類神經網路與癌症基因統計資訊應用於白血病醫學文件分類之研究
李俊宏 國立高雄應用科技大學 電機工程學系 E-mail: [email protected]摘 要
近年來生物資訊(Bioinformatics)以驚人速度成長,已成為專家學者相當重視的科學領域,其中以癌 症醫學相關研究特別受到矚目。目前癌症醫學的相關的文獻中潛藏著大量珍貴的癌症知識,可以透過文件 探勘技術將隱藏於文獻資料中癌症知識發掘出來。現階段於文件探勘領域裡,文件分類之技術已廣泛地應 用在基因名稱分析及醫學文件分類,本研究實驗主要以倒傳遞類神經網路(BP-NN)分類技術為基礎,結 合 Sanger 資料庫中的 Cancer Gene Census 資訊,針對 PubMed 資料庫類的癌症基因醫學文獻,發掘其癌症 疾病類別中子類別與基因之關聯性。本研究將以白血病作為主要探討之癌症疾病,依據 Cancer Gene Census 資訊,將白血病分成八個類別 (AML、ALL、CML、CLL、AML&ALL、AML&CML、ALL&CML、ALL&CLL)。白血病基因文獻將依 據此八個類別進行分類與效能評估,證實 BP-NN 之分類架構,有較精確的分類效能。
關鍵詞:Neural Nets, Data Mining, Entrez Gene, Leukemia, PubMed
1. 前 言
過去廿年生物資訊以驚人速度成長與發展,已經成為專家學者相當重視的科學領域。生物資訊所研究 範圍包含:分子生物、遺傳基因、生物醫學技術與應用、基因序列研究、蛋白質檢測、醫學影像、病歷分 析等。近年來由於數位化醫學資訊之建立,如:實驗報告、病歷、研究論文、病理影像,使醫學資訊資料 成長快速,致使醫學研究人員與醫學從業人員必須有效率地分析處理快速膨脹的醫學資訊。 有鑑於此,生物資訊系統必須提供足夠資料空間來儲存大量醫學資訊資料,以及運用該生物資訊系統 發掘重要之知識資訊。近年來知識管理(knowledge management)、資料探勘(data mining)、文件探勘(text mining)等技術,已廣泛應用於該領域中。 1.1 研究背景 文件探勘技術已經廣泛應用於醫學文獻分析的領域,生物醫學文件探勘主要由二個步驟所組成。第一 步驟,從文獻資料中確認所要探討之目標與知識概念,例如:於發掘基因與疾病之間的關係之前,必須從 文獻中基因與疾病名稱擷取出來。第二步驟,針對第一步驟所蒐集之目標與知識概念,進行關聯性之探討。 例如:發掘基因與蛋白質交互關係、蛋白質與蛋白質交互關係等。 目前許多研究實驗已經針對該問題進行探討,Fukuda [1]等人利用蛋白質文獻中的詞彙為線索,辨認蛋 白質與其他組成物的名稱之間有較高的關聯性。Tanabe 與 Wilbur [2]以 Naïve Bayesian model 辨別文章中基 因與蛋白質名稱。Hatzivassiloglou [3]等人比較 Naïve Bayesian model、decision trees 與 inductive rule learning 三種分類模組評估辨別基因與蛋白質之效能。類神經網路 [4]是源自於心理學(Psychology),主要目的是在發展出類似神經元的運算架構,方便了 解生物神經元訊息傳導之方式。換言之,類神經網路是透過內權重(weight)運算的方式來串連內部的處理
節點形成網路架構。其中最廣為使用之演算法則為倒傳遞演算法(Back-Propagation Algorithm)[5],並且為 本研究所應用之類神經網路演算法則。
1.2 研究動機與目的
近年來於基因與疾病相關醫學文件分類領域發展迅速,Adamic [6]等人以 Probabilistic model 分析文章 中基因與疾病出線機率,作為辨別基因與疾病之間關係的參考依據。Chiang [7]提出 GIS 生物醫學文件探勘 系統,結合類神經網路,發掘 PubMed 資料庫之基因文獻資料,以及文獻內基因名稱之間的關聯性。Xu [8] 等人利用類神經網路之技術,辨認醫學文獻中基因序列(DNA sequence)表示方式,以及與基因相關的蛋 白質序(protein sequence)列表示方式。Ritchie [9]等人於類神經網路加入基因演算的技術,提升人類基因 疾病研究資料內基因間交互作用之辨認與偵測。Miller [10]等人以貝氏分類器(Naïve Bayes Classifier)之技 術運用於醫學健康資訊的文章分類,將其文章分類成專業的資訊類別、入門專業的資訊類別以及大眾化等 類別,提供合適的醫學健康資訊給醫學專業人士、醫學方面的初學者、以及病人。Chen [11]等人依據癌症 的種類屬性,已經成功將決策樹(Decision Trees)分類技術應用於醫學文件分類上,而主要地目的是為了 輔助醫學研究人員,快速搜尋所需的癌症文件資訊,增進研究效率。 基因與疾病、基因與癌症相關醫學文件分類相關技術漸趨成熟,相對於癌症疾病類別中子類別與基因 之研究探討較少。有鑑於此,本研究將發掘癌症疾病類別中子類別與基因之關聯性,並對該類別之基因文 獻進行分類,將以白血病為主要討之癌症疾病。 1.3 白血病概述 白血病是一種組成血液或組織的細胞發生不正常變化之疾病,而白血病的分類方式主要依據下列兩種 方式來分類。一種是根據該疾病發生及惡化的速度而分,另一種則是根據所被影響的白血球細胞來分類。 1.3.1 依據疾病發生及惡化速度 可分為急性和慢性白血病。在急性白血病,體內的血液細胞為分化極不成熟的芽細胞,這些芽細胞 增加的速度非常迅速,以至於在短時間內便會出現嚴重的臨床症狀。在慢性白血病,體內的細胞是比急 性白血病分化較成熟的,可以完成它們部分的正常功能,且芽細胞的增加也比急性白血病慢。因此,慢 性白血病在初期可能不會出現任何症狀。 1.3.2 依所被影響的細胞來分類 白血病主要由兩種白血球所發生-淋巴性細胞或骨髓性細胞,當白血病發生在淋巴性細胞,就被稱 為淋巴性白血病。當發生在骨髓性細胞,這疾病就被稱為骨髓性細胞白血病或骨髓性白血病。 1.3.3 四種最常見的白血病 1.3.3.1 急性淋巴性白血病(ALL) 常見於年幼小孩的一類白血病,這類疾病同樣的會影響成人,特別是 65 歲以上的人。 1.3.3.2 急性骨髓性白血病(AML) 在成人和小孩都會發生此型的白血病,有時被稱為急性非淋巴性白血病(ANLL)。 1.3.3.3 慢性淋巴性白血病(CLL) 常發生於 55 歲以上的成人,有時也發生在年輕的成人。 1.3.3.4 慢性骨髓性白血病(CML) 主要是發生在成人,少部分小孩也同樣會產生此疾病。
