國立交通大學
生物資訊研究所
碩士論文
電腦化預測宿主微小核醣核酸在病毒基因組的靶位
點以及預測病毒微小核醣核酸 (初稿)
Computational prediction of host microRNA targets against virus
genome and identification of virus microRNAs (Draft)
研 究 生:林立人
指導教授:黃憲達 博士
電腦化預測宿主微小核醣核酸在病毒基因組的靶位點以及預測病毒微小核 醣核酸 (初稿)
Computational prediction of host microRNA targets against virus genome and identification of virus microRNAs (Draft)
研究生 : 林立人 Student : Li-Zen Lin
指導教授 : 黃憲達 博士 Advisor : Dr. Hsien-Da Huang
國立交通大學 生物資訊研究所
碩士論文
A Thesis
Submitted to Institute of Bioinformatics College of Biological Science and Technology
National Chiao Tung University In partial Fulfillment of the Requirements
For the Degree of Master
In Bioinformatics
June 2006
Hsinchu, Taiwan, Republic of China
電腦化預測宿主微小核醣核酸在病毒基因組的靶位點以及預測病
毒微小核醣核酸 (初稿)
學生 : 林立人 指導教授 : 黃憲達 博士 國立交通大學生物資訊所碩士班 中文摘要 近年來發現微小核醣核酸(microRNA)於哺乳動物、植物與昆蟲中,藉由 調控後轉錄作用產物,在發育、致癌及細胞凋亡上扮演重要的角色。值得 注意的是,微小核醣核酸不僅在真核生物中存在,病毒亦能產生微小核醣 核酸,且病毒感染宿主後,宿主細胞中的微小核醣核酸也具有調控病毒的 功能。因此,我們建置 ViTa 和 VirMiR 兩個資料庫來收集病毒產生的微小 核醣核酸,及探討病毒與宿主微小核醣核酸間的關係。資料的收集上,來 自病毒和四各物種中,已知的微小核醣核酸分別從 miRBase 和 miRNAMap 得來。病毒的資訊則收集自四各資料庫:NCBI、ICTVdB、VBRC 和 VirGen。 已知的宿主微小核醣核酸靶位點,則藉由瀏覽論文而得。另外,病毒上微 小核醣核酸的預測主要專注於染色體上的保存區段。預測宿主小核醣核酸 在病毒上的靶位點,則利用兩個軟體:miRanda 和 TargetScan 來提升預測準 確度。另外也提供相關的註解、被病毒感染的組織以及微小核醣核酸的組 織專一性等。最後,所有的資訊均整合以研究微小核醣核酸與病毒間的相 關性。 我們亦為使用者提供某些病毒間亞種的比較,像是流感病毒與肝癌病 毒,及同一種類中不同病毒株間的保存區段。為便利資料的取得及協助研 究,網站同時提供文字與圖形介面。目前這兩個資料庫 ViTa 和 VirMiR 已 分別建置在http://vita.mbc.nctu.edu.tw/ 和 http://virmir.mbc.nctu.edu.tw/。Computational prediction of host microRNA targets against virus genome and identification of virus microRNAs (Draft)
Student: Li-Zen Lin Advisor : Dr. Hsien-Da Huang
Institute of Bioinformatics National Chiao Tung University
Abstract
Recent work has indicated that microRNAs (miRNAs) play important roles on development, oncogenesis and apoptosis by binding to mRNAs to regulate the post-transcriptional level of gene expression in mammals, plants and insects. Notably, miRNAs also can be produced from virus, and the expression of virus genes are potentially regulated by host miRNAs after infecting host. In this thesis, two databases, VirMiR and ViTa, are developed to compile the virus miRNAs and the regulatory relationships between host miRNAs and virus genes, respectively. Known virus microRNAs in four species, such as humans, mouse, rat and chicken, were obtained from miRBase and miRNAMap. Virus information was collected from NCBI, ICTVdB, VBRC and VirGen. Experimentally validated host miRNA targets on viruses are identified via a literature survey. Then, miRanda and TargetScan are applied to identify the miRNA targets within virus genomes. The virus miRNAs were detected based on comparative sequence analysis. Finally, virus annotations, virus infected tissues from literatures and tissue-specificity of miRNAs from MIT are integrated, and the possible relationship between miRNAs and viruses are identified.
This work also provides comparisons between subtypes in some common viruses, such as influenza viruses, liver viruses, and conserved regions between different strains of the same virus species for users. Both textual and graphical web interfaces are provided to facilitate data retrieval from the two databases. The two databases can be available on the Internet at http://vita.mbc.nctu.edu.tw/ and http://virmir.mbc.nctu.edu.tw/.
Table of Contents
中文摘要... i Abstract ...ii Table of Contents...iii List of Tables ... v List of Figures ... vi Chapter 1 Introduction ... 1 1.1 Background... 11.1.1 miRNA Biogenesis and Function ... 2
1.1.2 Virus Infection and Propagation ... 3
1.1.3 The Relationship Between miRNAs and Viruses... 4
1.2 Motivation ... 6
1.3 Goal ... 7
Chapter 2 Related Works ... 8
2.1 Prediction of miRNAs ... 8
2.2 Prediction of miRNAs Targets... 11
Chapter 3 Materials and Methods... 13
3.1 Materials ... 13
3.1.1 The miRNAs Databases... 13
3.1.2 Virus Databases ... 14
3.1.3 Tools for Prediction ... 16
3.2 Methods ... 18
3.2.1 Overview ... 18
3.2.2 Prediction of Host miRNA Targets... 20
3.2.3 Prediction of Viral miRNAs ... 24
Chapter 4 Implementation ... 29
4.1 Data Preprocessing and Storage of Knowledgebase ... 29
4.2 Web Interface...30
Chapter 5 Results and Case Study... 36
5.1 Statistics of Results... 36
5.1.1 Result of Virus miRNAs Prediction ... 36
5.1.2 Result of Host miRNA Targets Prediction... 36
5.2 Case studies: Host miRNA Targeting to Viruses ... 43
5.2.2 Primate Foamy Virus Type 1 (PFV-1) versus Human miR-32 ... 44
5.2.3 Human Immunodeficiency Virus-1 (HIV) versus Human miRNAs ... 45
Chapter 6 Discussion and Conclusions ... 47
6.1 Discussion... 47
6.2 Future Works ... 48
6.3 Conclusions ... 48
List of Tables
Table 2.1 The miRNAs with reported functions [19]. ...10
Table 3.1 The statistic of microRNAs...14
Table 3.2 The list of the integrated external data sources...16
Table 3.3 The list of the integrated annotated tools in this work. ...17
Table 5.1 Statistics numbers of virus in the virus data warehouse. ...37
Table 5.2 The result of known virus targets predicted by miRanda and TargetScan...39
Table 5.3 Experimental miRNA targets on virus. ...39
Table 5.4 Statistics for predictive miRNA targets by miRanda. ...40
Table 5.5 Statistics for predictive miRNA targets by TargetScan...41
Table 5.6 Numbers of microRNAs versus liver viruses (wildtype)...41
List of Figures
Figure 1.1 The biogenesis and function of miRNAs [10]...3
Figure 1.2 The relationship between microRNAs and virus [17]...6
Figure 1.3 The location of known virus miRNAs on virus genome...7
Figure 2.1 Five microRNAs are produced from Rhesus Lymphocryptovirus...9
Figure 2.2 Location of known miRNAs produced from four viruses...10
Figure 2.3 Predicted target sites of human microRNAs on HUV-1 genome [16]. ...12
Figure 3.1 Putative miRNAs predicted by miRNAMap...14
