RNA序列實驗中檢測差異表現基因之統計方法 - 政大學術集成
28
0
0
全文
(2) 摘要 近年來,由基因之次世代定序 (next generation sequencing)科技所發展出的 RNA-Seq (RNA Sequencing)實驗隨著成本降低日益受到重視。該實驗利用高通量 定序技術來探討基因體的基因轉錄(transcriptomes),並以計數型態(count)的序列 資料來測量基因表現量。在考慮資料中之過度離散(overdispersion)的特性,我們 在此研究中採用負二項(negative binomial)分配假設,並以最大擬概似函數估計 (maximum pseudo-likelihood estimation)方法來估計基因之平均表現量。為了進一 步找出在兩組具有不同的外顯狀態(phenotype)受詴者間存在著差異表現量的基. 政 治 大 基因與外顯狀態相關程度之顯著性。我們利用統計模擬以驗證所提出的方法,最 立. 因(differentially expressed genes),我們運用上述估計量之 Wald 檢定統計量來檢定. ‧. ‧ 國. 學. 後也將此方法應用到真實範例資料。. sit. y. Nat. al. n. 定、RNA Seq. er. io. 關鍵字:負二項分配、過度離散、最大擬概似函數估計、差異表現基因顯著性檢. Ch. engchi. 1. i n U. v.
(3) 目錄 第一章 緒論.............................................................................................3 第二章 方法.............................................................................................5 第一節 序列資料..............................................................................5 第二節 參數估計..............................................................................5 第三節 假設檢定..............................................................................7. 政 治 大. 第三章 模擬研究與探討 ........................................................................9. 立. 第一節 模擬設計..............................................................................9. ‧ 國. 學. 第二節 模擬結果............................................................................ 11. ‧. 第四章 實證分析 ..................................................................................20. Nat. io. sit. y. 第五章 結論...........................................................................................24. n. al. er. 參考文獻...................................................................................................26. Ch. engchi. 2. i n U. v.
(4) 第一章. 緒論. 在過去的幾十年中,由於生物科技的進步,人類的基因已逐漸被解碼。近期 研究人員希望找出與重大疾病如癌症相關的基因。若能找出這些相關基因,則可 發展成本較低或較不侵入性的疾病檢測工具,則能有效早日偵測疾病的罹患。例 如由 Hall 等人於 1990 年所提出的 BRCA1,BRCA2 癌症基因檢測即為乳癌相關之癌 症篩檢。另一方面,若研究人員發現疾病治療方法的療效與特定的基因相關,例 如:基因表現量越高則標靶藥物療效越好,則未來就能給予個別病人適當的藥 物,為其打造量身定作的療程。像是治療非小細胞肺癌的標靶藥物艾瑞莎. 政 治 大 突變的癌細胞具有良好的治療功效。 立. (Iressa)當中的 Gefitinib 成分就由 Pao 等人於 2004 年提出對於帶有 EGFR 基因. ‧ 國. 學. 近二十年來,用以呈現基因表現的生物晶片實驗主要為微陣列實驗 (Microarray)。雖然其費用相對低廉,但在使用上有許多限制。它必須依賴已知. ‧. 的全基因體作為雜交 (hybridization)平台,然而雜交的方式在分析過程中會受. sit. y. Nat. 到背景干擾,進而影響表現量的差異。且在分析上是藉由螢光的顏色當作判斷,. al. er. io. 若基因表現量過高或過低皆將影響其測量之準確性。近年來,由於次世代定序. v. n. (next generation sequencing)的出現,進而發展出新技術 RNA Sequencing 簡. Ch. engchi. i n U. 稱 RNA-Seq,RNA-Seq 實驗是利用高通量定序技術來探討基因體的基因轉錄 (transcriptomes),以計數型態(count)序列數量 (reads)來測量基因表現量, 以藉此得到細胞或組織內 RNA 的表現情形。RNA-Seq 實驗之操作上不像微陣列序 列的限制,在生物體的全基因體未被定序下仍可進行實驗。由於科技進步,使得 其成本日益降低,目前該技術已逐漸取代微陣列序列分析,可參考 Wang 等人 (2009)所提出文獻,且該實驗被廣泛利用在生物標記,醫學研究,農業研究…等, 像是上述提到的乳癌基因篩檢及癌症標靶藥物。 本研究的目的為利用 RNA-Seq 實驗所產生的基因序列資料來偵測與生物外 顯狀態(phenotype)相關基因。實驗的方式是分析具有不同外顯狀況的受詴者樣. 3.
