國 立 交 通 大 學
應用化學系所
碩 士 論 文
比較 CH
3I 與 CH
3F 在 p-H
2間質中之紅外吸收光譜
Comparison of Infrared Spectra of CH
3I with CH
3F
in Solid para-Hydrogen
研究生:黃雅苓 (Ya-Ling Huang)
指導教授:李遠鵬 博士 (Dr. Yuan-Pern Lee)
i
摘要
本論文利用低溫間質隔離技術搭配霍氏轉換紅外光譜儀(FTIR) 比較在 500-5000 cm-1光區 CH 3I 與 CH3F 在 para- H2(p-H2)間質中的紅 外吸收光譜。CH3I 在 p-H2間質中之紅外吸收光譜並無像 CH3F 一樣 的單軸轉動之特性譜線,推論為碘(I)原子太大,因此 CH3I 佔了二個 晶格,使 CH3I 之 CH3遇到較大之阻力。且ν4~ν6光區中觀測到許多根 譜線,推論是由於 CH3I 在 p-H2間質中有二種可能之取代位置,且 CH3會有不同位向,因而有不同之吸收譜線位置。CH3F 和 CH3I 在 p-H2間質中所受到 o-H2影響之表現亦很不一樣,CH3F 之 C-F 伸張振 動模(ν3)光區有一系列隨著 o-H2濃度上升而產生的 CH3F〃(o-H2)n團 聚體譜線,且譜線紅位移約 0.7 cm-1,ν 4~ν6除了單軸轉動之譜線外, 僅觀測到單根譜線。而當 o-H2濃度上升時,CH3I 之 C-I 伸張振動模(ν3) 光區僅觀測到譜線半高寬由 0.3 cm-1變至 0.5 cm-1,並無觀測到可解 析之團聚體吸收譜線,但在 CH3搖擺振動模(ν6)則有一系列隨著 o-H2 濃度上升而產生的 CH3I〃(o-H2)n團聚體譜線,且譜線藍位移約 0.5 cm-1, 推論為 F 原子容易和 o-H2形成鍵結使得 C-F 伸張振動模(ν3)受 o-H2 之影響較大;但 CH3I 與 CH3F 之偶極矩相差不大,應不會有如此大 之差異,可能是 CH3I 之 CH3受到間質晶格之阻礙,使得 I 原子不易ii
接近 o-H2,因此 C-I 伸張振動模(ν3)受 o-H2之影響較小。又 CH3I 在
p-H2間質為雙取代,如果 CH3鄰近之晶格分子為 o-H2則其相互作用
iii
謝誌
將本論文獻給實驗室的所有成員:謝謝李遠鵬老師、月貴、Momo、 Barsia、韓姊姊、大學長、阿咩根、阿麥、海哥、魚飯、孔、皇上、 冏達、洋洋、傅龍、書毓、國翔、振孙、建亨,以及將不才的我領進 MIS 世界的瓊瑋學姊、Joerg Glatthaar 教授,還有謝謝 Ella 學姊、yy、 慧芬學姊、鴻菊學姊們的照顧。 二年的時光倏忽即逝,感謝李遠鵬老師願意收我這個程度欠佳的 學生當研究生,謝謝您給我機會。您對研究的熱忱以及教學的認真, 讓我持續充滿幹勁地追隨您的腳步向前行進;當我實驗遇到難題時, 您的提點與指引讓我能自我突破,並順利完成論文的研究與寫作,真 的很開心能當您的學生。 謝謝口詴委員王念夏老師和江素玉老師,讓我的論文更加地完整 有條理以及土屋莊次教授及松為宏幸教授的指導與鼓勵。 謝謝外表堅強、內心柔弱的韓姊姊,我不在妳身邊的時候不要太 寂寞啊!謝謝愛哼歌的棋文大學長,個性龜毛但是很會照顧人;謝謝 臉上總是掛著微笑的阿咩根,做事及想法都很有條理讓我學到很多東 西;謝謝好好先生的阿麥,外表木訥但內心卻是個很有梗的宅宅;因 為你們常常給我許多寶貴的建議,才有現在進步的我,我永遠不會忘
iv 記我的口詴投影片起死回生的事件。
謝謝愛切水果請大家吃的俞範,妳的笑容總是可以安定人心;聰 明但缺乏自信心的孔,謝謝你常和我討論實驗 and smoking Momo, thank you very much!
謝謝讓桃園和新竹近在咫尺的同學海哥、謝謝講話慢條斯理也具 有安定人心功效的皇上、謝謝和孔對話很好笑的勁達、謝謝讓生活充 滿驚奇的洋洋和傅龍、謝謝可愛又聰明的書毓。 謝謝莊媽、月貴、淑琳和建亨的幫忙和照顧,感謝之心無法言盡。 謝謝我親愛的同學兼室友筑鈞(牛牛)、好友若濃(阿紫),以及學 妹鈺芳(小雞)和郁芳(小阿芳),有妳們的陪伴一起歡笑和鼓勵,真好! 謝謝一年室友的郁娟(阿嬤)、書瑋、蘇瑋、亦蓉,謝謝妳們的陪伴。 謝謝我親愛的爸媽以及姊妹們的支持,每當我實驗遇到瓶頸的時 候,只要回家躺在軟軟的床上大睡、將媽咪煮的好料大吃以及和姊妹 們大聊一番,隔天就能精神飽滿的回到實驗室面對難題。謝謝你們! 雅苓 謹誌 99 年 8 月 新竹交大
v 目錄 第一章 緒論 ... 1 參考文獻: ... 10 第二章 實驗原理 ... 12 2.1 間質隔離法 (matrix-isolation method) ... 12 2.1.1 間質隔離法的原理 ... 12 2.1.2 光譜上的間質效應 ... 14 2.1.3 間質的選擇 ... 16 2.2 間質隔離法的新進展:以 para-hydrogen 為間質 ... 18 2.2.2 p-H2的發展 ... 20 2.2.3 p-H2的製備與保存 ... 22 2.2.4 p-H2的特性 ... 24 2.2.5 p-H2為間質的優點 ... 26 2.3 霍氏轉換紅外光譜儀 ... 28 2.3.1 麥克森干涉儀基本原理 ... 28 2.3.2 霍氏轉換紅外光譜儀裝置 ... 32 2.3.3 霍氏轉換紅外光譜儀之優點 ... 33 參考文獻... 48 第三章 實驗裝置與步驟 ... 50 3.1 實驗裝置簡介 ... 50 3.1.1 真空系統... 50 3.1.2 低溫系統... 51 3.1.3 p-H2轉換系統 ... 52 3.1.4 偵測系統... 52 3.2 實驗步驟... 53 3.2.1 樣品製備... 53 3.2.2 實驗步驟... 54 3.3 偏光實驗... 55 3.3.1 原理... 55 3.3.2 實驗步驟... 55 3.3.3 光譜分析... 56 第四章 結果與討論 ... 59 4.1 CH3I 與 CH3F 之紅外光吸收振動模 ... 59 4.2 CH3I 在 p-H2間質中之紅外吸收光譜 ... 60 4.2.1 C-H 對稱伸張振動模 ν1 ... 61 4.3.2 C-H 對稱彎曲振動模 ν2 ... 64 4.2.3 C-I 伸張振動模 ν3 ... 66
vi 4.2.4 C-H 不對稱伸張振動模 ν4 ... 67 4.2.5 C-H 不對稱彎曲振動模 ν5 ... 69 4.2.6 CH3搖擺振動模ν6 ... 70 4.2.7 CH3I 在 p-H2間質中的紅外吸收光譜之結論 ... 73 4.3 CH3I 和 CH3F 在 p-H2間質中的紅外吸收光譜比較 ... 76 4.3.1 C-H 對稱伸張振動模 ν1、C-H 對稱彎曲振動模 ν2和 C-F(I)伸張振 動模ν3之比較 ... 76 4.3.2 CH3I 與 CH3F 之 C-H 不對稱伸張振動模 ν4、C-H 不對稱彎曲振 動模ν5和 CH3搖擺振動模ν6之比較 ... 78 4.3.3 CH3F 之 C-F 伸張振動模 ν3及 CH3I 之 CH3搖擺振動模ν6比較 78 參考文獻... 127 第五章 結論 ... 128
1
第一章 緒論
由於 CH3Cl 可用來當作冷媒及噴霧劑之成份,但在高層大氣中光 解會釋出氯原子和臭氧進行催化分解反應,而使大氣中臭氧含量大量 降低,造成環境之危害[1]。為了降低此危害,近年來發展出環保冷 媒氫氟碳化合物(hydrofluorocarbons,HFCs)來取代 CH3Cl。而 CH3F 為 HFCs 的一種,可避免 CH3Cl 光解後釋出之氯原子對臭氧層之破壞 [2,3]。除了可用來當作環保冷媒之外,CH3F 在半導體之製程方面可 用來當作電漿薄膜蝕刻之氣體,因具有較佳之選擇性而不致蝕刻其它 材料之薄膜,而使生產良率提高[4,5]。 在間質隔離法中,由於待測分子被個別隔離在間質主體中, 降低其移動性,因此無法像在氣態下可以自由運動。由於甲烷(CH4, methane)是少數被證實可以在間質中轉動的分子[6,7],而 CH3F 是最 小的鹵甲烷分子,因此早期已有許多研究想探討 CH3F 是否像 CH4般 可以在低溫間質中轉動。1972 年 Barnes 等人[8]推測如果 CH3F 在 Ar 間質中可以轉動,則其 C-F 伸張振動模(ν3)在 20 K 之未解析之振轉譜 線半高寬約為 7 cm-1,但由於實驗觀察之 CH 4在 Ar 間質及 CH4譜帶 半高寬僅 1.4 cm-1,因此認為 CH 3F 在 Ar 及 CH4間質中並不會轉動。 在 1981 年 Gauthier-Roy 等人[9]利用自製之光柵光譜儀研究 200-20002 ppm 的12 CH3F 及其同位素12CD3F 和13CD3F 在 N2和 Ne、Ar、Kr 及 Xe 四種惰性氣體間質中在 1000 cm-1光區附近之紅外吸收光譜,觀測 到其譜線寬度並不會隨濃度的不同而變化,但會隨著溫度上升而變寬, 而認為 CH3F 及此三種分子可在 N2及此四種惰氣間質中轉動,並由
Devonshire’s theory 推得 CH3F 在 Ne、Ar、Kr 及 Xe 間質中之轉動能
