國 立 交 通 大 學
材料科學與工程學系
博 士 論 文
一維鎳奈米柱,鎳-氧化鎳、鎳-二氧化
鈦核殼奈米結構之製造與研究
Fabrication and Characterization of 1-D Ni nanorods,
Ni-NiO, Ni-TiO
2core-shell nanostructures
研 究 生
: 劉健民
指導教授: 陳 智 博士
一維鎳奈米柱,鎳-氧化鎳、鎳-二氧化鈦核殼奈米結構之製造與研究
Fabrication and Characterization of 1-D Ni nanorods, Ni-NiO, Ni-TiO2
core-shell nanostructures
研 究 生:劉健民 Student:Chien-Min Liu 指導教授:陳智 教授 Advisor:Prof. Chih Chen
國立交通大學
材料科學與工程學系博士班
博士論文
A Thesis
Submitted to Department of Materials Science and Engineering College of Engineering
National Chiao Tung University In partial Fulfillment of the Requirements
for the Degree of Doctor of Philosophy
In
Materials Science and Engineering July 2010
Hsinchu, Taiwan, Republic of China
一維鎳奈米柱,鎳-氧化鎳、鎳-二氧化鈦核殼奈米結構之製
造與研究
研究生:劉健民 指導教授:陳 智 教授 國立交通大學 材料科學與工程學系博士班摘 要
本 論 文 是 研 究 結 合 陽 極 氧 化 鋁 模 板(AAO) 及 無 電 鍍 技 術 (electroless plating)在矽基板上製作出規則排列的鎳奈米柱陣列(Ni nanorod array)。研究中發現 Ni nanorod array 之磁性表現與其微結構 有直接之關係。穿透式電子顯微鏡(TEM)的分析結果發現 Ni nanorod array 之結構為微晶結構(nanocrystalline)其平均晶粒尺寸 2~3nm,此微晶結構導致Ni nanorod array 表現出超順磁(superparamagnetic)的磁性
行為。Ni nanorod array 經 400℃退火後發現磁性表現由超順磁轉變為 鐵磁(ferromagnetic)行為,利用 TEM 觀察 Ni nanorod array 之暗視野 影像(dark field image)發現經退火後 Ni nanorod array 之微結構由微晶 轉變為片狀(laminar)之晶粒結構,由橫截面的暗視野影像發現片狀結
研究中的另一主題是將Ni nanorod array 置於大氣氣氛下退火後 製 作 出 鎳- 氧 化 鎳 之 核 - 殼 奈 米 柱 陣 列 (Ni-NiO core-shell nanorod array)。HRTEM 發現經大氣下退火後 Ni nanorod 表面生成多晶結構 (polycrystalline)之氧化鎳殼層,鎳柱與氧化鎳殼層之界面發現有第二 相析出。不同的退火條件造成氧化鎳殼層厚度及鎳柱微結構的改變而
此改進而影響Ni- NiO core-shell nanorod array 之電性及光感測的表
現。鎳-氧化鎳所形成的異質接面表現出整流特性,研究中發現此接 面在未施加外加偏壓情況下,紫外光(ultra-violet)照射時會有顯著的光 電流(short-circuit photocurrent)產生,此結果顯示 Ni- NiO core-shell nanorod array 有做為紫外光感測器的潛力。
研究中的最後主題是利用原子層沉積技術(ALD)在 Ni nanorod 上
沉積二氧化鈦(TiO2)進而製作出鎳-二氧化鈦之核-殼奈米柱陣列
(Ni-TiO2 core-shell nanrod array)。TEM 分析結果得知利用 ALD 所沉
積之二氧化鈦殼層為多晶結構且厚度相當均勻其厚度為 11nm。研究
中發現鎳-二氧化鈦(Ni-TiO2)之異質接面具有整流特性且在未施加外
加偏壓情況下,紫外光照射時會有顯著的光電流產生,此結果顯示
Ni-TiO2 core-shell nanrod array 有做為紫外光感測器的潛力。此外將
Ni-TiO2 core-shell nanrod array 與 TiO2 nanotube array 兩者做紫外光感
Fabrication and Characterization of 1-D Ni nanorods,
Ni-NiO,Ni-TiO
2core-shell nanostructures
Graduated Student:Chien-Min Liu Advisor: Prof. Chih Chen
Institute of Materials Science and Engineering National Chiao Tung University
Abstract
This dissertation investigates the fabrication and characterization of 1-D nanorods. This dissertation divided into three parts. The first part is study on the microstructures and magnetic properties of nickel nanorods using an anodic alumina oxide template and electroless deposition were investigated. The as-deposited nanorods were found to contain nanocrystalline grains with an average size of ~ 2-3 nm. The temperature-dependent magnetic hysteresis curves indicated a superparamagnetic behavior of the as-deposited rods as a result of the reduction of ferromagnetic crystallites. The superparamagnetic (SM) Ni nanorods transformed into ferromagnetic (FM) ones when annealed at 400 °C. Results from dark-field transmission electron microscopy (TEM) reveal that the microstructure of the rods tends to form a laminar structure with grain growth parallel to the long axis of the rods, together with the enhancement of ferromagnetic ordering along the same direction. The results suggest that the obtained SM-FM phase transition is
microstructure-driven.