(5) 本組,檢定在相異組之間存在著差異表現量的基因。常見的外顯狀態為特定疾病 的罹患與否,罹患疾病之嚴重程度,或特定治療的療效。文獻上已有多個差異性 檢定統計方法被發展及發表。例如 Robinson 和 Smyth 在 2007 年和 2008 年及 Robinson 等人於 2010 年提出的 edgeR 方法,以及 Li 等人於 2012 年提出的 PoissonSeq 方法。由於 RNA-Seq 實驗屬於個數型資料,所以他們分別假設資料 來自波氏 (Poisson)分配和負二項 (negative binomial)分配。又由於在實際資 料中經常發現過度離散 (overdispersion)的現象,所以 edgeR 方法利用納入負 二項分配解決過度離散的問題。而 PoissonSeq 方法則是採用資料轉換的方式來. 政 治 大 (sequence depth)的處理、差異性檢定以及錯誤發現率 (false discovery rate) 立. 克服過度離散問題。 除了分配假設不同,兩方法所運用的模型、序列深度. 校正也有差異。. ‧ 國. 學. 考慮到 RNA-Seq 基因序列資料大多數存在過度離散的問題,在此研究中我們. ‧. 假設資料來自負二項分配,並且將採用傳統推論方法—最大概似估計方法 (MLE). y. Nat. 來估計分配參數。但過程中發現負二項分配中的過度離散參數將使得數值計算變. er. io. sit. 得相當複雜,為了解決計算上的困難,我們引進了 Nakashima (1997)所提出的 擬概似方程式 (pseudo likelihood equation),並同時利用 Stirling’ s formula. al. n. v i n 近似原始之機率質量函數,最終獲得參數之最大擬概似估計量(maximum pseudo Ch engchi U. likelihood estimator),並以此得到基因差異性檢定之華德檢定統計量(Wald’ s statistic)。Nakashima (1997)文獻中提到該估計量在大樣本下漸近常態分配, 我們根據此性質得到檢定之近似 p 值以進一步找出存在顯著差異的基因。 本文之第二章將會詳細介紹資料、分配假設、以及參數之最大擬概似估計方 法,與基因差異性檢定之統計方法。在第三章模擬研究與探討中,我們將利用統 計模擬以驗證本方法,並且將納入其他方法與本方法比較。第四章為實證資料分 析,我們將本方法運用在兩組實際的 RNA-Seq 基因資料上。同樣的,我們將同時 利用本方法與另外兩法分析資料,並就獲得的結論進行比較。第五章的結論除了 概述本研究外,且將提出未來研究方向。 4.
(6) 第二章. 方法. 第一節 序列資料 假設由 RNA-Seq 實驗中獲得兩組獨立隨機樣本,樣本數各為 n i ,其中 i 1 為 控制組, i 2 為實驗組。樣本中每個受詴者的基因序列資料包含了 G 個基因。 我們定義 Yijg 為第 i 組樣本中第 j 個受詴者的第 g 個基因表現量,i=1,2; j=1,…,ni;g=1,…,G。已知序列資料為計數(count)型態,在統計分析中,一般 常 考 慮 波 氏 (Poisson) 分 配 。 但 實 證 上 發現 RNA-Seq 資 料 通 常有 過 度 離 散. 政 治 大 (negative binomial)分配,其平均數與變異數分別為: 立. (overdispersion)的現象,即變異數遠大於平均數,故我們假設 Yijg 來自負二項. ‧ 國. 學. 2 ijg mij ig,ijg ijg (1 ijg ig ) 。. ‧. 其中 m ij 為各樣本的序列深度(sequence depth),本文考慮利用該樣本的基因表 G. y. sit. g 1. Nat. 現量總和 y ij. y ijg 來估計 m ij,此為 RNA-Seq 資料常見的總數正規化處理方法. n. al. er. io. (total count normalization)。另一方面,參數 ig 為該組中第 g 個基因的相對. i n U. v. 表現量,而 ig 為第 g 個基因的過度離散參數。以下我們首先介紹參數 ' s與' s 的. Ch. engchi. 估計方法,另一方面,針對偵測差異表現的基因,我們也將提出相關統計檢定方 法。. 第二節 參數估計 在此研究中,我們考慮以最大概似估計方法(MLE)估計參數,故給定 Yijg ~ NB(μ ijg m ij λ ig , ig ), 0 λ ig 1,m ij 0,ig 0,i 1,2,j 1,..., n i,g 1,..., G,. 則 Yijg 的機率質量函數為:. 5.
(7) P(Yijg y ijg ) . Γ(y ijg -1ig ). (. ig m ijλ ig. y ijg ! Γ( ) 1 ig m ijλ ig -1 ig. y. ) ijg (. 1 - 1 ) ig , y ijg 0,1,2,... 。 1 ig m ijλ ig. 其中 (.)為伽瑪(gamma) 函數 當 y ijg 很大時,則機率質量函數中的伽瑪函數的計算 就會變得相當困難,所以我們利用以下的 Stirling’ s formula:考慮一固定 k,. 則當N , C kN ~. Nk ( N 1) ,即 ~ N k。 k! ( N 1 k ). 之後便可得以下近似機率質量函數:. (y ijg ig-1 1). lim yijg P(Yijg y ijg ) . ig-1 1. 立. ig m ij λ ig. ( ) 治 1 m λ (政 ) 大 -1 ig. ig. ij. y ijg. ig. 1 -1 ( ) ig 。 1 ig m ij λ ig. 實務上,當 yijg≧172 時,我們就採取上述近似結果。為求解參數 λ , 之最大概. ‧ 國. 學. 似估計量,假設每序列中基因的表現量之間獨立,則其概似函數 L 為以下連乘積: ni. G. ni. G. ln L ln P(Yijg y ijg ) 。. n. al. er. io. i 1 j1 g 1. y. j1 g 1. sit. 2. ‧. i 1. Nat. 對數概似函數為. 2. L P(Yijg y ijg ),. i n U. v. 將上列函數對 λ ig 偏微分,則可得 λ ig 之概似方程式(likelihood equation)為:. Ch. engchi. ln L ni y ijg m ij λ ig ,i 1,2; g 1,..., G。 (1) λ j1 λ (1 m λ ) ig ig ig ij ig . 另一方面, ig 之概似方程式則相對複雜。我們採用 Nakashima (1997)所提出的 下列擬概似方程式(pseudo likelihood equation)來替代: ni (y ~ mijλ ig ) 2 mijλ ig (1 ig mijλ ig ) ln L ijg 。 (2) j1 2(1 ig mijλ ig ) 2 ig . 則 λig ,ig 的最大擬概似估計量(maximum pseudo likelihood estimator)為(1)和 (2)式的零根,定義為 λˆ ig 和 ˆ ig 。由於我們的研究主要針對 λ ig 進行推論, 6.
