行政院國家科學委員會專題研究計畫 期中進度報告
全身性紅斑狼瘡患者單核性細胞的異常氧化還原狀態與其
免疫反應低下之間關係的研究(1/3)
計畫類別: 個別型計畫 計畫編號: NSC93-2314-B-002-085- 執行期間: 93 年 08 月 01 日至 94 年 07 月 31 日 執行單位: 國立臺灣大學醫學院分子醫學研究所 計畫主持人: 余家利 報告類型: 精簡報告 處理方式: 本計畫可公開查詢中 華 民 國 94 年 5 月 27 日
Reduced Redox Capacity, Impaired Mitochondria Functions, and Defective
Expression of Na
+and K
+Ion Transport-associated Molecules Predispose
Immune Hyporesponsiveness in Patients with Active Systemic Lupus
Erythematosus
Song-Chou Hsieh1, MD, PhD, Hsin-Su Yu2, MD, PhD, Ko-Jen Li1, MD, Ming-Chi Lu1,
MD, Chang-Youh3 Tsai, MD, PhD, Chia-Li Yu1, MD, PhD.
1Department of Internal Medicine, 2Department of Dermatology, National Taiwan
University Hospital, National Taiwan University College of Medicine, Taipei, Taiwan. 3Section of Allergy, Immunology & Rheumatology, Veterans General Hospital-Taipei,
Taipei, Taiwan
Address correspondence and reprint requests to Chia-Li Yu, MD, PhD. Department of Internal Medicine, National Taiwan University Hospital, #7 Chung-Shan South Road, Taipei, Taiwan 100.
TEL: +886-2-23123456 ext 5011 FAX: +886-2-23957801
E-mail: [email protected]
This work is supported by the grants from National Sciences Council (NSC93-2314- B002-085) and Yen Tijing Ning Medical Research Foundation (CI92-4-2)
ABSTRACT
Objective. Immune hyporesponsiveness of mononuclear cell (MNC) and polymorpho-
nuclear neutrophils (PMN) predisposes patients with active systemic lupus erythematosus (SLE) susceptible to infections. This study aimed to explore the pathogenetic factors for it.
Methods. Sixty-three patients in total fulfilling the 1982 revised ACR classification criteria
for SLE and the same number of sex- and age-matched normal individuals were studied. Reduction-oxidation (redox) capacity [glutathione (GSH) level, GSH peroxidase (GSH-Px) and GSH reductase (GSSG-R) activities], mitochondrial (mt) functions (mitochondrial potential, mitochondrial mass, ATP production and mtDNA 4977bp deletion), and activation-related parameters (membrane potential changes and expression of Na+ and K+-associated molecules) of MNC and PMN were compared in normal and SLE groups. In addition, the effects of anti-dsDNA autoantibodies (100IU/ml) purified from active SLE sera on GSH-Px activity and apoptosis was determined.
Results: The GSH level and GSH-Px activity in SLE-MNC and GSH level in SLE-PMN
are lower than the normal counterparts. The reduced redox capacity in SLE immune cells seems not relevant to the anti-SLE medications because the same tendency was also observed in 8 untreated Active SLE patients. The reciprocal expression of GSH-Px isomers in MNC and PMN was the same in normal and SLE patients in that dimer (50kDa) and tetramer (100kDa) are prominent in MNC whereas monomer (25kDa) and trimer (75kDa) are prominent in PMN. The defective redox capacity in SLE immune cells leads to
impaired mitochondrial functions with defective ATP production, decreased mitochondrial mass, reduced mitochondrial potentials, and increased mtDNA 4977bp deletion (18.2% in SLE vs. 4.3% in normal). The membrane potential of SLE-MNC and PMN was also diminished due to decreased expression of Na+-K+-ATPase, epithelial Na+ channel and renal outer medullary K+ channel. Anti-dsDNA antibodies (25-100IU/ml) purified from SLE sera suppressed GSH-Px activity and increasing normal cells apoptosis.
Conclusion. Reduced redox capacity, and defective Na+ and K+ transport-related molecule
expression are the predisposing factors for immune hyporesponsiveness in patients with active SLE. Anti-dsDNA autoantibodies play a role on reducing redox capacity in these patients.
Key words: systemic lupus erythematosus, immune hyporesponsiveness, redox capacity,
INTRODUCTION
Systemic lupus erythematosus (SLE) is an archetype of systemic autoimmune disorder. Susceptibility to common and opportunistic infections is a major cause of morbidity and mortality in these patients (1,2). It is recognized that functional defects in multiple SLE immune cells per se as well as the use of immunosuppressants are the important risk factors for the infections (3,4). Hyporesponsiveness of mononuclear cells (MNC) to phytomitogens, antigens, allogeneic and autologous cells and polymorphonuclear neutrophils (PMN) to chemokines in patients with SLE had been observed (5-8). Recently, a number of deranged antigen-receptor-mediated signaling in T and B lymphocyte was found in patients with active SLE (9). These signaling aberrations in SLE immune cells include TCR- or BCR-mediated protein tyrosine phosphorylation, cytoplasmic free Ca2+ level and TCR zeta chain deficiency that suggest the presence of cell pre-excitation in vivo (9). However, these defects are still unable to explain the immune hyporesponsiveness of SLE-MNC and -PMN to different stimulations in vitro. It is conceivable that many biological molecules important in cell signaling and gene expression are sensitive to reactive oxygen species at a concentration much lower than require inflict oxidative damage (10-12). These reduction-oxidation (redox)-sensitive target molecules include transcription factors (NF-κB, AP-1, NF-AT, PAX-8) (11-13), calcium metabolism modulators (mitochondrial permeability transition, adenyl cyclase, ryanodine receptor, L-type calcium channel) (14-16), ion transporters (small Cl- channel and K+ influx) (17,18), , cytokines (TNF-α,
IL-1, IL-2, IL-6, TGF-β) (19,20), cell growth-related genes (p21, ras-signaling) (21,22), protein kinase (23, 24), tyrosine phosphatase (25), iron regulatory proteins (IRP1/IRP2) (26) and heat shock proteins (27). Furthermore, the intracellular redox state can modulate immune functions such as lymphocyte proliferation and NK-mediated cytotoxicity (28). Based on these facts, we hypothesize that abnormal intracellular redox status may relate to immune hyporesponsiveness in SLE. Glutaredoxin-glutathione system composed of NADPH, glutathione (GSH), the flavoprotein glutathione peroxidase (GSH-Px) and glutathione reductase (GSSG-Rx) is one of the key players in redox regulation (29). Glutathione is present in all animal cells with high concentration and acts as the most important endogenous modulating molecule for redox, cell proliferation, DNA synthesis, immune response, and arachidonic acid metabolism (30,31).
