封面
國
立
交
通
大
學
應用化學系
碩
士
論
文
以紅外光
–真空紫外光游離光譜法研究
甲硫醇團聚體之紅外吸收光譜
研
究 生:傅 龍(Lung Fu)
指導教授:李遠鵬
教授(Prof. Yuan-Pern Lee)
書名頁
以紅外光
–真空紫外光游離光譜法研究
甲硫醇團聚體之紅外吸收光譜
Infrared Spectra of Methanethiol Clusters (CH
3SH)
nInvestigated with
the Infrared Depletion and Vacuum-ultraviolet Ionization Technique
研 究 生:傅 龍 Student:Lung Fu
指導教授:李遠鵬 Advisor:Yuan-Pern Lee
國 立 交 通 大 學 應 用 化 學 系
碩 士 論 文
A Thesis Submitted to M. S. Program, Department of Applied Chemistry
College of Science National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master in
Applied Chemistry June 2012
Hsinchu, Taiwan, Republic of China
i
摘要
吾人以紅外光削減–真空紫外光游離光譜法研究了甲硫醇團聚體 ((CH3SH)n,n = 2–5)之紅外吸收光譜。利用四波混頻產生之 132.5 nm 真 空紫外光游離分子射束中的甲硫醇團聚體,搭配飛行時間質譜儀可分別偵 測到不同大小團聚體之離子訊號。實驗時以可調變波長的OPO/OPA 雷射 作為紅外光源,掃描2470−2670 cm−1(S–H 伸張模光區)及 2800−3100 cm−1 (C–H 伸張模光區)之波長。若團聚體在被游離前吸收紅外光會造成預解 離,使其濃度減少,其相對應之離子訊號也會減少。記錄離子訊號隨波長 之變化可得到各團聚體之作用光譜,而根據分子射束中團聚體之分布以及 團聚體中分子間束縛能等資訊,可將作用光譜轉換為紅外吸收光譜。 甲硫醇單體之 S–H 伸張模(ν3)譜帶位於 2605 cm−1,而雙聚體之ν3譜 帶位於2601 cm−1,相較於單體僅有 4 cm−1之紅位移,顯示甲硫醇雙聚體間 應不以氫鍵鍵結。此外,在具氫鍵之甲醇雙聚體光譜中可觀察到因質子施 體與質子受體而造成各吸收峰分裂的現象,在本實驗中並無觀察到,亦為 甲硫醇雙聚體間不以氫鍵鍵結之證據。另一方面,三聚體、四聚體及五聚 體之ν3譜帶皆位於 2567 cm−1,相較於單體有38 cm−1之紅位移,吸收強度 也較單體強,顯示這些團聚體中甲硫醇分子間應以氫鍵互相鍵結。本實驗 結果證實了前人從理論計算結果得出甲硫醇雙聚體最穩定之結構並無氫鍵 鍵結,而三聚體皆為氫鍵鍵結之環狀結構的結論。ii
Abstract
We investigated IR spectra in the CH- and SH-stretching regions of size-selected methanethiol clusters, (CH3SH)n with n = 2−5, in a pulsed
supersonic jet by using the infrared (IR)-vacuum ultraviolet (VUV) ionization technique. VUV emission at 132.5 nm served as the source of ionization in a time-of-flight mass spectrometer. The tunable IR laser emission served as a source of predissociation. The variations of intensity of methanethiol cluster ions (CH3SH)n+ and CH3SH+ were monitored as the IR laser light was tuned across
the range 2470−3100 cm−1. In the SH-stretching region, the spectrum of
(CH3SH)2 shows a weak band near 2601 cm−1, only 4 cm−1 red-shifted from that
of the monomer. In contrast, all spectra of (CH3SH)n, n = 3−5, show a broad
band near 2567 cm−1 with much greater intensity. In the C−H stretching region,
absorption bands of (CH3SH)2 are locate near 2865, 2890, 2944, and 3010 cm−1,
red-shifted by 3−5 cm−1 from those of CH3SH. These red shifts increase slightly
for higher clusters and bands near 2856, 2884, 2938, and 3005 cm−1 were
observed for (CH3SH)5. The results indicate that the S−H···S hydrogen bonding
exists in clusters with n = 3−5, but not in (CH3SH)2, in agreement with
theoretical predictions. The absence of a band near 2605 cm−1 might indicate that the dominant stable structures of (CH3SH)n, n = 3−5, have cyclic
iii
謝誌
當年高三來新竹推甄面試的回憶如今還挺鮮明,轉眼間已是碩士論文 口試;口試結束的當下並沒有多少畢業的感覺,如今撰寫謝誌時,交大六 年的種種回憶突湧上心頭。 非常感謝李遠鵬老師的指導,從您那裡學到的不只是專業知識,還有 邏輯思考的能力,每次聽您講道理時雖都看您一派輕鬆,但對我來說句句 都受用無窮。另外,也要感謝大四升碩一暑假在溫哥華UBC 的指導教授 Ed Grant,那兩個月是非常難忘的回憶,在一流大學的實驗室學習,著實讓 我有見了世面的感覺。 感謝辛苦架設IR-VUV 系統的小韓學姊,因為妳的細心指導我才能順 利完成實驗,相信妳將來一定會是一位偉大的學者。感謝一起共同奮鬥了 快四年的林震洋與林書毓,尤其是坐我旁邊,得忍受我種種無理取鬧與無 聊玩笑的林震洋,我們培養出的深刻默契是無可取代的。感謝實驗室的夥 伴們,建亨、月貴、Momo、Prasanta、Barbara、Megan 學姊、俞範學姐、 棋文學長、皇上學長、菜哥、吳振宇、Wade、蘇育德、陳威宇、陳奕安。 感謝應化99 的大家,你們帶給我太美好的大學回憶。也要感謝不一定會看 到此篇謝誌的高中死黨們,期許十年後的我們都能小有成就。iv 團,你們的音樂帶給了我莫大的動力。 謝謝女朋友昀瑄,不只持續給我鼓勵,也稱職的扮演我的Office 小幫 手,助我順利完成論文。 最後感謝我的家人們,他們是我最大的支柱;最關心我進度,不時給 我方向的爸爸、每周末回家都準備佳餚的媽媽,以及時常與我交換文藝與 音樂資訊的弟弟,謹將此小小成就與你們分享。 傅龍 2012 年 夏
v
目錄
摘要 ... i Abstract ... ii 謝誌 ... iii 目錄 ... v 表目錄 ... vii 圖目錄 ... viii 第一章 緒論 ... 1 參考文獻 ... 11 第二章 實驗原理與技術 ... 13 2.1 紅外光–真空紫外光游離光譜技術簡介 ... 13 2.2.1 真空紫外光游離偵測–紅外光預解離技術(VUV-ID-IRPDS) ... 15 2.2.2 紅外光−真空紫外光游離光譜法(IR−VUV−PIS) ... 16 2.2 真空紫外光光源 ... 17 2.3 光參量共振與光參量放大技術 ... 21 2.3.1 光學參量震盪器 ... 21 2.3.2 光學參量放大 ... 25 2.4 分子射束法 ... 26 2.5 直線型飛行時間質譜儀 ... 30 參考文獻 ... 43 第三章 實驗裝置 ... 47 3.1 儀器架設 ... 47 3.1.1 真空紫外光光源 ... 47 3.1.2 可調式紅外光光源 ... 49 3.1.3 分子射束系統 ... 53 3.1.4 飛行時間質譜儀裝置 ... 54 3.1.5 訊號偵測及時序控制 ... 56vi 3.2 實驗步驟 ... 57 3.2.1 樣品配置 ... 57 3.2.2 對光步驟 ... 58 3.2.3 光譜擷取 ... 59 參考文獻 ... 71 第四章 結果與討論 ... 73 4.1 理論計算 ... 73 4.2 甲硫醇團聚體飛行時間質譜圖 ... 75 4.3 控制分子射束中團聚體分布 ... 79 4.4 甲硫醇及其團聚體紅外光吸收光譜之推導 ... 80 4.5 光譜指派與比較 ... 84 4.5.1 甲硫醇單體 ... 84 4.5.2 甲硫醇雙聚體 ... 86 4.5.3 甲硫醇三聚體 ... 89 4.5.4 甲硫醇四聚體及五聚體 ... 89 4.6 結論 ... 90 參考文獻 ... 114