(8) Nakashima (1997)證明出 λˆ ig 在大樣本下漸近常態分配,而且根據 λˆ ig 和 ˆ ig 可獲得. λˆ ig 之漸近變異數之估計值為:. SE 2 (λˆ ig ) . ni. m ij. j1. λˆ ig (1 m ij λˆ ig ˆ ig ). (. ) 1 ,. ni. 即當 n i 夠大時,. (λˆ ig λ ig ) d N(0,1), SE( λˆ ) ig. i 1,2; g 1,..., G。. 立. 政 治 大. ‧ 國. 學. 第三節 假設檢定. 我們的目的是找出在兩組具有不同的外顯狀態(phenotype)受詴者間存在著. ‧. 差異表現量的基因。針對這個問題,我們檢定下列統計假設以決定各個基因的顯. er. io. H 0g : λ1g λ 2g vs. H1g : λ1g λ 2g,. sit. y. Nat. 著性: 令 g=1,…,G,. al. n. v i n 則根據 ' s 的最大擬概似估計量,每個檢定的 Wald 檢定統計量為: Ch engchi U Zg . λˆ 1g λˆ 2g λˆ 1g λˆ 2g , 2 ˆ 2 ˆ SE( λˆ 1g λˆ 2g ) SE (λ1g ) SE (λ 2g ). 其中由於兩組資料之間之間為獨立,故上式中等式成立。已知在虛無假說之下, d. Zg N(0,1) ,則給定檢定統計量的觀測值 z g 時,可得其近似 p-值為: P value P( Z z g H 0 ) 1 - ( z g ) (- z g ).. 其中 為標準常態分配之累積機率。 一般來說,上述進行差異性檢定的基因個數動輒上萬,若檢定後沒有進行 p 值的校正,將會造成結論之整體型一誤差率膨脹。當考慮整體型一誤差率的校 7.
(9) 正,例如 Bonferroni 校正法,則結論會相當保守。所以根據這個問題,目前一 般考慮在錯誤發現率(false discovery rate)準則下對 p-值進行校正,兩個常 用的校正方法為 Benjamini 等人(1995)所提出 Benjamini-Hochberg (BH)方法和 Storey 等人 (2002)所提出的 q-value 方法來控制錯誤發現率。在下一章的模擬 研究與探討中,我們將根據這兩個方法對所獲得的 p-值作校正,我們採用 R 的 p.adjust 和 qvalue 封包來計算校正過的 p 值。以上我們提出的統計檢定方法命 名為 PMLE 法。. 立. 政 治 大. ‧. ‧ 國. 學. n. er. io. sit. y. Nat. al. Ch. engchi. 8. i n U. v.
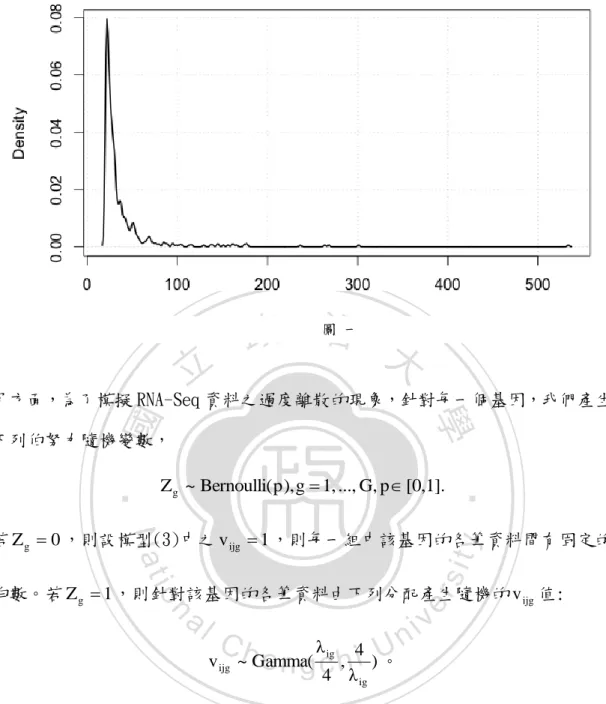
(10) 第三章. 模擬研究與探討. 第一節 模擬設計 以下利用電腦模擬來驗證我們的 PMLE 檢定方法在 RNA-Seq 資料上的表現, 本模擬的模型設定參考 Auer 等人 (2011)的文章,我們並同時將 Robinson 等人 (2010)提出的 edgeR 方法及 Li 等人 (2012)提出的 PoissonSeq 方法列入比較。 我們考慮一萬個基因,其中兩千個基因具有差異表現,各組樣本數則分別為五個 或十個。我們設計了三種過度離散的情境,第一種情境為所有基因皆沒有過度離 散現象存在,第二種情境為每個基因中有 50%的機會發生過度離散現象,第三種. 政 治 大. 情境為所有基因皆存在過度離散現象,各個情境皆模擬一百次。. 立. 學. ‧ 國. 我們由下列波氏分配產生資料:令 i 1,2, j 1,..., n i , g 1,...,10000,. Yijg ~ Poi(λ ig ν ijg ).. (3). sit. y. Nat. 當 g≦8000,. ‧. 我們首先考慮由柏拉圖(Pareto)分配產生亂數,並取其指數,作為 值。. io. al. n. 若 g>8000,. er. λ1g λ 2g ~ Exp(Pareto(location 3, shape 7)),. iid. Ch. engchi. i n U. v. λ1g , λ 2g ~ Exp(Pareto(location 3, shape 7)). 即前八千個基因為在組間無差異表現之基因,而第八千零一個至第一萬個基因則 在不同組間存在差異表現。圖一為樣本數五百個,位置參數為三,尺度參數為七 之柏拉圖分配,並將其取指數之分配圖,可由圖一中發現分配呈現右偏,且偏斜 程度與資料變異程度皆相當大。. 9.
