國立高雄大學應用化學系(研究所)
碩士論文
利用人工蜂群及基因演算法尋求甘胺酸-水溶劑分子團簇
的結構與 pK
a值的比較
Comparison of Using Artificial Bee Colony and Genetic
Algorithms to Search Glycine-Water Molecular Cluster
Structure and pK
aValues
研究生 : 楊佳蓉 撰
指導教授 : 莊曜遠 博士
I
謝誌
本論文承蒙吾師 莊曜遠老師在我碩士在學期間指導並且協助我完 成論文。從一開始對於計算化學的懵懂無知,到學習如何使用程式,查 詢手冊以及程式中指令的操作,加上運用程式遇到困難如何解決以及得 到最理想的結果,這些過程使我受益良多。也感謝 何永皓老師百忙抽空 之中為我指導論文必須注意的方向,以及 李頂瑜老師告訴我研究出來的 結果必須找到的價值以及觀念的重要。也謝謝 莊琇惠老師在我迷惘的時 候指引我必須努力的方向,也鼓勵我幫助我朝畢業方向邁進。 在就學期間,也感謝一路上協助我的學長、同學以及學弟妹。從一 開始彥翔與鴻昌學長教導的實驗技巧,以及陪伴我且跟我一起學習的智 皓、鈺翰、宗翔、心瑀同學,怡郡、欣怡學妹,不吝嗇的指導我以及協 助我學習上研究的討論以及論文的編排以及美編小技巧,也在碩士生涯 當中給予我一起歡樂一起讀書一起寫論文的時光。 就學期間常常一個人遠離台中的家鄉,但是常常在台中以及台北的 品親、韻靜、純毓、承樸,從中學一直到現在的好朋友願意撥空時間配 合我回去台中的時間,甚至還在去年的時候都在台中陪伴我度過美好的 生日,讓我覺得台中還是有個很溫暖的地方可以隨時歡迎我回去。即使 有的時候很忙碌沒有時間回去,也會傳訊息關切我當下的狀況,也會認 真聽我訴說我的心事,很多時候都很感謝你們都還在我身旁。II
目錄
第一章、溶劑模型(Solvation Model) ... 1
1-1. 微水合結構 (Microsolvation) ... 1
1-2. Continuum Solvation Models ... 2
1-3. Cluster-Continuum Solvation Models ... 4
1-4. 理論 pKa值計算 ... 7
第二章、蜂群演算法與基因演算法 ... 8
2-1. Artificial Bee Colony (人工蜂群演算法)(ABCluster) ... 8
2-1-1. ABC Algorithm in ABCluster ... 8
2-1-2. Atomic Cluster and Molecular Cluster in ABCluster ... 12
2-1-3. Rigid Molecular Clusters in ABCluster... 12
2-1-4. The Parameter of Glycine-Water Clusters in ABCluster... 13
2-2. Genetic Algorithm (基因演算法) ... 13
2-2-1. Procedure of Genetic Algorithm (基因演算法) ... 16
2-3. Ultrafast Shape Recognition (快速形狀識別) ... 18
2-4. 程序的計算方法 ... 20
2-5、論文目的 ... 21
第三章、實驗結果與分析結構 ... 22
3-1. 甘胺酸的人工算法形式的結構 ... 22
III 3-2-1. 甘胺酸水團簇作為剛性分子團簇(rigidmolecule)之計算 ... 28 3-2-2. 甘胺酸水團簇於 lego 程序之計算 ... 29 3-2-3. 甘胺酸水團簇於 lego 以及 rigidmolecule 程序之計算 ... 30 3-3. 甘胺酸以基因演算法(CLUSTER)程序計算之結構 ... 34 3-4. 人工算法與遺傳及蜂群演算法之最佳化甘胺酸與水團簇相對能量 比較 ... 36 3-5. 人工算法與遺傳及蜂群演算法之 pKa值計算 ... 48 第四章、總結 ... 52 第五章、未來展望 ... 54 附錄一、人工蜂群演算法之相對能量 E 與 USR 分布圖 ... 59 附錄二、基因演算法之相對能量 E 與 USR 分布圖 ... 62 附錄三、人工算法、蜂群與基因演算法之初始與最佳化結構以及相對能 量 E 比較圖... 64 附錄四、人工蜂群演算法使用 lego 程序之步驟流程圖 ... 86 附錄五、人工蜂群演算法使用 rigid 程序之步驟流程圖 ... 88 附錄六、基因演算法之步驟流程圖 ... 91
IV
圖目錄
圖 1.(a)Onsager 模型 (b)PCM 模型 ... 2 圖 2. 以人工蜂群演算法搜尋全域最小值之步驟 ... 11 圖 3. 中性 H∙(H2O)n (n=1-4)的團簇四種基本結構35 ... 14 圖 4. 以基因演算法搜尋全域最小值之步驟 ... 17 圖 5. 基因演算法中 Mating 之步驟(左圖)以及 Mutaion 之步驟(右圖)36 18 圖 6. 陽離子與陰離子的甘胺酸與三個 H2O 分子在 PCM 溶劑模型下,相 對能量與相似度之比較。 ... 22 圖 7. 陽離子甘胺酸與三個 H2O 分子在 PCM 溶劑模型下之最佳化結構 ... 23 圖 8.陰離子的甘胺酸與三個 H2O 分子在 PCM 溶劑模型下之結構 ... 24 圖 9. 兩性離子甘胺酸與一個 H2O 分子在 SMD 溶劑模型下,相對能量與 相似度 USR 之分布圖。 ... 25 圖 10. 兩性離子與甘胺酸與一個 H2O 分子在 SMD 溶劑模型下之結構 . 25 圖 11. 蜂群演算法初步典型結構 ... 29 圖 12. 甘胺酸與三個水分子團簇於人工蜂群演算法之 cube(左圖), rigid(中 圖)和 sphere(右圖)形式相對能量 E 與 USR 分布圖 ... 30 圖 13. 甘胺酸與三個水分子團簇於人工蜂群演算法之 cube 形式結構之比 較 ... 31 圖 14. 陽離子甘胺酸與三個水分子在蜂群演算法 rigid 形式之結構 ... 32 圖 15. 甘胺酸與三個水分子在蜂群演算法 sphere 形式結構 ... 33V 圖 16. 陽離子甘胺酸與 3 個 H2O 分子基因演算法下,相對能量 E 與相似 度 USR 之分布圖 ... 34 圖 17. 陽離子甘胺酸與 3 個 H2O 分子在基因演算法之最佳化結構 ... 35 圖 18. 以人工算法、蜂群與基因演算法之甘胺酸與一到二個 H2O 分子在 PCM 溶劑模型下最低能量結構進行最佳化計算,其相對能量與相似 度 USR 之比較。 ... 37 圖 19. 以人工算法、蜂群與基因演算法計算陽離子、兩性離子與陰離子 甘胺酸與三到六個 H2O 分子在 PCM 溶劑模型下之最佳化結構,相 對能量與相似度 USR 之比較。 ... 39 圖 20. 以三種演算法在 SMD 溶劑模型下最佳化陽離子、兩性離子與陰 離子甘胺酸與一到二個 H2O 分子得到相對能量與相似度 USR 之比較。 ... 42 圖 21. 以三種演算法在 SMD 溶劑模型下最佳化陽離子、兩性離子與陰 離子甘胺酸與三到六個 H2O 分子得到相對能量與相似度 USR 之比較。 ... 43 圖 22. 陽離子、兩性離子與陰離子的甘胺酸與一到六個 H2O 分子在 cube 形式下,相對能量 E 與相似度 USR 之比較。 ... 59 圖 23. 陽離子、兩性離子與陰離子的甘胺酸與一到六個 H2O 分子在 sphere 形式下,相對能量 E 與相似度 USR 之比較。 ... 60 圖 24. 陽離子、兩性離子與陰離子的甘胺酸與一到六個 H2O 分子在 rigid 形式下,相對能量 E 與相似度 USR 之比較。 ... 61
VI 圖 25. 陽離子、兩性離子與陰離子的甘胺酸與一到六個 H2O 分子在基因 演算法下,相對能量 E 與相似度 USR 之比較。 ... 62 圖 26. 兩性離子甘胺酸 xyz 檔 ... 86 圖 27. H2O 分子 xyz 檔 ... 86 圖 28. 兩性離子甘胺酸水分子團簇 cube 形式之 inp 檔 ... 86 圖 29. 兩性離子甘胺酸水分子團簇 sphere 形式之 inp 檔 ... 87 圖 30. 兩性離子甘胺酸水分子團簇之 cluster 檔 ... 87 圖 31. 人工蜂群演算法 glyh2o-LM 中得到能量的結構 ... 87 圖 32. 兩性離子甘胺酸之.str 檔案 ... 88 圖 33. 兩性離子甘胺酸之 toppar/par_all36_cgenff.rtf 檔案 ... 88 圖 34. 兩性離子甘胺酸之 xyz 檔 ... 89 圖 35. 水分子之 xyz 檔 ... 89 圖 36. 甘胺酸水團簇之 inp 檔 ... 90 圖 37. 甘胺酸水團簇之 cluster 檔 ... 90 圖 38. 人工蜂群演算法裡 glyh2o-LM 中得到能量的結構 ... 91 圖 39. 兩性離子甘胺酸與水團簇之.in 檔... 91 圖 40. 基因演算法之最低能量結構 ... 92
VII
表目錄
