序列組共同具有短序列樣式之詳盡搜尋演算法; Finding Motif by exhaustive algorithm
43
0
0
全文
(2) 基因序列定序資料的增加而增加,反而對生物學家而言大量的序列資 料產生,使得實驗研究經費及研究基因體功能所花費的時間都相對的 增加非常多。但也因為隨著大規模序列資料的日益增加,它的每一個 環節與資訊分析變得更緊密不可分,例如:測序儀的光密度採樣與分 析、鹼基讀出、載體標識與去除、拼接及填補序列間隙、重復序列標 識、讀框預測和 Motif 找尋的技巧等,每一步都是緊密依賴資訊技 術,也因此許多藉電腦技術來分析序列的工具也就因此因應而生。 Motif的找尋正是這龐大的資料中所要挖掘的其中一種重要的資 訊,motif是指生物體隨著不斷的演化突變,某些重要的基因片段依 然保留,並沒有隨著演化的過程中突變,反而形成存在生物體內的一 個重要構成部分。若能在這些龐大的序列資料中找到此段的資訊,生 物學家將能運用motif的訊息來尋找調控基因甚至藉此來了解基因的 功能及分類相似的物種,因此這將是一個急需解決的研究問題。. 二.問題陳述: 若將一條長度為 30 多億鹼基對的基因序列存成文字檔,大約需 要兩千片磁碟片才夠,若將人類基因體序列以一本 1,000 頁的百科全 書比喻(一頁約 3,000 字),總共會有 1,000 冊的書大約有 7-8 層樓之 高,另外加上 DNA 微陣列(micrroarray)基因表達的分析以及基因組 定序資料的成長,使人類基因組的定序速度目前呈現指數級數的速度 2.
(3) 成長,而許多有用的資料就隱藏在許多雜訊之中,而 motif 正是存在 於這許多雜訊中的一項有用的資訊。 Motif 是指一組序列中共同具有的短序列樣式,在生物中可用來 預測某種分子功能、結構的特性或是蛋白質家族的關係,在蛋白質、 DNA 或 RNA 序列中皆可測出 motif 的存在。Motif 在不同的序列可能 隨機出現在不同的位置,然而因為每個序列的上游(upstream)區並不 一定有足夠顯著的 motif 信號(signal),而且在 DNA 中存在許多其他 信號如:促進因子(promoters)、裂接點(splicing sites)等,這些問 題使得在找尋 motif 變得相當複雜。也因為找尋 motif 的問題如此複 雜,學者們也發展出許多演算策略來搜尋 motif。 然而在這許多發展的演算法中存在一個挑戰的問題(Challenge Problem),此問題是在 2000 年由 Pevzner PA 學者提出的,這個問題 是假設有一個固定但是卻未知的短序列片段(Motif)為 M 其長度為 L,在 T 個長度為 N 的序列中都有一個和 M 相似的片段,如何在 T 個 序列中找出 M,而這些和 M 相似的片段是允許有 d 個鹼基產生變異的 也就是構成(L ,d)-motif。 如圖 1.1 有四條序列,每條序列長度為 33bp,要在這四條序列 中尋找(8,2)的 motif,每條序列中都有一個和 M=ACAGGATC 相似的片 段,這些相似片段都與 M 有 2 個鹼基位置的不同。. 3.
(4) AGTTATCGCGGCACAGACTCCTTCTTTATAGCC ATGATAGCATCAACCTAACCCTAGATATGGGAT TTTTGGGATATATCGCCCCTACACTGGATGACT GGATATACATGAACACGGTGGGAAAACCCTGAC. ACAGGATC. 圖 1.1 Challenge Problem 範例. 三.研究問題 所謂的 Challenge Problem 則是在 20 個序列中,每條序列長度均 為 600bp 的鹼基,每個序列中內含有長度為 15 且允許有 4 個鹼基位 置產生變異的 motif,如何在這 20 條序列中找到(15,4)的 motif 正是這 問題的主要目標。 在Pevzner and Sze的研究中(Pevzner 2000)比較了 Consensus(Hertz 1999)、Gibbs sampling(Lawrence 1993)、 MEME(Bailey 1995)、WINNOWER(Pevzner 2000)、SP-STAR(Pevzner 2000)幾種演算法,在序列長度為100~1000的範圍內找尋 (15,4)-signals的執行係數(performance coefficients)如表1.1: 表 1.1 在不同序列長度下不同演算法的執行係數比較 Sequence length(N) 100 200 300 400 500 600 700 800 900 1000 CONSENSUS 0.92 0.94 0.53 0.31 0.29 0.07 0.15 0.09 0.01 0.04 GibbsDNA 0.93 0.96 0.51 0.46 0.29 0.12 0.09 0.34 0.00 0.12 MEME 0.91 0.78 0.59 0.37 0.17 0.10 0.02 0.03 0.00 0.00 4.
(5) WINNOWER(K=2) 0.98 0.98 0.97 0.95 0.97 0.92 0.58 0.02 0.02 0.02 WINNOWER(K=3) 0.98 0.98 0.97 0.94 0.97 0.92 0.90 0.93 0.90 0.88 SP-START 0.98 0.98 1 0.96 0.96 0.84 0.83 0.69 0.64 0.23 以Gibbs、CONSENSUS、MEME三種演算法,在尋找(15,4)的motif 的過程中當上游區的序列不長時或許有幫助,但隨著序列長度的增加 結果越不好(Sze.S 2002);另外雖然SP-STAR和WINNOWER在搜尋. (15,4)-motif可以得到不錯的結果,但SP-STAR在序列長度為1000bp 以及WINNOWER在序列長度大於1300bp時在搜尋(15,4)的motif也是不 盡理想的(Hertz, G. 1999)。 另外Buhler和TOMPA的研究中取了20個序列,每個序列長度為 600bp,來找尋不同類型的(l,d)的motif,比較如:Gibbs、WINNOWER、 SP-STAR、PROJECTION這幾種演算法的performance coefficients, 結果如表1.2(Sze 2002): 表 1.2 不同演算法找尋不同類型 motif 的執行係數比較 l 10 11 12 13 14 15 16 17 18 19. d Gibbs WINNOWER(k=2) SP-STAR PROJECTION 2 0.20 0.78 0.56 0.8 2 0.68 0.9 0.84 0.94 3 0.03 0.75 0.33 0.81 3 0.6 0.92 0.92 0.92 4 0.02 0.02 0.2 0.77 4 0.19 0.92 0.73 0.93 5 0.02 0.03 0.04 0.7 5 0.28 0.03 0.69 0.93 6 0.03 0.03 0.03 0.74 6 0.05 0.03 0.4 0.96 從上表可以發現如Gibbs、WINNOWER、SP-STAR在面臨如(14,4)、. 5.