(11) 政 治 大 圖 一. 立. 學. 下列伯努力隨機變數,. Zg ~ Bernoulli( p), g 1, ..., G, p [0,1].. ‧. ‧ 國. 另方面,為了模擬 RNA-Seq 資料之過度離散的現象,針對每一個基因,我們產生. sit. y. Nat. 若 Z g 0 ,則設模型(3)中之 v ijg 1 ,則每一組中該基因的各筆資料間有固定的. n. al. er. io. 均數。若 Z g 1 ,則針對該基因的各筆資料由下列分配產生隨機的 v ijg 值:. Ch. i n U. v. e n gλc4 h, λ4i ) 。. v ijg ~ Gamma(. ig. ig. 則受此隨機值之影響,使得該基因的各筆資料之均數間有變異,導致過度變異狀 況。我們的模擬考慮 p=1, 0.5 與 0 三種高、中、低度過度變異比例的情境。 已知我們比較的三個方法運用截然不同的資料處理、統計模型以及檢定方 法。在資料處理中有關序列深度的正規化上,PoissonSeq 法將不具顯著差異的 基因納入考量,而 PMLE 方法和 edgeR 方法,則是採用一般常見的總數正規法。 在統計模型方面,PoissonSeq 法採用波氏分配下的廣義線性模型,edgeR 方法採 用負二項分配,並引進條件加權概似函數 (conditional weighted likelihood) 估計過度離散參數,PMLE 方法亦採用負二項分配,但我們是利用擬概似函數 10.
(12) (pseudo likelihood)對參數進行最大概似估計法。而檢定方法方面,PoissonSeq 法採用 Score Test 為檢定統計量以避免複雜的參數估計問題,edgeR 方法中的 ,. ,. 檢定統計量為 Fisher s Exact Test,而 PMLE 方法檢定統計量為 Wald s Test。 另一個關鍵元素則是採用的 p 值校正法的影響。已知 edgeR 法建議採用 BH 校正 法,而 PoissonSeq 法則有其獨特的校正法。我們的 PMLE 模擬則考慮運用 BH 校 正法以及 q-value 值。我們期望透過以下的模擬來決定適當的校正法。. 第二節 模擬結果. 政 治 大 有 G 個基因,即 G 為檢定總數,其中 G 為無差異表現基因個數,G 為有差異表現 立 在這一節中,我們將運用模擬來估計各方法的真實錯誤發現率(FDR)。考慮 0. 1. R-V. G1-R+V. R. G-R. y. G0-V. sit. io. n. al. V. G0 G1 G. er. Nat. 對立假說為真. 不拒絕虛無假說. ‧. ‧ 國. 表 一. 拒絕虛無假說. 虛無假說為真. 學. 基因個數,則根據其真實狀態以及檢定結果可得以下的交叉表:. i n U. v. 其中 R 為拒絕虛無假說個數,即顯著個數;而 V 為錯誤拒絕虛無假說的個數。FDR. Ch. engchi. 的定義為 Benjamini 和 Hochberg(1995) 所提出: V FDR E 。 R. 在固定顯著個數下 R=r,1≦r≦G,在每次的模擬中可得知錯誤的拒絕個數 V=v 個,將 100 次的模擬 V 值取平均數,以此來估計 V 的期望值,即 E(V),則進而 估算出 FDR 水準為. Eˆ ( v) ,此為蒙地卡羅模擬(Monte Carlo simulation)的精神, r. 並以此繪製 FDR 曲線。當一檢定方法擁有較高的 FDR 曲線時,則代表其有較多的 錯誤顯著結論,故表現較差。另一方面,在每一次分析中,BH 校正法與 q 值皆 提供該組資料之 FDR 水準之估計,我們取這 100 次模擬結果的平均,計算出估. 11.
(13) 計 FDR 的平均數,藉由比較此平均 FDR 圖形與前述之真實水準估計,我們可得 知這些校正方法結論的適當性。若估計 FDR 曲線高於真實 FDR 估計曲線時,則 找到的顯著基因個數低於真實顯著個數,則代表其檢定結果較保守。反之,若估 計 FDR 曲線低於真實 FDR 估計曲線時,會找到偏多的顯著基因個數,則其檢定 結果過度樂觀,有較多的錯誤顯著結論。 圖二至圖四為樣本數為五個的相關圖型,而圖五至圖七則為樣本數為十的圖 型。其中圖二、圖五為每個基因無過度離散(p=0)現象,圖三、圖六為每個基因 有 p=50%的機會發生過度離散,而圖四、圖七則是每個基因皆發生過度離散情. 政 治 大 計真實 FDR 的曲線圖。圖(b)、圖(c)與圖(d)為三個方法的真實 FDR 估計曲線和 立 境。各情境中皆包括四個圖,(a)-(d)。其中圖(a)為三種檢定方法由 100 次模擬估. 其 100 次模擬結果的估計 FDR 曲線。其中 PMLE 法的估計 FDR 曲線採用 BH 校. ‧ 國. 學. 正 p-values 及 q-value 方法,edgeR 法則採 BH 校正 p-values,而 PoissonSeq 採用. ‧. 其自有的校正方法。. y. Nat. 由圖二(a)可發現 PoissonSeq 方法和 edgeR 方法兩者錯誤發現率幾乎相同。. er. io. sit. PMLE 法之 FDR 曲線則較其他兩法來得高,故我們的方法表現較差。圖二(b)可 發現我們提出的 PMLE 法採用 qvalue 方法和 BH 方法在 FDR 估計上都有高估的. al. n. v i n 現象,此高估會造成研究者將獲得較保守的檢定結果。其中 Ch engchi U. BH 方法比 qvalue. 方法來得更保守,故在各組樣本數五個,所有基因不存在過度離散狀況下,我們 建議 PMLE 法應利用 qvalue 校正法。圖二(c)發現 edgeR 方法所估計的 FDR 嚴重 高估其真實 FDR。圖二(d)可發現 PoissonSeq 之估計 FDR 估計結果和真實 FDR 相差不大,在估計 FDR 上,PoissonSeq 為三種中表現最好的方法。 在圖三中各組樣本數五個,每個基因有 50%機會發生過度離散現象。圖三(a) 發現三種方法錯誤發現率表現與圖二(a)不同,此時三條 FDR 曲線交錯。 PoissonSeq 在前半段由於容易偵測錯誤導致有很高的錯誤發現率,另外 edgeR 法 在尾段上有部分圖形超過 0.8,此代表該檢定的檢定力可能低於型一誤差率。圖 三(b)可發現當 PMLE 檢定利用 qvalue 和 BH 校正 p-value 對 FDR 的估計結果多 12.
