國
立
交
通
大
學
生物資訊及系統生物研究所
碩士論文
使用雙胜肽分數卡預測蛋白質在大腸桿菌表現
系統中之溶解度
Scoring Card of dipeptides for predicting solubility of
recombinant proteins in E. coli expression system
研 究 生:高德芬
指導教授:何信瑩 教授
使用雙胜肽分數卡預測蛋白質在大腸桿菌表現
系統中之溶解度
Scoring Card of dipeptides for predicting solubility of
recombinant proteins in E. coli expression system
研 究 生:高德芬 Student:Te-Fen Kao
指導教授:何信瑩 Advisor:Shinn-Ying Ho
國 立 交 通 大 學
生物資訊及系統生物研究所
碩 士 論 文
A Thesis Submitted to Institute of Bioinformatics and Systems Biology Department of Biological Science
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master in Bioinformatics
July 2011
Hsinchu, Taiwan, Republic of China
i
使用雙胜肽分數卡預測蛋白質在大腸桿菌表現
系統中之溶解度
學生:高德芬 指導教授:何信瑩
國立交通大學生物資訊及系統生物研究所碩士班
摘 要
蛋白質表現系統為一個非常實用的生物技術且普遍被利用在蛋白質相關的 研究上。而大腸桿菌則是最被常使用在蛋白質表現系統的宿主,因為大腸桿菌表 現系統具有簡單、快速,價格又低廉的優勢。但在此表現系統中有時會產生一個 嚴重的又難以解決的問題,表現時時有些蛋白質會形成一種結構不正確且無正確 生物功能的包涵體,所以無法被利用在接下來之研究上。所以生物學家都希望能 在蛋白質表現系統中盡可能得到可溶性之蛋白質,因可溶性蛋白質就代表結構正 確且擁有生物功能。而生物學家因此則利用改變各種實驗條件等方法,來使包涵 體能轉變為可溶性蛋白質,但目前此些改變實驗條件的方法都還是處於反覆詴驗 與不斷的嘗詴錯誤的階段,所以非常耗費材料、金錢與時間。 在目前的相關文獻中,許多研究都使用 SVM 等機器學習的方法來藉由蛋白 質的一級結構預測大腸桿菌表現後之蛋白質的溶解度。其中使用了許多與蛋白質 序列相關的特徵,包括了各種物化特性、胺基酸組合、雙胜肽或三胜肽組合等等 的特徵,不勝枚舉。然而相關文獻中所使用的分類方法對於生物學家幾乎都是屬 於黑盒子的分類法,難以了解其中分類過程的依據。所以在此篇研究中我們研究 了許多種特徵並挑選出了認為對此分類有效的雙胜肽特徵,於是提出了一個以雙 胜肽值來建立出的分數卡之方法來預測表現後的蛋白質之溶解度狀態。 本研究所提出之分數卡方法是一個簡單明瞭並可直接利用統計雙胜肽的方 式來達到預測之目的。每個欲測詴蛋白質都可從分數卡之計算得到一個分數,並 再藉由從驗證資料中計算出一個將兩類蛋白質由分數切開的臨界值。而為了更進 一步的強化分數卡的分類效果,之後我們又加入了智慧型基因演算法來調整由統 計產生之雙胜肽分數卡,其中在此問題中並以 ROC 曲線下之面積當做基因演算 法中的適應性函數值來判斷效能。在使用相同的資料下,智慧型基因演算法分數 卡的方法能得到 81.7%的準確率,高於使用 SVM 之 76.9%。並由比較 SVM、分 數卡方法與經過智慧型基因演算法調整後的分數卡方法之結果來證明基因演算 法的確可使此分類問題的準確率大幅提升。ii
Scoring Card of dipeptides for predicting solubility of
recombinant proteins in E. coli expression system
Student:Te-Fen Kao Advisor:Dr. Shinn-Ying Ho
Institute of Bioinformatics and Systems Biology
National Chiao Tung University
ABSTRACT
Protein expression system is a very common and useful experiment skill in protein studying. Nowadays, Escherichia coli (E. coli) are mostly universal hosts for cloning and expressing in a broad of researches with its fast and inexpensive
characters. However, there is a serious obstacle in protein expression system. Many proteins are produced in the form of insoluble aggregation that is a major obstruct for a lot of experiments, and the misfolded aggregation is called inclusion body.
Accordingly, researchers usually do their best to get the soluble form of protein via regulating experimental conditions, but the processes are still trial-and-error.
Many recent researches did their effort to predict the solubility of expressed proteins in E. coli via support vector machine (SVM). Existing methods applied a wide variety of primary structure feature sets, including physical chemical index and composition of amino acid, dipeptide and tripeptide. Generally, the prediction models and results using a black-box like method, such as SVM, are not easily interpretable. This study investigated several feature types and then proposed a scoring card method of dipeptides to predict the solubility of expressed proteins in E. coli.
The proposed scoring card is a very intuitive prediction method that uses dipeptide statistic to construct a scoring matrix. Every input sample can get a score according to this scoring matrix, and then a best cut-off value was chosen from the validation data. Furthermore, to improve the scoring card method, an intelligent genetic algorithm (IGA) is used to optimize the scoring matrix, in which it can get a better performance of ROC curve to promote the classification accuracy. The IGA-scoring card could yield an accuracy of 81.7%, higher than 76.9% of using an SVM method using the same dataset. Finally, the better accuracy and more efficient classification result could be confirmed by the comparison among SVM, scoring card and IGA-scoring card for this problem of classification between expressed proteins.
iii
誌 謝
非常幸運的我要感謝何信瑩老師有機會讓我加入 IClab 這個大家庭,並在這 兩年當中不斷的教導我許多知識與智慧,除了在實驗上的指導外,還從老師的身 上學習到了很多做人處事的道理與求學問的態度,我相信這些學問在我未來的生 涯裡都會非常有用。 而我也要感謝我的父母對我的培養與支持,讓我能在求學的這條路上無後顧 之憂地學習,而在此也將這些成就與榮耀歸功於我最親愛的父母。 此篇研究與論文要感謝黃慧玲老師參與的指導,與義雄學長、佳達學長、凱 迪學長與廖琴學姐等實驗室學長姐不吝嗇的提供他們寶貴的意見與幫助。特別是 Burnz 學長在程式的指導。還有感謝玉祥與泰欽兩位同學,總是互相幫忙彼此的 研究。 還有感謝實驗室當中所有的學長姐、同學與學弟妹們,讓我的研究所生涯充 滿了歡樂與鼓勵,也感謝你們對我的祝福,並希望未來的日子中大家能夠繼續保 持連絡互相幫助。iv
目 錄