(6) (16,5)、(18,6) motif的搜尋結果是不如預期的。而另一種演算法 PROJECTION(Buhler J 2002)可突破前面幾種的問題,能在20個序列每 個長度為2000bp中能成功的找到(15,4)的信號,然而在面臨尋找其他 不同類型的motif如:(9,2)、(11,3)、(13,4)、(15,5)、(17,6)仍有 待進一步的改進(Keich 2002)。. 四.研究目的: 目前的演算法中,傳統的方法包括了Gibbs、CONSENSUS、MEME、 TEIRESIAS(Rigoutsos 1998)演算法等等,後來又發展了另幾種演算。 如WINNOWER、SP-STAR、PROJECTION,這些演算法各有其優缺點。而 本研究主要目的是利用自行構思的詳盡式(exhaustive)搜尋來解決 Motif finding的問題,一般人認為詳盡搜尋的方式雖然可以準確的 找到motif,但是當基因序列和搜尋的motif長度過長的時候,在運算 時間容易呈指數的倍數成長,因此本研究透過一些輔助的技巧可以避 免因長度的增加使得運算時間的增長,又能夠兼顧效率及精準的方式 來找到最好的結果。另外嘗試針對目前搜尋motif的演算法所面臨的 問題加以突破解決,例如:突破PROJECTION在搜尋(9,2)、(11,3)、. (13,4)、(15,5)、(17,6)等信號其結果不佳的問題;以及解決當序列 長度(N)增加時所造成搜尋motif執行效率和準確度下降等問題。. 6.
(7) 第二章文獻探討 一. Motif 的類型(Gina 2000) 不同的搜尋方式可以找出不同類型的 motif,大部分一般的 motif 可分隔為:明確樣式(deterministic pattern)和機率樣式 (probabilistic pattern)。明確樣式是指給予一段 motif,在序列 中可以找到這段 motif,也可以沒有找到這段 motif,例如:TATA box 並非在所有序列中可以找的到,但可以確定的是 TATA box 屬於明確 樣式;而機率樣式指在序列中利用機率模式所獲取的樣式。. 1.明確樣式(deterministic pattern) 一般明確樣式的基因序列通常是簡單的形式,如:TATTATAT,然 而motif會根據不同的類型也有其他較為複雜的樣式,一般有下列三 種類型。 A 模糊的字元(Ambiguous character) 模糊的字元表示可能由任何字元所組成,例如:一個 motif A-[C、T]-G 由三個字元所組成,開始字元為 A,結束字元為 G,介 於 A 和 G 中間的可能為 C 或 T,因此組合可能為 ACG 或 ATG,[C、 T]就叫模糊字元。 B 隨意字元(Wild-card) Wild-card 為模糊字元的特殊類型,在蛋白質序列中以 X 表示, 7.
(8) 在核酸序列中以 N 表示,一般也有用「.」表示,當有一連串的 Wild-card 出現時則稱為 Gap。 C 彈性的間隙(Flexible gap) 所謂彈性間隙是指序列中 gap 的長度可以為變動的,例如: i 表 示序列中 gap 最低的長度,j 表示序列中 gap 最高的長度,則 x(i,j) 代表 gap 長度介於 i 和 j 兩者之間的序列都可以,另外一種形式 x(i). 定義為 gap 的長度為一固定數 i。例如:A-X(4)-T-X(1,3)-GC。. 2. 機率式樣式(probabilistic pattern) 明確樣式無法很輕易判斷隱藏在序列樣式中細微的資訊,因此需 要藉由機率模式來考慮序列中所有訊息,假設在一序列位置中可能出 現 A 的比率為 70%,而有 30%的機會出現 G,無法因為 G 比率較低而 忽略可能出現的機率,無論字元出現機率的強弱都必須合併考慮。此 代表性的類型如:位置權重矩陣(position-weight matrix)即屬於機 率式樣式類型。. 二. Motif Finding 的演算法 1.詳盡式搜尋(exhaustive search) 大部分較短的樣式(motif)都藉由詳盡式搜尋方式來達到解決問 題的目的,然而此種方式雖然可以得到不錯的結果,但是當序列長度 8.
(9) 增加時,運算時間相對會增加,且容易呈指數時間成長,所以需要其 他輔助技巧來降低運算時間,下列介紹幾個運用詳盡搜尋的演算法。. A.簡易列舉式搜尋(straightforward enumeration search) 主要是列舉(exhaustive)出所有可能的樣式(pattern),例如一 個樣式的長度為 4 則可能構成 44=256 種樣式的組合,再運用統計來 估計每種組合的顯著性,此種方式運用在短的樣式搜尋可以得到很好 的結果,但當 pattern 的長度增加時,相對的組合可能更多,例如 pattern 的長度為 10 則可能會有 1,048,576 種組合。 相關的研究如(van Helden 1998)利用許多已證實內含有調控特 徵(motif)的基因家族的上游序列以找尋一段長度約 4~9 的序列樣 式。此方法 pattern 的長度是可以隨使用者決定而變動,且不需藉由 反覆(iterative)精練來獲取最佳解,而且整個過程較啟發式 (heuristic)嚴謹(rigorous),但最主要此種方法能獲得詳盡 (exhaustive)準確的解,可全面找出序列中的轉錄訊號,其訊號若不 是轉錄訊號,也可利用找出的其訊號得知基因組中 DNA 全面結構 (global structual)的屬性(properties),加上只利用單一參數 (pattern 的長度)來做分析在速度上較非詳盡的演算方式要快許多。 然而此種方法利用統計方法產生結果,故在判斷 pattern 的顯著性會. 9.
(10) 有高估或低估的問題,例如:在 MET 基因家族中存在一個 CACGTG 的 motif 利用此方式所計算的顯著關係值為 sig=7.0,另外兩個強烈信 號(strongly)的重疊序列(overlapping sequences)TCACGT 其 sig=6.1 與 GTCACG 其 sig=0.7,這兩者皆是可能的信號,但卻有不同 的顯著結果。 同樣類似的研究(Tompa 1999)也是列舉出所有可能的 pattern 在 藉由 Z-sores 的計算來找尋最具有顯著性且沒有 gap 的短序列樣式, 樣本利用 H.influenzae 註冊基因的上游序列序列大約 1700 條每一條 長度約 20bp,以每個短序列以 5-mer 的發生次數來建構一個 table, 並運用下列參數:N 代表序列個數,Ps 表示在長度 L 的序列中 k-mer 至少發生一次的機率,Ns 表示 motif 實際發生次數,來估算 Z-sore= N s − NPs. NPs (1 − Ps ),以 Z-sore 來判斷每個短序列的顯著性。. B.修剪列舉樣式 (Pruning pattern enumeration) 一般簡易列舉式搜尋無法用於搜尋較長且不明確的motif,這時 必須運用樹枝(tree)來分解並列舉出所有可能的樣式,首先從找出較 短的pattern開始,接著再以所找到的較短pattern加以延伸,去尋找 出較長的pattern(Gina 2000)。例如:下列三個序列AAGA AGAA AAGG 以條件設定為support K=2(K:鹼基在序列中所發生的次數),首先運 用樹枝搜尋策略找出所有可能的樣式如圖2.1,在修剪掉無法提供 10.
(11) support的節點,保留support為2以上的如圖2.2。 Empty pattern. A Support 8. AA Support 3. G Support 4. AG Support 3. GG Support 1. GA Support 2. AAA Support 0. AAG Support 2. AGA Support 2. AGG Support 1. AAGA Support 1. AAGG Support 1. AGAA Support 1. AGAG Support 0. GAA Support 1. GAG Support 0. 圖 2.1 樹枝搜尋策略(1). Empty pattern. A Support 8. AA Support 3. G Support 4. AG Support 3. AAG Support 2. GA Support 2. AGA Support 2. 圖 2.2 樹枝搜尋策略(2) 這方面的演算法研究如 Pratt algorithm。此演算法可以讓使用 者自行定義樣式的長度和允許間隙(gap)的數目,此演算法整合了多 重序列演算法,並運用 Heuristic 和 Branch-and-Bound 方式來增加 搜尋 motif 的速度。另外 TEIRESIAS 演算法也有運用到此類型的方式。. 11.