Figure 3.2 The web interface of ICTVdB...15
Figure 3.3 The system flow...19
Figure 3.4 The flowchart of prediction host miRNA targets...20
Figure 3.5 The flowchart of prediction of miRNAs on viruses...25
Figure 3.6 Finding conserved secondary structures by RNAz. ...26
Figure 3.7 Prediction of mature miRNAs by mmiRNA. ...28
Figure 4.1 Browse (left) and search (right) of ViTa web interface...30
Figure 4.2 Interface of putative targeted viruses for user to browse putative host miRNA targets... 31
Figure 4.3 The conserved regions between virus and virus, and also show the relationship between conserved regions and microRNA target sites. ... 32
Figure 4.4 The expression profile of human microRNA and related viruses. ...32
Figure 4.5 The putative microRNAs shown in VirMiR...33
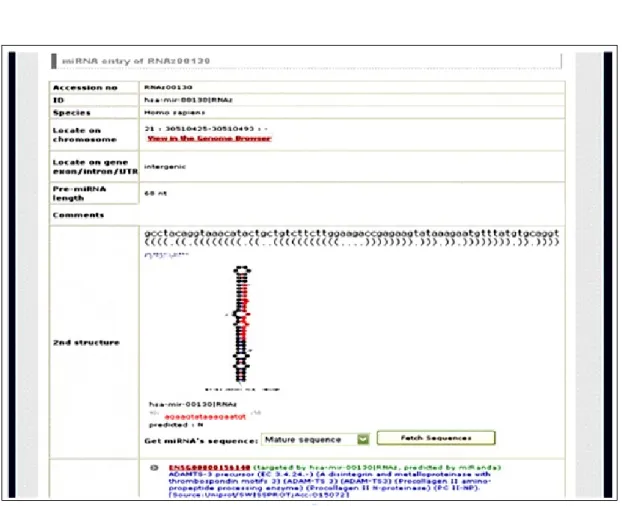
Figure 4.6 The detailed information of virus miRNA are provided for users. ...34
Figure 4.7 The VirMiR web interface supports multiple search for users...35
Figure 5.1 The known human miR-122a target sites on HCV were predicted...44
Figure 5.2 The known human miR-32 target sites on PFV-1 were predicted...45
Chapter 1 Introduction
1.1 Background
The microRNAs, which are small RNA molecules, are ~22 nt sequences that regulate gene expression by interfering the post-transcriptional level, resulting in degradation of mRNAs and repression of translation by the base pair to 3’ untranslated regions (3'-UTR) of the mRNAs. Many studies have investigated miRNAs. Ambros et al. developed a uniform system for identifying and annotating new miRNAs in numerous organisms [1]. The miRBase [2] supports the information for published miRNA genes and miRNA targets. The miRNAMap provides known, putative miRNA and miRNA targets to browse. Additionally, comparative sequence analysis, a fast and reliable method for identifying non-coding RNA structures [3], can be incorporated into the proposed resource to identify the conserved miRNA precursors in genomes of important in evolution, are developed by Washietl et al. In targets prediction, three tools, TargetScan [4], miRanda [5] and RNAHybrid [6] are commonly used to determine the most energetically most favorable hybridization sites of a small RNA to a large RNA. PicTar [7] is a computational method for identifying common targets of known miRNAs. Lu et al. developed an miRNA microarray for measuring the expression profiles of all known miRNAs in various normal tissues and tumors [8], which facilitate in accurately predicting miRNAs targets on viruses.
1.1.1 miRNA Biogenesis and Function
The microRNAs (miRNAs) are first discovered in C.elegan and regulate developmental stage. Currently, 44 species are found to produce miRNAs, not only in Eukaryotes, six virus species are also evidenced. More and more miRNAs are discovered, and have been shown to play important roles in a number of organisms at the level of development, apoptosis, and establishment of cell lineage. MicroRNA genes are one of the more abundant classes of regulatory genes in animals, estimated to comprise between 0.5 and 1 percent of the predicted genes in worms, flies, and humans, raising the prospect that they could have more regulatory functions than those uncovered to date [4]. The miRNAs are derived from precursor transcripts approximately 70–120 nts long sequences, which fold to form as stem-loop structures. These structures are believed to be recognized and taken out of nucleus by exportin 5. Pre-miRNA is then cleaved by Dicer (a ribonuclease III enzyme) to excise the mature miRNAs in the form of a small interfering RNA (siRNA) -like duplex, and asymmetrical assembly of the mature miRNA strands, which may be decided upon relative thermodynamic characteristics of the two 5 termini of strands, combining with the Argonaute proteins into effector complexes [9]. There are two ways to regulate gene expression: the common situation in plants, mRNA may be degraded when miRNA: mRNA perfectly complementary. In other situation,
always in animals, non-perfect complementary repress translation. The miRNAs appears to modulate methylation in chromatin level to silence chromatin, but this only occurs in yeast, some animals and plants. Figure 1.1 presents the biogenesis and functions of miRNAs.
Figure 1.1 The biogenesis and function of miRNAs [10].
1.1.2 Virus Infection and Propagation
In 1898, Friedrich Loeffler and Paul Frosch first found the nature of viruses, genetic entities that lie somewhere in the grey area between living and non-living states. Viruses are classified upon several characters, such as genome
type, shape, and life cycles depend on the host cells that they infect to reproduce. There are three steps of virus infection: attachment, penetration, and uncoating. When found outside of host cells, viruses exist as a protein coat or capsid, which encloses either DNA or RNA. Then it comes into contact with a host cell, a virus can penetrate, uncoat and insert its genetic material into host cell, literally taking over the host's functions. An infected cell produces more viral protein and genetic material instead of its usual products. Some viruses may remain dormant inside host cells for long periods, causing no obvious change in their host cells (a stage known as the lysogenic phase). But when a dormant virus is stimulated, it enters the lytic phase: new viruses are formed, self-assemble, and burst out of the host cell, killing the cell and going on to infect other cells. Viruses cause a number of diseases in eukaryotes. In humans, smallpox, the common cold, chickenpox, influenza, shingles, herpes, polio, rabies, Ebola, hanta fever, and AIDS are examples of viral diseases. Even some types of cancer - though definitely not all - have been linked to viruses.