障分別為 5.7、10.9、4.2 及< 2 cm-1。但在 1982 年,Jones 及 Swanson[10] 利用 FTIR 觀測 CH3F 及 CD3F 在 3-39 K 間 Ar 及 Kr 間質中的紅外吸 收光譜,指出由 Gauthier-Roy 等人[9]所指派之 CH3F 在 Ne 間質及 CD3F 在 Ar 間質之轉動譜線是 CH3F(或 CD3F)聚合物(polymer)之吸收 譜帶,而非 CH3F 及 CD3F 之轉動譜線。他們認為如果溫度升高而造 成的譜線增寬情形是由於轉動譜線所造成的,則所有 A1及 E 之對稱 性振動譜線應會有相同的情形,但其結果並無一致性,因此推論 CH3F 在 Ar 和 Kr 間質中並不轉動。在 1982 年,Apkarian 及 Weitz[11]利用 FTIR 觀測 CH3F 在 Kr 間質中之紅外吸收光譜,他們認為 Jones 及 Swanson[10]因觀測 CH3F 及 CD3F 在 Kr 間質中之溫度由 10 K 降為 3 K 時,譜線寬度變寬,而推測其在 Kr 間質中並無轉動之推論並不正確。 因他們認為其為不同溫度下轉動能階分佈的不同所造成之影響。並利 用 Devonshire’s model 與實驗觀測之 ν3振動模之光譜比較,證實 CH3F 在 Kr 間質中會有單軸之轉動,並推論其轉動能障為 80 cm-1。
3 近年來,以 para-hydrogen(p-H2)作為間質之研究漸漸發展出來 [12,13]。因為 p-H2的晶格零點振動幅度佔了其晶格常數約 20 % [14], 因此可提供間質客體柔軟的環境。由於 p-H2間質之柔軟性,使得圍 繞在客體分子周圍的 p-H2分子較易調整到能量最低之位置,因此每 個客體在 p-H2間質中之環境類似,而使得非勻相譜線增寬效應
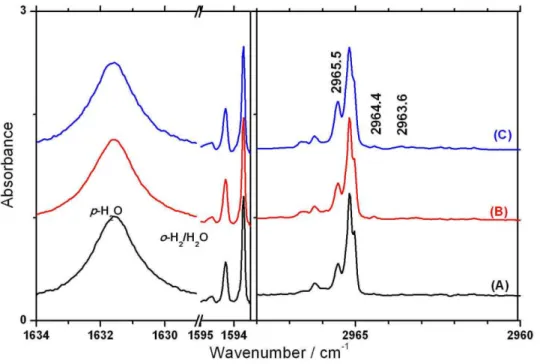
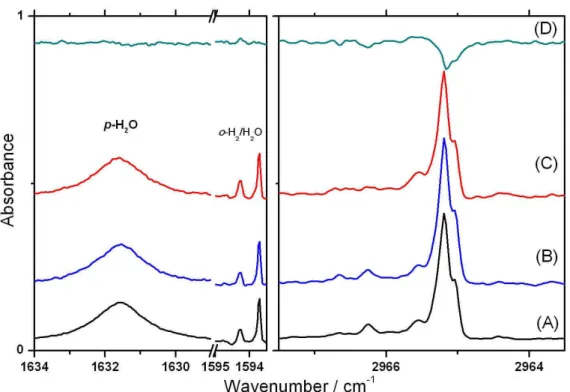
(inhomogeneous line broading)變小,因此譜線之寬度可以小於 0.01 cm-1 [14]。2003 年 Yoshioka 及 Anderson [16]利用 FTIR 觀測 CH3F 在
p-H2間質中的紅外吸收譜,他們沒有觀測到 CH3F 之轉動譜線,因此 認為在 p-H2間質中, CH3F 並無轉動運動。此外,他們並發現在 C-F 伸張振動模(ν3)光區有一系列隨著 o-H2濃度上升而產生之譜線,他們 將其指認為 CH3F(o-H2)n (n=0-12) 團聚體之吸收譜線。本實驗室吳孙 中學長在 2008 年研究 CH3F 在 p-H2間質中之紅外吸收光譜[17],觀 測到其簡併振動模ν4和ν6在較強的主要吸收譜線(A level)兩旁各有一 根微弱的吸收峰(E level),進而証實 CH3F 在 p-H2間質中具有單軸(C3 軸)轉動的特性。為了尋找另一個會在 p-H2間質中進行單軸轉動之例 子,本實驗探討在 p-H2間質中,CH3I 是否會和 CH3F 一樣,具有單 軸轉動的特性,同時也希望瞭解 o-H2對 CH3I 所產生之影響。 由於常被用於當作除草劑、殺蟲劑及滅火器之成分,或是用作土 壤消毒劑之 CH3Br 如果散逸到大氣中,在經太陽的紫外光照射同樣
4 也可以分解出 Br 原子而與臭氧進行催化分解反應,而減少大氣中臭 氧之含量。經研究,且溴原子對臭氧有更明顯的耗減作用,其破壞臭 氧之能力遠遠高於氯原子,大約為其 40~100 倍[18]。自 1987 年以來 蒙特婁條約制定後,溴甲烷(CH3Br)被禁止使用。碘甲烷(CH3I,methyl iodide,iodomethane) 曾被提議做為作為之溴甲烷的替代品,但也有 人研究發現 CH3I 光照後所形成的碘自由基也會造成對流層和帄流層 中臭氧的破壞[19],因此其對溴甲烷之取代性仍在評估。而在有機合 成方面,CH3I 之 CH3官能基因有較小之位阻效應(hindered effect),且 碘為良好的離去基,因此可作為甲基化詴劑[20]。在光化學反應中, CH3I 也常常被用來當做產生 CH3自由基的前驅物[21]。 CH3I 的氣態光譜,已被許多研究組觀測過。1965 年,Jones 及 Thompson [22]利用光柵光譜儀觀測 CH3I 氣態下各振動模之譜線並計 算其轉動參數。1971 年,Matsuura 及 Overend [25]利用以解析度為 0.03 cm-1的光柵光譜儀分析 CH3I 之 ν6和ν2 + ν6振動模,得到其轉動常數。 Matsuura 並於 1973 年 [28]分析高解析度光譜,考慮氣態下 CH3I 之 ν5
和ν3 + ν6 的 Fermi resonance 及 ν5和 ν2的 Coriolis coupling,並求得轉
動常數。1980 年,Das 等人[27]利用可調式二極體雷射光譜儀分析 CH3I
的ν6 振動模之吸收光譜。相較於 Matsuura 及 Overend [25]所觀測之 ν6
5 cm-1高解析度得以解析出六個 Q branch 之躍遷譜線,並得到更精確的 轉動參數。1983 年 Paso 等人[26]利用光柵光譜儀觀測 CH3I 的 ν1和ν2 + 2ν6振動模之躍遷譜線,解析度為 0.04 cm-1,並測得其轉動參數。他 們並於 1986 年 [24]利用 FTIR 觀測 ν4振動模之躍遷譜線,其解析度 為 5.4 x 10-3 cm-1,並且分析得到轉動參數。1989 年,Alanko 研究組 [22]利用 FTIR 觀測 CH3I 的 ν3振動模之躍遷譜線,其解析度為 0.004cm-1,並由觀測到之譜線計算得到轉動參數資訊。綜合以上之文 獻,CH3I 各振動模之基頻(fundamental band)及轉動常數如 表 1. 1 所示。 目前並無文獻報導觀測到 CH3I 在間質中之轉動資訊。Verderame 和 Nixon[29]觀察 CH3I 在 Ar 間質中的紅外吸收光譜,觀察到兩根譜 線分別位於 1431.5 cm-1 (ν5)和 1399.4 cm-1(ν3+ν6),並指派為 Fermi
resonance 所造成的譜線分裂。Ito 等人[30]利用 FTIR 觀測 CH3I 在 Ar
間質中的吸收光譜,其解析度為 0.5 cm-1,並藉由改變混合濃度及超 音波射束(supersonic jet)之停滯(stagnation)壓力,可觀察到單體 (monomer)和雙體(dimer)之間譜線的相對強度變化。Shida 研究組在 研究 p-H2中的 CH3I 的光解反應時,亦觀測到了 CH3I 在 p-H2間質中 ν1、ν2、ν4及ν5等譜線之吸收[31,32]。文獻上觀察 CH3I 在 Ar 及 p-H2 間質的吸收譜線位置,整理於表 1. 2 中。
6
除了單軸轉動是研究的課題之一,o-H2對客體光譜之影響也是另
一個重要的課題。一般在 p-H2間質中,仍存在少量的 o-H2。o-H2的
電荷密度分佈並不像 p-H2一樣為球形對稱,其分子仍然具有偶極矩,
因此相較於 p-H2而言,o-H2和具有偶極矩的待測分子間具有較大的
作用力。2003 年 Yoshioka 與 Anderson [16]利用 FTIR 觀測 CH3F(o-H2)
團聚體在 p-H2間質中的紅外吸收光譜,發現在 C-F 伸張振動模(ν3)光 區有一系列隨著 o-H2濃度上升而產生的團聚體譜線,推論是由於 o-H2 和氟原子之間容易形成鍵結,使得 C-F 伸張振動模相較於其他振動模 而言,所受 o-H2的影響較大,因此在 ν3光區的 CH3F(o-H2)叢聚物一 系列譜線具有較大的位移,他們依序將譜線指認為 CH3F(o-H2)n (n=0-12) 的吸收。Anderson 研究組認為 CH3F 可能取代 p-H2的單晶 格,如果依位移大小來看,一開始和 CH3F 作用之 o-H2會有較強之作 用力而有較大之譜線位移,而當有一半(n=6)以上的 o-H2和 CH3F 作 用後,其作用程度會變小,因此譜線位移也會變小。且觀測到最大之 譜線位移為 n=12,和 CH3F 在 p-H2間質之六方最密(hexagonal closest packed,HCP)晶格中相鄰的 p-H2分子之數目一致。由於 CH3F 與 CH3I 之偶極矩大小分別為 1.86 D 和 1.64 D[34],碘原子與氟原子之凡得瓦 爾半徑(van der Waals radius)分別為 198 pm 和 147 pm[4],所以 CH3F
7
別為 214 pm 和 138 pm [36]。因此,吾人希望藉由 CH3F 和 CH3I 在
p-H2間質中所受到 o-H2影響所產生之譜線變化相互比較,以推測
8 表 1. 1 氣態 CH3I 的六個振動模之基頻之光譜參數。 振動模 對稱性 Gas (cm-1) ν0 A” A' B” B' Ref. ν1 A1 2971.25 5.173 5.122 0.2502 0.2501 [26] ν2 A1 ν3 A1 533.22 5.173 5.166 0.2502 0.2484 [22] ν4 E 3060.08 5.173 5.144 0.2502 0.2503 [24] ν5 E 1434.69 5.173 5.127 0.2499 0.2502 [28] ν6 E 882.68 5.173 5.207 0.2500 0.2490 [25] 882.68 5.171 5.206 0.2502 0.2494 [27]
9 表 1. 2 CH3I 在 Ar 及 p-H2間質中的六個振動模之吸收譜線位置。括號 中的為強度較弱之吸收譜線。 振動模 對稱性 Solid Ar (cm-1) [30] Solid p-H2 (cm-1) [31] ν1 A1 2966 2954 (2950、2951) ν2 A1 1245 1248 (1250、1252) ν3 A1 ν4 E 3054 (3057、3058) ν5 E 1432 1429 (1432) ν6 E 882
10 參考文獻:
