doi:10.1152/ajpheart.00417.2004
289:H220-H227, 2005. First published 21 January 2005;
Am J Physiol Heart Circ Physiol
Chang
Tsung-Ming Lee, Mei-Shu Lin, Tsai-Fwu Chou, Chang-Her Tsai and Nen-Chung
hypertrophy in spontaneously hypertensive rats
Effect of pravastatin on development of left ventricular
You might find this additional info useful...
43 articles, 24 of which can be accessed free at: This article cites
http://ajpheart.physiology.org/content/289/1/H220.full.html#ref-list-1 5 other HighWire hosted articles
This article has been cited by
[PDF]
, August 24, 2008; .
QJM
J. Sánchez-Quiñones, F. Marín, V. Roldán and G.Y.H. Lip
The impact of statin use on atrial fibrillation
[PDF] [Full Text] [Abstract]
, November , 2008; 101 (11): 845-861.
QJM
J. Sánchez-Quiñones, F. Marín, V. Roldán and G.Y.H. Lip
The impact of statin use on atrial fibrillation
[PDF] [Full Text] [Abstract]
, August 19, 2009; 54 (3): 591-597.
Hypertension
Chang, Dae-Won Sohn, Byung-Hee Oh and Young-Bae Park
Sung-A Chang, Yong-Jin Kim, Hye-Won Lee, Dae-Hee Kim, Hyung-Kwan Kim, Hyuk-Jae
in Hypertensive Heart With Established Left Ventricular Hypertrophy
Effect of Rosuvastatin on Cardiac Remodeling, Function, and Progression to Heart Failure
[PDF] [Full Text] [Abstract]
, November 30, 2009; .
Nephrol. Dial. Transplant.
Eun Hui Bae, In Jin Kim, Jeong Woo Park, Seong Kwon Ma, Jong Un Lee and Soo Wan Kim
salt hypertensive rats −
Renoprotective effect of rosuvastatin in DOCA
[PDF] [Full Text] [Abstract]
, April , 2010; 25 (4): 1051-1059.
Nephrol. Dial. Transplant.
Eun Hui Bae, In Jin Kim, Jeong Woo Park, Seong Kwon Ma, Jong Un Lee and Soo Wan Kim
salt hypertensive rats −
Renoprotective effect of rosuvastatin in DOCA
including high resolution figures, can be found at: Updated information and services
http://ajpheart.physiology.org/content/289/1/H220.full.html
can be found at: AJP - Heart and Circulatory Physiology
about Additional material and information
http://www.the-aps.org/publications/ajpheart
This infomation is current as of April 7, 2011.
ISSN: 0363-6135, ESSN: 1522-1539. Visit our website at http://www.the-aps.org/.
Physiological Society, 9650 Rockville Pike, Bethesda MD 20814-3991. Copyright © 2005 by the American Physiological Society. intact animal to the cellular, subcellular, and molecular levels. It is published 12 times a year (monthly) by the American
lymphatics, including experimental and theoretical studies of cardiovascular function at all levels of organization ranging from the publishes original investigations on the physiology of the heart, blood vessels, and AJP - Heart and Circulatory Physiology
on April 7, 2011
ajpheart.physiology.org
Effect of pravastatin on development of left ventricular hypertrophy
in spontaneously hypertensive rats
Tsung-Ming Lee,1Mei-Shu Lin,2Tsai-Fwu Chou,3Chang-Her Tsai,4and Nen-Chung Chang5
1Cardiology Section, Department of Medicine, Taipei Medical University and Chi-Mei Medical Center, Tainan;2National
Taiwan University and Department of Pharmacy, National Taiwan University Hospital, Taipei;3Department of Surgery,
Municipal Jen-Ai Hospital, Taipei;4Cardiology Section, Department of Surgery, National Taiwan University Hospital,
Taipei; and5Cardiology Section, Department of Medicine, Taipei Medical University and Hospital, Taipei, Taiwan
Submitted 4 May 2004; accepted in final form 19 January 2005
Lee, Tsung-Ming, Mei-Shu Lin, Tsai-Fwu Chou, Chang-Her Tsai, and Nen-Chung Chang. Effect of pravastatin on development
of left ventricular hypertrophy in spontaneously hypertensive rats.
Am J Physiol Heart Circ Physiol 289: H220 –H227, 2005. First
published January 21, 2005; doi:10.1152/ajpheart.00417.2004.—En-dothelin (ET)-1 has been implicated in the development of cardiac hypertrophy. We investigated the effect of pravastatin on development of ventricular hypertrophy in spontaneously hypertensive rats (SHR) and whether the attenuated hypertrophic effect was via reduced ET-1 expression. Normolipidemic SHR were treated with one of the fol-lowing therapies for 8 wk: vehicle, the nonselective ET receptor antagonists bosentan, pravastatin, mevalonate, hydralazine, or combi-nation of pravastatin⫹ mevalonate from the age of 8 wk at the very early stage of cardiac hypertrophy. Treatment with bosentan and pravastatin significantly decreased left ventricular mass index for body weight and cardiomyocyte sizes isolated by enzymatic dissoci-ation. The myocardial ET-1 levels and preproET-1 mRNA assessed using real-time quantitative RT-PCR were significantly higher (both
P ⬍ 0.001) in the SHR compared with Wistar-Kyoto rats. The
increased tissue ET-1 levels can be inhibited after pravastatin admin-istration. Immunohistochemical analysis confirmed the changes of ET-1. Left ventricular mass index for body weight correlated posi-tively with tissue ET-1 levels (P⫽ 0.0004). A dissociation between the effects of blood pressure and cardiac structure was noted, because pravastatin and hydralazine reduced arterial pressure similarly. Prav-astatin-induced effects were reversed by the addition of mevalonate. In conclusion, these results suggest a crucial role of cardiac endothelin system in the early development of ventricular hypertrophy in the SHR. Pravastatin is endowed with cardiac antihypertropic properties that are independent of its hemodynamic and hypolipidemic effects and appear to be related to their capacity to decrease cardiac ET-1 levels, which is linked to mevalonate metabolism.
cardiomyocytes; endothelin-1; immunohistochemistry
EPIDEMIOLOGICAL STUDIES have demonstrated that increased left
ventricular (LV) mass is a risk factor of cardiac morbidity and mortality in patients with hypertension (24). Previous data revealed that LV mass regression reduced cardiovascular com-plications (11). Hypertrophied myocardium has been shown to generate arrhythmias more readily than normal tissue. Agents that cause the regression of ventricular hypertrophy have been shown to decrease the susceptibility of ventricular arrhythmias (2). Thus the use of LV mass to stratify risk and target antihypertensive therapy has been proposed. To more effec-tively prevent cardiac hypertrophy and more successfully apply therapeutic interventions, it is important to better understand
the factor(s) involved in ventricular growth at the early stage of cardiac hypertrophy, rather than at the established stage, so that therapeutic interventions can be more successfully applied.