(14) 數都有低估的現象,低估的結果會造成研究者對檢定結果較樂觀,且 qvalue 方 法比 BH 方法來的更樂觀,故當基因有中度的過度離散時,建議使用較保守的 BH 校正方法在 PMLE 檢定上。圖三(c)可發現 BH 方法估計 FDR 結果為嚴重高 估。圖三(d)中 PoissonSeq 之估計 FDR 估計結果和真實 FDR 相近。 在圖四中,各組樣本數為五個,而所有基因皆存在過度離散現象。圖四(a) 三種方法錯誤發現率表現與圖二(a)大致相同,同樣的 PoissonSeq 在前端部分較 容易有錯誤偵測。圖四(b)中 PMLE 法兩種校正法低估 FDR,且低估程度更甚半 數基因存在過度離散的狀況,見圖三(b)。圖四(c)BH 方法估計 FDR 結果亦為高. 政 治 大 圖五、圖六與圖七圖形趨勢大致上與圖二、圖三與圖四相似,其中唯一有差 立. 估。圖四(d),同樣地 PoissonSeq 之估計 FDR 估計結果和真實 FDR 比較相近。. 異的地方可以從圖五、圖六與圖七中的(b)圖發現 PMLE 法的估計 FDR 曲線較靠. ‧ 國. 學. 近真實 FDR 估計曲線,所以當樣本數增加,可以使 PMLE 法在估計 FDR 上更加. ‧. 準確。. y. Nat. 總結以上結果,我們發現 PMLE 法的錯誤發現率較另外兩個方法差。當無. er. io. sit. 過度離散時,則 PMLE 法採用 q-value 校正將提供較不保守的結論。當過度離散 發生時,則兩種校正方法都提供過度樂觀的結論。而 edgeR 法採用的 BH 校正法. al. n. v i n 則普遍的提供過度保守的結論。PoissonSeq 的結論之錯誤率則最靠近其真實水 Ch engchi U. 準。值得一提的是,當過度離散發生時,拒絕個數少的時候,PoissonSeq 結論的 FDR 值偏高。. 13.
(15) (a). 立. 政 治 大. (b). ‧. ‧ 國. 學. n. er. io. sit. y. Nat. al. Ch. engchi. (c). i n U. v. (d) 圖 二. 圖二:各組樣本數五個,所有基因不存在過度離散現象。(a):三種方法真實 FDR 曲線圖。(b):PMLE 真實 FDR 和由 BH 方法及 q-value 方法所估計之 FDR。(c):edgeR 真實 FDR 以及由 BH 方法所估計之 FDR。(d):PoissonSeq 真實 FDR 以及其估計 FDR。. 14.
(16) (a). 立. (b). 政 治 大. ‧. ‧ 國. 學. n. er. io. sit. y. Nat. al. Ch. engchi. (c). i n U. v. (d) 圖 三. 圖三:各組樣本數五個,有一半存在過度離散現象。(a):三種方法真實 FDR 曲線圖。(b):PMLE 真實 FDR 和由 BH 方法及 q-value 方法所估計之 FDR。(c):edgeR 真實 FDR 以及由 BH 方法所估計之 FDR。 (d):PoissonSeq 真實 FDR 以及其估計 FDR。. 15.
(17) (a). (b). 立. 政 治 大. ‧. ‧ 國. 學. n. er. io. sit. y. Nat. al. (c). Ch. engchi. i n U. v. (d). 圖 四. 圖四:各組樣本數五個,所有基因存在過度離散現象。(a):三種方法真實 FDR 曲線圖。(b):PMLE 真實 FDR 和由 BH 方法及 q-value 方法所估計之 FDR。(c):edgeR 真實 FDR 以及由 BH 方法所估計之 FDR。(d):PoissonSeq 真實 FDR 以及其估計 FDR。. 16.
(18) (a). (b). 立. 政 治 大. ‧. ‧ 國. 學. n. er. io. sit. y. Nat. al. (c). Ch. e n圖g五c h i. i n U. v. (d). 圖五:各組樣本數十個,所有基因不存在過度離散現象。(a):三種方法真實 FDR 曲線圖。(b):PMLE 真實 FDR 和由 BH 方法及 q-value 方法所估計之 FDR。(c):edgeR 真實 FDR 以及由 BH 方法所估計之 FDR。(d):PoissonSeq 真實 FDR 以及其估計 FDR。. 17.
