dysregulate the metabolic harmony, which implicate to increase the rates of
obesity-associated disease, such as type 2 diabetes. The etiological factors of type 2 diabetes are typical insulin resistance, and accompanied with pancreatic β cell
deterioration, in which previous studies have demonstrated the crucial role of pancreatic β cell in the pathogenesis of diabetes to seize the debate of its contribution
(7, 10, 64).
Pancreatic β cell is a fundamental element for the development of diabetes, and
its unique secretory capacity of insulin manipulates individual energy balance.
Impairment of glucose-stimulated insulin secretion (GSIS) might cause from the
deadaptation of peripheral insulin resistance or chronic exposure of high glucose condition. Chronic overloaded metabolic demand crashes β cell in response to glucose,
which could lead to a progressive deterioration of cell function (65). Once glucose transporters facilitate the glucose entry to pancreatic β cell, and then the glycolysis
process responses to generate ATP considered as a factor in secretory mechanism.
Previous studies have shown that mitochondrial dysfunction linked to β cell
dysregulation (66, 67), hence in the Fig. 1 results, Rutin and EGCG has the effect on elevation of cellular ATP production under short-tem glucose stimulation or
29
incubation of long-term high glucose condition. The maintenance of ATP productive efficiency revealed the preservation of glucose sensing ability by the actions of Rutin and EGCG. Long term actions of glucose dramatically decrease the insulin secretory capacity to cause the insulin insufficiency; nevertheless, Rutin and EGCG both effectively increase the secretory activity in response to glucose stimulation, even after the long-term high glucose incubation.
The insulin secretory machinery in glucose-stimulated response, leading to
activate the IRS2 signaling through the autocrine mechanism. IRS2 signaling controls the function and proliferation of pancreatic β cell, thus disrupted the pathway could
lead to lose the insulin action and cell failure. Rutin and EGCG have the effect on enhancement of insulin secretion, furthermore, they increase the activity of IRS2 pathway (Fig.2). Previous studies have shown the role of IRS2 in diabetes (14, 16, 68), the effect of Rutin and EGCG to increase the IRS2 expression level and activate its signaling via Tyr phosphorylation, which might attenuate the high glucose-induced toxicity.
The major downstream signaling of IRS2 is Akt/FoxO1, in which Akt has been reported involved in pancreatic β cell proliferation and insulin secretion ability (18, 57,
69, 70). The effects of Rutin and EGCG on enhance the Akt activation are observed in 6-12 h, however, FoxO1 is rapidly phosphorylated before the activated Akt
30
enhancement. FoxO1 as a critical factor in insulin-regulated gene expression, such as inhibition of hepatic glucose production via suppress the expression of PEPCK, and repression the transcription factor PDX-1 expression in pancreatic β cell.
Phosphorylated FoxO1 is excluded from nucleus that has been suggested to involve in pancreatic β cell compensation to peripheral insulin resistance (21). The actions of
Rutin and EGCG have the improvement of Akt activity, and FoxO1 nuclear exclusion via Ser phosphorylation, however, FoxO1 is excessive phosphorylated earlier then the
effect on Akt, retaining other signalings involved in this mechanism, such as MEK/ERK signaling.
The treatment of Rutin or EGCG could increase and preserve the glucose sensing ability of pancreatic β cell, then enhance the insulin secretory machinery in response
to glucose stimulation. The efficient secreted insulin has much effect on activation of IRS2 signaling, following to deliver it to the downstream signals which maintain the cell function and viability. PDX-1 is a vital transcription factor in β cells which regulates the expression levels of insulin, GLUT2, GK, and its function is affected by the cellular localization (23, 71). Glucotoxicity or lipotoxicity would lead to reduce the expression of PDX-1 or block the nuclear translocation, however, the treatment of Rutin or EGCG both promoted the nuclear translocation under high glucose incubation, which could lead to activate its regulation of responsive genes expression.
31
The effect on PDX-1 from Rutin and EGCG is diverse in its expression level. The difference between Rutin and EGCG to alter the expression, which could correlate with the effect on IRS2 signaling, in which EGCG could stimulate the IRS2/Akt/FoxO1 signaling better than Rutin did.
The synergism of disorder of glucose and lipid metabolism would accelerate the deterioration of pancreatic β cell, which could result from long term action of glucose.