摘要... i Abstract... ii 誌謝... iii 目錄... iv 表目錄... vi 圖目錄... vii 第一章 緒論... 1 1.1 研究動機... 1 1.2 研究目的... 1 1.3 研究題目定義... 1 1.4 章節概要... 2 第二章 重組蛋白之蛋白質表現系統介紹... 3 2.1 重組 DNA 技術... 3 2.2 大腸桿菌蛋白質表現系統... 3 2.3 凝膠體電泳法(SDS-PAGE)... 4 2.4 包涵體(inclusion body)... 5 第三章 背景知識與方法... 6 3.1 智慧型基因演算法(IGA)... 6 3.1.1 基因演算法... 6 3.1.2 直交交配法... 7 3.2 繼承式雙目標基因演算法(IBCGA-SVM)... 8 3.2.1 支援向量機 SVM... 8 3.2.2 染色體編碼... 9 3.2.3 繼承式雙目標基因演算法實驗流程... 10 3.3 相關研究介紹... 10 第四章 研究方法... 12 4.1 研究資料... 12 4.2 使用特徵... 15 4.2.1 相關研究使用過的特徵介紹... 15 4.2.2 雙胜肽(dipeptide)... 16 4.2.3 物化特性... 16v 4.3 繼承式雙目標基因演算法實驗流程設計... 17 4.4 雙胜肽分數卡(scoring card)... 18 4.4.1 分數計算... 18 4.4.2 分類方法... 19 4.5 IGA-scoring card... 20 4.5.1 初始族群設定... 21 4.5.2 適應度計算... 21 4.5.3 直交交配法... 22 4.5.4 IGA-scoring card 演算法流程... 22 4.5.4 視窗型臨界值實驗... 23 第五章 實驗結果與討論... 25 5.1 726dataset 做 IBCGA 的結果與討論... 25 5.2 Sd957 做 IBCGA 的結果與討論... 26 5.3 Scoring card 結果與討論... 27 5.4 IGA-scoring card 結果與討論... 28 5.4.1 長條圖分數分析... 31 5.4.2 視窗型臨界值實驗結果... 32 5.5 Scoring card 與文獻結果之比較... 33 5.6 Scoring card 與 SVM 之比較... 33 5.7 雙胜肽與相關文獻之生物特性探討... 35 第六章 結論與展望... 40 6.1 結論... 40 6.2 未來展望... 40 參考文獻... 42
vi
表目錄
表 1 L 8 (2 7 )直交表與主效果範例 ... 7 表 2 617 特徵與物化特性(PCP)經 IBCGA-SVM 結果 ... 25 表 3 617 特徵與物化特性(PCP)經 IBCGA-SVM 挑選後統計出現頻率超過一半的 特徵 ... 26 表 4 整合後特徵經過 IBCGA 挑選出出現頻率高的特徵 ... 26表 5 957dataset 做 IBCGA 與選出後特徵做 SVM-grid 的結果... 27
表 6 Scoring card 之結果 ... 28
表 7 IGA-scoring card 之結果 ... 30
表 8 文獻方法、scoring card 與 IGA-scoring card 的結果比較 ... 33
表 9 SVM、scoring card 與 IGA-scoring card 的結果比較 ... 34
表 10 IGA-scoring card 依照胺基酸平均之結果 ... 36
表 11 Scoring card 中的個別胺基酸分析 ... 36
vii
圖目錄
圖 1 重組 DNA 技術之過程 ... 3 圖 2 帶有特定質體的大腸桿菌大量複製... 4 圖 3 基因演算法流程圖... 6 圖 4 SVM 示意圖 ... 8 圖 5 染色體編碼範例示意圖... 10 圖 6 研究資料來源示意圖... 15 圖 7 實驗資料流程示意圖... 18 圖 8 IGA-scoring card 實驗流程示意圖 ... 20圖 9 IGA 應用在 scoring card 的流程圖。 ... 21
圖 10 IGA-scoring card 演算法之流程圖 ... 23
圖 11 Scoring card 的 heat map 表示法 ... 29
圖 12 IGA-scoring card 的 heat map 表示法 ... 30
圖 13 Scoring card 之 independent test data 的分數分佈長條圖 ... 31
圖 14 IGA-scoring card 之 independent test data 的分數分佈長條圖 ... 32
圖 15 視窗型界值的結果... 33
圖 16 Scoring card 的 ROC curve ... 34
1
第一章 緒論
1.1 研究動機
蛋白質表現是利用基因工程技術將目標蛋白質的 DAN (Deoxyribonucleic acid)序列與載體(vector)進行重組後,轉殖入宿主細胞,藉由宿主細胞大量表現 出目標蛋白質的方法,此種表現白質的技術已廣泛應用在許多生物實驗、醫學、 製藥產業等方面。而大腸桿菌(Escherichia coli,E. coli)的基因序列已被人類充分 了解,且具有生長快速、產量高、成本低等優點[1, 2],所以目前以大腸桿菌做 為宿主細胞之表現系統最為普遍使用,但利用大腸桿菌為表現系統時有一個常見 的缺點,就是蛋白質在表現時容易形成不溶狀態的包涵體(inclusion body)。 形成包涵體的蛋白質並沒有進行正確的折疊而產生錯誤的結構,在錯誤的結 構下缺乏了此蛋白質原本的功能,所以生物活性極低或不具有生物活性。如何不 讓蛋白質形成包涵體一直是生物實驗上渴望解決的問題,但蛋白質在表現的過程 中形成包涵體的機制尚未明瞭。以往生物學家會利用很多方法,例如:附加一段 增加溶解度的融和蛋白(fusion protein)、降低表現溫度或誘導物濃度,以減緩表 現速率等的方法,為了得到結構正確的可溶性蛋白質(soluble protein)。在過去的 研究中指出,蛋白質胺基酸(amino acid)序列的單點突變(point mutation)就可以讓 在相同表現條件下的蛋白質改變可溶性[3-9],造成蛋白質狀態從包涵體變成可溶 性蛋白,所以代表著胺基酸的序列也扮演著影響著蛋白質溶解度的角色。1.2 研究目的
本篇研究主要有兩個目的,第一為利用蛋白質一級結構(primary structure)預 測在 37℃下大腸桿菌表現系統中所表現出的蛋白質為可溶性蛋白質或包涵體, 可以讓生物學家先知道在正常表現條件下蛋白質表現出的可能狀況,再加以對表 現條件做適度的調整,可省去大量需從未知的情況下開始測詴實驗條件的時間與 資源。 目前對於蛋白質形成可溶性蛋白質或包涵體的機制還尚未明白,第二個目的 是希望可以找出蛋白質一級結構的特性中有哪些會影響在大腸桿菌表現系統中 蛋白質的溶解度,而又是如何影響的,得以更進一步了解蛋白質在被表現出來到 形成特定結構的機制。1.3 研究題目定義
本篇研究主要為提出一個新的方法,利用統計雙胜肽的方式來建立出 scoring card,並經過智慧型基因演算法(IGA)調整後,使之用在分類蛋白質在大腸桿菌 表現系統中所表現出的溶解狀態。因雙胜肽在此研究的過程中被發現為分類此問2 題的重要特徵,所以進一步以雙胜肽特徵來做延伸的研究方法,而發展出雙胜太 分數卡的方法,以 400 個分數當作權重來算出待測蛋白質的分數,再以一個臨界 值區分此蛋白質的類別。 且此區分類別之臨界值能擴張為一個範圍,只計算為在範圍值兩端的蛋白質 預測結果,如此一來便可提升整體準確率,也可讓使用者只需對準確率有超過某 個值以上的蛋白質拿來做後續的研究。
1.4 章節概要
本篇論文第一章主要介紹此篇研究的動機與目的,和之前與此論文主題相關 的研究。 第二章在說明蛋白質表現系統的機制,因為研究中是利用蛋白質序列來做預 測,所以此章從 DNA 之重組技術介紹起,與表現後的蛋白質是如何來測得其為 溶性蛋白質或包涵體。 第三章為現有方法介紹,在敘述先前已經存在並被使用在本研究中的的一些 研究方法,會較偏向簡單的理論介紹。 而第四章為本研究為了解決此研究中之分類問題所提出的方法。為本論文之 重點部分,一開始即介紹此篇研究所使用的 dataset,而後則說明實驗中之各個方 法的應用與描述了本研究中所提出來解決問題的新方法。 第五章為實驗結果與討論,一一介紹每個實驗方法之結果並討論分析此些結 果。而此張之最後一節為對結果做較詳盡的生物分析。 第六章為最後一個章節,為對與此篇研究所做之結論與位來展望的探討。3
第二章 重組蛋白之蛋白質表現系統介紹
2.1 重組 DNA 技術
在整個蛋白質表現的實驗中,最先要做的事情就是將我們欲表現的目標蛋白 質基因與載體結合,而這個結合的過程就稱為重組 DNA。重組 DNA 技術 (recombination DNA technology)就是一般常聽到的基因工程。如圖 1,首先以相 同的內切酶(endonucleases)在質體(plasmid)與外源基因上切出相同的切位點,被 切開的質體與切成片段的外源基因再以連接酶(ligase)結合在一起,及形成重組的 DNA。 圖 1 重組 DNA 技術之過程 資料來源: 詴驗方案(http://www.51protocol.com/) 說明: 利用相同的內切酶(此圖範例為 EcoRI)在質體與外源基因上切出相同的切 口(此圖範例為 TTAA 與 AATT),再利用連接酶連接切開的質體和外源基因片段, 重新組合成新的基因序列。
2.2 大腸桿菌蛋白質表現系統
將重組好的 DNA plasmid 轉殖入大腸桿菌後,先經過抗生素(antibiotic)的篩 選,可去除沒有轉殖成功的大腸桿菌,再將剩下帶有重組 DNA 的大腸桿菌大量 複製,如圖 2。