vii
表目錄
表 1-1 理論計算得到甲硫醇雙聚體與三聚體的 SH 伸張模相對於單體之紅位移及吸收 強度(參考文獻25 中 Table 3、5、6),各結構之作用能亦列於表中。 ... 10 表 4-1 實驗中觀測到甲硫醇單體及團聚體 ν3之吸收與理論計算結果之比較。 ... 110 表 4-2 本實驗所觀測到不同大小之甲硫醇團聚體在 C–H 伸張模光區之吸收與理論計算 結果之比較(其中振動頻率單位為cm−1,吸收強度單位為km mol−1)。 ... 111 表 4-3 甲硫醇單體、雙聚體、三聚體及四聚體中,質荷比為 M+1 及 M+2 之質譜訊號 積分面積相對於質荷比為M 之質譜訊號積分面積之比例,實驗所得之值(Expt.) 以及依碳及硫原子同位素比例計算出之值(Calc.)皆列於表中。比例小於 0.01% 之離子未列於表中。 ... 112 表 4-4 前人理論計算所得之甲硫醇團聚體解離能(dissociation energy)(單位為 eV)。viii
圖目錄
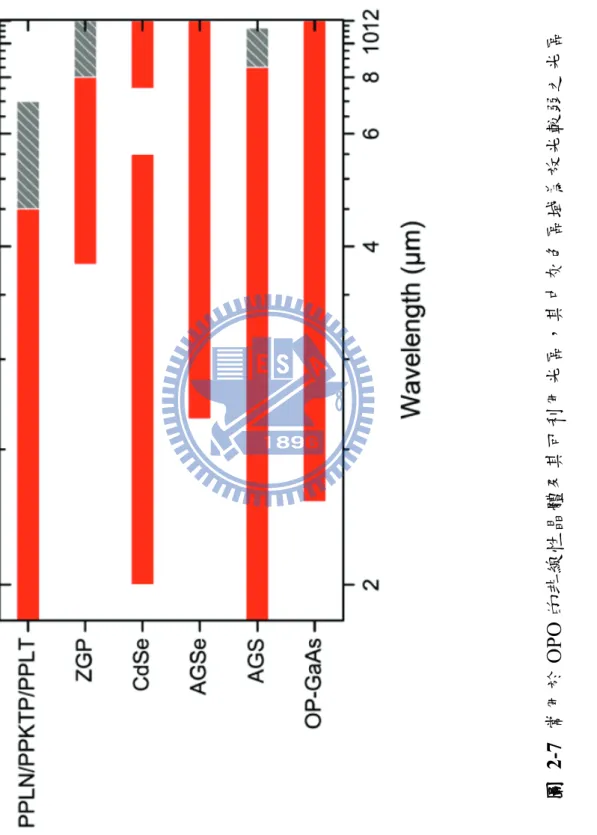
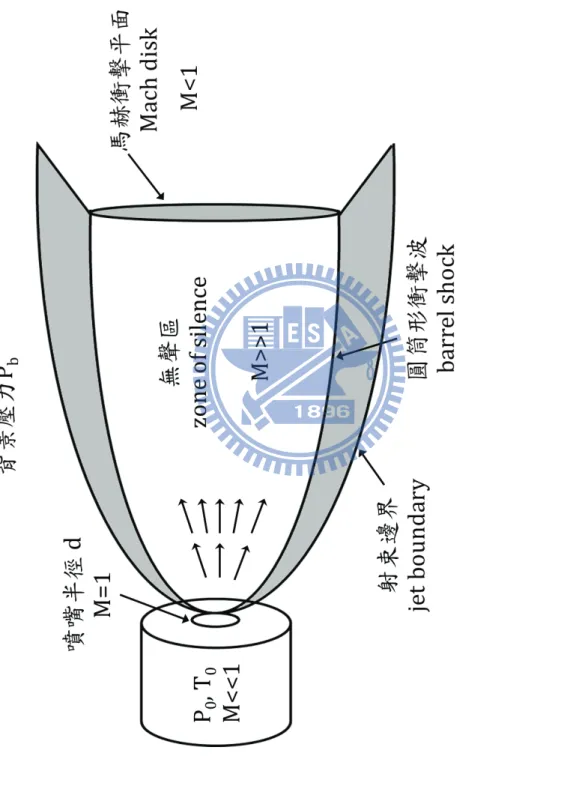
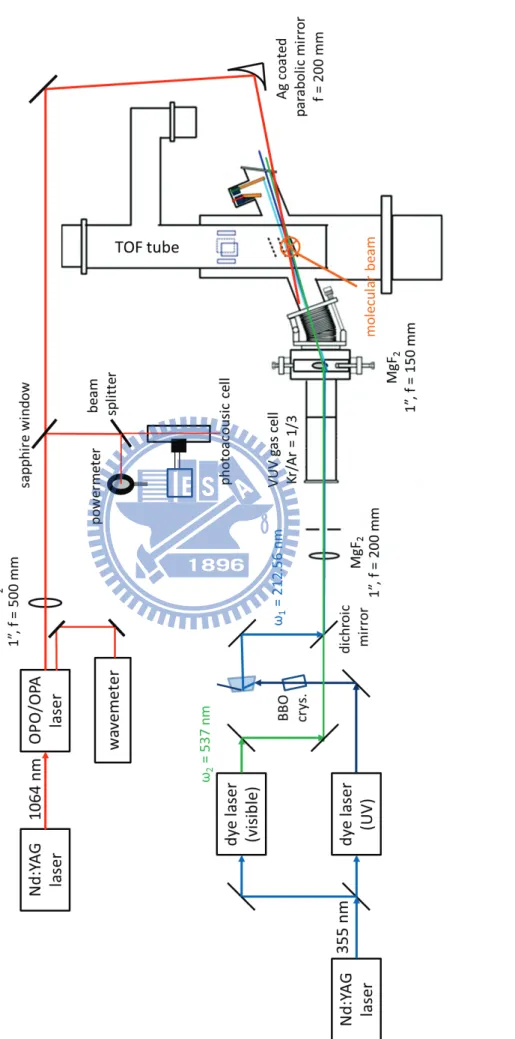
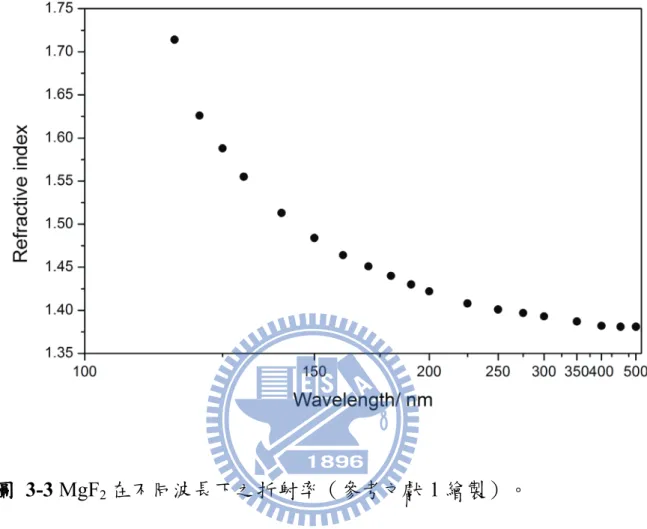
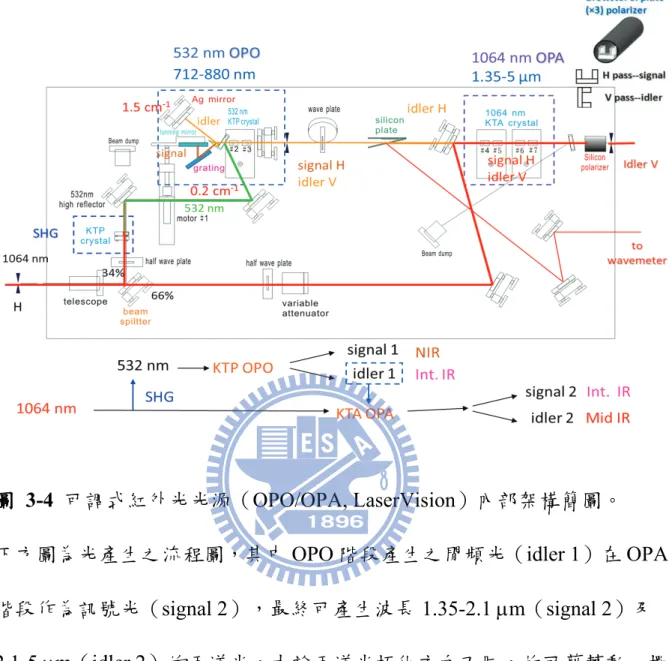
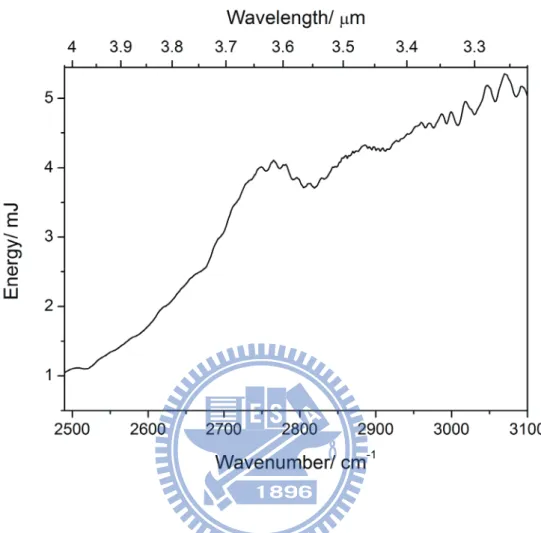
圖 1-1 Bakó和 Pálinkás 經理論計算所得之兩種甲硫醇雙聚體穩定結構(摘自參考文獻 25)。 ... 8 圖 1-2 Cabaleiro-Lago 和 Rodríguez-Otero 理論計算所得最佳化之甲硫醇團聚體結構。 . 9 圖 2-1 兩種不同的 IR−VUV 光游離偵測機構。 ... 33 圖 2-2 共振四波和頻(ω𝑉𝑈𝑉 = 2ω1+ ω2)及差頻(ω𝑉𝑈𝑉 = 2ω1− ω2)之示意圖。 ... 34 圖 2-3 相位匹配參數隨 bΔk 值的變動(假設b/L=0,𝑓/𝐿 = 0.5)(參考文獻 28 繪製)。 ... 35 圖 2-4 使用 Hg, Xe, Kr, Ar 等非線性氣體作共振加強四波和頻或差頻之能階示意圖,及 可產生的真空紫外光範圍(參考文獻27 繪製)。 ... 36 圖 2-5 一些常見利用非線性晶體產生可調變光源的示意圖。 ... 37 圖 2-6 OPO 及 OPG 中相位匹配之示意圖(參考文獻 34 繪製)。 ... 38 圖 2-7 常用於 OPO 的非線性晶體及其可利用光區,其中灰色區域為放光較弱之光區 (摘自參考資料33)。 ... 39 圖 2-8 OPA 中相位匹配之示意圖(參考文獻 34 繪製)。 ... 40 圖 2-9 超音波射束之示意圖。 ... 41 圖 2-10 雙電場加速直線型飛行時間質譜儀示意圖。 ... 42 圖 3-1 IR-VUV 光游離光譜法裝置簡圖。 ... 61 圖 3-2 真空紫外光光源裝置簡圖。 ... 62 圖 3-3 MgF2在不同波長下之折射率(參考文獻1 繪製)。 ... 63 圖 3-4 可調式紅外光光源(OPO/OPA, LaserVision)內部架構簡圖。 ... 64 圖 3-5 以 520 mJ 之 Nd:YAG雷射激發 OPO/OPA系統後所能得到的紅外光能量示意圖。 ... 65 圖 3-6 在本實驗掃描光區內測得之紅外光能量。 ... 66 圖 3-7 紅外光光徑及光聲效應訊號擷取示意圖,光聲效應使用之電路亦示於圖中。 . 67 圖 3-8 Even-Lavie 分子射束閥及相關裝置(驅動器、電源供應器、數位脈衝產生器) 接線示意圖 ... 68 圖 3-9 飛行時間質譜儀架構圖,其中 MCP 偵測器內部架構圖示於左上方。 ... 69 圖 3-10 實驗中使用 BNC575 產生之觸發脈衝時脈示意圖。 ... 70 圖 4-1 甲硫醇分子(CH3SH)之結構。 ... 92 圖 4-2 MP2/aug-cc-pVDZ 理論計算所得之五種雙聚體之穩定結構。 ... 93ix 圖 4-3 Cabaleiro-Lago 及 Rodríguez-Otero 理論計算所得之五種三聚體穩定結構(摘自參 考文獻3)。 ... 94 圖 4-4 甲硫醇分子射束飛行時間質譜圖 ... 95 圖 4-5 使用 9.4520 eV 真空紫外光作為游離光源所得到的質譜圖。 圖中可觀察到質荷 比M+1 及 M+2 之訊號,各波峰皆以 Gauss 函數適解,以計算其面積比例。 ... 96 圖 4-6 觀察質譜中質荷比為 48、49 及 50 的訊號隨真空紫外光能量增強之變化。 ... 97 圖 4-7 使用不同分子射束閥驅動電流所觀測到的質譜圖。 ... 98 圖 4-8 比較不同條件下團聚體離子訊號之大小。 ... 99 圖 4-9 控制分子射束中最大物種為雙聚體(文中條件 A)所取得之 (A)飛行時間質譜 圖,(B)紅外光作用光譜。飛行時間質譜圖中標示*之離子為殘留在分子射束閥或 飛行時間質譜儀中的二甲基二硫(DMDS,m/z = 94)。 ... 100 圖 4-10 控制分子射束中雙聚體佔 80 %以上,且最大物種為五聚體(文中條件 B)所 取得之 (A)飛行時間質譜圖,(B)紅外光作用光譜。 ... 101 圖 4-11 控制分子射束中最大物種約為二十聚體(文中條件 C)所取得之 (A)飛行時間 質譜圖,(B)紅外光作用光譜。 ... 102 圖 4-12 比較條件 B 中之實線為四聚體之作用光譜,虛線為若假設五聚體全部解離為四 聚體,照質譜圖中之訊號大小比例(1:0.25)將五聚體之作用光譜與四聚體之 作用光譜相加所得之光譜。 ... 103 圖 4-13 不同條件下雙聚體作用光譜之比較。 ... 104 圖 4-14 比較五聚體、六聚體、七聚體及八聚體在 C–H 伸張吸收光區之作用光譜。 105 圖 4-15 甲硫醇及其團聚體((CH3SH)n,n = 2–5)之紅外吸收光譜。 ... 106 圖 4-16 以 IR-VUV 光譜法取得之 CH3SH 紅外吸收光譜。以 SpecView 軟體對 ν3及ν2 振動模模擬所得之光譜亦列於圖中。 ... 107 圖 4-17 甲硫醇分子在室溫下之氣態 FT-IR 光譜。 ... 108 圖 4-18 三聚體、四聚體及五聚體在 S–H 伸張吸收光區光譜之比較。 ... 109
1
第一章
緒論
科學家一直都對分子團聚體(cluster)的研究有很大的興趣,因為了解 團聚體之特性有助於了解分子在固態或液態下之作用力。團聚體中單體間 通常靠微弱的凡得瓦力(van der Waals force)或較強的氫鍵(hydrogen bond) 相吸引,其大小範圍從兩個單體大至數千、數萬個單體。光譜學的研究通 常最能了解團聚體的結構與性質,隨著超音波分子射束(supersonic molecular beam)及雷射技術的進步,科學家發展出許多具有團聚體大小選 擇性(size-selectivity)的光譜技術 [1–4],而得以從光譜的變化研究從小 團聚體發展至大團聚體時結構排列與鍵結強度等等變化。這些在氣態無碰 撞(collision-free)條件下所獲得的實驗結果有助於從微觀分子角度來了解 分子間(inter-molecular)或分子內(intra-molecular)的作用,進而解釋巨 觀下凝態分子的種種現象。 在團聚體的研究中以含有氫鍵之團聚體研究最為廣泛 [5– 9]。氫鍵為許 多凝態分子,如:水、醇類等,重要的分子間作用力。氫鍵也在生物學中 扮演舉足輕重的角色,構成DNA雙股螺旋的鹼基透過氫鍵配對,生物體內 的各種高分子也透過氫鍵形成二級、三級或四級結構。近年來有許多研究 組結合超音波射束及多樣的雷射光譜技術研究DNA鹼基、胺基酸及其團聚 體,並得到氣態下的振動光譜、游離能等等資訊 [10,11]。