2. 文獻探討
類神經網路[4]是源自於心理學(Psychology),主要目的是在發展出類似神經元的運算架構,方便了解 生物神經元訊息傳導之方式。換言之,類神經網路是透過內權重(weight)運算的方式來串連內部的處理節 點形成網路架構。 而 將 類 神 經 網 路 應 用 於 分 類 問 題 上 有 下 列 幾 點 優 勢 , 類 神 經 網 路 對 於 雜 訊 資 料 有 較 高 的 容 忍 度 (tolerance),其次類神經網路能藉由學習與訓練的方式來完成資料分類,此外近來已發展出多種演算法則 應用於類神經網路上。其中最廣為使用之演算法則為倒傳遞演算法(Back-Propagation Algorithm),並且為 本研究所應用之類神經網路演算法則。 2.1 倒傳遞演算法 倒傳遞演算法[12]是建構在多層認知器(Multilayer Perceptron)上的分類方法,從學習過程中可分成正 向傳遞(forward)與反向傳遞(backward)二部份來探討。所謂正向傳遞係指輸入訊號從輸入層(Input layer) 進入倒隱藏層(Hidden layer)然後傳遞至輸出層(Output layer),其中每個神經元(neuron)只負責將前一 層之輸出訊號傳播至下一層之神經元,以產生最終輸出訊號。而返向傳遞係指將輸出誤差值從輸出層反饋 (feedback)至隱藏層,之後依據輸出誤差值從新分配隱藏層內部權重,因此倒傳遞演算法是透過修正全種 植的方式使輸出值更接近問題的解。2.1.1 正向傳遞演算法(Propagation the inputs forward)
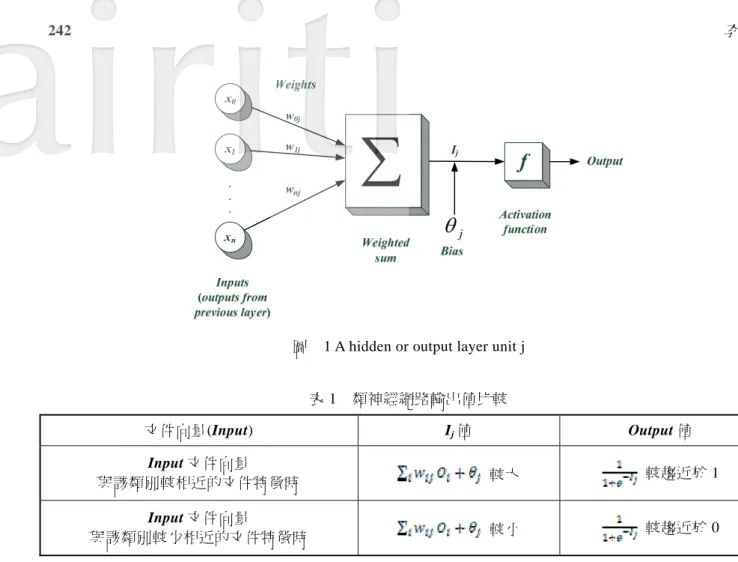
首先將訓練資料置於輸入層進行運算,以類神經網路內神經元j(Unit j)為例,如圖 1所示,將前 一層之輸出值作為該層之輸入值,進行權重運算如方程式(1)錯誤! 找不到參照來源。所示,其中Oi代表
前一層神經元i之輸出值,wij表示神經元i到神經元j的權重,θj代表偏移量,Ij為權重運算後之輸出值。其
次將經由轉換函數之計算,調整Ij輸出範圍,產生輸出Oj值向下一層神經元傳遞。而類神經網路所採用之 轉換函數,通常具有正向收斂與負向收斂型態之函數,其中以Log-Sigmoid轉換函數較廣泛使用。而 Log-Sigmoid轉換函數表示方式如方程式(2)錯誤! 找不到參照來源。所示,當輸入變數趨近於無限大時, 則輸出值會趨近於 1,相反地,當輸入變數趨近於負無限大時,則輸出值會趨近於 0。而在白血病基因文 件分類的部分,以AML類別為例,當測試文件的輸出值趨近於 1 時,表示該文件內擁有與AML類別較相 近的文件特徵,因此該文件屬於AML類別,反之輸出值趨近於 0 時,表示該文件內擁有與AML類別較少 相近的文件特徵,因此該文件不屬於AML類別,如表 1所示。 (1) (2)
∑
j
θ
圖 1 A hidden or output layer unit j
表 1 類神經網路輸出值比較 文件向量(Input) Ij值 Output 值 Input 文件向量 與該類別較相近的文件特徵時 較大 較趨近於 1 Input 文件向量 與該類別較少相近的文件特徵時 較小 較趨近於 0
2.1.2 反向傳遞演算法(Back-propagation the error)
反向傳遞是將訓練過程中輸出值與實際值有落差時,進行誤差值計算,作為之後調整權重運算之依 據。若神經元 j 位於輸出層內,其誤差值計算如方程式(3)所示,其中 Oj為神經元 j 輸出值,Tj為實際值。 (3) 而位於隱藏層內部神經元 j 之誤差值計算,則須加入該神經元與下一個神經元的權重運算數值以及下 一個神經元的誤差值,表示方式如方程式(4)所示,其中 wjk為神經元 j 到神經元 k 之權重值,Errk為單位 k 之誤差值,Oj為單位 j 輸出值。 (4) 類神經網路之學習訓練過程是藉由修正權重的方式,使得輸出值更接近實際值。權重重新修正方式 如方程式(5)、(6)所示,Δwij表示經過一個學習循環的誤差程度,wij表示神經元 i 到神經元 j 的權重,l 表 示學習速率(learning rate),其數值通常介於 0~1 的範圍內,當數值越大時,則每次權重之修正量就越大。 Errj為神經元 j 之誤差值,Oi為神經元 i 輸出值。 (5)
(6) 偏移量重新修正方式如方程式(7)、(8)所示,Δθj表示經過一個學習循環的偏移量改變程度,θj表示單 位 j 的偏移量。 (7) (8) 由於在文件分類的實驗過程中所得到的輸出值不一定為 0 或 1,必須藉由誤差值計算的方式,重新分 配類神經網路內部的權重,調整輸出的範圍成為 0 或 1。 2.2 貝式分類器
貝氏定理(Bayesian Theorem)是由十八世紀英國數學家 Thomas Bayes 從條件機率推導出來的,因此 貝氏分類器(Naïve Bayes classifier)係指以貝氏定理(Bayesian Theorem)為基礎計算出該資料與各類別之 間的機率,以機率方式來推薦分類結果[13]、[14]、[15]、[16]。而貝氏分類器是採用監督式學習方式,因此 於分類之前須事先了解分類型態,藉由訓練學習的方式,有效地處理未來欲分類的資料。貝氏分類器數學 模組如方程式(9)表示。 (9) dj:單一篇文件,j = 1 ~ m Ci:單一個類別,i = 1 ~ n Wkj:關鍵字 Wk 在 dj文件中的權重 以下將以 AML 為例,說明貝氏分類演算法應用於醫學文件分類上。在蒐集之 100 篇文獻資料中,屬於 AML 類別有 5 篇文件(以下該類別將以 C1表示),非 AML 類別之文件有 6 篇(以下該類別將以 C2表示)。 假設一文件 D 內存在關鍵字詞 Tk1、Tk2、Tk3,其中 Tk1於文件 D 中出現 4 次,整體文獻資料中出現 25 次, Tk2於文件 D 中出現 2 次,整體文獻資料中出現 8 次,Tk3於文件 D 中出現 3 次,整體文獻資料中出現 15 次。 2.2.1 文件向量空間權重值 TFIDF Tk1(TFIDF) = 4*log(100/25) = 2.4 Tk2(TFIDF) = 2*log(100/8) = 2.2 Tk3(TFIDF) = 3*log(100/15) = 2.8 2.2.2 分屬於 C1與 C2類別關鍵字詞之機率 P(Tk1|C1) = 2.4/5 = 0.48 P(Tk2|C1) = 2.2/5 = 0.44 P(Tk3|C1) = 2.8/5 = 0.56