1. F. S. Rowland, Roy. Swed. Acad. Sci. 19, 281 (1990).
2. J. S. Francisco and M. M. Maricq, Acc. Chem. Res. 29, 391 (1996).
3. S. R. Wilson, K. R. Solomon, and X. Tang, Photochem. Photobiol. Sci. 6, 301 (2007).
4. Y. Iijima, Y. Ishikawa, C. Yang, M. Chang, and H. Okano, Jpn. J. Appl. Phys. 36, 5498 (1997).
5. L. Chen, L. Xu, D. Li, and B. Lin, Microelec. Eng. 86, 2354 (2009).
6. A. Cabana, G. B. Savitsky, and D. F. Hornig, J. Chem. Phys. 39, 2942 (1963). 7. F. H. Frayer and G. E. Ewing, J. Chem. Phys. 48, 781 (1968).
8. J. Barnes, H. E. Hallam, J. D. R. Howells, and G. F. Scrimshaw, J. Chem. Soc. Faraday Trans. 2 69, 738 (1973).
9. B. Gauthier-Roy, C. Alamichel, A. Lecuyer, and L. Abouaf-Marguin, J. Mol. Spectrosc. 88, 72 (1981).
10. L. H. Jones and B. I. Swanson, J. Chem. Phys. 76, 1634 (1982). 11. V. A. Apkarian and E. Weitz, J. Chem. Phys. 76, 5796 (1982). 12. M. E. Fajardo and S. Tam, J. Chem. Phys. 108, 4237 (1998).
13. T. Yoshioka, P. L. Raston, and D. T. Anderson, Int. Rev. Phys. Chem. 25, 469 (2006).
14. J. V. Kranendonk, Solid hydrogen (Plenum, New York, 1983).
15. S. Tam, M. E. Fajardo, and H. Katsuki, J. Chem. Phys. 111, 4191 (1999). 16. K. Yoshioka and D. T. Anderson, J. Chem. Phys. 119, 4731 (2003).
17. Y.-P. Lee, Y.-J. Wu, and Jon T. Hougen, J. Chem. Phys. 129, 104502 (2008). 18. N. Washida, T. Imamura, and H. Bandow, Bull. Chem. Soc. Jpn. 69, 535
(1996).
19. N. Bell, L. Hsu, D. J. Jacob, M. G. Schultz, D. R. Blake, J. H. Butler, D. B. king, J. M. Lobert, and E. Maier-Reimer, J. Geophys. Res. 107, 4340 (2002). 20. J. G. Avila-Za´rraga and R. Martı´nez, Synth. Commun. 31, 2177 (2001). 21. T. Momose, M. Miki, M. Uchida, T. Shimizu, I. Yoshizawa, and T. Shida, J.
Chem. Phys. 103, 1400 (1995).
22. S. Alanko, V.-M. Horneman, and J. Kauppinen, J. Mol. Spectrosc. 135, 76 (1989).
23. E. W. Jones and H. W. Thompson, the Royal Society of London. Series A, Mathematical and Physical Sciences 288, 1412 (1965).
24. R. Anttila and R. Paso, J. Mol. Spectrosc. 119, 190 (1986).
25. H. Matsuura and J. Overend, Spectrochim. Acta. 27, 2165 (1971).
26. R. Paso, V.-M. Horneman, and R. Anttila, J. Mol. Spectrosc. 101, 193 (1983). 27. P. P. Das, V. M. Devi, and K. N. Rao, J. Mol. Spectrosc. 84, 305 (1980). 28. H. Matsuura, J. Chem. Phys. 59, 1449 (1973).
29. F. D. Verderame and E. R. Nixon, J. Chem. Phys. 45, 3476 (1966). 30. F. Ito, T. Nakanaga, Y. Futami, S. Kudoh, M. Takayanagi, and M. Nakata,
Chem. Phys. Lett. 343, 185 (2001).
31. T. Momose, M. Uchida, N. Sogoshi, M. Miki, S. Masuda, and T. Shida, Chem. Phys. Lett. 246, 583 (1995).
32. M. Fushitani, N. Sogoshi, T. Wakabayashi, T. Momose, and T. Shida, J. Chem. Phys. 109, 6346 (1998).
33. M. D. Marshall and J. S. Muenter, J. Mol. Spectry. 83, 279 (1980).
11
401 (1989).
35. A. Bondi, J. Phys. Chem. 68, 441 (1964).
36. W. Gordy, J. W. Simmons, and A. G. Smith, Microwave spectra. 74, 243 (1948).
12
第二章 實驗原理
2.1 間質隔離法 (matrix-isolation method) 西元1954年Porter研究組[1]及Pimentel研究組[2]分別發展出間質 隔離技術,其後Pimentel及其他研究組致力發展相關技術及其應用, 使其成為研究不穩定分子的重要技術,研究對象涵蓋離子、不穩定物、 有機和無機金屬化合物。以下對低溫間質隔離法的原理、光譜上的間 質效應及間質的選擇等加以說明。 2.1.1 間質隔離法的原理 間質隔離法是將少量待測分子,又稱為客體(guest)與做為間質 的大量惰性原(分)子,又稱為主體(host)在氣態下均勻混合後, 共同沉積於低溫(3-30 K)的樣品靶面(sample target)上。此時主體 因低溫冷凍形成固體晶格,而客體濃度因相較於主體極低而被稀疏的 鑲嵌在晶格中。藉由晶格的穩固性可將客體個別隔離在間質主體中, 降低其移動性,使其無法像在氣態下自由運動。又因主體為惰性原(分) 子,與客體間作用力小,也不易發生反應,且低溫下主體能有效地弛 緩客體的內能,即使是不穩定的分子或活性強的自由基,也能長時間 保存在間質中,以利進一步的光譜測定。此外,樣品能夠長時間的沉13 積,以達足夠吸收強度供光譜儀測定,不僅可提升光譜的訊雜比,對 於吸收強度較弱的振動模式也有較佳的觀測結果。在樣品使用量方面, 間質隔離法使用的樣品量遠較氣態實驗少,可以使用昂貴的化合物或 同位素進行實驗。 在間質中,除了少數小分子,如 HCl、H2O、NH3 等,大部分的 待測分子會被固定在間質中無法轉動,而無轉動能階,因此在光譜中 觀測到的大多是不含轉動只有振動吸收之狹窄單峰。再者,依據波茲 曼分佈(Boltzmann distribution)原理,低溫下(3-30 K)待測分子大 多分佈於振動基態(v=0),故主要觀測到的是基頻(fundamental)、 泛頻(overtone)和結合吸收帶(combination band),而不受熱譜帶 (hot band)干擾,因此較氣態下的紅外光譜簡化且易於分析。 待測分子處於間質中,經適當的光源光解後可能產生不穩定的分 子碎片(photofragment),若分子碎片所獲得的動能不足以克服晶格 對其之束縛能,則光解產物會留在原晶格中。因此,產物可經由各種 不同的途徑再繼續反應或是形成原來的分子,或是其結構異構物 (structural isomer)或其他產物,此即以間質隔離法進行光化學的一 個重要性質,稱為「晶格效應(cage effect)」。利用此晶格效應(cage effect),可產生在一般氣態或溶液態中不易產生的新穎分子,這些分 子常在大氣化學或星際化學扮演重要的角色。
14 2.1.2 光譜上的間質效應 由於間質主體對於待測客體造成的微擾,使得待測客體在間質主 體中的光譜與氣態光譜相較,仍有些微的差異。這種因為間質環境所 造成光譜不同於氣態光譜的差異,稱為「間質效應」(matrix effect)。 以下討論幾種在光譜學上常見的間質效應︰ (1)間質位移: 當待測分子被獨立鑲在晶格中時,可能會與間質主體作用,而使 得間質隔離光譜與氣態光譜的吸收頻率有所差異 Δν,此差異即為「間 質位移」(matrix shift),定義為 Δν ≣ νgas-νmatrix。根據 Jacox[3]比較