Endothelin (ET)-1 gene expression is enhanced in heart in spontaneously hypertensive rats (SHR) (36), an experimental model of genetic hypertension. In SHR, pressure overload initially developed at 8 wk, followed by an active phase of hypertrophic growth between 16 and 20 wk (3). ET-1, a potent growth-promoting peptide derived from endothelial cells, is also produced by cardiac myocytes (30). ET-1 acts as a key autocrine/paracrine mediator to trigger the hypertrophic signal-ing pathways by activation of extracellular signal-regulated kinase in myocardium via activation of ETA/Breceptor (43).
ET receptor blockade attenuates ventricular hypertrophy (9). Thus treatment with ET antagonists appears to be an attractive alternative to attenuate ventricular hypertrophy.
Pravastatin, an inhibitor of 3-hydroxy-3-methyglutaryl-co-enzyme A reductase (statin), blocks the mevalonate pathway, reducing cholesterol biosynthesis. Epidemiological studies have demonstrated that the benefit of statin treatment extends to patients with normocholesterolemia (41). Statins exert pleio-tropic properties and interfere with signal pathways for hyper-trophy, effects that may contribute to their beneficial effects on ventricular hypertrophy (41). Statins have been shown to inhibit ET-1 and to subsequently prevent cellular hypertrophy (41). There are many kinds of statins used clinically that show differences in pharmacological lipophilicity, structure, and solubility. Because lipophilic statins may enter cardiomyo-cytes, there was a difference in their potency to inhibit meva-lonate synthesis in extrahepatic cells (42), which could lead to a different effect on cardiac hypertrophy between lipophilic and hydrophilic statins. Thus, although lipophilic statins have been shown to attenuate ventricular hypertrophy (1, 12), the effects cannot be extrapolated to hydrophilic statins. Whether pravastatin, a hydrophilic statin, has an effect on ventricular hypertrophy remains unknown in SHR. The purpose of this study was to investigate the effect of pravastatin on cardiomy-ocyte sizes and the role of circulating and cardiac ET systems at the early hypertrophic phase of SHR. To further confirm the role of chronic endothelin activation in the prevention of pravastatin-induced ventricular hypertrophy, we assessed the effect of the nonselective ETA/Breceptor antagonist bosentan.
We also explored the downstream functional significance of reduced ventricular hypertrophy by ventricular pacing.
Address for reprint requests and other correspondence: N.-C. Chang, Car-diology section, Dept. of Medicine, Taipei Medical Univ. and Hospital, 252 Wu-Hsing St., Taipei, Taiwan (E-mail: ncchang@tmu.edu.tw).
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. First published January 21, 2005; doi:10.1152/ajpheart.00417.2004.
on April 7, 2011
ajpheart.physiology.org
METHODS
Experimental animals. Male normocholesterolemic SHR had free
access to food and water. At 8 wk of age at the very early stage of cardiac hypertrophy, SHR were randomly allocated to one of six groups and treated for 8 wk with vehicle, the nonselective ET receptor antagonist bosentan (10 mg/kg per day), pravastatin (5 mg/kg per day), mevalonate (50 mg/kg per day), hydralazine (10 mg/kg per day), or a combination of pravastatin and mevalonate. The dose of prava-statin used in this study was derived from previous experiments in which pravastatin restored the infarct size-limiting effect of ischemic preconditioning in hyperlipidemic rabbits (39). Because pravastatin has a blood pressure-lowering effect, hydralazine was used to deter-mine the degree to which this decrease in blood pressure is respon-sible for attenuating cardiac hypertrophy. To further confirm the role of chronic ET activation in the progression of ventricular hypertrophy, we used the nonselective ETA/Breceptor antagonist bosentan at a dose
of 10 mg/kg per day (Actelion Pharmaceuticals, Allschwil, Switzer-land). The therapeutic efficacy of this dose has been previously demonstrated without hypotensive effects (27). The drugs were dis-solved in drinking water, and the concentration was adjusted for the daily water intake and body weight to obtain the target dosage. In each treated group, drugs were withdrawn⬃24 h before the experiments to eliminate their pharmacological actions. One control group of age-matched normotensive Wistar-Kyoto (WKY) rats received no treat-ment. All the procedures were approved by the Institutional Animal Care and Use Committee and the Institutional Review Board of Chi-Mei Medical Center.
Hemodynamics and induced arrhythmias. Functional parameters
were measured in anesthetized rats at the end of the study. Using a 2-F micromanometer-tipped catheter (model SPR-407; Miller Instru-ments, Houston, TX) inserted through the right carotid artery, we measured LV systolic and diastolic pressure as the mean of measure-ments of five consecutive pressure cycles. The maximal rate of contraction (⫹dP/dt) and relaxation (⫺dP/dt) was measured. Next, the heart was rapidly excised and suspended for retrograde perfusion with a Langendorff apparatus. Each heart was perfused with a noncircu-lating modified Tyrode solution containing (in mM) 117.0 NaCl, 23.0 NaHCO3, 4.6 KCl, 0.8 NaH2PO4, 1.0 MgCl2, 2.0 CaCl2, and 5.5
glucose, equilibrated at 37°C and oxygenated with a 95% O2-5% CO2
gas mixture. The perfusion medium was maintained at a constant temperature of 37°C with a peristaltic pump at a constant flow of 4 ml/min. Epicardial electrograms were recorded using an atraumatic unipolar electrode placed on the epicardial surface of the right atrium and anterior LV wall 2 mm below the circumflex artery. A bipolar pacing electrode was placed near the apex of the heart on the anterior epicardial surface of the right ventricle. Atrial and ventricular epicar-dial electrocardiograms were continuously displayed on a Gould recorder at a chart speed of 500 mm/s and a Hewlett-Packard oscil-loscope (model 54503A) at a sweep speed of 100 mm/s.
After isolation, the hearts were observed for 20 min to allow stabilization of hemodynamics. During the period, spontaneous ar-rhythmias were recorded. Induced arar-rhythmias were effected using an electrical Bloom stimulator. Stimulation intensity was twice the threshold, and stimulus length was 5 ms. The protocol for pacing and an arrhythmia scoring system were used as previously described (22).
Immunohistochemical analysis of ET-1. To investigate the spatial
distribution of ET-1, we performed immunohistochemical staining on LV muscle. At completion of the electrophysiological tests, the heart was then rapidly divided into right and left atria and right and left ventricles. Each tissue was then weighed individually. LV muscle was snap frozen in liquid nitrogen and embedded in OCT compound (Tissue-Tek), and cryosections were prepared at a thickness of 7m. The slides containing the sectioned tissues were rehydrated in 0.01% sodium bicarbonate at pH 7.4. Sections were blocked with 0.1 mM L-lysine in PBS containing 0.1% Triton X-100 for 45 min. Tissues were incubated with a rabbit polyclonal anti-ET-1 antibody
(Immuno-Biological Lab, Gunma, Japan) at 1:200 dilution in 0.5% BSA in PBS overnight at 37°C. Immunostaining with ET-1 antibodies was per-formed using a standard immunoperoxidase technique (N-Histofine Simple Stain MAX PO kit; Nichirei, Tokyo, Japan). The antibody used had been tested for specificity in the rat. Negative controls were performed by omitting the primary antibody.