4 圖 2 帶有特定質體的大腸桿菌大量複製 資料來源: 科學月刊三十卷十二期, 1999-基因工程的衝擊。 說明: 利用抗生素可篩選出含有帶重組 DNA 的大腸桿菌,再將帶有重組 DNA 的大腸桿菌大量複製。 經過篩選內部含有重組 DNA 的大腸桿菌接下來會被進行培養,通常是以正 常溫度 37℃在 LB broth (Luria-Bertani broth)中培養數個小時。經由加入誘導物可 啟動大腸桿菌內重組 DNA 所攜帶之外源基因的表現,而在培養的數個小時中, 藉由大腸桿菌的資源與運作,外源基因會轉錄成 RNA (ribonucleic acid),再轉譯 成蛋白質,此表現出的蛋白質就是實驗人員想要的目標蛋白質。而目標蛋白質存 在於大腸桿菌內部,所以培養結束後再使用超音波震盪(sonication)、溶解(lisys) 或冷凍-解凍等方法打破菌體以萃取蛋白質。
2.3 凝膠體電泳法(SDS-PAGE)
凝膠體電泳法(SDS-PAGE)為利用界面活性劑 SDS (sodium dodecyl sulfate) 使蛋白質變性(denature),並附在變性的蛋白質的表面,藉由通過電流讓蛋白質 上本身攜帶負電荷的 SDS 往正極移動,而膠體的密度,即內部孔洞大小對不同 分子量蛋白質有篩選效果,分子量越小的蛋白質在移動時的速率越快,相反的, 大分子量的蛋白質移動速率越慢,所已經過一段時間後,小分子量的蛋白質會跑 在膠體的前端,而大分子量的蛋白質會留在後端,再經過分子量的比對,則可得 知實驗中的目標研究蛋白質為哪一個。 將打破後的大腸桿菌經過低溫高速離心會分成上清液與沉澱物兩層,上清液 所包含的為大腸桿菌內部正常的蛋白質;而沉澱物則是由細胞碎片與蛋白質聚集 在一起大團分子。如果此次表現出的蛋白質為可溶性,則此蛋白質會存在於上清 液的部分,反之,如果蛋白質為不溶性的包涵體,則會存在於底部之沉澱物,拿 取上清液與沉澱物分別去跑 SDS-PAGE 後,藉由分子量的比對可看出表現出的 目標蛋白是存在於上清液還是沉澱物中,由此可判斷此次表現的蛋白質為可溶性 蛋白或是包涵體。
5
2.4 包涵體(inclusion body)
從大腸桿菌表現出的外源蛋白質,有些是折疊(folding)正確且有正常功能或 酵素活性,萃取後可以直接拿來做後續的實驗,但有些表現出的蛋白質並非如此, 許多蛋白質在表現的過程中會聚集(aggregate)成包涵體,這些包涵體沒有正確的 折疊結構,也沒有正常的功能與酵素活性,且不溶於水,所以這些形成包涵體的 蛋白質並沒有任何功用,不能拿來應用在後續的實驗當中,而這個問題長久以來 一直困擾的生物學家。 許多生物學家會用降低表現時的溫度或是降低誘導物的濃度等方式[10],讓 大腸桿菌新陳代謝速率減低,表現目標蛋白質的速率也減緩,避免在折疊的過程 中速率太快導致過多的疏水性胺基酸任意鍵結,形成不對的結構;或是在欲表現 的蛋白質之前端或後端添加一段融合蛋白(fusion protein)[11, 12],以提高整體的 親水性;另外也還有其他方式例如:調整培養液的成分、選擇其他大腸桿菌品種 或其他表現系統等。但這些改變實驗條件以得到可溶性蛋白質的方法目前都還是 需耗費很多時間與資源的反覆測詴(Trial-and-error),沒有特定的依據,只能逐一 去測詴每個實驗條件改變後的結果,且不管是從新製作重組 DNA 或是重新培養 大腸桿菌,都需要又再花費數小時至數天的功夫。而能夠有系統性指標出現的話, 將會讓科學家對於此機制有更多了解。 但是如果改變了許多實驗條件都無法得到可溶性蛋白質,那生物學家只好將 包涵體進行變性(denature),先打斷蛋白質中所有的鍵結,讓蛋白質打開形成一 及結構,再藉由低溫緩慢的重新折疊(refolding),讓蛋白質能折疊回原本正確有 生物活性的結構。但是變性與重新折疊的過程都需要花費非常久的時間,也會消 耗多餘的資源,且不是所有包涵體經過變性與再折疊都可以變回有生物活性的蛋 白質,大約只有三成的產量才具有生物活性,甚至更低[13],而就算經過變性-重新折疊但沒有生物活性的蛋白質,也是沒有任何功用,無法在後續的實驗中被 使用。所以生物學家在做蛋白質表現實驗時,都會盡量以能直接取得可溶性蛋白 質為主,而花費許多時間與資源在反覆測詴表現時的實驗條件,如果能發展出一 套預測蛋白質在大腸桿菌表現系統中的溶解度工具,可以大幅減低實驗人員時間 與資源的耗費。6
第三章 背景知識與方法
3.1 智慧型基因演算法(IGA)
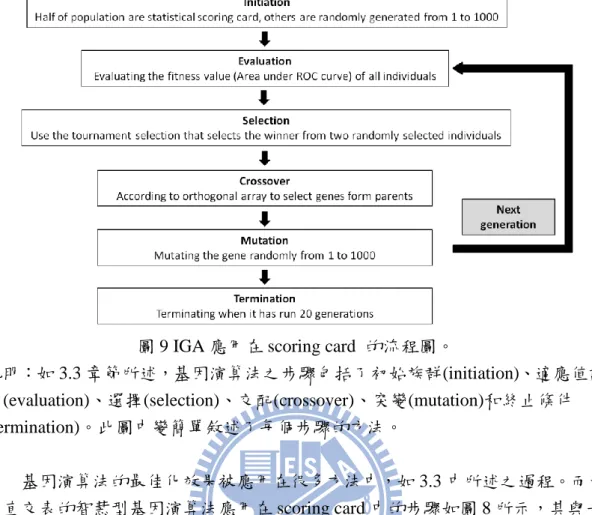
不同於一般的基因演算法,智慧型基因演算法在其中交配(crossover)之步驟 加入了直交表(orthogonal arrays,OA)的機制,使可以有效率的來挑選出好的參 數,所以能夠克服參數量大的問題。適應性函數(fitness function)是用來評斷每 組基因組合之實驗的好壞,而適應性值越高的基因組合,則可被當作子代保留遺 傳至下一個世代。下文中會先介紹一般的基因演算法再比較智慧型基因演算法與 一般基因演算法的不同處。3.1.1 基因演算法
基因演算法(genetic algorithm , GA)是目前廣為應用的最佳化方法之一,其原 理為仿效自然界的生物中之物競天擇的方式,從族群中經過交配(crossover)與突 變(mutation)等過程,挑選出較好的母代遺傳至下一個世代,反覆經過多個世代 後,較好的基因就可被保留至最後。GA 是屬於平行運算,能同時考慮空間中的 多個點,而不會使答案陷入局部最佳解。 GA 的演化流程如圖 3 所示,起初先隨機產生 N 個初始族群,然後評估每個 每個個體的適應函數值,再經由適應函數值每次來挑選兩個親代進行交配,也就 是互相交換彼此的基因,可分為單點交配、多點交配、隨機均勻交配等多種方法。 交配後產生與親代相同數目的子代再依據突變機率來決定是否做進一步的突變, 去單一的改變個別基因,然後這些個體再進入下一個世代,再回到評估是應函數 值步驟,去評估每個個體的適應函數值,直到滿足終止條件為止,就會停止並達 到最佳化。 圖 3 基因演算法流程圖 說明:最開始產生初始族群,經過評估適應函數值決定是否到達終止條件,如果
7
沒有到達終止條件,就會進行挑選、交配、突變等步驟,產生下一代群體,重複 此些過程直到達到終止條件。
3.1.2 直交交配法
相較於全因子參數實驗(complete factorial experiment)會去對每個參數排列 組合後產生的所有可能性做評估,使得實驗過為龐大,耗費大量時間,IGA 利用 直交表來作部分因子參數實驗(fractional factorial experiment),設計有效率的實 驗參數組合,使得在最少的實驗次數中可得到最佳的結果。 直交表中的每一行(row)代表每一個參數,也就是染色體中的每個基因,而 每一列(column)代表一條染色體,也就是等於一次實驗組合。每個參數的主效果 (main effect)為不考慮其他參數影響的情況下,單一參數對結果的影響,所以因 此可以依照主效果來對所有的參數做影響力的排序。 兩水準直交表的設計為一個 L M (2 M-1 )的表格,包含了 M 列和 M-1 行,也 就是 M 次實驗與 M-1 個因子所組成,如表 1 的範例為一個 L 8 (2 7 )的直交表, 每組實驗都會得到一個適應性指標值(fitness),每一個因子都會可計算出 Sj1與 Sj2, 其分別代表每一個因子在水準 1 與水準 2 的情況下之適應性指標值總和,而主效 果分析(MED)則是將 Sj1與 Sj2的值相減後取絕對值,而從 MED 我們可以得知哪 些因子對得到較好的適應值有較大的貢獻,在表 1 中就可由 MED 得知影響力最 大的為第六個因子,因為它的 MED 是七個因子裡面最高的。 表 1 L 8 (2 7 )直交表與主效果範例 資料來源:擷取自[14]並加以修飾。 說明:以 L 8 (2 7 )直交表來當作範例,解釋直交表的應用與主效果分析。
8
3.2 繼承式雙目標基因演算法
繼承式雙目標基因演算法(Inheritable bi-objective genetic algorithm, IBCGA)[15]為一種尋找最佳化參數的工具,可以在大量的參數中挑選最少的參數 並得到最大的適應性(fitness)。IBCGA 主要由 IGA 智慧型基因演算法(Intelligent genetic algorithm, IGA) [16]與繼承機制構成。
適應性函數(fitness function)是用來評斷 IBCGA 的結果好壞的指標,在本實 驗中,此適應性指標是每個染色體經過SVM 分類後的準確率,所以下面會先介 紹SVM 之原理。IBCGA 可以同時尋找出最少量且有影響力的特徵與 SVM 中的 C 和 γ 參數之組合。
3.2.1 支援向量機 SVM
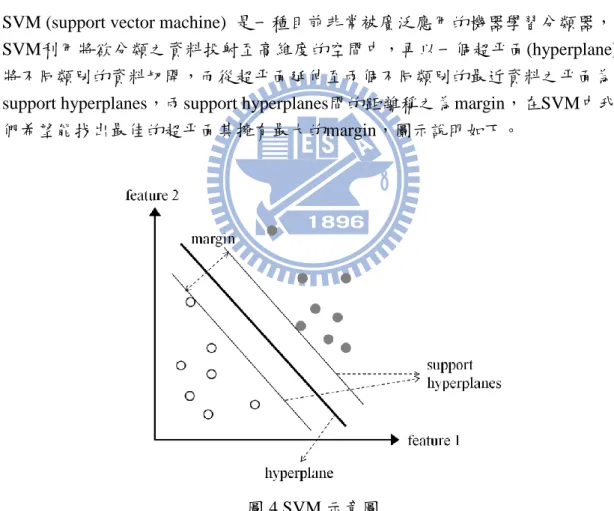
SVM (support vector machine) 是一種目前非常被廣泛應用的機器學習分類器, SVM利用將欲分類之資料投射至高維度的空間中,再以一個超平面(hyperplane) 將不同類別的資料切開,而從超平面延伸至兩個不同類別的最近資料之平面為 support hyperplanes,兩support hyperplanes間的距離稱之為margin,在SVM中我 們希望能找出最佳的超平面其擁有最大的margin,圖示說明如下。
圖 4 SVM 示意圖
說明: 以二維空間為例子,利用資料在 feature 空間的分布,在其中找到一個使 margin 最大的超平面,使兩類資料可以分開至最大距離。
假設training dataset共有n個資料點,分別為{xi , yi},i =1, 2,...,n, xi ∈ Rd,
資料點類別yi ∈ {1, -1},而我們希望找到一條直線 f(x)=wTx-b 對於兩組資料點
9 +δ與wTx=b-δ,再將其做scaling則可簡化為wTx=b+1與wTx=b-1。兩個 support hyperplanes間的距離為2/||w||,所以希望得到最大的margin就可以衍生 出下列公式: ε ε 其中b為常數,εi為第i個資料點如果是位在與其類別不同側時的誤差值,C 為花費參數,用來決定懲罰分類錯誤時的權重。所以在這個公式中我們希望同時 得到最大的margin與最小的誤差值。