230 組雙原子分子在氣態與惰性間質中的振動頻率,顯示帄均間質位 移小於 2%。一般而言,不同間質主體造成分子譜線位移大小依序為 Ne<Ar<Kr<N2。至於間質位移是藍位移(blue shift)或紅位移(red
shift),則視客體與間質主體的作用力而定,目前仍無簡單準則可循。 (2)多重晶格位置(multiple trapping sites):
待測分子鑲嵌在間質主體的模式有很多種,可能取代一個或多個 主體的位置(substitutional site)、鑲嵌在四面體或八面體的間隙中 (interstitial site),或以錯排(dislocation)的方式插在間質晶格中。 待測分子處於不同晶格位置,所受到周遭間質主體的作用不同,使得 待測分子具有些微不同的能階,其間質光譜因而出現較寬之譜線或多
15 重吸收峰的現象。此類多重譜線的特徵為譜線之相對強度不隨待測分 子的濃度而變化,反而隨沉積條件而改變;通常可藉由回火(anneal), 提高間質溫度使晶格重新排列,或改變沉積條件,形成較穩定的晶格 結構,以減少多重譜線。 表 2. 1 列出一些鈍氣及 H2的固態晶格參數及游離能[4]。根據結 晶學理論,惰性氣體在低溫中通常以面心立方(face-centered cubic, fcc)結構形成晶格。Meyer 等人提出以惰性氣體當間質主體時,若含 有少量的 O2、N2及 CO 等分子,會使部份面心立方結構轉換為六方 最密堆積結構(hexagonal close-packed,hcp)[5]。Winn 在紅外吸收 光譜及理論研究中指出,待測分子在 Ar 間質中有結晶格位置(crystal cage site)和非結晶格位置(non-crystalline cluster site or amorphous cluster site)等兩種堆積位置,且形成非最密堆積的結構[6]。因間質 氣 體 急 速 冷 卻 成 固 體 所 得 到 的 非 帄 衡 態 晶 體 , 會 呈 現 多 孔 性 (porosity)、錯排(dislocation)及空位(vacancy)的現象。通常可 藉提高間質溫度使晶格重新排列,以形成更穩定的晶格結構。 (3)聚合現象(aggregation): 待測分子濃度過高或分子間作用力強時,於氣態中易形成雙體 (dimer)、多體(polymer)甚至是叢聚物(cluster)共同沉積於同一 晶格中,使間質光譜中出現多重吸收峰,此類譜線可藉由稀釋樣品濃
16 度而減弱或消除。另外,當間質不夠穩固時,照光給予能量或提升靶 面溫度後,客體經擴散(diffusion)作用也會發生聚合現象。 (4)選擇率(selection rule): 由於待測分子會受到間質環境的微擾(perturbation),破壞其波函 數的對稱性,增加原本在氣相中為禁制(forbidden)躍遷的躍遷機率 (transition probability),使得在氣態中不易觀測到的譜線,可在間質 中被觀測到,例如:O2 氣態分子在紅外光區的對稱伸張模是禁制的, 但 O2直接沉積於低溫的樣品靶面時,其分子本身會形成晶格,而受 間質環境干擾使對稱性被破壞,故可觀測其吸收譜線[7]。於不同溫 度下,O2分子有 α、β 及 γ 晶形,其吸收譜線分別位於 1549、1595 及 1557cm-1。 2.1.3 間質的選擇 由於間質隔離法是利用大量鈍性間質主體來隔離待測分子,若能 選擇適當的間質主體,則可減低主體與客體之間的相互作用,有助於 光譜的分析。在選擇間質主體時頇考慮下列幾項因素: (1)間質純度: 待測分子與間質主體的混合比例通常在 1:100 以上,甚至高達 1:10000,若間質主體的純度不高,所含雜質之量可能高於待測分子 之量,不僅對光譜造成干擾,甚至影響待測分子的物理和化學性質。
17 因此,一般使用純度 99.9%以上,甚至 99.9995%以上之氣體做為間 質主體。 (2)間質鈍性: 為了使間質主體對待測分子的干擾降至最低,應選用化性較低, 不易與待測分子發生反應的間質主體;又因間質主體與待測分子間的 作用力會造成間質效應,而使間質光譜的光譜參數與氣態光譜有差異, 宜選用微擾最小的分(原)子做為間質。一般常用鈍氣做為間質主體, 其與待測分子間只存在微弱的凡得瓦力(van der Waals force)。此外, 因凡得瓦力與分子大小成正比,故宜選用質量數較小的鈍氣做為間質 主體,將其對待測分子的干擾降到最低。 (3)間質堅固性: 間質主體的溫度決定了間質晶格的堅固性,其溫度至少需低於熔點的 二分之一,晶格的穩定性才能抑制擴散現象發生。表 2. 2 列出一般常 用間質之擴散溫度、熔點及沸點,當溫度低於擴散溫度時,才能有效 抑制擴散現象。 (4)光區透明性: 間質主體在欲研究的光區內不能有吸收,否則將會遮蔽待測分子 的吸收,進而影響光譜的分析。表 2.3 列出常用間質的吸收波長,由 表得知間質主體的吸收範圍多在真空紫外光區,因此對於波長大於
18 200 nm 的光區範圍無影響。 考慮上述四個因素,最常使用惰性氣體(如:Ne、Ar 和 Kr)或 氮氣(N2)作為間質主體。而在電子態躍遷的研究中,因 Ne 間質對 電子能態的微擾作用最小,常使用 Ne 作為間質主體;但從事紅外光 譜學研究時,Ar 的擴散溫度高於 Ne 擴散溫度,且 Ar 間質所造成 的間質位移不大,故實驗常用 Ar 作為間質主體。 2.2 間質隔離法的新進展:以 para-hydrogen 為間質 近年來,由於低溫技術的普及和致冷技術的進步,因此發展出有 別於傳統間質隔離法的技術。當溫度略低於氦的臨界溫度(transition temperature, 2.12 K)時,氦會轉變成超流體(superfluid),以此特性 發展出的技術稱為 superfluid helium nanodroplet[8];而利用氫分子轉 動量子數 J=0 的 para-hydrogen (p-H2)當作間質,利用其量子固體 (quantum solid)的優點[9],而展現出與傳統間質不同的特性。以下 即對 p-H2 的基本性質、發展、製備與保存、特性及其作為間質的優 點加以簡介。 2.2.1 p-H2的基本性質 分子的完整波函數包含電子態、振動、轉動及核自旋波函數。根 據庖利不相容原理(Pauli exclusion principle),當分子具有核自旋不
19 為 零 的 兩 個 以 上 原 子 時 , 此 分 子 有 核 自 旋 異 構 物 ( nuclear spin isomers),而這些異構物的差異就在於核自旋量子數的不同。以氫分 子為例,氫原子的核自旋量子數 I=1/2,屬於費米子(fermion),必頇 符合 Fermi-Dirac 的統計方式:粒子交換符合反對稱的波函數性質。 而氫分子具有封閉殼層(closed-shell),電子態波函數Ѱe或振動態波 函數 Ѱv在交換過程中皆對稱,因此由轉動波函數Ѱr和核自旋波函數 Ѱs 來決定整個波函數的對稱性。氫分子的轉動波函數經交換運算子 (exchange operator, P12)運算可表示為:P12YJM (θ,φ) = (-1)J YJM (θ,φ), 其中 YJM為轉動波函數,J 為轉動量子數,θ 和 φ 為三度空間球座標 表 示 法 中 的 角 度 。 當 J 是 奇 數 , 轉 動 波 函 數 是 反 對 稱 的 (antisymmetric);當 J 是偶數,轉動波函數是對稱的(symmetric)。 為了符合粒子交換後是反對稱的波函數性質,轉動量子數 J 為奇數時, 必頇對應到對稱核自旋(I = 1),稱為 ortho-H2(o-H2);轉動量子數
J 為偶數時,必頇對應到反對稱核自旋(I = 0),稱為 para-H(p-H2 2)。
室溫下,氫分子在奇數 J 和偶數 J 的分佈(population)相近,但對 稱核自旋有三個簡併數(degeneracy = 3),而反對稱核自旋則為非簡 併(non-degenerate),所以室溫下的氫氣(normal-H2,n-H2)其 o-H2
和 p-H2的比例約為 3:1。在極低溫(~4 K)下,轉動能階分佈會移
20 其比例仍維持 3:1,並不符合波茲曼分佈的規範 J=0 應佔大多數。 為了產生 p-H2,必頇在低溫下使用磁性催化劑促進核自旋反轉,才能 將 o-H2轉換成能量較低的 p-H2。 2.2.2 p-H2的發展 80年代中期芝加哥大學Oka教授研究組發現固態p-H2光譜的特殊 性質:由p-H2形成的固態晶體在紅外光區之吸收譜線(v=0←0,J=
6←0)半高寬(full width at half maximum, FWHM)窄至0.006 cm-1
[10]。 在Raman光譜的研究中,p-H2的譜線寬度更窄至0.0003 cm-1[11],相較 於一般凝態下所量測的光譜半高寬窄了許多,因此推測固態p-H2分子 間作用力極小,或許可作為間質使用。但由於氫分子的沸點為20 K, 三相點為13.6 K,即使在4.2 K下仍有很高的蒸汽壓(~10-6 -10-7 torr) [12],而當時常用之壓縮機式冷卻系統只能達到10 K,無法使氫分子 直接沉積,故常使用液氦。 日本京都大學 Momose 和 Shida 研究組發展以封閉式樣品槽 (enclosed-cell)結晶方式將 p-H2 作為間質主體[13]。他們利用液氦為 冷卻工具,並在銅製腔體兩端的光學鏡片與腔體之間,放置導熱性佳 的銦(In)作為墊片形成真空腔體,其裝置如圖 2. 1 所示。實驗時待 測分子與 p-H2以小於 100 ppm 的濃度比例預先混合收集於鋁瓶中, 再以 300 mmol h-1的流速流入溫度保持在 7 K 的銅製腔體,約經過 3