表 1. 人工蜂群演算法中甘胺酸與水形成剛性分子團簇之參數 ... 28 表 2. 陰離子甘胺酸與六個 H2O 分子在 PCM 溶劑模型下初始結構與最佳 化結構比較 ... 45 表 3. 陰離子甘胺酸與六個 H2O 分子在 SMD 溶劑模型下初始結構與最佳 化結構比較圖 ... 46 表 4. 甘胺酸與一到六個水團簇使用 ωB97XD/6-31+G(d, p) 基組在 PCM 以及 SMD 溶劑模型下計算水溶液自由能差異∆G1和∆G 2值 ... 49 表 5. 甘胺酸與一到六個水團簇使用 ωB97XD/6-31+G(d, p)基組在 PCM 以 及 SMD 溶劑模型下計算 pKa1和 pKa2值 ... 50 表 6. 陽離子甘胺酸與一個 H2O 分子在 PCM 溶劑模型下初始結構與最佳 化結構比較圖 ... 64 表 7. 陽離子甘胺酸與一個 H2O 分子在 SMD 溶劑模型下初始結構與最佳 化結構比較圖 ... 65 表 8. 陽離子甘胺酸與二個 H2O 分子在 PCM 溶劑模型下初始結構與最佳 化結構比較圖 ... 66 表 9. 陽離子甘胺酸與二個 H2O 分子在 SMD 溶劑模型下初始結構與最佳 化結構比較圖 ... 67 表 10. 陽離子甘胺酸與三個 H2O 分子在 PCM 溶劑模型下初始結構與最 佳化結構比較圖 ... 68 表 11. 陽離子甘胺酸與三個 H2O 分子在 SMD 溶劑模型下初始結構與最VIII 佳化結構比較圖 ... 69 表 12. 陽離子甘胺酸與六個 H2O 分子在 PCM 溶劑模型下初始結構與最 佳化結構比較圖 ... 70 表 13. 陽離子甘胺酸與六個 H2O 分子在 SMD 溶劑模型下初始結構與最 佳化結構比較圖 ... 71 表 14. 兩性離子甘胺酸與一個 H2O 分子在 PCM 溶劑模型下初始結構與 最佳化結構比較圖 ... 72 表 15. 兩性離子甘胺酸與一個 H2O 分子在 SMD 溶劑模型下初始結構與 最佳化結構比較圖 ... 73 表 16. 兩性離子甘胺酸與二個 H2O 分子在 PCM 溶劑模型下初始結構與 最佳化結構比較圖 ... 74 表 17. 兩性離子甘胺酸與二個 H2O 分子在 SMD 溶劑模型下初始結構與 最佳化結構比較圖 ... 75 表 18. 兩性離子甘胺酸與三個 H2O 分子在 PCM 溶劑模型下初始結構與 最佳化結構比較圖 ... 76 表 19. 兩性離子甘胺酸與三個 H2O 分子在 SMD 溶劑模型下初始結構與 最佳化結構比較圖 ... 77 表 20. 兩性離子甘胺酸與六個 H2O 分子在 PCM 溶劑模型下初始結構與 最佳化結構比較圖 ... 78 表 21. 兩性離子甘胺酸與六個 H2O 分子在 SMD 溶劑模型下初始結構與 最佳化結構比較圖 ... 79
IX 表 22. 陰離子甘胺酸與一個 H2O 分子在 PCM 溶劑模型下初始結構與最 佳化結構比較圖 ... 80 表 23. 陰離子甘胺酸與一個 H2O 分子在 SMD 溶劑模型下初始結構與最 佳化結構比較圖 ... 81 表 24. 陰離子甘胺酸與二個 H2O 分子在 PCM 溶劑模型下初始結構與最 佳化結構比較圖 ... 82 表 25. 陰離子甘胺酸與二個 H2O 分子在 SMD 溶劑模型下初始結構與最 佳化結構比較圖 ... 83 表 26. 陰離子甘胺酸與三個 H2O 分子在 PCM 溶劑模型下初始結構與最 佳化結構比較圖 ... 84 表 27. 陰離子甘胺酸與三個 H2O 分子在 SMD 溶劑模型下初始結構與最 佳化結構比較圖 ... 85
X
利用人工蜂群演算法及基因演算法尋求甘胺酸-水溶劑
分子團簇的結構與 pK
a值的比較
指導教授 : 莊曜遠 教授 國立高雄大學應用化學系 碩士班 學生 : 楊佳蓉 國立高雄大學應用化學系 碩士班 摘要 本研究主要為比較三種不同算法進行甘胺酸與水分子的結構搜索,尋找最低能量 結構。其三種算法分別為人工算法、蜂群演算法以及基因演算法。利用三種算法來探 討其相對能量大小,以及與 Ultrafast Shape Recognition (USR)系統來定義結構的相似 度,了解三種算法得到不同形狀的結構。藉由不同演算法計算後所產生的結構以及利 用 USR 的差異與結構的關聯性,來判斷其 USR 對於分子團簇結構的敏感程度,可以 發現在不同能量下且相似度接近的結構會使 USR 較靈敏,甘胺酸與水團簇中的 H2O 分子上的 H 原子方向不同,USR 就會差異很大。第二部分則使用 ωB97XD / 6-31 + G(d, p)將上述部分求得分子團簇結構進行最佳化與振動頻率的計算,並計算 pKa值。根據 pKa值計算得到的結果可以發現在水分子較少的條件下,利用蜂群演算法的 cube 與 rigid 形式可以得到良好的 pKa值,而水分子較多的條件下,基因演算法較其他方法 進行搜尋最低能量結構更較有優勢。此外我們也證實了 Schlegel26與 Galano27的結論, 分別為羧酸端與氨基端的甘胺酸與三個 H2O 分子結構,在 SMD 溶劑模型下可以到良 好的結果。以及我們參考 Bachrach5放置 H2O 分子的結構以及在氫鍵相互作用力較大 的結構下也可以計算出良好的 pKa值。 關鍵字 : 蜂群演算法、基因演算法、微水合結構、pKa計算XI
Comparison of Using Artificial Bee Colony and Genetic
Algorithms to Search Glycine-Water Molecular Cluster
Structure and pK
aValues
Advisor: Dr. (Professor) Yao-Yuan Chuang Department of Applied Chemistry National University of Kaohsiung
Student: Jia-Rong Yang Department of Applied Chemistry
ABSTRACT
This study mainly compares three different methods to search Glycine-Water molecule cluster and find the lowest energies of structures. Three methods are artificial method, Artificial Bee Colony algorithm and Genetic Algorithms. We explored the relative energy magnitude, and the similarity with the ultra-fast shape recognition (USR) system to define the structures understanding the three algorithms to obtain different shape structures. The structures we searched combined with USR to determine the sensitivity of the USR with the molecular cluster structure. It is found that USR more sensitive by the case of the structures with different energies but USR value were close, the H orbital of H2O molecule in Glycine-Water molecule cluster had different direction to let USR values large discrepancy. In the second part, we optimized and vibration frequency calulated the lowest energies of three methods atωB97XD/ 6-31 + G (d, p) level in PCM and SMD solvation model, and reserved pKa values. According to the calculated pKa value, it can get a good pKa value with the cube and rigid type of Artificial Bee Colony algorithm under the condition of less water molecules, and Genetic Algorithms under condition of more water molecules was more advantageous. In addition, we also confirmed the conclusions of Schlegel26 and Galano27 that H2O molecule in carboxylic acid and amino acid can calculate the good pKa values in SMD solvent model. We also found good pKa value by referring to the structure of Bachrach’s study.5
Keywords: Artificial Bee Colony Algorithm, Genetic Algorithm, Microsolvation, pKa Calculation.
1
第一章、溶劑模型(Solvation Model)
1-1. 微水合結構 (Microsolvation)