P(Tk1|C2) = 2.4/6 = 0.4 P(Tk2|C2) = 2.2/6 = 0.37 P(Tk3|C2) = 2.8/6 = 0.47 2.2.3 最後得到 C1與 C2總機率分別 P(D|C1) = 0.48*0.44*0.56 = 0.12 P(D|C2) = 0.4*0.37*0.47 = 0.07 由於 P(D|C1) = 0.12 > 0.07 = P(D|C2),因此將文件 D 歸類為 AML 類別。 2.3 決策分類器 目前決策樹演算法已是相當成熟的分類技術,其中以澳洲雪梨大學的教授 Ross Quinlan 所開發一系列 以亂度(Entropy)為基礎之決策樹演算法,最為知名並且廣泛應用於分類問題上,從最早期的 ID3(Iterative Dichotomiser 3)到後來的 C4、C4.5、C5,非常受到資料探勘領域人員歡迎。 2.3.1 C4.5 演算法
C4.5 演算法為 Ross Quinlan 所提出的決策樹演算方法[17]、[18]、[19],是從 ID3(Iterative Dichotomiser 3)演進而來。C4.5 演算法主要可從兩個部份來探討,第一為分類的標準,分類的標準是依據 Gain Ratio 來計算,依據 Gain Ratio 計算可以建構完整的決策樹,其次則為剪裁的標準,修剪的標準是依據 Error Based Pruning(EBP)來計算,經由 EBP 計算剪裁決策樹內部不需要的分支,以提高分類的準確性。以下概述 Gain Ratio 與 Error Based Pruning 的運算方式。
2.3.1.1 Gain Ratio 運算方式
Gain Ratio 主要是將計算出來之 Information Gain 正規化(normalize),運算方式如方程式(10)所示:
(10)
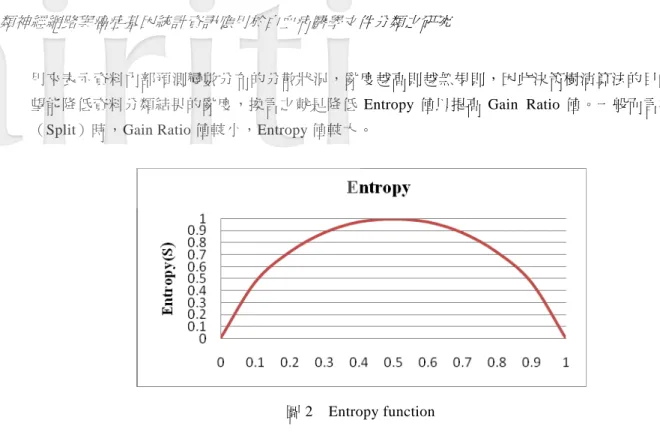
在 ID3 演算法中 Information Gain 主要是用於計算資料集在測試前與測試後的差異情況,運算之方 式是將「測試前的資訊」減去「測試後的資訊」。倘若測試之後資料集內的亂度越小時,所計算出來的 Information Gain 將越大,如此一來對於決策樹的建構將會越有利。Information Gain 運算方式如方程式 (11)所示: (11) Entropy在計算機科學領域最初主要是用於亂度(impurity)的計算,當資料集合內的亂度最高時(資 料集合內的亂度達到最亂時),Entropy的值將會為 1,如圖 2所示。Entropy運算方式如方程式(12)所示: (12) 在 Entropy 運算方式中,P+代表正類別資料集合在正、負類別資料集合中所佔的比例,P-代表負類 別資料集合在正、負類別資料集合中所佔的比例,S 為所要計算的資料集合。Entropy 在資料探勘中是
用來表示資料內部預測變數分布的分散狀況,亂度越高則越無規則,因此決策樹演算法的目的就是希 望能降低資料分類結果的亂度,換言之就是降低 Entropy 值以提高 Gain Ratio 值。一般而言於分岔點 (Split)時,Gain Ratio 值較小,Entropy 值較大。
圖 2 Entropy function
2.3.1.2 Error Based Pruning 運算方式
節點的錯誤率的方式,尋找出導致整棵決策樹錯誤率升高的節 點,
Error Based Pruning 是以計算出每個
將這些節點修剪掉以提升決策樹的準確率。而錯誤率即表示該節點所有的正、負類別資料中,非 正、負類別資料與所有類別資料的比值,由於決策樹的建構過程目的是將全部正、負類別資料能夠迅 速地分類完成。Error Based Pruning 運算方式如方程式(13)所示:
(13) q 為該節點或是該分枝的錯誤率,Sv 為非主要類別資料的數量,S 為該節點的資料總合,e 為所定 義的 2.4 醫學文獻資料庫 訊技術發展迅速,醫學文件數量以驚人地速度成長,以 PubMed(MEDLINE)為例, 每週 2.4.1 PubMed 資料庫系統 ]的建立是為了方便存取 MEDLINE 資料庫內文獻資料的搜尋技術,MEDLINE 資料 錯誤率。決策樹技術應用於文件分類上的方式,是將各文件內部之關鍵字(Key Words)視為屬性 值,作為判斷決策樹向下分支之依據。利用各個類別訓練集合所訓練出來的決策樹規則,針對未知類 別文件集合進行分類。 近年來由於生物資 以 1500 篇的速度增加,因此建立一套有效管理、分類醫學文獻的資料庫系統,提供學生、專業人士、 研究人員專業資料的查詢與蒐集,成為一件重要且刻不容緩的事。NCBI(National Center for Biotechnology Information)是現今生物醫學專業領域文獻蒐集最完整且一般研究人員最普遍使用的資料庫查詢系統,而該 資料庫系統也針對不同研究領域,設立不同搜尋引擎與搜尋法則。
PubMed 資料庫系統[20
2.4.1.1 OLDMEDLINE 是指於 1966 年之前的 MEDLINE 文獻資料,早期的醫學文獻資料主要以紙本的方式蒐集管理,直 到近廿年間醫學文獻資料才以電子文獻的方式管理與蒐集,PubMed 也提供 OLDMEDLINE 文獻檢索與 文獻引用資料之存取。 2.4.1.2 典藏文獻的管理與蒐集 其中包含重要科學期刊、化學期刊、生物及醫學期刊文獻檢索與文獻引用資料,以及部份電子期 刊全文資料。
2.4.1.3 MeSH(Medical Subject Headings)[21]文獻檢索存取
MeSH 是 NLM 用以分析生物醫學方面之期刊文獻、圖書、視聽資料、電子資源等資源的主題內容 之控制語彙表。
而現今生物醫學文獻探勘大多都是透過 PubMed 搜尋引擎,來蒐集研究所需的文獻資料。Miller [10]、 Polavarapu [22]、Liu [23]、Chen [11]、Krauthammer [13]等均透過 PubMed 搜尋引擎獲得研究所需的文獻 資料。
2.4.2 Entrez Gene