21 小時沉積可形成長 6 cm、寬與高約 3 cm 的長方晶體。待結晶完成後, 將溫度降至 5 K 量測光譜。此結晶方式得到之晶體為穩定的六方最密 堆積(hexagonal close-packed,hcp)結構,待測分子能階會受晶體對稱 性(D3h)影響而分裂,並產生不同位置的譜線。在結晶過程中,因待 測物對 p-H2溶解度低,易形成團聚物,雖可將濃度控制在 100 ppm 以 下以減少團聚物,卻不易觀測待測分子較微弱的吸收譜線。此外,此 法的樣品槽設計使得光束受限特定方向,僅有一個光軸可運用,實驗 上並不方便。 於90年代中期,美國空軍實驗室的Fajardo研究組發展出與傳統間 質隔離法類似的快速沉積法(rapid vapor deposition)[14]:在實驗系 統上直接裝置p-H2轉換系統,於沉積前將n-H2轉換成p-H2,其裝置如 圖 2. 2所示。由於p-H2在沉積到金屬靶前經過了預冷的過程,且他們 對液氦抽氣使金屬靶溫度可降至2.2 K,在此溫度下H2的蒸汽壓僅10-13 torr,故可以流量(~200 mmol h-1)直接快速沉積大量樣品。 綜觀以上兩種方法,封閉式樣品槽(Enclosed-cell)結晶方式缺點為 沉積的方式受限制,濃度僅可控制在100 ppm以下,且只有一個光軸 可運用,實驗上之廣用度較低;而快速沉積法的低溫系統結構雖與傳 統間質隔離系統相似,且可以大流量沉積,但缺點是使用液氦抽氣冷 卻的花費極為昂貴。因此,此兩種技術皆不若以Ar為間質的隔離法般
22 被廣泛應用。 由於上述兩種技術對於進行以p-H2為間質的實驗並不容易,而當 初市售封閉循環式三段壓縮系統,最低溫僅可達5 K,本實驗室遂發 展出脈衝式沉積法[15]。將預先配置好的待測分子與p-H2混合氣體由 脈衝噴閥(General Valve)噴出至低溫樣品靶沉積,由於噴出的瞬間 壓力大於H2在5 K下的蒸汽壓(10-4 torr),因此可有效的在低溫靶上 沉積,只要維持沉積速率大於揮發速率即可。比較此法與Fajardo研究 組[16]利用快速沉積法取得CH4在p-H2間質中得到的光譜,幾乎完全 相同。此方法使得擁有5 K封閉循環式三段壓系統之實驗室可以進行 以p-H2為間質之實驗,但缺點為在5.4 K下,H2之蒍氣壓約為1×10-4 Torr,其間質只能維持4小時後便會開始揮發。現今商業化的低溫冷 卻系統已能降至3.2 K,在此溫度下H2的蒸汽壓僅有10-10 torr,如同一 般間質隔離實驗,可直接沉積p-H2,不僅免除使用液氦所需的昂貴花 費,且產生的間質能維持數天以上,可從事長時間之實驗。 2.2.3 p-H2的製備與保存 即使在極低溫下(~4 K),若無磁作用力幫助核自旋反轉,處 在J=1的o-H2並無法弛緩到J=0的p-H2。因此,欲得到高純度的p-H2, 需在低溫下將n-H2通過催化劑促使核自旋翻轉,其轉換效率取決於轉 換時催化劑的溫度和o-H2對催化劑的吸附效率。目前文獻中發表過三
23 種磁性催化劑,第一種為APACHI[17,18],由順磁性的鎳包覆特殊矽 膠所組成,因其有效催化面積大,具有良好的轉換效率;第二種為 Fe(OH)3[19],為本實驗室先前所使用之催化劑;第三種則為Fe2O3[20], 因其表面積較大且粒子大小相同,而具有良好的轉換效率,是目前本 實驗室所使用的。Fajardo研究組在不同溫度下做p-H2轉換,並計算H2 的Q1(0)與Q1(0)+S0(1)的吸收譜線面積比,求得o-H2與p-H2的比例[21]。 如圖 2. 3所示,當轉換溫度為15 K時,o-H2約只有0.01%,隨著轉換 溫度提高,o-H2的濃度越來越高;在37 K 時,o-H2的濃度高達8%。 由於氫分子J=1與J=0的能階差為117.9 cm-1,根據波茲曼分布,在37 K時其分布比例應為0.01:1,即o-H2的濃度應為1%,顯示轉換效率 不只與溫度有關,也與o-H2吸附在催化劑及去吸附的效率有關,並非 僅由波茲曼分佈來決定。 在催化劑的前處理部份,本實驗室利用1/8”的銅管,填充約3 g的 Fe2O3,並在催化劑前後兩端填入玻璃棉,防止在轉換時催化劑被高 壓的n-H2氣體衝出。再將銅管纏繞在直徑為2 cm、高3 cm之導熱效果
佳的無氧銅(oxygen free high conductivity copper, OFHC)圓柱體上,如 圖 2. 4所示。並利用銅片包覆銅管及圓柱體,降低圓柱體與銅管間之 空隙,增加溫度傳導之效果。並利用螺絲及墊片將其固定於冷卻頭上, 且在墊片及圓柱體間、圓柱體和冷卻頭間,分別加入導熱性佳的銦作
24 為墊片,以提高熱傳導率。且將連接H2進氣口的那端銅管設置於低溫 系統的冷卻頭的尾端,而將出氣口設於靠近低溫系統冷卻頭的地方, 可確保出氣口位於冷卻頭最低溫的部位。裝填催化劑後,將加熱帶纏 繞至銅管及圓柱體上,以160 °C的溫度烘烤活化磁性催化劑三天並去 除水份後,以高純度的H2氣體流過催化劑8小時,再流入Ne氣體約8 小時用以除去吸附在催化劑上的水氣與空氣。因為催化劑上若吸附太 多的水氣與雜質則會降低轉換效率。 圖 2. 5為J=1的o-H2與J=0的p-H2的波茲曼分佈和溫度的關係圖, 由此可知,製備高純度的p-H2需要將催化劑溫度維持在15 K以下。但 圖 2. 6顯示在15 K時H2的飽和蒸汽壓約只有100 torr,即轉換後的p-H2 無法以高壓的方式儲存在收集瓶中。故本實驗室將轉換器的末端管路 直接導入間質隔離的低溫系統,將轉換得到的p-H2直接沉積於低溫靶 面,也可將銅腔體的壓力保持在H2飽和蒸汽壓下。 p-H2接觸磁性物質會漸漸轉換成 o-H2,所以轉換後的 p-H2頇儘 量避免接觸不鏽鋼或鐵磁性物質。p-H2轉換系統間的管路也儘量以銅 管連接,必要時可使用鋁、銅或玻璃製的收集瓶儲存,但儲存容器內 的雜質含量頇減到最低,避免催化 p-H2轉變為 o-H2。 2.2.4 p-H2的特性 (1)低溫下僅佔據一個轉動能態:
25 氫分子的轉動常數為 59.3 cm-1,J=0 至 J=2 的能階差為 354.4 cm-1(4.28 kJ mol-1)[9],在 4 K 時,J=2 能階之佈居數小於 1.5×10-55。 因此, p-H2在低溫下(1-14 K)形成固體後,轉動量子數幾乎全部 分佈在 J=0。此外,J=1 至 J=3 的能階差為 587.1 cm-1(7.05 kJ mol-1), 因此,在低溫下,p-H2和 o-H2的轉動量子數分別表示為 J=0 和 J=1。 (2)球形的電荷分佈對稱性質: 固態 p-H2(v=0,J=0)的電荷分佈為球形對稱,因此分子間沒 有任何多重偶極矩作用力,僅存在微弱的分散力[16]。 (3)固態氫分子幾乎能自由轉動: 氫分子固體間的鍵結為長距離作用力(long-rang interaction),因 此氫分子形成固體後依然可以自由轉動[22]。Balasubramanian 等人[23] 指認在 940.0 和 1173.5 分別為 S0(0)+ S0(1)和 S0(1)+ S0(1)之純轉動躍 遷譜線。 (4)晶格常數(lattice constant): 原(分)子間形成固體晶格之最小單位晶格(unit cells)間的距離為 晶格常數,而 substitutional hole 則為原(分)子間的距離。對具有相同 晶格結構的原(分)子而言,通常晶格常數愈大,則原(分)子間距離愈 大,如表 2.1 所示。p-H2分子間的相互作用力很弱,形成固體晶格後, 分子間距離大小為 3.78 Å ,較 Ne 晶格(3.16 Å )更大,而和 Ar 晶格
26 (3.75 Å )相似。
(5)大幅度的零點運動(zero-point amplitude motion):
雖然 p-H2形成固體晶格後,分子間的距離(3.78 Å )和 Ar 原子 間的距離(3.75 Å )相差不大,但由於 p-H2分子間僅存在微弱的分散 力,且質量數較一般的鈍氣原子小,因此具有較大的零點振動幅度。 比較量子固體(H2,D2)與一般鈍氣固體(Ne,Ar)的方均根位移 (root-mean-square amplitude)與晶格常數的比值,H2=0.18,D2=0.14, Ne=0.09,Ar=0.05,可知 p-H2晶格的零點振動幅度約為晶格常數的 20%左右[21,24],因此可提供間質客體柔軟的環境。 (6)極大的導熱性(thermal conductivity): 高純度的 p-H2結晶體之導熱係數為 0.72 W cm-1 K-1[9],略大於鐵 的 0.68 W cm-1 K-1,而 Ar 之導熱係數為 0.04 W cm-1 K-1[25]。實驗用 的無氧銅導熱係數約為 4.10 W cm-1 K-1。 2.2.5 p-H2為間質的優點 (1)待測分子有較大機會在間質中轉動: 由於 p-H2可以提供間質客體柔軟的環境,因此鑲嵌在其中的待測 分子有較大的機會可轉動[26]。根據 Pauling 在 1930 年的假設[27], 分子欲在固體晶格中轉動頇滿足下式:
27 max 1 1 2 V B (2. 1) 其中Vmax為分子在固體晶格中轉動時遭遇的能障, B 為分子的轉動常 數。若其數值小於 1,分子可在晶格中轉動;反之,則作擺動運動 (libration)。由於 p-H2間質較其它間質柔軟,Vmax相對於其它間質小, 分子有較大的機會在 p-H2間質中轉動,進而得到分子的轉動資訊。 (2)無晶格效應: 如前所述,在鈍氣間質中的待測分子經光解或其他方式得到能量 後,若產物無足夠能量逸出晶格外,會再度與晶格中其他光裂解物結 合,形成原來的分子或是其異構物,不易產生自由基物種。而在 p-H2 間質中,因晶格束縛能較小,光解產物有較大機會逸出此晶格,剩下 的光裂解產物形成自由基被穩定的保存在間質中,有利於研究光解後 所產生的自由基物種。Shida 研究組[28]用 UV 光源照射 p-H2 中的 C2H5I,觀測到 C2H5自由基及其它產物的吸收譜線;用波長 253.7 nm 的光源照射 p-H2中的 CH3I,觀測到 CH3自由基的吸收譜線[29,29]。 (3)高解析光譜: 由於 p-H2間質之柔軟性,使得 p-H2分子都能調整到能量最小的 位置,因此每個客體在 p-H2 間質中之環境類似,而使得非勻相譜線
增寬效應(inhomogeneous line broading)變小。故在 p-H2間質下可
28
2.3 霍氏轉換紅外光譜儀