Accumulation of lipids in pancreatic β cell have been shown to precede the onset of
dysfunction, and even associated with insulin resistance in other tissues, such as liver and muscle (72, 73). Profound effects of declined AMPK activity not only involved in fuel sensing ability, but have been considered to link with insulin resistance and upregulate the cellular lipogenesis. Several developed anti-diabetic drugs, such as metformin and TZDs (Thiazolidinediones), have been proved to activate AMPK and effectively treat insulin resistance and disorders associated with the metabolic syndrome.
Rutin and EGCG act as activators of AMPK and rapidly enhance the activity via Thr phosphorylation against high glucose incubation, which presents a reciprocal effect on FAS expression. Chronic elevated glucose could initiate the activation of cellular lipogenesis, in which lipogenic genes expression would be upregulation. The effects on AMPK activity of Rutin and EGCG suppressed the FAS expression, and
32
depressed the activity of ACC via Ser phosphorylation; moreover, they interrupted the SREBP1 maturation or generation in causing the inhibition of lipid biogenesis (Fig.4).
AMPK activation protects cells against insulin resistance and cellular dysfunction in regulation of energy homeostasis; therefore Rutin and EGCG play crucial roles in
maintaining the manners of glucose and lipid metabolism caused from chronic exposure of high glucose condition in pancreatic β cell.
SREBP1, a lipogenic transcription factor, has been shown as a instrumental factor in the development of pancreatic β cell dysfunction (53). Overexpression of SREBP1 would lead to β cell dysfunction, which increases the lipogenic gene
expression, furthermore, it targets multiple genes implicated in cell growth and survival. After adaptation of insulin resistance, pancreatic β cell goes through the
decompensation process and into the onset of progressive deterioration of cell function, and massive cell failure in causing apoptosis. Induction of SREBP1 would cause defects in genes implicated in glucose metabolism and cellular function, such as
GLUT2, glucokinase, PDX-1, IRS2, p21, and BAX. IRS2 and PDX-1 as important factors to maintain β cells growth and survival, which could be suppress the
expression via SREBP1 overexpression, however, p21 and BAX would be elevated in provoking cell cycle arrest and facilitating apoptosis to occur (74).
The impact of Rutin and EGCG on pancreatic β cell would suppress the
33
elevating expression of p21 and Bax with the high glucose incubation. To meet the chronic overloaded metabolic demand, the cell mass is essential for preservation of insulin sufficiency in response of glucose action. Cyclin D1 and Bcl-2 are regard as critical factors in cell proliferation and expansion (75-78), therefore to effective maintain the cell population could be argument against decompensation (Fig.5). The effects of Rutin and EGCG affected the cell viability via the expression levels of Cyclin D1 and Bcl-2 under high glucose incubation, which implicated that Rutin and EGCG could prolong the adaptation to delay the onset of progression.
Type 2 diabetes is caused from multiple abnormalities of metabolism, in order to find out the cure candidate, scientists have investigated the underlying molecular mechanism for decades. Natural herbs contain countless chemical compounds that have been used in traditional Chinese medicine provided abundant sources for
validation (79, 80). The tea polyphenol EGCG and the citrus flavonoid glycoside Rutin in this study have been observed the effects on pancreatic β cell whether they
could be candidates for improving cell function and viability against chronic
metabolic demand. Broad effects of Rutin and EGCG are elucidated the roles in manipulating the glucose and lipid metabolism on β cells, which effectively keep the
order of energy homeostasis under long term actions of glucose. The current study highlights the novel function of two natural occurring compounds called Rutin and
34
EGCG might be agents for protection of pancreatic β cell against glucotoxicity.
35
References
1. Stumvoll M, Goldstein BJ, van Haeften TW 2005 Type 2 diabetes: principles of pathogenesis and therapy. Lancet 365:1333-1346
2. Murphy R, Ellard S, Hattersley AT 2008 Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab 4:200-213
3. Shih DQ, Stoffel M 2002 Molecular etiologies of MODY and other early-onset forms of diabetes. Curr Diab Rep 2:125-134
4. Zimmet P, Alberti KG, Shaw J 2001 Global and societal implications of the diabetes epidemic. Nature 414:782-787
5. Lazar MA 2005 How obesity causes diabetes: not a tall tale. Science 307:373-375
6. Kahn SE, Hull RL, Utzschneider KM 2006 Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444:840-846
7. Prentki M, Nolan CJ 2006 Islet beta cell failure in type 2 diabetes. J Clin Invest 116:1802-1812
8. Rhodes CJ 2005 Type 2 diabetes-a matter of beta-cell life and death? Science 307:380-384
9. Lingohr MK, Buettner R, Rhodes CJ 2002 Pancreatic beta-cell growth and
36
survival--a role in obesity-linked type 2 diabetes? Trends Mol Med 8:375-384
10. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC 2003 Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes.