The second part relate to the fabrication of highly ordered Ni-NiO core-shell arrays were fabricated by directly annealing Ni arrays at a desired temperature, naturally forming a nano-size semiconductor (NiO)-metal (Ni) heterojeuction at the surfaces of the nanorods. High resolution TEM reveal the developing mechanism of core shells, showing an involvement of a second phase segregation at Ni/NiO interface upon annealing. Results suggest that the increase of annealing time leads to the modifications of microstructure that are responsible for the electrical and photoconductive properties of the devices. The developed nanojunction enables the device’s functionalities including current-rectifying and short-circuit photocurrent generating, showing an environmentally friendly nature. These make the device worthy of being considered an alternative for a UV photodetector.
The third part relate to the fabrication of well-aligned Ni-TiO2
core-shell nanorod arrays on Si substrate by electroless deposition using anodic alumina oxide and atomic layer deposition technique. Results from TEM and high-resolution TEM indicated that the as-prepared samples were a vertically well-aligned Ni-TiO2 core-shell nanorod array,
and the outer TiO2 shell was polycrystalline anatase phase with a
thickness about 11nm. The asymmetry of the current-voltage curve reveled that a schottky barrier formed between the Ni core and TiO2 shell.
The enhanced separation of photogenerated holes and electrons was demonstrated by photoluminescence and photocurrent measurement. Under UV light irradiation, the short-circuit photocurrent of the Ni-TiO2
core-shell nanorod arrays was eight times large than that of the TiO2