Mitochondrial DNA (mtDNA) is much more vulnerable to oxidative damage from mitochondrial environment than nuclear DNA. Berneburg et al. (32) demonstrated that mtDNA 4977bp depletion is a useful biomarker for oxidative damage in human cells exposed to UV irradiation or singlet oxygen produced by thermo-decomposition of an organic endoperoxidase. The deletion mutation of mtDNA impairs the mitochondrial permeability transition and respiratory function for bioenergy production (14, 33). Nagy et al. (34) demonstrated that GSH-Px effectively protected cells from the toxic effects of xenobiotics and environmental damages by removal of metabolic products such as hydrogen peroxides, lipid peroxides and organic hydroperoxides. It is possible that the
pre-excitation of SLE immune cells in vivo by autoimmune signaling defects alters intracellular redox status and enhances oxidative damage of mtDNA in the cells. Whether these pathological mechanisms predispose SLE immune hyporesponsiveness have not been explored in the literatures. On the other hand, membrane potential change is one of the early events of cell activation that require exchanges of Ca2+, K+ and Na+/H+ in the intracellular compartments (35, 36). Whether defects in these cationic ion transport relating to immune hyporesponsiveness of active SLE is discussed.
PATIENTS AND METHODS
Patients and controls
Sixty-three patients fulfilling the 1982 ACR Revised Classification Criteria of SLE were enrolled in the present study. The same number of age and sex-matched normal individuals were the control. All of the SLE patients were in active status judging from the SLEDAI (37). Eight of them were newly diagnosed and did not receive glucocorticoids or immunosuppressants before study were regarded as disease control group. The daily medications (mean+s.d.) of 63 patients group were 17.6+10.7mg of prednisolone and 71.6+22.7mg of azathioprine. Besides, 10 bronchial asthma (BA) patients with acute exacerbation who received intravenous methylprednisolone 20mg twice daily for at least 3 days were drug control group. All participants signed informed consent to joint this study approved by the Institutional Review Board of National Taiwan University Hospital.
Isolation of mononuclear cells and polymorphonuclear neutrophils from peripheral
blood
Heparinized venous blood obtained from active SLE patients and controls was mixed with one-quarter volume of 2% dextran solution (mol. wt. 500,000 Daltons) and incubated at 37°C for 20 min. The leukocyte-rich supernatant was collected and diluted with the same volume of Hanks’ balanced salt solution. The suspension was layered over Ficoll-Hypaque density gradient cushion (specific gravity 1.077) and centrifuged at 150g for 30min, the MNC were aspirated from the interphase whereas the PMN were obtained from the bottom.
The viability and purity of both MNC and PMN were greater than 95% confirmed by trypan blue dye exclusion and Wright’s stain, respectively.
Measurement of GSH concentration
Soluble cellular GSH concentration was measured with BIOXYTECH GSH-400TM Colorimetric Assay Kit (OXIS International Inc. Portland, OR, USA). The detailed procedures are described in the manufacturer’s instruction booklet. Briefly, the concentration of MNC and PMN was adjusted to 1x107 cells/ml and were sonicated immediately at 100w for 60sec. The GSH measurements include only soluble cellular form (GSH + glutathione disulfide). The detection limit of the assay is 0.5µm/ml.
Determination of GSH-Px activity
We used BIOXYTECH GPx-340TM Colorimetric Assay Kit (OXIS International Inc.) for measuring GSH-Px activity. One milliunit (mU) of GSH-Px activity is defined as the activity that catalyzed the oxidation of 1 nmol NADPH/ml/min using an extinction molar coefficient of 6.22 x 106 M-1 cm-1 for NADPH.
Determination of GSSG-R activity
We used BIOXYTECH GR-340TM Colorimetric Assay Kit (OXIS International Inc.) for measuring GSSG-R activity. The definition of mU of GSSG-R enzymatic activity is of the same as in GSH-Px activity
Detection of GSH-Px mRNA expression in MNC and PMN by RT-PCR
primers as following:
GSH-Px: 5’-GGG GCC TGG TGG TGC TCG GCT-3’ (sense) 5’-CAA TGG TCT GGA AGC GGC GGC-3’ (anti-sense) G3PDH: 5’-ACC ACA GTC CAT GCC ATC AC-3’ (sense)
5’-TCC ACC ACC CTG TTG CTG TA-3’ (anti-sense)
The amplified PCR products were 354bp for GSH-Px and 452bp for G3PDH (as internal control).
Western blot analysis of GSH-Px isomers
MNC and PMN at a concentration of 5x106/ml were lysed and electrophoresized in 10% SDS-PAGE. The expression of GSH-Px isomers was probed by monoclonal anti-human GSH-Px antibody (MBL International, Woburn, MA. USA) and detected by enhanced chemiluminescence protein detection kit (Amersham International Plc. Chalfont, Bucking- Hamshire, UK) after electro-transferred to nitrocellulose membrane.