Diabetes 52:102-110
11. Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, Sharma A 2001 Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 50 Suppl 1:S154-159
12. Fujimoto S, Nabe K, Takehiro M, Shimodahira M, Kajikawa M, Takeda T, Mukai E, Inagaki N, Seino Y 2007 Impaired metabolism-secretion coupling in pancreatic beta-cells: role of determinants of mitochondrial ATP production. Diabetes Res Clin Pract 77 Suppl 1:S2-10
13. MacDonald PE, Joseph JW, Rorsman P 2005 Glucose-sensing mechanisms in pancreatic beta-cells. Philos Trans R Soc Lond B Biol Sci 360:2211-2225
14. Brady MJ 2004 IRS2 takes center stage in the development of type 2 diabetes. J Clin Invest 114:886-888
15. White MF 2003 Insulin signaling in health and disease. Science 302:1710-1711 16. Hennige AM, Burks DJ, Ozcan U, Kulkarni RN, Ye J, Park S, Schubert M, Fisher TL, Dow MA, Leshan R, Zakaria M, Mossa-Basha M, White MF 2003 Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes.
37
J Clin Invest 112:1521-1532
17. Kim SJ, Winter K, Nian C, Tsuneoka M, Koda Y, McIntosh CH 2005 Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. J Biol Chem 280:22297-22307
18. Bernal-Mizrachi E, Fatrai S, Johnson JD, Ohsugi M, Otani K, Han Z, Polonsky KS, Permutt MA 2004 Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest 114:928-936
19. Martinez SC, Cras-Meneur C, Bernal-Mizrachi E, Permutt MA 2006 Glucose regulates Foxo1 through insulin receptor signaling in the pancreatic islet beta-cell. Diabetes 55:1581-1591
20. Nakae J, Biggs WH, 3rd, Kitamura T, Cavenee WK, Wright CV, Arden KC, Accili D 2002 Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 32:245-253
21. Okamoto H, Hribal ML, Lin HV, Bennett WR, Ward A, Accili D 2006 Role of the forkhead protein FoxO1 in beta cell compensation to insulin resistance. J Clin
38
Invest 116:775-782
22. Wang H, Maechler P, Ritz-Laser B, Hagenfeldt KA, Ishihara H, Philippe J, Wollheim CB 2001 Pdx1 level defines pancreatic gene expression pattern and cell lineage differentiation. J Biol Chem 276:25279-25286
23. Brissova M, Shiota M, Nicholson WE, Gannon M, Knobel SM, Piston DW, Wright CV, Powers AC 2002 Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J Biol Chem 277:11225-11232
24. Kushner JA, Ye J, Schubert M, Burks DJ, Dow MA, Flint CL, Dutta S, Wright CV, Montminy MR, White MF 2002 Pdx1 restores beta cell function in Irs2 knockout mice. J Clin Invest 109:1193-1201
25. Johnson JD, Ahmed NT, Luciani DS, Han Z, Tran H, Fujita J, Misler S, Edlund H, Polonsky KS 2003 Increased islet apoptosis in Pdx1+/- mice. J Clin Invest 111:1147-1160
26. Poitout V, Hagman D, Stein R, Artner I, Robertson RP, Harmon JS 2006 Regulation of the insulin gene by glucose and fatty acids. J Nutr 136:873-876
27. Ruderman N, Prentki M 2004 AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov 3:340-351
28. Prentki M, Joly E, El-Assaad W, Roduit R 2002 Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta-cell adaptation and failure in the
39
etiology of diabetes. Diabetes 51 Suppl 3:S405-413
29. Long YC, Zierath JR 2006 AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest 116:1776-1783
30. Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D 2003 LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13:2004-2008
31. Ferre P, Azzout-Marniche D, Foufelle F 2003 AMP-activated protein kinase and hepatic genes involved in glucose metabolism. Biochem Soc Trans 31:220-223 32. Foretz M, Carling D, Guichard C, Ferre P, Foufelle F 1998 AMP-activated protein kinase inhibits the glucose-activated expression of fatty acid synthase gene in rat hepatocytes. J Biol Chem 273:14767-14771
33. Eto K, Yamashita T, Matsui J, Terauchi Y, Noda M, Kadowaki T 2002 Genetic manipulations of fatty acid metabolism in beta-cells are associated with dysregulated insulin secretion. Diabetes 51 Suppl 3:S414-420
34. Brunham LR, Kruit JK, Verchere CB, Hayden MR 2008 Cholesterol in islet dysfunction and type 2 diabetes. J Clin Invest 118:403-408