致 謝
首先感謝指導教授陳智老師悉心的教導,使我得以一窺利用無電 鍍技術與氧化鋁膜模板結合於矽基材上製備鎳奈米柱並對於其異質 接面研究領域的深奧,不時的討論並指點我正確的方向,對我的耐心 指導與經驗傳授,使我在這些年中獲益匪淺,老師對學問的嚴謹更是 我輩學習的典範。 本論文的完成另外亦得感謝交通大學材料系曾院介老師以及南 台科技大學光電工程學系的鄭錫恩老師以對我研究上的指教評點與 原子層化學氣相沉積系統儀器的協助。國科會貴重儀器經費上的支持 及實驗中給予任何協助和指導之先進,也因為有你們使得本論文能夠 更完整。 本論文的完成另外亦得感謝詠煌學長、元蔚、佳凌的經驗傳承, 給予無數寶貴的意見與實驗上大力協助。因為有你們幫忙,不厭其煩 的指出我研究中的缺失,且總能在我迷惘時為我解惑,使得本論文能 夠更完整而嚴謹。,也感謝、筱芸、翔耀、宗寬、漢文、佑俊、誠風、 永昌、旻峰、宗憲、明慧、瑋安、建志、岱霖、若薇實驗室的大家們 當然也不能忘記,你們的幫忙及搞笑我銘感在心。四年多的日子裡, 實驗室裡共同的生活點滴,學術上的討論、言不及義的閒扯、讓人又 愛又怕的宵夜、趕報告的革命情感,感謝眾位學長姐、同學、學弟妹的共同砥礪,你/妳們的陪伴讓五年的研究生活變得絢麗多彩。 感謝我的雙親及女朋友雪芬對我的關懷與支持。讓我可以保持樂 觀和衝勁,來完成學業。
目錄
中文摘要 ··· . Ⅰ 英文摘要 ··· . Ⅲ 誌謝 ··· VI 目錄 ··· VIII 表目錄 ··· XI 圖目錄 ··· XII 第一章 研究動機 ··· 1 第二章 文獻回顧 ··· 2 2.1 奈米科技 ··· 2 2.1.1 奈米材料之簡介 ··· 2 2.1.2 奈米材料特質 ··· 4 2.2 一維奈米磁性金屬線 ··· 10 2.3 一維奈米材料之合成方式 ··· 14 2.4 一維磁性奈米金屬線文獻回顧··· 18 2.5 多孔性陽極氧化鋁膜 ··· 29 2.5.1 多孔性氧化鋁之生長機制··· 33 2.5.2 多孔性陽極氧化鋁膜之應用 ··· 382.6.1 無電鍍原理 ··· 39
2.6.2 無電鍍鎳反應機制 ··· 40
2.7 原子層化學氣相沉積 (Atomic Layer Deposition, ALD) ··· 41
2.8 一維奈米線之異質接面 ··· 43 2.9 參考文獻 ··· 45 第三章 實驗流程與方法 ··· 50 3.1 一維鎳奈米柱陣列(1-D Ni nanorod arrays) ··· 50 3.2 一維鎳-氧化鎳之核-殼奈米柱陣列(1-D Ni-NiO core-shell nanorod arrays) ··· 57 3.3 一維鎳-二氧化鈦之核-殼奈米柱陣列(1-D Ni-TiO2 core-shell nanorod arrays) ··· 61
第四章 結果與討論 ··· 64 4.1 一維鎳奈米柱陣列 ··· 64 4.1.1 一維鎳奈米陣列之形貌分析 ··· 64 4.1.2 一維鎳奈米陣列之微結構分析 ··· 65 4.1.3 一維鎳奈米陣列之磁性分析 ··· 66 4.2 一維鎳-氧化鎳之核-殼奈米柱陣列 ··· 77
4.2.1 一維鎳-氧化鎳之核-殼奈米柱陣列之形貌微及微結 構分析 ··· 77 4.2.2 一維鎳-氧化鎳之核-殼奈米柱陣列之 PL 分析 ··· 79 4.2.3 一維鎳-氧化鎳之核-殼奈米柱陣列之電性分析 ··· 80 4.2.4 一維鎳-氧化鎳之核-殼奈米柱陣列之 UV 光感測 分析 ··· 80 4.3 一維鎳-二氧化鈦之核-殼奈米柱陣列 ··· 90 4.3.1 一維鎳-二氧化鈦之核-殼奈米柱陣列與二氧化鈦奈 米管之形貌及微結構分析 ··· 90 4.3.2 一維鎳-二氧化鈦之核-殼奈米柱陣列與二氧化鈦奈 米管之PL 分析 ··· 91 4.3.3 一維鎳-二氧化鈦之核-殼奈米柱陣列與二氧化鈦奈 米管之電性分析 ··· 91 4.3.4 一維鎳-二氧化鈦之核-殼奈米柱陣列與二氧化鈦奈 米管之UV 光感測分析 ··· 92 4.4 參考文獻 ··· 101 第五章 結論 ··· 106