2 氫鍵發生在已經以共價鍵與X原子鍵合的氫原子與另一個原子Y之間 (X–H…Y),其中X及Y原子通常都是電負度較強的原子,如氮(N)、 氧(O)及氟(F)等等。電負度相對較弱的原子,如硫(S)、氯(Cl) 也可能形成氫鍵,但鍵能較低。就硫而言,硫氫基(sulfhydryl group)是 否能形成氫鍵曾是具有爭議的 [12]。凝固點降低測定法(cryoscopy)無法 觀察到硫醇類分子具有其他含氫鍵之分子間會發生的自結合 (self-association)現象 [13],而硫醇類的沸點相對於醇類低很多,亦為硫 醇類分子間作用力較弱之證據。然而,隨著紅外光譜法和核磁共振光譜法 (nuclear magnetic resonance spectroscopy, NMR)技術的漸臻成熟,再加上 計算化學的興起,科學家陸續提出許多有力證據來證實S–H…S類型氫鍵的 存在。以結構和水分子相仿的硫化氫(hydrogen sulfide, H2S)為例,間質 隔離(matrix isolation)光譜中可以觀測到以氫鍵鍵結的硫化氫雙聚體(dimer) ((H2S)2)之振動吸收 [14],科學家亦利用理論計算所得之結構和束縛能 等資訊證實硫化氫分子間會形成氫鍵 [15,16]。除了S–H…S類型的氫鍵, 科學家近年也以光譜技術證實O–H…S類型的氫鍵 [17]與N–H…S類型氫 鍵 [18]的存在;此兩種類型的氫鍵常見於生物體內含硫的分子,如半胱胺 酸(cysteine)及蛋胺酸(methionine)這兩種胺基酸,與體內其他分子的 鍵結 [19],故研究這些含硫的氫鍵有助於我們了解人體內蛋白質的摺疊機
3 制。硫氫氫鍵的重要性早已不可同日而語,是故越來越多的研究組投入研 究各種含硫之氫鍵。 紅外光譜法為辨別氫鍵是否存在最有力的方法之一,參與氫鍵鍵結的 官能基,其振動吸收峰頻率通常都會有明顯的紅位移(red-shift),亦即其 頻率會較無氫鍵鍵結時為低,且吸收強度較無氫鍵鍵結時的強度增加。但 是相較於O–H 及 C–H 等等伸張躍遷而言,由於 S–H 伸張躍遷通常為較弱 的吸收,所以需要雷射光譜法等具有較高靈敏度的技術來研究硫醇分子的 光譜。 本論文研究的分子為硫醇(thiols)類中分子最小的甲硫醇(methanethiol, CH3SH)。甲硫醇存在於人類與其他動物的血液、大腦,以及植物組織中 [20],但相較於與其結構類似的甲醇(methanol, CH3OH),有關於甲硫醇 及其團聚體的研究非常的少。 Barnes等人在間質隔離光譜中除了觀測到甲硫醇單體在 2603 cm−1之吸 收外,亦觀測到甲硫醇的團聚體吸收 [21],並將在 2576 cm−1的吸收指派 為甲硫醇開環雙聚體(open-chain dimer)之吸收。他們並藉由和甲醇團聚 體的光譜比較紅位移,將2550 cm−1的吸收指派為環狀四聚體(cyclic tetramer)之吸收。
Odutola等人利用分子束電場偏轉(molecular beam electric deflection, MBED)技術發現甲硫醇雙聚體與三聚體(trimer)都為極性分子 [22],相
4 對於甲醇雙聚體為極性,而三聚體而非極性,可以推論甲硫醇三聚體與甲 醇三聚體有不同的分子排列結構。其中甲醇三聚體的結構已經得到實驗及 理論計算的驗證為環狀結構,且所有O−H…O的氫鍵都位於同一平面上, 因此為非極性分子 [6]。 理論計算方面,Pecul和Janoschek曾使用SCF-MO-LCGO (self-consistent field-molecular orbital-linear combination of Gaussian orbitals)計算方法計算 出甲硫醇雙聚體之結構 [23],並計算出其相對於甲硫醇單體之作用能 (interaction energy)為−5.9 kJ mol−1。此外,他們也模擬出甲硫醇雙聚體 在溫度為20 K時,S–H伸張模區域的振動光譜,其中雙聚體的吸收為 2312 cm−1,相較於計算出的單體吸收 2324 cm−1有12 cm−1的紅位移。他們認為, 雖然計算出之振動頻率與Barnes等人 [21]得到的光譜間有約 280 cm−1之差 距,但整體而言光譜中吸收譜帶之分布相類似。 近十年來,隨著電腦硬體以及計算方法的進步,科學家可以利用更複 雜且更完善的方法來進行理論計算,科學家也發現早期一些利用簡單計算 方法得出的結果往往都與實驗值有很大的偏差。Sum和Sandler曾利用 HF/6-31G**計算法得出甲硫醇雙聚體、三聚體以及四聚體(tetramer)的最 佳化結構 [24],並利用更高階的MP2 計算方法搭配aug-cc-pVDZ基底函數 組算出團聚體的作用能、團聚體形成的平衡常數等等資訊。他們將甲硫醇 的計算結果和甲醇、乙醇以及正丙醇的結果比較,指出甲硫醇沒有表現如
5
醇類的協同現象(cooperativity effect),而得出甲硫醇間不會形成氫鍵的 結論。其後,有兩個研究組對甲硫醇團聚體進行理論計算研究而得到和Sum 和Sandler不同的結果 [25,26]。Bakó和Pálinkás [25]利用B3LYP密度泛函理 論(density function theory)及MP2 計算方法搭配 6−31+G**或 6−311+G** 基底函數組對甲硫醇雙聚體結構做最佳化,得出兩種不同的甲硫醇雙聚體 穩定結構,示於圖1-1。其中一種結構(T1)與Sum和Sandler [24]得到的結 構相類似,單體間具有S−H···S形式的鍵結,而以MP2/6−311+G**計算出之 作用能為−2.3 kJ mol−1。在Bakó和Pálinkás得到的另外一種結構(T2)中, 分子間不具有S−H···S形式的鍵結。T2 結構中兩個分子的CSH平面互相垂 直,雖然不具備類似氫鍵鍵結的結構,但其作用能卻較強,以 MP2/6−311+G**計算出之作用能為−4.4 kJ mol−1。他們以B3LYP計算方法算 出T1 結構在SH伸張模的紅位移有約 50 cm−1,使用MP2 計算方法算出之紅 位移也有約20 cm−1;但在兩種計算方法中,T2 結構的紅位移只有約 1–4 cm−1,顯示了S−H…S形式的鍵結對紅位移大小的影響。 另一方面,Cabaleiro-Lago和Rodríguez-Otero指出Sum和Sandler [24]直 接使用HF/6-31G**最佳化所得的結構來進行MP2 運算得出的結果不盡精 確。他們認為HF計算方法沒有考慮分子間的分散作用力(dispersion interactions),而在鍵結較弱的團聚體中,分散作用力往往是影響其結構 的重要因子,若使用HF等較低階的計算方法會得到過短的分子間距離。事
6 實上,對甲硫醇團聚體這類弱鍵結的分子來說,最少需要MP2,甚至CCSD(T) 等級的高階計算方法並搭配一定程度大小的基底函數組才能夠得到較準確 的結構 [27]。Cabaleiro-Lago和Rodríguez-Otero使用MP2 計算方法搭配 aug-cc-pVDZ基底函數組對甲硫醇雙聚體和三聚體做結構最佳化,並在雙聚 體和三聚體的位能面上分別找到了五個不同結構的能量最低點,這些不同 的穩定結構分別圖示於圖1-2 (A)和圖 1-2 (B)。他們所計算出雙聚體的五種 結構(圖1-2 (A)中 2A–2E)裡只有兩種結構具有氫鍵鍵結(2A及 2E結構), 而最穩定(作用能最強)的結構2B則不具有氫鍵鍵結的結構,其作用能為 −11.2 kJ mol−1,較其他四種結構強約0.7–2.8 kJ mol−1不等。此外他們指出, 在甲醇雙聚體中可以觀察到質子施體(proton donor)甲醇分子的O–H鍵鍵 長因氫鍵鍵結而增加,但在甲硫醇雙聚體中卻沒有觀察到S–H鍵的增長。 更重要的是,在S–H伸張模振動波數的理論計算中,並沒有在最穩定的 2B 結構之吸收頻率觀察到明顯的紅位移,僅在具有氫鍵鍵結的2A及 2E中分 別觀察到55 cm−1及41 cm−1之紅位移(參見表 1-1)。至於甲硫醇三聚體 而言,該實驗組計算出的五種穩定結構(圖1-2 (B)中 3A–3E)皆具有S–H…S 鍵結,且偶極矩皆不為0,與MBED實驗結果吻合 [22]。其中具有最強作 用能的結構為3C,其作用能為−28.1 kJ mol−1。而在三聚體的振動波數計算 方面,五種穩定結構在S–H伸張模皆有明顯的紅位移,在 67–87 cm−1之間, 且吸收峰的強度較單體增加了30 倍以上(參見表1-1)。基於這些計算結
7 果,Cabaleiro-Lago和Rodríguez-Otero認為甲硫醇雙聚體不具有氫鍵鍵結, 而三聚體具有氫鍵鍵結。 至今仍無實驗組發表氣態下甲硫醇團聚體之光譜,無法證實理論計算 之結果。對團聚體之光譜研究而言,使用超音波分子射束雖有利於團聚體 的生成,亦可以簡化振動光譜,但在分子射束中通常都會有多種不同大小 之團聚體存在,這些團聚體通常在光譜中之吸收區域相近,使得光譜重疊 難以辨析。飛行時間質譜儀(time-of-flight mass spectrometer, TOF-MS)可 藉由飛行時間的差異分離游離後不同質荷比(質量和帶電荷的比值)的離 子,搭配靈敏度高的雷射光譜技術,為研究團聚體適合之選擇。本實驗室 先前已成功利用紅外光−真空紫外光游離光譜技術搭配飛行時間質譜儀研 究小型甲醇團聚體在2650–3750 cm−1光區之吸收,並依照其解離機構以作 用光譜推導出其紅外吸收光譜 [28]。從吸收光譜可證實甲醇雙聚體結構為 O–H…O氫鍵直線排列的的開環結構(open-chain structure),而三聚體、 四聚體、五聚體(pentamer)及六聚體(hexamer)皆為環狀結構。在本論 文中,吾人將同樣的技術應用於甲硫醇團聚體上,首次觀測到(CH3SH)n(n = 1–5)在氣態中的光譜,光譜顯示雙聚體並不具氫鍵鍵結,而三、四、五 聚體皆為具氫鍵鍵結之結構。