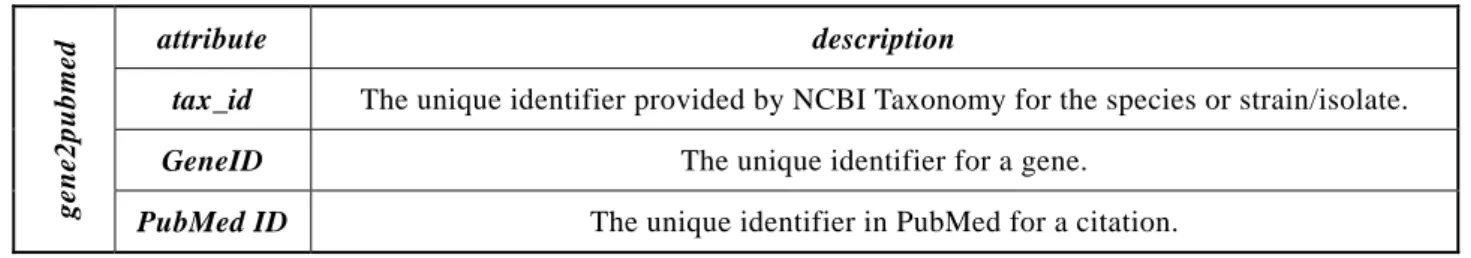
Entrez Gene(Wheeler 2005)[24]為NCBI醫學文獻資料庫專門用於提供基因文獻資料與基因、染色體化 學結構資訊的搜尋引擎資料庫,所提供之資訊包含:基因名稱(nomenclature)、染色體位址(chromosomal localization)、基因生成物及其屬性(gene products and their attributes)、顯性特徵(phenotypes)、交互作用 (interactions)等資訊。此外該資料庫也提供gene2pubmed、gene2go、gene2refseq等檢索檔,讓使用者更直 接地、更便利地存取MEDLINE資料庫內文獻資料。以gene2pubmed為例檔案描述如表 2。
表 2 The description of gene2pubmed
attribute description
tax_id The unique identifier provided by NCBI Taxonomy for the species or strain/isolate.
GeneID The unique identifier for a gene.
gene2pubmed PubMed ID The unique identifier in PubMed for a citation.
2.4.3 Sanger Cancer Genome Project
由於長期暴露於電磁波的環境中、攝取有致癌風險的食物以及不正常作息習慣,容易造成基因突變、 變異,導致癌症疾病的發生。因此 Sanger Institute 針對這類議題進行研究,Sanger Institute 是專門研究人 類的基因組成結構、基因變異、癌症基因之研究中心,並且積極整合極推動多領域的大型研究計畫,如 Human Genome Project、Cancer Genome Project、Ensembl(The Ensembl Genome Browser)、Pathogen Sequencing 等。
本研究實驗採用上述 Sanger Institute 提供的癌症基因統計資訊(Cancer Gene Census)與 Entrez Gene 資料庫中的 gene2pubmed 檢索資料作為醫學文件分類之特徵選取重要依據,並且以傳遞類神經網路作為 白血病基因文件分類主要的分類技術,最後與 Ross Quinlan 所提出的 C4.5 決策樹分類器、Naïve Bayes 貝氏分類器評估分類效能。
3. 研究方法
本研究目的是為了發掘癌症疾病子類別與基因之關聯性,並對該類別之基因文件進行分類,將以白血 病為主要討論之癌症疾病。白血病基因文件分類實驗流程如圖 3所示,於實驗設計上分成兩階段,第一階段 分類器的設計與訓練,如圖 3實線部分流程所示,第二階段白血病基因文件分類測試,如圖 3虛線部分流程 所示。 圖 3 白血病基因文件分類實驗流程圖 3.1 癌症基因文件特徵選取 Sanger Institute是專門研究人類的基因組成結構、基因變異、癌症基因之研究機構,本研究將利用該網 站上所提供的癌症基因統計資訊(Cancer Gene Census),作為特徵選取之參考依據,並且運用Entrez Gene 資料庫中的gene2pubmed檢索資料輔助特徵之選取,癌症基因文件特徵選取之流程如下,圖示如圖 4。3.1.1 Step1
首先將癌症基因統計資訊(Cancer Gene Census)與 gene2pubmed 檢索資料篩選出白血病基因的表示名 稱,分別建立 Gene and cancer symbol list 與 Gene symbol list。
3.1.2 Step2
其次將之前篩選出白血病基因的表示名稱(Gene and cancer symbol list 與 Gene symbol list)進行基因與 癌症名稱統計分析,並且建立癌症基因表示名稱清單。
3.1.3 Step3
圖 4 癌症基因文件特徵選取之流程圖
3.2 白血病基因名稱資料分析結果
從Cancer Gene Census資訊與gene2pubmed檢索資料分析得知,白血病(leukemia)可分成四個類別:急 性骨髓性白血病(以下以AML簡稱之)、急性淋巴性白血病(以下以ALL簡稱之)、慢性骨髓性白血病(以 下以CML簡稱之)、慢性淋巴性白血病(以下以CLL簡稱之)。此外Cancer Gene Census資訊與gene2pubmed 檢索資料分析結果亦發現,在部份類別之白血病基因資料擁有相同的基因表示名稱,本研究將此交集之基 因名稱獨立成一類別,以提升分類的準確性。最後將統計分析得到的基因名稱、基因ID、腫瘤類型紀錄於 表格內,而各類別分析結果如表 3、表 4所示。透過此八個類別癌症基因特徵清單,保留下該文件基因特徵, 以確保爾後特徵維度篩選時,該基因特徵不會被刪除。
表 3 AML、ALL、CML、CLL 基因名稱清單
Symbol Gene ID Tumor Symbol Gene ID Tumor
ABL2 27 AML AF1Q 10962 ALL AF15Q14 57082 AML AF3p21 51517 ALL ARHGEF12 23365 AML AF5q31 27125 ALL ARNT 405 AML CDK6 1021 ALL CEBPA 1050 AML EWSR1 2130 ALL CREBBP 1387 AML FBXW7 55294 ALL
Symbol Gene ID Tumor Symbol Gene ID Tumor
D10S170 8030 CML BCL3 602 CLL HOXA11 3207 CML BTG1 694 CLL MSI2 124540 CML CCND2 894 CLL FSTL3 10272 CLL MYC 4609 CLL TCL1A 8115 CLL
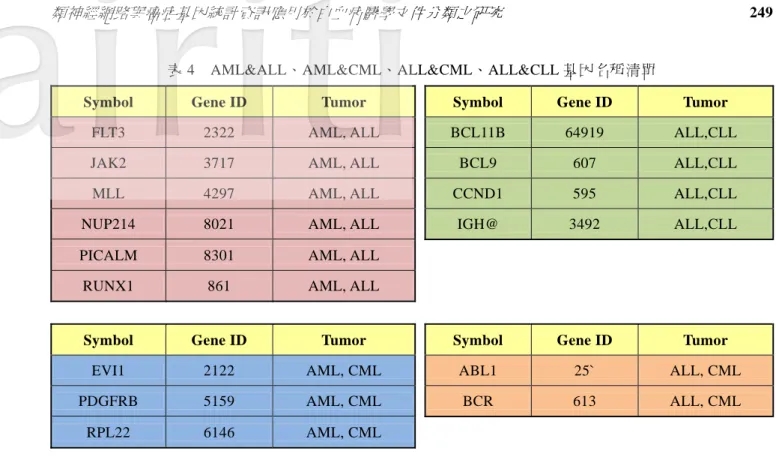
表 4 AML&ALL、AML&CML、ALL&CML、ALL&CLL 基因名稱清單
Symbol Gene ID Tumor Symbol Gene ID Tumor