表目錄
表2-1 奈米晶材料與其非晶質及結晶材料之性質比較 ··· 4
表2-2 銅奈米粒子之粒徑與表面能及其於總能量中所占比例之關係 ··· 7
表2-3 金屬奈米粒子之熔點變化 ··· 8
圖目錄
圖2-1 奈米粒子之大小與原子分布粒子表面比例之關係 ··· 7 圖2-2 界面張力與粒子相對大小之關係 ··· 9 圖2-3 電子於一維導線中之散射(diffusive)與彈射(ballistic)傳導 現象,λF 為導電電子之費米波長,而 λ 則為其平均自由徑 ··· 12 圖 2-4 不同直徑之鎳金屬奈米線於垂直(●)或平行(□)線材方向 之磁滯曲線 ··· 13 圖2-5 以聚碳酸酯製備之多孔模板,孔徑約為 1μm··· 15 圖2-6 (A)鎳與(B)鈷奈米線陣列其於垂直(實心圓點)與平行(空心圓點) 奈米線外加磁場下之矯頑磁力對直徑之關係圖 ··· 19 圖2-7 (a)約 500 層之鈷(10 nm) / 銅(10 nm)奈米線之明視野穿透電子 顯微鏡相片,(b)為放大圖,圖中顏色較淡者為鈷層,顏色較深 者為銅層,(c)為溫度 4.2 K 下之 MR%為 19 %,(d)於 290 K 之 MR%為 15 % ··· 21 圖2-8 鈷(5 nm)/ 銅(5 nm)之磁電阻與磁滯曲線奈米碳管的石墨平面 ··· 22圖2-9 為鈷/銅之磁電阻曲線 ··· 23 圖2-10 為 80 nm 鎳奈米金屬線之磁電阻曲線 ··· 24 圖 2-11 於 77K 所量測 MR 分別實線為鎳鐵/銅奈米線及虛線鈷(10 nm)/銅(5nm)奈米線,縮圖為 4.2K 鎳鐵/銅奈米線之 MR ··· 24 圖 2-12 鎳奈米線陣列其矯頑磁力對直徑(擴孔時間)之關係圖,圖中 之磁滯曲線由左至右分別為直徑8 nm、18 nm 以及 21 nm 之奈米線量測結果 ··· 26 圖 2-13 由上至下分別為長寬比為 3、36 與 67 鎳奈米線陣列之磁滯 曲線圖,線之直徑分別為180 nm、50 nm 及 18 nm ··· 27 圖2-14 (a)為以60 nm 陽極氧化鋁膜與聚酯膜之鎳與鈷磁電阻值,(b) 為以60 nm 陽極氧化鋁膜與聚酯膜之鈷/銅磁電阻值 ··· 28 圖2-15 (a)為以 60 nm 陽極氧化鋁膜合成鐵 8 nm(鈷 1.5 nm/銅 4 nm/ 鈷2.5 nm)之磁電阻值,與(b)改變銅層對磁電阻之影響 ··· 29 圖2-16 多孔性氧化鋁膜之製備概略圖 ··· 32 圖2-17 多孔性氧化鋁膜之示意圖 ··· 32
圖2-18 孔洞大小與外加電壓之關係 ··· 33 圖2-19 氧化鋁表面形成局部電場集中之過程示意圖 ··· 34 圖2-20 陽極氧化鋁孔洞形成之機制 ··· 35 圖2-21 藉由聚焦離子束得到有序之孔洞與形狀之製作流程圖 ··· 36 圖2-22 藉由聚焦離子束得到有序之孔洞與形狀之 SEM 上視圖 ··· 36 圖2-23 藉由 SiC 模版於鋁箔上壓模得到有序之孔洞之製作流程圖 ··· 37 圖2-24 藉由 SiC 模版於鋁箔上壓模得到有序之孔洞之 SEM 側視圖 ···· 37 圖3-1 為一維鎳奈米柱陣列之實驗流程圖 ··· 51 圖3.2 AAO 製具與試片示意圖 ··· 53 圖3.3 兩階段陽極氧化處理過程示意圖 ··· 53 圖3-4 一維鎳奈米柱陣列之製程示意圖 ··· 56 圖3-5 一維鎳-氧化鎳之核-殼奈米柱陣列之製程示意圖 ··· 57 圖3-6 為一維鎳-氧化鎳之核-殼奈米柱陣列之實驗流程圖 ··· 58
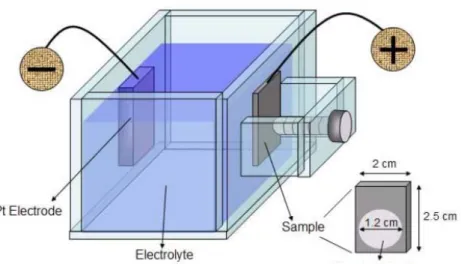
圖3-7 一維鎳-氧化鎳之核-殼奈米柱陣列之電性量測示意圖 ··· 59
圖3-8 一維鎳-氧化鎳之核-殼奈米柱陣列之 UV 光感測示意圖 ··· 60