8
ΔE = −2.3 kJ mol−1 ΔE = −4.4 kJ mol−1
圖 1-1 Bakó和 Pálinkás 經理論計算所得之兩種甲硫醇雙聚體穩定結構(摘 自參考文獻25)。
該實驗組以MP2/6−311+G**方法計算出之作用能(相對於單體之能量)列 於結構圖下方。
9 (A) (B) 圖 1-2 Cabaleiro-Lago 和 Rodríguez-Otero 理論計算所得最佳化之甲硫醇團 聚體結構 (A)甲硫醇雙聚體,(B)甲硫醇三聚體(摘自參考文獻 26)。以 MP2/aug-cc-pVDZ 計算方法所得之作用能標示於各結構上方。
ΔE = −9.6 kJ mol−1 ΔE = −11.2 kJ mol−1
ΔE = −8.4 kJ mol−1
ΔE = −10.5 kJ mol−1 ΔE = −10.3 kJ mol−1
ΔE = −24.8 kJ mol−1 ΔE = −28.1 kJ mol−1
ΔE = −28.1 kJ mol−1
10 表 1-1 理論計算得到甲硫醇雙聚體與三聚體的 SH 伸張模相對於單體之紅 位移及吸收強度(參考文獻26 中 Table 3、5、6),各結構之作用能亦列 於表中。 a 相對於單體之波數位移,負值代表紅位移。 b 吸收強度相較於單體之倍數。 B3LYP/aug-cc-pVDZ MP2/aug-cc-pVDZ ΔE (kJ) Δω (cm−1)a I/I 0b ΔE (kJ) Δω (cm−1)a I/I0b 2A −4.9 −64.6 2.8 23.3 0.4 −9.6 −54.6 −6.6 79.1 0.2 2B −3.9 −7.6 −6.8 2.1 0.5 −11.2 −6.1 −4.3 2.7 0.5 2C −3.3 −1.7 −1.7 1.9 0.0 −8.4 −4.6 −4.5 0.0 1.9 2D −3.7 −4.4 −4.1 0.2 1.7 −10.5 −2.8 −2.1 0.4 0.7 2E −4.7 −62.4 2.8 23.5 0.6 −10.3 −40.7 −6.2 30.9 1.4 3A −11.4 −73.2 −67.0 −67.0 0.0 32.7 32.7 −24.8 3B −13.0 −85.3 −75.6 0.1 33.8 42.5 −0.5 −28.1 3C −13.5 −87.2 −67.5 −58.8 24.6 27.0 27.3 −28.1 3D −11.7 −70.4 −63.2 −1.8 19.2 31.2 1.1 −26.8 3E −12.3 −86.7 −75.3 −2.4 32.2 37.8 0.6 −26.9
11
參考文獻
[1] T. S. Zwier, Annu. Rev. Phys. Chem. 47, 205 (1996). [2] B. Brutschy and P. Hobza, Chem. Rev. 100, 3861 (2000). [3] J. M. Lisy, J. Chem. Phys. 125, 132302 (2006).
[4] Y. Matsuda, N. Mikami, and A. Fujii, Phys. Chem. Chem. Phys. 11, 1279 (2009).
[5] T. Ebata, A. Fujii, and N. Mikami, Int. Rev. Phys. Chem. 17, 331 (1998). [6] U. Buck and F. Huisken, Chem. Rev. 100, 3863 (2000).
[7] R. Ludwig, Angew. Chem. Int. Edit. 40, 1808 (2001).
[8] A. D. Buckingham, J. E. Del Bene, and S. A. C. McDowell, Chem. Phys. Lett. 463, 1 (2008).
[9] H. M. Lee, A. Kumar, M. Kolaski, D. Y. Kim, E. C. Lee, S. K. Min, M. Park, Y. C. Choi, and K. S. Kim, Phys. Chem. Chem. Phys. 12, 6278 (2010).
[10] L. Belau, K. R. Wilson, S. R. Leone, and M. Ahmed, J. Phys. Chem. A 111, 7562 (2007).
[11] M. S. de Vries and P. Hobza, Annu. Rev. Phys. Chem. 58, 585 (2007). [12] M. R. Crampton, The Chemistry of the Thiol Group, edited by S. Patai
(John Wiley & Sons, Bristol, 1974) Vol. 1, pp. 379 [13] E. N. Lassettre, Chem. Rev. 20, 259 (1937).
[14] H. Tsujii, K. Takizawa, and S. Koda, Chem. Phys. 285, 319 (2002). [15] G. de Oliveira and C. E. Dykstra, Chem. Phys. Lett. 243, 158 (1995). [16] J. M. Hermida-Ramon, E. M. Cabaleiro-Lago, and J. Rodriguez-Otero, J.
Chem. Phys. 122, 204315 (2005).
[17] H. S. Biswal, S. Chakraborty, and S. Wategaonkar, J. Chem. Phys. 129, 184311 (2008).
[18] H. S. Biswal and S. Wategaonkar, J. Phys. Chem. A 113, 12763 (2009). [19] L. M. Gregoret, S. D. Rader, R. J. Fletterick, and F. E. Cohen, Proteins:
Struct., Funct., Bioinf. 9, 99 (1991).
[20] Hazardous Substances Data Bank [Internet]. Bethesda (MD): National Library of Medicine (US); Methyl Mercaptan; Hazardous Substances
12
Databank Number: 813; Available from:
http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?HSDB
[21] A. J. Barnes, H. E. Hallam, and J. D. R. Howells, J. Chem. Soc., Faraday Trans. 68, 737 (1972).
[22] J. A. Odutola, R. Viswanathan, and T. R. Dyke, J. Am. Chem. Soc. 101, 4787 (1979).
[23] K. Pecul and R. Janoschek, Theor. Chem. Acc. 36, 25 (1974). [24] A. K. Sum and S. I. Sandler, J. Phys. Chem. A 104, 1121 (2000). [25] I. Bakó and G. Pálinkás, Theochem-J. Mol. Struct. 594, 179 (2002).
[26] E. M. Cabaleiro-Lago and J. Rodríguez-Otero, J. Phys. Chem. A 106, 7440 (2002).
[27] C. Morgado, M. A. Vincent, I. H. Hillier, and X. Shan, Phys. Chem. Chem. Phys. 9, 448 (2007).
[28] H.-L. Han, C. Camacho, H. A. Witek, and Y.-P. Lee, J. Chem. Phys. 134, 144309 (2011).