在甘胺酸研究中,因為甘胺酸中具有 Neutral 與 Zwitterion 型式, 所 以有些文獻中會探討這兩種形式哪一個較為穩定?再來探討以 Neutral 與 Zwitterion 兩種型式中添加水分子後哪個會比較穩定? Microsolvation 中最複雜的問題為當進行溶質與溶劑分子的結構最佳 化(geometry optimization)的過程中,當每一個溶劑分子加入時,結構的自 由度會急速增加。例如每加一個水分子時,整個結構的自由度會增加 9, 所以甘胺酸和水的微水合結構進行計算時會產生以下幾個問題: (1)當以 兩性離子形式的異構體存在時需要多少水分子來穩定結構? (2)需要多少 的水分子才能達到中性與兩性離子具有相同能量。 為了探討甘胺酸在 microsolvation 計算方法的應用,於是,在 1995 年,Jensen 等人3使用 MP2 / DZP ++ // HF / DZP 方法來討論這兩個問題。 表明兩性離子甘胺酸因為不穩定而不存在於氣相中。而在水溶液中則需 要至少兩個水分子來穩定兩性離子甘胺酸,表示兩性離子甘胺酸增加兩 個水分子形成微水合結構時,得到結構改變但能量變化卻不大的結果且 甘氨酸分子的扭曲提供了與兩個水分子更強的氫鍵相互作用。Wang 等人4也重新審視 Glycine,他們利用 B3LYP,PBE1PBE,X3LYP 方法以及 MP2 方法來最佳化甘胺酸與一個水分子的結構。發現缺乏極化 函數基組的 B3LYP 和 MP2 方法最佳化無法找到局部最小值,因為兩性 離子氨基端上的質子會轉移至羧酸端上形成中性結構。研究發現需要極
2 化函數與正電子相關,以便正確評估甘胺酸與一個水分子的能量,也發 現使用 aug-cc-pVTZ 基組在所有單點計算中採用完全平衡(CP)方法,可 以降低基組疊加誤差(BSSE)。 Bachrach5在 2007 年報導了使用 PBE1PBE / 6-311 + G(d, p)計算探索 中性和兩性離子甘氨酸的溶劑效應。基於在水分子之間以及在水和甘氨 酸之間建立的氫鍵網絡來分析團簇的結構。並使用 B3LYP / 6-31G *計算 兩性離子與單一水分子結構,發現使用 PBE1PBE / 6-311 + G(d, p)再次最 佳化時會形成中性結構,證實了與 Wang 等人4的結果一致。此研究也為 Jensen 和 Gordon3等人對於第二個問題提出了答案,透過在添加七個水分 子的條件下,中性與兩性離子的甘胺酸僅差異 0.01 kcal/mol。
1-2. Continuum Solvation Models
當一個溶質存在於極性溶劑中,其溶質的電荷分布會受到溶劑的影 響。因為 microsolvation 需要較多的計算資源,所以有時我們將溶劑分子 視為一個連續反應場(reaction field)來表示。在此結構中,溶劑可以藉由 介電常數項(dielectric constant, ε)、表面張力和分散力來描述。
Continuum Solvation Model 中最簡單模型為 Onsager6模型,將溶
子放置於半徑為 R 的球形腔中,如 圖 1 (a)所示
3
圖 1.(a)Onsager 模型 (b)PCM 模型
Gordon7於 2000 年提出了以有效片段電位(EFP)與連續 Onsager 溶劑
模型以及 ab initio 與 Onsager 模型結合的兩種方法,用於中性與兩性離子 甘胺酸的溶劑效應。可以發現對於裸甘胺酸異構體兩者得出的結果表現 不佳,誤差約-4 至-5 pKa單位。但以等密度可極化連續體模型(IPCM) 或溶劑化模型 5.42R(SM5.42R)提供了改進的結果,誤差約至 1.1pKa 單位。且甘胺酸中性分子與兩性分子的能量誤差降低 0.83 kcal/mol。 Kim8在 2011 年提出利用 CPCM 連續體模型中結合各種不同空腔模 型(UA0,UAHF,UAKS,UFF,BONDI 和 PAULING)來計算中性(N)與 兩性離子(Z)甘胺酸與八個水分子形成的團簇產生結構與相對能量。發現 在 B3LYP/6-311+G(d, p) 的理論水平下可以得到 ΔGZ-N為-7.3kcal/mol,與
先前計算 BONDI(-7.2kcal/mol)和 PAULING 模型(-8.2kcal/mol)所得到的 實驗值較一致。 1981 年,Tomasi 等人9開發了一種極化連續場模型(Polarized continuum model, PCM),如 圖 1(b)所示。其作法是以溶質分子的各原子為中心,使用原子球殼 堆疊方式產生孔腔。而在 2008 年,Cramer 和 Truhlar10等人提出 SMD 溶 劑模型,將標準狀態水合自由能分為兩項。第一,根據非均相帕松方程 式來處理靜電貢獻項,藉由實際空間電子密度計算。第二,根據表面積 (Analytical surface area, ASA)算法,計算溶質與溶劑分子於第一溶劑化殼 層的短程作用力。在 SMD 模型中,目前可執行的模型有兩種: 分別是 IEFPCM11-14和 CPCM15,16。
4
但 Continuum Solvation Model 計算上低估了溶劑分子與溶劑之間可 能存在的短程作用力,如氫鍵作用力和電荷傳遞等。於是 Riveros17、 Tomasi18和 Cui19提出 Cluster-Continuum Solvation Model,此部分詳見第 1-3 節。
1-3. Cluster-Continuum Solvation Models
Cluster-Continuum Solvation Model 中的離子在第一溶劑化殼層中 可以引入分子與溶質-溶劑內部強烈且特殊的相互作用,這可以降低了離 子在溶劑模型中產生溶劑化自由能貢獻的差異。
一旦建立模型,必須專門解決兩個問題:(a)如何使用該模型正確計 算水合自由能 (b)應在 Cluster-Continuum Solvation Model 中包括多少溶 劑分子
於是,Pliego20,21在 2002 年提出了使用 MP2 / 6-311 + G (2df,2p),
將 Cluster-Continuum Solvation Model 方法和 Continuum Solvation Model 結合 SM5.42R22與 PCM23方法應用於具有不同官能團的幾種有機物質的
pKa計算。發現 Cluster-Continuum Solvation Model 優於 Continuum
Solvation Model,其方均根誤差(rms)為 2.2 pKa單位,對於 SM5.42R22和
PCM23方法則誤差為 7 pKa單位。結果顯示包含溶劑分子相對於
Continuum Solvation Model 可以顯著降低誤差,可歸因於溶質 - 溶劑相 互作用的因素。
Kelly24,25等人也在 2006 年利用開發的 SM6 連續模型與
5 SM6T 溶劑模型,來計算在 298K 時 H,C 和 O 組成的化合物的單質子酸 的自由能,其 MUE 值結果誤差僅為 0.08 kcal/mol。此外,該模型與無溫 度依賴性的模型相比 MUE 降低了 6.5 倍。為了進一步探討溶劑分子與溶 質之間所產生的短程相互作用力,研究結果發現尤其在當共軛鹼 CO3 2-與三個水分子形成團簇時,誤差小於 0.1 kcal/mol 以內,與實驗預期結果 相似。 Schlegel26等人在 2017 年計算與硫結合的氨基酸化合物以及具有羥 基(R-OH)以及羧基(R-OOH)官能基的 72 種有機化合物,在三個顯性水分 子的 SMD 溶劑化模型的條件下,使用 6-31+ G(d, p)和 6-311 ++ G(d, p) 計算之 pKa值產生平均誤差分別為-0.11±0.50 和+0.15±0.58 pKa單位。發 現 SMD 隱式溶劑化模型雖然低估了水溶液中硫醇鹽的穩定性,但是在硫 的周圍包含三個顯性水分子的結構下,使用不同功能(B3LYP 和ωB97XD) 可以將計算出的 pKa值降低至 1 pKa單位內。 Galano27等人於 2012 年提出準確預測質子化胺類的 pKa值,將熱力 學循環與 cluster 連續溶劑模型結合使用,利用密度泛函理論(DFT)和 MP2 中九種不同方法進行計算,並且推薦 F2 的反應方案[HA+ (H 2O) + 3H2O = A +H3O+ (3H2O) ],結合 M05-2X / 6-311 ++ G (d, p)與 SMD 溶劑模型進行 計算。結果顯示此方案的優勢除了可得到最低的平均無符號誤差(MUE) 值 0.54 kcal/mol 以外,也對於較大結構的胺類如三甲基胺(HT1+)具有可 行性,其幾何結構平均差異為≤ 0.014Å,代表計算溶劑化能量時可以不考 慮鬆弛效果且不依賴於質子的實驗值以及擬合校正。
6 此外,在 2014 年,Galano 等人28再次提出評估未知酚類的 pK a值, 使用擬合參數 FP 方法,將 PBE 和 PBE0 基組與 SMD 溶劑的模型組合進 行計算。並且推薦 E3[HA + OH-(3H 2O) = A-(3H2O) + H2O]和 EN3[HA(H2O) + OH-(3H2O) = A-(3H2O) + 2H2O]兩種方案。結果發現直接用 SMD 計算溶 劑化能比用熱力學循環和 PCM 計算更好,並且與 PBE 和 PBE0 基組結合 會得到 MUE 值小於 1pKa單位,即為 0.77 kcal/mol 。 Ho 等人29於 2010 年發表質子交換方案 * ΔG - soln
-(aq) (aq) (aq) (aq)
HA +Ref HRef +A ( 1 ) 其中 Ref 為參考的氨基酸,以甘胺酸為例的參考酸為丙胺酸(Alanine)。 Ho 表示在直接或絕對方法雖然取決於溶劑化模型,但精準度仍然不高。 質子交換方案在 CPCM-UAKS 下可產生良好的結果,以及在 IPCM 的連 續溶劑模型下,所得到的 MUE 值會小於 2 單位誤差。 此外,Ho30等人也於 2014 年提出使用高水平計算(MP2 / GTMP2Large 和 G3 (MP2) - (RAD (+))與在 SMD 溶劑化模型中直接計算直 接在連續模型相比,可獲得誤差只有 0.5 kcal/mol 以內非常相似的結果。 表示即使在氣相與液相結構不同的條件下,可以運用直接方法來改善結 果,這樣的結論可能是因為高能量校正集中在解離過程中有涉及到實際 的溶液相物質的關係。 然而,在 2016 年 Ho 等人31擴大研究範圍,以 Cluster-Continuum Model 與 SMD-M062X,SMD-HF,CPCM-UAKS 和 CPCM-UAHF 幾種常見的 溶劑型態於各種中性,自由基和離子反應進行 pKa計算。發現使用
7