1.1.3 The Relationship Between miRNAs and Viruses
Recently, the relationship between miRNAs and viruses appears to be interesting. Besides animals, insects and plants, miRNAs were also recently discoveredin viruses. Pfeffer et al [11] performeda small RNA profile of human cells infected by Epstein-Barrvirus to study therole that RNA silencing can play
during viral infection in animals. Besides host miRNAs, they identified five miRNAs originatingfrom the virus. Experimental verification of predictions in herpesviruses resulted in identification of several novel miRNAsfrom Kaposi sarcoma-associated virus, mouse gamma-herpes virus and human cytomegalovirus, SV40. Rhesus Lymphocryptovirus also encodes 16 microRNAs [12]. Additionally, virus miRNAs appear to have no homology between each other or with human miRNAs. Positions of miRNAs within different virus genomes were alsonot conserved in some viruses [13]. Although the function of most viral miRNAs seems unclear, the potential role in modulating virus activity is worthy to search. The miRNAs produced from viruses are considered to play a part in existence of virus by regulating virus own activity or host gene expression to escape immunity system. Besides, host miRNAs have been shown to target to viruses, such as PFV-1 [14], hepatitis C virus [15] and HIV [16], after they enter a host. Host miRNAs appear to cause different result in effecting virus. For example, experimental verification says that human miR-122a assists HCV replication in a host by targeting to the 3’ UTR on the HCV genome. However, HIV is suppressed by targeting several genes by human microRNAs.
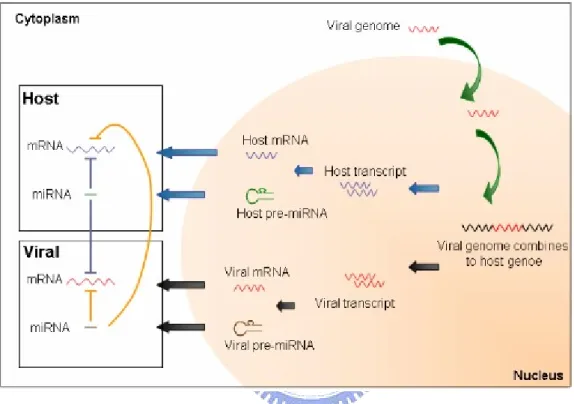
These indicate two roles of virus in host: one is miRNAs can be produced from viruses, the other is that virus can be regulated by host microRNAs (Fig. 1.2). The relationship between viruses and host miRNAs is more various and worthy of further research. Research investigating miRNAs and viruses is at
initial stages, and the multi-relationships between viruses and miRNAs are valuable to investigate.
Figure 1.2 The relationship between microRNAs and virus [17].
1.2 Motivation
In fact, virus data is not easy to obtain, and data format is not identical and enough. Construct a complete virus database by integrating other databases for users is one of our aim. Besides, the miRNAs seems to be important for viral exist. To research the role of miRNAs in viral infection, disease and cancer, this work focuses on prediction of miRNAs and host miRNA targets to facilitate the research of miRNAs in virology, develops databases, and confer the relationship between viruses, miRNAs and diseases. The host miRNA targets within virus
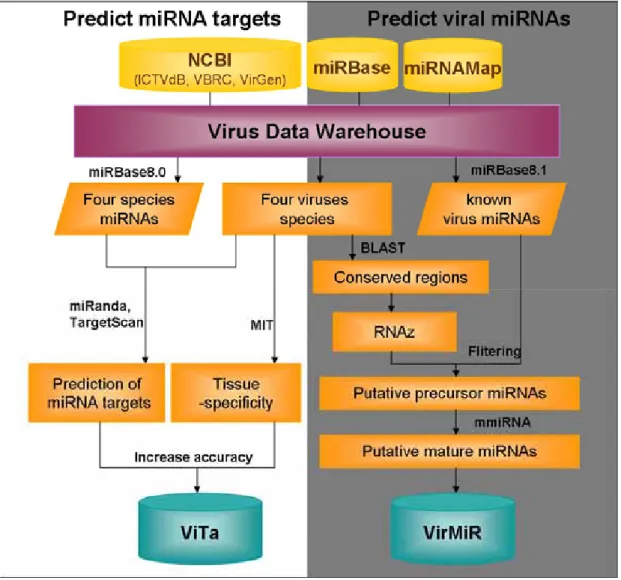
genomes are predicted both by miRanda [5] and TargetScan [4], and the relationships between host miRNAs and targeted viruses are identified and integrated by ViTa database. And the miRNA candidates are predicted and stored in VirMiR. The possible circular cycle pathway is shown in Fig.1.3.
Figure 1.3 The location of known virus miRNAs on virus genome.
1.3 Goal
This work assists the research in virology by constructing a virus data warehouse, including processing data and identical format, and by establishing the computational relationship between the miRNAs and viruses, including host miRNAs versus viruses, and miRNAs produced from viruses. Furthermore, this work provides effective annotations, including those for human miRNA expression, virus infected tissues and annotations of viruses, comparisons
between viruses. Additionally, various functions and a graphical web interface are presented that help users in investigating the microRNA roles in viruses, and facilitate the data access for biology. Thus, our aim includes not only provides miRNAs (VirMiR) and host miRNA targets (ViTa), expression profiles of known human miRNAs, virus annotation and cross-links to other biological databases and comparison between viruses, such as influenza viruses, hepatitis B, C virus, and combination of miRNAs. The role of miRNAs in cancer research is also supported. This work contains the relationship between host miRNA and viruses, and also miRNAs produced from viruses.
Chapter 2 Related Works
2.1 Prediction of miRNAs
Prediction of miRNAs in mammals, insects and plant is already well developed, such as miRBase [5], miRNAmap [18], but in viruses is at initial stages. There are several microRNAs produced from six viruses. Pfeffer S. et al. [11] firstly discovered five miRNAs on Epstein-Barr virus, a large DNA virus of the Herpes family that preferentially infects human B cells, and identified viral regulators of host and/or viral gene expression. Then, they found ten miRNAs in the Kaposi sarcoma-associated virus (KSHV or HHV8), nine miRNAs in the mouse gammaherpesvirus 68 (MHV68) and nine miRNAs in the human
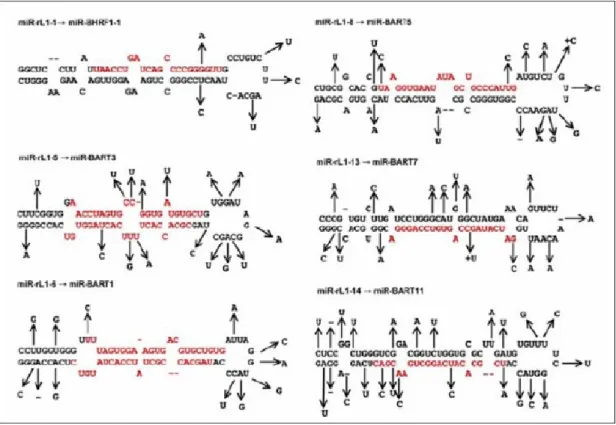
cytomegalovirus (HCMV or HHV5) [11]. Rhesus Lymphocryptovirus (rlcv), which is separated from EBV by 13 million years of evolution, expresses at least 16 distinct miRNAs, seven of which are closely related to EBV miRNAs [12] (Fig. 2.1). These miRNA genes are expressed individually or in clusters from either polymerase (pol) II or pol III promoters, and share no substantial sequence homology with one another or with the known human miRNAs. This causes prediction of miRNAs difficult. Figure 2.2 shows known miRNAs located on four species viruses.