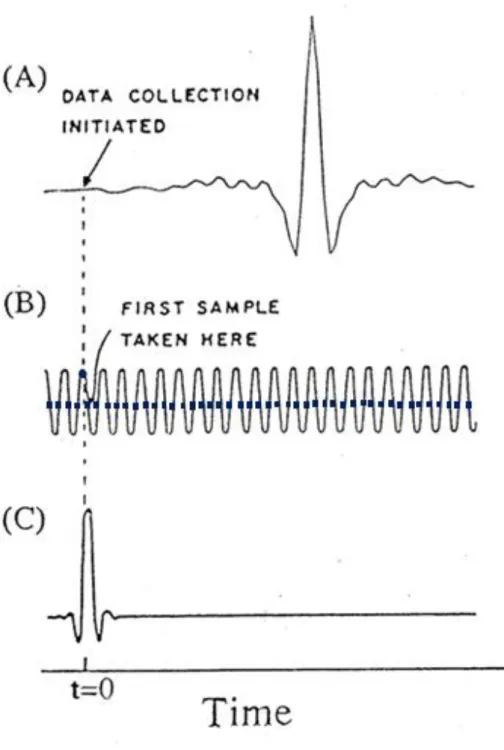
西元 1891 年 Michelson 發明干涉儀(interferometer)[31],利用 干涉現象及理論推算,將干涉圖譜轉換成一般光譜,得到傳統頻域之 光譜資訊;礙於當時技術有限,故僅能解析原子線光譜的細微結構 (find structure)。直到 1950 年代,Fellgett [32]和 Jacquinot [33]等人提 出霍氏轉換光譜儀(Fourier Transform Spectrometer),首次經由計算 方式將干涉圖譜轉換成傳統光譜,短時間內可得到比傳統用光柵或稜 鏡分光的光譜儀靈敏度更高的光譜。西元 1965 年 Cooley 和 Tukey [34] 提出快速霍氏轉換(Fast Fourier Transform,FFT)演算法,並結合當 時新進之微電腦處理系統,大幅降低轉換時間。近年來,隨著微電腦 處理系統與氦氖雷射製造的突破,讓霍氏紅外光譜儀幾乎已完全取代 傳統之紅外光譜儀。 霍氏紅外光譜儀 [35]主體為麥克森干涉儀,搭配電腦系統做霍氏 轉換及數據擷取和處理。以下分別介紹麥克森干涉儀的基本原理及霍 氏轉換紅外光譜儀的基本裝置和優點。 2.3.1 麥克森干涉儀基本原理 麥克森干涉儀主要由分光片(beam splitter)、移動鏡(moving mirror)及固定鏡(fixed mirror)所組成,其架構如圖 2. 7 (A)所示。
29 光源經聚光後,形成帄行光進入干涉儀,再經分光片分成強度幾近相 等的兩道光束,其中一道光束穿透分光片到達固定鏡,另一光束則經 由分光片反射至依光軸方向作等速移動的移動鏡。兩道光束分別由固 定鏡及移動鏡反射後於分光片上混合,每一束光有一半會被導回光源 處,另一半則穿過待測樣品,再由光學鏡組聚焦至偵測器。若移動鏡 及固定鏡分別與分光片相距 m及l,則抵達偵測器的兩束光線會產生 光程差=2(m−l),或稱遲滯(retardation),而發生干涉現象。若光源 為單色光(monochromatic),當光程差為單色光波長λ 之整數倍時會 產生建設性干涉(constructive interference),此時混合光束的強度最 強;當光程差為單色光半波長 λ/2 之奇數倍時會產生破壞性干涉 (deconstructive interference),此時混合光束的強度最弱。若移動鏡 以等速率移動,兩束光線光程差大小隨移動鏡位置變化而作線性改變, 並呈現週期性的建設性與破壞性干涉,如圖 2. 7 (B)所示。相較於傳 統光譜是量測光強度隨波長或頻率之變化,干涉儀所產生的干涉光譜 (interferogram)是測量光強度隨光程差的變化,干涉光譜與傳統光 譜可藉由霍氏轉換互相變換:
2 d i B I e
(2. 2) 其中I
為干涉光譜(光強度對光程差的函數),B
為傳統光譜(光30 開成:
cos 2
d
cos 2
d B I i I
(2. 3) 若只考慮實數部分且理想的干涉光譜應為左右對稱,上式可寫成:
0 2 cos 2 d B I
(2. 4) 數學上,B
稱為I
的霍氏餘弦轉換,I
稱為B
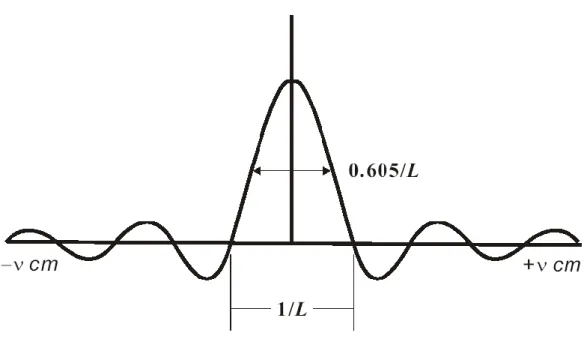
的逆霍氏餘 弦轉換。只要將得到的干涉光譜作霍氏餘弦轉換即可得到傳統光譜。 由式 (2. 4)可知,要將干涉光譜作霍氏轉換頇從 0積分到 , 才能得到傳統光譜。但實際上移動鏡的移動距離有限,光程差無法達 到無限大,僅能在=L至L間掃描,因此若移動鏡移動至L,測得之 光譜如同完整的干涉光譜在光程差L處被截斷(truncated),可視為在式(2. 4)的積分式中乘上一匣式截斷函數(boxcar truncation function)
D :
1 D 當 L L (2. 5)
0 D 當 L (2. 6)即IL
I D 。根據霍氏分析卷積定理(the convolution theoremof Fourier analysis),即兩個函數之乘積(multiplication,數學符號為“”) 的霍氏轉換為此兩個函數個別之霍氏轉換的卷積(convolution,數學 符號為“”),即:
31
L
F I F I D G (2. 7)
F I F D B f (2. 8)
' ' d '
G B f B f
(2. 9)
D 經霍氏轉換可得到一個 sinc 函數 f
,稱為儀器譜線形狀函數(instrument line shape function):
2 sin 2
2 sinc 2
2 L L f L L L (2. 10) 當單色光頻率為1時,式(2.10))可表示為:
2
1 sinc 2
1
G LB L (2. 11)
G 即是截斷後之干涉光譜經霍氏轉換後得到的實際光譜。 如圖 2. 8 所示,經截斷的干涉光譜轉換成傳統光譜後,在主峰兩 邊會產生微小的測波(side lobe),易與其附近之微弱吸收訊號混淆。 為除去匣式截斷函數所造成的側波干擾,可用其他函數取代匣式截斷 函數,其作用彷彿削去主峰旁的足部一樣,故稱此類函數為削足函數 (apodization function)。雖然削足函數可以降低側波之干擾,但也導 致主峰的頻寬增加。因此,若頻寬不是重要的考量,則可選擇側波最 大振幅值 Hs(side lobe amplitude maximum,SLAM)與主峰高度 Hm的比值較小的削足函數;反之,若頻寬為主要的考量因素,則可選用 s m H H 較大的削足函數。本實驗所使用的削足函數名稱為 Hamming,方程式為 0.54+0.46
cos D
,其中D=光程差
/最大光程差
L ;經 Hamming32 削足後,主峰的半高寬變為原本的 91%,側波最大振幅為主峰極高 值的 0.69%。 2.3.2 霍氏轉換紅外光譜儀裝置 一般的霍氏轉換紅外光譜儀有三組不同光源之干涉儀,三組 干涉儀共用分光鏡和移動鏡。三組光源包括連續波長的紅外光源、氦 氖雷射以及連續白光光源,分別做為偵測樣品光譜、測量取樣之相對 光程差、定義零光程差位置之用途。氦氖雷射可提供頻率極為穩定之 單色光源(波長=632.8 nm),故其干涉圖譜為一餘弦函數,如圖 2. 9(B) 所示,餘弦波每段波長有兩個零交叉點,其間隔為 316.4 nm。霍氏轉 換紅外光譜儀以氦氖雷射干涉圖譜的零交叉點(zero-crossing)做為定 位點,建立一個固定光程差的量度法,並以之作為取樣的間隔。由於 此餘弦波之頻率與移動鏡速率成正比,若移動鏡速率稍有變動時,則 餘弦波之頻率亦隨之改變。因此,電腦不能依固定時間間隔取樣,而 必頇依固定之光程差(即:He-Ne 雷射干涉圖譜之零交叉點)取樣,才 能確保每一個取樣點之光程差的準確性。由於氦氖雷射只能定位移動 鏡位移每段距離的相對位置,故利用白光光源干涉圖譜的最高點作為 零光程差位置的訂定。如圖 2. 9(C)所示,連續波長的白光,其干涉 圖譜在 0時,為完全建設性干涉,強度最大;在 0時其強度迅 速減弱,故可產生一個強而窄的訊號,而以此定位取樣的起始點。
33 2.3.3 霍氏轉換紅外光譜儀之優點 霍氏轉換光譜儀相較於傳統分光光譜儀有下列之優點: (1)高光通量之優點(throughput advantage): 由於傳統分光儀的解析度受到狹縫開口的限制,通過之光量只佔 光源之一小部分,而干涉儀無頇使用光狹縫,只是利用光圈來限制光 的發散角,因此光通量遠高於傳統分光儀。相較於傳統分光儀,偵測 器所能量測到的訊號強度較大,對吸收物質的偵測靈敏度較高,也有 更好的訊雜比(signal to noise ration,S/N)。此高光通量(high throughput)優點由 Jacquinot 提出,又稱 Jacquinot 優點[36]。
(2)多重波數之優點(multiplex advantage): 分光式光譜儀是利用光柵將多色光分散開,並調整光柵角度使欲 觀測的單一波長投影在出口狹縫上,因此一次只能作單一波長的測量; 但干涉儀可同時偵測到所有頻率的入射光,不頇分光掃描,不僅相較 傳統光譜節省很多時間,也可避免單一波長掃描時受到系統(如:光 源、吸收分子濃度)不穩定之影響。此外,在固定偵測時間內,霍氏 紅外光譜儀可取得多次干涉光譜並加以帄均,使訊雜比大為提升,並 提高對微弱吸收的偵測靈敏度。此優點由 Fellgett 提出,亦稱 Fellgett 優點[37]。
34 傳統分光式光譜儀無法直接精確測量光柵的轉動角度來準確地 量測光束實際的波數,必頇利用標準樣品產生的已知譜線來校正光譜 儀的波數讀數。而霍氏紅外光譜儀則是使用頻率穩定的氦氖雷射來標 定光程差,經霍氏轉換干涉圖譜後,可得到波數準確度達 0.001 cm-1 的傳統光譜,並不需要另外進行波數的校正工作。此優點由 Connes 提出,又稱 Connes 優點[38]。 (4)抑制散逸光之優點(stray-light control): 使用傳統分光式光譜儀時,波長與單光儀設定不同波長之光子穿 越入口狹縫後,理論上不應由出口狹縫射出,但因為光學零件之不完 美,極少數之光子仍可由出口狹縫散逸而被偵測器偵測到,此稱為散 逸光(stray-light)。對於傳統分光儀而言,欲降低此散逸光並不容易。 然而就干涉儀而言,對於每一波數為 的單色光源,若移動鏡以等速 移動,則偵測器可測得餘弦干涉訊號頻率為 f 2之,即干涉儀掃 描光譜時,已藉由移動鏡速度對訊號加以調頻(modulation)。對非單 一波長組成的光束,移動鏡雖以固定速度掃描,卻是用不同頻率對不 同波長之光線做調頻,如果選用適當的電子濾波器將其他頻率範圍的 訊號過濾除去,便可有效抑制特定波段的散逸光。
(5)高解析度之優點(high resolution advantage):