(12) C. TEIRESIAS (Rigoutsos 1998): TEIRESIAS也是一個類似完整式搜尋的方法,它運用類似修剪列 舉樣式的概念,將所有可能的短pattern列出,再將這些短pattern 結合為符合所需長度的pattern。此演算法可以用來尋找內含隨意字 元(wild character)的pattern,一般都以一個(L,W)的pattern表示 之,例如:L=3 and W=5, 一短序列「CD..E」即屬於(3,5)的pattern。 TEIRESIAS演算法對參數有幾個定義: 1. Pattern的開頭和結尾必須是一般字元,而不能是隨意字元。 2. Pattern必須具有L個不含有隨意字元的子字串,且子字串的長 度不可超過W。 3. 另外至少需要有k個序列內含有(L,W)的Pattern。 舉例來說如圖2.3:在下序四個不同的序列中以k=2、L=3、W=4的 條件找尋,也就是必須條件為每條子字串的長度不可超過4,每個子 字串內需含有3個一般字元的可能pattern,則可發現AAC及GT.G這兩 個樣式符合條件。 AA C G T C G A. AAC 和 GT.G. C C C AA C C G. AST. T G G C AA C T. AST. GTG GATAT. GT.G. 圖 2.3TEIRESIAS 演算法範例一 12.
(13) 另一個TEIRESIAS原理是假設P為一個(L,W)的pattern,而且在k 個序列中都有出現的話,則此P的子字串也會是一個(L,W)pattern, 而且也會在k個序列中出現。只要將這些短的pattern 慢慢的加長, 最後就可以得到一個長度符合需要的pattern。例如圖2.4:有三個 Elementary patterns分別為A.AC、ACT和CTC,三個pattern最後可以 構成A.ACTC。 A.AC A.ACT ACT. A.ACTC. CTC. Elementary patterns 圖 2.4 TEIRESIAS 演算法範例二. 然而TEIRESIAS演算法仍有一些問題,例如:執行時間會隨著 pattern組合的增加而呈指數的成長,加上因為TEIRESIAS只允許 mismatches為隨意字元(Wild-card)故對含有模糊字元(ambiguous character)的pattern則無法進行比對,另外TEIRESIAS並無法處理 Gap的長度可以彈性的變動。. D. WINNOWER (Pevzner 2000) WINNOWER是一種搜尋所有可能的pattern組合,並建構一個圖. 13.
(14) 形,運用圖形理論來描繪出motif,圖形內的頂點(vertices)及邊界 (edges)對應到相似的l-mers,兩個相連結的l-mers彼此距離不可超 過2d,若以一個(l,d)的motif為例,(15,4)的motif彼此連結的距離 最 大 到 8 , 另 外 WINNOWER 也 是 算 是 一 種 反 覆 演 算 法 (iterative algorithm)藉由反覆過濾(Filtering)出謬誤(spurious)的邊界來找 出大量的群體(Clique),這種過濾的方式主要有三種模式: 1. Filtering weak vertices (k=1):在K=1的模式中,每個點 (vertex)代表一個Clique,若一點(Clique)與任何區塊中 都至少有一點彼此相關連,則稱此點為擴展的群體 (expandable clique),反之若在圖形G中若該點與周圍鄰 近的點(neighbor)不相關,則此點為謬誤(spurious)的需 將此點過濾。然而此種方式並不是適當的,很容易造成謬 誤無法產生好的結果。 2. Filtering weak edges (k=2):在K=2的模式中Clique為點 與點所形成的邊界(edge)例如:(X,Y)兩點形成的邊界稱為 Clique,假設有另一個點W,W可以和點X與點Y形成邊界, 這三個點可以構成一個循環(cycle)則稱此為expandable clique,利用Clique過濾移除不相關的邊界,這方式其執 行的結果比CONSENSUS, GibbsDNA and MEME要來的好。. 14.
(15) 3. Filtering weak triangles (k=3):此方式能得到最好的結 果,相對的也較複雜,需同時考慮頂點與邊界,在n個點中 n − 2 個expandable clique,也就是每一條 k − 2. 必須至少要有 . 邊界至少必須包含(n–2)個extendable triangles,舉例 來說假設有三個點A、B、C可以構成(A,B)、(A,C)、(B,C) 3 − 2 = 1 個 3 − 2. 三 條 邊 界 , 在 此 模 式 下 每 條 邊 界 必 須 包 含 expandable clique。. 以 K=1為 例 , WINNOWER利 用 建 構 圖形來 過 濾 出 與周 圍鄰 近 的 pattern不相關的謬誤pattern,如下圖:虛線的為謬誤的pattern,而 TTCGT、CGCGT、AGCGT、TGCCT可以集合而成Clique如圖2.4。 TTCGT TTCGT. AACGT. CGCGT. CTCAT. TTACT. 刪除謬誤 patern. CGCGT. AGCGT. AGCGT. TGCCT. TGCCT. 圖 2.4 WINNOWER 演算法範例一. 舉例來說想從下列四個序列中找出(15,4)-motif,首先先標出所 有可能的點(pattern),並將相關的點彼此間連成邊界,再過濾出與 周圍鄰近的點不相關的謬誤的點如下圖,虛線的為謬誤的點,實線代. 15.
(16) 表可以形成 Clique 的點,最後可以找出一個 Expandable clique 如 圖 2.5。 ATGACCGGGATACTGATAGAAGAAAGGTTGGGTATAATGGAGTACGATAA ATGACTTCAATAAAACGGCGGGTGCTCTCCCGATTTTGAGTATCCCTGGG GCAATCGCGAACCAAGCTGAGAATTGGATGTCAAAATAATGGAGTGGCAC GTCAATCGAAAAAACGGTGGAGGATTTCAAAAAAAGGGATTGGACCGCTT. Expandable clique. AGAAGAAAGGTTGGGT. CAATAAAACGGCGGG CAAAATAATGGAGTG. CAAAAAAAGGGATTG. 圖 2.5 WINNOWER 演算法範例二 WINNOWER此種演算法經由排除謬誤訊號的模式來尋找真正的信 號,因此在尋找的過程中謬誤資訊太多不容易判斷,加上一個large 的clique中往往只能找出很少量的signal,所以有時候最後的結果中 常找不到signal(empty no signal)。. 2. 非詳盡式搜尋(non-exhaustive search): 非詳盡搜尋的方式,一般藉由計分函數、機率模式、背景比率等 16.