Figure 2.1 Five microRNAs are produced from Rhesus Lymphocryptovirus.
Most viral microRNAs are found by both experiment and prediction. Searchers predict microRNAs only in conserved regions or specific regions, such as 3’ UTR, because conserved regions seem to be more significant and play
a critical role in virus life cycle. Prediction of miRNAs on whole viral genome appears to cause high false positive, thus finding miRNAs on conserved regions is opted, even though where some known viral miRNAs are not located (Table 2.1).
Table 2.1 The miRNAs with reported functions [19].
2.2 Prediction of Host miRNAs Targets
Host miRNAs are evidenced to influence viral life cycle after virus infecting host. Lecellier et al [14] first demonstrated that in human cells, a cellular microRNAs effectively restricts accumulation of the retrovirus primatefoamy virus type 1 (PFV-1), a complex retrovirus (akin to human immunodeficiency virus) that, in addition to the Gag, Pol, and Env proteins,produces two auxiliary factors, Bet and Tas, from the internalpromoter (IP). Conversely, PFV-1 also encodes a protein, Tas, that suppresses microRNA-directed functions in mammalian cells and displays cross-kingdom anti-silencingactivities. Another virus is proved to be regulated by human miRNAs. Hepatocellular carcinoma (HCC) is a common disease caused by hepatitis C virus [15]. Analysis of the genetic interaction between human miR-122a and the 5’ non-coding region of the HCV genome via mutation of the predicted microRNA binding site and ectopic expression of miR-122 molecules containing compensatory mutations indicated that human miR-122a is likely to facilitate replication of the viral RNA, and may present a target for antiviral intervention. HIV is also predicted as miRNA targets by four predictive tools [16]. Human microRNAs that are expressed in T-cells, the natural site of infection by HIV-1 infection, may target HIV-1 genes and related clade sequences at highly conserved target sites. These studies imply that human miRNAs potentially affect expression of HIV-1 genes (Fig. 2.3) and may, in the future, be utilized in developing therapies to inhibit
HIV-1. Therefore, through fortuitous recognition of foreign nucleic acids, cellular microRNAs have direct antiviral effectsin addition to their regulatory functions.
Chapter 3 Materials and Methods
3.1 Materials
3.1.1 The miRNAs Databases
miRBase is the new home for microRNA data, incorporating the database and gene naming roles previously provided by the miRNA Registry, and including the new miRBase Target database. There are 73 viral miRNAs included by six virus species - Epstein Barr virus (EBV), Human cytomegalovirus (HCMV), Kaposi sarcoma-associated herpesvirus (KSHV), Mouse gammaherpesvirus 68 (MGHV68), Simian virus 40 (SV40), Rhesus Lymphocryptovirus (rlcv) [2]. miRNAMap [18] is a database including two portions: miRNAs and miRNA targets, and focuses on four species, human, mouse, rat and dog. miRNAMap collects published miRNAs from the microRNA registry [20] and predict putative miRNA precursors (Fig. 3.1) by RNAz [3] which is based on genome-wide mapping of conserved RNA secondary structures from UCSC clustering [21], and also predicts the mature miRNAs by an algorithm which is based on MDD (maximal dependence decomposition), all of the known mature miRNAs have been detected their targets by miRanda [5] and TargetScan [4] results. The statistic of the materials is given in Table 3.1.
Table 3.1 The statistic of microRNAs.
Species Human Mouse Rat Chicken Virus Total
Number 328 266 238 125 73 1030
Figure 3.1 Putative miRNAs predicted by miRNAMap.
3.1.2 Virus Databases
ICTVdB [22], VBRC [23] and VirGen [24] are all virus databases. ICTVdB is a database of viruses, whose hosts are classified into Algae, archaea, bacteria, fungi, invertebrates, plants, protozoa and vertebrates (Fig. 3.2). The VBRC provides comprehensive web-based genomics resources to the scientific community conducting basic and applied research on microorganisms selected from the NIH/NIAID Category A, B, and C priority pathogens that are regarded
as possible bioterrorist threats or as emerging or re-emerging infectious diseases. The VirGen employs the sequenced-based bioinformatics approaches to annotate viral genome records and to identify the ‘alternative names’ of viral proteins, where available. It is the first resource to provide phylogenetic trees of viral species computed using whole genome sequence data, and houses predicted B-cell epitopes of known viral antigenic proteins in an attempt to link the genome to its vaccinome. Table 3.2 lists the integrated databases in this work.
Table 3.2 The list of the integrated external data sources. Integrated
Databases Description Literature
miRBase 8.1 Known microRNAs and microRNA targets [2] miRNAMap microRNA and miRNA targets of human, mouse,
rat, and dog [18]
ICTVbB Virus information [22]
VirGen Virus information [24]
VBRC Virus information [23]
3.1.3 Tools for Prediction
miRanda [5] is an algorithm for finding genomic targets for microRNAs and performs a Dynamic Programming local alignment between the query miRNA sequence and the reference sequence where G:U wobble is allowed, generating scores for the alignments. The thermodynamic stability of the high scoring pairs is calculated using the Vienna RNA package [25] and scores are generated. TargetScan is also a prediction tool which combines thermodynamics-based modeling of RNA:RNA duplex interactions with comparative sequence analysis to predict miRNA targets conserved across multiple genomes [4]. ClustalW [26] is a general purpose multiple sequence alignment program for DNA or proteins. It produces biologically meaningful multiple sequence alignments of divergent sequences and calculates the best match for the selected sequences, and lines them up so that the identities, similarities and differences can be seen. Evolutionary relationships can be seen via viewing other tools packaged. RNAz is program for predicting structurally conserved and thermodynamic stable RNA
secondary structures in multiple sequence alignments. It can be used in genome wide screens to detect functional RNA structures, as found in noncoding RNAs and cis-acting regulatory elements of mRNAs. Accurately predict location of mature microRNAs in the miRNA precursors remains a challenging problem, this work use mmiRNA [18] here. Mature miRNA is most likely to be present in particular sequences, rather than randomly. As presented in Fig. 3.7, both 5’-flanking and 3’-flanking sequences (10~14 nts) are used to train computer models, which are then used to identify the sequences of mature miRNA. The aim is to use two flanking models to identify the mature miRNAs. The model accurately locates mature miRNA within the miRNA precursor. Table 3.3 givens the analyzing tools used in this work.