在SVM中最常被使用的核心方程式為放射基底方程式(radial basis function), 在本研究中也是使用此核心方程式,其公式為: k(xi, xj)= exp(−γ∥xi−xj∥),γ> 0 (2) γ 為核心參數,將資料點映射到高維度的特徵空間。在 SVM 當中,使用者 則藉由調整 C 與 γ 參數來找到一個最佳的超平面。而在多類別的分類問題中, SVM 為兩類分類器,所以可分為一對多(One-against-all,OAA)與一對一 (One-against-one, OAO)兩種方式,一對多是將某個類別當做一類,而非此類 別的其他資料則當做是另一類,此方式分類的時間較短;一對一則是需要訓練 N(N-1)/2 個二分類分類器,使每一個類別一一做比較,所已花費的時間也較長。 本實驗是應用Chih-Jen Lin 實驗室所研發出之 LIBSVM[17],其所包含了完整的 套件如最佳 C 與 γ 參數的尋找工具。
3.2.2 染色體編碼
IBCGA 中的染色體,是使用二維化 (binary) 來挑選特性基因,加上兩個 SVM 分類器中的參數 C 和 γ所組成的 GA 染色體 (GA-chromosome)。例如 aaindex 有 531 個物化特性要去挑選,前面 531 個基因就分別決定哪些特性要被 挑選出來,而挑選的機制如圖 5 的範例中所示,如果 genei = 0,則第 i 個特性就 不挑選出來當作 SVM 分類的參數,但如果 genei = 1,第 i 個特性就包含在 SVM 分類器的參數中。最後兩個基因包含了兩個 4 位元(4-bit)的 GA 基因來調整 SVM 中的 C 和 γ 參數,而 C 和 γ 參數則是由 16 個值下去作挑選,分別為{2 -7 , 2 -6 …, 2 8 }。所以每組染色體都包含了特定的特性與配合此組特性的 SVM 中之 C 和 γ 參數。10 圖 5 染色體編碼範例示意圖 資料來源:擷取自[14] 說明:使用 aaindex 531 個物化特性來當作繼承式雙目標基因演算法之染特體編 碼的範例。
3.2.3 繼承式雙目標基因演算法實驗流程
先將資料進行前處理,將資料轉換成 IBCGA 可讀取的檔案格式,因為 IBCGA 中是以 SVM 分類出的準確率來當作適應性函數值,所以此輸入的檔案格式也與 libsvm 輸入的檔案格式相同。 而在一開始時,即設定好 GA 染色體中之編碼為 1 個個數與終止條件的編碼 為 1 之個數,設定好各參數值後 IBCGA 就會開始照以下流程進行: 1. 初始化(Initiation):隨機產生一組起始的群組(population),其中包含了 N 條 染色體(individuals)。在染色體中的 n 個基因中包含了在參數設定時有幾個 1 的基因。 2. 評估(Evaluation):利用 SVM 分類的準確率來評估每條染色體的適應性函數 值。 3. 選擇(Selection):利用傳統的比較方法,隨機比較兩條染色體,並將有較好 結果的染色體放到交配池(mating pool)中。 4. 交配(Crossover):將直交實驗運用到交配過程中,不以隨機的方式做交配, 而是經由直交表來選出較優良的基因遺傳至子代中。 5. 突變(Mutation):隨機選擇 p m ×N 個子代染色體來進行交換式突變(swap mutation)以產生新的子代染色體,為 p m 突變發生率。在突變的過程中, 為了避免最好的結果被消滅掉,因此 IBCGA 會先把最好的子代染色體挑出 來,使其不參與突變步驟。 6. 結束測詴(Termination test):如果參數解滿足停止條件,則輸出一組最好的 染色體參數解,如沒有達到停止條件時,則回到步驟 2。3.3 相關研究介紹
利用胺基酸序列預測蛋白質溶解度的模型最早是由 Wilkinson 和 Harrison 在 1991 年時建立的[18],當時他們只使用了 81 個蛋白質與六個特徵,且在此 81 個 蛋白質中有很多蛋白質包含了融合標幟(fusion tag)或其它外加的蛋白質序列,最 後得到的準確率值為 88%。11
而後再 1999 時,Davis 等人修改了 Wilkinson 和 Harrison 的辨別分析模式 (discriminant model),並發現 Wilkinson 和 Harrison 所使用的六種特性中,轉折形 成胺基酸(turn forming residues)其包括 Asn,Gly,Pro 和 Ser 與平均電荷(average charge)兩種特性是決定性的影響因子[19]。
2005 年 Idicula-Thomas 和 Balaji 也利用了辨別分析(discriminant analysis)預測 了一組新的 170 個蛋白質資料與特性[20],並得到 62%的準確率。隔年他們又增 加了一些蛋白質資料,並改用 SVM(Support vector machine)當做分類器,準確率 也大幅提高至 72%[21]。
2007 年時 Smialowski 等人發展了一套 PROSO 系統,為一個結合 SVM 和 Naive Bayes 的兩層分類器,並使用了 14200 個來自 TargetDB 與 PDB 資料庫的 蛋白質資料,得到 71.7%的準確率[22]。並使用 Wilkinson-Harrison 的模型來測詴 此 14200 個蛋白質資料指得到 56.2%的準確率,也使用了 Idicula-Thomas 2005 的 的模型來測詴 14200 個蛋白質資料並也只得到 53.1%的準確率。
2009 年,Magnan 因為了想解決蛋白質資料太少而收集了許多資料庫與文獻, 建立了含有 17408 個蛋白質的資料庫,其來源包含 PDB、SwissProt、TargetDB 和先前文獻,並使用了兩層式 SVM(two-stage support vector machine)[23]。
隔年,Diaz 提出了之前都無人使用的邏輯迴歸(logistic regression)分析了 212 個蛋白質[24]。最近一篇相關研究是出自於台灣中研院的 Wen-Ching Chan,這篇 的特色為其蛋白質資料包含不只有目標蛋白質,因為以往的研究中都認為如果目 標蛋白質接上其他會干擾溶解度結果的蛋白質序列,就會影響預測過程,而此篇 文獻中的蛋白質資料也包含了不同載體上攜帶的融和蛋白(fusion protein)[25],與 以往研究中只預測目標蛋白質不同,考慮到了在真實生物實驗情況中,通常表現 出的不只有單一的目標蛋白質,而會依照實驗需要附加不同的融和蛋白;而此文 獻使用的是 libsvm 的方法並在兩類的分類中得到 83.5%之準確率,其中此篇作者 有嘗詴將 libsvm 做修改,但結果都沒有原始的 libsvm 來得好。
12
第四章 研究方法
4.1 研究資料
在這篇研究中的資料主要有三篇相關文獻:1) SOLpro: accurate
sequence-based prediction of protein solubility[23];2) Prediction of Protein Solubility in Escherichia coli Using Logistic Regression[24];3) Learning to predict expression efficacy of vectors in recombinant protein production[25]。以下會分別一一介紹各篇 中所使用的資料與來源。
(1) SOLpro: accurate sequence-based prediction of protein solubility
這篇研究使用了非常龐大的資料量,總共 17408 個蛋白質序列,分別來自於 不同的線上資料庫,包括 PDB (Protein Data Bank)、SwissProt 和 TargetDB,而除 了從資料庫收集外,也納入了先前文獻所使用的蛋白質資料。 (a)PDB PDB 目前含有約七萬多個蛋白質結構資料,作者使用了資料庫中的註解功 能,作者先利用註解“EXPRESSION_SYSTEM:ESCHERICHIA COLI”,選 出蛋白質表現系統是使用大腸桿菌的蛋白質,再從其中選出註解為 “EXPRESSION_SYSTEM_VECTORTYPE:PLASMID”,為使用質體當來做 載體的蛋白質。符合上述註解的蛋白質共 44450 筆,並納入作者的資料庫, 作者將此部分蒐集的蛋白質全部歸於可溶性蛋白質。 (b) SwissProt 作者認為所有大腸桿菌中本身的酵素(enzyme)理所當然在大腸桿菌中會是 可溶的、結構正確、有生物活性的,所以作者在 SwissProt 中搜尋有註解 “E.coli”, “Enzyme”和“Reviewed”的蛋白質,共找到 3306 筆資料,之 後將這些所有屬於大腸桿菌的酵素蛋白全部歸在可溶性蛋白類別。 (c) TargetDB 在 TargetDB 中每筆蛋白質資料都包含了蛋白質在製造過程中的狀態,包 括 ”cloned”, “expressed”, “soluble”和”purified”等。作者提到在這 些狀態的註解中,對搜尋與此篇研究相關的蛋白質有兩個嚴重的不足,一是 這些狀態中並沒有明確指出蛋白質是屬於不溶性蛋白質,在這個資料庫中, 可溶性蛋白質有被標註為”soluble”,但不溶性蛋白沒有特別標註;另一則 是在所有蛋白質的狀態中,並沒有標註蛋白質是從哪一種表現系統中得到的 註解,但作者提出蛋白質在其他表現系統中所表現的溶解度,會與在大腸桿 菌表現系統中表現出的狀態大至相同,所以最後作者蒐集有”Cloned”和 “Expressed”註解的蛋白質 76503 筆,再從其中將有標註為”Soluble”的 蛋白質歸到可溶性蛋白類別,而沒有標註”Soluble”的蛋白質則歸類為包涵 體類別。
13
(d)先前文獻