13
第二章
實驗原理與技術
2.1 紅外光–真空紫外光游離光譜技術簡介 使用超音波分子射束冷卻產生的團聚體通常不適合以如傅氏轉換紅外 光譜法(FTIR)等的傳統紅外光譜法研究,主要是由於不同大小的團聚體 分子之光譜極為相似,如系統中含有不只一種大小之團聚體,則光譜相互 重疊,導致光譜難以分析。因此,具有選擇性的光譜技術便成了研究團聚 體最適合的選擇。共振加強多光子游離(REMPI, resonance-enhaced multiphoton ionization) 係一具選擇性的團聚體偵測技術 [1]。其原理為,團聚體分子從其電子基態 吸收m個光子後躍遷至穩定中間態(intermediate state),再吸收n個光子後 游離,搭配飛行時間質譜儀可依抵達偵測器時間之不同分別觀察不同大小 團聚體之訊號。REMPI中最常用於團聚體偵測之技術為m及n皆為 1 的共振 雙光子游離(R2PI, resonant two-photon ionization)[2,3],使用雷射波長通 常在可見光及紫外光範圍,由於不同大小的團聚體分子皆具有相同之吸光 團(chromophore)官能基,吸光波長相近,故通常單一雷射便可偵測所有 不同大小之團聚體。將R2PI結合紅外光雷射,可以取得團聚體之紅外吸收 光譜,此方法被稱為紅外光削減共振雙光子游離(IR-R2PI, IR-depletion resonant two-photon ionization)[3],其原理為,若施加一道紅外光雷射使團
14 聚體分子躍遷至振動激發態,則團聚體會因振動預解離(vibrational predissociation)而使數量變少,在數十或數百奈秒後,以R2PI技術偵測團 聚體,便可觀察到有吸收紅外光之團聚體離子訊號減少。IR-R2PI已被廣泛 用於研究各種具有氫鍵之團聚體在N–H及O–H光區之吸收光譜 [4–7]。然而, IR-R2PI雖具有選擇性,卻無法應用於芳香族等等不具有可吸收紫外光而躍 遷至穩定中間態的吸光團之分子,導致其應用有限。且雙光子游離的吸收 截面(cross section)很小,需要高強度雷射才能進行雙光子游離,並且容 易產生大量離子碎片。 直接用真空紫外光(VUV)游離分子可克服前述的限制。VUV 雷射直 接游離分子有諸多優點,如: (a) 許多分子及其團聚體的游離能量皆在真空紫外光範圍 [8],分子經 VUV光照射後直接游離,不受中間激發態生命期的限制。
(b) VUV為軟性游離的光源(soft ionization source),較不會有分子碎 片的產生 [9]。
(c) VUV 之吸收截面較雙光子游離者大,游離效率較高。
多年來,VUV光游離光譜法已經被應用於研究分子光解以及分子反應動 態學研究 [10– 16]。如Ng研究組所發展出的紅外光−真空紫外光游離光譜法 (IR-VUV photoionization spectroscopy, IR-VUV-PIS) [10–13],以及Fujii 研究組發展出之真空紫外光游離偵測紅外光預解離技術(VUV-ionization
15
detected-IR predissociation spectroscopy, VUV-ID-IRPDS) [14,15] 和光誘導 拉曼真空紫外光光游離光譜法(stimulated Raman-VUV-photoionization spectroscopy)[16]等等。有關許多不同的紅外光−真空紫外光光譜技術及其 應用於團聚體研究之實例,Matsuda等人有對此做詳細的回顧 [17],在此不 加詳述,僅對吾人所採用的兩種不同技術簡單介紹。 2.2.1 真空紫外光游離偵測–紅外光預解離技術(VUV-ID-IRPDS) Matsuda等人曾利用VUV-ID-IRPDS研究氨的團聚體以及甲醯胺與水的 團聚體 [17],此一光譜法的機制如圖 2-1(A)所示。從超音波分子射束產生 的團聚體被一道能量略高於其游離能之真空紫外光游離,單光子游離會產 生相對應的離子,且此離子不會再進一步解離,而在通過飛行時間質譜儀 後可分別偵測到不同大小團聚體之離子訊號。若不同大小團聚體之游離效 率皆相近,離子訊號的強弱分布便與團聚體分子在基態的數量成正比。若 在真空紫外雷射光照射分子射束前大約數十或數百奈秒先照射一道紅外雷 射光,則當紅外光的頻率與團聚體分子之振動能階能量相同時,團聚體會 躍遷至振動激發態,如果此能量高於團聚體之解離能,則會產生解離,因 而造成其濃度減少,亦即離子訊號的減少。故掃描紅外光波長並觀察不同 大小團聚體離子訊號的減少,便可以得知其紅外吸收光譜。但因含有n個單 體的團聚體解離時會產生具n−1 個單體甚至更小的團聚體,而造成較小團聚 體離子訊號的增加,故在將團聚體之預解離光譜轉換為吸收光譜時需考慮
16 較大團聚體對其數量的貢獻 [18]。此光譜技術的優點為分子不需具有吸光 團便可偵測,但缺點為不適用於在躍遷至振動激發態後不會解離之分子或 團聚體,亦無法研究較低頻率之振動模,因能量不足以使分子解離。 2.2.2 紅外光−真空紫外光游離光譜法(IR−VUV−PIS) IR-VUV-PIS 為 Ng 實驗組所發展出之光譜技術,該實驗組曾利用此光 譜技術研究如三氯乙烷、氨及丙炔等等分子的高解析紅外光譜 [13],圖 2-1 (B)為此光譜法之示意圖。與 VUV-ID-IRPD 光譜法不同的地方在於,實驗 中使用之真空紫外光雷射的能量略低於欲研究分子的游離能,故只照射真 空紫外光時無法觀測到其相對應的離子訊號。但若分子吸收紅外光並躍遷 至振動激發態,此時紅外光加上真空紫外光的能量高過其游離能,離子可 被游離,便可偵測到離子訊號,而離子訊號隨掃描紅外光波長的增加程度 即可對應為分子之吸收光譜。IR-VUV−PIS 最大的優點在於,若紅外光的波 長不符合分子之振動躍遷時幾乎觀測不到離子訊號,亦即沒有背景訊號的 干擾,故此光譜法的靈敏度非常高。此光譜技術亦有分子不需具有吸光團 之優點,然而在研究某些分子時,其振動激發態與離子態之結構相差較大, 造成游離之Franck-Condon 重疊較低,離子訊號較弱,此類分子較不適用於 此光譜技術。
17
2.2 真空紫外光光源
目前較常見的真空紫外光源可分為兩種,一種為利用電子加速所產生的 同步輻射(synchrotron radiation),經單光儀(monochromator)等儀器區 分出實驗所需頻率的真空紫外光。另外一種則是利用共振加強四波混頻 (resonance enhanced four-wave mixing)的方法產生。
四波混頻為一種非線性光學現象,主要概念為三道入射光波交互作用於 物質上,經由介質之第三階非線性磁化率χ(3)(non-linear susceptibility)引 導出第三階非線性極化P(3) ,產生第四道雷射光。非線性介質被一或多道雷 射光照射時,雷射光會對此介質施加一電場E(ω),產生一非線性極化的現 象,其強度P(ω)取決於入射雷射電場的強度。其作用可用多項式展開成多 階形式如下: P(ω) = N(χ(1)𝐄(ω) + χ(2)𝐄(𝛚)𝐄(𝛚) + χ(3)𝐄(𝛚)𝐄(𝛚)𝐄(𝛚) + ⋯ ), (1) 其中N 為介質的密度,χ(1)為物質的線性磁化率,χ(n)為介質的第n 階非線性 磁化率。在通常的弱電場條件下,高階項因為係數很小而可以忽略,極化 的程度和電場強度可近似成線性的正比關係。但是在強雷射場作用下,極 化強度的高階項強度變為不可被忽略。 線性磁化率χ(1)的大小通常和介質的折射和吸收現象有關,而第二階非 線性磁化率χ(2)則和二倍頻產生(second harmonic generation, SHG)或是光 學參量產生(optical parametric generation, OPG)等現象有關。這些非線性
18 光學現象的極化強度與和入射的兩道光的電場強度乘積成正比。SHG及 OPG現象被廣泛應用於雷射技術中,基本上從遠紅外光至UV光都可以靠 SHG及OPG產生。但具有高χ(2)的非線性晶體如BBO等,通常都會吸收如真 空紫外光等短波長的光,而無法作為真空紫外光的光源。科學家於是利用 如惰性氣體 [19− 23]或金屬蒸氣 [24−26]等等的介質來產生真空紫外光。然 而由於這些介質具有反轉對稱性(inversion symmetry),其χ(2)值為0,故 科學家利用更高次的χ(3)來產生真空紫外光。通常χ(3)值較χ(2)小,且氣體的密 度也較晶體低,故通常需使用較高能量的雷射光,並聚焦於氣體腔體中。 另外,若入射的雷射頻率和作為介質的氣體的吸收頻率符合的話,便能夠 因共振現象而顯著提升轉換效率,是為共振加強四波混頻。Yamanouchi及 Tsuchiya曾對共振加強四波混頻做完整的回顧 [27],以下就其原理做簡單介 紹。 共振加強四波混頻可分為和頻產生及差頻產生,如圖2-2 所示,使用兩 道頻率分別為ω1及ω2的雷射光並將兩者都聚焦在裝有介質氣體的腔體中, 便可得到一頻率為ωvuv的真空紫外光,ωvuv值可由式(2)算出: 𝜔𝑣𝑢𝑣 = 2𝜔1± 𝜔2。 (2) 若使用的兩道雷射光聚焦點重合,且為橫模(transverse mode)皆相同的高 斯光束(Gaussian beam),產生的真空紫外光能量可以下式計算出: 𝑃𝑉𝑈𝑉 ∝ 𝑁2�𝜒(3)�2𝑃𝜔12 𝑃𝜔2𝐹(𝑏∆𝑘), (3)
19
其中Δk 為波向量(wave vector)之不匹配,可以下二式計算出:
∆k = 𝑘𝑣𝑢𝑣− (2𝑘1+ 𝑘2) (適用於和頻混波)、 (4)
∆k = 𝑘𝑣𝑢𝑣− (2𝑘1− 𝑘2) (適用於差頻混波), (5)
b 值為共聚焦參數(confocal parameter),或稱景深(depth of focus),其 定義為 b = 2𝜋𝑤02 𝜆 = 2𝜋𝑤02𝑛 𝜆0 , (6) 其中w0為雷射在聚焦處之光點半徑,亦即光腰(beam waist),其值等於 𝑓𝜆 𝜋𝑤1,f 為焦距,w1則是聚焦鏡上的光點大小,λ為光在非線性介質的波長,n為介 質之折射率,而λ0為光在真空中的波長。在強烈聚焦的情形下,b值會遠小 於氣體腔體的長度L,而聚焦的區域不超出氣體腔體之範圍,即b ≤ f ≤ L-b。 對和頻混波來說,此時的相位匹配(phase-matching)參數F(bΔk)可以下式 表示 [28]: 𝐹𝑠𝑢𝑚(𝑏∆𝑘) = 𝜋2(𝑏∆𝑘)2𝑒𝑥𝑝 �𝑏∆𝑘2 � (∆𝑘 < 0) 𝐹𝑠𝑢𝑚(𝑏∆𝑘) = 0 (∆𝑘 ≥ 0)。 (7) 對差頻混波來說,F(bΔk)值為 𝐹𝑑𝑖𝑓𝑓(𝑏∆𝑘) = 𝜋2exp (−𝑏|∆𝑘|)。 (8) 圖2-3 描繪了若我們假設在強烈聚焦的情形下,𝑏 𝐿 = 0, 𝑓 𝐿 = 0.5,相位匹配 參數F(bΔk)隨bΔk值的變動。圖 2-3(A)中可以看到對和頻混波來說,相位匹 配參數的最大值發生在bΔk = −4 時,由圖我們可以得知,對和頻混波來說,