Table 3.3 The list of the integrated annotated tools in this work. Integrated
Tools Description Literature cited
miRnada Predicting of miRNA targets [5] TargetScan Predicting of miRNA targets [4] RNAz Identifying non-coding structural RNAs [3] RNAfold RNA structural analyses [25] mmiRNA Identifying the mature miRNA in miRNA
precursor [18]
Mfold Drawing the miRNA secondary structures [27] ClustalW Multiple sequence alignment [26]
3.2 Methods
3.2.1 Overview
To facilitate the research of miRNA and viruses in Virology, this work focuses on two points – the prediction of virus miRNAs and the identification of host miRNA targets against viruses. First, this work collects virus data to construct a virus data warehouse system, which including viruses assigned 23 families and six genome types (dsDNA virus, ssDNA virus, dsRNA virus, (+) ssRNA virus, (-) ssRNA virus, retro-transcripting virus) are obtained from NCBI and other databases. The known microRNAs, such as human, mouse, rat, chicken and six viruses species (EBV, HCMV, KSHV, MGHV68, SV40, RLCV) are obtained from miRNAMap [18] and miRBase (8.1) [2]. Experimentally validated host miRNA targets against virus and virus infected are both identified by surveying the literature, and expressive tissue of miRNAs are obtained from MIT [8]. After collecting data, sequences are preprocessed upon life cycle of viruses. Conserved regions of virus are obtained by BLAST to confer the relationship between evolution and miRNAs, and for virus miRNA prediction. Furthermore, similarity among sequences is compared with subtypes in every species, and conserved regions between different strains of the same species are found. This warehousing system supports material for our search. The system flow of this work is shown in Fig. 3.3. The flow divides into two parts: the prediction of virus miRNAs (right-hand part) and the identification of host
miRNA targets (left-hand part).
3.2.2 Prediction of Host miRNA Targets
DATA GENERATION
Figure 3.4 presents a flowchart of data generation for the prediction of host miRNA targets. The flowchart comprises the following three main steps: (1) collect data from databases and the literature; (2) mine information from collective data and handle sequences; (3) predict host miRNA targets on virus genomes. Each step is described in detail below.
Collect data from databases and the literature
Known virus, including six species (EBV, HCMV, KSHV, MGHV68, SV40, RLCV) and chicken miRNAs are obtained from miRBase (8.0), and known human, mouse, rat miRNAs are obtained from miRNAMap. Known host miRNA targets on viruses are collected from literature. Collecting virus data is difficult as the NCBI only releases genomes of influenza viruses for download now, and non-classifiable, non-identical virus data packaged that can be directly obtained via an FTP site. Additional data must be obtained from other databases that are also linked to NCBI. Most data are from four databases - NCBI, ICTVdB, VBRC, and VirGen - and some from surfing websites. The virus warehouse contains 26870 records (total 2108 species), including genomes and transcripts. For human-related viruses, there are 15357 records (446 species), mice (414 species), rat (37 species), chicken (2529 species), 10333 records are other species. To provide more virus information and accurate the result of prediction, this work will maintain the collection of data.
Mine information from collective data and handle sequences
After data are collected, the second step involves mining information. Because virus data is non-identical, i.e., virus name, strain, serotype, hosts and virus infected tissue are not enough. To resolve this problem, other virus databases and related literature are accessed to unite virus data. And the ViTa
website provides dual classifications: genome type and biological classification (order, family, subfamily, genus, species) to facilitate user research. Then, to identify hosts and virus infected tissues, various sources, such as databases, literatures, websites and books are accessed. Prior to predict host miRNA targets, sequences must be preprocessed upon life cycle of virus. Decide what stage of virus life cycle could be as miRNA targets is hard. In some situations, predict both reverse and not reverse transcript, i.e., negative ssRNA viruses is rever-transcripted.
Predict host miRNA targets on virus genomes
In this work, miRanda [5] and TargetScan [4] are both used to identify host (human, mouse, rat and chicken) miRNA targets, which are the most probable miRNA targets of the antivirus miRNAs. The predictive parameters, including miRanda Minimum Free Energy (MFE) and miRanda score, are altered for the miRNA target prediction by comparing results of known miRNA/targets data and referring to miRNAMap. However, variability of viruses causes another problem in setting prediction parameters. Based on known miRNA target (Table 5.2), mfe is at <=-14 and score is at >=140 for prediction of known host miRNA targets. To prevent loss of real targets and provide all possible answers for users, the lowest parameter is adopted, which is lower than that adjusted from known data. For human viruses, mfe sets at <=-10, and score sets at >=120. Thus, users can choose their favorite parameters when searching.
The relationships between the host miRNAs and the targeted viruses are determined in reference to the annotations and the organization of viruses. The ViTa database supports expression profiles of the known miRNAs, different strains of the same virus species, comparison of subtypes, annotations and various links to biological databases. Virus annotations were obtained from databases and websites. The conserved regions in each species in the database are obtained by BLAST. The expression profiles of the miRNAs obtained from MIT are useful in elucidating the regulatory roles of miRNAs, and the tissue-specificity of known miRNAs, which can also be used to accurately identity the relationship between miRNAs and viruses, both of which exist in the same tissue.
3.2.3 Prediction of Viral miRNAs
Virus data of four infected host (human, mice, rat, chicken) are obtained from our warehousing system. To predict microRNAs from viruses, there are main four steps: (1) Find conserved regions on genomes in different strains of the same species by BLAST, because microRNAs are considered to be located in conserved regions. Then, stable and long enough stem-loop structures are the essential condition in forming microRNA precursors. (2) Prediction of conserved secondary structures by RNAz [3]. (3) Find miRNA precursors and mature miRNAs by our develop tool-mmiRNA, which is implemented and evaluated using experimentally validated vertebrate mature miRNAs, accurately locates mature miRNA within the miRNA precursor. Then, a database - VirMiR established, including known and putative miRNAs from viruses, analysis and statistic of results, annotation of microRNAs to promote the investigation of miRNAs located on viruses. Like ViTa, a proposed web interface are also designed and developed to facilitate the data access, including known and putative miRNAs from viruses, information of miRNAs, i.e., location, sequence, structure. The flowchart of VirMiR is shown in Fig. 3.5.
Figure 3.5 The flowchart of prediction of miRNAs on viruses.
Find conserved regions on genomes in different strains of the same species by BLAST
Because miRNA are considered to be located on conserved regions. The conserved regions are found by BLAST, identity and length of which >=70% and <=200, are switched into ClustalW format to run RNAz. Besides, sequences are switched with window slide =15, length =200 nts if identity >=70% and length >=200 nts.
Prediction of conserved secondary structures by RNAz
After finding conserved regions, conserved secondary structures are predicted by RNAz [3], which is a computational prediction tool for non-coding RNA structures based on the comparative sequence analysis. Figure 3.6 shows the process of finding conserved RNA secondary structure. Consensus structures are predicted from multiple alignment sequence with ClustalW format by RNAz.
Figure 3.6 Finding conserved secondary structures by RNAz.