圖3-9 一維鎳-二氧化鈦之核-殼奈米柱陣列之製程示意圖 ··· 61
圖3-10 一維鎳-二氧化鈦之核-殼奈米柱陣列之實驗流程圖 ··· 62
圖4-1-1 (a) AAO 之平視野 SEM 影像。(b) Ni nanorod arrays 之平視野 SEM 影像。(c) Ni nanorod arrasys 之平視野 SEM 影像 ··· 70
圖4-1-2 Ni nanorod arrays TEM 之(a)橫截面影像。(b)平視野影像 ··· 71
圖4-1-3 Ni nanorod arrays 之 HRTEM 影像。圖中虛線白框所圈選的區 域為微晶晶粒。(a)Ni nanorod arrays 之根部與 Si 基板間有一 層原生氧化層存在。(b)Ni nanorod arrays 支柱狀結構由維晶所 組成。右上角為Ni 微晶{111}平面之繞射影像 ··· 72
圖4-1-4 (a)各種退火條件下 Ni nanorod arrays 之室溫 M-H 曲線。(b) 不同量測溫度下Ni nanorod arrays 之 M-H 曲線。量測時磁場 施加方向與 nanorod arrays 長軸平行。Y 軸所表示的 M/Ms 對於(a)之 Ms 為 400℃-2min,(b)之 Ms 為 2K ··· 73
圖4-1-5 不同退火條件下 Ni nanorod arrays 之 Ms/Ms(2 min)對退火時間
之作圖 ··· 74
圖4-1-6 Ni nanorod arrays 400℃退火 1min 之 TEM 暗視野影像,(a)
平行 nanorod 長軸 (b)垂直 nanorod 長軸。(c)為對應圖(a)之
選區繞射影像,繞射點的拉長方向垂直圖(a)晶粒之長軸 ··· 75
圖4-1-7 Ni nanorod arrays 400℃下退火(a)30s、(c)1min、(e)2min 之 TEM 平視野影像,(b)、(d)、(f)為對應 30s、1min、2min 之 TEM 暗試也影像 ··· 76
圖4-2-1 (a)300℃-30min (b)300℃-3hour 之 Ni-NiO core-shell nanorod arrays 橫截面 TEM 影像 ··· 83
圖4-2-2 Ni-NiOcore-shell nanorod arrays 之 HRTEM 影像
(a)300℃-30min (b)300℃-3hour。圖(a)之右上角影像為圈選
NiO 區域作 FFT 轉換之繞設圖形 ··· 84
圖4-2-3 Ni-NiO core-shell nanorod arrays 之 HRTEM 影像 (a) 250°C 15-min (b) 250°C 50-min and (c) 250°C 4-hr ··· 85 圖4-2-4 (a)Ni-NiO core-shell nanorod arrays 兩種條件(300℃-30min、
果 ··· 86
圖4-2-5 ITO/Ni-NiO (300°C 30-min)/Si、 ITO/Ni-NiO (300°C 3-hr)/Si 和 ITO/Ni/Si 三種試片之 I-V 曲線 ··· 87
圖4-2-6 ITO/Ni-NiO (300°C 30-min)/Si、 ITO/Ni-NiO (300°C 3-hr)/Si 和 ITO/Ni/Si 三種試片之 UV 感測圖譜,量測時並無施加外 加偏壓V=0 ··· 88
圖4-2-7 Ni-NiO 異質接面之能帶圖,UV 光由 NiO 端照射,藍色區域
為空乏區 ··· 89
圖4-3-1 TiO2 nanotube arrays 之 TEM 橫截面影像,圖中右上角為 TEM
平視野影像 ··· 94
圖4-3-2 Ni-TiO2 core-shell nanorod arrays 之 TEM 橫截面影像 (a)整體
影像 (b)局部放大影像 ··· 95