Measurement of ATP levels in MNC and PMN
Intracellular ATP levels were determined using ATP Determination Kit (Molecular Probes, Eugene, OG, USA).
Flow cytometric determination of mitochondrial potential and mitochondrial mass
We followed the methods described by Marchetti et al. (38) and Mancini et al. (39). The fluorescein probes were 3,3’-dihexyloxacarbocyanine [DiOC6 (3)] (Molecular Probes; 40nM in PBS, pH 7.2) for mitochondrial potential and 10-n-nonyl acridine orange (NAO)
(Molecular Probes) for mitochondrial mass with 488nm excitation.
Detection of mtDNA 4977bp and 7436bp deletion
Isolation of mitochondrial DNA and detection of mtDNA 4977bp and 7436bp deletion mutation in MNC and PMN were conducted by the method developed by Liu et al. (40). The paired primers were obtained from Bio Basic, Inc. (Ontario, Canada) as shown below:
L1 (amplified position: 3304-3323): 5’-AAC ATA CCC ATG GCC AAC CT-3’ L3 (amplified position: 8251-8270): 5’-GCC CGT ATT TAC CCT ATA GC-3’ L5 (amplified position: 8531-8550): 5’-ACG AAA ATC TGT TCG CTT CA-3’ H1 (amplified position: 3836-3817): 5’-GGC AGG AGT CCT CAG AGG TG-3’ H2 (amplified position: 13650-13631): 5’-GGG GAA GCG AGG TTG ACC TG-3’ H5 (amplified position: 16255-16236): 5’-CTT TGG AGT TGC AGT TGA TG-3’
(a) The size of the PCR product amplified from mtDNA with the length mutation (bp) by L1-H1 is 533bp that represents an internal standard of total mtDNA.
(b) The size of the PCR product amplified from mtDNA with the length mutation (bp) by L3-H2 is 423bp that represents the detection of the 4977bp deletion in mtDNA.
(c) The size of the PCR product amplified from mtDNA with the length mutation (bp) by L5-H5 is 289bp that represents the detection of 7436bp deletion in mtDNA
Na+-K+-ATPase, ENaC and ROMK1 on MNC and PMN
We followed the method of Shapiro et al. (41) for the estimation of surface membrane potential of the individual cells. The indicator dye used was 3.3’-dipentyl-oxacarbo- cyanine iodide [DiOC5 (3)]. The monoclonal antibodies against human Na+-K+-ATPase (Santa Cruz Biotechnology, Inc. CA. USA), ENaC (Alpha Diagnostic International Inc. Tx. USA) and ROMK1 (Alomone Labs. Ltd. Jerusalem, Israel) were used to stain the respective protein molecules expressed on the MNC and PMN detected by flow cytometry with 488nm excitation.
Affinity-purified polyclonal anti-dsDNA autoantibodies from the sera of patients with
active SLE
We used dsDNA-conjugated Sepharose 4B affinity column to purify polyclonal anti-dsDNA autoantibodies from active SLE sera as in our previous report (42). The dsDNA binding capacity of the purified antibodies was assayed by an anti-dsDNA ELISA kit (BioHyTech Co., Ramat Gan, Israel).
Morphological observation of cell apoptosis
Briefly, MNC or PMN at a concentration of 2x106 cells/ml were incubated with different concentrations of affinity–purified anti-dsDNA from 25 to 100IU/ml for 24, 72 and 120h. After incubation, the cell suspension was cytocentrifuged, 2% paraform- aldehyde-fixed, and stained with H & E solution. The morphology of the cells with karyopyknotic/karyorrhexic nuclei and cell volume shrinkage were considered as apoptosis
under light microscopic observation.
Statistical analysis
The results in the whole study are represented by mean+standard deviation. The statistical significance was assessed by non-parametric Wilcoxon signed rank test.
RESULTS
Comparison of redox capacity in different normal blood cells and plasma
Glutathione is one of the most important endogenous molecules with high concentration for modulating redox state in all animal cells (29-31). At first, we measured the intracellular GSH levels in different blood cells including MNC, PMN, RBC, platelets and plasma of normal individuals. We noticed that GSH is most abundant in the plasma and decrease in different blood cells by the order of MNC>PMN>RBC>platelet (Fig 1-A). Biologically, the reduced-form GSH is generated by the action of GSH-Px after reduction of the oxidative-form GSSG (44). Accordingly, we then measured the GSH-Px activity and found MNC and plasma exhibited much more GSH-Px enzymatic activity than the other blood cells (Fig.1-B). Although the mRNA expression of GSH-Px in normal MNC and PMN was not different (2 of the 4 cases are presented in Fig.1-C), 10% SDS-PAGE analysis of normal MNC and PMN revealed a total difference in the distribution of GSH-Px isomers in the two cells. As shown in Fig.1-D, MNC contains mainly dimer (50kDa) and tetramer (100kDa) but less monomer (25kDa) and trimer (75kDa) of GSH-Px isomers. In contrast, PMN contains mainly monomer and trimer, but less dimer and tetramer of GSH-Px isomers. These results are compatible with Misso et al. (43) in that normal neutrophils contain mainly monomer and trimer of GSH-Px isomers. Since the GSH-Px activity of MNC is much greater than PMN, it seems possible that dimer and tetramer of GSH-Px possess more potent anti-oxidant activity than monomer and trimer.