20 相位匹配的條件較嚴謹,因為產生和頻混波時必須要是負色散(negative dispersion),意即新生成的真空紫外光之波向量要小於原本入射的光波之 波向量和(∆𝑘 < 0)。但對差頻混波而言,在正色散(positive dispersion) (∆𝑘 > 0)或負色散的條件下皆可以產生真空紫外光,如式(8)及圖 2-3(B)中 所示。在強聚焦情況下,高斯光束會在聚焦處產生一相位滑動(phase slip), 此一相位滑動通常稱為Gouy phase [29,30],其大小為tan-1ξ,其中𝜉 = 𝑧𝜆
𝜋𝜔02, z為光波的位置。對於和頻混波而言,入射光有3 tan-1ξ的相位滑動,而新 生成的真空紫外光有tan-1ξ的相位滑動,兩者間的差為2 tan-1ξ。對於差頻 混波而言,入射光的相位滑動為tan-1ξ,而新生成的光亦有tan-1ξ的相位 滑動,兩者間的差為0。這些相位滑動可以利用波向量的不匹配來補償,以 得到最佳的出光,補償相位滑動後的最佳Δk值Δkopt可表示如下: ∆𝑘𝑜𝑝𝑡 ≅ �− 2.2 𝑏⁄ (適用於和頻混波) 0 (適用於差頻混波), (9) 故對和頻混波而言,最佳的bΔk 值為 bΔkopt = −2.2,而對差頻混波而言,最 佳的bΔk 值為 bΔkopt = 0。通常可藉由調整聚焦的強弱程度來改變 b 的大小, 而改變介質氣體的壓力或是在腔體內添加緩衝氣體(buffer gas)可以調整 Δk 的大小,進而使bΔk 值接近 bΔkopt。 圖2-4 列出了幾種常用於四波混頻的非線性介質,以及可用來進行差頻 或和頻混波的能階。以本實驗使用的氪(krypton, Kr)為例,若使用 212.556 nm之ω1和其 4p−5p [1/2,0]躍遷共振,再搭配另一出光波長範圍為 220–740
21 nm之雷射光,可以產生範圍在 124–206 nm的真空紫外光。使用氪生成真空 紫外光時,可在腔體中添加氬(argon, Ar)以利相位匹配,Marangos等人發 現若調整氣體的壓力比為 𝑃𝐴𝑟 𝑃𝐾𝑟 = 3 時可達到最佳的相位匹配 [31]。 2.3 光參量共振與光參量放大技術 2.3.1 光學參量震盪器
光學參量震盪器(optical parametric oscillator, OPO)提供可調變的紅外 波長輸出,在紅外光譜技術中扮演相當重要的角色。在紅外光區中有許多 雷射光源的選擇,如摻有稀土金屬(如Tm、Ho、Er等等)的晶體或纖維, 利用金屬之電振躍遷(vibronic transition)發光的固態雷射(如Fe或Cr), 或是異質介面(heterojunction)的雷射二極體,以及最近興起的量子級聯 雷射(quantum cascade laser)等等,都是實驗室常用的紅外光源。這些光 源中,唯獨OPO具有可調變波長範圍大、輸出能量強等等優點,使得OPO 成為近數十年實驗中主流的紅外光源 [32,33]。 若使用一道雷射光入射一個第二階非線性磁化率χ(2)值不為0 的晶體, 而光強度足以達到所謂的參量增益(parametric gain)時,便會放出參量螢 光(parametric fluorescence)。參量螢光與固態雷射放大器(amplifier)中 會發生的自發輻射(spontaneous emission)現象類似,但只會發生在滿足 相位匹配的方向,故通常出光方向與入射光相同,利用參量螢光相位匹配
22
條件的不同產生不同波長光的技術稱為光學參量產生(optical parametric generation, OPG)。入射的激發光(pump beam)經由 OPG 會轉換為兩道 較低能量的光,分別稱為訊號光(signal beam)及閒頻光(idler beam), 如圖2-5(A)所示,其中訊號光定義為兩道出光中頻率較高的光。因能量守 恆,入射光的頻率ωp等於新生成的訊號光頻率ωs及閒頻光頻率 ωi和, 𝜔𝑝 = 𝜔𝑠 + 𝜔𝑖。 (10) 在滿足相位匹配條件時,入射光的動量kp亦等於訊號光動量ks及閒頻光動 量ki之和, 𝑘𝑝 = 𝑘𝑠 + 𝑘𝑖。 (11) 而因為動量可轉換成下列形式: k = 2𝜋𝜆 = 𝑛(𝜔)𝜔𝑐 , (12) 其中λ 為波長,n(ω)為光在介質中之折射率,c 為光速,可得知, 𝑛�𝜔𝑝�𝜔𝑝 = 𝑛(𝜔𝑠)𝜔𝑠 + 𝑛(𝜔𝑖)𝜔𝑖。 (13) 根據式(10)及式(13): 𝑛�𝜔𝑝� = 𝑛(𝜔𝑠) = 𝑛(𝜔𝑖)。 (14) 從式(14)可得知,若要滿足相位匹配條件,激發光、訊號光及閒頻光在非 線性晶體中的折射率要相同,故須選用具備雙折射性(birefringent)的晶 體。所謂的雙折射性是指不同極化方向的光在晶體中具有不同的折射率, 其中依其極化方向的不同可分為常態光(ordinary wave)及非常態光
23 (extraordinary wave),兩道光電場互相垂直。雙折射性晶體組成分子在 晶體中排列之對稱軸稱為光軸(optical axis),常態光的極化方向與光波前 進的方向以及晶體的光軸垂直,其折射率不因其光波前進方向而改變。非 常態光的折射率則與光波前進方向與晶體光軸的夾角有關。常態光折射率 no與非常態光折射率ne與光軸夾角 θ 的關係可以下式表示: 1 𝑛𝑒2(𝜃) = sin2𝜃 𝑛𝑒2(90°)+ cos2𝜃 𝑛𝑜2 。 (15) 由上式可得知,旋轉非線性晶體的角度可以調整訊號光及閒頻光的能量比, 藉以調變出光的波長。 在OPG及後述的OPO中,相位匹配可以入射光與生成光極化方向的不 同而區分為第一類相位匹配(type-I phase-matching)和第二類相位匹配 (type-II phase-matching)兩種情形 [34]。第一類相位匹配中,訊號光與閒 頻光的極化方向相同,而激發光的極化方向與該二道光不同,如圖2-6(A) 所示。第二類相位匹配中,訊號光與閒頻光的極化方向不同,而激發光的 極化方向與閒頻光相同,如圖2-6(B)所示。 若在非線性晶體外加上一共振腔,使OPG 生成的光波可以在晶體來回 反射加強能量,稱為光學參量震盪(optical parametric oscillation, OPO), 如圖2-5(B)所示。選擇共振腔端面反射鏡(end mirror)及輸出耦合鏡(output coupler)的材質使其會在共振腔內共振,OPO 可以此分類為只有訊號光或 閒頻光在腔內共振的單共振OPO(singly resonant OPO)、訊號光及閒頻光
24
都會共振的雙共振OPO(doubly resonant OPO),以及激發光、訊號光以 及閒頻光都會共振的三重共振OPO(triply resonant OPO)。
選用不同的非線性晶體材料可以輸出不同範圍的波長,從1 μm 到 10 μm 的範圍都可以涵蓋,如圖2-7 所示。對於一般波長較短的中紅外光
(mid-infrared)範圍(2 μm–5 μm),週期性極化的鐵電氧化物(periodically poled ferroelectric oxides)如 LiNbO3、KTP、KTA、LiTaO3 等等為合適的 選擇,這些物質皆可使用出光波長1 μm 的固態雷射作為激發光源,且具有 準相位匹配(quasi-phase-matching, QPM)的特性。QPM 為藉由在晶體內 部製造週期性的結構所達到的相位匹配;因為晶體內的非線性週期性的改 變,產生一個新的波向量(2𝜋 λ),因而補償了波動量的不匹配,達到動量 守恆。QPM 提供了許多優點,如激發光、訊號光以及閒頻光可以在同樣的 光徑上傳遞而不會有行進方向偏離波向量方向的情形發生,角度可調範圍 大而不會受到相位匹配條件的限制等等。PP KTP 及 PP KTA 為兩種被廣泛 使用於OPO 的材質,其損傷臨界值較其他常見之晶體高,且折射率較低, 光通過晶體時產生的偏折較小。目前最常見的OPO 使用 Q-switched Nd:YAG 脈衝雷射作為激發光源,可以發出近紅外光(near-infrared)及中 紅外光區的紅外光,而其出光能量在毫焦耳(millijoule)等級。 對於波長較長的IR 光(5–12 μm)而言,一般的鐵電氧化晶體本身會 吸收此光區的光,故需要使用其他非線性晶體材料,如ZGP(ZnGeP2)、
25
CdSe、AGSe(AgGaSe2)等等,這些材料一般需要波長在 2 μm 附近的激
發光源,且其轉換效率通常較鐵電氧化晶體差。
2.3.2 光學參量放大
光學參量放大(optical parametric amplification, OPA)中,當一道訊號 光伴隨著頻率較高的激發光入射非線性晶體時,激發光的光子會轉換為與 訊號光頻率相同的光子,使訊號光之強度增強,同時並產生一道閒頻光, 如圖2-5(C)所示。閒頻光之頻率為激發光與訊號光之差,意即
ω𝑖 = 𝜔𝑝 − 𝜔𝑠, (16)
和OPG 及 OPO 一樣,OPA 的相位匹配可分為二類:在第一類相位匹 配中,訊號光與新生成的閒頻光極化方向相同,但與激發光極化方向不同, 如圖2-8(A)所示 [34]。第二類相位匹配中,新生成的閒頻光與激發光極化 方向相同,而訊號光極化方向與該二者不同,如圖2-8(B)所示。
OPA 的作用和一些常見雷射的放大器作用類似,惟 OPA 可以產生一頻 率不同的額外出光。通常會將OPG 或 OPO 的晶體與 OPA 晶體串接,利用 OPG 或 OPO 所產生之訊號光或閒頻光作為 OPA 的訊號光,此時不僅前一 階段的放光可以得到加強,亦可以產生一頻率更低的光。本實驗所使用的 可調變紅外雷射(LaserVision)即是串接 OPO 及 OPA 以得到中紅外光出 光,其內部架構詳述於3.1.2 節。
26 2.4 分子射束法 室溫下,氣相的分子有足夠的內能分布在能量較高的振動與轉動能階。 對能階單純的小分子而言尚不致於造成偵測的光譜譜線過於擁擠,但對大 分子而言,振動模數目增加和振轉能階間複雜的偶合現象,容易造成光譜 譜線的增加和重疊,甚至成為很寬的譜帶,無法解析。降低溫度雖可使分 子內能減少,測得的光譜簡化,但靜態或穩定流動的樣品在溫度太低時其 分子會凝結成液體或固體,又增添了光譜的複雜性。分子射束技術的發展, 使分子能保持在低內能溫度及低碰撞頻率而近乎孤立狀態的環境。以此技 術結合各種光學與非光學的偵測方法,成為研究氣態原子、分子或離子的 光譜和化學動力學的最佳方法之一 [35− 40]。 分子射束形成的過程是利用高壓氣體由噴嘴(nozzle)向高真空區域噴 射的快速絕熱膨脹(adiabatic expansion)過程中,將沿著噴射方向之氣體 分子的內能(internal energy)轉變成分子傳動能(translational energy), 而此傳動能之分布極窄。此急速膨脹冷卻之過程具有溫度不平衡之現象, 可在分子間不凝結,且分子的振動能不完全消除的情況下,得到非常低的 轉動及徑向傳動溫度。由於低溫使得可進行之分子躍遷減少,可達到簡化 光譜、增強訊號的目的。 圖2-9 為分子射束膨脹的示意圖,射束的核心區域在膨脹過程中進行了 可逆的絕熱膨脹,通常被稱為等熵區(isentropic region)。由於此區氣體不