(17) 技巧盡可能縮小比對範圍,透過反覆精練來達到搜尋的目標,非詳盡 搜尋可以快速的找到所要的答案,但此答案未必是最精確的解,下列 介紹幾個演算方式。. A. CONSENSUS (Hertz 1999): CONSENSUS 有點類似貪婪(greedy)演算法,主要使用 entropy 來計 算 motif 得分把其視為 ungapped 的 patterns,並運用計分矩陣來收 集越來越多的 Patterns instance,並以得分最高的矩陣不斷循環建 構出最有可能的 motif。例如:有三個序列 S1=ACTGA、S2=TAGCG、 S3=CTTGC 若以 4-mer 為一組假設首先利用 S1 中 ACTG 來建構矩陣, 在以得分最高的矩陣來持續進行 pattern 收集。 A 1 0 0 0. A C T G. A C T G T A G C A C T G. 1 0 1 0. 1 1 0 0. 0 0 1 1. T 0 0 1 0. G 0 0 0 1. A C T G A G C G A C T G. 0 1 0 1. A C T G. C 0 1 0 0. A A C 2 0 0 0. C G T 0 1 0 1. 2 0 0 0. T C T 0 1 1 0. G G G 0 0 0 2. 0 1 0 1. 0 1 1 0. A C T G C T T G. 0 0 0 2. A A T 2 0 0 0. A C T G 17. A C T G. 1 1 0 0. 0 1 1 0. C G T 0 1 0 1. T C G 0 1 1 0. G G C 0 0 0 2. 圖 2.6 Consensus 演算法範例. 0 0 2 0. 0 0 0 2. A C T G T T G C A C T G. 1 0 1 0. 0 1 1 0. 0 0 1 1. 0 1 0 1.
(18) Consensus 需利用兩個構面的參數(pattern 長度和期望發生數)來 搜尋,相較於只運用一個構面(pattern 的長度)的方式在執行速度上 要來的慢了許多。. B. Gibbs sampler (Lawrence 1993): Gibbs的做法主要概念是利用反覆式啟發演算法(Iterative heuristic method)(Claverie 1986)配合位置比重矩陣(position weight matrix)來計算每個子字串(subsequence)出現在整個序列的 機率來找到一個最接近的答案。首先使用隨機(randomized)的方式選 擇在各序列上的開始位置,重複去改善起始的Motif,最後再使用位 置比重矩陣(position weight matrix)來計算每個子字串 (subsequence)出現在整個序列的機率,經由反覆的計算來讓位置比 重矩陣趨近最佳結果。因為是運用隨機的方式,所以每次執行時都有 可能會產生不同的結果,通常選擇執行結果為最好的一次。另有一些 Gibbs sampler是採用一些特殊物種的背景分佈(background distributions)像是酵母菌(yeast)來幫助尋找更精確的motif。然而 因為Gibbs sampler所找出來的解趨近於區域(local)最佳化,所以並 無法保證能找出最佳的解,因此若有許多重要的pattern再序列中發 生的頻率不高,那將會有極大的可能會忽略此調控特徵。. 18.
(19) C. MEME(Bailey 1995): MEME 對於找尋 motif 是一種比較常用且流行的方法,它是一種 統計的程序用來預測遺漏的值,可以自動搜尋 motif 長度和估計候選 motif 統計上的顯著,MEME 演算法基本上是假設資料中至少須有一條 相近的子序列(subsequence)藉由這子序列找尋 pattern,方式如下: 1. 設立一個位置權重矩陣(position-weight matrix)的起始模 式(initial model),每一條子序列利用起始模式將序列中 每一個鹼基所對應的一個位置給予一個機率 P(此機率介於 0.5~0.8 間)。 2. 將每個起始的模式運用執行反覆的 EM 演算法來計算相似得 分,再選擇相似得分最高的模式將其運用於反覆 EM 演算法。 MEME運用一種學習演算方式稱為EM(expectation-maximization) 模式的演算法,根據所給的不同的序列利用這種方式計算出在不同位 置出現的機率,方法是首先利用一個motif,這個motif未必是好的, 再經由下列兩步驟反覆精練來獲取更好的motif: 1. E Step:計算出 pattern 在序列中每個位置出現的機率。之後的 計算都根據先前所算出的機率進行修改,最後得出一個位置比重 矩陣。但若是在未知的情況下計算時,所有的參數通常都是使用 亂數產生。. 19.
(20) 2. M Step:根據在 E Step 中位置比重矩陣所算出的機率值,利用 這些機率值做出一個新的機率分布模型,用來算出 pattern 的出 現機率。. D. SP-STAR (Pevzner 2000): 主要是以設計得分函數(Sum of pairs scoring)來找尋motif。例 如,假設得分方式為:match得分+1 ; mismatch得分-1,則序列AAGAT 得分為score(A,A)+score(A,G)+score(A,A)+score(A,T)+score(A,G) +score(A,A)+score(A,T)+score(G,A)+score(A,T)=1-1+1-1-1+1-11-1=-3。再利用得分函數存取收集到的候選motif,從這些當中找出 最好的instance,這些收集到的instance將成為一個起始(initial) 的 motif , 在 利 用 啟 發 (heuristic) 區 域 改 良 方 式 (local improvement),來改良所找到的起始(initial) motif。 然而此種演算法當長度越長無法有個很好的得分估計,舉列來說: 假設match得分+1 ; mismatch得分-1,則AAA和AAG兩者分別得分為3 和-1兩者相差4分,當序列增長後如AAAAA和AAAAG則分別得分為10和2 兩者相差8分,雖然兩種情形都只有一個變異,但在得分卻有所不同。. E. Random projection Approach(Buhler 2002):. 20.
(21) 為 了 增 進 執 行 效 能 Buhler and Tompa’s 發 展 了 另 一 種 演 算 法 PROJECTION,此種方式就可以在序列長度超過1300bp又能精準的找到 Motif。首先先隨機均勻選擇一個k個位置的projection,在一個序列 中以每l-tuple長度為一組,用hash的方式並依照所選擇的k個位置的 字母相同的將其納入bucket,再從bucket中找尋motif,步驟如下: 步驟一如圖2.7:首先選個長度l-tuple,l = 7 (motif size),隨機 選擇k = 4 (projection size)的位置(1,2,5,7),用hash方式每ltuple為一組搜尋序列中可能的motif放入Buckets Input Sequence ...TAGACATCCGACTTGCCTTACTAC.... Buckets. ATCCGAC. GCCTTAC. GCTC. ATGC. 圖 2.7 Random projection 步驟一 步驟二如圖2.8:每個bucket至少有s個序列,假設s=4,計算出矩陣 在利用EM或Gibbs sampler反覆精練(refinement)出最好的motif. ATCCGAC ATGAGGC. A T . C G. 1. 0. . 25. .5. 0. .5. 0 0. 0 0. . 25 .5. . 25 0. 0 1. 0 . 25. 0. 1. 0. . 25. 0. . 25. Frequency matrix. ATAAGTC. EM algorithm. ATGTGAC. Refined matrix ATGC. 圖 2.8 Random projection 步驟二 21. 0 1 0 0 .
(22) F. MULTIPROFILER(Keich 2002): 而在2002年學者針對PROJECTION做了改善發展了MULTIPROFILER演 算法,MULTIPROFILER能夠找尋與PROJECTION發現相同的Motif並達到 99%的準確度,另外可在序列長度為3000中發現約98%的Motif。. 22.
