§ 3-2 CO 和 H 2 O 分子的吸附結構和吸附能探討
為了探討各個中間產物吸附於表面時的吸附能大小及結構變 化,我們首先計算了這些中間產物於單一氣態分子時的結構與能量,
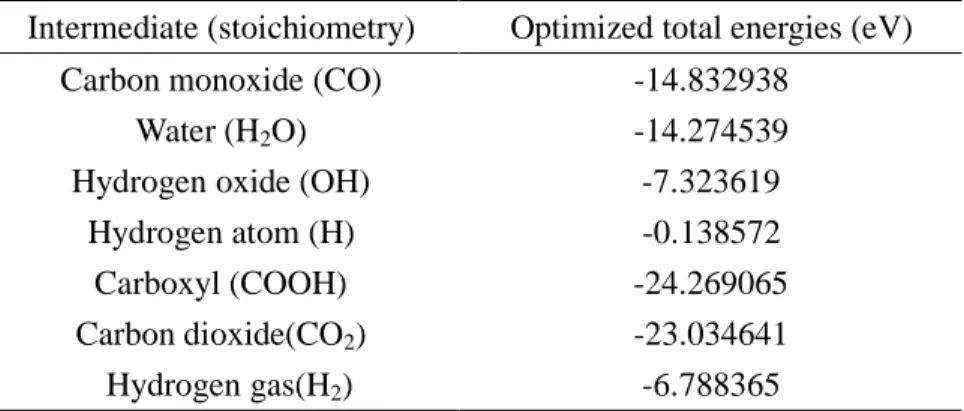
經過幾何優選的結果呈現在圖 4 中(包括 species 1-6)。計算後的系統 能量記錄於表 4 當中。VASP 計算程式一般使用於計算週期性的大分 子系統,將其應用在模擬單一氣態分子時,我們的作法是先將單位晶 格擴大到 25×25×25 Å 3 ,以避免分子間的作用力;其他的收斂條件則相 同。將我們的結果比較文獻上的實驗值或計算值 46-48 發現,無論在鍵 長或是鍵角都能夠非常的吻合,與文獻的差值皆少於±0.02 Å 和±1.0 degree。因此在隔離的系統中(isolate system),VASP 程式也能夠得到 相當可信的模擬結果。
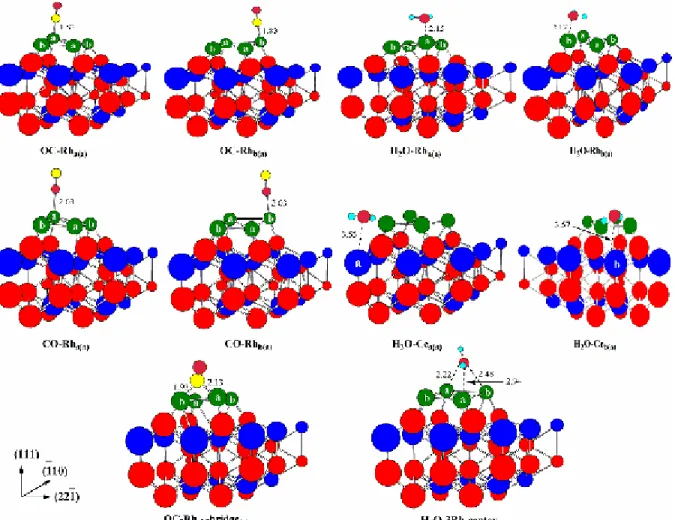
進一步,我們探討一氧化碳和水分子吸附於 4Rh/CeO 2 (Rh-cluster) 表面的穩定結構,經過幾何優選計算後的結果描繪於圖 5 中;檢視 4Rh/CeO 2 (Rh-cluster)表面,我們可以把表面的 Rh 原子視其所在環境 的對稱性分為兩種,分別標示為 Rh a 和 Rh b 。各個吸附位置其鍵長、
鍵角和吸附能表列於表 5 中。對於一氧化碳的吸附而言,我們計算了
各種可能的吸附結構,包括:一氧化碳分子以碳端或氧端接 Rh 原子
(Rh 或 Rh ),或是一氧化碳分子橋接在 Rh 和 Rh 原子間,這些結構
19
C
C C
O
O
O
O O O O
H H
H
H H
1.14 H
0.97 0.97
104.7
0.99
126.1 1.19
1.35 108.1
0.98 1.17 1.17 0.75
(1).CO (2).H 2 O (3).OH
(4).COOH (5).CO 2 (6).H 2
圖 4 氣態分子經 VASP 計算幾何優選結構圖。(1) carbon monoxide, (2) water, (3) hydrogen oxide, (4) carboxyl, (5) carbon dioxide, (6)
hydrogen gas。鍵長單位為 Å。
表 4 水氣轉移反應中反應物,產物和中間產物的氣態分子絕對能量計算值
a
。 各分子的結構紀錄於圖 4 中。Intermediate (stoichiometry) Optimized total energies (eV) Carbon monoxide (CO) -14.832938
Water (H
2
O) -14.274539 Hydrogen oxide (OH) -7.323619Hydrogen atom (H) -0.138572 Carboxyl (COOH) -24.269065 Carbon dioxide(CO
2
) -23.034641 Hydrogen gas(H2
) -6.788365a.
All calculations were performed with the VASP package.