Comparison of redox capacity (GSH levels, GSH-Px and GSSG-R activity) in MNC
and PMN of normal individuals and SLE patients
The reduced-form GSH levels in SLE-MNC and –PMN (Fig.2-A), and the GSH-Px activity of SLE-MNC are significantly less than the normal counterparts (Fig.2-B). However, the expression of GSH-Px isomers in SLE cells is not different from the normal counterparts (Fig.2-C). The GSSG-R activity, another redox modulating enzyme containing active dithiol moieties for protection and repair of protein sulfhydryls in an oxidative stress situation (44), was also not different among normal and SLE cells (Fig.2-D). For fear of the side effects from glucocorticoids or immunosuppressants on redox capacity of active SLE, 8 untreated active SLE patients (as disease activity control)) and 10 active BA patients on medium-dose prednisolone treatment at least 3 days (40mg methlprednisolone daily, as drug control) were also compared. We found a tendency of reduced redox capacity in the non-treated active SLE group and normal redox capacity in methyprednisolone-treated group (data not shown). We believe that the reduced redox capacity and the other defects found in active SLE mainly in the study originated from the disease activity rather than the side effects from anti-SLE medications.
Defective ATP production, reduced mitochondrial mass, decreased mitochondrial
potential and increased mtDNA 4977bp deletion in SLE cells
Mitochondria are the pivotal sites for ATP production. These organelles are quite vulnerable to oxidative damage because of lacking protection from DNA-binding proteins
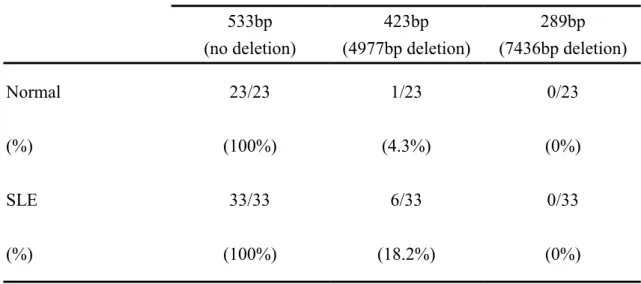
and introns. Since the redox capacity in SLE-MNC and -PMN is deficiency, increase mitochondrial DNA damage would occur especially in the sustained pre-excitation status in active SLE in vivo (9). Our results clearly demonstrated that decreased ATP production by SLE-MNC (Fig.3-A) was found due to impaired mitochondria functions with reduced mitochondrial mass (3 of 6 cases are presented in Fig.3-B), and decreased mitochondrial potential (3 of 6 cases are presented in Fig.3-C). Decreased mitochondria mass and potential, but not ATP production, were also noted in SLE-PMN (Fig.3-A, B, C). The frequency of mtDNA 4977bp, but not 7436bp, deletion in SLE mitochondria (6/33, 18.2%) is higher than control cells (1/23, 4.4%) (Fig.3-D and Table 1).
Decreased surface membrane potential and expression of Na+ and K+ ion transport-
associated molecules on SLE-MNC and -PMN
Membrane potential change is one of the early events in cell activation that requires intracellular Ca2+, K+, and Na+/H+ exchange (35, 36). We found the membrane potential of SLE-MNC and –PMN decreased compared to control cells (2 of the 4 cases are presented in Fig.4-A). This defect may derive from the decreased expression of Na+-K+-ATPase (Fig.4-B), ENaC (Fig.4-C) and ROMK1 (Fig. 4-D) on SLE cells. We deduce that the multiple membrane receptor expression defects related to cationic ion transport render SLE immune cells not only difficulty in activation but hyporesponsiveness to stimulation.
The effects of anti-dsDNA autoantibodies on GSH-Px activity and cell apoptosis of
Polyclonal anti-dsDNA autoantibodies (25~100IU/ml) significantly suppressed GSH-Px activity on normal MNC, but not PMN, after 24h incubation (Fig.5-A). The unique autoantibodies also enhanced apoptosis of normal MNC after 72h incubation (Fig.5-B) and PMN after 8h incubation (Fig.5-C). These results suggest that polyclonal anti-dsDNA autoantibodies in SLE serum are pathogenetic factors for decreasing redox capacity and subsequently elicit diverse immune abnormalities in patients with SLE.
DISSCUSSION
Systemic lupus erythematosus is characterized by the presence of a diverse spectrum of autoantibodies and autoreactive T lymphocytes in vivo. Defective cell-mediated immunity is documented in that anergic reaction of MNC to phytomitogens, specific antigens, and allogeneic/autologous lymphocyte stimulation in vitro (3-6, 8), and cutaneous reaction to common antigens in vivo (9) in active SLE patients. The innate immunity in SLE is also defective in that decreased phagocytosis in vivo and hyporesponsiveness to bacterial products and chemokine stimulators of PMN in vitro (1, 2, 4, 7). These defects in innate and adaptive immunities render SLE patients susceptible to common and opportunistic infections (1, 2). The present study aims to explore the precipitating factor(s) for the immune hyporesponsiveness of MNC and PMN in patients with active SLE. Several original observations were derived: (a) Decrease intracellular GSH concentration in MNC and PMN (Fig.2-A), and decreased GSH-Px activity (Fig.2-B), but not GSSG-R (Fig.2-D) in MNC. (b) No difference in the expression of GSH-Px isomers between SLE and normal cells in that dimer and tetramer are major isoforms in MNC whereas monomer and trimer are major isomers in PMN. (c) Defective bioenergetics in SLE-PMC with decreased ATP production (Fig.3-A), decreased glucose uptake (data not shown), and increased basal lactate level (data not demonstrated) seem due to decreased mitochondria mass (Fig.3-B) and mitochondria potential (Fig.3-C). (d) Increase mtDNA 4977bp deletion rate in SLE. (e) Defective expression of Na+ and K+ ion transport-associated molecules (Na+-K+-ATPase,
ENaC and ROMK1) (Fig.4-B, C, and D) leads to decreased membrane potential in both SLE-MNC and SLE-PMN. (f) Anti-dsDNA antibodies suppress GSH-Px activity of normal immune cells via enhancing cell apoptosis. These multiple defects in active SLE immune cells seem irrelevant to the use of glucocorticoids or immunosuppressants although 87.3% (55/63) of our patients received anti-SLE medications. For clarifying the roles of these medications on reduced redox capacity, two disease control groups were selected for comparison; (a) 8 untreated active SLE patients, and (b) 10 bronchial asthmatics with acute exacerbation and received a short-term methyprednisolone 40mg daily. We found (a) group showed a tendency of reduced redox capacity as in other treated SLE patients despite that glucocorticoids possesses a catabolic effect to facilitate the protein degradation and immunosuppressants inhibit the protein and DNA synthesis (data not shown). In addition, we noticed the abnormal redox capacity restored gradually to normal level after effective treatment. One the other side, we did not find reduced redox capacity in the methyl- prednisolone-treated BA patients. These results let us believe that the SLE disease activity