G3(MP2)-RAD(+)得到的誤差分別為 0.4, 1.0, 0.5,和 1.1 pKa單位。
1-4. 理論 pK
a值計算
將水溶液中的有機酸物質稱為 HA,其中 HA 解離可以寫成
+
-(aq) (aq) (aq) ΔG (aq) HA H A ( 2 ) 其中酸度解離常數 Ka為一個分離的量度,通常表示為 + -a [H ][A ] = [HA] K ( 3 ) a 10 a pK = -log K ( 4 ) 其 pKa值可以從酸解離的水溶液自由能得出 (aq) a ΔG p = ln10RT K ( 5 ) 上述式中∆G(aq)為質子溶劑化自由能,實驗值是根據 Camaioni 和 Schwerdtfrger32所提出的參考值,其溶劑化自由能實驗值為ΔG aq,solv(H+) = -265.9 kcal/mol。Schlegel26表示水相中質子的自由能須將 1atm 轉換為 1M 的狀態下進行計算,計算實驗值的公式為Gaq* (H+) = o g G (H+) + ∆ aq,solv(H+) + ∆G1atm→1M,而水相質子自由能為 * aq G (H+) = -270.29 kcal/mol。 目前 pKa值理論計算中,其水合自由能誤差精確度於± 2 kcal/mol 內, 相當於 pKa值誤差為 1.47 pKa單位。
8
第二章、蜂群演算法與基因演算法
2-1. Artificial Bee Colony (人工蜂群演算法)(ABCluster)
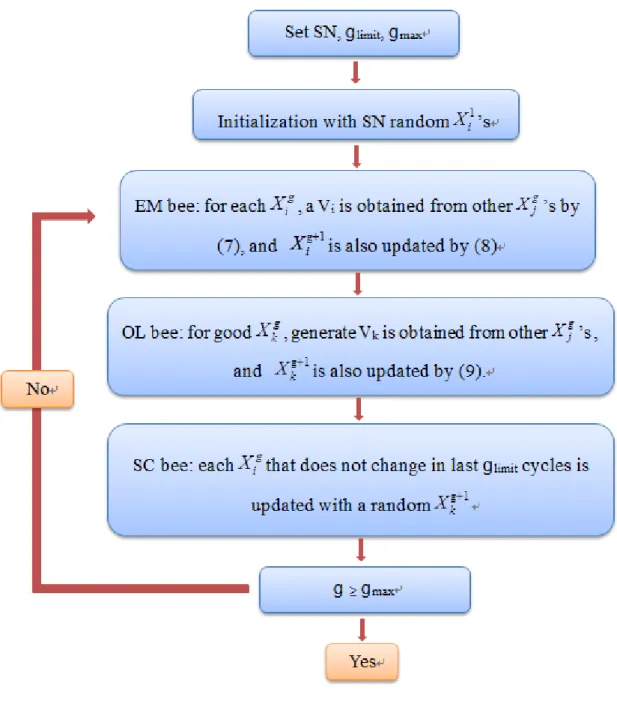
人工蜂群(ABC)算法是由 Karaboga 於 2005 年提出,用於團簇的 global minimum (GM)搜索領域33。這種方法的靈感主要來自蜜蜂蜂群的 覓食行為。為了尋找到最好的蜂蜜來源,蜜蜂們會專門針對不同的任務, 採取有效率的分工執行。以增加花蜜量到最大值。ABC 算法一直在針對 約束最佳化的問題進行延伸。 而 Zhang34提出一種新的無偏差,基於群體智能的方法,將 ABC 算 法引入最小群及搜索領域中。與其他方法相比只需要三個參數: 團簇大小 (SN),偵測極限( glimit)和最大循環數(gmax)。
2-1-1. ABC Algorithm in ABCluster
蜂群是用三種蜜蜂類型所組合而成: employed bees (僱工蜜蜂), onlookers bees(旁觀者蜜蜂),scout bees(偵查蜜蜂)。然而每隻蜜蜂都可以 找到花蜜並估算其質量。更重要的是,它可以通過例如搖擺舞來與其他 蜜蜂分享這些信息。 在一個搜索週期中,假設以團簇中使用蜜蜂的數量等於找尋到食物 來源的數量。首先,生成隨機分布的初始團簇,利用僱工蜜蜂自己既有 的記憶中以及其他蜜蜂的知識來修改食物源的位置以及尋找新的食物源, 並選擇最接近的食物來源。其次,旁觀者蜜蜂與僱工蜜蜂進行特定的舞 語。旁觀者蜜蜂評估其花蜜量後會在“好”的蜜蜂周圍尋找新的蜜源。 如果新花蜜量高於舊花蜜量的話,僱工蜜蜂會記住新花蜜量的位置而忘
9 記舊的花蜜量的位置。而偵查蜜蜂記錄先前搜尋的食物來源,將確定為 低質量的部分丟棄,並由偵查蜜蜂發現新的食物來源而取而代之。經過 幾個循環後,最終選擇了“最佳”花蜜量來源。 在群集最佳化的背景下,其中以 X 是花蜜源,潛在平滑勢能 U(X) 是質量。該簇的大小為 N,估計最大座標值 L,以及其他潛在的參數。 使用 ABCluster 生成的隨機分佈的溶液初始團簇的三種參數: SN 為蜂 群體大小以及偵查限制 glimit和最大循環數 gmax。 對於較大型的群集,以不同的初始估計執行多次最佳化來比較最終 結果為更好的方法。而對於具有短程或多峰相互作用,又或是具有多個 相互作用的位點的團簇的條件下,則需要非常大的 glimit和 SN,才能獲得 較好的結果。 搜尋結構第一步,人口初始化: 1 1 X ,…,X1SN。初步猜測一個位置為 1 Xi, 1 Xi每個分量從範圍[0, L]中隨機取得。如果在較困難的情況下則使用播種 猜測,通常為大小 N-1 團簇的 GM 定義隨機一個原子。再藉由 the limited memory-Broyden–Fletcher–Goldfarb–Shanno (L-BFGS)算法47進行局部最 佳化。 第二步,僱工蜜蜂建模: 定義週期為 g,在週期 g 中,每個Xig (i=1,…,SN)其新試驗方案 Vi會根據其他 X g j ( j = 1,…,SN; j≠i)使用蜜蜂搜 索共享訊息所產生。這裡使用三角變異算子49: 1 1 2 2 3 3 1 2 3 2 1 3 2 1 3 1 ( ) ( )( ) 3 ( )( ) ( )( ) g g g g g i k k k k k g g g g k k k k V X X X p p X X p p X X p p X X ( 6 ) 其中 k1, k2, k3為{1,…,SN}的隨機數且 k1≠k2≠k3≠i
10 1 2 3 4 1 2 3 4 X (X X X X ) { X (X X X X ) g g g g g k k k k k k g g g g g best k k k k F V F (m=1, 2, 3) ( 7 ) U 為勢能變化函數,其中U(X)min :{ (X)}U ,可由 the L-BFGS 算法 47局部最佳化最小值得到。式(7)可以非常有效的交換信息,再使用下列 式(8)更新此試驗方案。 1 { i g i g i V X X 如果
U
(Vi) <U
( g i X )則相反 ( 8 ) 第三步,旁觀者蜜蜂建模: 使用僱工蜜蜂搜索後,旁觀者蜜蜂將會選 擇一個好的來源為 g k X ,並根據其他的Xgj 來再次生成新的試驗方案解 Vk。 要使用Xkg為“好”的信息,我們基於Xgk 生成 Vk,即為式(9) 1 2 3 4 1 2 3 4 X (X X X X ) { X (X X X X ) g g g g g k k k k k k g g g g g best k k k k F V F 如果 η < 0.5 則相反 ( 9 ) 其中 k1,k2,k3和 k4是{1,…,SN}的隨機整數且 k1≠k2≠k3≠k4≠k。F 和 η是[0, 1]中的隨機數。在式(8)兩個表達式由“ABC / current / 2”和“ABC / best / 2”表示,代表試驗解決方案是通過 2 個向量差異的當前最佳方案。 而Xkg也選擇式(8)來更新。 第四步,偵查蜜蜂建模: 在旁觀者蜜蜂搜索後,檢查每個Xig (i=1,…,SN)。如果Xgi 在最後的 glimit循環中沒有改變,則會被偵查蜜蜂所 搜尋的隨機試驗解決方案Xig1取代,無論是否優於Xig。如果沒有偵查蜜 蜂即會被收斂至 LM。偵察蜜蜂搜索對於保持解決方案群體的多樣性以及 圍繞 LM 跳出 funnel 非常重要。 第五步,如果 g ≥ gmax則計算完成,如果沒有即返回至第二步直到計 算完成。
11
12
2-1-2. Atomic Cluster and Molecular Cluster in ABCluster
Zhang34於 2015 年提出人工蜂群演算法運用於幾種電位計算原子團 簇,建議實踐中設置 SN 在 50 到 100 之間。而對於雙體電位如 Coulomb– Born–Mayer, Lennard-Jones, Morse, Z 電位 glimit可以設置為 5,對於多體
電位如 Gupta 電位 glimit可以設置為 2。對於短程作用力的電位或大型團 簇的條件下 gmax應設置非常大,約 15000 至 30000 間以避免停滯於局部 最小值中。 Zhang35於 2016 年也利用人工蜂群演算法計算分子團簇。該算法以 TIP4P 水分子團簇(H2O)N (N ≤ 20)為基準,應用於 10 個微水團簇、4 個甲 醇微水合團簇、4 個非極性團簇和兩個離子芳香族團簇,發現計算結果與 理論非常相似,例如在(H2O)10團簇下僅誤差約 0.07 KJ/mol。
2-1-3. Rigid Molecular Clusters in ABCluster
假設團簇中的分子皆為剛性分子,其剛性分子利用在最佳化期間的 內部自由度,例如鍵長、鍵角、二面角等皆保持不變的情況下,對於小 分子團簇來說,可以求得相當好的近似值。因此團簇中每個分子都可以 用六個外部自由度來描述。幾何中心的座標 R{X, Y, Z}和三個歐拉角 Ω {α, β, γ},相對於預先定義的坐標系。 假設剛性的分子團簇的勢能可以以下列表示 2 12 6 1 0 U(Q)= ( 4 (( ) ( ) )) 4 I J I J I J I J I J I J I J I J N N i j i j i j i j I I J i I j I i j i j i j q q e r r r
( 10 ) 建立剛性分子除了建立原子分別在 xyz 軸座標以外,也需建立其分 子的力場(CHARMM)。建立方式是根據其式(10)中能量 q, ε (KJ/mol)和 σ13 (Å )所組成。而其中能量 q 可以從 CGENFF49中找到,將能量 q 值參考 MacKerel50中得到的 ε 和 σ 分別由下列公式導出 ABCluster CHARMM ε =ε ( 4.184) ( 11 ) 5 6 ABCluster CHARMM σ =σ ×2 ( 12 )
其中式(10)與式(11)中的 εCHARMM和σCHARMM由 MacKerel50找到的值
帶入計算,其詳細內容參考附錄四。
2-1-4. The Parameter of Glycine-Water Clusters in ABCluster
Zhang35在此文獻中利用 rigid 程序建議設置 SN 和 glimit可以設置為
60 和 4,而對於微水團簇以及甲醇微水團簇 gmax應設置較大,可以為 30000
至 35000,對於非極性團簇與芳香團簇的條件下,gmax可以設為 5000,