Real-time RT-PCR. Real-time quantitative RT-PCR was
per-formed from LV samples obtained with the TaqMan system (Prism 7700 sequence detection system; PE Biosystems) as described previously (23). For ET-1, the primers were 5 ⬘-TGCTGTTTGTG-GCTTTCCAA-3⬘ and 5⬘-CAAGGATCGCTTAGACCTAGAAGG-3⬘. For glyceraldehydes-3-phosphate-dehydrogenase (GAPDH), the primers were 5⬘-CTTCACCACCATGGAGAAGGC-3⬘ and 5⬘-GGC-ATGGACTGTGGTCATGAG-3⬘. For quantification, ET-1 expres-sion was normalized to the expressed housekeeping gene GAPDH. Reaction conditions were programmed on a computer linked to the detector for 40 cycles of the amplification step.
Plasma and tissue levels of ET-1. Because of a local release of
ET-1 and a poor correlation between plasma and tissue ET-1 levels, blood samples from the aortic root and the tissue from the LV were obtained for measurements of systemic and local ET-1 levels at the end of the study. For the measurement of cardiac ET-1 levels, the apical two-thirds of the LV was immediately homogenized with a Polytron homogenizer for Triton-X, boiled for 7 min, and centrifuged at 20,000 g for 30 min at 4°C. After measurement of the protein concentration (in pg/mg), the supernatant was stored at⫺70°C until use. Plasma ET-1 concentration was measured by collecting 4 ml of blood in test tubes containing 2% EDTA (80 l/ml blood). Blood samples were immediately centrifuged at 3,000 g for 10 min, and the plasmas were stored at⫺70°C until further analysis was performed. ET-1 was measured using immunoassay (R&D Systems, Minneapolis, MN). Plasma (1 ml) was acidified with 3 ml of 4% acetic acid, and ET-1 was extracted with a Sep-Pak C-18 cartridge. The detection limit was 1 pg/ml for ET-1. Intra-assay and interassay coefficients of variation were 4.5 and 6.6%, respectively.
Cell isolation. Because cardiac hypertrophy is a combination of
reactive fibrosis and myocyte hypertrophy, we measured cardiomyo-cyte sizes from the LV in addition to using myocardial weight to avoid the interference of nonmyocytes. Because immunohistochemical anal-ysis does not permit quantitation of cardiomyocyte sizes, additional groups of rats (n⫽ 5 in each group) were used for measurement of cell sizes. Myocytes were enzymatically isolated according to previ-ously described techniques (23). Briefly, the rats were heparinized, and heart was excised and perfused at a constant flow of 8 ml/min by using a modified Langendorff technique at 37°C with a nominally Ca2⫹-free, oxygenated Tyrode solution (pH 7.4) containing (in mM) 137 NaCl, 5.4 KCl, 1.1 MgCl2, 11 dextrose, and 10 HEPES. After 5
min of equilibration, the perfusion was changed to the same solution containing 0.34 mg/ml collagenase (type II; Sigma Chemical, St. Louis, MO). After 8 –10 min of digestion, the residual enzyme-containing solution was cleaned by 5 min of perfusion with 0.2 mM Ca2⫹Tyrode solution. The heart was then removed from the cannula, and the LV was mechanically dispersed. Random high-power fields of the rodlike relaxed myocytes with clear striations were selected in phase-contrast illumination mode of confocal microscopy (LSM-410 Invert; Zeiss) to eliminate selection bias. At least 100 cells from each section were selected for measurement of cell area, length, and width, and mean values were used as an individual value for each section.
Statistical analysis. Results are presented as means⫾ SD.
Differ-ences among the groups of rats were tested using two-way ANOVA. Subsequent analysis for significant differences between the two groups was performed with a multiple comparison test (Scheffe´’s method). Electrophysiological data (scoring of programmed electrical stimulation-induced arrhythmias) were compared using a Kruskal-Wallis test followed by a Mann-Whitney test. An interaction term of pravastatin and mevalonate effects was incorporated into the model. Correlation between the degree of attenuated cardiomyocyte
hyper-H221
PRAVASTATIN AND LV MASS
AJP-Heart Circ Physiol•VOL 289 • JULY 2005 •www.ajpheart.org
on April 7, 2011
ajpheart.physiology.org
trophy and the tissue ET-1 levels was assessed using Pearson’s correlation coefficient. The significant level was assumed at a value of
P⬍ 0.05.
RESULTS
Pravastatin does not lower serum cholesterol in rats, consis-tent with the notion that compensatory increases in hepatic enzyme production were observed in rats treated with statins. These data indicate the nonlipid effect of pravastatin on ven-tricular hypertrophy.
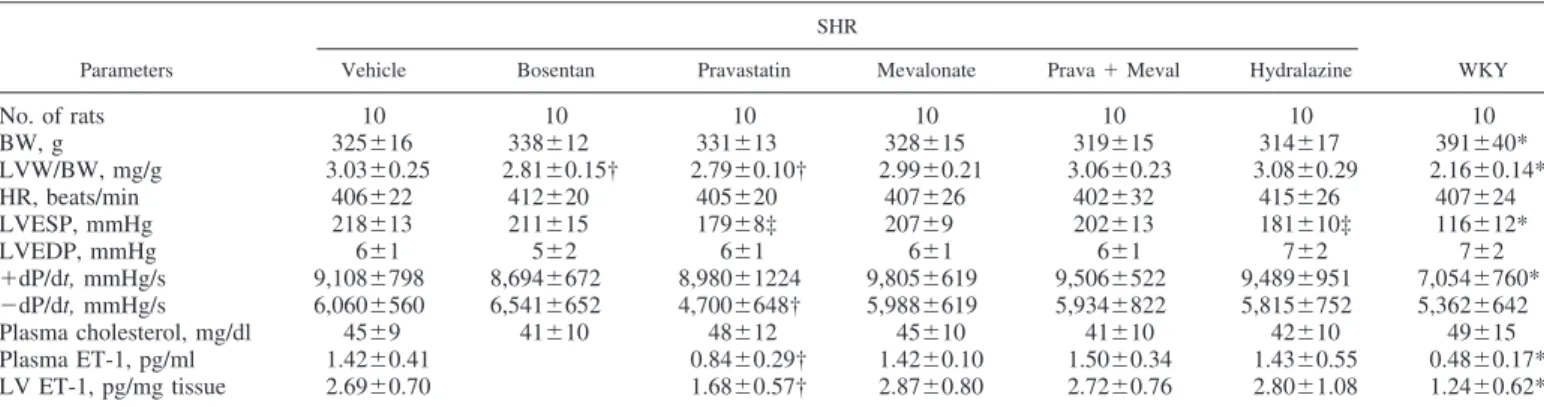
Hemodynamic data. During the 8-wk treatment period,
prav-astatin decreased systolic blood pressure by 39 mmHg without, however, normalizing it (Table 1). Chronic antihypertensive treatment for 8 wk with hydralazine led to a similarly lower systolic blood pressure compared with pravastatin-treated SHR. Heart rate did not differ among all groups. The maximal rate of LV pressure rise (⫹dP/dt) was significantly increased in SHR compared with WKY rats.