per se elicits immune cell pre-excitation in vivo and later reduces redox capacity of the cells,
but not the side of anti-SLE medications. However, more detailed and long-term dynamic study is necessary to confirm it. Among these defects of SLE immune cells, reduced redox capacity seems to be a pivotal factor for immune hyporesponsiveness since many biological molecules critically important in cell signaling and gene expression, such as ion- transporters, mitochondria-mediated Ca2+ metabolism, cytokine production (IL-1, IL-2,
IL-6, IL-8, TGF-β, TNF-α), and cell growth-related genes are sensitive to reactive oxygen species (10,17-22). Clinically, redox imbalance in some diseases is related to immune defects. The abnormal redox state in blood cells or body fluid of rheumatoid arthritis (44-46), cardiovascular disorders (47 48), and atopic asthma (43, 49), is derived from imbalanced glutathione reductase or glutathione peroxidase. The manipulation of cell redox state may provide an important strategy for the management of some forms of cancer (31). Tarp et al. (44) reported that the abnormal redox state in severe rheumatoid arthritis seems correlated with decreased selenium concentration. However, the supplementation of the element failed to restore anti-oxidative capacity in PMN of the patients. Maurice et al. (45) demonstrated the role of altered redox state in hyporesponsiveness of rheumatoid synovial T cells. Supplement of GSH with glutathione precursor, N-acetyl-L-cysteine, enhances mitogen-induced proliferative responses and IL-2 production of synovial T lymphocytes. However, patients with atopic asthma (43, 49), ischemic heart disease (47) or stroke (48) do not exhibit distinct immune hyporesponsiveness to external stimuli. Although reduced redox capacity exists in these diseases, it is clear that factors other than imbalanced redox capacity involve in T cell hyporesponsiveness in autoimmune disorders. Our finding of multiple defects in immune cells including redox capacity, bioenergetics, mitochondrial mtDNA deletion, cationic ion transport and also immune signaling (9) obviously contribute to the immune hyporesponsiveness in active SLE patients.
mechanisms. Undoubtedly, the alterations in bioenergetics may occur after long-term cell activation in SLE. In our preliminary study, elevated basal lactate level in SLE-MNC and PMN reflects increased anaerobic metabolism of the cells after sustained cell activation (data not shown). Similarly, the decreased ATP production (Fig.3-A) by SLE-MNC also reflects impaired mitochondria functions from reduced mitochondrial potential (Fig.3-B) and mitochondria mass (Fig.3-C). Mancini et al. (39) reported that paradoxical mitochondrial membrane depolarization occurs after cell apoptosis in certain cell lines. Our finding of SLE immune cell mitochondrial depolarization may suggest increased cell apoptosis by activation-induced cell death. But the real mechanism for it needs further investigation. Since mtDNA is quite vulnerable to oxidative damage from mitochondrial environment, defective redox capacity in the cells can easily impair mitochondrial functions and consequently mtDNA damage. mtDNA 4977bp deletion mutation , a common hotspot of mtDNA damage, deranges motochondrial respiratory function and bioenergy production (33). Our results suggest that 18.2% of SLE mtDNA exhibit 4977bp deletion (Table 1) that enough to shift glucose metabolism from aerobic to anaerobic pathway and increase basal lactate level in SLE cells (data not shown). Whether abnormal bioenergetics of SLE cells entirely derived from direct adverse effects of reduced redox capacity or genetic predisposition needs further investigation.
Multiple surface membrane receptor deficits such as complement receptor (type I), IgG-FcR (I, II and III), and cytokine/chemokine receptors on SLE-MNC and -PMN cause
florid immune abnormalities in these patients (50). Our finding of defective expression of Na+ and K+ transport-associated molecules (Na+-K+-ATPase, ENaC and ROMK1) on SLE-MNC and -PMN has not been reported in the literature. The Na+ and K+ ion transport determine membrane potential changes and is regarded as one of the early events in cell activation (35, 36). Increased cellular K+ triggers key metabolic pathway and initiate DNA synthesis (51). Na+-K+-ATPase directly couples the hydrolysis of ATP to the vectorial transport of 3Na+ out and 2K+ in crossing the plasma membrane to regulate transmembrane electrical potential (52). Defective expression of Na+ and K+ ion transport-associated molecules on SLE cells not only retard cell activation but cell proliferation in stimulation. Direct measurement of the intracellular concentration of [Na+] and [K+] in MNC and PMN of normal and SLE patients by patch-clump technology to measure the functions of these molecules is now under progression. Whether the multiple receptor expression defects on SLE immune cells are congenital or acquired is not solved in this study.
Many authors had demonstrated that anti-dsDNA autoantibodies are found exclusively in the serum of patients with active SLE and are clinically correlated with disease activity, and lupus nephritis. In our previous study, we reported that anti-dsDNA cross-reacts with human acidic ribosomal phosphoproteins P0, P1 and P2 expressed on different cells to exert a cytostatic effects (53). This unique autoantibody also exhibited a suppressive effect on GSH-Px activity of normal cells and accelerated cell apoptosis as shown in Fig.5-A, B and C. This novel activity of anti-dsDNA autoantibodies has not been reported in the literature.