21
圖 5 CO 和 H
2
O 分子吸附於 4Rh/CeO2
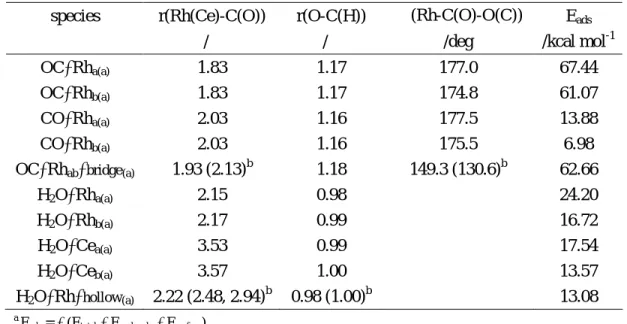
(Rh-cluster)表面的可能結構經由 VASP 計算後,幾何優選結構示意圖。圖中藍色表示 Ce,綠色 表示 Rh,紅色表示 O,黃色表示為 C,淡藍色為 H。鍵長的單位為 Å。表 5. H
2
O 分子和 CO 分子在 4Rh/CeO2
(Rh–cluster)表面的鍵長鍵角和吸附能 Eads
(kcal/mol)a
的計算值。經由計算後的結構呈現於圖 5。a
E
ads= – (E
total– E
molecule– E
surface)
b
對於另一個不同位相的鍵長和鍵角計算值
species r(Rh(Ce)-C(O)) /Å
r(O-C(H)) /Å
∠(Rh-C(O)-O(C)) /deg
E
ads
/kcal mol-1
OC–Rh
a(a)
1.83 1.17 177.0 67.44OC–Rh
b(a)
1.83 1.17 174.8 61.07CO–Rh
a(a)
2.03 1.16 177.5 13.88CO–Rh
b(a)
2.03 1.16 175.5 6.98OC–Rh
ab
–bridge (a)
1.93 (2.13)b
1.18 149.3 (130.6)b
62.66H
2
O–Rha(a)
2.15 0.98 24.20H
2
O–Rhb(a)
2.17 0.99 16.72H
2
O–Cea(a)
3.53 0.99 17.54H
2
O–Ceb(a)
3.57 1.00 13.57H
2
O–Rh–hollow (a)
2.22 (2.48, 2.94)b
0.98 (1.00)b
13.0823
分別標示為 OC-Rh a(a) ,OC-Rh b(a) ,CO-Rh a(a) ,CO-Rh a(a) 和 OC-Rh ab -
bridge (a) 。對於其他可能的吸附位置,如 : 一氧化碳吸附於 Ce 原子的
正上方或吸附於表面 O 原子的正上方…等等,雖然這些結構的結果
沒有呈現於本篇論文中,但我們還是做了一系列的計算探討;計算結
果顯示,一氧化碳以這些吸附模式附著於表面時,僅為物理吸附;吸
附能與吸附於 Rh 原子上比較,大約少了 30~55 kcal/mol,顯示當一
氧化碳與表面 Ce 和 O 原子接觸時並不能穩定吸附。在反應需要高溫
的條件下,容易因吸附物本身具有高動能而無法成功吸附於表面。這
樣的結果和 Yang 等作者 31 的發現相同。根據其利用理論計算模擬的
結果,不論一氧化碳利用碳端或是氧端與純的 CeO 2 (111)表面(沒有
Rh 金屬)作用,吸附能皆小於 3.92 kcal/mol。而 Gorte 等作者 49 也在
文獻中提到,在 Rh/CeO 2 系統中,一氧化碳分子較容易吸附於 Rh 原
子上。觀察五種列於表 5 的可能吸附結構和吸附能可知,一氧化碳能
與表面 Rh 金屬形成強烈的鍵結,且鍵結形式為利用碳端和 Rh 原子
相接。這種一氧化碳以其碳端而非氧端吸附於表面的性質,可以
coorperative effect 解釋 : 當一氧化碳分子以其碳端提供(donate)電子
與表面相接時,氧端上的電子易轉移至碳端而產生一同向之協同作
用,稱為 coorperative effect,利用氧端與表面作用時則無此效果。因
此,一氧化碳的碳端有較強的提供(donate)電子能力,使得吸附能大
於以氧端鍵結的情形。而一氧化碳與過渡金屬元素的鍵結機制則可以 用 Blyholder model 50 來說明 (Scheme 2) : 當一氧化碳以碳端與過渡 金屬元素鍵結時,一氧化碳利用 5σ 軌域提供電子給予表面 Rh 原子,
表面的 Rh 原子則利用其 d 軌域電子反向提供(back donation)電子到一 氧化碳分子的 2π*軌域,因此可形成穩定的 OC-Rh 鍵結。此外,我 們可以由 CO 分子的振動頻率來證實 Rh 原子 d 軌域的反向提供性 質。由表 6 中的數據可以發現,當 CO 分子吸附於表面 Rh 原子上時,
其振動頻率會有紅位移(red shift)的現象產生,這是由於 Rh 電子反向 提供於 CO 分子的 antibonding(2π*)中,造成 CO 鍵長的變長而使得振 動頻率降低。另外值得一提的是,我們所計算的頻率數值能夠和實驗 值相當吻合。
水分子與 4Rh/CeO 2 (Rh-cluster)表面的吸附結構,計算結果呈現 於圖 5 中,吸附能大小則表列於表 5 下半部。經由計算的結果發現,
C O
e - C
O
e -
CO to Rh metal σ donation
metal Rh to CO π* backdonation
Rh Rh
25
表 6 CO 分子在氣態和在 4Rh/CeO
2
(111)表面上的振動頻率實驗值和理論值species frequency
CO
(g)
2126.71 cm-1
CO-Rh
a(a)
1989.64 cm-1
CO-Rh
b(a)
1983.39 cm-1
exp (gas phase) 2143 cm
-1
exp