(23) 第三章: 研究設計與方法 一. 研究樣本 一般運用電腦演算法在長度為數百至數千的 DNA 序列中找尋 Motif,會有兩種設計情況: 1. 一個則是給予一組序列已知其中有 motif,去找其共同的特徵, 並在每個序列中找出其可能位置。 2. 另一是蒐集一些已知的調控上游區,而這些上游區以確定其內含 有調控因子,嘗試是否能從中找出內涵的 motif。. 在此我們採用兩種情況分別取不同的樣本: 1.研究樣本一 根據Pevzner (2000)所提的找尋(15,4)的問題,首先從基因資 料中隨機挑取20條序列,每條序列長度為1000bp,在將預先設計好 的motif長度為15bp植入每條序列中隨機的位置,在植入的同時將 設計好的motif隨機改變4個位置以產生變異,以此設計的樣本再利 用本研究的方式來找出真正的解。同樣的,以相同的樣本設計方 式,隨機挑取20條序列,每條序列長度為600bp的設計來嘗試找出. (9,2)、(11,3)、(13,4)、(15,5)、(17,6)這些不同類型的motif。. 23.
(24) 2.研究樣本二 一般使用真實的生物資料去發現真核基因的上游轉錄調控成 分,大都是使用同源同功性的序列,做法是從許多有機體的基因上游 區域去進行測試,我們的做法也是利用一些同源同功性序列,這些序 列都是已發表含有轉錄調控因子的序列: 1. preproinsulin 基因的上游區: preproinsulin 訊號有二類,一 類是從 TRANSFAC 資料庫的訊號(Wingender 1996) ,另一類是 CT II 成分(Boam 1990)。 2. dihydrofolate reductase (DHFR) 基因的上游區: DHFR 的已 知 motif 為非 TATA 轉錄起始訊號(non-TATA transcription start signal)( McInerny 1997)。 3. metallothioneins 基因的上游區: metallothionein 的已發表 motif 則是有三種分別是 MREa 促進子(promoter)、MREd 促進 子、MREf 促進子(Andersen 1987)。 4. c-fos 基因上游區: 3 端 c-fos serum response 成分(3’end of c-fos serum response element)( Means 1990)。 5. 酵母菌 S. cerevisiae 基因 SWI4, CLN3, CDC6, CDC46, CDC47 的促進子區域: ECB 成分,是已知包含有細胞週期獨立的促進 子(cell-cycle-dependent promoter) (McInerny 1997)。. 24.
(25) 二.研究方法: 以詳盡式搜尋方式來尋找motif雖可以得到完美的結果,但相 對的需要付出更多的運算時間,因為運算時間會隨著motif長度的增 加而成指數倍數的成長。因此,我們為了能利用詳盡式搜尋的優點, 又必須減少運算時間上的花費時間,所以自行設計比對的模式並運用 一些輔助的技巧來增加執行效率和減少儲存空間如下: 1. 將資料轉換數據化來處理,除了可減少記憶空間外,在進行 pattern比對上特有的運算方式相對也增加其比對速度。 2. 當pattern長度為6則可能會有46種組合,若又加上變異的組合如 (6,1)就會有更多的組合需要考慮,本研究的方式不需要評估所 有可能的pattern,只須針對序列中內含的pattern進行分析。 3. 在進行pattern篩選時,藉由判斷各序列pattern總數的多寡,以 及各序列中pattern所擁有的GC出現比率來判斷兩序列中的結構 差異程度,利用這兩種輔助方式將有助於過濾刪除一些謬誤的 pattern,以減少比對謬誤pattern的來增加整體比對的速度。 4. 為了避免忽略較不敏感的motif,故允許有2d個變異位置,最後 再配合每個pattern的match次數須有N-1次的條件來做最後過濾 出謬誤pattern的動作,這樣將可以非常準確的找出符合的答案。. 25.
(26) 1.理論步驟: 首先將序列資料做資料轉換,再對每一DNA序列分割成長度為k 的Pattern,每次分割平移1個鹼基,於是每條DNA序列就形成由長度 為k的Pattern所形成的集合,並計算這些小集合位於DNA序列中的位 置與出現次數,在根據這些所獲得的資訊進行搜尋,簡單步驟如下。 步驟一: 首先以(莊振村 2003)的方式將各序列用函數轉換資料 型態,將 Si i=1,2…,N 定義長度為 L 且內含有 Motif 的 DNA 序列,將 Pk 定 義為長度 k 的 pattern,Pk={w1w2…wK},wK ∋ {A、T、G、C},K 可以根據 使用者所需來自訂長度如:(15,4)-motif 則 K=15,則 Si 中最多可能包 含(L-K+1)個 pattern。 步驟二:將序列 Si 以每 k 個為一長度分割成許多 Pattern,以雜 湊(hash)的方式每次分割平移 1 個鹼基,並分別紀錄所有可能 pattern 在序列中的出現次數(count)與出現位置(position)。出現 的次數(count)代表 pattern 在序列中出現的次數,出現位置 (position)代表 pattern 在序列中分佈的位置。每一條序列 Si 只需要 執行一次即可獲得各序列中所有 K 個長度的 pattern 出現次數及位 置,若序列中有 pattern 重複出現,則將其直接歸到先前相同的 pattern。 步驟三:計算各序列 Si 中所有出現的 pattern 總數目,並另外分. 26.
(27) 別計算每個序列中所有的 pattern 內含的 G 與 C 出現比率。 例如:序列 AATCG 若 k=3,則 pattern 可切割為:AAT、ATC、TCG, 此序列中所有 pattern 內含的 GC 比為 3/(3*3)=3/9。 GC 比率=所收集到樣式內含的 GC 出現次數/所收集到的樣式個數*K 步驟四:首先將 pattern 總數目最少的序列與總數次少的序列兩 兩比對,若遇到 pattern 總數相同時,則運用所計算出來的 GC 比率 來判斷序列之間彼此的相似程度,將差異性越大的先挑出來比對,若 無法利用 GC 比來判斷彼此的相似程度時(如:兩序列的 GC 比率相 同),則從序列中隨機挑選來比對。再將所找出的可能的 pattern 逐 步與其他序列中的 pattern 比對,當可能的 pattern 無法在下一序列 中找出有符合(l,d)的 pattern 時,則刪除它,持續到所有序列比對 完成。 在每一次的比對過程除了收集符合條件的 pattern 外,另需建立 一個 Pattern Match Table 來放置收集到符合條件的 pattern,在收 集符合條件的 pattern 過程中,若有因不符合條件的 pattern 則直接 從 Pattern Match Table 刪除,另外並紀錄每一個 pattern 在比對的 過程中與其他 pattern 所累計的 match 次數,若在比對過程中出現相 同的 pattern 時,則將兩相同 pattern 的 match 次數相加並刪除掉其 中一個 pattern。. 27.
(28) 完成所有比對後,再以累計 match 的次數需要大於 N-1(N:序列 (Si)的個數)為條件,這條件主要是假設每個序列中彼此都一定會有 至少一段相似的 pattern,所以每個收集到的 pattern 必須在序列中 與其他 pattern match 的次數 ≥ N-1 次,若有不符合此條件者將其刪 除。. 2.Xor 運算 在兩兩 pattern 比對的過程中我們運用特有的 Xor 運算表 3.1 找 出兩序列中符合(l,d)規則的 pattern,利用 Xor 比對兩序列中同位 置的位元,以"互斥-或"的邏輯來得到結果,並允許最多 2d 個位置產 生變異,收集所符合的 pattern。 [result =] Sequence Si i=1..n 的 pattern Xor Sequence Si i=1..n 的 pattern 表 3.1 Xor 運算. 28.