In conclusion, we are the first authors to investigate the factors involving the immune hyporesponsiveness of active SLE patients. Reduced redox capacity, impaired mitochondrial functions and defective expression of Na+ and K+ ion transport-associated molecules are important precipitating factors for it. Anit-dsDNA autoantibodies exert a suppressive effect on GSH-Px activity and therefore, play a pathological role in immune hyporesponsiveness of patients with active SLE.
REFERENCES
1. Ginzler E, Diamond H, Kaplan D, Weiner M, Schlesinger M, Seleznick M. Computer analysis of factors influencing frequency of infection in systemic lupus erythematosus. Arthritis Rheum 1979; 21: 37-44.
2. Staples PJ, Gerding DN, Decker JL, Gorden RS. Jr. Incidence of infection in systemic lupus erythematosus. Arthritis Rheum 1994; 17:1-10.
3. Rosenthal CJ, Franklin EC. Depression of cellular-mediated immunity in systemic lupus erythematosus. Related to disease activity. Arthritis Rheum 1975; 18:207-13. 4. Landry M. Phagocyte function and cell-mediated immunity in systemic lupus erythema-
tosous. Arch Dermatol 1977; 113:147-54.
5. Suciu-Foca N, Buda JA, Theim T, Reemtsma K. Impaired responsiveness of lymphocytes in patients with systemic lupus erythematosus. Clin Exp Immunol 1974; 18:295-313.
6. Kuntz M, Innes JB, Weksler M. The cellular basis of the impaired autologous mixed lymphocyte reaction in patients with systemic lupus erythematosus. J Clin Invest 1979; 63:151-9.
7. Hsieh SC, Tsai CY, Sun KH, Yu HS, Tsai CY, Wang JC, et al. Decreased spontaneous and lipopolysaccharide stimulated production of interleukin 8 by polymorphonuclear neutrophils of patients with active systemic lupus erythematosus. Clin Exp Rheumatol 1994; 12:627-33.
8. Malave I, Layrisse Z, Layrisse M. Dose-dependent hyporeactivity to phyto- hemagglutinin in systemic lupus erythematosus. Cell Immunol 1995; 15:231-6.
9. Tsokos GC, Liossis S-N. Immune cell signaling defects in lupus: activation, anergy and death. Immunol Today 1999; 20: 119-24.
10. Sen SK. Redox signaling and the emerging therapeutic potential of thiol antioxidants. Biochem Pharmacol 1998; 55: 1747-58.
11. Sen CK, Roy S, Packer L. Involvement of intracellular Ca2+ in oxidant-induced NF-κB activation. FEBS Lett 1996; 385: 58-62.
12. Sen SK, Packer L. Anti-oxidant and redox regulation of gene transcription. FASEB J 1996; 10: 709-20.
13. Kambe F, Nomura Y, Okamoto T, Seo H. Redox regulation of thyroid-transcription factors, Pax-8 and TTF-1, is involved in their increased DNA-binding activities by thyrotropin in rat thyroid FRTL-5 cells. Mol Endocrinol 1996; 10:801-12.
14. Bindoli A, Callegaro MT, Barzon E, Benetlic M, Rigobello MP. Influence of the redox state of pyridine nucleotides on mitochondrial sulfhydryl groups and permeability transition. Arch Biochem Biophys 1997; 342: 22-8.
15. Zable AC, Fevero TG, Anramson JJ. Glutathione modulates ryanodine receptor from skeletal muscle sacroplasmic reticulum. Evidence for redox regulation of the Ca2+ release mechamism. J Biol Chem 1997; 272:7069-77.
in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothrols. J Gen Physiol 1996; 108: 277-93.
17. Kourie JI. A redox O2 sensor modulates the SR Ca2+ countercurrent through voltage and Ca 2+-dependent Cl- channels. Am J Physiol 1997; 272: C324-332.
18. Sen CK, Kolosova I, Hanninen O, Orlov SN. Inward potassium transport systems in skeletal muscle derived cells are highly sensitive to oxidant exposure. Free Radic Biol Med 1995; 18:795-800.
19. Schenk H. Vogt M, Droge W, Schulze-Osthoff K. Thioredoxin as a potent costimulus of cytokine expression. J Immunol 1996; 156:765-71.
20. Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-β1. Mol Endocrinol 1996; 10:1077-83.
21. Epsosito F, Cuccovillo F, Vanonic M, Cimino F, Anderson CW, Appella E, et al. Redox mediated regulation of p21 expression involves a post-transcriptional mechanism and activation of the mitogen activated protein kinase pathway. Eur J Biochem 1997; 245: 730-7.
22. Irani K, Xia Y, Zweier JL, Scollott SJ, Der CJ, Fearson ER, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblast. Science 1997; 275:1649-52. 23. Goldstone SD, Hunt NH. Redox regulation of the mitogen-activated protein kinase
pathway during lymphocyte activation. Biochim Biophys Acta 1997; 1355:355-60. 24. Nakamura K, Hori T, Sato N, Sugie K, Kawakami T, Yodoi J. Redox regulation of a
src family protein tyrosine kinase p56lck in T cells. Oncogene 1993; 8:3133-9.
25. Zipser Y, Piade A, Kosower NS. Erythrocyte thiol status regulates band 3 phosphotyrosine level via oxidation/reduction of band 3-associated phosphotyrosine phosphatase. FEBS Lett 1997; 406:126-30.
26. Pantopoulos K, Mueller S, Atzberger A, Ansorge W, Stremmel W, Hentze MW. Differences in the regulation of iron regulatory protein-1 (IRP-1) by extra- and intracellular oxidative stress. J Biol Chem 1997; 272: 98028.