圖4-3-3 Ni-TiO2 core-shell nanorod arrays 之 HRTEM 影像 ··· 96
圖4-3-4 Ni-TiO2 core-shell nanorod arrays 與 TiO2 nanotube arrays 之 PL
圖4-3-5 ITO/Ni-TiO2/Si 與 ITO/TiO2/Si 兩種試片之 I-V 曲線 ··· 98
圖4-3-6 ITO/Ni-TiO2/Si 與 ITO/TiO2/Si 兩種試片之 UV 光感測圖譜,
量測時並無施加外加偏壓V=0 ··· 99
圖 4-3-7 (a)Si-TiO2 P-N 接面之能帶圖 (b)Ni-TiO2 異質接面之能帶
第一章 研究動機
當材料尺寸逐漸縮小至奈米尺度時具有相當特殊的化學及物理 性質,因此奈米材料近年來被廣泛的研究。在奈米材料的研究領域 中,如何利用簡易的製程製作奈米材料以及規則化的製作在所要的基 板上是相當重要的研究課題。由於現今的元件製程絕大部分是利用矽 晶片做為基板,因此如何將奈米材料的製程與矽基板做整合並且規則 化的製作在矽基板上是奈米材料要實際運用時必須面對的主要問 題。因此在本研究中使用陽極氧化鋁(anodic alumina oxide, AAO) 在 矽 晶 片 上 作 為 模 板 並 結 合 簡 易 的 無 電 鍍 沉 積 技 術 (electroless plating),在矽晶片上製作出規則之鎳奈米陣列(nickel nanorod array)。由於元件尺寸日亦縮小,在奈米材料的研究領域中,有關奈 米元件的製程在近年來也受到極大的重視。以往元件製程中由於元件 尺寸較大,所以接點(junction)面積所佔比例相對較小,而在元件 尺寸縮小至奈米尺度時,接點所佔的面積比例急遽上升,而這也使接 點研究在奈米材料的研究中成為重要的課題之ㄧ。因此在本研究中利 用簡單的退火處理及原子層沉積技術(atomic layer deposition, ALD)在鎳奈米陣列的表面上製作出 Ni-NiO 及 Ni-TiO2兩種不同的異質接
第二章 文獻回顧
2.1 奈米科技
2.1.1 奈米材料之簡介
奈米技術為20世紀80年代末始誕生並迅速崛起之技術,最早將此 一術語用至技術上為日本於1974 年,其應用原子與分子創造新物質 之技術;研究尺寸範圍於1~100 nm 之間物質組成;此極其微小之空 間,恰乃為原子與分子之尺寸範圍,亦為它們相互作用之空間。奈米 技術之故會受至如此之重視,乃因當其物質小至奈米之尺度時,因量 子效應、物質之局域性、巨大之表面與界面效應,使物質之多種性能 發生變化。此些變化應用至各個工業領域後,將引導出一輪嶄新之工 業革命【1】。 一奈米為十個氫原子並排在一起之長度,十奈米約為人類頭髮直 徑之三萬五千分之一,而目前最先進之積體電路其最窄線寬約為四十 五奈米。當粒子直徑、晶粒尺寸、孔隙直徑、薄膜厚度或電子元件中 之導線寬度等特徵長度若小至1-100奈米之間,則定義為奈米材料 (nanomaterials)。現今,廣義地,奈米材料為指在三維空間中至少存 在一維處於奈米尺度範圍或經此作為基本單元構成之材料。若依照維 數,奈米材料之基本單元即可分為零維(zero dimension ; 0D)、一維(onedimension ;1D)以及二維(two dimension ; 2D)三類。 其中零維,指於空間三維尺度均於奈米尺度,如奈米尺度顆粒 (nanoparticle)、原子團簇等,具奈米孔隙之材料亦歸類為零維材料; 一維,指於空間有兩維處於奈米尺度,如奈米線(nanowire)、奈米棒、 奈米管(nanotube)等;二維,指於三維空間中有一維於奈米尺度,如 奈米薄膜,奈米塗層; 超晶格等。於奈米尺度下,一般傳統科學理論、 技術均已不適用,因於此尺度之物理、化學性質與較大尺寸(如微米) 所呈現者截然不同,亦即當物質尺寸小至1 nm 至100 nm 範圍,其物 理、化學與生物性質可能將與較大結構尺寸時大相逕庭。例如,燒結 溫度與熔點可能大幅下降、反應性與觸媒特性可能大幅提升、不導電 且易脆之陶瓷材料可能變得既導電又具延展性、導電金屬之導電度可 能下降、油溶性之藥物可能變成水溶性、不透明之材料可能變得透 明、半導體光電材料之吸光波長會往短波長偏移等。此些性質之改 變,並非因化學組成改變所致,純屬結構尺寸縮小所造成。如表2-1 所示,以下即分別舉例說明其特殊性質。