(29) 3.比對技巧. 尋找序列中 pattern 總 數最少的序列(S1). 判斷 pattern 總數最少的序 列是否只有一 條序列. YES. NO. 以各序列所有 pattern 內含的 GC 比率來找出兩條相似度 差異大的序列. 尋找下一條 pattern 總數最少的序列. YES. 判斷 pattern 總數最少的序 列是否只有一 條序列. NO. YES. 計算 S1 中其內含 的 G 和 C 出現比率 兩序列比對. 兩序列比對. 將剩餘序列所有 pattern 內含的 GC 比與 S1 的 GC 比來找出一條相似 度差異大的序列. YES. 兩序列比對. 是否能找出 一條與 S1 結構 差異大的序列. 是否能找出兩 條結構差異大 的序列. NO. 以隨機方式 挑選兩條序列. 兩序列比對. NO. 以隨機方式 挑選一條序列. 兩序列比對. 收集比對後 符合條件的 所有 pattern. 圖 3.1 比對技巧一. 29.
(30) 收集比對後符 合條件的所有 pattern. 計算符合條件的 pattern 在比對的過程中與其他 pattern 所 match 的次數. 尋找下一條 pattern 總數最少的序列. YES. 判斷 pattern 總數最少的序 列是否只有一 條序列. NO. 計算收集到的所有 pattern 內含的 GC 出現比率 與所有收集 到的 pattern 比對. 將剩餘序列所有 pattern 內含的 GC 比與其比較 來找出一條相似度差異 大的序列. 是否能找出 一條結構差異 大的序列. YES. NO. 以隨機方式 挑選一條序列. 與所有收集 到的 pattern 比對. 與所有收集 到的 pattern 比對. 找出新的符合條件的 pattern,並收集所有符 合條件的 pattern. 將最終所收集到的 pattern 計算其所累計的 match 的次數. 保留此 pattern. Yes. 判斷 pattern 累計 的 match 次數是否 大於 N-1. 圖 3.2 比對技巧二 30. NO. 刪除此 pattern.
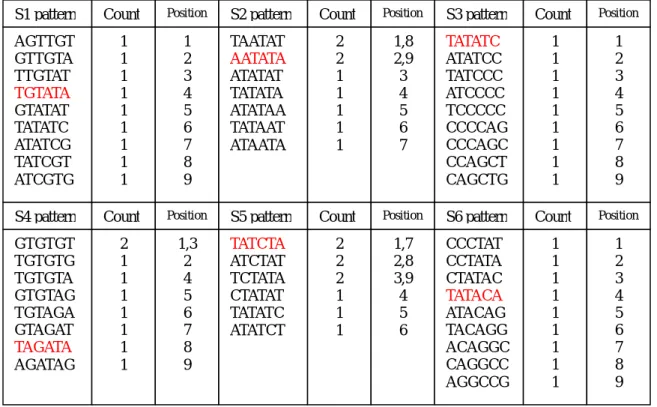
(31) 4.研究方法實例: 例如:假設有 6 條序列每條序列長度為 14bp,分別為 S1=AGTTGTATATCGTG、S2=TAATATATAATATA、S3=TATATCCCCAGCTG、 S4=GTGTGTGTAGATAG、S5=TATCTATATCTATA、S6=CCCTATACAGGCCG,在這 6 條序列中分別植入一個 motif 為 TATATA,每次植入隨機給予一個位 置的變異構成(6,1)-motif,如何從序列中找尋這些(6,1)的 motif。 首先將資料轉換為所要格式,因目標是尋找(6,1)的 motif 故以 k=6 為切割單位,將四條序列分割成許多小的 pattern,並紀錄 pattern 出現次數及位置如表 3.2。 S1 pattern. Count. Position. S2 pattern. Count. Position. S3 pattern. Count. Position. AGTTGT GTTGTA TTGTAT TGTATA GTATAT TATATC ATATCG TATCGT ATCGTG. 1 1 1 1 1 1 1 1 1. 1 2 3 4 5 6 7 8 9. TAATAT AATATA ATATAT TATATA ATATAA TATAAT ATAATA. 2 2 1 1 1 1 1. 1,8 2,9 3 4 5 6 7. TATATC ATATCC TATCCC ATCCCC TCCCCC CCCCAG CCCAGC CCAGCT CAGCTG. 1 1 1 1 1 1 1 1 1. 1 2 3 4 5 6 7 8 9. S4 pattern. Count. Position. S5 pattern. Count. Position. S6 pattern. Count. Position. GTGTGT TGTGTG TGTGTA GTGTAG TGTAGA GTAGAT TAGATA AGATAG. 2 1 1 1 1 1 1 1. 1,3 2 4 5 6 7 8 9. TATCTA ATCTAT TCTATA CTATAT TATATC ATATCT. 2 2 2 1 1 1. 1,7 2,8 3,9 4 5 6. CCCTAT CCTATA CTATAC TATACA ATACAG TACAGG ACAGGC CAGGCC AGGCCG. 1 1 1 1 1 1 1 1 1. 1 2 3 4 5 6 7 8 9. 表 3.2 方法實例 pattern 紀錄表. 31.
(32) 步驟三:6條序列中以S5的pattern數最少為6個,其次分別為S2 的pattern總數為7個和S4的pattern數為8個,其餘的序列皆為9個 pattern數。另外計算各序列中GC比率分別為:S1=15/54、S2=0、 S3=33/54、S4=18/48、S5=6/36、S6=27/54。 步驟四:依照所設計的比對方式,首先從序列中挑選pattern總數 最少的序列,分別為S5與S2來比對找出可能符合(6,1)的pattern,在 比對過程中允許擁有2d的位置變異,也就是只要在6個位置中只要有4 個位置相同即符合條件。收集符合(l,d)並將不符合的pattern刪除, 並紀錄每個符合的pattern在比對的過程中與其他pattern所match的 次數如圖3.3。 S5 pattern TATCTA ATCTAT TCTATA CTATAT TATATC ATATCT. S2 pattern. Pattern Match Table. TAATAT. 符合條件 的 Pattern. 累計 Match 次數. TATCTA ATCTAT TCTATA CTATAT TATATC ATATCT TAATAT AATATA ATATAT TATATA ATATAA TATAAT. 2次 2次 2次 3次 2次 2次 1次 2次 3次 3次 3次 1次. AATATA ATATAT TATATA ATATAA TATAAT ATAATA. 圖 3.3 方法實例 S5 與 S2 比對後 把 S2 與 S5 比對後所收集到的 pattern 與下一條 pattern 總數少 的序列 S4 做比對,若先前所收集到的 pattern 在與 S4 比對過程中遭. 32.