35. van Herpen NA, Schrauwen-Hinderling VB 2008 Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav 94:231-241
36. Kao YH, Chang HH, Lee MJ, Chen CL 2006 Tea, obesity, and diabetes. Mol
40
Nutr Food Res 50:188-210
37. Lin CL, Huang HC, Lin JK 2007 Theaflavins attenuate hepatic lipid accumulation through activating AMPK in human HepG2 cells. J Lipid Res 48:2334-2343
38. Lin CL, Lin JK 2008 Epigallocatechin gallate (EGCG) attenuates high glucose-induced insulin signaling blockade in human hepG2 hepatoma cells. Mol Nutr Food Res
39. Kawa JM, Taylor CG, Przybylski R 2003 Buckwheat concentrate reduces serum glucose in streptozotocin-diabetic rats. J Agric Food Chem 51:7287-7291 40. Stanley Mainzen Prince P, Kamalakkannan N 2006 Rutin improves glucose homeostasis in streptozotocin diabetic tissues by altering glycolytic and gluconeogenic enzymes. J Biochem Mol Toxicol 20:96-102
41. Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV 1996 PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122:983-995
42. Ruderman NB, Saha AK, Vavvas D, Witters LA 1999 Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol 276:E1-E18
43. Brun T, Roche E, Kim KH, Prentki M 1993 Glucose regulates acetyl-CoA carboxylase gene expression in a pancreatic beta-cell line (INS-1). J Biol Chem
41
268:18905-18911
44. Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI 1999 Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 42:113-116
45. Jacob S, Machann J, Rett K, Brechtel K, Volk A, Renn W, Maerker E, Matthaei S, Schick F, Claussen CD, Haring HU 1999 Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes 48:1113-1119
46. Ryysy L, Hakkinen AM, Goto T, Vehkavaara S, Westerbacka J, Halavaara J, Yki-Jarvinen H 2000 Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes 49:749-758 47. Prentki M, Corkey BE 1996 Are the beta-cell signaling molecules malonyl-CoA and cystolic long-chain acyl-CoA implicated in multiple tissue defects of obesity and NIDDM? Diabetes 45:273-283
48. Mulder H, Lu D, Finley Jt, An J, Cohen J, Antinozzi PA, McGarry JD, Newgard CB 2001 Overexpression of a modified human malonyl-CoA decarboxylase blocks the glucose-induced increase in malonyl-CoA level but has no impact on
42
insulin secretion in INS-1-derived (832/13) beta-cells. J Biol Chem 276:6479-6484 49. El-Assaad W, Buteau J, Peyot ML, Nolan C, Roduit R, Hardy S, Joly E, Dbaibo G, Rosenberg L, Prentki M 2003 Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 144:4154-4163 50. Pick A, Clark J, Kubstrup C, Levisetti M, Pugh W, Bonner-Weir S, Polonsky KS 1998 Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 47:358-364
51. Unger RH 2002 Lipotoxic diseases. Annu Rev Med 53:319-336
52. Buteau J, Accili D 2007 Regulation of pancreatic beta-cell function by the forkhead protein FoxO1. Diabetes Obes Metab 9 Suppl 2:140-146
53. Wang H, Maechler P, Antinozzi PA, Herrero L, Hagenfeldt-Johansson KA, Bjorklund A, Wollheim CB 2003 The transcription factor SREBP-1c is instrumental in the development of beta-cell dysfunction. J Biol Chem 278:16622-16629
54. Shimano H, Amemiya-Kudo M, Takahashi A, Kato T, Ishikawa M, Yamada N 2007 Sterol regulatory element-binding protein-1c and pancreatic beta-cell dysfunction. Diabetes Obes Metab 9 Suppl 2:133-139
55. Wild S, Roglic G, Green A, Sicree R, King H 2004 Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care
43
27:1047-1053
56. Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM 2000 Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med 6:998-1003
57. Park S, Dong X, Fisher TL, Dunn S, Omer AK, Weir G, White MF 2006 Exendin-4 uses Irs2 signaling to mediate pancreatic beta cell growth and function. J Biol Chem 281:1159-1168
58. Yki-Jarvinen H 2004 Thiazolidinediones. N Engl J Med 351:1106-1118
59. Anton S, Melville L, Rena G 2007 Epigallocatechin gallate (EGCG) mimics insulin action on the transcription factor FOXO1a and elicits cellular responses in the presence and absence of insulin. Cell Signal 19:378-383