FLT3 2322 AML, ALL BCL11B 64919 ALL,CLL JAK2 3717 AML, ALL BCL9 607 ALL,CLL MLL 4297 AML, ALL CCND1 595 ALL,CLL NUP214 8021 AML, ALL IGH@ 3492 ALL,CLL PICALM 8301 AML, ALL
RUNX1 861 AML, ALL
Symbol Gene ID Tumor Symbol Gene ID Tumor
EVI1 2122 AML, CML ABL1 25` ALL, CML PDGFRB 5159 AML, CML BCR 613 ALL, CML
RPL22 6146 AML, CML
3.3 訓練與測試文件集合
在訓練與測試文件集合部分,將透過 Entrez Gene 資料庫蒐集 AML、ALL、CML、CLL、AML&ALL、 AML&CML、ALL&CML、ALL&CLL 等八個類別文件資料作為訓練集合。其次文件分類測試部分,以 ARNT、EWSR1、HOXA11、CCND2、RUNX1、PDGFRB、BCR、CCND1 等八種基因文件資料來測試多類 別文件分類效能,並且與貝氏分類演算法、決策樹演算法比較其分類效能。 表 5代表實際訓練文件資料蒐集部分,起初設定文件蒐集數量之目標為:各個類別文件資料均 500 篇,但 在文件蒐集過中,發現CML類別在PubMed資料庫中僅有 128 篇,未達原先設定數量。因此為了讓參與訓練 分類器的初始數值相同,利用重複抽取CML類別文件資料進行訓練,達到各類別相同的文件資料數量,如表 7所示。調整之後之訓練集合如表 8所示。表 6代表實際測試文件資料蒐集部分,起初設定文件蒐集數量之 目標為:各個類別文件資料均 100 篇,所蒐集基因文件數量均超過 100 篇之數量,只需經過篩選將可以得 到所需測試文件資料,如表 9所示。 表 5 各類別蒐集文件數目
Tumor Types ALL AML CLL CML ALL,CLL ALL, CML AML, ALL AML, CML No. of doc. 532 512 538 128 475 500 521 468
表 6 各基因類別蒐集文件數目
Gene EWSR1 ARNT CCND2 HOXA11 CCND1 BCR RUNX1 PDGFRB No. of doc. 161 175 200 101 180 223 181 195 Tumor Types ALL AML CLL CML ALL,CLL ALL, CML AML, ALL AML, CML
表 7 各類別詳細文件數目
Symbol Tumor Types doc. No. Symbol Tumor Types doc. No. AF1Q ALL 17 BCL11B ALL, CLL 49 AF3p21 ALL 3 BCL9 ALL, CLL 36 AF5q31 ALL 13 CCND1 ALL, CLL 190
CDK6 ALL 300 IGH@ ALL, CLL 200 EWSR1 ALL 110 ABL1 ALL, CML 156 FBXW7 ALL 89 BCR ALL, CML 300 ABL2 AML 5 FLT3 AML, ALL 150 AF15Q14 AML 3 JAK2 AML, ALL 100 ARHGEF12 AML 4 MLL AML, ALL 100 ARNT AML 200 NUP214 AML, ALL 19
CBL AML 200 PICALM AML, ALL 2 CREBBP AML 100 RUNX1 AML, ALL 150
BCL3 CLL 153 EVI1 AML, CML 180 BTG1 CLL 76 PDGFRB AML, CML 240 CCND2 CLL 200 RPL22 AML, CML 48 FSTL3 CLL 20 MYC CLL 79 TCL1A CLL 10 D10S170 CML 19 HOXA11 CML 101 MSI2 CML 8 表 8 調整後各類別蒐集文件數目
Tumor Types ALL AML CLL CML ALL,CLL ALL, CML AML, ALL AML, CML No. of doc. 500 500 500 500 500 500 500 500
表 9 各基因類別蒐集文件數目
Gene EWSR1 ARNT CCND2 HOXA11 CCND1 BCR RUNX1 PDGFRB No. of doc. 100 100 100 100 100 100 100 100 Tumor Types ALL AML CLL CML ALL,CLL ALL, CML AML, ALL AML, CML
3.4 分類效能評估之方式 分類效能評估可從幾個面向著手,第一為分類速度(speed),可利用量測時間的方式解分類器之執行速 度。其次為強健程度(robustness),考量分類器是否因為雜訊資料的加入,而影響其分類的準確度。第三為 可 擴 充 性 ( scalability), 評 量 該 分 類 器 是 否 能 夠 有 效 率 處 理 越 來 越 大 的 資 料 集 合 。 最 後 為 可 闡 明 程 度 (interpretability),其目的為了瞭解分類器內部的複雜程度,如:決策樹內部的節點數、類神經網路隱藏層 內部的單位數等。 目前較常用於分類器正確率之評估方式是以 Precision、Recall、F1 measure 等三種方式,其表示方式如 方程式(14)、(15)、(16)。 (14) (15) (16) t_pos:正確分類之文件集合 f_pos:不符合分類要求,但被分類器納入分類類別之文件集合 f_neg:符合分類要求,但未被分類器納入分類類別之文件集合
由於 F1 measure 評估方式均採用 Precision、Recall 的評估數值,因此本研究實驗將主要以 F1 measure 來評估分類要能,並且與貝氏分類演算法、決策樹演算法比較其分類效能。
4. 分類實驗結果