(33) 到刪除則 Pattern Match Table 中的 pattern 也刪除,並加入新發現 的 pattern 到 Pattern Match Table 中如圖 3.4。 符合條件 的 Pattern. S4 pattern. TATCTA. GTGTGT. 符合條件 的 Pattern. 累計 Match 次數. ATCTAT. TGTGTG. TCTATA. TGTGTA. CTATAT. GTGTAG. TATATC. TGTAGA. ATATCT. GTAGAT. TAATAT. TAGATA. TATCTA TCTATA CTATAT TATATC AATATA ATATAT TATATA ATATAA TGTGTA TGTAGA GTAGAT TAGATA AGATAG. 4次 5次 4次 3次 3次 5次 6次 4次 3次 1次 2次 5次 2次. AATATA. Pattern Match Table. AGATAG. ATATAT TATATA. 圖 3.4 方法實例 S5 與 S2 S4 比對後. ATATAA TATAAT. 再將 S2、S5 與 S4 比對後所收集到的 pattern 與下一條 pattern 總數 少的序列做比對,然而因為 S1、S3 與 S6 的 pattern 總數相同,故必須 比較彼此 GC 出現比率來找出相似性差距大的序列,所以將比對 S2、 S4 與 S5 所收集到符合(6,1)的 pattern 計算出 C 和 G 比率為 13/78, 將此值與 S1=15/54 和 S3=33/54 以及 S6=27/54 比較,結果發現 S3 的差 距較大,故比對順序為 S3-S6-S1。 在與S3比對的過程中發現有出現相同的pattern-TATATC,則將比對 之後match的次數相加並刪除掉後來者如圖3.5,之後在比對S6如圖. 33.
(34) 3.6。 符合條件的 Pattern. TATCTA. Pattern Match Table S3pattern. TCTATA. TATATC. CTATAT. ATATCC. TATATC. TATCCC. AATATA. ATCCCC. ATATAT. TCCCCC. TATATA. CCCCAG. ATATAA. CCCAGC. TGTGTA. CCAGCT. TGTAGA. CAGCTG. 符合條件 的 Pattern. 累計 Match 次數. TATCTA TCTATA TATATC AATATA ATATAT TATATA ATATAA TAGATA ATATCC TATCCC. 6次 6次 5+6 次 4次 6次 7次 5次 5次 2次 2次. GTAGAT TAGATA. 圖 3.5 方法實例 S5 與 S2 S4 S3 比對後. AGATAG. 符合條件的 Pattern. TATCTA TCTATA TATATC AATATA ATATAT TATATA ATATAA TAGATA ATATCC TATCCC. Pattern Match Table. S6 pattern CCCTAT CCTATA CTATAC TATACA ATACAG TACAGG ACAGGC. 符合條件 的 Pattern. 累計 Match 次數. TATCTA TCTATA TATATC AATATA ATATAT TATATA ATATAA TAGATA CTATAC TATACA ATACAG. 7次 7次 12 次 5次 7次 8次 7次 6次 2次 6次 2次. CAGGCC AGGCCG. 圖 3.6 方法實例 S5 與 S2 S4 S3 S6 比對後. 34.
(35) 在與S1比對的過程中發現有出現相同的pattern-TATATC,則將比對之 後所match的次數相加並刪除掉後來者如圖3.7。 符合條件的 Pattern. Pattern Match Table S1 pattern. TATCTA TCTATA TATATC AATATA ATATAT TATATA ATATAA TAGATA CTATAC TATACA. AGTTGT GTTGTA TTGTAT TGTATA GTATAT TATATC ATATCG TATCGT ATCGTG. 符合條件 的 Pattern. 累計 Match 次數. TATCTA TCTATA TATATC AATATA ATATAT TATATA ATATAA TAGATA CTATAC TATACA ATACAG TGTATA GTATAT ATATCG TATCGT. 10 次 9次 14+7 次 7次 9次 10 次 9次 8次 3次 8次 3次 7次 3次 3次 1次. ATACAG. 圖 3.7 方法實例 S5 與 S2 S4 S3 S6 S1 比對後 最後計算所有收集到符合(l,d) pattern的累計match次數。並以每個 pattern所累計的match次數為6-1=5次為篩選條件,將不符合條件的 pattern刪除如圖3.8。 Pattern Match Table 符合條件 的 Pattern. 累計 Match 次數. TATCTA TCTATA TATATC AATATA ATATAT TATATA ATATAA TAGATA TATACA TGTATA. 10 次 9次 21 次 7次 9次 10 次 9次 8次 8次 7次. 圖 3.8 方法實例最後結果 35.
(36) 最後所得的結果中自然包含了當初植入的TGTATA、AATATA、 TATATC、TAGATA、TATCTA、TATACA這些motif序列。但是由於是舉例 的資料中,難免的會發生所產生的資料,包含了一組以上的motif。 因此也可能搜尋到可能成為其他motif類型的序列如ATATAT和 ATATAA。. 36.
(37) 第四章:結論 Motif Finding 這個問題引起許多人的興趣,因此也造就了許多搜 尋演算法的產生。例如:完整式搜尋可以對這類搜尋的問題獲取較佳 的結果,但相對的需要增加運算的時間。另外非完整式搜尋做法總是 去盡量想辦法縮小可能比對範圍,一方面藉此來增加速度又可得到一 個可能的解,然而因為獲得的是一可能的解所以在準確性上相較於完 整式搜尋來的低。 而不管是何演算法最終目的都希望可以得到一個快速且能正確 的找出答案,並且又不會因為序列長度的增加、motif長度和變異的 訊號的干擾使得搜尋上更增加困難度。然而面對這問題要如何做到能 兼顧運算的效率,又不會為失去其原有的準確度,這正是目前能不斷 思考改進的地方。 本研究以完整搜尋的方式,將冗長的DNA序列以量化的型態表示, 對每一DNA序列分割成長度為k的Pattern,每次分割可以根據使用者 所設定來平移S個鹼基,於是每條DNA序列就形成由長度為k的 Pattern所形成的集合,並將這K-mer的出現頻率及位置建構成一個 table,利用這table將DNA序列以另一種資料形式呈現,再依據不同 功能需求來配合不同的輔助技巧來達到目的。 此種演算方式利用序列全面的屬性(properties)進行分析,將序列. 37.
(38) 結構中所有可能構成的pattern組合進行比對來找出局部最感興趣的 特徵,這樣的方式較為詳盡並且嚴謹。在使用者方面不需要事先預設 許多條件,例如:pattern的期望發生的次數,使用者只需自行定義 pattern的長度(k)這樣的方式可以減少使用的複雜程度,並且可以針 對不同類型的motif如:(9,2)、(11,3)、(13,4)、(15,5)等來進行精 確的比對搜尋。另外不需反覆的去精鍊結果只需要將所有序列比對過 一次即可得精確的答案,更不會因為序列長度的增加造成精準度的下 降。因此這樣的演算方式改善了完整式搜尋的缺點,可以節省許多不 必要的時間又能夠確保答案的準確性。此外,因為以編碼轉換的方式 來儲存相關序列,因此整個所需要的系統資源也大幅降低。. 38.