60. Hale PJ, Horrocks PM, Wright AD, Fitzgerald MG, Nattrass M, Bailey CJ 1989 Xiaoke tea, a Chinese herbal treatment for diabetes mellitus. Diabet Med 6:675-676
61. Reaven GM 1988 Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37:1595-1607
62. Reaven G 2002 Metabolic syndrome: pathophysiology and implications for management of cardiovascular disease. Circulation 106:286-288
63. Ruderman N, Chisholm D, Pi-Sunyer X, Schneider S 1998 The metabolically
44
obese, normal-weight individual revisited. Diabetes 47:699-713
64. Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, Reinecke M 2005 Mechanisms of beta-cell death in type 2 diabetes. Diabetes 54 Suppl 2:S108-113
65. Grill V, Bjorklund A 2000 Dysfunctional insulin secretion in type 2 diabetes:
role of metabolic abnormalities. Cell Mol Life Sci 57:429-440
66. Lowell BB, Shulman GI 2005 Mitochondrial dysfunction and type 2 diabetes.
Science 307:384-387
67. Maechler P, Wollheim CB 2001 Mitochondrial function in normal and diabetic beta-cells. Nature 414:807-812
68. White MF 2006 Regulating insulin signaling and beta-cell function through IRS proteins. Can J Physiol Pharmacol 84:725-737
69. Dickson LM, Rhodes CJ 2004 Pancreatic beta-cell growth and survival in the onset of type 2 diabetes: a role for protein kinase B in the Akt? Am J Physiol Endocrinol Metab 287:E192-198
70. Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC 1999 Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A 96:7421-7426 71. Marshak S, Benshushan E, Shoshkes M, Havin L, Cerasi E, Melloul D 2000
45
Functional conservation of regulatory elements in the pdx-1 gene: PDX-1 and hepatocyte nuclear factor 3beta transcription factors mediate beta-cell-specific expression. Mol Cell Biol 20:7583-7590
72. McGarry JD 2002 Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 51:7-18
73. Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH 1994 Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc Natl Acad Sci U S A 91:10878-10882
74. Ide T, Shimano H, Yahagi N, Matsuzaka T, Nakakuki M, Yamamoto T, Nakagawa Y, Takahashi A, Suzuki H, Sone H, Toyoshima H, Fukamizu A, Yamada N 2004 SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat Cell Biol 6:351-357
75. Rabinovitch A, Suarez-Pinzon W, Strynadka K, Ju Q, Edelstein D, Brownlee M, Korbutt GS, Rajotte RV 1999 Transfection of human pancreatic islets with an anti-apoptotic gene (bcl-2) protects beta-cells from cytokine-induced destruction.
Diabetes 48:1223-1229
76. Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, Patane G, Boggi U, Piro S, Anello M, Bergamini E, Mosca F, Di Mario U, Del Prato S,
46
Marchetti P 2002 Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated.
Diabetes 51:1437-1442
77. Zhang X, Gaspard JP, Mizukami Y, Li J, Graeme-Cook F, Chung DC 2005 Overexpression of cyclin D1 in pancreatic beta-cells in vivo results in islet hyperplasia without hypoglycemia. Diabetes 54:712-719
78. Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF 2005 Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol 25:3752-3762
79. Wolfram S, Wang Y, Thielecke F 2006 Anti-obesity effects of green tea: from bedside to bench. Mol Nutr Food Res 50:176-187
80. Zheng G, Sayama K, Okubo T, Juneja LR, Oguni I 2004 Anti-obesity effects of three major components of green tea, catechins, caffeine and theanine, in mice. In Vivo 18:55-62
47
Figures
Figure 1. Enhancement of Glucose Stimulated Insulin Secretion and preservation of glucose sensing ability during the high glucose incubation in the action of Rutin or EGCG
Fig. 1A
Fig. 1B
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
Control 0.1 μM Rutin 10 μM Rutin 0.1 μM EGCG 10 μM EGCG ATP(μM)
***
** *** *
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
Control 0.1 μM Rutin 10 μM Rutin 0.1 μM EGCG 10 μM EGCG ATP(μM)
** ** **