Morphometric studies. Chronic drug treatment did not
mod-ify the increase of body weight with age. Table 1 shows LV weight-to-body weight ratios of animals from each group. Before treatment, the SHR groups had equivalent baseline body weights. At the end of the treatment period, body weights were similar in the SHR groups. Untreated SHR showed LV hypertrophy, with a significant increase of 40% in the ratio of LV weight to body weight compared with WKY rats. Compared with untreated SHR, bosentan and pravastatin decreased heart weight-to-body weight ratios by 7 and 8%, respectively (P⫽ 0.02 and 0.01). Despite producing similar antihypertensive effects, hydralazine given to SHR showed a significant increased LV weight compared with those treated with pravastatin.
To characterize the cardiac hypertrophy on a cellular level, we isolated cardiomyocytes from different treated groups (Ta-ble 2). The cells isolated from untreated SHR group were significantly larger by 26% compared with those from WKY hearts (3,803⫾ 213 m2in SHR group vs. 3,010⫾ 192 m2
in WKY group, P ⬍ 0.0001). Bosentan and pravastatin re-duced cell areas 19 and 17%, respectively, compared with the untreated SHR group (both P⬍ 0.0001). The cell width of the bosentan- and pravastatin-treated myocytes was significantly smaller than that of vehicles (8 and 13%, respectively, both
P ⬍ 0.05). Conversely, the rats to which mevalonate was
administered developed cardiomyocyte hypertrophy greater than that in the pravastatin-treated group alone.
Electrophysiological stimulation. To further elucidate the
physiological effect of attenuated cardiomyocyte hypertrophy, we performed ventricular pacing. Arrhythmia scores in WKY rats were very low (0.2⫾ 0.4). In contrast, ventricular tachy-arrhythmias consisting of ventricular tachycardia and ventric-ular fibrillation were inducible by programmed stimulation in SHR (Fig. 1). Bosentan and pravastatin treatment showed a significant reduction in arrhythmia scores compared with those treated with the vehicle, mevalonate, hydralazine, and a com-bination of pravastatin and mevalonate.
Circulating and myocardial ET-1 levels and preproET-1 mRNA. Circulating and myocardial ET-1 levels are shown in
Table 1. There were significantly higher circulating and myo-cardial ET-1 levels in SHR compared with WKY rats (1.42⫾ 0.41 vs. 0.48 ⫾ 0.17 pg/ml for circulating ET-1 levels, P ⬍ 0.0001; 2.69 ⫾ 0.70 vs. 1.24 ⫾ 0.62 pg/mg tissue for myo-cardial ET-1 levels, P ⫽ 0.0001). Pravastatin administration attenuated the increased ET-1 concentrations, which were reversed after the addition of mevalonate.
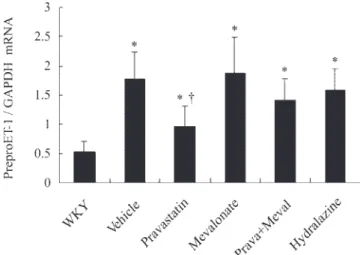
As shown in Fig. 2, myocardial levels of preproET-1 mRNA were on average 3.3-fold higher in SHR than in WKY rats (P ⬍ 0.0001). This overexpression was attenuated by prava-statin administration. Thus the mRNA levels of preproET-1 changed in parallel to the tissue peptide levels, implying that the production of preproET-1 is a critical regulation step for its local activation.
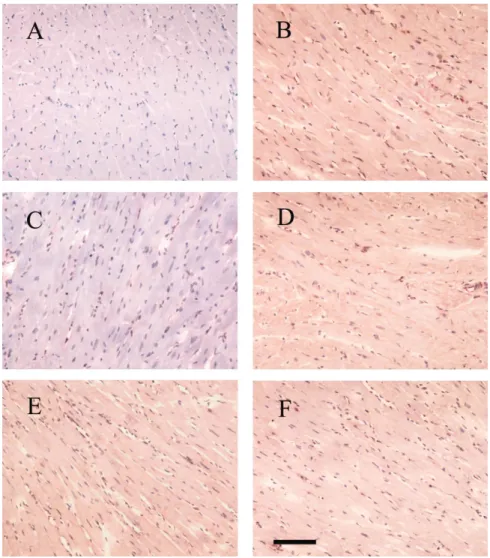
Immunohistochemical analyses. To reveal the localization of
cells containing the mature ET-1 peptide, we performed im-munohistochemistry. Immunohistochemical analysis of the LV revealed the presence of ET-1 immunoreactivity in the myo-cardial tissue (Fig. 3). Stronger ET-1 signals in vehicle-treated SHR were observed than in the same region of WKY rats. The intensity of the immunoreaction was reduced in pravastatin-treated groups compared with that in the vehicle group.
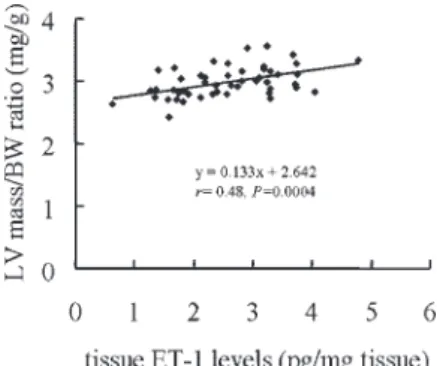
Correlation. As shown in Fig. 4, a significant reverse
cor-relation was found between LV weight-indexed body weight and regional ET-1 levels [LV weight/body weight (mg/g) ⫽ 0.133⫻ tissue ET-1 (pg/mg tissue) ⫹ 2.642; r ⫽ 0.48, P ⫽ 0.0004]. No correlation was detected between LV weight-indexed body weight and blood pressure.