Find miRNA precursors and mature miRNAs
The putative miRNA precursors are filtered from the non-coding RNA structures by applying the following constraints: (i) the height of the stem of the non-coding RNA structure is at least 25 nts and the length of the loop must <20 nts; (ii) the RNAz z-score of the structure is smaller than -3.5. (iii) consensus mfe is between -20 and -30 kcal/sec. Finally, it resulted that 54 putative miRNA precursors are filtered out. The parameter are referring from miRNAMap and
[28].
miRNA of the putative miRNA genes should be determined. Therefore, mmiRNA, a machine learning method for computationally determining the mature miRNA in the miRNA precursors and developed by miRNAMap, is applied. The mature miRNA is most likely to be present in particular sequences, rather than randomly. As presented in Fig. 3.7, both 5’-flanking and 3’-flanking sequences (10~14 nts) are used to train computer models, which are then used to identify the sequences of mature miRNA. mmiRNA is to use two flanking models to identify the mature miRNAs and combines the 5’-HMMs-group and 3’-HMMs-group to determine the position of mature miRNAs. An instance of mature miRNA is detected if the predicted 5’-flanking sites and the predicted 3’-flanking sites are located in a single window, with sizes from 18 nts to 24 nts, which is the possible length of the known mature miRNAs. Furthermore, the distance constraint imposed by the two models can increase their specificity. The detailed process of mature miRNA detector is described in miRNAMap.
Chapter 4 Implementation
4.1 Data Preprocessing and Storage of Knowledgebase
Because NCBI only release genome of influenza viruses for download, there is no classifiable, identical of other virus data packaged to be directly obtained from FTP. Collection and preparing virus data is not easy. What problem meets first is few data obtained. Obtain more data from other databases and websites is necessary. After collection, virus data isn’t identical and enough, i.e., virus name, strain, serotype, host and infective tissue of many viruses are also not recorded. To solve problems, other virus databases and related–literatures are referred, and virus data must be united. To facilitate research, dual classifications are provided: genome type and biological classification (order, family, subfamily, genus, species). Then, non-recorded hosts and infecting tissues is identified from various aspects, like databases, literatures, websites and books. The next step, sequences must be preprocessed based on life cycle before prediction. The relationship between miRNAs and viruses by predicting host miRNA targets and virus miRNAs is identified.
4.2 Web Interface
Two web interfaces are developed to facilitate the data access. The proposed web interface of ViTa provides multilateral searching and browsing (Fig. 4.1), such as conserved regions between strains of virus species (Fig. 4.3), detailed information about targeted viruses (Fig. 4.2), and further statistics of results to facilitate miRNA: virus research, expression profile of miRNAs (Fig. 4.4). When browsing miRNA targets, the ViTa database provides two modes, genome types and biological classification (left portion of Fig. 4.1). The ViTa also supports multiple type of search for users, such as keywords, infected tissue, disease and virus names. Searching the database by submitting favorable parameters assists users in finding results with increased accuracy (right portion of Fig. 4.1).
Figure 4.2 presents a graphical visualization tool used to present targeted viruses. All miRNAs and target sites are also provided in text format, stating the genomic locations of target sites, the MFE of miRNAs, target sites sequences, and alignment of hybridization structures. Explicate cooperative or combinatorial control of viruses by a group of miRNAs are provided. The tutorial documentation for the ViTa and VirMiR web site describes in detail how to use the web interfaces.
Figure 4.2 Interface of putative targeted viruses for user to browse putative host miRNA
Figure 4.3 The conserved regions between virus and virus, and also show the relationship
between conserved regions and microRNA target sites.
The web interface of VirMiR not only provides known virus microRNAs, but also putative miRNA precursors browsing (Fig. 4.5) and their information, such as secondary structure, minimum free energy, linkage to view more information. The VirMiR database also supports known virus miRNAs, including the sequence, mature miRNA, genomic location, and citations in literature. The diagram illustrating the stem-loop RNA structure of the miRNA precursor (Fig. 4.6), which is folded using RNAfold [25], is generated graphically by mfold [27]. The VirMiR also supports multiple type of search for users (Fig. 4.7). Various search criteria can be used to search.
Chapter 5 Results and Case Study
5.1 Statistics of Results
5.1.1 Result of Virus miRNAs Prediction
Number of sequence in one virus species up to 6 is not easy to collect. And prediction of viral miRNA in our aim only focuses on conserved regions up to 6 sequences, some known miRNAs, such as EBV, HIV, can’t be predicted in this work. There are 73 viral known miRNAs from miRBase (8.1) and 54 putative miRNA precursors in VirMiR.
5.1.2 Result of Host miRNA Targets Prediction
The ViTa database currently stores 2156 virus species from NCBI, of which 446 are human virus (Table 5.1), and nine known host miRNA targets separately from HCV, HIV and PFV-1.
Table 5.1 Statistics numbers of virus in the virus data warehouse.
Viruses Human-related virus Mouse Rat Avian Other
species Total Liver-related viruses 285 Influenza viruses 12137 Other disease virus 3135 Numbers 15357 (446) 414 37 2529 10333 28670 (2156)
The minimum free energy (MFE) of the miRNA–target duplex was determined by miRanda [5] and TargetScan [4] to identify host miRNA target sites. Low MFE values of the miRNAs–target sites indicate probable hybridizations between the miRNAs and the targeted viruses. Table 5.2 presents the statistics for the miRanda-predicted known human miRNA targets extracted from literature (Table 5.3). The known human miRNA targets on HCV can be identified when the miRanda MFE threshold and miRanda score threshold are set at -10 kcal/mol and 120, respectively. However, these parameters are likely to grossly over-predict the number of miRNA targets per virus. Alternatively, the proposed database allows users to utilize a set of parameters that is more stringent and obtains fewer false positives that does the thresholds. All predictive targets are determined and stored in the proposed ViTa database when the MFE is ≦-10 kcal/mol and score for miRNA targets is ≧120. For instance,
when the MFE threshold is set at -20 kcal/mol and the miRanda score threshold is set at 120, the average number of targeted viruses for each miRNA is 83.65 (Table 5.4). The average number of distinct miRNAs targeting each virus is 64.50. Similarly, when the score threshold is set at 160 and the MFE is set at -10 kcal/mol, the average number of the targeted viruses for each miRNA is 50.64. The average number of the distinct miRNAs targeting each virus is 33.75. Table 5.5 presents the results obtained with TargetScan, and the relationship between miRNAs and common virus, such as liver viruses and influenza viruses, are presented in Table 5.6 and Table 5.7.
Table 5.2 The result of known virus targets predicted by miRanda and TargetScan.