(19) (a). 立. 政 治 大. ‧. ‧ 國. 學. n. al. er. io. sit. y. Nat (c). (b). Ch. engchi. i n U. v. (d). 圖 六. 圖六:各組樣本數十個,一半基因存在過度離散現象。(a):三種方法真實 FDR 曲線圖。(b):PMLE 真 實 FDR 和由 BH 方法及 q-value 方法所估計之 FDR。(c):edgeR 真實 FDR 以及由 BH 方法所估計之 FDR。 (d):PoissonSeq 真實 FDR 以及其估計 FDR。. 18.
(20) (a). (b). 立. 政 治 大. ‧. ‧ 國. 學. n. er. io. sit. y. Nat. al. (c). Ch. engchi. i n U. v. (d). 圖 七. 圖七:各組樣本數十個,所有基因存在過度離散現象。(a):三種方法真實 FDR 曲線圖。(b):PMLE 真 實 FDR 和由 BH 方法及 q-value 方法所估計之 FDR。(c):edgeR 真實 FDR 以及由 BH 方法所估計之 FDR。 (d):PoissonSeq 真實 FDR 以及其估計 FDR。. 19.
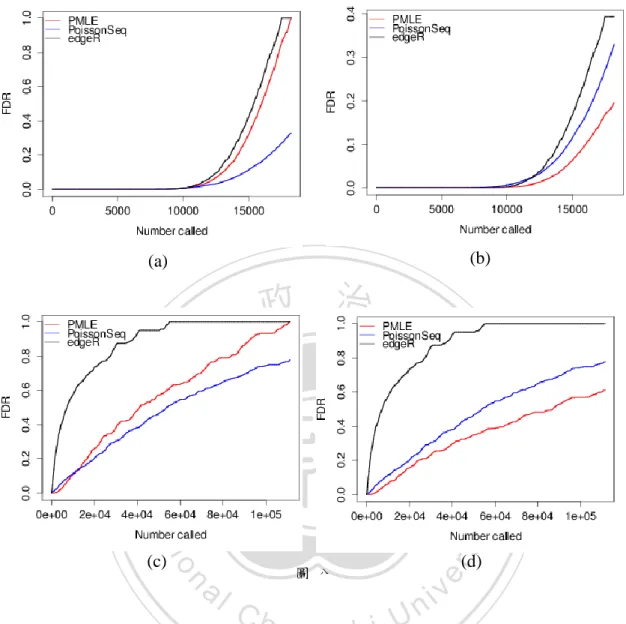
(21) 第四章. 實證分析. 我們將 PMLE 方法運用到 Marioni (2008)的 RNA-Seq 資料和’ t Hoen (2008) 的 Tag-Seq 資料上。Marioni (2008)的資料來自一人類屍體中的肺和腎臟,各個 器 官 上 由 Illumina NGS platform 各 自 重 複 檢 測 五 次 , 此 即 為 技 術 複 製 (technical replicate)樣本,故此資料包含了五個人類腎臟資料和五個肝臟資 料,基因個數為 18,226 個。另外’ t Hoen (2008)的資料,主要為兩群野生老鼠和 轉錄型老鼠小腦海馬體資料,兩群樣本數目各為四個,基因個數為 111,809 個。 圖八(a)(b)為 Marioni(2008)資料的分析結果,其中 PMLE 方法與 edgeR 方. 政 治 大 至 0.4,其他三圖則為 0 立 至 1。我們發現 PMLE 與 edgeR 方法,當採用 BH 方法所. 法分別為(a)採用 BH 方法和(b)q-values 方法進行校正。在圖(b)中 FDR 範圍為 0. ‧ 國. 學. 估計 FDR 皆比 PoissonSeq 所估計 FDR 來的保守。但當採用 q-value 校正時,其 保守結論將被修正,其中相較之下 edgeR 的修正效果較少,而 PMLE 方法的結論. ‧. 之 FDR 將大幅降低,甚至將得到較 PoissonSeq 低的 FDR 估計水準。圖八(c)(d). sit. y. Nat. 為’ t Hoen (2008)資料的分析結果,趨勢大致與(a)(b)相近。在此資料中,edgeR. n. al. er. io. 方法採用兩種不同校正方法並未產生不同 FDR 估計曲線。. Ch. engchi. 20. i n U. v.
(22) (b). (a). 立. 政 治 大. ‧. ‧ 國. 學. n. al. 圖 八. Ch. engchi. (d). er. io. sit. y. Nat (c). i n U. v. 圖八:PMLE、PoissonSeq 和 edgeR 估計之 FDR,上方資料來自 Marioni (2008),下方 資料來自’t Hoen (2008)。(a)和(c)中 PMLE 和 edgeR 採用 BH 方法校正。(b)和(d)中 PMLE 和 edgeR 採用 q-values 方法校正。. 21.