Table 1. Cardiac morphology, hemodynamics, cholesterol, and plasma and tissue ET-1 concentrations among groups
Parameters
SHR
WKY Vehicle Bosentan Pravastatin Mevalonate Prava⫹ Meval Hydralazine
No. of rats 10 10 10 10 10 10 10 BW, g 325⫾16 338⫾12 331⫾13 328⫾15 319⫾15 314⫾17 391⫾40* LVW/BW, mg/g 3.03⫾0.25 2.81⫾0.15† 2.79⫾0.10† 2.99⫾0.21 3.06⫾0.23 3.08⫾0.29 2.16⫾0.14* HR, beats/min 406⫾22 412⫾20 405⫾20 407⫾26 402⫾32 415⫾26 407⫾24 LVESP, mmHg 218⫾13 211⫾15 179⫾8‡ 207⫾9 202⫾13 181⫾10‡ 116⫾12* LVEDP, mmHg 6⫾1 5⫾2 6⫾1 6⫾1 6⫾1 7⫾2 7⫾2 ⫹dP/dt, mmHg/s 9,108⫾798 8,694⫾672 8,980⫾1224 9,805⫾619 9,506⫾522 9,489⫾951 7,054⫾760* ⫺dP/dt, mmHg/s 6,060⫾560 6,541⫾652 4,700⫾648† 5,988⫾619 5,934⫾822 5,815⫾752 5,362⫾642 Plasma cholesterol, mg/dl 45⫾9 41⫾10 48⫾12 45⫾10 41⫾10 42⫾10 49⫾15 Plasma ET-1, pg/ml 1.42⫾0.41 0.84⫾0.29† 1.42⫾0.10 1.50⫾0.34 1.43⫾0.55 0.48⫾0.17* LV ET-1, pg/mg tissue 2.69⫾0.70 1.68⫾0.57† 2.87⫾0.80 2.72⫾0.76 2.80⫾1.08 1.24⫾0.62* Values are means⫾ SD. BW, body weight; ET-1, endothelin-1; HR, heart rate; LVEDP, left ventricular end-diastolic pressure; LVESP, left ventricular end-systolic pressure; LVW, left ventricular weight; Meval, mevalonate; Prava, pravastatin; WKY, Wistar-Kyoto rats. *P⬍ 0.05 compared with spontaneously hypertensive rat (SHR) groups. †P⬍ 0.05 compared with vehicle-, mevalonate-, combination-, and hydralazine-treated groups. ‡P ⬍ 0.05 compared with vehicle-, bosentan-, mevalonate-, and combination-treated groups.
on April 7, 2011
ajpheart.physiology.org
DISCUSSIONS
The present study demonstrates for the first time that prav-astatin prevents ongoing cardiac hypertrophy at the early hy-pertrophic phase of SHR, independently of hemodynamic and lipid changes. The present study demonstrates three novel findings through combined use of molecular, biochemical, and morphological methods. First, ET-1 levels are responsible for cardiac remodeling in SHR. In this study, width and two-dimensional cell surface area of myocytes in SHR were sig-nificantly greater than those in WKY rats. Cell width was significantly greater in SHR cardiomyocytes, indicative of the pressure pattern of hypertrophy that is consistent with previous studies (5). Hypertrophic changes in the heart of SHR were observed at the cellular level as well as the organ levels. SHR, acting through the corresponding increase of ventricular pre-proET-1 mRNA and ET-1 protein levels, developed ventricular hypertrophy. Our results were consistent with those of a recent study showing that the density of ETB receptor is increased
during active hypertrophy at the age of 16 –24 wk in SHR (21). This notion was further confirmed by directly administering an ET receptor blocker. Second, the beneficial effects of prava-statin on the prevention of ventricular hypertrophy may be associated with reduced expression of preproET-1 mRNA and ET-1 protein levels independently of hemodynamic changes. Hydralazine did not reduce any of the parameters despite similar inhibition of blood pressure. Third, the inhibitory effect of pravastatin is specifically prevented by the addition of exogenous mevalonic acid, indicating that the inhibitory ef-fects of the drug are caused by blockade of the pathways leading to isoprenoid synthesis. These combined results
indi-cate that pravastatin prevents progression of ventricular hyper-trophy through attenuation of mevalonate-dependent tissue ET-1 levels in SHR.
Mechanisms. The present study suggests that pravastatin
affects myocyte function (attenuated pacing-induced arrhyth-mias) and growth (attenuating myocardial hypertrophy). The mechanisms by which pravastatin attenuates cardiac hypertro-phy remain to be defined. Clearly, hemodynamics did not play a major role in the process. Various statins have been shown to reduce blood pressure in patients (10) and animals (37). Glo-rioso et al. (10) have shown hemodynamic improvement after 16-wk therapy with pravastatin in hypertensive patients. How-ever, the mechanism by which pravastatin prevents cardiac hypertrophy is probably not due to its antihypertensive effect alone. In this study, we found that chronic treatment of SHR with hydralazine did not prevent ongoing cardiac hypertrophy even though it was as effective as pravastatin in lowering blood pressure in SHR. Thus, although we demonstrated hyperkinetic circulation in relatively young SHR, hemodynamics did not play a major role in attenuation of cardiac hypertrophy in pravastatin-treated SHR. Further evidence of this is our finding that LV weight correlates with tissue ET-1 levels but not with blood pressure. Thus some factors other than high blood pressure may contribute to the pathogenesis of cardiac hyper-trophy in SHR. These findings support the notion that prava-statin has direct tissular effects dissociated from its hemody-namic systemic effects.
Fig. 1. Inducibility quotient of ventricular arrhythmias by programmed elec-trical stimulation. *P ⬍ 0.05 compared with Wistar-Kyoto (WKY) rats, bosentan-treated spontaneously hypertensive rats (SHR), and pravastatin-treated SHR. Meval, mevalonate; Prava, pravastatin.
Fig. 2. Left ventricular (LV) preproendothelin-1 (preproET-1) mRNA levels in WKY rats and vehicle-treated, pravastatin-treated, mevalonate-treated, prav-astatin ⫹ mevalonate (combination)-treated, and hydralazine-treated SHR. Each mRNA level was corrected for an mRNA level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Values are means⫾ SD. *P ⬍ 0.05 compared with WKY rats. †P⬍ 0.05 compared with vehicle-, mevalonate-, combination-, and hydralazine-treated SHR.
Table 2. Characteristics of isolated cardiomyocytes
Parameters
SHR
WKY Vehicle Bosentan Pravastatin Mevalonate Prava⫹ Meval Hydralazine
No. of animals 5 5 5 5 5 5 5
Myocyte length,m 139⫾27 138⫾20 135⫾20 138⫾25 137⫾21 138⫾16 134⫾15
Myocyte width,m 24⫾3 22⫾2* 21⫾2* 26⫾4 26⫾3 25⫾2 21⫾3*
Measured myocyte areas,m2 3,803⫾213 3,098⫾146* 3,170⫾157* 3,647⫾229 3,561⫾207 3,872⫾102 3,010⫾192* Values are means⫾ SD. *P ⬍ 0.05 compared with vehicle-, mevalonate-, combination-, and hydralazine-treated groups.
H223
PRAVASTATIN AND LV MASS
AJP-Heart Circ Physiol•VOL 289 • JULY 2005 •www.ajpheart.org
on April 7, 2011
ajpheart.physiology.org
Our observations showed that reduced pravastatin-related ET-1 levels might play crucial roles in attenuation of myocyte hypertrophy. The role of ET-1 in pathogenesis of myocyte hypertrophy was further confirmed by directly blocking ET receptors. Attenuated cardiac hypertrophy accompanied by reduced cardiac ET-1 indicated that autocrine/paracrine ET-1 secretion pathways could be a factor in the proliferative pro-cess blunted by pravastatin. The addition of mevalonate to pravastatin-treated rats impaired not only their ability to atten-uate cellular hypertrophy but also their ability to suppress ET-1 levels. Thus blocking the mevalonate pathway is a critical step in the mechanism of pravastatin-induced attenuation of ET-1 levels. The molecular mechanism of noncholesterol effects of statins is the inhibition of the isoprenoid intermediates of the cholesterol pathway. Isoprenoids are essential for the function of signal transduction molecules of the Rho family (20). Regulation of Rho activity by statins is separate from that of statins on lipids. Inhibition of Rho signaling by statins can activate peroxisome proliferator-activated receptors (6), which in turn suppress ET-1 secretion (34). In addition, decreases in regional ET-1 concentrations may result from decreases in ET-1 production and/or increased ET-1 clearance. The produc-tion of ET-1 begins with the cleavage of the translaproduc-tional product preproET-1. Our results show inhibition of pravastatin on preproET-1 mRNA at the transcription levels. Thus
de-creases in ET-1 production may play a major role in regional ET-1 changes. Together, both secretion and synthesis of ET-1 may be inhibited after the administration of pravastatin reduces tissue ET-1 levels, which attenuated progression of cardiomy-ocyte hypertrophy.