35 制,一般傳統分光儀解析度不易優於 0.1 cm-1。而干涉儀的理論解析 度是 1/2L,其中 L 為移動鏡的最大移動距離,因此,解析度與兩倍 的移動鏡距離成反比。雖然光學元件的對正(alignment)、移動鏡移 動時的對正、光學鏡面的帄整度、光照孔徑(aperture)大小以及使 用的削足函數等,都是影響解析度的因素,但就一般常用的解析度而 言,其影響不是很大。一般霍氏紅外光譜儀可輕易達到 0.2 cm-1以上 的解析度,甚至可以達到 0.001 cm-1 , 遠較於傳統分光儀高出許多。 (6)靈活且應用廣泛(versatile)之優點: 只要選擇適當的光源、分光鏡及偵測器等光學元件,即可將霍氏 轉換光譜儀應用在遠紅外光、中紅外光、近紅外光、可見光或是紫外 光區的測量。此外,若將光譜儀搭配其他元件,例如:氣相層析儀(GC)、 液相層析儀(HPLC)、質譜儀(MS)、顯微鏡(microscope)、衰減式全 反 射 (ATR) 、 拉 曼 光 譜 儀 (Raman spectroscopy) 、 光 聲 光 譜 儀 (photoacoustic spectroscopy)與多重反射吸收槽(White cell)等,即可應 用於其他定量、定性的分析。
36
圖 2. 1 京都大學 Momose 研究組的封閉式樣品槽(enclosed-cell)實驗 裝置圖[39]。
37
圖 2. 2 美國空軍實驗室 Fajardo 研究組快速沉積法的實驗裝置示意 圖[40]。
38 圖 2. 3 p-H2在不同轉換溫度下o-H2 濃度(以百分比列於第二個數 字)沉積於低溫樣品靶上的紅外吸收光譜。(a)15 K,0.01%,(b) 28 K, 2%,(c) 37 K,8%,(d)52 K,25%和(e)135 K,75%。其中Q1(0)為p-H2 的ν=1←0,J=0←0之禁制躍遷,因為p-H2間質中的o-H2會誘發此吸 收,可用此譜線與Q1(0)+S0(0)譜線之吸收面積比值估算o-H2在間質中 的比例。19
39
圖 2. 4 轉換器內層腔體示意圖。銅圈管內填充著磁性催化劑,其圍 繞著的中圓柱材質為具高熱傳導係數的無氧銅,而外圍之銅片可減少 銅圈與圓柱之間的縫隙。
40
41
42 (A)
(B)
圖 2. 7 (A)麥克森干涉儀(Michelson interferometer)基本架構示意圖; (B)單色光強度 I()與光程差關係之干涉圖譜。
43
圖 2. 8 當移動鏡位移有限,其最大光程差為 L 時,偵測器測得光程 差從-L 到+L 之間的單色光干涉光譜進行霍氏轉換後之圖譜,其波 形為 sin x/x,即 sinc x 函數。
44
圖 2. 9 霍氏紅外光譜儀中三組干涉儀的干涉光譜。(A)碳化矽紅外 光源照射待測樣品,(B)氦氖雷射,圖中實心圓點為零光程差點, 實心方格為零交叉點,(C)白光。
45
表 2. 1 鈍氣分子及 H2的固態晶格參數及游離能[4,41]。
Ne Ar Kr Xe H2
Space group fcc fcc fcc fcc fcc hcp Atomic radius/ Å 0.71 0.98 1.12 1.31 0.74* Lattice parameter/ Å 4.47 5.31 5.65 6.13 5.312 a:3.761
c:6.105 Substitutional hole/ Å 3.16 3.75 3.99 4.34 3.789† Octahedral hole/ Å 1.31 1.56 1.65 1.80 -- Tetrahedral hole/ Å 0.71 0.85 0.90 0.97 -- Ionization energy/ eV 21.56 15.8 14.0 12.1 15.4 *: covalent bond length
46 表 2. 2 一般常用間質的熱性質參數[4]。 間質 擴散溫度(Td)/K 熔點/K 沸點/K Ne 10 24.5 27.1 Ar 35 83.9 87.4 Kr 50 116.6 120.8 Xe 65 161.3 166.0 N2 30 63.3 77.4 CH4 45 90.7 109.2 CF4 -- 123.0 144.0 CO 35 68.1 81.7 CO2 63 212.6 194.6 NO -- 109.6 184.7 SO2 -- 197.6 263.1 SF6 -- 222.7 209.4
47 表 2. 3 鈍氣分子最低能量非禁制電子躍遷之波長及波數。 氣體 波長/nm 波數/104 cm-1 He 58.43 17.114 Ne 73.59 13.589 74.37 13.446 Ar 104.82 9.550 106.67 9.375 Kr 116.49 8.585 123.58 8.092 Xe 129.56 7.718 146.96 6.804
48 參考文獻
1. I. Norman and G. Porter, Nature, 74, 508 (1954).
2. E. Whittle, D. A. Dows, and G. C. Pimentel, J. Chem. Phys. 22, 1943 (1954). 3. M. E. Jacox, J. Mol. Struct. 157, 43 (1987).
4. I. R. Dunking, Matrix-Isolation Techniques:a practical approach (Oxford University Press, New York, 1998).
5. L. Meyer, C. S. Barrett, and P.Jaaser, J. Chem. Phys. 40, 2744 (1964). 6. J. S. Winn, J. Chem. Phys. 94, 5275 (1991).
7. B. R. Cairns, and G. C. Pimentel, J. Chem. Phys. 43, 3432 (1965).
8. S. Grebenev, J. P. Toennies, and A. F. Vilesov, Science 279, 2083 (1998). 9. I. F. Silvera, Rev. Mod. Phys. 52, 393 (1980).
10. M. Okumura, M. C. Chan, and T. Oka, Phys. Rev. Lett. 62, 32 (1989). 11. T. Oka, Annu. Rev. Phys. Chem. 44, 299 (1993).
12. T. J. Lee, J. Vac. Sci. Tech. 9, 257 (1972).
13. T. Momose, M. Miki, and M. Uchida, J. Chem. Phys. 103, 1400 (1995). 14. M. E. Fajardo and S. Tam, J. Chem. Phys. 108, 4237 (1998).
15. Y. J. Wu, X. Yang, and Y.-P. Lee, J. Chem. Phys. 120, 1168 (2004). 16. S. Tam, M. E. Fajardo, and H. Katsuki, J. Chem. Phys. 111, 4191 (1999). 17. I. F. Silvera, Rev. Mod. Phys. 52, 393 (1980).
18. T. C. Nast and I. C. Hsu, Adv. Cryog. Eng 29, 723 (1984). 19. B. AN, M. VI, and P. AN, Instrum. Exp. Tech. 35, 607 (1992).
20. B. A. Tom, Y. Miyamoto, S. Bhasker, T. Momose, and B. J. McCall, Rev. Sci. Inst. 80, 016108 (2009).
21. S. Tam and M. E. Fajardo, Rev. Sci. Inst. 70, 1926 (1999). 22. J. V. Kranendonk, Solid hydrogen (Plenum, New York, 1983).
23. T. K. Balasubramanian, C. H. Lien, J. R. Gaines, K. Narahari Rao, E. K. Damon and
R. J. Nordstrom, J. Mol. Spectrosc. 92, 77 (1982).
24. T. R. Koehler, in Dynamical Properties of Solids, edited by G. K. Horton and A. A. Maradudin (North-holland, Amsterdam, 1975).
25. G. K. White and S. B. Woods, Phil. Mag. 3, 785 (1958).
26. H. Hoshina, T. Wakabayashi, T. Momose, and T. Shida, J. Chem. Phys. 110, 5728 (1999).
27. L. Pauling, Phys. Rev. Lett. 36, 430 (1930).
28. N. Sogoshi, T. Wakabayashi, T. Momose, and T. Shida, J. Phys. Chem. A 101, 522 (1997).
49
29. T. Momose, M. Uchida, N. Sogoshi, M. Miki, S. Masuda, and T. Shida, Chem. Phys. Lett. 246, 583 (1995).
30. M. Fushitani, N. Sogoshi, T. Wakabayashi, T. Momose, and T. Shida, J. Chem. Phys. 109, 6346 (1998).
31. A. A. Michelson, Philos. Mag. 31, 256 (1891). 32. P. Fellgett, J. Phys. Radium 19, 187 (1958). 33. P. Jac quinot, Rep. Prog. Phys. 23, 267 (1960).