27 受背景壓力的干擾,故又稱無聲區(zone of silence)。在超音波膨脹中, 可以馬赫數(Mach number, M)來描述射束的性質。M 之定義為分子流的 速度u 和以該氣體為介質之音速 vs的比值: M = vu s, (17) 其中vs可由下列算式算出 vs= �γRTm � 1 2, (18) 其中m 為氣體的分子量,γ 代表等壓熱容(CP)和等體積熱容(CV)的比 值,R 為理想氣體常數,T 為氣體溫度。根據熱力學第一定律,我們可以列 出: h+u22 = h0, (19) 其中h0及 h 分別為噴嘴內外氣體單位質量的焓(enthalpy)。在氣體膨脹時, 焓下降而平均速度上升。對理想氣體而言,焓和熱容量的關係可表示為: dh = CpdT, (20) 由式(19)和(20)和可得到分子射束的平均速度及溫度的關係如下: u2 = 2�h 0-h� = 2 ∫ CTT0 pdT, (21) 對於理想氣體,Cp不隨溫度改變, Cp = γ-1γ mR, (22) 又分子射束經膨脹及能量轉移後,內能溫度會降得很低,即T ≪ T0,故可利 用式(21)及(22)計算分子射束的終極速度為:
28 u∞ = �γ-12γRTm0� 1 2。 (23) 由上式可看出,氣體的分子量越小,可到達的終極速度就越快。因此,在 實驗中可將質量大的分子稀釋在質量小的氣體分子中,以增加分子射束的 終極速度。對於理想氣體混合物而言,熱容量和分子量可由組成分率xi取 平均值,即 Cp = ∑ xi iCpi= ∑ xi i( γi γi-1) R m, (24) 而m = ∑ 𝑥𝑖 𝑖𝑚𝑖,將此平均分子量及熱容量值代入式(23)即可估算混合氣體 的終極速度。 在超音波膨脹的過程中,M值會持續上升,在過度膨脹後,前緣會產生 壓縮的現象而形成衝擊波前;因該波前呈一碟狀,故慣稱為馬赫碟域(Mach disk)。越過馬赫碟域後,分子射束的性質會受到衝擊波擾亂或背景壓力影 響使其性質難以掌握,故通常會使用一金屬製的圓錐(skimmer)設置於馬 赫碟域前,來排除非無聲區的氣體。Bier和Schmidt [41]曾推導出馬赫碟域 在分子射束中形成的位置XM為: XM ≅ 0.67d �PP0b� 1 2, (25) 其中d 為噴嘴的直徑,P0為噴嘴內壓力,Pb為噴嘴外壓力。d 通常遠大於高 壓氣體在噴嘴內的平均自由徑,由此式可知噴嘴內外壓力會影響馬赫碟域 形成的位置。 超音波膨脹的過程中會形成團聚體,在超音波膨脹的過程中氣體的壓力
29 及溫度快速的下降,而當下降到一定程度時,若分子間之作用力不可忽略 且分子濃度較高,氣體分子會互相聚集,並發生成核(nucleation)現象並 形成團聚體,這樣的成核現象通常僅需數個微秒即可完成,其過程尚未被 科學家詳細解釋 [42]。我們可定義一Γ值來描述超音波膨脹後團聚體的組 成: Γ = n0dqT0��2-γ�q-2�/2�γ-1�, (26) 其中n0為氣體的密度,T0為氣體的溫度,而 q 則為一介於 0.5 到 1 的經驗 參數,不同氣體具有不同的q 值。具有相同 Γ 值的不同超音波膨脹條件下 會得到相同的團聚體組成。Hagena 在 1981 年使用另一參數 Γ*藉以比較不 同氣體進行超音波膨脹所得到的團聚體組成 [42]。他定義出特性溫度 Tch (characteristic temperature)及特性半徑 rch(characteristic radius)兩個參數, 其中Tch被定義為 Tch = ∆Hk0, (27) 其中ΔH0為氣體在0 K 時的的昇華焓(sublimation enthalpy),k 為波茲曼 常數。而rch定義為 rch = �mρ� 1 3, (28) 其中ρ 為物質在固態下的密度。Γ*可由下式導出: Γ* = nrch3 �rdch� q �T0 Tch� ��2-γ�q-2�/2�γ-1� 。 (29) 從Γ*的值可以判斷超音波膨脹後會得到的團聚體組成分布,Hagena 將其歸
30 納成三種情形: (a) Γ* ≦ 200 時,無團聚體生成。 (b) 200 < Γ* ≦ 1000 時,通常形成較小的團聚體,大小不超過 100 個單 體。 (c) Γ* > 1000 時,形成較大的團聚體,大小超過 100 個單體。 2.5 直線型飛行時間質譜儀 飛行時間質譜儀為Cameron和Eggers在 1948 年發展出之質譜技術 [43]。 當中性的分子被游離時,以一靜電場加速帶電荷的離子,進入一段自由飛 行的區域。由於同電荷之離子所獲得之動能相同,質荷比之不同造成各種 離子具有不同之初速度,因此經過一定距離的飛行後到達偵測器的時間亦 不相同,藉此區分不同質荷比之離子物種。由於飛行距離(L)為已知,精 確記錄離子的飛行時間(t),即可得到離子的速度(v = 𝐿 𝑡)。而因為離子 的動能E為已知,從E = 1 2𝑚𝑣2即可得到離子的質量。簡單地說,測得離子 飛行時間即可得到原子或分子的質量。 與四極式(quadrupole)、磁控式(magnetic sector)等其他質譜技術相 比,飛行時間質譜儀的解析度較低,這是因為具有相同質量的離子因為初 速的不同,以及其在游離區域的空間分布不是一點所造成的。初速不同的 問題可以使用如延遲脈衝萃取(delayed pulse extraction)[44]、反射型架構 (reflectron)[45]或靜電磁極(electrostatic sectors)[46]等等改良式的飛行
31
時間質譜儀解決。而分子空間分布的問題可以多重電場加速型的飛行時間 質譜儀解決。Wiley和McLaren在 1955 年發明的雙電場加速直線型飛行時間 質譜儀即為一例 [44]。
雙電場加速直線型飛行時間質譜儀通常在游離區有三片電極,分別為 推斥電極(repel plate)、萃集柵極(extraction grid)以及接地柵極(ground grid),如圖2-10 所示。假設推斥電極電壓為 VA1,萃集柵極電壓為VA2, 此二電極之電壓差為Vp(= VA2 − VA1),其距離為d1,則在此游離區之電 場為𝑉𝑝 𝑑1。對於一個帶有電荷q 的離子,其所感受到的電場 𝑉𝑝 𝑑1 = 𝑚𝑎1 𝑞 ,其中m 為離子之質量,則我們可以推導出離子在游離區的加速度a1 = 𝑉𝑝𝑞 𝑚𝑑1。飛行 時間t1可以下式導出: t1 = �2𝑠𝑎1 = (2𝑠𝑚𝑑𝑉𝑝𝑞1) 1 2 = (2𝑚)12(𝑠𝑑1 𝑉𝑝𝑞) 1 2 , (30) 而在通過萃集柵極時的速度為 𝑣1 = 𝑎𝑡11 = (2𝑠𝑉𝑚𝑑𝑝1𝑞) 1 2。 (31) 假設從萃集柵極與接地柵極間之距離為d2,而離子從萃集柵極飛行至接地 柵極之時間為t2,則根據如下式的動能守恆: 動能= q ∙𝑉𝑝 𝑑1 ∙ 𝑠 + 𝑞 ∙ 𝑉𝐴2 = 1 2𝑚𝑣2, (32) 其中q ∙𝑉𝑝 𝑑1 ∙ 𝑠為推斥電極及萃集柵極間得到的動能,𝑞 ∙ 𝑉𝐴2為萃集柵極和接 地柵極間得到的動能,可以導出離子在通過接地柵極時之速度v2為 𝑣2 = (2𝑞𝑚 (𝑑𝑉𝑝1+ 𝑉𝐴2)) 1 2。 (33)
32 而利用如下式的動量守恆: 動量= q𝑉𝐴2 𝑑2 𝑡2 = 𝑚(𝑣2− 𝑣1), (34) 可推導出從萃集柵極飛行至接地柵極之時間t2為: 𝑡2 = (2𝑚𝑞 ) 1 2∙ (𝑑2 𝑉𝐴2) �� 𝑠𝑉𝑝 𝑑1 + 𝑉𝐴2� 1 2 − (𝑠𝑉𝑝 𝑑1) 1 2�。 (35) 由於自由飛行區之電場為0,故離子感受到的加速度為 0,離子飛行的速度 保持在v2,其在自由飛行區的飛行時間t3可以下式算出: 𝑡3 =𝑣𝐿2 = 𝐿 ∙ [2𝑞𝑚�𝑠𝑣𝑑1𝑝� + 𝑉𝐴2]− 1 2。 (36) 根據式(30)、(35)及(36),可以得知分子從游離區至偵測器的飛行時間 tf為: 𝑡𝑓 = �2𝑚𝑞 � 1 2��𝑠𝑑1 𝑉𝑝� 1 2 + �𝑑2 𝑉𝐴2� �� 𝑠𝑉𝑝 𝑑1 + 𝑉𝐴2� 1 2 − �𝑠𝑉𝑝 𝑑1� 1 2�� + 𝐿 �2𝑞𝑚�𝑠𝑣𝑝𝑑1�+𝑉𝐴2� 1 2。 (37)
33 (A) (B) 圖2-1 兩種不同的 IR−VUV 光游離偵測機構。 (A)真空紫外光游離偵測−紅外光預解離光譜法(VUV-ID-IRPD),(B)紅外 光−真空紫外光游離光譜法(IR−VUV−PI)。
34
圖 2-2 共振四波和頻(ω𝑉𝑈𝑉 = 2ω1 + ω2)及差頻(ω𝑉𝑈𝑉 = 2ω1 − ω2) 之示意圖。
35 (A) (B) 圖 2-3 相位匹配參數隨 bΔk 值的變動(假設𝑏 𝐿 = 0, 𝑓 𝐿 = 0.5)(參考文獻 28 繪製)。 (A)下半圖為和頻混波中 F(bΔk)值的變化,上半圖為 F(bΔk)之一次微分, bΔk=−4 時為 F(bΔk)之最大值;(B)差頻混波中 F(bΔk)值的變化,最大值出 現於bΔk=0 時。
36
圖 2-4 使用 Hg, Xe, Kr, Ar 等非線性氣體作共振加強四波和頻或差頻之能 階示意圖,及可產生的真空紫外光範圍(參考文獻27 繪製)。
37
(A)
(B)
(C)
圖 2-5 一些常見利用非線性晶體產生可調變光源的示意圖。
(A)光學參量產生(optical parametric generation, OPG),(B)光學參量震盪
(optical parametric oscillation, OPO),(C)光學參量放大(optical parametric amplification, OPA)。
38 (A) (B) 圖 2-6 OPO 及 OPG 中相位匹配之示意圖(參考文獻 34 繪製)。 (A)第一類相位匹配(type-I phase-matching),(B)第二類相位匹配(type-II phase-matching)。其中 ωp、ωs、ωi分別為激發光、訊號光及閒頻光。
39 圖 2 -7 常用 於 O PO 的 非 線性 晶體及其 可利用光 區,其中 灰色區域 為放光較 弱之光區 ( 摘自參考 資料 33 )。
40 (A) (B) 圖 2-8 OPA 中相位匹配之示意圖(參考文獻 34 繪製)。 (A)第一類相位匹配(type-I phase-matching),(B)第二類相位匹配(type-II phase-matching)。其中 ωp、ωs、ωi分別為激發光、訊號光及閒頻光。
41 圖 2 -9 超音 波射束之 示意圖。
42 圖 2 -10 雙 電場加速 直線型飛 行時間質 譜儀示意 圖。
43
參考文獻
[1] C. E. H. Dessent and K. Müller-Dethlefs, Chem. Rev. 100, 3999 (2000). [2] T. S. Zwier, Annu. Rev. Phys. Chem. 47, 205 (1996).