而 amplitude 設置為 4.0。而對於 lego 程序則建議 SN 需設置為 1,glimit
可以設置為 2,其 gmax可根據自訂用戶來定義。
在 lego 程序中 SN 和 glimit分別設置為 50 和 3,而 gmax則設置為 50。
2-2. Genetic Algorithm (基因演算法)
Genetic Algorithms 程序是基於所謂的遺傳性基因演算法(GA)來尋找 原子或分子簇的全局最小值。這種方法通常涉及透過利用結構訊息或是 遺傳數據來將初始物種群體變成連續幾代的候選結構之後,逐步地最佳 化固定大小的結構團簇。再增加團簇系統轉移中所保留的有利結構所有 特徵的可能性,以及包含所有可能的全局最小值。 Alexandrova 等人於 2005 年開發的梯度嵌入式遺傳基因算法(GEGA), 結合了相對能量與幾何第一原理精確度以及快速收斂速度。最初這個方
14 法只應用於 Li 原子團簇。利用芳香性與金屬團簇共振高對稱性和平面形 狀並在分子軌道中含有 4N+2 的結構來個別作基因結合,形成穩定的 C2v 與 D3h幾何結構,並且能找到全局與局部最小值36。 Alexandrova 等人37後期也運用於分子團簇上。在 2010 年時發表應 用於中性H∙(H2O)n (n=1-4)的團簇中,其中 H∙ (H2O)n (n=1-4)的團簇具有 四種基本結構,其結構如圖 3 所示: 圖 3. 中性 H∙(H2O)n (n=1-4)的團簇四種基本結構37 其中圖 3 中類型分別為: (I). H 自由基與一個水分子的氧原子弱配位 鍵 (II). H 自由基與一個水分子的 H 原子弱配位鍵 (III). 由 H2和 OH 自 由基以及 n-1H2O 分子組成 (IV).由 H3O+和 n-1H2O 組成。而類型 I 和 II 皆有全局最小值,因為兩者類型在解離過程中可以完全解離出 H 離子, 水團簇能量僅高 3 kcal/mol。而類型 III 和 IV 的團簇大約比全局最小值高 10 kcal/mol。 而 Kanters38等人介紹了一種結構搜索方法,該方法為類似 GEGA 的方法,但可以避免大量初始隨機創造結構,也可以增加額外的靈敏度。 結合強鍵結的 Si-Li 團簇和 ZnF2團簇與弱相互作用的水三聚體團簇來確
15 定能量最小值。結果發現,在強相互作用的 n 個分子單元聚體的情況下, 使用 n-1 的分子片段與原先的原子版本結合,可以允許有更大的靈敏度 以及更好的潛在能量表面。而弱相互作用的水三聚體團簇條件下會有許 多豐富的局部最小值,必須通過使用 H2O 單體構建兩個 1D,5 個 2D 和 20 個 3D 簇進行 5 次初始搜索,然後使用 MOPAC 的 PM7 半經驗方法進 行 30 次交配,關鍵字為 PRECISE,δR 為 1 Hartree。再使用 LDA / TZP-(large)方法和 1 Hartree 的 δR 再次最佳化得到的九種結構作為候選者 進行配對。
16
2-2-1. Procedure of Genetic Algorithm (基因演算法)
第一步、建立結構: 在建立結構之前會有兩種步驟,一種為直接被重 新最佳化,第二種為使用之前組合成的結構來跟定義的結構作為一種新 建或是重新最佳化的候選者。第二種的部分涉及1D,2D或3D集群構建過 程。1, 2或3反映了用於定位每個片段的隨機位移矢量的維數,該隨機位 移矢量圍繞其中心隨機旋轉。其中1D為僅沿著x軸的片段中心建構候選者 的數量,2D的部分為沿著x與y平面的候選者數量,3D的部分則為沿著x、 y與z平面建構候選者的數量。 第二步、幾何最佳化的定義: 幾何的標準在於核排斥能量 R。將一 個量(R)將指定的團簇中以 N 個帶電核的庫侖排斥量總合為: i N i j i l i j ij q q R r
( 13 ) 其中 qi和 qj是原子 i 和 j 的核電荷,rij是兩個原子之間的距離。如 果兩個值分別在指定的分辨率值δE 和δR 內,其靜止點會被認為是重複 的,並且會被添加到候選庫中。如果 E1> E2 且 E1-E2 < δE 以及| R1-R2 | < δR,則兩個候選者中,候選 1 就會被歸類認為是候選 2 的副本。 第三步、定義候選者片段組成: 其片段可以為原子或分子。在定位 片段之前會隨機旋轉構建成一個片段,其順序也是隨機的。沿著位移矢 量的平移距離來使原子片段可以接觸但不穿透先前放置的一個或多個原 子片段之中,這種策略是避免原子彼此之間的距離太小。此外,也可以 在片段中指定額外的半徑,來定義元素的原子半徑或平移向量的因子。 第四步、每個親代皆以每個片段點為中心,然後親代會隨機繞著該17 中心旋轉。親代起初必須要有兩個。而後代會複製親代 1 的 xy 平面上方 的部分, 然後再將每個剩餘的片段來取代親代 2 的 xy 平面下方的原子。 如果上方與下方為不同的片段,則必須再次進行隨機旋轉與平移,直到 最佳化至最先預定到最大次數後,可以選擇替代性的 3D 候選者或是終止 程序。 第五步、產生初始群集後,每次交配後產生適合的候選者就會被加 到群集中。如果認為重複則可以利用現有的候選者替換,但是如果新物 種是獨特的,就會被列為新的候選者而被添加到群體中以進行配對。 圖 4. 以基因演算法搜尋全域最小值之步驟
18 圖 5. 基因演算法中 Mating 之步驟(左圖)以及 Mutaion 之步驟(右圖)38 然而,在各種全局最小值的結構比較能量的情況下,我們必須以一 個固定的系統來辨別他們相似性質,所以我們以最低能量結構的質心為 中心定義出一個指數,將其他相對性能量結構也做相對性的指數來判定 他們彼此之間結構相似的程度,此部分在第 2-3 節討論。
2-3. Ultrafast Shape Recognition (快速形狀識別)
Richards 等人提出了超快速形狀識別(USR)的分子形狀比較方法 39, 利用分子數據庫中搜索與給定查詢分子形狀的最相似化合物進行比較。 其模擬了篩選的分子與大分子受體對接的過程,配體分子形狀與大分子 受體上相應結合位點的互補性。 USR 是根據觀察到分子內每個原子形狀的相對位置來確定分子的形 狀。將分子視為顆粒(原子)系統,而不是固體的常規處理。原子在分子的 相對位置可以完全由所有原子間距離集合決定,這樣可以消除了平移與 對齊的需要。 Richards 等人利用 USR 可以找到形狀相似但原子數不同的分子,儘 管使用明顯不同數量的原子計算,兩個得到的形狀描述符號的矢量非常
19 相似,相對位置也非常相似。代表著 USR 提供了一個高靈敏度的概念。 且辨別了疊加方法與非疊加方法的不同,前者需要類似形狀的分子以及 正確的排序方法作為比較,後者則發現每個分子的形狀訊息編碼皆獨立 於形狀描述符號的矢量。 Cannon 等人擴展了 Ballester 開發超快速形狀識別技術40,用於計算 這四組原子距離形成四個力矩。並獲得 16 個描述符號 M,其中 j 值為 1,j 16。以定義相似性指數 ζ 可以由曼哈頓距離得到: 16 1 0, 1 1 (t) (1 | ( ) |) 16 j j j M M t
( 14 ) 第一步、建構這些描述符號: 先透過選擇合適的原子間距離集合,考 慮了所有原子距離集合的四個分子位置: (1) 分子質心(ctd) (2) 最接近 ctd 原子(cst) (3) 離 ctd 遠的原子(fct) (4) 離 fct 距離最遠的原子(ftf)。 第二步、基於參考坐標計算四組原子距離測量值 : 第一個描述符號 1(
ctd)
對應於距離分子質心的原子距離分佈的第一個時刻,({
d
kctd})
Nk1, 其中 N 是原子數) 它是到幾何中心的平均原子距離,因此提供了估計分 子的大小。第二個描述符號 2(
ctd)
是相同分佈的第二個矩,它是這些原子 距質心的變異數,因此它與分子的緊密程度有關。第三個描述符號 3(
ctd)
是同一分佈的第三個矩,其為偏角。它估計其不對稱性,從而估計原子 是否與質心的平均原子位置相近或遠。第四個描述符號 4(
ctd)
是同一分佈 的第四個矩,又稱為峰值。高峰值意味著更多的變異數是由於較不頻繁 但更極端的偏差,又為計算更頻繁的較小偏差。 第四步、定義四個力矩和四個參考坐標產生了 16 個矩陣描述符號20 Mj (j=1,16),以歸一化的分數函數來量化這些分子之間的相似程度。其中 值 1 對應於最大相似度,0 對應於最小相似度。 16 1 0, 1 1 (t) (1 | ( ) |) [0,1] 16 j j j M M t
( 15 )2-4. 程序的計算方法
除了人工蜂群演算法中 rigid 程序是以 CHARMM(力場)搜尋結構以 外,主要利用 MOPAC 的程序來進行計算,建立輸入檔。MOPAC (Molecular Orbital PACkage)是一種基於 Dewar 和 Thiel 的 NDDO 近似的 半經驗量子化學的程序,用於研究研究涉及分子,離子和線性聚合物的 化學反應。以 Hamiltonians MNDO41, AM142, PM343, PM6, RM1,MNDO-d41,42, 和 PM7 的半經驗法用於計算電子的部分,來獲得分子軌道、 分子能量以及分子幾何的衍生物。並在完全集成的程序中組合了振動光 譜,熱力學量,同位素替代效應和力常數的計算。 我們使用 PM6-DH+ PRECISE 指令操作 MOPAC 程序,PM7 為 PM6 的 D2 形式最佳化指令,我們使用 PM6-DH+ PRECISE COSMO 指令進行 單點及最佳化的計算。 而 COSMO 為溶劑篩選模型,是將溶劑分子平均分布於均勻的電介 質中。其中可以包括非介電性表面積的函數化: non-elst
E f(ε)(CAV0+CAV1 area) ( 16 )
其中
(ε) (ε -1) / (ε )
f x ( 17 )
21 質函數(其默認值為 CAV0 = 0.0,CAV1 = 0.0067639)。 COSMO 又可以稱為一種連續介電模型。可以計算能量導數,以及 幾何優化、頻率計算等等46-48。