There are controversies as to the role of ET-1 in the patho-genesis and maintenance of ventricular hypertrophy in SHR. Some reports have shown that the ET system does not appear to play an important role in ventricular hypertrophy in SHR (25). However, others have suggested a causal role for ET in the development of hypertension in SHR (29). These discrep-ancies may be due to differences in ages of hypertensive rats that were associated with different pathological and physiolog-ical conditions during exposure to different developmental periods. It has been suggested that different stages of hyper-tension may be differentially regulated in SHR (3). Ito et al. (14) have proposed that cardiac ET-1 may act as an initiating hypertrophic factor during the early stage of pressure overload but that other factors, such as the renin-angiotensin system, may take over as maintaining factors during the late stage of pressure overload. Our studies were performed in quite young animals, i.e., 16-wk old at the end of treatment, in which the development of hypertrophy is at its early stages. Our data are consistent with the notion that early ET-1 production acts as a triggering factor to hypertrophy. Indeed, our results are
com-Fig. 3. Immunohistochemical microscopy of ET-1 (magnification ⫻200). Positive staining for ET-1 (brown) was distributed in the myocardium. The inten-sity of ET-1 was significantly lower in pravastatin-treated SHR (C) than in SHR pravastatin-treated with vehicle (B), mevalonate (D), pravastatin ⫹ mevalonate (E), and hydralazine (F). A: WKY rats. Bar, 100m.
on April 7, 2011
ajpheart.physiology.org
patible with those of previous studies showing that cardiac ET-1 levels were significantly higher at 8 wk of age in SHR compared with WKY rats (16) and that increased cardiac ET-1 activity plays an important role in triggering cardiac hypertro-phy at the development phase of hypertension in SHR (13). Our results also are compatible with those of previous studies suggesting that ET-1 could act at the developmental rather than at the established phase of cardiac hypertrophy (8).
Tanabe et al. (35) have shown that hydralazine (20 mg/kg per day) can attenuate cardiac ET-1 mRNA expression for 8 wk, from the age of 4 wk, in SHR-SP/Izm rats. The latter findings were not consistent with ours. The differences could be the different species used, different doses used, and different age at the start of treatment. Furthermore, because there were great content variations of ET-1 mRNA expression in cardiac chambers (36) and no mention of which chamber was exam-ined in the latter study, comparisons are difficult. Thus it is possible that ET-1 signaling pathways remain in an activated state with hydralazine treatment, resulting in a failure to prevent cardiomyocyte hypertrophy.
Other mechanisms. In addition to the reduction of ET-1,
pravastatin may have altered other mechanisms involved in the antihypertropic effect. On the basis of the correlation shown in Fig. 4, tissue ET-1 levels explain only 23% of the variation in LV mass. Therefore, pravastatin-induced attenuated LV mass cannot be attributed solely to cardiac tissue ET-1 levels, which stresses the importance of other factors. First, blockade of free radicals alleviated the development of cardiac hypertrophy, and pravastatin was shown to inhibit production of free radicals (22). Second, blunting by pravastatin of antihypertropic effect may also result from attenuation of nitric oxide inhibition. Liu et al. (26) have shown increased LV mass in endothelial nitric oxide synthase knockout mice. Third, Benderdour et al. (4) have shown that NADP-isocitrate dehydrogenase adducts in the heart have significantly increased at the prehypertensive stage in 7-wk-old SHR compared with WKY rats. Mitochon-drial NADP-isocitrate dehydrogenase activity is crucial for cardiac hypertrophy development. Statins may prevent cardiac hypertrophy by attenuating the enzyme activities (40). Finally, concerning the mechanisms involved in the attenuation of pravastatin-induced ET-1, it also is attractive to consider a potential role of angiotensin II, which may interact with statins. In vitro studies have demonstrated cross talk between the angiotensin system and the ET-1 system (15). Angiotensin II
has been shown to induce ET-1 synthesis in cardiomyocytes in vitro (31). Lovastatin has been shown to inhibit activity of angiotensin II (31), thus accounting for the downregulation effect on ET-1 protein expression (7). Although pravastatin has little effect on cardiac angiotensin activity (19), we cannot rule out the possibility of decreased cardiac angiotensin activity secondarily to an inhibition of ET-1 exerted by the agent. Complex interactions among angiotensin II, free radical, nitric oxide, and NADP-isocitrate dehydrogenase activity could af-fect cardiac hypertrophy. Therefore, a variety of upstream regulators could interfere with hypertrophic signaling path-ways. The failure to completely abrogate the hypertrophic process was not surprising in view of the underlying complex hypertrophic process, which is unlikely to be amendable to one therapeutic intervention. Extensive investigation of such path-ways and the mechanisms by which pravastatin attenuates cardiac hypertrophy require additional studies.
Arrhythmias. LV hypertrophy is associated with structural,
hemodynamic, and electrophysiological abnormalities. Our results showed that attenuated ventricular hypertrophy after pravastatin administration has benefits in the incidence of ventricular arrhythmias. Myocyte hypertrophy may cause a lengthening of action potential duration, which may generate arrhythmias, via downregulation of the transient outward cur-rent (Ito) as well as the delayed and background rectifier current
(Ik, Ik1) (18). Rials et al. (33) have shown that regression of
cardiac hypertrophy leads to normalization of the prolonged action potential. Furthermore, ET-1 has been shown to directly inhibit potassium channel and act as an important mediator of arrhythmogenesis (17), which was consistent with the antiar-rhythmic effect of pravastatin by attenuation of ET-1 levels. In fact, our results were consistent with those of Matsumoto et al. (28), showing that long-term treatment with ET-1 blockers inhibits electrical remodeling and suppresses ventricular ar-rhythmias. Third, SHR have been shown to have increased sympathetic tone (38), a key trigger of fatal arrhythmias. Statins may decrease ventricular vulnerability by normalizing sympathetic control (32). Together, regardless of the relative importance of each of these factors, all of the changes caused by pravastatin are compatible with our understanding of ben-eficial effects on induction of ventricular arrhythmias.