Table 5.3 Experimental miRNA targets on virus.
miRNAs Target virus References
Hsa-miR-122a Hepatitis C Virus 1a strain H77c (AF011751) [15] Hsa-miR-122a Hepatitis C Virus 1b strain HCV-N (AF139594) [15] Hsa-miR-29a HIV-1 isolate BRU (K02013) [16] Hsa-miR-29b HIV-1 isolate BRU (K02013) [16] Hsa-miR-149 HIV-1 isolate BRU (K02013) [16] Hsa-miR-324-5p HIV-1 isolate BRU (K02013) [16] Hsa-miR-378 HIV-1 isolate BRU (K02013) [16] Hsa-miR-378 HIV-1 isolate ELI (K03454) [16]
Hsa-miR-32 PFV-1 [14]
miRanda TargetScan
miRNAs Target virus References
Score MFE (kcal/mol) MFE (kcal/mol)
Hsa-miR-122a HCV 1a strain H77c (AF011751) [15] 146.00 -14.30 -20.10 -24.10 Hsa-miR-122a HCV 1b strain HCV-N (AF139594) [15] 148.00 -15.70 -20.10 Hsa-miR-29a HIV-1 isolate BRU
(K02013)
[16]
175.00 -22.10 -29.20 Hsa-miR-29b HIV-1 isolate BRU
(K02013)
[16]
179.00 -23.00 -31.30 Hsa-miR-149 HIV-1 isolate BRU
(K02013)
[16]
194.00 -26.60 -32.10 Hsa-miR-324-5p HIV-1 isolate BRU
(K02013)
[16]
191.00 -24.50 -32.70 Hsa-miR-378 HIV-1 isolate BRU
(K02013)
[16]
177.00 -25.30 -31.10 Hsa-miR-378 HIV-1 isolate ELI
(K03454)
[16]
177.00 -25.30 -31.10 Hsa-miR-32 PFV-1 [14] 120.00 -9.91 No result
Table 5.4 Statistics for predictive miRNA targets by miRanda. MFE (kcal/mol) Average number of targeted
virus for each miRNA
Average number of distinct miRNAs for each targeted transcript Average targetsite for microRNA score ≥ 120 ≤ -10 229.13 260.93 33754.86 ≤ -15 188.96 188.47 11693.65 ≤ -20 83.65 64.50 1755.98 ≤-25 19.13 9.66 182.86 score ≥ 140 ≤ -10 170.40 156.92 6174.33 ≤ -15 132.66 113.22 3524.48 ≤ -20 56.19 38.77 819.14 ≤-25 14.06 6.63 117.03 score ≥ 160 ≤ -10 50.64 33.75 620.39 ≤ -15 45.21 29.52 537.10 ≤ -20 22.37 12.88 213.28 ≤-25 8.41 3.29 61.24 score ≥ 180 ≤ -10 5.81 2.03 44.48 ≤ -15 5.81 2.03 44.48 ≤ -20 5.53 1.93 37.68 ≤-25 3.91 1.49 28.58 score ≥ 200 ≤ -10 3 1 6.33 ≤ -15 3 1 6.33 ≤ -20 3 1 6.33 ≤-25 3 1 6.33
Table 5.5 Statistics for predictive miRNA targets by TargetScan.
MFE (kcal/mol)
Average number of targeted virus for each
miRNA
Average number of distinct miRNAs for each targeted
transcript
Average targetsite for each microRNA
≤ -10 87.48 108.45 1056.20
≤ -15 83.34 101.27 972.19
≤ -20 54.30 59.80 522.72
≤-25 18.03 14.90 125.69
Table 5.6 Numbers of microRNAs versus liver viruses (wildtype). MFE (kcal/mol) HAV (M14707) HBV (X98077) HCV (NC_004102) HDV (NC_001653) HEV (NC_001434) HGV (U44402) Run by miRanda ≤ -10 129 117 132 82 125 135 ≤ -15 79 78 113 44 102 116 ≤ -20 24 20 58 15 34 61 ≤-25 3 6 11 5 3 13 Run by TargetScan ≤ -10 33 0 53 0 25 57 ≤ -15 31 0 48 0 25 57 ≤ -20 19 0 31 0 14 40 ≤-25 6 0 14 0 3 11
Table 5.7 Average numbers of microRNAs v.s. influenza viruses. MFE
(kcal/mol) InfluenzaA InfluenzaB InfluenzaC
Run by miRanda ≤ -10 102.96 174.65 107.79 ≤ -15 46.94 70.02 37.75 ≤ -20 7.16 9.61 6.63 ≤-25 0.40 0.19 0.44 Run by TargetScan ≤ -10 0 0 0 ≤ -15 0 0 0 ≤ -20 0 0 0 ≤-25 0 0 0
5.2 Case studies: Host miRNA Targeting to Viruses
To validate the result of prediction, three published papers are tested. And the results predicted by miRanda and TargetScan are given in Table 5.2.
5.2.1 Hepatitis C Virus (HCV) versus Human miR-122a
Jopling, Yi., et al (2005) discovered that miR-122 is likely to facilitate replication of the viral RNA by targeting to 3’ UTR on HCV genome [15]. Although the result of miRNAs targeting assist virus replication, it suggests that miR-122 may present a target for antiviral intervention, and there may have a more complicated mechanism. Here, the target sites of human miR-122a on HCV are predicted by ViTa (Fig 5.1).
Figure 5.1 The known human miR-122a target sites on HCV were predicted.
5.2.2 Primate Foamy Virus Type 1 (PFV-1) versus Human miR-32
Lecellier et al (2005) showed that a cellular microRNA effectively restricts the accumulation of the retrovirus primate foamy virus type 1 (PFV-1) in human cells, and PFV-1 also encodes a protein, Tas, that suppresses microRNA-directed functions in mammalian cells and displays cross-kingdom anti-silencing activities [14]. Therefore, through fortuitous recognition of foreign nucleic acids, cellular microRNAs (hsa-miR-32) have direct antiviral effects in addition to their regulatory function. Hsa-miR-32 is also predicted to target to PFV-1 and
showed in the ViTa web interface (Fig 5.2).
Figure 5.2 The known human miR-32 target sites on PFV-1 were predicted.
5.2.3 Human Immunodeficiency Virus-1 (HIV) versus Human
miRNAs
Manoj Hariharan et al (2005) suggest that miRNAs, in principle, can contribute to the repertoire of host pathogen interactions during HIV-1 infection [16]. They used consensus scoring approach, and high scoring miRNA-target pairs were selected which were identified by four well-established target prediction tools. Hsa-mir-29a and 29b can target the nef gene, hsa-mir-149 targets the vpr gene, hsa-mir-378 targets env, and hsa-mir-324-5p targets the vif gene. The microRNA expression profiles show that the five human miRNAs are expressed in T-cells, the normal site of infection of HIV-1 virus. The authors first report of human microRNAs which can target crucial HIV-1 genes. The
ViTa also predicts that human miRNAs target to HIV and show the results below (Fig 5.3).
Chapter 6 Discussion and Conclusions
6.1 Discussion
Although the current experimental evidence of virus encoded miRNAs is restricted to just three groups of viruses, computational approaches have suggested the existence of miRNAs in other viruses such as poxviruses and adenoviruses but not in most of the RNA viruses. However, there are several examples of modulation of viruses by host-encoded miRNAs. The best recent example is the remarkable positive regulation of replication of hepatitis C virus by the liver-specific miR-122. Impaired translation of target sequences from primate foamy virus 1 (PFV) by miR-32 has also been reported. The interaction of HIV with host T cells might also be modulated by miRNAs. Finally, latency type III EBV infections are associated with induction of miR-155 in B cells (M. Zavolan, unpublished). This miRNA is also highly expressed in Hodgkin’s, primary mediastinal and diffuse large cell lymphomas, which raises the question of whether miR-155 induction might, in fact, be responsible for some of the malignant disorders associated with EBV. This work predicts putative virus miRNAs on all viruses, not only restrict to just three groups of viruses.