在先前文獻中所使用的蛋白質資料,大部分都已經在 PBD 與 TargetDB 的資 料庫出現過了,唯一例外的是在 Idicula-Thomas 的” Understanding the relationship between the primary structure of proteins and its propensity to be soluble on overexpression in Escherichia coli”[20]這篇文獻中所使用的蛋白 質資料,所以作者也納入其中的 175 筆蛋白質資料自作者的資料庫。在 Idicula-Thomas 這篇研究中的蛋白質資料來源為從 PubMed 中找尋有關於蛋 白質表現的實驗相關文獻,並經過嚴格篩選,去除狀態不明或文獻中敘述不 清楚的蛋白質資料。 在收集完上述四個來源的蛋白質資料後,作者分別將不屬於下列三種特性的 蛋白質刪除:1)在蛋白質的狀態中有「膜蛋白」之註解或經過 TMHMM[26]的預 測為膜蛋白者,因為膜蛋白在蛋白質表現系統中如果沒有加入其他融合蛋白,是 不可能表現為可溶性的。2)在蛋白質之胺基酸序列中含有兩個或兩個以上連續的 未知胺基酸者。3)胺基酸長度超過 10 或 2000 者。 在這篇文獻中蒐集了許多來源,也建立出龐大的資料庫,但本篇研究只保守 的採用其中來源為「先前文獻」的蛋白質資料,因為在 PDB 來源中,作者將在 PDB 中選出的所有蛋白質都認定為可溶性蛋白質,但本篇研究認為 PDB 內包含 大量的已知結構蛋白質,但並非其中所有的蛋白質在表現的過程中都是以可溶性 蛋白質,也會有許多是包涵體再經過變性-重新折疊、測詴有生物活性後的蛋白 質,所以本篇研究認為將此部分所有蛋白質歸類為可溶性蛋白類別有些不妥。從 SwissProt 所挑選出的蛋白質在作者的設定中為大腸桿菌本身的酵素蛋白質,蛋 是從表現外源蛋白質的角度去看蛋白質表現系統,較少實驗會利用大腸桿菌表現 系統表現大腸桿菌本身的蛋白質,且在分類學的角度,如果大腸桿菌本身內部的 蛋白質佔所有資料的分量過大,會造成分類器將所有蛋白質分類為屬於或是不屬 於大腸桿菌的本身蛋白質,而就不是在分類外源蛋白質在表現時的可溶或不可溶 性質。最後,在 TargetDB 中選取的資料,作者在文獻中本身就有提到關於此資 料庫搜尋此研究需要資料的兩個缺點,一為註解裡無法確定蛋白質為不溶性的包 涵體,二為也無法確定表現系統的宿主為大腸桿菌,本篇研究認為如此之蛋白質 狀態不確定性太高,也並不納入本篇研究的實驗資料。 所以在慎選蛋白質資料的角度下,本篇研究只選用先前文獻證實過的蛋白質 資料。
(2) Prediction of Protein Solubility in Escherichia coli Using Logistic Regression
此篇文獻中總共蒐集了 212 個蛋白質,包括 160 個包涵體與 52 個可溶性蛋 白質資料。作者蒐集蛋白質資料的方式為先前文獻搜尋,搜尋條件為利用大腸桿 菌為表現系統,在 37℃下表現且不含任何融合蛋白或伴隨蛋白(chaperon)之資料, 並在從其中去除膜蛋白的部分。在分泌性蛋白(secretory protein)或穿膜蛋白
14
(transmembrane protein)的 N 端通常會有一段疏水性的訊息序列(signal sequence), 而藉由訊息序列可讓細胞分辨蛋白質該送往何處,而最後都會被切除。所以在此 篇文獻中,作者去除蛋白質資料中的訊息序列,因為訊息序列並不屬於蛋白質結 構的一部分,訊息序列被去除後也不會影響蛋白質溶解度的預測結果。 此篇文獻作者藉由 SDS-PAGE 的實驗方式將 212 個目標蛋白質歸類為屬於 可溶性蛋白或包涵體。由於此篇文獻使用的資料並不是直接從網路上的蛋白質資 料庫搜尋得到,而是從先前文獻擷取出來,並經過作者利用 SDS-PAGE 等實驗 方式證明其溶解度,所以可以證明在此篇文獻中的蛋白質資料可信度,也將予以 採用在本實驗當中。
(3) Learning to predict expression efficacy of vectors in recombinant protein production 這篇文獻中蛋白質資料的來源為台灣中研院之基因體核心設施(core facility), 其使用高通量(high-throughput)之方式得到較多較完整的蛋白質資料。此篇文獻 中所使用了相當廣泛物種之蛋白質資料,包括病毒、細菌、老鼠至人類等種族, 而目標蛋白質的長度為 48 到 1054 個胺基酸。 每筆蛋白質資料都不只包涵了目標蛋白質的序列,且更包涵了一段屬於載體 上的序列,此載體上的序列為一段融合標幟(fusion tag),此篇文獻中使用了六種 不同的融合標幟,分別為:(1) calmodulin-binding peptide (CBP)、(2) glutathione S-transferase (GST)、(3) N utilization substance A (NusA)、(4) Histidine (His)、(5) maltose-binding protein (MBP)和(6) thioredoxin (Trx)。不同的融合標幟能夠協助不 同的目標蛋白增加其溶解度,或是用來當作後續實驗中方便辨認的標幟,但並非 只需要某種特定之融合標幟就足以應付各種的目標蛋白質的應用,所以在真實的 實驗當中,可能會將一個目標蛋白質組合至不同的融合標幟,並將每種組合都放 置蛋白質表現系統中,再去測表現出的蛋白質之溶解度,此方法也是非常費時與 資源的反覆測詴(Trial-and-error)方式。 此篇文獻中共包涵 121 種不同的目標蛋白質與 6 種不同的融合標幟,經過所 有的排列組合後,最後共得到 726 條不相同的蛋白質序列資料。作者並將其分為 兩類與三類,在三類中共分為 231 個可溶性蛋白、 236 個包涵體和 259 個不表 現蛋白;作者為了與先前文獻的分類結果做比較,而在其他先前文獻中都只有將 蛋白質分成可溶性蛋白質與不溶的包涵體,所以作者也將 726 個蛋白質資料分成 兩類的型態,在兩類的分類中,將包涵體與不表現之蛋白質一起組合成不溶蛋白 類別,即可分類可溶性蛋白與不溶蛋白。 因此篇文獻中所有的蛋白質都是經過真實實驗後得到的狀態,所以 726 個蛋 白質也全部納入本實驗的資料中。比較值得注意的是,對於利用胺基酸序列來做 機器學習之部分,此 726 條蛋白質序列中,相同的一個目標蛋白質共重複了 6 次;而相同的一個融合標幟也重複了 121 次,但這樣序列上的重複,並不會造成 機器學習分類器辨別的困擾,因為不同的目標蛋白組合不同的融合標幟質後,經
15 過將胺基酸序列轉換成許多特徵(feature),投射至非線性高維度的特徵空間 (feature space)中,再進行資料分類,而即使相同目標蛋白與六種不同融合標幟所 轉換出的特徵也是完全不一樣的。 統和三篇文獻中所挑選出的蛋白質資料,共有 980 條蛋白質序列,包含可溶 性蛋白質 289 個與不溶性蛋白質 672 個,再經過刪除重複的蛋白質序列後,最後 剩下 957 筆資料,包含可溶性蛋白質 285 個與不溶性蛋白質 672 個,圖示化過程 由圖 6 表示,並將此 957 筆資料以下稱為 Sd957。 圖 6 研究資料來源示意圖 說明:以圖表之方式表達此研究之研究資料的來源與篩選過程。
4.2 使用特徵
4.2.1 關研究使用過的特徵介紹
最早利用胺基酸序列預測蛋白質溶解度的模型是由 Wilkinson 和 Harrison 在 1991 年時建立的,其使用了六種特徵包括: 平均電荷(charge average)、半胱胺酸 分數(cysteine fraction)、脯胺酸分數(proline fraction)、疏水性指標(hydrophilicity index)、胺基酸總數 (total number of residues)和轉折形成胺基酸分數(turn-forming residue fraction)。後續文獻所使用的一級結構特徵中,許多篇都有使用 20 個胺基酸的組成 (amino acid composition, AAC),也有幾篇嘗詴使用 400 個雙胜肽(dipeptide)與 8000 個三胜肽(tripeptide)[20, 21, 25],分子量(molecular weight)與電荷(charge)也是常 被使用的特徵,而與溶解度相關的領域知識(domain knowledge)包括疏水性 (hydrophobicity)也在很多文獻中可以看到[21, 23, 24]。在 2005 年 Idicula-Thomas S 的文獻中另外使用了脂肪族胺基酸(aliphatic index)與不穩度指數(instability index),脂肪族胺基酸包括丙胺酸(Alanine)、異亮胺酸(Isoleucine)、亮胺酸(Leucine) 和纈胺酸(Valine)。2006 年 Idicula-Thomas S 與 2009 年 Magnan CN 的文獻中都使
16
用了可以降低特徵個數的 reduced alphabet,將單一胺基酸依照某些特性予以做分 類,讓特性相同的胺基酸屬於同一個類別。
也有幾篇文獻中有使用到二級結構相關的特徵,例如在最早 1991 年
Wilkinson 和 Harrison 的文獻中有使用到 turn-forming 胺基酸特徵,並且也在 1999 年經由 Davis 證實 Wilkinson-Harrison 模型中的有用的特徵是 turn-forming 胺基酸 和平均電荷(average charge)。在 2010 年 Diaz 使用的邏輯迴歸(logistic regression) 模型當中也使用了α-helix propensity、β-sheet propensity、α-helix
propensity/β-sheet propensity 與 turn-forming residue fraction。
不同與以往只使用胺基酸相關之特徵,比較特別的是在最近一篇台灣中研院 的 Wen-Ching Chan 發表之文獻中,使用了核甘酸相關的特徵,包括核甘酸序列 長度、核甘酸組合、Guanine-cytosine content(GC content)與 Codon Adaptation Index (CAI),因在此篇文獻中的分類分為可溶性蛋白、包涵體與不表現蛋白,作 者認為蛋白質是否表現應考慮 mRNA 表現時的穩定度與 codon 的偏愛性等。
4.2.2 雙胜肽(dipeptide)