(39) 第五章:未來發展 以本研究的方法為基礎的模式,所思考出未來可擴展的部分如 下:. 一.串聯式重複序列(Tandem Repeat) 串聯序列即連續兩個以上pattern的重複串聯成較長的片段屬於 另一種搜尋pattern的問題,通常是由於生物在演化過程所產生的一 種特殊變異現象。在DNA序列上有著相當高的比例會出現連續的重 複,這些序列重複比率程度和疾病有關目前已知的疾病有甘乃迪氏症 又稱為脊髓球肌萎縮症(Kennedy's disease)、杭丁頓氏症 (Huntington’s Disease)及小腦脊髓運動失調症候群 (Spinocerebellar ataxia type I)等。 而利用演算法所建構的 table 將 DNA 序列以另一種資料形式呈 現,此方式亦可推展於 Tandem repeat 的尋找,只需在部分參數上進 行修正,在針對序列中各 pattern 出現的頻率和位置進行紀錄,並在 序列中標示出 pattern 連續出現的位置即可快速判斷出 Tandem Repeat 的位置。 若是要找出影響疾病的序列片段只需利用兩組樣本對照,例如: 將患有 Huntington’s Disease 疾病的人與正常人的基因進行 Tandem Repeat 特徵分析的研究,即可判斷兩群體不同的基因成分及位置所. 39.
(40) 在,藉此找出致病基因。. 二.蛋白質分類 蛋白質分類需根據相似度與胺基酸所共同擁有的pattern,其中 胺基酸共同的pattern,必須要看蛋白質經由比較是否有相同生物醫 學的功能或是其他分子活動的重要特徵的胺基酸共同pattern被保留 下來。然而針對每個新的蛋白質去做分類是一件極為浩大的工程,目 前著名的資料庫Prosite catalog (Bairoch 1991),蛋白質序列間就 是以共同胺基酸(motif)的發生次數來分類,藉由搜尋蛋白質序列中 共同具有的Motif作為各種蛋白質分類的依據,若此蛋白質的序列中 能與過去已分類好的蛋白質家族中有相似的共同序列(motif),則此 蛋白質自然可以歸類到此蛋白質家族中,若是無法任何有相似的 motif的存在,表示此蛋白質序列可能擁有新的功能。 初步上,首先先將現有蛋白質資料庫轉換成自己的資料型態並建 構成資料庫,在根據各個蛋白質家族中所具有的共同序列(motif)建 立索引(index),當有新的蛋白質被發表後只需與索引中的motif比 對,利用若有符合的即可歸類;若當此蛋白質無法對應一個相似的 motif,則可能屬於尚未歸類的蛋白質家族,因此需另作歸類。. 40.
(41) 研究限制 本演算設計的方式目前只能針對 motif 的長度固定,且未含有 Gap 的 motif 進行比對;另外當序列長度越長則需較多的記憶空間來 紀錄所切割出的可能 pattern。. 41.
(42) 參考文獻 1.. 2.. 3. 4.. 5. 6. 7. 8. 9. 10. 11.. 12.. 13.. 14.. Andersen, R.D., Taplitz, S.J., Wong, S., Bristol, G., Larkin, B., and Herschman, H.R. Metal-dependent binding of a factor in vivo to the metal-responsive elements of the metallothionein 1 gene promoter. Molecular and Cellular Biology 7, 3574–81. 1987. Bailey, T., and Elkan, C. Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Machine Learning; 21:51-80. 1995. Bairoch A. PROSITE: A dictionary of sites and patterns in proteins. Nucleic Acids Res. 19, 2241–2245 1991. Boam, D.S.W., Clark, A.R., and Docherty, K. Positive and negative regulation of the human insulin gene by multiple trans-acting factors. J. Biological Chem. 265, 8285–96. 1990. Buhler J. and Tompa M.. Finding motifs using random projections. J Comput Biol,;9(2):225-42. 2002. Claverie, J. M. & Bougueleret, L., Heuristic informational analysis of sequences, Nucl. Acids Res. 14, 179-196, 1986. Gina Holguin, Cheryl Patten.Finding Patterns in Biological Sequences.Project report for CS798g, Fall 2000. Hattori M., Fujiyama A., Taylor T. D., et al. The DNA sequence of human chromosome 21. Nature, 405, 311-319. 2000. Hertz, G., and Stormo, G. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics;15:563-577, 1999. Keich U. and Pevzner P.A. Finding motifs in the twilight zone. Bioinformatics,;18(10):1374-81. 2002. Lawrence, C.; Altschul, S.; Boguski, M.; Liu, J.; Neuwald, A.; and Wootton, J. Detecting subtle sequence signals: a Gibbs sampling strategy for mul-tiple alignment. Science..262:208-214. 1993. McInerny, C.J., Partridge, J.F., Mikesell, G.E., Creemer, D.P., and Breeden, L.L. A novel Mcm1-dependent element in the SWI4, CLN3, CDC6, and CDC47 promoters activates M/G1-speci. c transcription. Genes and Development 11, 1277–88 1997 Means, A.L., and Farnham, P.G. 1990. Transcription initiation from the dihydrofolate reductase promoter is positioned by hip1 binding at the initiation site. Mol. Cell. Biol. 10, 653–61 1990 Natsan, S., and Gilman, M. 1995. YY1 facilitates the association of serum response factor with the c-fos serum response element. Mol. Cell. Biol. 15, 42.
(43) 15. 16.. 17. 18.. 19.. 20.. 21.. 5975–82 1995. Pevzner PA, Sze SH. Combinatorial approaches to finding subtle signals in DNA sequences Proc Int Conf Intell Syst Mol Biol.;8:269-78 2000. Rigoutsos, I. and Floratos, A. Combinatorial pattern discovery in biological sequences: The TEIRESIAS algorithm. Bioinformatics, Published erratum appears in Bioinformatics. 14(1):55~67 1998. Sze.S. Gelfand.M. and Pevzner.P. Finding weak motifs in DNA sequences. In proceedings of Pacitic Symposium on Biocomputing.;235-246 2002. Tompa, M. An exact method for finding short motifs in sequences, with application to the ribsomoe binding site problem. In proceedings of the 7th International Conference on Intelligent Systems for Molecular Biology (ISMB), page: 262-271, 1999. van Helden, J., Andre,B., and Collado-Vides, J. Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. Journal of Molecular Biology, 281(5):827~832. 1998. Wingender, E., Dietze, P., Karas, H., and Knüppel, R. TRANSFAC: A database on transcription factors and their DNA binding sites. Nucl. Acids Res. 24, 238–41 1996. 莊振村、楊鎮嘉、黃梅芬 “人類第22對染色體核苷酸各種序列組合頻率統 計分析”慈濟醫學雜誌 2004 16(3). 43.
(44)
數據
Outline
相關文件
sort 函式可將一組資料排序成遞增 (ascending order) 或 遞減順序 (descending order)。. 如果這組資料是一個行或列向量,整組資料會進行排序。
所以 10 個數字 個數字 個數字 個數字 pattern 就產生 就產生 就產生 就產生 10 列資料 列資料 列資料 列資料 ( 每一橫 每一橫 每一橫
Tai Po Old Market Public School (Plover Cove)
• 問你當前的序列裡的第 k個人是誰 (區間詢問?). •
• 問你當前的序列裡的第 k個人是誰 (區間詢問?). •
也就是設定好間隔時間(time slice)。所有的 程序放在新進先出的佇列裡面,首先CPU
[r]
求出 Select Case 運算式之值,並逐一與 Case 運算式值串列比對,若符合則執行該 Case 之後的敘述區段。1. 如果所有的