27. McDuffee AT, Senisterra G, Huntley S, Lepock JR, Sekhar KR, Meredith MJ, et al. Proteins containing non-native disulfide bonds generated by oxidative stress can act as signals for the induction of the heat shock response. J Cell Physiol 1997; 171:143-51.
28. Viora M, Quaranta MG, Straface E, Vari R, Massella R, Malorni W. Redox imbalance and immune functions: opposite effects of oxidized low-density lipoproteins and N-acetylcysteine. Immunology 2001; 104: 431-8.
29. Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989; 264:13963-6. 30. Holmgren A. Glutathione-dependent synthesis of deoxyribonecleotide. Characterization
of the enzymatic mechanism of Escherachia coli glutaredoxin. J Biol Chem 1979; 254:3672-8.
31. Sen CK. Nutritional biochemistry of cellular glutathione. J Nutr Biochem 1997; 8: 660-72.
32. Berneburg M, Grether-Beck S, Kurten V, Ruzicka T, Brivida K, Sies H, et al. Singlet oxygen mediates the UVA-induced generation of the photoaging-associated mitochondrial common deletion. J Biol Chem 1999; 274: 15345-9.
33. Marchetti P, Decaudin D, Macho A, Zamzami N, Hirsch T, Susin SA, et al. Redox regulation of apoptosis: impact of thiol oxidation status on mitochondria function. Eur J Immunol 1997; 27:289-96.
34. Nagy G, Koncz A, Perl A. T cell activation-induced mitochondrial hyperpolarization is mediated by Ca2+- and redox-dependent production of nitric oxide. J Immunol 2003; 171: 5188-97.
35. Sachs HG, Stambroook PJ, Ebert JD. Changes in membrane potential during cell cycle. Exp Cell Res 1974; 83:362-6
36. Lazzari KG, Proto PJ, Simons ET. Simultaneous measurement of stimulus induced changes in cytoplasmic Ca2+ and in membrane potential of human neutrophils. J Biol Chem 1986; 261:9710-30.
37. Bombardier C, Gladman DD, Urowitz MB, et al. Derivation of the SLEDAI: A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum 1992; 35: 630-.
38. Marchetti P, Decandin D, Macho A, Zamzami N, Hirsch T, Susin SA, et al. Redox regulation of apoptosis: impact of thiol oxidant status on mitochondrial function. Eur J Immunol 1977; 27: 289-96
39. Mancini M, Anderson BO, Caldwell E, Sedghinasab M, Paty PB, Hockenbery DM. Mitochondrial proliferation and paradoxical membrane depolarization during terminal differentiation and apoptosis in a human colon carcinoma cell line. J Cell Biol 1997; 138; 449-69.
40. Liu CS, Kao SH, Wei YH. Smoking associated mitochondrial DNA mutations in the human hair follicle. Environ Mol Mutagen 1997; 30:47-55.
41. Shapiro MM, Natale PJ, Kamentsky LA. Estimation of membrane potentials of individual lymphocytes by flow cytometry. Proc Natl Acad Sci USA. 1979; 76: 5728-30.
42. Sun KJ, Liu WT, Tsai CY, Tang SJ, Han SH, Yu CL. Anti-dsDNA antibodies cross-react with ribosomal P proteins expressed on the surface of glomerular mesangial cells to exert a cytotoxic effect. Immunology 1995; 85:262-9.
43. Misso NLA, Peroni DJ, Watkins DN, Stewart GA, Thompson PJ. Glutathione peroxidase activity and mRNA expression in eosinophils and neutrophils of asthmatic and non-asthmatic subjects. J Leukoc Biol 1998; 63: 124-30.
44. Tarp U, Strengard-Pedersen K, Hansen JC, Thorling EB. Glutathione redox cycle enzymes and selenium in severe rheumatoid arthritis: lack of antioxidative response to selenium supplementation in polymorphonuclear leukocytes. Ann Rheum Dis 1992; 51:1044-9.
Evidence for the role of an altered redox state in hyporesponsiveness of synovial T cells in rheumatoid arthritis. J Immunol 1997; 158:1458-65.
46. Bazzichi I, Ciompi ML, Betti L, Rossi A, Melchiorre D, Fiorini M, et al. Impaired glutathione reductase activity and levels of collagenase and elastase in synovial fluid of rheumatoid arthritis. Clin Exp Immunol 2002; 20:761-6,.
47. Porter M, Pearson DJ, Suarez-Mendez VJ, Blann AD. Plasma, platelet and erythrocyte glutathione peroxidases as risk factors in ischaemic heart disease in man. Clin Science 1992; 83:343-5,.
48. Isshibashi N, Prokopenko O, Reuhl KR, Mirochnitchenko O. Inflammatory response and glutathione peroxidase in a model of stroke. J Immunol 2002; 168:1926-33.
49. Comhair SAA, Bhathena PR, Farver C, Thunnissen FBJM, Erzurum SC. Extracellular glutathione peroxidase induction in asthmatic lung: evidence for redox regulation of expression in human airway epithelial cells. FASEB J 2001; 15: 70-8.
50. Fyfe A, Holme ER, Zoma A, Whaley K. C3b receptor (CR1) expression on the polymorphonuclear leukocytes from patients with systemic lupus erythematosus. Clin Exp Immunol 1987; 67:300-8.
51. Segal GB, Lichtman MA. Potassium transport in human blood lymphocytes treated with phytohemagglutinin. J Clin Invest 1976; 58:1358-69.
52. Musch MW, Clarke LL, Mamah D, Gawenis LR, Zhang Z, Ellsworth W, et al. T cell activation causes diarrhea by increasing intestinal permeability and inhibiting
epithelial Na+/K+-ATPase. J Clin Invest 2002; 110: 1739-47.