雙胜肽為由兩個單一的胺基酸所組成的分子,但在蛋白質序列特徵中的雙胜 肽並不是指單一的雙胜肽分子,而是蛋白質序列中利用滑動窗(sliding window) 使每兩個胺基酸為一組的特徵,而雙胜肽的組合為 20 種不同的氨基酸所組合, 所以共有 20×20 個樣式。本實驗雙胜肽特徵的計算方法如下: 其中 x 是 400 個雙胜肽中的其中一組,N 為蛋白質的長度,也就是胺基酸數 目。單一胺基酸組成(amino acid composition, AAC)與雙胜肽是最簡單、最直觀, 也是廣泛被應用的特徵。而雙胜肽特徵不只提供了蛋白質中胺基酸的組成資訊, 也提供了胺基酸的局部順序(local order),所以比起 AAC,雙胜肽提供了較完整 的資訊。雙胜肽也被證實在很多預測中扮演重要的角色,如蛋白質在細胞內的位 置(subcellular localization)[27]、蛋白質折疊(folding)預測[28]或細胞核受體 (nuclear receptor)的分類[29]等等。4.2.3 物化特性
本實驗中所使用的物化特性特徵是由 aaindex (amino acid indices) database 中 得到的資訊,在 aaindex 中包含了許多已經發表的胺基酸之物化特性,再經由胺 基酸與其對應的物化特性值來將整個蛋白質轉換成各種物化特性數值。在最新版 本的 aaindex 中總共含有 544 個物化特性[30],而其中有些特性的並沒有某些胺
17 基酸的值,其顯示為 NA,將含有 NA 的物化特性刪除後,共剩下 531 個。 利用篩選物化特性之特徵來預測,可以很明白的了解是甚麼樣的特性在影響 著蛋白質的分類,此部分可以搭配被挑選出的雙胜肽來交叉做分析,因為被挑選 出的雙胜肽也應該是帶有某些特性而影響著本身的蛋白質。蛋白質序列轉換成物 化特性的步驟如下: 1. 從 aaindex 的資料庫中,取得 531 個物化特性對應到每個胺基酸的值。 2. 將每個蛋白質中的胺基酸轉換成物化特性值。 3. 以平均的方式,將其中一種物化特性之每個胺基酸對應值相加,在除以 序列長度。 4. 一個蛋白質資料可得到 531 個物化特性值。
4.3 繼承式雙目標基因演算法實驗流程設計
在此章第一節中有介紹本實驗中使用的 dataset,經篩選整理過後共有 957 個蛋白質資料,其中包括可溶性蛋白質 285 個與不溶性蛋白質 672 個。此次實驗 流程設計分為兩層,在最開始先將整個 dataset 隨機的分成五份,並重複此步驟 十次當作十重複,此五份為第一層,其中一份要拿來當作最後的 independent test, 如圖 7 中最右邊之咖啡色箭頭處,而這一份資料是完全不會出現在任何訓練與找 尋特徵的過程;另外的四份則進入尋找特徵的步驟。這四份再新分為五份,每個 第一層的重複都再重複十次,所以在第二層中共有一百個重複,新的五份中的一 份拿來當作 IBCGA 實驗中的 independent test,如圖 7 中之紫色箭頭處,而新的 五份中的四份則用來當作 IBCGA 實驗中的 training data,分別進行十重複之 400 個雙胜肽與 531 的物化特性之 feature selection。經過 IBCGA 實驗,分別從雙胜肽與物化特性中挑選出較有影響力的特徵後, 再將其組合,之後使用組合後的特徵來訓練原本第一層中五分之四的 training data,並用第一層中留下來五分之一的 independent test 來測詴最後的結果,如此 最後會得到 10 個 independent test 結果,並將之平均。
此種兩層實驗資料分法是為了要將從 400 個雙胜肽與 531 個物化特性中挑選 出來的特徵加以整合,而最後測詴整合後特徵的資料是不能出現在特徵挑選的過 程中的。
18
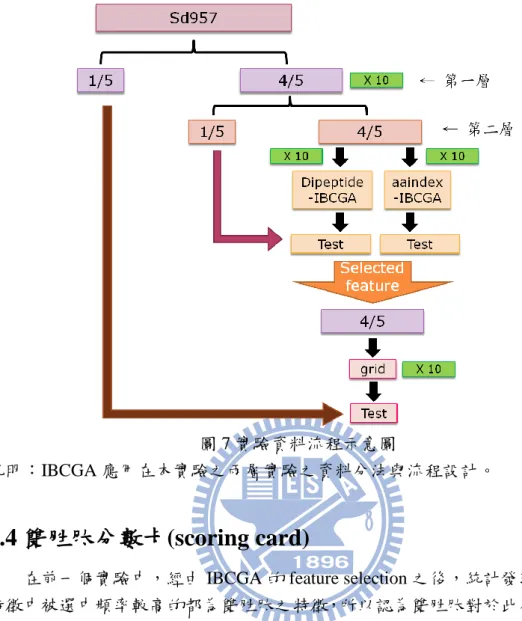
圖 7 實驗資料流程示意圖
說明:IBCGA 應用在本實驗之兩層實驗之資料分法與流程設計。
4.4 雙胜肽分數卡(scoring card)
在前一個實驗中,經由 IBCGA 的 feature selection 之後,統計發現挑選出的 特徵中被選中頻率較高的都為雙胜肽之特徵,所以認為雙胜肽對於此分類問題應 該有較大的影響力。此部分之實驗為利用雙胜肽特徵做進一步的研究。在此我們 提出一個既簡單效果又好的方法,並且可以直接的從以容易明瞭之統計方式,將 蛋白質序列轉換成 400 個雙胜肽值,來做成一個 20×20 的 scoring card,再依據 此 scoring card 與臨界值(threshold)的挑選來做為蛋白質的分類。
4.4.1 分數計算
此方法為利用兩組類別中雙胜肽的差異程度建造出一個含有 400 個雙胜肽 分數的 scoring card,再將預測詴的蛋白質序列中之雙胜肽個數乘上分數卡中相 對應的雙胜肽分數,之後每一個預測詴的蛋白質都可以得到一個分數。雙胜肽分 數卡之計算與詳細的實驗步驟如下: 1. 將 training data 分為 0 組(可溶性蛋白質)與 1 組(不溶性蛋白質),再分別 計算兩組中每個雙胜肽之個數。(例如:AA 在 0 組有 1067 個;AA 在 1 組有 1833 個)。19 2. 因為 0 與 1 兩組內的蛋白質資料個數不相同,每個蛋白質的長度也不同, 所已將 0 組的每個雙胜肽個數分別除以 0 組的總雙胜肽數;將 1 組的每 個雙胜肽個數分別除以 1 組的總雙胜肽數。(例如:0 組的總雙胜肽數為 97147;1 組的總雙胜肽數為 217263。0 組的 AA 數除以總數等於 0.01098; 1 組的 AA 除以總數等於 0.0084)。 3. 將 0 組的某個雙胜肽個數減 1 組的相同之雙胜肽個數(預設為可溶性蛋 白質的雙胜肽為+1 分;不可溶性蛋白質的雙胜肽為-1 分)。(例如:兩組 的 AA 相減為 0.002547)。 4. 將兩組相減之雙胜肽數值都各乘上 1000,建立出包含 400 個雙胜肽的 scoring card。(例如:0.002547 乘上 1000 等於 2.547)。 5. 此時在 scoring card 中每個值為有正負號之小數點,為了減低複雜度, 所以又將每個分數 scale 成 0~1000 的數值。(scoring card 的 AA 經過 scale 成 0~1000 後變成 792)。 6. 計算出 test data 中每個蛋白質序列之 400 個雙胜肽個數,將在此蛋白質 的雙胜肽個數乘上分數卡中相對應的雙胜肽數值(例如:第一條測詴蛋 白質的 AA 有 1 個,所以 1×792 等於 792,以此類推,如果此蛋白質沒 有某個 dipeptide,那乘出來的結果就是 0)。 7. 將欲測詴蛋白質乘完分數卡後的 400 個雙胜肽數值相加,加總後的值再 除以本身蛋白質的胺基酸個數。(將上述步驟所計算出的每個分數從 AA+AC+AD....YY,再將此分數除以蛋白質長度)。 8. 最後 test data 中每個蛋白質都得到一個分數。
4.4.2 分類方法
藉由分數卡來計算出每個蛋白質的分數,需再以此分數為依據來判斷此蛋白 質屬於可溶性或不可溶性之類別,而如何挑選一個好的臨界值(threshold)來區分 兩類資料就變得非常重要,且會大大影響到結果的好壞。 因在計算時 0 組(可溶性蛋白質)的雙胜肽得分為+1 分,而 1 組(不可溶性蛋 白質)的雙胜肽得分為-1 分,所以選定某個臨界值後,分數大於此臨界值的蛋白 質將歸類為可溶性蛋白質,而小於此臨界值的蛋白質就歸類為不可溶性蛋白質。 在 training 的實驗中,已經從 training data 中分出五分之一來當做 validation data, 而找到最佳臨界值的方法是利用從 validation data 中最小的蛋白質分數,每次增 加 2 當作臨界值來區分 0 組與 1 組,直到最大且不超過 validation data 中最大的 蛋白質分數,並計算每次的準確率(accuracy)。利用 training data 中所分出的 random validation data 來找出使 accuracy 達到最高的臨界值後,再以此臨界值來 分類 test data。20
4.5 IGA-scoring card
此方法為使用 IGA(intelligent genetic algorithm)加以改良雙胜肽分數卡,調整 其中之雙胜肽分數,並藉由 ROC 的 AUC(area under curve)來判斷調整過後的分 數卡之效能。在基因演算法中,即挑選 AUC 最高的個體遺傳至下一代,使經過 每一代的演化後,最後能找到效能最高的 scoring card。下圖為 IGA-scoring card 的實驗分法與流程。
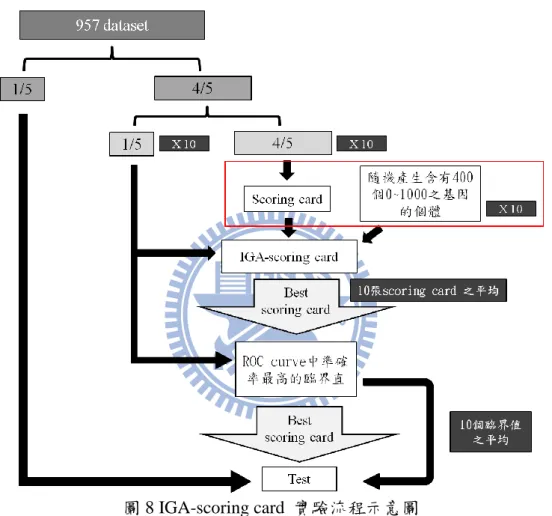
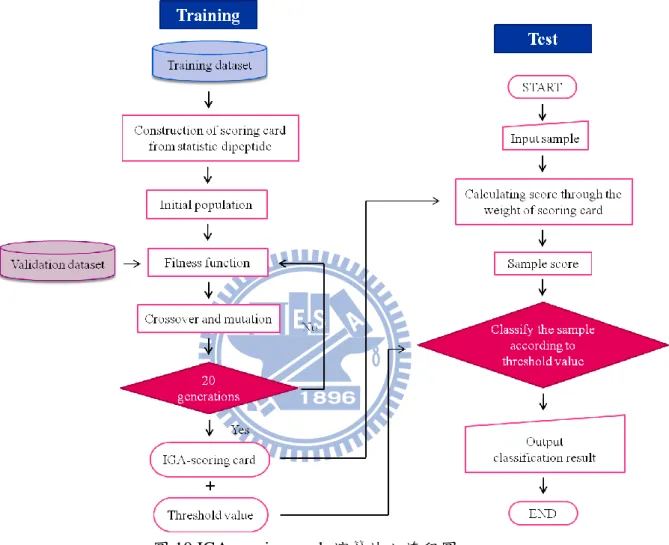
圖 8 IGA-scoring card 實驗流程示意圖
說明:以整個 dataset 的 4/5 來建立 IGA-scoring card 的模型。在初始族群(圖中紅 色框框內)設定中 10 個為真實統計的 scoring car,另外 10 個為隨機產生之個體, 並用此 4/5 中之 1/5 的 data 來進行最佳適應度之 scoring card 的搜尋,最後將 10 個結果平均得到一組適應度最高的 scoring card,並用剩餘且沒有參與建立模型 過程中的 5/1 dataset 來測詴調整後的 scoring card。
21
圖 9 IGA 應用在 scoring card 的流程圖。
說明:如 3.3 章節所述,基因演算法之步驟包括了初始族群(initiation)、適應值評 估(evaluation)、選擇(selection)、交配(crossover)、突變(mutation)和終止條件 (termination)。此圖中變簡單敘述了每個步驟的方法。 基因演算法的最佳化效果被應用在很多方法中,如 3.3 中所述之過程。而加 了直交表的智慧型基因演算法應用在 scoring card 中的步驟如圖 8 所示,其與一 般基因演算法差異較大的步驟包括初數族群之設定、適應度計算與最佳化的交配 方法,所以下文中會再對此幾個步驟做詳細的說明。