Study limitations. A potential problem with the present study
is whether cardiomyocytes contribute to increased ET-1 ex-pression. Although immunohistochemical analysis of the LV revealed the presence of ET-1 immunoreactivity in this study, a number of other cell types not normally present in the myocardial tissue to any substantial degree may also produce ET-1 and may contribute to the increased expression during ventricular hypertrophy. These include vascular endothelial cells, smooth muscle cells, and fibroblasts (30). Because we did not perform a double staining for ET-1 and a specific myocyte linage, we cannot rule out the possibility that non-myocytes may provide an alternative source of ET-1 to initiate ventricular hypertrophy.
In conclusion, the results of the present study show that SHR at the early stage of hypertension have an increase of pre-proET-1 mRNA and the mature ET-1 peptide. The finding can be rationalized mechanistically, suggesting a pathogenetic role of regional ET-1 expression in the development of myocardial hypertrophy. Early intervention with pravastatin can reduce the inducibility of ventricular arrhythmias as a result of attenuated
Fig. 4. ET-1 concentrations of the left ventricle in relation to the ratio of LV mass to body weight from data in SHR treated with vehicle, pravastatin, mevalonate, pravastatin⫹ mevalonate, and hydralazine. A significant corre-lation was found between the 2 variables (r⫽ 0.48, P ⫽ 0.0004).
H225
PRAVASTATIN AND LV MASS
AJP-Heart Circ Physiol•VOL 289 • JULY 2005 •www.ajpheart.org
on April 7, 2011
ajpheart.physiology.org
ventricular hypertrophy, probably through the ET-1 pathway, which is linked to mevalonate metabolism. The pharmacolog-ical profile of pravastatin gives new perspectives in the early treatment of ventricular hypertrophy.
ACKNOWLEDGMENTS
We thank In-Ping Cheng, Li-Lan Chien, Shu-Ping Sun, and Fang-Chin Lee for excellent technical assistance and Yu-Wei Chao for performing real-time RT-PCR analysis.
GRANTS
This work was supported by Chi-Mei Medical Center Grants CMFHT 9201, CMFHR9303, CMFHR9307, and CM-TMU9305. Bosentan was a generous gift from Actelion Pharmaceuticals Ltd., Allschwil, Switzerland. Pravastatin was a generous gift from Sankyo Pharmaceuticals Ltd., Tokyo, Japan.
REFERENCES
1. Bauersachs J, Galuppo P, Fraccarollo D, Christ M, and Ertl G. Improvement of left ventricular remodeling and function by hydroxym-ethyglutaryl coenzyme A reductase inhibition with cerivastatin in rats with heart failure after myocardial infarction. Circulation 104: 982–985, 2001. 2. Belichard P, Savard P, Cardinal R, Nadeau R, Gosselin H, Paradis P,
and Rouleau JL. Markedly different effects on ventricular remodeling
result in inducibility of ventricular remodeling. J Am Coll Cardiol 23: 505–523, 1994.
3. Bell D, Kelso EJ, Argent CC, Lee GR, Allen AR, and McDermott BJ. Temporal characteristics of cardiomyocyte hypertrophy in the spontane-ously hypertensive rat. Cardiovasc Pathol 13: 71–78, 2004.
4. Benderdour M, Charron G, DeBlois D, Comte B, and Des Rosiers C. Cardiac mitochondrial NADP⫹-isocitrate dehydrogenase is inactivated through 4-hydroxynonenal adduct formation: an event that precedes hy-pertrophy development. J Biol Chem 278: 45154 – 45159, 2003. 5. Brooksby P, Levi AJ, and Jones JV. The electrophysiological
charac-teristics of hypertrophied ventricular myocytes from the spontaneously hypertensive rat. J Hypertens 11: 611– 622, 1993.
6. Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC,
Najib J, Duriez P, and Staels B. Peroxisome proliferator-activated
receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res 85: 394 – 402, 1999.
7. D’Uscio LV, Shaw S, Barton M, and Luscher TF. Losartan but not verapamil inhibits angiotensin II-induced tissue endothelin-1 increase: role of blood pressure and endothelial function. Hypertension 31: 1305– 1310, 1998.
8. Fareh J, Touyz RM, Schiffrin EL, and Thibault G. Endothelin-1 and angiotensin II receptors in cells from rat hypertrophied heart. Receptor regulation and intracellular Ca2⫹modulation. Circ Res 78: 302–311, 1996. 9. Fraccarollo D, Hu K, Galuppo P, Gaudron P, and Ertl G. Chronic endothelin receptor blockade attenuates progressive ventricular dilation and improves cardiac function in rats with myocardial infarction. Possible involvement of myocardial endothelin system in ventricular remodeling.
Circulation 96: 3963–3973, 1997.
10. Glorioso N, Manunta P, Filigheddu F, Troffa C, Stella P, Barlassina
C, Lombardi C, Soro A, Dettori F, Parpaglia PP, Alibrandi MT, Cusi D, and Bianchi G. Effect of the HMG-CoA reductase inhibitors on blood
pressure in patients with essential hypertension and primary hypercholes-terolemia. Hypertension 34: 1281–1286, 1999.
11. Haider AW, Larson MG, Benjamin EJ, and Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol 32: 1454 –1459, 1998.
12. Hayashidani S, Tsutsui H, Shiomi T, Suematsu N, Kinugawa S, Ide T,
Wen J, and Takeshita A. Fluvastatin, a 3-hydroxy-3-methyglutaryl
coenzyme A reductase inhibitor, attenuates left ventricular remodeling and function after experimental myocardial infarction. Circulation 105: 868 – 873, 2002.
13. Iemitsu M, Miyauchi T, Maeda S, Sakai S, Kobayashi T, Fujii N,
Miyazaki H, Matsuda M, and Yamaguchi I. Physiological and
patho-logical cardiac hypertrophy induces different molecular phenotypes in the rat. Am J Physiol Regul Integr Comp Physiol 281: R2029 –R2036, 2001. 14. Ito H, Hiroe M, Hirata Y, Fujisaki H, Adachi S, Akimoto H, Ohta Y,
and Marumo F. Endothelin ETA receptor antagonist blocks cardiac
hypertrophy provoked by hemodynamic overload. Circulation 89: 2198 – 2203, 1994.
15. Iwanaga Y, Kihara Y, Inagaki K, Onozawa Y, Yoneda T, Kataoka K,
and Sasayama S. Differential effects of angiotensin II versus endothelin-1
inhibitions in hypertrophic left ventricular myocardium during transition to heart failure. Circulation 104: 606 – 612, 2001.
16. Iyer RS, Singh G, Rebello S, Roy S, Bhat R, Vidyasagar D, and Gulati
A. Changes in the concentration of endothelin-1 during development of
hypertensive rats. Pharmacology 51: 96 –104, 1995.
17. James AF, Ramsey JE, Reynolds AM, Hendry BM, and Shattock MJ. Effects of endothelin-1 on K⫹ current from rat ventricular myocytes.
Biochem Biophys Res Commun 284: 1048 –1055, 2001.
18. Kleiman RB and Houser SR. Outward currents in normal and hypertro-phied feline ventricular myocytes. Am J Physiol Heart Circ Physiol 256: H1450 –H1461, 1989.