[3] B. Brutschy, Chem. Rev. 100, 3891 (2000).
[4] T. Ebata, A. Fujii, and N. Mikami, Int. Rev. Phys. Chem. 17, 331 (1998). [5] R. Brause, H. Fricke, M. Gerhards, R. Weinkauf, and K. Kleinermanns,
Chem. Phys. 327, 43 (2006).
[6] H. Fricke, G. Schafer, T. Schrader, and M. Gerhards, Phys. Chem. Chem. Phys. 9, 4592 (2007).
[7] K. Bartl, A. Funk, and M. Gerhards, J. Chem. Phys. 129, 234306 (2008). [8] S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Lerin, and W.
G. Mallard, J. Phys. Chem. Ref. Data Suppl. 17 (1988)
[9] F. Dong, S. Heinbuch, J. J. Rocca, and E. R. Bernstein, J. Chem. Phys. 124, 224319 (2006).
[10] H. K. Woo, P. Wang, K.-C. Lau, X. Xing, C. Chang, and C. Y. Ng, J. Chem. Phys. 119, 9333 (2003).
[11] M.-K. Bahng, X. Xing, S. J. Baek, and C. Y. Ng, J. Chem. Phys. 123, 84311 (2005).
[12] M.-K. Bahng, X. Xing, S. J. Baek, X. Qian, and C. Y. Ng, J. Phys.Chem. A
110, 8488 (2006).
[13] C. Y. Ng, Frontiers of Molecular Spectroscopy, edited by L. Jaan (Elsevier, Amsterdam, 2009), pp. 659.
[14] Y. Matsuda, M. Mori, M. Hachiya, A. Fujii, and N. Mikami, Chem. Phys. Lett. 422, 378 (2006).
[15] D. Sakai, Y. Matsuda, M. Hachiya, M. Mori, A. Fujii, and N. Mikami, J. Phys. Chem. A 112, 6840 (2008).
[16] Y. Matsuda, M. Hachiya, A. Fujii, and N. Mikami, Chem. Phys. Lett. 442, 217 (2007).
44
(2009).
[18] H.-L. Han, C. Camacho, H. A. Witek, and Y.-P. Lee, J. Chem. Phys. 134, 144309 (2011).
[19] R. Wallenstein, Frontiers of Laser Spectroscopy of gases, edited by A. C. P. Alves, J. M. Brown, and J. M. Hollas, NATO Advanced Study Institute Series C 234, 53 (1988).
[20] R. G. Tonkyn, J. W. Winniczek, and M. G. White, Chem. Phys. Lett. 164, 137 (1989).
[21] H. H. Fielding and T. P. Softley, Chem. Phys. Lett. 185, 199 (1991).
[22] G. Hilber, A. Lago, and R. Wallenstein, J. Opt. Soc. Am. B 4, 1753 (1987). [23] W. Kong, D. Rodgers, and J. W. Hepburn, Chem. Phys. Lett. 203, 497
(1993).
[24] R. T. Hodgson, P. P. Sorokin, and J. J. Wynnc, Phys. Rev. Lett. 32, 343 (1974).
[25] W. Jamroz and B. P. Stoicheff, Prog. Optics 20, 327 (1983).
[26] C. R. Vidal, Tunable Lasers, Topics in Applied Physics, edited by L. F. Mollenauer and J. C. White (Springer, Heidelberg, 57, 1988).
[27] K. Yamanouchi and Soji Tsuchiya, J. Phys. B: At. Mol. Opt. Phys. 28, 133 (1995).
[28] G. Bjorklund, IEEE J. Quantum Electron. 11, 287 (1975). [29] L. G. Gouy, C. R. Acad. Sci. Paris 110, 1251 (1890).
[30] A. E. Siegman, Lasers (University Science Books, Mill Valley, 1986)
[31] J. P. Marangos, N. Shen, H. Ma, M. H. R. Hutchinson, and J. P. Connerade, J. Opt. Soc. Am. B 7, 1254 (1990).
[32] Solid-state mid-infrared laser sources, edited by I. T. Sorokina and K. L. Vodopyanov (Springer-Verlag, 2003).
[33] A. Godard, C. R. Phys. 8, 1100 (2007).
[34] Laser Chemistry: Spectroscopy, Dynamics and Applications, H. H. Telle, A. González Ureña, and R. J. Donovan (Wiley, Chichester, U.K., 2007 ) [35] D. H. Levy, Annu. Rev. Phys. Chem. 31, 197 (1980).
[36] Atomic and Molecular Beam Methods, edited by G. Scoles (Oxford University Press, Oxford, U.K., 1998).
45
[37] Atomic and Molecular Beams, edited by R. Campargue (Springer, Berlin, 2001).
[38] T. A. Miller, Science 223, 545 (1984). [39] Y. T. Lee, Science 236, 793 (1987).
[40] P. C. Engelking, Chem. Rev. 91, 399 (1991).
[41] K. Bier and B. Schmidt, Z. Angew. Phys. 13, 493 (1961). [42] O. F. Hagena, Surf. Sci. 106, 101 (1981).
[43] A. E. Cameron and J. D. F. Eggers, Rev. Sci. Instrum. 19, 605 (1948). [44] W. C. Wiley and I. H. McLaren, Rev. Sci. Instrum. 26, 1150 (1955). [45] B. A. Mamyrin, V. I. Karataev, D. V. Shmikk, and V. A. Zagulin, Sov.
Phys. JETP 37, 45 (1973).
47
第三章
實驗裝置
3.1 儀器架設 本實驗之儀器架設如圖3-1 所示,主要包括真空紫外光光源、可調式紅 外光光源、分子射束系統及飛行時間質譜儀裝置,以下分別說明之: 3.1.1 真空紫外光光源 本實驗中使用之真空紫外光係由共振加強四波差頻(ωvuv = 2ω1 − ω2) 產生。系統中設計一腔體裝填氪氣與氬氣(壓力比1:3,總壓 60 torr), 連接於飛行時間質譜儀裝置上,並使用一道波長為ω1的雷射光,與氪之 4P6(1S 0)→4P5(2P03/2)5P(2[1/2]0)躍遷(能階差 94092.86 cm−1)行雙光子(2ω1) 共振,並與另一道頻率較低的雷射光(ω2)行非線性光學作用後產生真空 紫外光,其架設圖如圖3-2 所示,並詳細說明於下。前述之ω1及ω2分別由兩台染料雷射(2E-OG, Scanmate, Lambda Physik) 產生,此二台染料雷射皆使用一內建三倍頻晶體之Nd:YAG 雷射(GCR-270, Spectra Physics,能量 230 mJ,出光波長 355 nm,垂直極化)作為激發光源。 其中一台染料雷射使用Stilbene 420 染料(Exciton,別稱 LC4200 或 Stilbene 3,可放光範圍 412–444 nm),輸出波長設定為 425.112 nm,其輸出雷射光 極化方向和激發光源同為垂直極化,光束大小約為3×3 mm2。在雷射出口 處設有一由兩面直角石英稜鏡組成的光延遲裝置(optical delay line),增加
48
雷射光的行經距離以確保ω1及ω2到達氣體腔體的時間相同。此道雷射光隨 後被導入一BBO 晶體(Castech, type I,晶體編號:2-43203-0001),二倍 頻後生成波長212.556 nm,水平極化之 ω1。該BBO 晶體放置於一以電腦操 控的可旋轉平台(SGSP-60YAW, Sigma Koki)上,可藉由旋轉其水平擺放 角度達到最佳相位匹配條件。雷射光在通過BBO 晶體後被導入一 Pellin-Broca 稜鏡,藉以將 425.112 nm 之基頻光與新生成的 212.556 nm 倍頻 光分離;因兩道光在稜鏡中折射率不同,212.556 nm 的光會以與原本光徑 夾90°角的路徑射出稜鏡,425.112 nm 的光徑則以一大於 90°之角度射出稜 鏡。此Pellin-Broca 稜鏡放置的角度為 212.556 nm 雷射光的 Brewster 角度, 降低因反射而造成的能量損失。另外一台染料雷射裝填LC5400 染料 (Exciton,別稱 C153,可放光範圍 516–575 nm),波長設定為 537 nm, 出光極化方向為垂直極化。在雷射出口端將光束導入一自製的透鏡組(−100 mm, +250 mm)以將光束大小放大至5×5 mm2,隨後進入一半波片(half-wave plate),將雷射光極化方向由垂直極化轉換為與 ω1相同的水平極化,以利 後續四波混頻。 系統中以一可穿透ω2、反射ω1的雙色鏡(dichroic mirror) (LWP-45-R210-TP308-PW-1025-UV, CVI),使兩道原本行進方向相垂直 之雷射光重合,接著以一凸透鏡(MgF2, f = 200 mm)將兩道光聚焦於氣體 腔體中。由於經非線性作用生成的真空紫外光與ω1、ω2之行進路線相同,