本實驗將訓練出 AML、ALL、CML、CLL、AML&ALL、AML&CML、ALL&CML、ALL&CLL 等八 個白血病基因分類器,透過 Entrez Gene 資料庫蒐集 AML、ALL、CML、CLL、AML&ALL、AML&CML、 ALL&CML、ALL&CLL 等八個類別文件資料作為訓練集合。其次文件分類測試部分,以 ARNT、EWSR1、 HOXA11、CCND2、RUNX1、PDGFRB、BCR、CCND1 等八種基因文件資料來測試多類別文件分類效能, 並且與貝氏分類演算法、決策樹演算法比較其分類效能。 4.1 特徵維度篩選 各分類器之特徵維度篩選,將依據實驗過程所設定之 TFIDF 門檻值(threshold)來調整各分類器之特 徵維度,如:當 TFIDF 值為 10 時,則刪除該類別字詞特徵向量空間 TFIDF 值低於 10 之字詞。藉由調整各 分類器之特徵維度,尋求各分類器最佳的分類情形本實驗將 TFIDF 值分成 1, 5, 10…45 等十個範圍進行探討。
4.2 各分類器效能評估 依據八個類別分類器測試結果得知(如表 10所示),而大部份分類器最佳分類情形(F1 值最大者),其 特徵維度大都落於 250 上下,僅CML類別之特徵維度落於 355。而CML類別之特徵維度不同於其他類別, 主要受到先前重複抽取CML類別文件資料進行訓練,達到各類別相同的文件資料數量之影響,造成訓練過 程中,CML類別文件TFIDF數值提高,使得特徵維度高於其他類別。 表 10 各類別最佳情形測試結果
Category TFIDF dim precision recall F1
ALL 40 256 0.9151 0.9857 0.9491 AML 40 223 0.8937 0.9762 0.9331 CLL 35 253 0.9011 0.9883 0.9427 CML 35 355 0.9213 0.9891 0.9540 ALLCLL 40 225 0.8949 0.9910 0.9405 ALLCML 40 224 0.8607 0.9719 0.9129 AMLALL 35 284 0.8873 0.9864 0.9342 AMLCML 35 271 0.8872 0.9981 0.9338 圖 5為各類別於不同TFIDF範圍F1 measure評比結果,大部份分類器最佳分類情形,其TFIDF範圍大都落於 35、40 區間(特徵維度落於 250 上下)。 圖 5 各類別於不同 TFIDF 範圍之 F1 數值 4.3 基因文件資料分類效能評估 類別基因文件分類結果如表 11所示,ANN演算方法在EWSR1、ARNT、CCND2、HOXA11、CCND1、 RUNX1 基因文件類別,比BP演算方法與DT演算方法有較佳的F1 Measure值,而DT演算方法則於BCR、 PDGFRB基因文件類別有較佳的分類效能。
其次ANN演算方法未能於BCR、PDGFRB基因文件類別獲得較佳的分類效能,可以從兩個方向來探討。 第一、從白血病基因文件集合之結構進行探討,在各類別文件集合內容中,ALL、AML、CLL、AMLCLL 由六種基因 文件所組成 ,ALLCLL由四種基因 文件所組成 ,CML、AMLCML由 三 種基因文件 所組成, ALLCML由二種基因文件所組成,如表 7所示。明顯地發現ALLCML、AMLCML訓練文件類別內,主要由 ABL1、BCR與EVI1、PDGFRB、RPL22 基因文件所組成,少於其他類別基因文件所組成,致使在特徵選取 過程中,擷取出特徵字彙與ALLCML、AMLCML文件類別關聯性較低,降低分類效能。第二、從表 7得知, ALLCML、AMLCML訓練文件類別內BCR、PDGFRB有較高的文件數量,使得ALLCML、AMLCM分類器 習慣該訓練文件類別字彙結構,一旦出現與訓練文件類別字彙結構不同的BCR、PDGFRB文件,不易歸類 至正確的類別。 此外在 CML 文件資料類別部分,其資料結構與 ALLCML、AMLCML 文件資料類別相似,但由於 CML 文件資料類別於 PubMed 資料庫內僅有 128 篇醫學文件,而且本研究實驗均將此 128 篇文件資料納入訓練 集合內,因此 CML 類別分類效能不但未下降,並且為本研究實驗分類效能最佳的文件類別。 表 11 ANN、BP、DT 分類之 F1 measure 數值 Classifier F1 measure ANN BP DT EWSR1 0.9531 0.9258 0.9343 ARNT 0.9323 0.9119 0.9237 CCND2 0.9576 0.9343 0.9412 HOXA11 0.9751 0.8423 0.9478 CCND1 0.9453 0.9117 0.9132 BCR 0.9053 0.9137 0.9122 RUNX1 0.9257 0.9147 0.9152 Gene doc. PDGFRB 0.8957 0.8973 0.9153
5. 結論與未來工作
5.1 研究結論特性 醫學文件分類於文件探勘相關研究領域裡已受到專家學者相當重視與探討的議題,特別針對如何協助 醫生與醫學從業人員,從癌症疾病文件資料中,萃取出隱藏於該文件裡重要的蛋白質、染色體、基因等相 關訊息。於知識管理層面而言,有效率地分類取得與保存癌症疾病基因文件資料,以及發掘潛藏於該文件 資料中的重要訊息,為提升知識與工作效率之基礎。本研究實驗以倒傳遞類神經網路(Back-Propagation Neural Network)分類技術為基礎,結合 Sanger 資 料庫中的 Cancer Gene Census 資訊與 Entrez Gene 資料庫中的 gene2pubmed 檢索資料,針對 PubMed 資料庫 類的癌症基因醫學文獻,發掘其癌症疾病類別中子類別與基因之關聯性,依據該關聯性對白血病基因文獻 資料進行文件分類。最後與 Ross Quinlan 所提出的 C4.5 決策樹分類器、Naïve Bayes 貝氏分類器評估分類效 能,並且證實以倒傳遞類神經網路分類技術之分類架構,有較精確的分類效能。
對白血病基因文獻資料進行文件分類之研究,研究過程察覺到下列幾點特性。 5.1.1 Cancer Gene Census 資訊輔助有效提升白血病基因文件分類正確性