34. J. W. Cooley, and J. W. Tukey, Math. Comput. 19, 297 (1965).
35. P. R. Griffiths, and J. A. De Haseth, Fourier Transform Infrared Spectrometry. (John Wiley & Sons, New York, 1986).
36. P. Jacquinot, ΧVⅡème Congrès du G.A.M.S., Paris (1954). 37. P. B. Fellgett, Symposium Ohio State Univ. (1952).
38. J. Connes, J. Phys. Radium 19, 197 (1958).
39. T. Momose, M. Miki, M. Uchida, T. Shimizu, I. Yoshizawa, and T. Shida, J. Chem.
Phys. 103, 1400 (1995).
40. M. E. Fajardo and S. Tam, J. Chem. Phys. 108, 4237 (1998). 41. R. L. Mills and A. F. Schuch, Phys. Rev. Lett. 15, 722 (1965).
50
第三章 實驗裝置與步驟
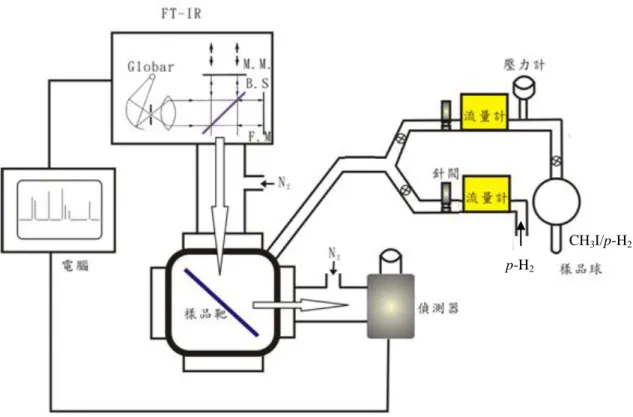
3.1 實驗裝置簡介 本實驗裝置是由真空系統、低溫系統、p-H2轉換系統,以及偵測 系統四部份所組成。真空系統是利用機械幫浦配合分子渦輪幫浦抽氣, 而使系統壓力維持在 3.0×10-6 torr 以下。低溫系統為封閉循環式兩段 壓縮系統,啟動後約 40 分鐘可由室溫降至 3.2 K。p-H2轉換系統可製 造純度約 99.99%的 p-H2,末端連接至真空系統,如前 2.2.3 節所述。 偵測系統則是霍氏紅外轉換光譜儀(Bomem DA8),原理與特性如前 2.3 節所述。 3.1.1 真空系統 真空系統包含間質隔離腔體及腔體前端樣品球之氣體管路二部 份。利用機械前置幫浦(Edwards RV-12,抽氣速率為 237 L min-1)及分子渦輪幫浦(Varian turbo-V301 Nav,抽氣速率為 280 L s-1)可
達成及維持系統的真空度在 3.0×10-6
torr 以下。壓力計分別使用電容 式真空計(MKS,model 223B)和離子式真空計(ion gauge,Ulavc GI-TL2),量測範圍分別為 103-10-1 torr 和 10-3-10-8 torr。
51 3.1.2 低溫系統 間質隔離低溫系統是利用密閉循環式二段壓縮系統(Sumitomo CSW-71),包含壓縮機組、膨脹器、高壓連結管路及溫度控制器 (Lakeshore 331)。當間質隔離腔體壓力下降至 3.0×10-6 torr 以下時, 啟動壓縮機組,壓縮機內部氦氣壓力由靜置時的 1.69 MPa(245 psi) 壓縮至約 2.65 MPa(384 psi),此高壓氦氣經高壓連結管傳送至膨脹 器,由膨脹器內的閥門馬達控制此高壓氦氣進入第一級及第二級之置 換器內部,氦氣在此因膨脹帶走熱量而產生降溫作用。膨脹後的低壓 氦氣(約 100 psi)經由低壓管路導回壓縮機而完成一次的制冷循環。 藉由重複多次的壓縮及膨脹循環過程而達到降溫目的。約 40 分鐘後 可由室溫降至 3.2 K。用於沉積間質樣品的低溫樣品靶則安裝在膨脹 器的最末端,其材質為無氧銅表面鍍上金,而溫度則由矽二極體溫度 計(silicon diode thermometer)測量,並由溫度控制器讀取,且可視 實驗需要由控制器提高溫度加熱樣品靶。低溫裝置之真空外腔底部為 四面窗型,依實驗需要分別裝上溴化鉀(KBr)鹽片及石英(quartz) 片,以利實驗中進行紅外光譜偵測或雷射光解時使用。此外,膨脹器 和真空腔外管之間裝有兩個 O 型環,使膨脹器得以在保持真空狀態 下自由轉動,而將樣品靶分別轉向樣品沉積口及紅外光譜偵測方向, 或是雷射光解的光窗。
52
3.1.3 p-H2轉換系統
p-H2 轉 換 系 統 包 含 低 溫 系 統 及 內 外 層 腔 體 。 低 溫 系 統 是 由
Advanced Research Systems(DE204A)運作,包含壓縮機組、膨脹器、 高壓連結管路及溫度控制器(Lakeshore 331)。而內層腔體連接低溫 系統的冷卻頭,啟動壓縮機組約 45 分鐘後可使低溫系統由室溫降至 10 K。利用溫度控制器可以調整實驗所需要的轉換溫度。外層真空腔 體用來隔絕熱傳導,所需之真空度利用機械式前置幫浦(Edwards RV-12,抽氣速率為 237 L s-1)及分子渦輪幫浦(Varian turbo-V70, 抽氣速率為 68 L s-1)達成及維持系統的真空度在 5.0×10-4 torr 以下。 壓力計分別使用電容式真空計(MKS,model 223B)和熱對流式真空 計(convectron gauge,Ulavc GI-TL2),量測範圍分別為 103
-10-1 torr 和 1-10-4 torr。 3.1.4 偵測系統 於本實驗中,霍氏紅外光譜儀是以碳矽棒(globar)為紅外光源(放 射波長範圍為 200-10000 cm-1),以溴化鉀(KBr)為分光片(450-5000 cm-1),偵測器為 MCT(光區為 400-5000 cm-1),光圈大小選用 2.5 mm, 光譜解析度設定為 0.1 或 0.05 cm-1,移動鏡移動速度選用 1.5 cm s-1, 帄均掃描次數 400 次,於此條件下擷取光譜時間約為 40 分鐘,解析
53 度為 0.05 cm-1時,擷取光譜時間約 80 分鐘。 此外,FT-IR 至偵測器之間,曝露於大氣的部份皆通以氮氣, 以除去光徑中大氣所含的 CO2及 H2O,降低其在紅外光區的吸收所造 成的光譜干擾。 3.2 實驗步驟 3.2.1 樣品製備 實驗中樣品的沉積是採用預先配好特定比例之主客體混合氣體 玻璃球,以針閥控制適當流速,並同時和客體(p-H2)共同沉積於樣品 靶上,而獲得隔離狀態較好的光譜。而樣品的製備是先將樣品球及真 空管路抽至 1.0×10-6 torr,接著關閉通往幫浦的閥門,通入 1.0 torr 的 樣品氣體至樣品球中,待壓力帄衡後再通入 p-H2 氣體,稀釋至待定 比例。依照實驗需要不同之 o-H2濃度而設定不同之 p-H2轉換系統之 溫度,而不同之轉換系統溫度所得到的 p-H2壓力也不同。在 10.5 K、
11.5 K、12.3 K 及 13 K 之 p-H2壓力分別為 8.5 torr、18.5 torr、32.0 torr
及 45.0 torr。將其通入內含 1.0 torr 氣體之樣品球中稀釋二至四次可得 到預混合之氣體樣品濃度約為 1/ 500~1/100。連接至沉積口之氣體管 路及樣品的配置如圖 3. 1 所示,包括樣品玻璃球、流量計(MKS, 10 sccm)、針閥等。沉積條件是 p-H2流量 2~12 sccm,氣體樣品 CH3I/
54 p-H2流量分別為 0.3~0.8 sccm,沉積的樣品間質濃度為 CH3I/ p-H2= 1/ 20000~1/250,沉積時間為一至六小時。 3.2.2 實驗步驟 進行CH3I與p-H2間質之實驗時,當真空系統壓力降至3.0×10-6 torr 時啟動壓縮機開始降溫,待靶面溫度達3.2 K時,取光區為450-5000 cm-1的參考光譜。啟動p-H2轉換系統之壓縮機降溫,外層腔體以真空 幫浦維持在5.0×10-4 torr以下。待溫度降至10.0 K左右後,依實驗需要 將溫度控制在10.0至20.0 K間的特定溫度。先將主低溫系統的靶面轉 開,打開內層腔體至主低溫系統之沉積靶面間的閥門,再打開氫氣鋼 瓶至內層腔體間的閥門。由於剛開始通過轉換器之p-H2壓力還未帄衡, 轉換之p-H2效率並不高,因此等待一段時間使其壓力帄衡後,打開裝 有CH3I/ p-H2的樣品球之氣體管路閥門,並以針閥(needle valve)控制樣 品流量。使CH3I與p-H2在氣體管路中混合後再共同沉積於3.2 K的靶面 上,沉積步驟約進行一到六小時。 待沉積步驟結束後依下列條件擷取紅外吸收光譜: 1. 在3.2 K擷取剛沉積之光譜。 2. 將樣品靶升溫至4.5 K 並維持在該溫度40分鐘後擷取光譜。 3. 樣品靶溫度降回3.2 K擷取光譜。 4. 在3.2 K靜置12小時~134小時後擷取光譜。
![圖 2. 1 京都大學 Momose 研究組的封閉式樣品槽(enclosed-cell)實驗 裝置圖[39]。](https://thumb-ap.123doks.com/thumbv2/9libinfo/8552336.188220/43.892.149.756.187.730/圖21京都大學Momose研究組的封閉式樣品槽enclosedcell實驗裝置圖39.webp)


![圖 4. 40 則為 CH 3 I 取代不同層之 p-H 2 晶格示意圖。其中 I、C 及 H 原 子之凡得瓦爾半徑(van der Waals radius)分別為 198、170 及 120 pm [5],](https://thumb-ap.123doks.com/thumbv2/9libinfo/8552336.188220/82.892.142.625.113.501/則為I取代不同層之晶格示意圖其及原子之凡得瓦爾半分別及.webp)