(23) 下表為在 FDR 水準在 5% 與 10% 下個方法所偵測出的顯著基因個數。在 Marioni (2008)資料上,不論 FDR 水準為 0.05 或 0.1,PMLE 方法所偵測出顯著 差異表現量個數會在 12,117~14,549 個,約佔總數之三分之二。PoissonSeq 的顯 著差異表現量為 13,238,故 PoissonSeq 和 PMLE 在此組資料的結論相近。edgeR 方法所偵測顯著差異個數略低於其他兩法。 在’ t Hoen (2008)資料上,則 PMLE 找出的顯著差異表現量個數明顯多於其 他兩方法,而 edgeR 所找出顯著差異表現量為三方法中最少的。且三方法所偵測 出顯著差異表現量個數皆差距甚大,在此組資料之表現皆不相同。另外 PMLE 法. 政 治 大. 的結論與所採行的校正方法密切相關,在採用 q-value 方法下增加近三分之一至 二分之一之顯著個數。. 立. 表 二. ‧ 國. Marioni(2008). edgeR(BH) edgeR(q-value) PoissonSeq PMLE(BH) PMLE(q-value) edgeR(BH) edgeR(q-value) PoissonSeq. 11,638 12,839 13,238 13,030 16,015 12,533 13,909 14,690. io. al. n. 0.1. Ch. engchi. 22. y. 12,117 14,549. sit. PMLE(BH) PMLE(q-value). er. 0.05. ‧. 方法. Nat. FDR. 學. 顯著基因個數. i n U. v. ’ t Hoen(2008) 6,053 8,317 375 375 3,919 9,041 14,075 789 789 8,415.
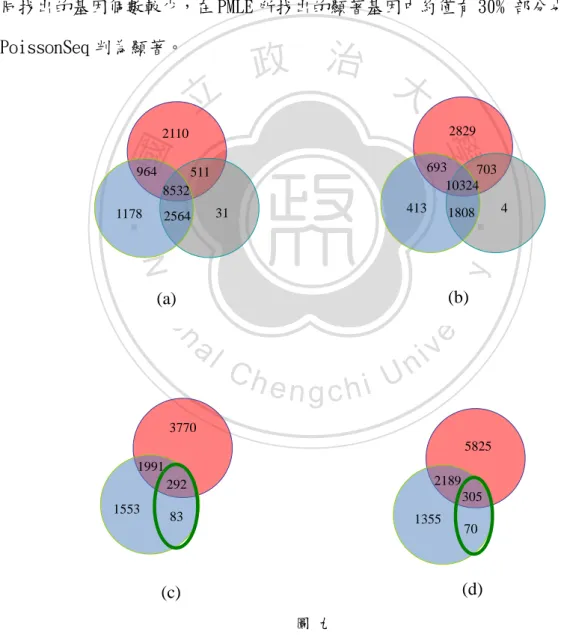
(24) 圖九為三個方法在 FDR 水準為 0.05 下所找出基因集合,其中紅色為 PMLE 方法,藍色為 PoissonSeq,灰色及綠色框線為 edgeR。圖九上為 Marioni (2008) 資料,分別採用(a)BH 方法和(b)q-value 校正。從這些圖中可發現三方法共同找 出的基因個數很多,分別為 8,532 個和 10,324 個,PMLE 所找出的基因中,共有 80% 和另兩個方法相同。圖九下為’ t Hoen (2008)資料,分別採用(c)BH 方法和 (d)q-value 校正。在(c)(d)中得知 edgeR 找出的基因數遠少於另二法,而且找 出的基因全包含在另二法的顯著基因集合內。另一方面,PMLE 和 PoissonSeq 共 同找出的基因個數較少,在 PMLE 所找出的顯著基因中約僅有 30% 部分也同時被. 政 治 大. PoissonSeq 判為顯著。. 立. 2829 693 703 10324. 511. 8532. 1178. 413. 31. 2564. 1808. ‧. ‧ 國 964. 學. 2110. Nat. sit. y. 4. (b). io. n. al. er. (a). Ch. engchi. i n U. v. 3770 5825 1991 2189. 292. 305 1553. 83. 1355. 70. (d). (c) 圖 九. 圖九:PMLE、PoissonSeq 和 edgeR 在 FDR 為 0.05 之基因個數,上方資料來自 Marioni (2008),下方資料來自’t’ Hoen (2008)。(a)和(c)中 PMLE 和 edgeR 採用 BH 方法校正。(b) 和(d)中 PMLE 和 edgeR 採用 q-values 方法校正。 23.
(25) 第五章. 結論. 在此研究中,針對兩組具有不同的外顯狀態受詴者,為了偵測兩組間具有差 異表現的基因,我們提出 PMLE 統計檢定方法。此處的基因資料產生自 Rna-Seq 實驗,其屬於計數型的資料,而且實務上該資料大多會有過度離散的問題,所以 我們假設資料來自負二項分配,並考慮以最大概似估計方法來估計參數。由於在 機率質量函數的數值計算上遇到困難,所以我們利用 Stirling’ s formula 得到 近似機率質量函數。另一方面由於負二項分配中的過度離散參數在參數估計上也 相當困難,我們採用擬概似方程式來替代並估計參數,得到最大擬概似估計量,. 政 治 大 為 PMLE 方法。最後並在錯誤發現率準則下對 p-值進行校正,我們採用兩個常用 立 並根據其大樣本漸進常態的性質找出檢定差異表現基因之 Wald 檢定統計量,稱. ‧ 國. 學. 的校正方法為 BH 方法和 q-value 方法來控制錯誤發現率。. 在第三章的模擬研究中,考慮固定顯著基因個數下,我們發現一般而言 PMLE. ‧. 法的錯誤發現率傾向高於其他兩個方法。而考慮固定 FDR 水準,當基因不存在過. sit. y. Nat. 度離散時,PMLE 採用兩種校正方法皆提供較保守的結論。當基因存在過度離散. al. er. io. 時,則兩種校正方法都提供過度樂觀的結論。但增加樣本數有助於改善 PMLE 法. v. n. 的結論。而從第四章實證資料分析中我們發現在 Marioni (2008)資料上 PMLE 方. Ch. engchi. i n U. 法所偵測出的顯著差異表現基因個數和其他兩方法相近,三方法所偵測出基因重 疊性高,故三方法在此組資料上分析表現差距不大。然而在’ t Hoen (2008)資料 分析上,我們發現三方法共同偵測出的顯著基因相對少,且 PMLE 方法所找出的 顯著基因個數比其他兩方法來得多。 上述提到我們提出的 PMLE 統計檢定法是根據大樣本漸進常態的性質來計算 p-value,但由於 RNA-Seq 實驗費用考量,樣本數都相當少,其大樣本漸進性質 的適用性存疑,且我們從第四章實證資料分析中發現 PMLE 方法比其他兩法偵測 出的顯著基因數還多,傾向提供過度樂觀的結論,這也許是因為大樣本分配的不 適當,造成型一誤差率膨脹所造成的結果。未來可以尋求小樣本適用的無母數方. 24.