19. Koga T, Shimada Y, Kuroda M, Tsujita Y, Hasegawa K, and
Yamazaki M. Tissue-selective inhibition of cholesterol synthesis in vivo
by pravastatin sodium, a 3-hydroxy-3-methyglutaryl coenzyme A reduc-tase inhibitor. Biochim Biophys Acta 1045: 115–120, 1990.
20. Laufs U, Kilter H, Konkol C, Wassmann S, Bohm M, and Nickenig G. Impact of HMG-CoA reductase inhibition on small GTPases in the heart.
Cardiovasc Res 53: 911–920, 2002.
21. Lee GR, Bell D, Kelso EJ, Argent CC, and McDermott BJ. Evidence for altered ETBreceptor characteristics during development and progres-sion of ventricular cardiomyocyte hypertrophy. Am J Physiol Heart Circ
Physiol 287: H425–H432, 2004.
22. Lee TM, Chou TF, and Tsai CH. Association of pravastatin and left ventricular mass in hypercholesterolemic patients: role of 8-iso-prosta-glandin F2␣formation. J Cardiovasc Pharmacol 40: 868 – 874, 2002. 23. Lee TM, Chou CF, and Tsai CH. Effect of pravastatin on cardiomyocyte
hypertrophy and ventricular vulnerability in normolipidemic rats after myocardial infarction. J Mol Cell Cardiol 35: 1449 –1459, 2003. 24. Levy D, Garrison RJ, Savage DD, Kannel WB, and Castelli WP.
Prognostic implications of echocardiographically determined left ventric-ular mass in the Framingham Heart Study. N Engl J Med 322: 1561–1566, 1990.
25. Li JS and Schiffrin EL. Effect of chronic treatment of adult spontane-ously hypertensive rats with an endothelin receptor antagonist.
Hyperten-sion 25: 495–500, 1995.
26. Liu YH, Xu J, Yang XP, Yang F, Shesely E, and Carretero OA. Effect of ACE inhibitors and angiotensin II type 1 receptor antagonists on endothelial NO synthase knockout mice with heart failure. Hypertension 39: 375–381, 2002.
27. Marano G, Palazzesi S, Bernucci P, Grigioni M, Formigari R, and
Ballerini L. ETA/ETBreceptor antagonist bosentan inhibits neointimal development in collared carotid arteries of rabbits. Life Sci 63: PL259 – PL266, 1998.
28. Matsumoto Y, Aihara H, Yamauchi-Kohno R, Reien Y, Ogura T,
Yabana H, Masuda Y, Sato T, Komuro I, and Nakaya H. Long-term
endothelin A receptor blockade inhibits electrical remodeling in cardio-myopathic hamsters. Circulation 106: 613– 619, 2002.
29. Miyauchi T, Ishikawa T, Tomobe Y, Yanagisawa M, Kimura S,
Sugishita Y, Ito I, Goto K, and Masaki T. Characteristics of pressor
response to endothelin in spontaneously hypertensive and Wistar Kyoto rats. Hypertension 14: 425– 434, 1989.
30. Miyauchi T and Masaki T. Pathophysiology of endothelin in cardiovas-cular system. Annu Rev Physiol 61: 391– 415, 1999.
31. Oi S, Haneda T, Osaki J, Kashiwagi Y, Nakamura Y, Kawabe J, and
Kikuchi K. Lovastatin prevents angiotensin II-induced cardiac
hypertro-phy in cultured neonatal rat heart cells. Eur J Pharmacol 376: 139 –148, 1999.
32. Pliquett RU, Cornish KG, Peuler JD, and Zucker IH. Simvastatin normalizes autonomic neural control in experimental heart failure.
Circu-lation 107: 2493–2498, 2003.
33. Rials SJ, Wu Y, Ford N, Pauletto FJ, Abramson SV, Rubin AM,
Marinchak RA, and Kowey PR. Effect of left ventricular hypertrophy
and its regression on ventricular electrophysiology and vulnerability to induce arrhythmia in the feline heart. Circulation 91: 426 – 430, 1995. 34. Satoh H, Tsukamoto K, Hashimoto Y, Hashimoto N, Togo M, Hara
M, Maekawa H, Isoo N, Kimura S, and Watanabe T.
Thiazolidine-diones suppress endothelin-1 secretion from bovine vascular endothelial cells: a new possible role of PPAR␥ on vascular endothelial function.
Biochem Biophys Res Commun 254: 757–763, 1999.
on April 7, 2011
ajpheart.physiology.org
35. Tanabe A, Naruse M, Adachi C, Seki T, Yoshimoto T, Takagi S,
Naruse K, and Takano K. Hydralazine decreases blood pressure and
endothelin-1 mRNA expression in tissues but not cardiac weight in SHR-SP/ Izm rats. J Cardiovasc Pharmacol 36, Suppl 1: S176 –S178, 2000. 36. Thibault G, Arguin C, and Garcia R. Cardiac endothelin-1 content and
receptor subtype in spontaneously hypertensive rats. J Mol Cell Cardiol 27: 2327–2336, 1995.
37. Tonolo G, Melis MG, Formato M, Angius MF, Carboni A, Brizzi P,
Ciccarese M, Cherchi GM, and Maioli M. Additive effects of
simva-statin beyond its effects on LDL cholesterol in hypertensive type 2 diabetic patients. Eur J Clin Invest 30: 980 –987, 2000.
38. Trippodo NC and Frohlich ED. Similarities of genetic (spontaneous) hypertension. Man and rat. Circ Res 48: 309 –319, 1981.
39. Ueda Y, Kitakaze M, Komamura K, Minamino T, Asanuma H, Sato
H, Kuzuya T, Takeda H, and Hori M. Pravastatin restored the infarct
size-limiting effect of ischemic preconditioning blunted by
hypercholes-terolemia in the rabbit model of myocardial infarction. J Am Coll Cardiol 34: 2120 –2125, 1999.
40. Varghese S and Oommen OV. Long-term feeding of dietary oils alters lipid metabolism, lipid peroxidation, and antioxidant enzyme activities in a teleost (Anabas testudineus Bloch). Lipids 35: 757–762, 2000. 41. Vaughan CJ, Murphy MB, and Buckley BM. Statins do more than just
lower cholesterol. Lancet 348: 1079 –1082, 1996.
42. Vliet AK, Thiel GC, Huisman RH, Moshabe H, Yap SH, and Cohen
LH. Different effects of 3-hydroxy-3-methyglutaryl coenzyme A
reduc-tase inhibitors on sterol synthesis in various human cell types. Biochim
Biophys Acta 1254: 105–111, 1995.
43. Yue TL, Gu JL, Wang C, Reith AD, Lee JC, Mirabile RC, Kreutz R,
Wang Y, Maleeff B, Parsons AA, and Ohlstein EH. Extracellular
signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy.
J Biol Chem 275: 37895–37901, 2000.
H227
PRAVASTATIN AND LV MASS
AJP-Heart Circ Physiol•VOL 289 • JULY 2005 •www.ajpheart.org
on April 7, 2011
ajpheart.physiology.org