4.5.1 初始族群設定
在 IGA-scoring card 的族群中(population),每個個體(individual)的基因編碼 為 400 個基因(gene),就是由 400 個雙胜肽分數構成的 scoring card。
在圖 7 的紅色框框中,初始族群個數設定為 20 個,其中 10 個個體為利用 10 個 training data 所統計出的 400 個雙胜肽分數,統計方法如 3.5.1 中所述,每 張卡中的分數都是經過 scale 成 0~1000 之範圍內的整數。而另外的 10 個個體則 是隨機產生 400 個分數,此分數的最大值與最小值分別為 0 與 1000。
4.5.2 適應度計算
在從 validation data 來每次增加 2 分當作臨界值來區分 0 組與 1 組時,會同 時利用真陽性率(TPR)和假陽性率(FPR)來畫出 ROC(Receiver Operating Characteristic) curve。TPR 與 FPR 的計算公式如下:22 TPR = TP/ (TP+FN) (4) FPR = FP/ (FP+TN) (5) TP 為真陽性、TN 為真陰性、FP 為假陽性、FN 為假陰性。每個個體的適應 度計算方法為將測詴蛋白質的雙胜肽個數乘上分數卡中對應的分數,相加後每個 蛋白質可得到一個分數,而分數卡在此實驗中就是每個基因演算法中的每個個體。 再從 validation data 中找尋一個適當的臨界值,而從找尋臨界值的過程中,可以 藉由準確率、真陽性率和假陽性率來畫出 ROC curve,而計算每個個體的 AUC 就是每個個體的適應度。AUC 的值越大,代表此個體的適應度越好。
在此圖 7 的實驗設計中,原本 4/5 的 training data 又被分為 1/5 的 validation data 與 4/5 的 scoring card 統計資料,所以總共有 10 個不同的 validation data。每 個 validation data 都會參與一次的 IGA 的調整來得到一張適應度最高的 scoring card,因此最後會有 10 張由 10 個不同 validation data 得到的 scoring card,在最 後將 10 張卡片中每個雙胜肽數值予以平均,就會產生如圖 7 中箭頭符號所代表 的一張最好的 scoring card (Best scoring card)。將 10 組從 validation data 得到的 準確率最高的 threshold 做平均,之後得到一個 threshold 的值再拿來用在分類 independent test 的資料上。
4.5.3 直交交配法
在此 IGA-scoring card 的實驗中,因為每個個體都含有 400 個雙胜肽的數值, 所以在直交交配法中產生一組可以容納 400 個因子的直交表,每一個雙胜肽值都 為一個因子。每個直交表共含有 512 個實驗次數,每個實驗都要計算一次適應度, 所以每組實驗都要經過一次 validation data 的測詴,如 3.3.2 章節所述,所以此步 驟會花去較多的時間。而後由主效果分析(MED) (在 3.3.2 中有詳述此方法)來判 斷哪個因子較具有影響力,MED 越大代表此因子越有影響力,而此因子即被保 留在要進入下一個世代的一個子代中,而另外一個子代則是挑選其中一個 MED 最小,也就是最沒有影響力的因子,來交換成另一個親代的因子,如此一來兩個 親代經過直交交配法後也會得到數目相同的兩個子代,而此兩個子代也需經由 validation data 的測詴來計算出適應函數值。 最後由兩個親代、512 組實驗與兩個子代中再挑選適應度最高的兩個進入下 一個世代,但通常是由兩個經過直交表篩選後的子代會擁有最高的適應函數值。 在交配過程中加入了直交表來使交配後的結果能得到最佳化,如此經過幾個世代 很快的就可以得到最佳化因子組和的個體,也就是適應函數值最高的個體。4.5.4 IGA-scoring card 演算法流程
此節為介紹 IGA-scoring card 方法的完整演算法流程,流程圖如圖 10 所示。23
綜合上述所介紹之 IGA-scoring card,在訓練的流程中,一開始由 training dataset 與 validation dataset 來建立出雙胜肽分數卡來經過智慧型基因演算法之最佳化調 整(如圖 10 左半部),並得到最佳化過後的分數卡與最佳之臨界值。而在測詴的 流程中每個輸入的蛋白質 sample 藉由分數卡的計算來得到一個分數,再由臨界 值來判斷此蛋白質 sample 是屬於可溶或是不可溶類別。
圖 10 IGA-scoring card 演算法之流程圖
說明:此流程圖主要分為 training 與 test 兩個主要流程。在 training 部分一開始 為利用 training dataset 來建立出分數卡,再將這些分數卡與 validation dataset 經 由 IGA 的最佳化來得到最佳的分數卡與臨界值;而在 test 的流程中,輸入的蛋 白質 sample 藉由最佳化後的分數卡來計算出一個分數,再依據臨界值來分類此 蛋白質的類別。
4.5.5 視窗型臨界值實驗
在利用 validation data 找到一個使準確率最高的臨界值後就可以用來區分 test data,但 scoring card 的實驗還可以利用 window threshold 的方法來將準確率 再提高。
24
視窗型臨界值實驗(Window threshold)為從最佳臨界值開始,每回合都加減 1 分,直到到達 test data 最邊界的分數。因為在分數計算時 soluble 蛋白質為正分, insoluble 蛋白質為負分,所以兩種類別的蛋白質會由分數高低而往兩邊被區分開, 然而將臨界值從單一數值慢慢拉大成一個範圍時,在此範圍內的蛋白質被認為是 不明確(unknown)的蛋白質,不予以計算此範圍內蛋白質的準確率,而超過這範 圍兩邊的蛋白質就越來越會是屬於本身的類別,如此準確率就會隨著 window threshold 的範圍增大而慢慢提高。
25
第五章 實驗結果與討論
5.1 726 dataset 做 IBCGA 的實驗結果與討論
在最初時使用從中研院 Chan, W.C.發表在 2010 的文獻[25]中相同的資料,此 份資料敘述如 3.1 中的(3),並共包含 617 中特徵。此篇文獻的作者提到他們使用 LIBSVM 中提供的 feature selection 套件後,其中一個最高的準確率從 87.84%降 至 45.95%,所以最後他們決定不使用 feature selection 的結果,將所有 617 個特 徵都使用來分類。 將與[25]相同的 617 資料經過隨機分類成十份的 1/10 的 test 與 9/10 的 training, 此與文獻中的資料分法相同,training 則用在 IBCGA-SVM 來挑選出重要的特徵。 表 2 為十次的統計結果,在表 2 中使用與[25]相同的 617 個特徵經過 IBCGA 挑 選後得到的準確率平均為 80.72,比文獻中使用 617 全部的特徵進行 SVM 分類 的結果略低,文獻中的準確率為 83.51。 表 2 617 特徵與物化特性(PCP)經 IBCGA-SVM 結果 說明:617 為文獻中作者使用的 617 個特徵;PCP 為從 aaindex 取得的 531 個物 化特性之特徵。兩者除特徵使用不同外,其他實驗方法步驟都相同。在 training- validation 中使用 10-fold,test 資料為玩全沒有出現在 training 過程中的 independent test。表格內的結果為經 SVM 分類後的準確率。 統計每次 IBCGA 所挑選出的特徵數量,再分別選出 617 與 PCP 兩組中十重 複的實驗裡面哪些特徵出現次數超過一半,這些特徵列表如表 3。 在 617 個特徵中包含了 87 個核甘酸相關特徵、71 個轉譯後修飾 (post-translational modification,PTM)、459 個蛋白質相關特徵,其中在蛋白質相 關特徵中有 400 個為雙胜肽。由表 3 可明顯看出在 617_IBCGA-SVM 這組中, 經過 feature selection 後選出 17 個出現頻率超過一半以上的特徵,而其中大部分 都是雙胜肽特徵,其他少部分為轉譯後修飾相關的特徵。 在 PCP 中出現頻率超過一半以上的總共有 14 個物化特性特徵。因 617 個特 徵經過 feature selection 後出現頻率高的幾乎都是雙胜肽,於是接下來我將 617 與 PCP 中出現頻率超過一半的特徵整合,形成 31 個新的特徵組合。並想藉由此 新的組合讓 IBCGA 挑選,因為其中幾乎一半包含了物化特性中影響力高的特徵, 如果此部分挑選出的大部分依然是雙胜肽的話,就代表雙胜肽在此分類中扮演了
26
重要的角色。
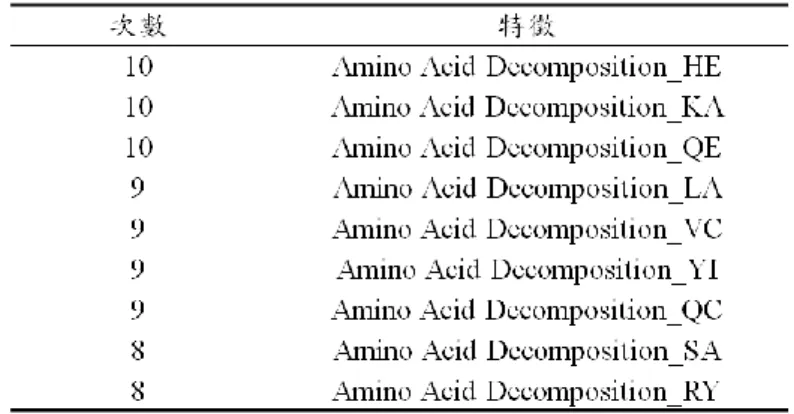
表 3 617 特徵與物化特性(PCP)經 IBCGA-SVM 挑選後統計出現頻率超過一半的 特徵
說明:617_IBCGA-SVM 與 PCP_IBCGA-SVM 兩組各別經過 feature selection 後 再選出出現頻率超過一半以上的特徵。
表 4 是整合後包含 617 與 PCP 的 31 個特徵,實驗步驟與上述實驗相同,經 過 IBCGA 的 feature selection 後選出出現頻率高的特徵,其因為整個特徵資料只 有 31 個,所以增加了每個特徵被挑選出的機率,所以此部分最後挑選十次中出 現 8 次以上的特徵顯示出來。
表 4 整合後特徵經過 IBCGA 挑選出出現頻率高的特徵
說明:由表 3 為經 IBCGA 挑選出的 31 個特徵的組合,再經過 IBCGA 的挑選。 由表 4 的結果可看出,在十次隨機分 training 與 test 的實驗中,出現 8 次以