在白血病基因文件特徵萃取部分,透過 Cancer Gene Census 資訊之篩選與過濾處理,可以有效過濾 非該文件特徵之字詞,有效提升文件分類的正確性。 5.1.2 特徵維度於 250 維上下有較佳的分類效能 各分類器之特徵維度篩選,將依據實驗過程所設定之 TFIDF 門檻值(threshold)來調整各分類器之 特徵維度,如:當 TFIDF 值為 10 時,則刪除該類別字詞特徵向量空間 TFIDF 值低於 10 之字詞。藉由調 整各分類器之特徵維度,尋求各分類器最佳的分類情形。大部份分類器最佳分類情形(F1 值最大者),其 特徵維度大都落於 250 上下,僅 CML 類別之特徵維度落於 355。 5.1.3 倒傳遞類神經網路、貝氏分類器與決策樹分類器之分類效能評比 ANN 演算方法在 EWSR1、ARNT、CCND2、HOXA11、CCND1、RUNX1 基因文件類別,比 BP 演 算方法與 DT 演算方法有較佳的 F1 Measure 值,而 DT 演算方法則於 BCR、PDGFRB 基因文件類別有較 佳的分類效能。 5.2 研究結論貢獻 本研究貢獻在於提供一個完整白血病基因醫學文件分類架構,於字彙特徵選取部分,匯入癌症基因統 計資訊(Cancer Gene Census)與 gene2pubmed 檢索資料,作為字彙特徵選取參考的重要依據。於分類技術 部分,系採用傳遞類神經網路、C4.5 決策樹分類器、Naïve Bayes 貝氏分類器三種常用分類技術,用以評估、 比較其分類效能,分類結果於此三種分類技術上,均有不錯的分類效能。本研究之貢獻敘述如下。
5.2.1 發掘不同白血病文件類別,文件內存在著相同的基因名稱
從 Cancer Gene Census 資訊與 gene2pubmed 檢索資料得知,目前與白血病相關之基因名稱有 124 種, 並且在癌症醫學文件、Cancer Gene Census 資訊與 gene2pubmed 檢索資料分析結果發現,在部份類別之白 血病基因文件資料中,有相同的基因表示名稱,如:AML 與 ALL 文件資料內,均存在 RUNX1 基因名稱。 因此本研究將此交集之基因名稱獨立成一類別,以提升文件特徵選取過程中,關鍵字彙篩選的正確性。 5.2.2 白血病基因名稱清單的建立
本研究實驗從 124 種白血病基因名稱中,篩選出部分較常見的白血病基因文件,進行文件分類實驗, 之後以統計分析得到的基因名稱、基因 ID、腫瘤類型紀錄於表格內。AML 類別篩選出 ABL2、AF15Q14、 ARHGEF12、ARNT、CEBPA、CREBBP 基因表示名稱,ALL 類別篩選出 AF1Q、AF3p21、AF5q31、CDK6、 EWSR1、FBXW7 基因表示名稱,CML 類別篩選出 D10S170、HOXA11、MSI2 基因表示名稱,CLL 類別 篩選出 BCL3、BTG1、CCND2、FSTL3、MYC、TCL1A 基因表示名稱,AML ALL 類別篩選出 FLT3、JAK2、 MLL、NUP214、PICALM、RUNX1 等六個交集基因名稱,AML CML 類別篩選出 EVI1、PDGFRB、RPL22 等三個交集基因名稱,ALL CML 類別篩選出 ABL1、BCR 等二個交集基因名稱,ALL CLL 類別篩選出 BCL11B、BCL9、CCND1、IGH@等四個交集基因名稱。 5.2.2.1 白血病基因階層架構 主要的目的是為了之後白血病基因清單的建立,其次是方便研究人員了解白血病基因的階層關 係。本研究將針對白血病進行探討,將白血病基因依據其統計分析結果之類別屬性,建立白血病基因 階層關係,使非醫學背景的研究人員,清楚了解白血病基因的屬性。 5.2.2.2 提升文件之特徵字彙篩選得正確性 由於在分類類別設定時,已經預先將相同基因名稱之白血病類別,獨立成為另一新類別,使得分
類器在分類實驗過程中,不易受相同基因名稱的影響,減少分類錯誤的發生。 5.2.3 提供常見文件分類技術之比較 本研究實驗主要以倒傳遞類神經網路、C4.5 決策樹分類器、Naïve Bayes 貝氏分類器作為常見文件 分類技術之比較,而此三種分類演算法於白血病基因醫學文件分類上,其輸出結果 Precision 與 F1 Measure 數值差異不大,均有不錯的分類效能,其中 BP-NN 演算方法在 EWSR1、ARNT、CCND2、 HOXA11、CCND1、RUNX1 基因文件類別,比 NB 演算方法與 DT 演算方法有較佳的 F1 Measure 值, 而 NB 演算方法則於 BCR 基因文件類別有較佳的分類效能,DT 演算方法則於 PDGFRB 基因文件類別 有較佳的分類效能。 5.3 未來工作 本研究僅針對白血病作為癌症基因文件分類的雛型架構,實驗的文件資料也僅以部分基因文件來訓練 與測試。而未來不僅針對癌症基因文件進行分類,可以結合其他醫學資料,如:醫學影像資訊、維陣列資 訊、蛋白質基因序列資訊,更全面性地對癌症基因資料進行分類,下列幾點是值得期待的發展方向。 5.3.1 結合 Gene Ontology 資訊 基因或蛋白質從 GO 的角度看來,可從三個層面來註解;因此 GO 也以此三個層面來做三大獨立本 體的建立,此三大層面分別是:biological process, molecular function 及 cellular component。研究 Gene ontology 的科學家(Gene ontology researcher)也試著收集各真核生物(如 SGD, MGI, FlyBase..)的基因 或蛋白質,利用已知的資料及序列比較資訊為基礎,將所有的真核生物的基因或蛋白質都基於在此系統 (Gene ontology)下作註解(annotation)與分類(classification)。因此可利用 Gene Ontology 資訊,發覺 基因、蛋白質、水解酶之間的關連性。
5.3.2 加入 Medicine Online 資訊
Medicine Online 網站提供結腸癌(Colon Cancer)、白血病(Leukemia)、肺癌(Lung Cancer)、愛滋 病(HIV/AIDS)等腫瘤疾病之藥物處方資訊,可將其資訊應用於治療與醫學教育上。因此透過 Medicine Online 內部藥物處方資訊,瞭解現階段治療癌症藥物之相關訊息,並且結合癌症基因醫學文獻資訊,運 用資料探勘的技術,發掘癌症疾病、基因、癌症藥物處方三者之間的關聯性。
參考文獻
[1] Fukuda et al. “Toward information extraction: identifying protein names from biological papers.”, In Proceedings of the 3rd Pacific Symposium on Bio-computing, 1998.