2-5、論文目的
本論文研究目的為利用團簇-連續溶劑模型計算 pKa值。第一步則使 用人工蜂群演算法與基因演算法協助半經驗法和力場進行全域最佳化以 提供可能的候選者。第二步,再使用快速形狀識別(USR)分析第一步的候 選者,建立良好的初始結構。最後將初始結構進行密度泛函理論(DFT) 最佳化以計算 pKa值。22
第三章、實驗結果與分析結構
3-1. 甘胺酸的人工算法形式的結構
我們以原先 Bachrach 等人5得到的結構做為參考,以甘胺酸與一到 六個 H2O 分子使用 ωB97XD/6-31+G(d, p) 基組進行最佳化計算,得到最 低能量結構的比較,以下是由以甘胺酸與三個水分子為例在 PCM 溶劑模 型所得到的結果 : a) Cation/3 H2O b) Anion/3 H2Oa) Relative Energy = E(mPXn-mPXn-1) (m=Artificial Method, n=1~6, XS1,XS2; X=C, Z, A, P=PCM solvation model) 圖 6. 陽離子與陰離子的甘胺酸與三個 H2O 分子在 PCM 溶劑模型下,相 對能量與相似度之比較。 我們取其中最低能量的結構分別命名為 mPC3-1和 mPA3-1,其他不是 最低能量結構根據順序分別命名為 mPC3-2、mPC3-3,其陰離子以及兩性離 子也根據此命名法。以陽離子甘胺酸與三個水的分布圖及結構的例子進 行分析,提出最低結構 mPC3-1、相對能量較低但 USR 差異較大的 mPC3-5 以及相對 mPC3-1結構以及相對能量差異較大的 mPC3-4,其結構如下:
23 mPC3-1[1.0, 0.0] mPC3-5[0.4, 0.01] mPCS1[0.46, 2.29] mPC3-4[0.45, 8.59] a) mPXn-1[USR, ERelative]=[0, 1] 圖 7. 陽離子甘胺酸與三個 H2O 分子在 PCM 溶劑模型下之最佳化結構 我們可以看出 mPC3-1與 mPC3-5 的 USR 彼此相差為 0.34, H-Bond 距離差異不大,三個 H2O 分子結構也皆分別放置於氨基端與兩個羧酸端, 可歸因於編號 1 到 3 的 H2O 分子上的 H 原子角度不同,而導致 mPC3-1與 mPC3-5產生相似度的差異。然而 mPC3-1與 mPC3-4兩者的 USR 差異為 0.55, 可以看出兩個結構所放置編號 1 到 3 的 H2O 分子位置上皆不同,以及 mPC3-4沒有形成環狀結構,故能量上以及相似度差異相對較大。 1.697Å 1.547Å 1.737Å 1.862Å 1.868Å 1.761Å 1.768Å 1.692Å 1.547Å 1.863Å 1.737Å 1 2 3 1 2 3 1 2 3 1.507Å 1.733Å 1.902Å 1.907Å 1 2 3
24
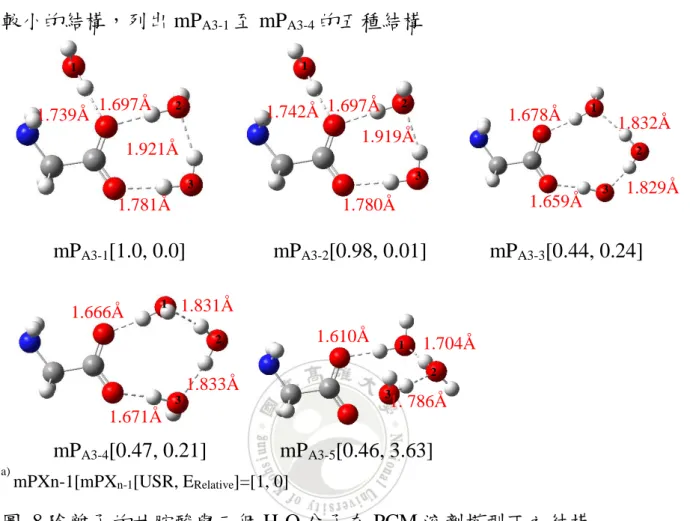
以陰離子甘胺酸與三個 H2O 為例,可以探討 USR 以及相對能量差異
較小的結構,列出 mPA3-1至 mPA3-4的五種結構
mPA3-1[1.0, 0.0] mPA3-2[0.98, 0.01] mPA3-3[0.44, 0.24]
mPA3-4[0.47, 0.21] mPA3-5[0.46, 3.63]
a)
mPXn-1[mPXn-1[USR, ERelative]=[1, 0]
圖 8.陰離子的甘胺酸與三個 H2O 分子在 PCM 溶劑模型下之結構
從分布圖來看,可以發現 mPA3-1和 mPA3-2以及 mPA3-3和 mPA3-4為兩
種 USR 以相對能量較接近的結構,其 USR 相差分別為 0.1 以及 0.03。 mPA3-1和 mPA3-2差異在於標號 1 的 H2O 分子的 H 原子位置不同而有所差 異,另外兩個 H2O 相對位置以及 H-Bond 距離皆差異不大。 而 mPA3-3和 mPA3-4則是標號 1 的水分子的 H 原子分別一個朝內一個 朝外的方向而產生些微 USR 差異約 0.03。根據 H2O 分子上的 H 原子可 以看出,USR 的差異性非常靈敏。 而在 SMD 溶劑模型下得到的結果,以兩性離子甘胺酸與一個水分子 1.697Å 1.739Å 1.921Å 1.781Å 1.742Å 1.697Å 1.780Å 1.919Å 1.678Å 1.832Å 1.829Å 1.659Å 1.666Å 1.833Å 1.671Å 1 2 3 1 2 3 1 2 3 1 2 1.610Å 1.704Å 1.831Å 1. 786Å 1 2 3 3
25
為例,最低能量結構,命名為 mSZ1-1。其他陽離子與陰離子命名也依此
類推。其分布圖如下:
a) Relative Energy = E(mSXn-mSXn-1) (n=1~6, XS1,XS2; X=C, Z, A,S=SMD solvation model) 圖 9. 兩性離子甘胺酸與一個 H2O 分子在 SMD 溶劑模型下,相對能量與 相似度 USR 之分布圖。 mSZ1-1[1.0, 0.0] mSZ1-1[1.0, 0.0] mSZ1-5[0.92, 0.02] mSZ1-3[0.82, 0.02] mSZ1-7[0.36, 0.03] mSZ1-6[0.47, 7.85] a) mSXn-1[mSXn-1[USR, ERelative]=[1, 0] 圖 10. 兩性離子與甘胺酸與一個 H2O 分子在 SMD 溶劑模型下之結構 1.761Å 1.761Å 1.762Å 1.785Å 1.770Å 1.764Å
26 以 mSZ1-1作為基準來看,與 mSZ1-1最相似的結構為 mSZ1-5。兩者的 差異在於從氨基端上方視角上可以看出位於 mSZ1-1的 H2O 分子較偏向兩 性離子甘胺酸的右端,而 mSZ1-5的 H2O 分子則是在兩性離子甘胺酸的偏 左端。由此可見,根據角度上不同,也會造成 USR 的差異。 而與 mSZ1-1第二相似結構為 mSZ1-3。其 H2O 分子在不同羧酸端上且 方向也不相同。而與 mSZ1-1最不相似的結構為 mSZ1-7,其 H2O 分子放置 於氨基端上。能量差異最大為 mSZ1-6的結構,mSZ1-6的 H2O 分子放置於 氨基端上,且氨基端上的 H 原子也因 H2O 分子放置的位置而有所改變。 胺基端的兩個 H 原子朝外,一個 H 原子則是朝向羧酸端的方向。我們可 以根據氨基端的結構來推斷出可能為 mSZ1-6相對能量差異較大的原因。 根據圖 8 可以發現在 PCM 溶劑模型條件下,每一個資料點會與最 低能量的點(也就是基準點)相對能量以及差異性皆很大。然而大部分兩性 離子甘胺酸會比其陽離子與陰離子的部分較聚集在某一個地方。 而根據圖 9 中甘胺酸與一個 H2O 分子在 SMD 溶劑模型的條件下的 分布情況是最理想的,因為我們希望可以搜尋出各個差異性較大但是能 量上差異卻不大的結構。 我們可以看出無論 PCM 或是 SMD 溶劑模型下,所得出最低結構大 部分都會將 H2O 分子放置羧酸端的結構較於其他的穩定。然而在兩性離 子的情況下可能會有將 H2O 皆放置在氨基端與羧酸端上方的位置,這是 因為兩性離子的氨基端具有部分正電荷,而羧酸端會帶部分負電荷,以 分子必須為穩定的分子架構來說,利用 H2O 分子上的 O 以及 H 分別與兩
27 性離子的部分正負電荷鍵結形成氫鍵是合理的。 而在我們搜索這些結構中可以發現,除了 H2O 分子放置羧酸端較穩 定的情況之外,可以看出 H2O 分子與甘胺酸分子形成環狀或是在較多 H2O 分子的條件下形成球狀的部分得到的能量較低且較穩定也較有利於 到全域最小值的結構。
28
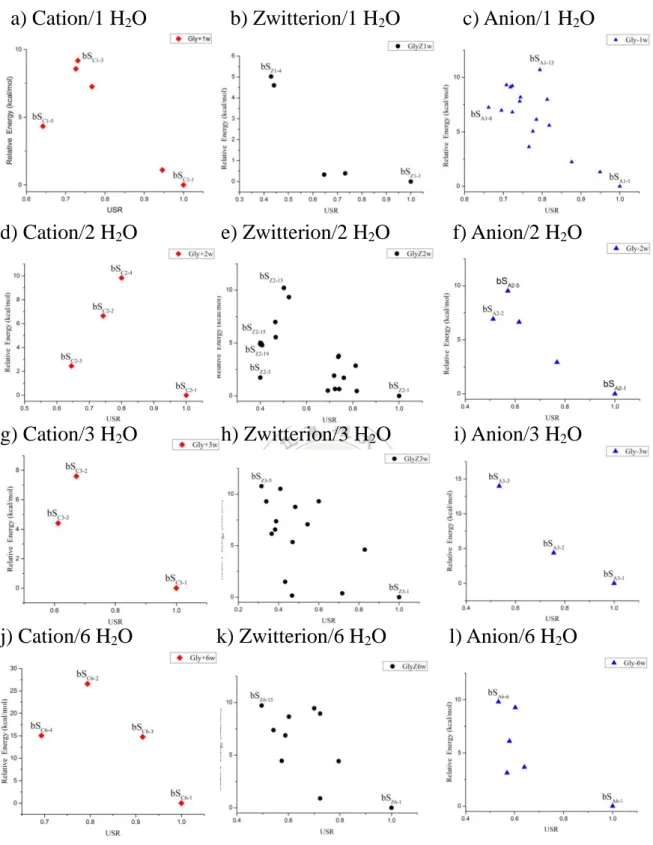
3-2. 甘胺酸水團簇以人工蜂群演算法計算之結構
3-2-1. 甘胺酸水團簇作為剛性分子團簇(rigidmolecule)之計算
根據 2-1-3 節 Zhang 等人所提供的建議來調整甘胺酸在甘胺酸一到六 個水分子團簇中之剛性分子計算上之參數。在 rigid 程序中根據得到最大 能量與最低能量誤差最小的結果,來設置甘胺酸與水團簇設置 SN, glimit, gmax和 amplitude 參數如下: 表 1. 人工蜂群演算法中甘胺酸與水形成剛性分子團簇之參數SN gmax glimit amplitude GlyC1w 150 150 1 0.5 GlyC2w 100 100 1 1.5 GlyC3w 100 100 2 3.0 GlyC6w 50 50 3 4.0 GlyZ1w 150 150 1 3.0 GlyZ2w 100 150 3 4.0 GlyZ3w 150 100 2 4.0 GlyZ6w 100 100 3 4.0 GlyA1w 150 150 1 1.5 GlyA2w 150 150 1 2.5 GlyA3w 150 50 3 3.0 GlyA6w 50 50 3 4.0
29 此不同的參數是根據找出參考點(最低點)與能量最高的分布點之能 量差異「最小」的條件下而定。由上述可以看出,水分子愈多的情況下, 其偵測極限(g limit)以及 amplitude 需設置更大的參數,而團簇大小(SN)和最 大循環數(g max)根據水分子多的情況下,反而需要較少參數即得能量低的 結構。得到五十種最低能量的結構並作相對能量 E 與 USR 分布圖。
3-2-2. 甘胺酸水團簇於 lego 程序之計算