There are some questions in prediction of viral miRNAs. Because the conserved regions of virus genome appear to play a critical role in maintaining virus life cycle, prediction of miRNAs only focuses on conserved region in spite
of the location of microRNAs is still uncertain. Besides, few sequences can be available for every virus species, it cause only some species, not including virus which produced known microRNAs, can be predicted. These two questions are worthy to think and discuss.
The interaction between viruses and their hosts seems to involve host- and virus-encoded miRNAs, which continue to expand our knowledge about fundamental aspects of gene regulation. It will be fascinating to follow how the understanding of virus–host interactions is shaped by the discovery of the targets of these small RNAs.
6.2 Future Works
The database will be further developed as follows. (i) The database will support miRNA annotations for miRNA genes and miRNA targets on viruses for hosts other than four species, human, rat, mice and chicken, and provide more data types; (ii) Data will be further analyzed to support miRNAs involved in combinatorial control of virus expression. (iii) Complete relationships between viruses and diseases will be added, and (iv) The virus data warehouse will annotate more species and keep update to provide new information.
6.3 Conclusions
between miRNAs, virus and cancer, and stored putative, known host miRNA targets in ViTa, putative, known miRNAs in VirMiR, and also provided the regulatory relationships between miRNAs and viruses. The database can provide sufficient information to support any virus-related works. For example, miRNAs may contribute to cancer, miRNA-mediated tumorigenesis results from either down or up-regulating virus activity. For disease, such as influenza viruses and liver viruses, our work predicts host microRNA targets, result analysis and data statistics are also utilized to provide multi-directional research for users. Tissue data for viruses and miRNAs improves the accuracy of results, and consolidate the relationship between miRNAs and targeted viruses. The relationship between viruses and miRNAs participating in cancer cell regulation can be systematically identified for further experimental verification.
References
1. Ambros, V., et al., A uniform system for microRNA annotation. Rna, 2003. 9(3): p. 277-9.
2. Griffiths-Jones, S., et al., miRBase: microRNA sequences, targets and gene
nomenclature. Nucleic Acids Res, 2006. 34(Database issue): p. D140-4.
3. Washietl, S., I.L. Hofacker, and P.F. Stadler, Fast and reliable prediction of noncoding
RNAs. Proc Natl Acad Sci U S A, 2005. 102(7): p. 2454-9.
4. Lewis, B.P., et al., Prediction of mammalian microRNA targets. Cell, 2003. 115(7): p. 787-98.
5. John, B., et al., Human MicroRNA targets. PLoS Biol, 2004. 2(11): p. e363.
6. Rehmsmeier, M., et al., Fast and effective prediction of microRNA/target duplexes. Rna, 2004. 10(10): p. 1507-17.
7. Krek, A., et al., Combinatorial microRNA target predictions. Nat Genet, 2005. 37(5): p. 495-500.
8. Lu, J., et al., MicroRNA expression profiles classify human cancers. Nature, 2005.
435(7043): p. 834-8.
9. Bartel, D.P., MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, 2004. 116(2): p. 281-97.
10. He, L. and G.J. Hannon, MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet, 2004. 5(7): p. 522-31.
11. Pfeffer, S., et al., Identification of virus-encoded microRNAs. Science, 2004.
304(5671): p. 734-6.
12. Cai, X., et al., Epstein-Barr Virus MicroRNAs Are Evolutionarily Conserved and
Differentially Expressed. PLoS Pathog, 2006. 2(3): p. e23.
13. Berezikov, E. and R.H. Plasterk, Camels and zebrafish, viruses and cancer: a
microRNA update. Hum Mol Genet, 2005. 14 Spec No. 2: p. R183-90.
14. Lecellier, C.H., et al., A cellular microRNA mediates antiviral defense in human cells. Science, 2005. 308(5721): p. 557-60.
15. Jopling, C.L., et al., Modulation of hepatitis C virus RNA abundance by a
liver-specific MicroRNA. Science, 2005. 309(5740): p. 1577-81.
16. Hariharan, M., et al., Targets for human encoded microRNAs in HIV genes. Biochem Biophys Res Commun, 2005. 337(4): p. 1214-8.
17. Schutz, S. and P. Sarnow, Interaction of viruses with the mammalian RNA interference
pathway. Virology, 2006. 344(1): p. 151-7.
18. Hsu, P.W., et al., miRNAMap: genomic maps of microRNA genes and their target
genes in mammalian genomes. Nucleic Acids Res, 2006. 34(Database issue): p.
19. Sullivan, C.S., et al., SV40-encoded microRNAs regulate viral gene expression and
reduce susceptibility to cytotoxic T cells. Nature, 2005. 435(7042): p. 682-6.
20. Griffiths-Jones, S., The microRNA Registry. Nucleic Acids Res, 2004. 32(Database
issue): p. D109-11.
21. Karolchik, D., et al., The UCSC Genome Browser Database. Nucleic Acids Res, 2003.
31(1): p. 51-4.
22. Buchen-Osmond, C., The universal virus database ICTVdB. COMPUTING IN
SCIENCE & ENGINEERING, 2003. 5(3): p. 16-25.
23. Lefkowitz, E.J., et al., Poxvirus Bioinformatics Resource Center: a comprehensive
Poxviridae informational and analytical resource. Nucleic Acids Res, 2005. 33(Database issue): p. D311-6.
24. Kulkarni-Kale, U., et al., VirGen: a comprehensive viral genome resource. Nucleic Acids Res, 2004. 32(Database issue): p. D289-92.
25. Hofacker, I.L., Vienna RNA secondary structure server. Nucleic Acids Res, 2003. 31(13): p. 3429-31.
26. Thompson, J.D., D.G. Higgins, and T.J. Gibson, CLUSTAL W: improving the
sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res, 1994. 22(22): p. 4673-80.
27. Zuker, M., Mfold web server for nucleic acid folding and hybridization prediction.
Nucleic Acids Res, 2003. 31(13): p. 3406-15.
28. Pfeffer, S., et al., Identification of microRNAs of the herpesvirus family. Nat Methods, 2005. 2(4): p. 269-76.
![Figure 1.1 The biogenesis and function of miRNAs [10].](https://thumb-ap.123doks.com/thumbv2/9libinfo/8754911.206669/11.892.130.724.294.872/figure-biogenesis-function-mirnas.webp)
![Figure 1.2 The relationship between microRNAs and virus [17].](https://thumb-ap.123doks.com/thumbv2/9libinfo/8754911.206669/14.892.130.763.251.671/figure-relationship-micrornas-virus.webp)
![Table 2.1 The miRNAs with reported functions [19].](https://thumb-ap.123doks.com/thumbv2/9libinfo/8754911.206669/18.892.125.794.369.1070/table-the-mirnas-with-reported-functions.webp)
![Figure 2.3 Predicted target sites of human microRNAs on HUV-1 genome [16].](https://thumb-ap.123doks.com/thumbv2/9libinfo/8754911.206669/20.892.133.751.298.514/figure-predicted-target-sites-human-micrornas-huv-genome.webp)