(26) 法,例如:重抽方法,以改善 p-value 的估算。另外本文提出的 PMLE 方法適用 於比較兩組具有不同外顯狀態樣本,未來我們可以運用變異數分析的方法運用在 至三組以上樣本的比較。. 立. 政 治 大. ‧. ‧ 國. 學. n. er. io. sit. y. Nat. al. Ch. engchi. 25. i n U. v.
(27) 參考文獻 [1] Auer, P. L. and Doerge, R. W. (2011) A Two-stage Poisson Model for Testing RNA-Seq Data, Statistical Applications in Genetics and Molecular Biology, 10, 1–26. [2] Benjamini, Y. and Hochberg, Y. (1995) Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing, J. Roy. Statist. Soc. Ser. B, 57, 289-300. [3] Hall, J.M., Lee, M.K., Newman, B., Morrow, J.E., Anderson, L.A., Huey, B., King, M.C.(1990) Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science, 250, 1684–1689.. 政 治 大 and False Discovery Rate 立 Estimation for RNA-sequencing Data, Biostatistics, 13,523-538.. [4] Li, J., Witten, D. M., Johnstone, I. M. and Tibshirani, R. (2012) Normalization, Testing,. ‧ 國. 學. [5] Marioni, J. C., Mason, C.E., Mane, S. M., Stephens, M. and Gilad, Y. (2008) Rna-seq: an Assessment of Technical Reproducibility and Comparison with Gene Expression. ‧. Arrays,Genome Res., 18, 1509-1517.. sit. y. Nat. [6] Nakashima, E. (1997) Some Methods for Estimation in a Negative-Binomial Model, Ann.. n. al. er. io. Inst. Statist. Math., 49, 101-105.. i n U. v. [7] Pao, W., Miller, V., Zakowski, M., Doherty, J., Politi, K., Sarkaria, I., Singh, B., Heelan,. Ch. engchi. R., Rusch, V., Fulton, L., Mardis, E., Kupfer, D., Wilson, R., Kris, M. and Varmus, H. (2004) EGF Receptor Gene mutations are Common in Lung Cancers from Never Smokers and are associated with Sensitivity of Tumors to Gefitinib and Erlotinib, Proceedings of the National Academy of Sciences of the United States of America, 101, 13306–13311. [8] Robinson, M. D., McCarthy, D. J. and Smyth, G. K. (2010) edgeR: a Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data, Bioinformatics, 26,139-140. [9] Robinson, M. D. and Smyth, G. K. (2007) Moderated Statistical Tests for Assessing Differences in Tag Abundance, Bioinformatics, 23, 2881-2887.. 26.
(28) [10] Robinson, M. D. and Smyth, G. K. (2008) Small-sample Estimation of Negative Binomial Dispersion, with Applications to SAGE Data, Biostatistics, 9, 321-332. [11] Storey, J. D. (2003) The Positive False Discovery Rate: a Bayesian Interpretation and the q-value, Annals of Statistics, 31, 2013-2035. [12] ’t Hoen, P. A. C., Ariyurek, Y., Thygesen, H. H., Vreugdenhil, E., Vossen, R. H., De Menezes, R. X., Boer, J. M., Van Ommen, G. J. and Den Dunnen, J. T. (2008) Deep Sequencing-Based Expression Analysis Shows Major Advances in Robustness, Resolution and Inter-lab Portability over Five Microarray Platforms. Nucleic Acids. 政 治 大 [13] Wang, Z., Gerstein, M. and Snyder, M. (2009) RNA-Seq: a Revolutionary Tool for 立 Research, 36, e141.. Transcriptomics,Nat. Rev. Genet., 10, 57-63.. ‧. ‧ 國. 學. n. er. io. sit. y. Nat. al. Ch. engchi. 27. i n U. v.
(29)
數據
相關文件
柯西不等式、 排序不等式、 柴比雪夫不等式、 布奴利不等式、 三角不等式、 詹森不等 式、 變數代換法、 數學歸納法、 放縮法、 因式分解法、 配方法、 比較法、 反證法、
以不有 (實無) 之有 (幻有) 而成於有 (能有) 有 (所有) 之宗 (妙有) 以不空 (頑空) 之空 (真空) 而現 於空 (能空) 空 (所現) 之境
在這一節裡會提到,即使沒辦法解得實際的解函數,我們也 可以利用方程式藉由圖形(方向場)或者數值上的計算(歐拉法) 來得到逼近的解。..
(二)受補助單位申請支付款項,應本誠信原則對所提出支用單據
設計了正立方體框架的組合,在計算方塊個數與框架的差異性可透過等差數列的概念作 結合;而作品「腳踏實地」
隨機實驗是一種過程 (process),是一種不能確定預知會
• 田口方法 (Taguchi method) 的意義為利用損 失函數的概念評估品質,採用實驗設計的 方法使產品不易受到不想要或無法控制因 子
按照湛然「萬法是真如,由不變故,真如是萬法,由隨緣故」[20]