首先我們先設置甘胺酸與 H2O 分子的結構,利用 3-1 節所推論的理 想結構來建構,所以我們以 cube 與 sphere 兩種主要的形式作為基準。Cube type Sphere type 圖 11. 蜂群演算法初步典型結構 首先我們先使用 PM6-DH+ PRECISE COSMO 進行單點計算,搜尋 出五十種結構,去除一些不合理的結構,例如 H2O 分子距離甘胺酸太遠 或是結構上的原子放置到其他周圍,然後定義相對能量在 1~10 kcal/mol 之間,尋找最低至最高能量結構。取 cube 形式,以原子彼此距離設定為 3.5Å ,其邊界長寬高分別定義為 3x3x3 個原子分布進行搜尋結構。 然而,我們取第二種為 sphere 形式,定義以甘胺酸為中心作半徑為 3.5Å,使用 PM6-DH+ PRECISE COSMO 進行單點計算,得到 50 種結構, 去除不合理的結構後,作相對能量 E 與 USR 之分布圖。
30
3-2-3. 甘胺酸水團簇於 lego 以及 rigidmolecule 程序之計算
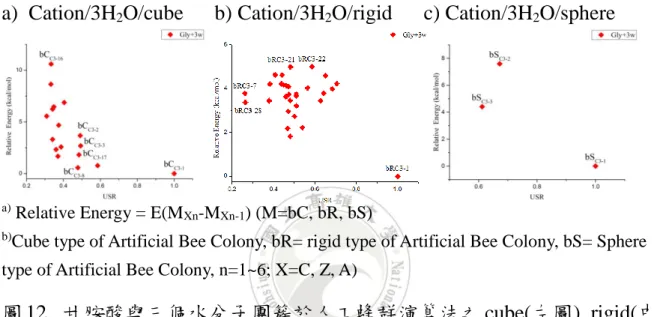
人工蜂群演算法以 rigid, cube 和 sphere 三種形式得到甘胺酸與水團 簇形成的結構,以陽離子甘胺酸與三個水分子團簇為例進行相對能量 E 與 USR 作以下分析:
a) Cation/3H2O/cube b) Cation/3H2O/rigid c) Cation/3H2O/sphere
a) Relative Energy = E(MXn-MXn-1) (M=bC, bR, bS)
b)Cube type of Artificial Bee Colony, bR= rigid type of Artificial Bee Colony, bS= Sphere type of Artificial Bee Colony, n=1~6; X=C, Z, A)
圖 12. 甘胺酸與三個水分子團簇於人工蜂群演算法之 cube(左圖), rigid(中 圖)和 sphere(右圖)形式相對能量 E 與 USR 分布圖
根據圖 12 中依序左圖為 cube 形式,中圖為 rigid 形式,右圖為 sphere 形式之人工蜂群演算法分布圖。可以看出在 cube 形式的條件下,所產生 的分布點較呈垂直分布,大致上為相似度 USR 差異較小但相對能量 E 差 異較大的情況。我們取其中四個相似的分布點 bCC3-2和 bCC3-3以及 bCC3-8
31 bCC3-2[0.491, 3.68] bCC3-3[0.493, 2.69] bCC3-17[0.484, 1.82] bCC3-8[0.478, 0.57] a) mSXn-1[mSXn-1[USR, ERelative]=[1, 0] 圖 13. 甘胺酸與三個水分子團簇於人工蜂群演算法之 cube 形式結構之比 較 根據圖 13 可以看出 bCC3-2和 bCC3-3的相似度 USR 僅差 0.002,但結 構上三個水分子的位置皆不相同。而 bCC3-17和 bCC3-8的 USR 分別為 0.478 與 0.484,其兩者的結構中 H2O 分子只剩下 bCC3-17上的一個 H2O 分子較 接近羧酸端,故 bCC3-17的 USR 會比 bCC3-8略高一些。其四種結構中可以 看出 bCC3-8是最不相似的,因為 bCC3-8的水分子放置於羧酸端皆與其他三 個不同,所以相似度 USR 差異最大。
32 而在圖 12 中,rigid 形式得出的相對能量 E 和 USR 分布圖的資料點 均集中於相對能量 E 約 2 到 5 kcal/mol 之間且相似度 USR 分布落在 0.5 左右。這些資料點的相對能量 E 和 USR 都與最低能量結構 bRC3-1均相差 較大。 bRC3-1 [1.0, 0.0] bRC3-7 [0.262, 3.77] bRC3-28 [0.264, 3.37] bRC3-21 [0.481, 4.98] bRC3-22 [0.586, 4.99] a) mSXn-1[mSXn-1[USR, ERelative]=[1, 0] 圖 14. 陽離子甘胺酸與三個水分子在蜂群演算法 rigid 形式之結構 在圖 14 中,最低能量結構為 bRC3-1,最不相似的結構為 bRC3-7和 bRC3-28,能量差異最大為 bRC3-21和 bRC3-22。可以看出 rigid 形式之結構上 的水分子均較偏向放置氨基端上的結構。bRC3-21和 bRC3-22的結構相對能 量 E 較大,可以歸因於在羧酸端的水分子形成的氫鍵距離比起 bRC3-7和 bRC3-28較遠,故相對能量差異較大。 而在 sphere 形式的情況下,可以看出資料點明顯較少,原因可歸咎 於起初半徑設置距離最小為 3.5Å 的關係,會使放置水分子時沒有特定的
33 範圍內,使得水分子會放置至離甘胺酸較遠的情況下,較成為不合理的 結構。因此大多資料點搜尋只能找到距離最低能量結構 bSC3-1較近的結構, 其結果如下 bSC3-1[1.0, 0.0] bSC3-2[0.672, 7.59] bSC3-3[0.612, 4.41] a) mSXn-1[mSXn-1[USR, ERelative]=[1, 0] 圖 15. 甘胺酸與三個水分子在蜂群演算法 sphere 形式結構 可以看出對於最低能量結構 bSC3-1來說,相對能量較小的 bSC3-3 以 及相對能量較大 bSC3-2結構均會環繞於甘胺酸其周圍,根據 sphere 形式 可以達到搜尋出的結構均傾向於形成球狀的結構。
根據結果我們可以討論 cube 及 sphere 形式的結構差異。cube 形式的 部分,起初設定的閾值我們是使用距離 3.5Å 及 3x3x3 的晶格定義,即晶 格中長寬高階只能放置 3 個原子且原子與原子彼此間距離為 3.5 Å 的範圍, 所以放置 H2O 分子必須固定於晶格內中。與 cube 形式相比之下,sphere 形式的方式就比較不那麼侷限於同一空間,sphere 形式的定義是基於將 甘胺酸視為一個圓心,而 H2O 分子的部分則是以半徑定義為 3.5Å 的距離 放置。故 sphere 形式的擺放空間比較大,相似度也會差異較大。 1.759Å
34 而在 rigid 形式上來說,雖然並非沒有完全找到與最低能量結構之相 對能量差異較低的結構,但是對於 rigid 形式來說根據不同水分子的條件 下設定參數的結果可以搜尋到比 cube 及 sphere 形式相對能量更低的結構。 對於在不同水分子的條件下,使用 rigid 形式搜尋不同較低能量的結構上 來說,是具有優勢的。
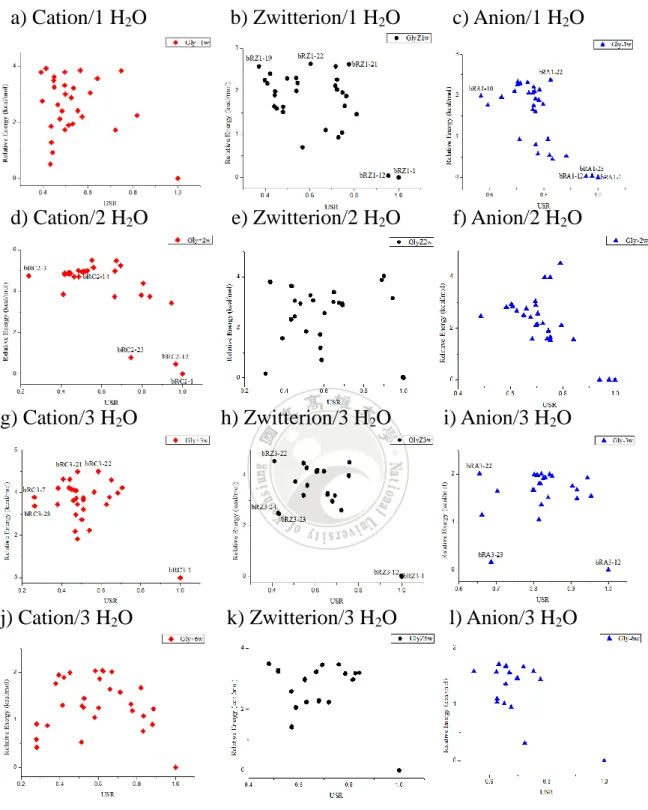
3-3. 甘胺酸以基因演算法(CLUSTER)程序計算之結構
另一個程序是使用基因演算法,將甘胺酸的片段輸入執行檔中進行 不同低能量結構的搜索。我們使用 PM6-DH+ PRECISE COSMO 方法做 最佳化尋找最低能量結構。搜索出 150 個以能量低到高的結構,程序上 會先自動過濾重複的結構,所以能量上差異不大,檢查出來能量較高的 結構都沒有距離太遠而刪除的問題。再將剩下的候選物做快速形狀識別 USR 以及相對能量 E 作圖。 為了與蜂群演算法作比較,我們也以陽離子甘胺酸與三個 H2O 分子 的結構部分進行探討。 圖 16. 陽離子甘胺酸與 3 個 H2O 分子基因演算法下,相對能量 E 與相似 度 USR 之分布圖35 根據圖 16 中 GAC3-2為 GAC3-1能量差異最小但不相似結構,GAC3-10 為 GAC3-1最不相似之結構,而 GAC3-15則為 GAC3-1最不相似且能量差距最 大結構 GAC3-1(0.0) GAC3-2(0.17) GAC3-10(2.59) GAC3-15(7.23) a) mSXn-1[mSXn-1[USR, ERelative]=[1, 0] 圖 17. 陽離子甘胺酸與 3 個 H2O 分子在基因演算法之最佳化結構 可以看出 GAC3-2與 GAC3-1在氨基端一側形成環能量會較低,而在氨 基端一側形成環的 GAC3-10結構,則會高出 2 kcal/mol。最不相似的 GAC3-15 結構雖然能量差距最大,其 3 個 H2O 分子分布於不同官能基上方,但其 結構仍以環狀為主。 1.549Å 1.697Å 1.927Å 1.421Å 1.511Å 1.726Å 1.807Å 1.601Å 1.562Å 1.724Å 1.814Å 1.727Å 1.517Å 1.674Å 1.810Å 1.793Å
36
3-4. 人工算法與遺傳及蜂群演算法之最佳化甘胺酸與水團簇相
對能量比較
根據上述使用蜂群演算法與基因演算法的尋找各種不同結構的結果, 將此兩種演算法中取出最低能量結構,使用ωB97XD/6-31+G(d, p) 基組 在 PCM 與 SMD 溶劑模型下進行最佳化的計算。再與我們原先人工算法 最佳化的結果與其他兩種演算法的最低能量結構進行能量的比較,來探 討其結構對能量 E 與相似度 USR。 其結構命名 bC 即為蜂群演算法 cube 形式結構,bR 為蜂群演算法 rigid 形式結構,bS 即為蜂群演算法 sphere 形式之結構,GA 則為基因演 算法結構,S1 及 S2 分別以 Galano27與 Schlegel26於文獻中提出的結構, 再進行最佳化計算,而人工算法部分則以 M 作為命名。 此外,在人工算法我們也提出了在六個 H2O 分子的條件下以 Galano27 與 Schlegel26放置 H 2O 分子方法的概念,將三個水分子分別放置於氨基端 與羧酸端形成六個 H2O 分子的結構,也將此結構與蜂群以及基因演算法 搜索出的六個 H2O 分子之結構作為比較。其結果如下:37 a) Cation/1 H2O b) Cation/2 H2O c) Cation/3 H2O d) Cation/6 H2O 圖 18. 以人工算法、蜂群與基因演算法之陽離子甘胺酸與一、二、三和 六個 H2O 分子在 PCM 溶劑模型下最低能量結構進行最佳化計算,其相 對能量與相似度 USR 之比較。
38 a) Zwitterion/1 H2O b) Zwitterion/2 H2O c) Zwitterion/3 H2O d) Zwitterion/6 H2O 圖 19. 以人工算法、蜂群與基因演算法計算陰離子甘胺酸與一、二、三 和六個 H2O 分子在 PCM 溶劑模型下之最佳化結構,相對能量與相似度 USR 之比較。