[2] Tanabe, L. and Wilbur, W.J. “Tagging gene and protein names in biomedical text.”, National Center for Biotechnology Information, NLM, NIH, Bethesda, Maryland 20894, USA, 2002.
[3] Hatzivassiloglou et al. “Columbia Multi-Document Summarization: Approach and Evaluation.”, In Proceedings of the Document Understanding Conference, 2001.
[4] Anderson, J.A. “An Introduction to Neural Networks.”, MIT Press, Cambridge, MA, 1995.
[5] Chauvin, Y. and Rumelhart, D.E. “Backpropagation: Theory, Architectures, and Applications.”, Erlbaum, Mahwah, NJ, 1995.
[6] Adamic, L.A., Wilkinson, D., Huberman, B.A. and Adar, E. “A Literature Based Method for Identifying Gene-Disease Connections.”, Bioinformatics Conference, 2002. Proceedings. IEEE Computer Society.
[7] Chiang, J.H., Yu, H.C. and Hsu, H.J. “GIS: a biomedical text-mining system for gene information discovery.”, Bioinformatics Oxford University Press 2004, Vol. 20 no. 1 2004, pages 120-121.
[8] Xu, Y., Mural, R.J., Einstein, J.R., Shah, M.B. and Uberbacher, E.C. “GRAIL: a multi-agent neural network system for gene identification.”, Proceedings of the IEEE Volume: 84, Issue: 10 On page(s): 1544-1552, 1996.
[9] Ritchie, M.D., White, B.C., Parker, J.S., Hahn, L.W. and Moore, J.H. “Optimization of neural network architecture using genetic programming improves detection and modeling of gene-gene interactions in studies of human diseases.”, BMC Bioinformatics 2003, Volume 4.
[10] Miller, T., Leroy, G., Chatterjee, S., Fan, J. and Thoms, B. “A Classifier to Evaluate language Specificity of Medical Documents.”, accepted for 40th IEEE HICSS, 2007.
[11] Chen, S.N. and Wen, K.C. “An Integrated System for Cancer-Related Genes Mining from Biomedical Literatures.”, International Journal of Computer Science & Applications, 2006.
[12] Werbos, P. “The Roots of Backpropogation: From Ordered Derivatives to Neural Networks and Political Forecasting.” Wiley, New York, 1994.
[13] Krauthammer, M. and Nenadic, G. “Term identification in the biomedical literature.”, J Biomed Inform., 2004, 37(6):512-26.
[14] Lewis, D. D. “An evaluation of phrasal and clustered representations on a text categorization task.” In Proceedings of SIGIR-92, 15th ACM International Conference on Research and Development in Information Retrieval, Copenhagen, Denmark, 1992, pp.37–50.
[15] Lewis, D. D. AND Gale, W. A. “A sequential algorithm for training text classifiers.”, In Proceedings of SIGIR-94, 17th ACM International Conference on Research and Development in Information Retrieval, Dublin, Ireland, 1994, pp.3–12. [16] Robertson, S. E. and Harding, P., (1984), “Probabilistic automatic indexing by learning from human indexers.”, Journal of
Document, 1984, Vol.40, No.4, pp.264–270.
[17] Cheng, S.C., Huang, Y.M., Chen, J.N., and Lin, Y.T. “Automatic Leveling System for E-learning Examination Pool Using Entropy-based Decision Tree.”, Proceedings of 4th International Conference on Web-based Learning (ICWL2005), 2005. [18] Esposito, F., Malerba, D., and Semeraro, G. “A Comparative Analysis of Methods for Pruning Decision Trees.”, IEEE
transactions on pattern analysis and machine intelligence, vol. 19, No. 5, May 1997.
[19] Rokach, L. and Maimon, O. “Top-Down Induction of Decision Trees Classifiers -- A Survey.”, IEEE transactions on systems ,man ,and cybernetics-PART C:Application and Reviews, October 2004.
[20] http://www.ncbi.nlm.nih.gov/sites/entrez?db=pubmed&cmd=search&term= [21] http://www.nlm.nih.gov/mesh/
[22] Polavarapu, N., Navathe, S.B., Ramnarayanan, R.ul, Haque, A., Sahay, S. and Liu, Y. “Investigation into biomedical literature classification using support vector machines.”, Computational Systems Bioinformatics Conference, 2005, Proceedings., 2005 IEEE.
[23] Liu, Y. (2005), "Text Mining Biomedical Literature for Genomic Knowledge Discovery.", Georgia Institute of Technology.
[24] Wheeler D.L., Benson,D.A., Bryant,S., Canese,K., Church,D.M., Edgar,R., Federhen,S., Helmberg,W., Kenton,D., Khovayko,O. et al. “Database resources of the National Center for Biotechnology Information: Update.”, Nucleic Acid Res, 2005, 33, D39–D45.