27 上的特徵都是雙胜肽。由此可推測雙胜肽的影響力非常大。而此上的實驗都是先 以[25]此篇文獻的 dataset 來做的初步實驗。而從此些實驗結果可看得出來 feature selection 的結果被挑選出來的幾乎都是雙胜肽之特徵。所以在此認為雙胜肽對於 soluble 與 insoluble 的分類具有一定的影響力,而後也將對雙胜肽之特徵做進一 步的研究。
5.2 Sd957 做 IBCGA 的結果
此部分的實驗資料不同於 5.1 章節的資料,此為整合 4.1 章節所述之三篇文 獻的資料。資料分法與流程如圖 7 所示,分為兩層,第一層的 test 為最後的 independent test,並不參與在任何的訓練與特徵挑選的過程中,而 IBCGA 的 feature selection 之實驗過程則由第二層來做,此實驗所使用的特徵為 400 個雙胜 肽與 531 aaindex 物化特性。第一層將資料隨機的分成五份十次,而其中每份 4/5 的資料在進入第二層時又隨機的分了十次,所以第二層共統計 100 個實驗之結 果。表 5 前兩個結果為第二層之 IBCGA 做 feature selection 後的準確率結果,400 個雙胜肽的準確率略高於 531 個物化特性,所以從此推測雙胜肽的特徵對於蛋白 質溶解度的分類比物化特性之特徵還較有影響力。而從兩組 IBCGA 的結果中各 選出出現次數超過 15 次以上的特徵,dipeptide 的 feature selection 後選出 41 個, 而 aaindex 中選出 9 個,並將其相加起來,共 50 個組合的特徵,然後再將這 50 個特徵直接做 SVM-grid,直接挑選 SVM 的 C 與 γ 參數。此結果如表 5 的第三 組準確率值,利用挑選後的特徵組合起來的結果原本的結果好出許多,也較穩定。 此可以證明IBCGA 從大量特徵中所挑選出之特徵是真的對分類結果有較具影響 力的。並由 50 個組合的特徵中可得知其中雙胜肽佔了 41 個,為大多數,所以此 結果可再次證明雙胜肽在 soluble 與 insoluble 的分類中所佔的重要性。
表 5 957dataset 做 IBCGA 與選出後特徵做 SVM-grid 的結果
說明:957dataset 之兩層實驗結果。前兩者為第二層實驗中雙胜肽與物化特性使 用 IBCGA 的準確率,最後一個為將從雙胜肽與物化特性所挑選出的特徵結合後 直接做 SVM-grid 的準確率。
28
5.3 Scoring card 結果與討論
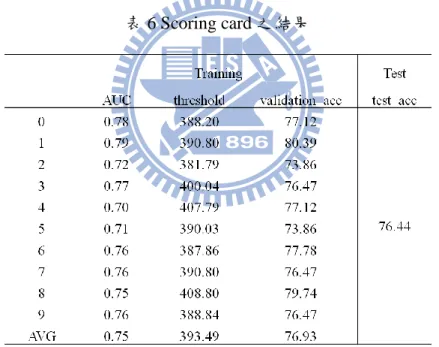
因先前的實驗結果中顯示雙胜肽為分類蛋白質溶解度的重要特徵,且雙胜肽 與其他物化特性等的特徵比起來明顯為較簡單的序列特徵,所以在這邊我們提出 了一個簡單又能有效分類蛋白質溶解度的方法,且讓生物學家可以輕而易舉的知 道每個雙胜肽對於分類蛋白質溶解度的比重,不會像 SVM 等黑盒子(black box) 的方法,無法直覺式的判斷蛋白質的分類。 此實驗的資料分法與圖 7 所示相同,也是將資料先分出一組 independent test, 其他的資料再分為 validation data 與用來統計 scoring card 的部分。十組的結果如 表 6 所示,AUC 為每組由 validation data 所計算出的 area under curve,AUC 的值 越大代表分類能力越好。而表 6 中的 threshold 為 validation data 中使準確率最高 的臨界值,validation_acc 為 validation data 中最高的準確率。Test 只有一組是如 圖 7 所分出的 independent test,使用了 training 後的十張 scoring card 的平均,所 以最後會只有一張有 400 個值的 scoring card,再以此 scoring card 來測詴 test, 並使用平均過後的臨界值之值來分類 test data 中的蛋白質。表 6 Scoring card 之結果
說明:Scoring card 的結果。Training 中包含了 validation 的準確率與最高準確率 之臨界值與每組 validation data 的 AUC。一組的 test 則由十組 scoring card 與十組 臨界值之平均來做分類。
下面的圖為使用 heat map 的方式來表現將平均過後的 scoring card,接下來 也會以此方式來表達 IGA-scoring card。圖 9 利用 scoring card 中不同分數範圍來 用不同的顏色表示,而越極端的值代表權重越重,也越有影響力,也就是如果雙 胜肽為紅色或黑色就代表在 scoring card 越為極端值。
29
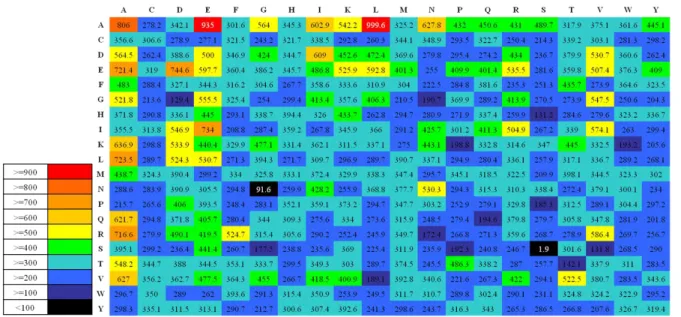
圖 11 Scoring card 的 heat map 表示法
說明:為包含 400 個值的雙胜肽 scoring card,並以 heat map 的形式表現,每間 隔 100 分即為不同顏色。雙胜肽的組合先看行再看列。
由圖 9 可得知在純粹使用統計的 scoring card 中偏向極端值的包括 LA、EA、 AA、DG、GN 和 SS,代表這幾個雙胜肽在 soluble 與 insoluble 的蛋白質中的數 量相差最多。此部分實驗是以單純的統計雙胜肽來達到分類的目的,但雖然使用 的是較簡單的方法,卻也比使用挑選過後的物化特性或雙胜肽來做 SVM 的準確 率還要高,所以此方法有簡單快速又容易明白且能達到優越分類效果的優勢。
5.4 IGA-scoring card 結果與討論
不同於 scoring card,IGA-scoring card 加入了智慧型基因演算法來調整 scoring card 中的分數,使整張 scoring card 能得到更好的分類效果。下表為 IGA-scoring card 的結果,資料分法如圖 7 所示,統計出的十張 scoring card 被用 來當做 IGA 的初始族群,經過十組 validation data 的調整後再將十組 scoring card 做平均來並測詴 independent test。
在表 7 的結果中,IGA-scoring card 中的 AUC 值比 scoring card 的 AUC 高出 許多,平均為 0.84,且其實如果不是將十組 scoring card 做平均,各別由 IGA 調 整過後的 validation data 之 AUC 幾乎都可以到達 0.9 以上甚至幾乎接近 1 的分數, 由此可證明智慧型基因演算法的效力,且有達到以 AUC 來做為 IGA 在調整時的 適應性函數。而在 independent test 的準確率也比沒有經過 IGA 調整的結果高出 5 個百分點左右。
30
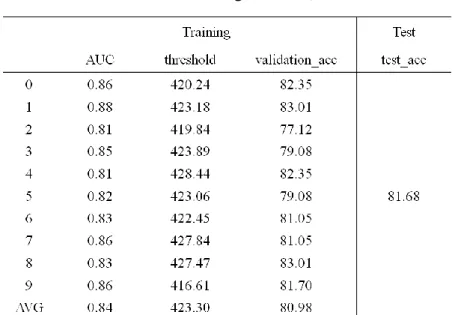
表 7 IGA-scoring card 之結果
說明:IGA-scoring card 的結果。Training 中包含了 validation 的準確率與最高準 確率之臨界值與每組 validation data 的 AUC。一組的 test 則由十組 scoring card 與十組臨界值之平均來做分類。
圖 12 IGA-scoring card 的 heat map 表示法
說明:為包含 400 個值的雙胜肽 scoring card,並以 heat map 的形式表現,每間 隔 100 分即為不同顏色。雙胜肽的組合先看行再看列。
但從圖 10 的 heat map 來看,400 個雙胜肽的極端值稍為被拉近了,經過 IGA 調整後的 scoring card 的極端值變得較少且較不極端,且總觀而言,所有分數值 幾乎都往上提升,整張 heat map 的顏色有從藍色轉變為綠色與黃色的趨勢。但
31
與 scoring card 相同之處就是 LA、GN 和 SS 仍是處於整個 400 個值得最極端值。 而經過調整後的 scoring card 改變最多的雙胜肽包括 EA、VR、LA、AA、 TP、MC 和 SD 等,其中較令人注意的是,EA 在原本的 scoring card 中第二高的 值,而經過 scoring card 的調整後卻變得不是那麼極端,反而是由 AE 跟 AL 取代, 推測 AE 與 AL 在 soluble 的蛋白質類別中也扮演了具有影響力的角色。而不同於 soluble 類別的蛋白質,insoluble 蛋白質類別中仍然以 GN 與 SS 佔優勢,
5.4.1 長條圖分數分析
因為每個蛋白質 sample 被轉換成一個數值,再從中選擇某個臨界值來將兩 類分開。圖 11 為以長條圖的方式來表示 scoring card 之 test 資料中蛋白質的分佈, soluble 蛋白質在計算時為+1 分,所以會偏往高分區,相對的,insoluble 為-1 分,所以會往低分區偏向。但從圖 11 來看,未經過 IGA 最佳化的 scoring card 計算出的分數中兩類蛋白質之重疊率相當大,所以分類準確率當然也不會太高。
圖 13 Scoring card 之 independent test data 的分數分佈長條圖
說明:此為 scoring card 實驗中 test 資料的分佈圖,在此 test 資料中分數最高值 為 441.49,最低為 300.2,X 軸為分數,以每 10 分為一個間隔,Y 軸為此分數範 圍內蛋白質 sample 的數量。藍色的 bar 代表 soluble 蛋白質,紅色為 insoluble 蛋 白質。圖中黃色框框處的範圍為臨界值的切點,此臨界值為 393.49。
而圖 12 為經過 IGA 最佳化後的 IGA-scoring card 的 test 資料中蛋白質的分 佈,與圖 11 比較起來,可以很明顯的看出兩類蛋白質有往兩極區分開的趨勢, 此可以證明 IGA 最佳化的效果是真的能將蛋白質依照雙胜肽的分數來區分溶解 度。
從圖 12 也可看出以 dipeptide scoring card 來分析蛋白質的溶解度,的確是可 以將兩類蛋白質依照此分數來分開,在長條圖中形成兩個雙峰。圖中可看出在