39 a) Anion/1H2O b) Anion/2 H2O c) Anion/3 H2O d) Anion/6 H2O 圖 20. 以人工算法、蜂群與基因演算法計算陰離子甘胺酸與一、二、三 和六個 H2O 分子在 PCM 溶劑模型下之最佳化結構,相對能量與相似度 USR 之比較。
40 根據在 PCM 溶劑模型下所產生相對能量的比較,可以看出陽離子甘 胺酸在一到六個水分子的情況下最大相對能量差約 6 至 8 kcal/mol,差異 上不大。然而對於兩性離子甘胺酸結構可以看出在兩個水分子與六個水 分子差異變化較大,約 8 至 10 kcal/mol,差異最小可至 0.9 kcal/mol 內。 而在陰離子甘胺酸的條件下,與六個水分子的情況得到的相對能量差至 15 kcal/mol。而在相似度 USR 的方面來說,每個條件之相似度 USR 皆落 在 0.3 至 1.0 之間都具有不同結構。
41 為了與 PCM 溶劑模型比較,下方也列出為人工算法、蜂群與基因演 算法使用ωB97XD/6-31+G(d, p) 基組在 SMD 溶劑模型下進行最佳化的 計算。相對能量的比較,其結構與相似度 USR 之分布圖得到的結果: a) Cation/1 H2O b) Cation/2 H2O c) Cation/3 H2O d) Cation/6 H2O 圖 21. 以人工算法、蜂群與基因演算法計算陰離子甘胺酸與一、二、三 和六個 H2O 分子在 PCM 溶劑模型下之最佳化結構,相對能量與相似度 USR 之比較。
42 a) Zwitterion/1 H2O b) Zwitterion/2 H2O c) Zwitterion/3 H2O d) Zwitterion/6 H2O 圖 22. 以三種演算法在 SMD 溶劑模型下最佳化兩性離子甘胺酸與一、 二、三到六個 H2O 分子得到相對能量與相似度 USR 之比較。
43 a) Anion/1 H2O b) Anion/2 H2O c) Anion/3 H2O d) Anion/6 H2O 圖 23. 以三種演算法在 SMD 溶劑模型下最佳化陽離子、兩性離子與陰 離子甘胺酸與三到六個 H2O 分子得到相對能量與相似度 USR 之比較。 與 PCM 溶劑模型相比,在 SMD 溶劑模型下得到的相對能量 E 的結 果可以發現除了陽離子甘胺酸在三個水分子的條件下最大相對能量差皆 6 kcal/mol 以外,其他條件下的最大相對能量差的差距明顯降低。在 SMD 溶劑模型下大致上較可以找到能量較低的結構。 然而以 SMD 溶劑模型得到的相似度 USR 與 PCM 溶劑模型相比,可 以發現相似度 USR 的部分都較集中,例如陽離子甘胺酸兩個水團簇在
44 SMD 溶劑模型下皆落在 0.4 至 0.5 的範圍內,且在兩性離子甘胺酸與兩 個水團簇得到相似度 USR 從 0.7 至 1.0 的範圍內。 在 PCM 與 SMD 溶劑模型的分布圖可以歸納出,PCM 溶劑模型得到 的資料點大致傾向於相似度 USR 與相對能量 E 差異較大的結構。而 SMD 溶劑模型的資料點則偏向於得到相似度 USR 與相對能量 E 差異較小的結 構。其原因可歸因於 SMD 溶劑模型下本身具有分散力的因素,所以最佳 化的情況下,會傾向於得到較低能量的結構。 我們也列出 PCM 與 SMD 溶劑模型中,陰離子甘胺酸與六個水分子 團簇之比較,其結果如下:
45
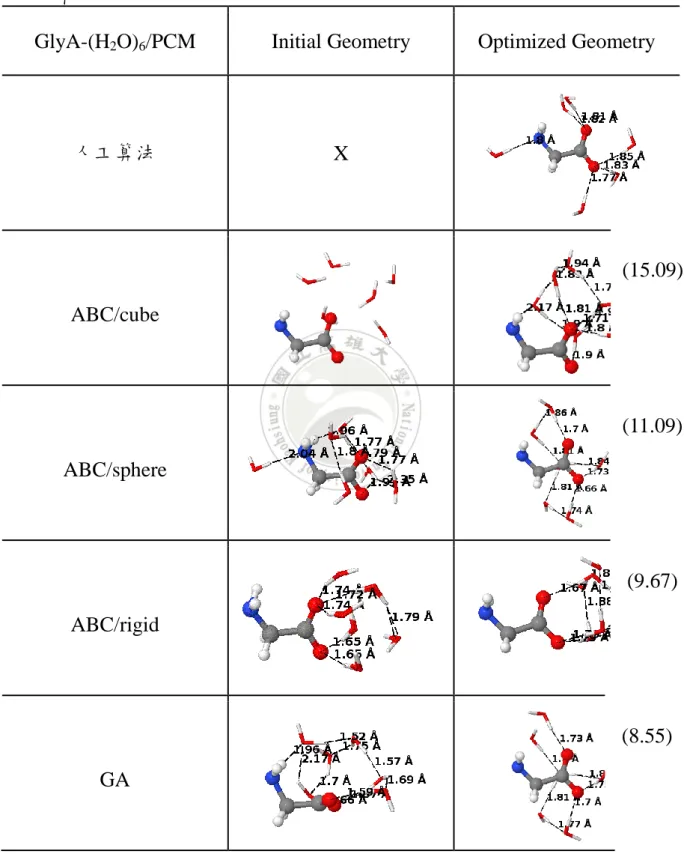
表 2. 陰離子甘胺酸與六個 H2O 分子在 PCM 溶劑模型下初始結構與最佳
化結構比較
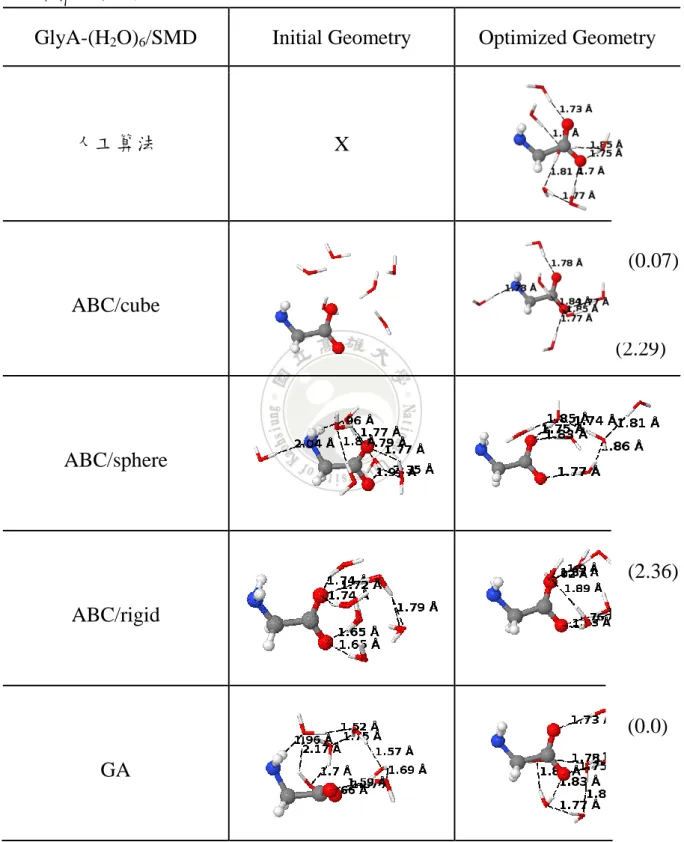
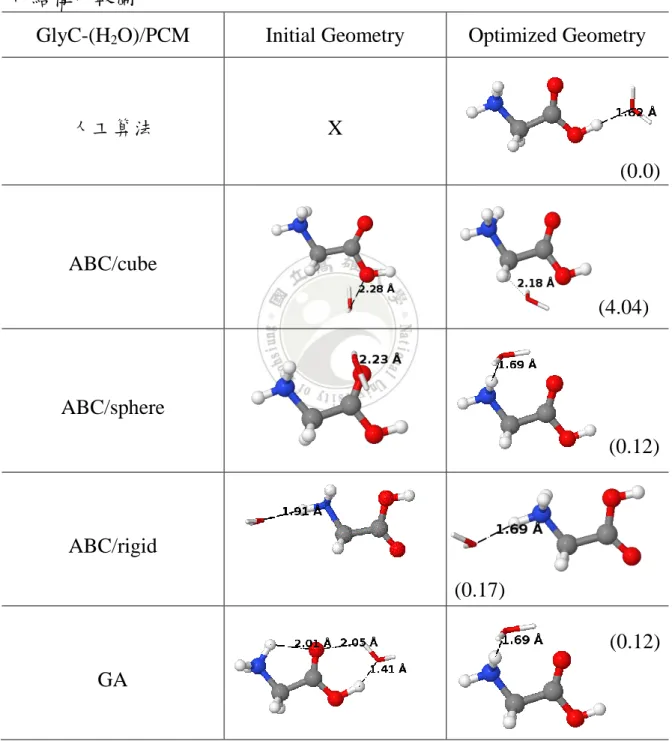
GlyA-(H2O)6/PCM Initial Geometry Optimized Geometry
人工算法 X
ABC/cube
ABC/sphere
ABC/rigid
GA
a)ABC 為人工蜂群演算法,GA 為基因演算法,其中括弧內為相對能量 E(kcal/mol)
(15.09)
(11.09)
(9.67)
(8.55)
46
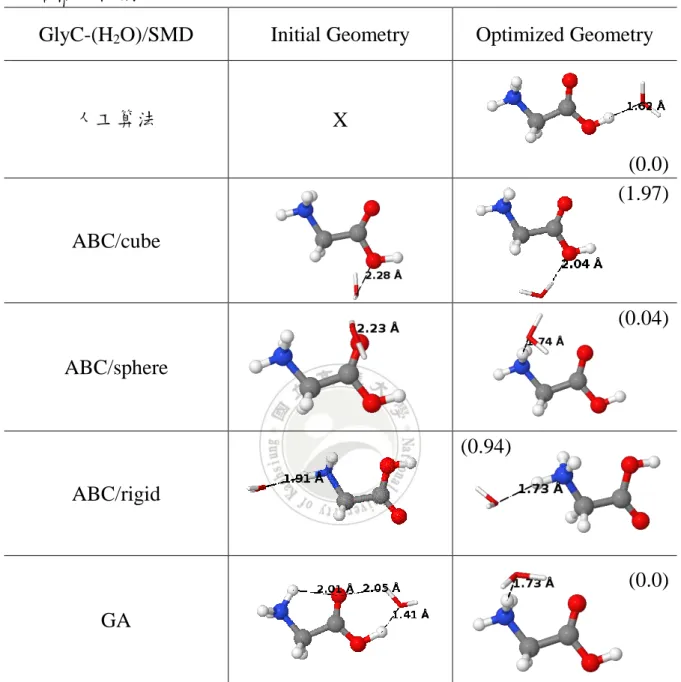
表 3. 陰離子甘胺酸與六個 H2O 分子在 SMD 溶劑模型下初始結構與最佳
化結構比較圖
GlyA-(H2O)6/SMD Initial Geometry Optimized Geometry
人工算法 X ABC/cube (2.29) ABC/sphere ABC/rigid GA
a)ABC 為人工蜂群演算法,GA 為基因演算法,其中括弧內為相對能量 E(kcal/mol)
(0.07)
(2.36)
(0.0)
47 根據 PCM 與 SMD 得到最佳化結構的結果,可以看出差異最多的為 人工蜂群演算法 sphere 形式與基因演算法的結構。而對於最低能量結構 可以發現在 PCM 溶劑模型下基因演算法為最低能量結構,其呈現在羧酸 端行程之鏈狀的結構,相對 SMD 溶劑模型的情況下,以人工蜂群演算法 rigid 形式可以得到最低能量結構,呈現在羧酸端放置水分子形成環狀的 結構。 可以發現兩者溶劑模型得到的最佳化結構大多是將水分子放置羧酸 端以達到最穩定的結構。不同的是,在 PCM 溶劑模型下傾向於水分子延 伸出支鏈後再與其他水分子形成環狀的結構以達到穩定狀態,而 SMD 溶 劑模型下則是將水分子以甘胺酸的兩側羧酸端水分子形成環狀以達到穩 定的狀態。可以推敲出 SMD 溶劑模型的結構因與甘胺酸形成氫鍵作用力 較強,整體結構較緊密,得到的能量上會比 PCM 溶劑模型來得較低。
48
3-5. 人工算法與遺傳及蜂群演算法之 pK
a值計算
以人工算法、遺傳演算法和蜂群演算法所得到甘胺酸與一到六個 H2O 分子在 PCM 與 SMD 溶劑模型下最佳化結構。取這三種不同方法中
49 表 4. 甘胺酸與一到六個水團簇使用 ωB97XD/6-31+G(d, p) 基組在 PCM 以及 SMD 溶劑模型下計算水溶液自由能 差異∆G1和∆G 2值 Solvation Model GCation (Hartree) GZwitterion (Hartree) GAnion
(Hartree) ∆G1(kcal/mol) ∆G2(kcal/mol) PCM -284.825 -284.379 -283.911 0.44 14.04 PCM-1W -361.171 -360.734 -360.299 3.57 2.95 PCM-2W -437.579 -437.163 -436.696 -9.20 22.65 PCM-3W -513.990 -513.556 -513.104 2.03 13.30 PCM-6W -743.210 -742.785 -742.359 -3.05 -3.48 SMD -284.846 -284.396 -283.925 1.94 17.86 SMD-1W -361.193 -360.755 -360.302 4.57 13.89 SMD-2W -437.604 -437.163 -436.911 3.17 16.85 SMD-3W -514.015 -513.556 -513.124 4.18 14.97 SMD-6W -743.243 -742.785 -742.357 1.42 16.42 a) ∆G1=(Gcation-GZwitterion)*627.5096 + * aq G (H+) b) * aq G (H+) = -270.29 kcal/molc) + ΔG1 +
(aq) (aq) (aq)
Gly GlyZ +H ΔG2 - +
(aq) (aq) (aq) GlyZ Gly +H
50
表 5. 甘胺酸與一到六個水團簇使用 ωB97XD/6-31+G(d, p)基組在 PCM 以及 SMD 溶劑模型下計算 pKa1和 pKa2值
Solvation
Model ∆G1(kcal/mol) ∆G2(kcal/mol) pKa1 pKa2 ∆pKa1 ∆pKa2 PCM 0.44 14.04 0.32 10.30 -2.02 0.70 PCM-1W 3.57 2.95 2.62 2.16 0.28 -7.44 PCM-2W -9.20 22.65 -6.75 16.61 -9.09 7.01 PCM-3W 2.03 13.30 1.49 9.75 -0.85 0.15 PCM-6W -3.05 -3.48 -2.24 -2.55 -4.58 -12.15 SMD 1.94 17.86 1.42 13.10 -0.92 3.50 SMD-1W 4.57 13.89 3.35 10.18 1.01 0.58 SMD-2W 3.17 16.85 2.33 12.06 -0.01 2.46 SMD-3W 4.18 14.97 3.06 10.98 0.72 1.38 SMD-6W 1.42 16.42 -1.97 13.12 -1.30 2.44 Expt. 2.34 9.60 a) + ΔG1 +
(aq) (aq) (aq)
Gly GlyZ +H ΔG2 - +
(aq) (aq) (aq)
51 理論 pKa值絕對誤差是根據表 5 下方提出的理論 pKa1以及 pKa2值為 基準,而從表 5 得到的結果可以發現甘胺酸與三個 H2O 分子在 PCM 溶 劑模型下,pKa1和 pKa2值皆可獲得良好的結果。而對於 pKa1值來說,在 SMD 溶劑模型下誤差皆在 1.5 pKa單位內,尤其是在甘胺酸與兩個 H2O 分子的情況下精確至 0.01 pKa單位內。在 pKa2值的情況下,SMD 溶劑模 型除了在甘胺酸與兩個 H2O 分子與六個 H2O 分子誤差稍微較大以外,基 本上皆可以得到良好的準確結果。