53. Sun KH, Liu WT, Tang SJ, Tsai CY, Hsieh SC, Wu TH, et al. The expression of acidic ribosomal phosphoproteins on the surface of different tissues in autoimmune and normal mice which are the target molecules for anti-double-stranded DNA antibodies. Immunology 1996; 87: 362-71.
Table 1. Comparison of mitochondrial DNA 4977bp and 7436bp deletion in normal and SLE patients
mitochondrial DNA length mutation (deletion)
533bp (no deletion) 423bp (4977bp deletion) 289bp (7436bp deletion) Normal 23/23 1/23 0/23 (%) (100%) (4.3%) (0%) SLE 33/33 6/33 0/33 (%) (100%) (18.2%) (0%)
FIGURE LEGENDS
Fig. 1: Detection of reduced-form glutathione (GSH) level, glutathione peroxidase (GSH-Px) activity and mRNA expression, and expression of GSH-Px isomers in different normal blood cells and plasma. (A) Intracellular GSH level was compared among mononuclear cells (MNC), polymorplonuclear neutrophils (PMN), red blood cells (RBC), platelets, and plasma of normal individuals by commercial GSH Colorimetric Assay Kit. (B) Comparison of GSH-Px activity among different blood cells and plasma of normal individuals by commercial GSH-Px Colorimetric Assay Kit. (C) Detection of GSH-Px mRNA expression in MNC and PMN of two normal individuals by RT-PCR method: lane 1: G3PDH (452bp, as internal control), lane 2: GSH-Px (354bp). The same experiment was conducted in another two normal individuals with a similar tendency. (D) Detection of GSH-Px isomers expression in normal MNC and PMN by Western blot. Different volumes (2µl in lanes 1 and 4; 4µl in lanes 2 and 5; 10µl in lanes 3 and 6) of MNC (lanes 1-3) and PMN (lanes 4-6) cell lysates (protein concentration 26mg/ml) were analyzed in 10% SDS-APGE probed by anti-human GSH-Px antibody. Four protein isomers are identified as monomer (25kDa), dimer (50kDa), trimer (75kDa) and tetramer (100kDa).
(GSH-Px) activity, expression of GSH-Px isomers, and glutathione reductase (GSSG-R) activity in MNC and PMN of normal and SLE patients. (A) GSH level in both SLE-MNC and -PMN, is significantly lower than normal group. (B) The GSH-Px activity of SLE-MNC was significantly lower than normal MNC whereas no difference between SLE-PMN and normal PMN. (C) Western blot analysis of GSH-Px isomer expression in two MNC and PMN of normal and SLE patients reveals both MNC contain mainly dimer (50kDa) and tetramer (100kDa), and less monomer (25kDa) and trimer (75kDa). In contract, both PMN contain mainly monomer and trimer, and dimmer and tetramer. Lane 1 and lane 2 are different cases of PMN. Lane 3 and lane 4 are different cases of MNC. (D) Glutathione reductase (GSSG-R) activity in MNC and PMN was not different in normal and SLE groups detected by commercial GSSG-R Colorimetric Assay Kit.
Fig. 3: Comparison of bioenergetics in MNC and PMN of normal and SLE patients. The defective bioenergetics in SLE-MNC and -PMN was reflected by decreased ATP production that is caused by reduced mitochondrial mass, decreased mitochondrial potential, and increased rate of mitochondrial DNA 4977bp deletion. (A) Intracellular ATP level in SLE-MNC, but not SLE-PMN, was significantly lower than normal MNC. (B) Decreased mitochondrial mass in both
MNC and PMN of three SLE patients was shown compared to normal by flow cytometry. The same experiment was conducted in another 3 normal and 3 SLE patients with a similar tendency. (C) Reduced mitochondrial potential in both MNC and PMN of three SLE patients was shown compared to normal ells by flow cytometry. The same experiment was conducted in another 3 normal and 3 SLE patients and showed a similar tendency. (D) Detection of mitochondrial DNA 4977bp and 7436bp deletion in 3 normal and 4 SLE patients by PCR amplification products. The appearance of 533bp product indicates no deletion (lane 1) whereas appearance of 423bp (lane 2) and 289bp (lane 3) suggest deletion of 4977bp and 7436bp, respectively. The deletion rate of mtDNA in 23 normal and 33 SLE is demonstrated in Table I.
Fig. 4: Comparison of membrane potential changes and expression of Na+ and K+ ion transport-associated molecules on MNC and PMN of 2 normal and 2 SLE patients by flow cytometry. (A) Decreased membrane potential of both SLE-MNC and -PMN was noted compared to normal cells. The same experiment was conducted in another 2 normal and 2 SLE and showed a similar tendency. The decreased membrane potential in SLE cells is the combination results of defective expression of Na+-K+-ATPase (B), epithelial sodium channel (ENaC) (C) and renal outer medullary potassium channel 1 (ROMK1) (D) on SLE cells.
Fig. 5: The effects of polyclonal anti-dsDNA autoantibodies (anti-dsDNA) purified from active SLE sera on GSH-Px activity and cell apoptosis of normal MNC and PMN. (A) Suppression of GSH-Px activity on both MNC and PMN of normal individuals is found by anti-dsDNA (100IU/ml), but not by human non-specific IgG (IgG, 10mg/ml), after incubation for 24 h. (B) Anti-dsDNA (100IU/ml) induced much more MNC apoptosis after 72h and 120h incubation than medium or non-specific human IgG (10mg/ml) (C) Anti-dsDNA (100IU/ml) induced much more PMN apoptosis after 8h incubation than medium or non-specific human IgG (10mg/ml). The arrows indicate MNC and PMN with karyopyknotic/karyorrhexic nuclei, a typical change of cell apoptosis (original magnification 1000x in all pictures).
![HPSH [ 氧化數平衡反應式係數 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)