表2-1 奈米晶材料與其非晶質及結晶材料之性質比較【1】。 性質 材料 奈米晶 非晶質 結晶 熱膨脹係數(10-6/K) Cu 31 18 16 熱容(298K,J/gk) Pd 0.37 -- 0.24 飽和磁化強(4K,emu/g) Fe 130 215 222 超導臨界溫度(K) Al 3.2 -- 1.2 擴散活化能(eV) Ag in Cu Cu in Cu 0.51 0.64 -- 2.0 2.04 密度(g/cm3) Fe 6 7.5 7.9
2.1.2 奈米材料特質
當固體粒徑逐漸減小時,其光、電、磁、熱、催化與機械力學等 特質均將隨之改變,以下將簡述奈米材料之基本特質: (a) 光學性質 不同粒徑粉末對光透明度,即其對光遮蔽力隨光之波長而改變, 對可見光(波長400~700 nm)而言,遮蔽力最強之粒徑應為波長之 1/2,即200~350 nm 。例如,於此粒徑範圍內之TiO2 即有很高遮蔽 力。但若TiO2 粒徑降至15~50 nm 時,則對可見光反射能力減弱, TiO2 粉將變成透明,但對波長更短之紫外線則具有較強之遮蔽力, 可作為抗紫外線玻璃之成分。(b)電導性質 金屬粒子中原子間距將隨粒徑變小而變小,使其密度將大幅增 加,如此金屬中自由電子之平均徑將減小,電子碰撞頻率驟增,故電 導率會降低。 (c) 磁學性質 粒徑為20~30 nm 之Fe-Co-Ni 合金具有強磁性可作為紀錄器, 其信號與雜訊比極高。如奈米金屬粒子因其小尺度使其具塊材並未具 備之磁特性如超順磁性、極高之矯頑磁力、較低之居禮溫度與飽和磁 化率等,而磁性金屬奈米線其於不同方向之矯頑力存在相當大之差 異,當奈米線之長寬比增加時,其與長端平行之矯頑磁力將隨之增大 等。此外,塊材磁化率與溫度關係不大。但當磁性物質粒徑變小時, 其磁化率將隨溫度降低,甚至降至為零,而成為絕緣體。 (d)熱導性質 奈米粒子於低溫時之熱阻趨近於零,1997 年法國研究人員藉由 此一性質,利用70 nm 燒結之銀顆粒作為低溫導熱材料,使稀釋致冷 機之溫度從300×10-3K 降到3×10-3K。 (e) 催化與化學性質 因當粒子縮小至奈米尺度後,表層之原子數比例很高,而可充分 發揮吸附作用,且表面原子分布為一階梯狀結構,含有許多未飽和鍵
結,因而增加其熱與化學不穩定性,使具有極高之表面能,加強其化 學反應能力,使得奈米觸媒的反應活性及選擇性大為提昇。此時分布 於表面之原子鍵結狀態十分不穩定,極易使外來原子與其產生反應或 鍵結而組成新結構,例如與原本無法混合之金屬或聚合物形成合金 等。銅奈米粒子,顆粒由10 nm降至1 nm 時,表面原子分布比例則由 20%驟增為99%,如圖2-1 所示,而表面能之數值亦迅速增加,且其 於總能量中所占之比例亦隨之增加,若銅奈米粒子之粒徑由100 nm 降至5 nm,其表面能增加接近20 倍,其於總能量中所占之比例亦增 加20 倍,如表2-2 所示。此種現象因與維度有關,故於三維度均為 奈米尺寸之奈米粒子中最為明顯。因奈米粒子具此種高表面能特性, 故為製備觸媒之最佳材料,如Pt 奈米粒子即於燃料電池中作為甲醇 催化之用途,而奈米TiO2 粒子則因具光觸媒之效果,常被塗佈於器 壁或房舍外牆,使其具自潔之效用。
圖2-1 奈米粒子之大小與原子分布粒子表面比例之關係【2】。 表2-2 銅奈米粒子之粒徑與表面能及其於總能量中所占比例之關係 【3】。 粒徑 (nm) 全表面積 (cm2) 表面能量 (erg) 表面能量/ 總能量 (%) 5 nm 8.54 x 107 1.88 x 1011 5.51 10 nm 4.27 x 107 9.40 x 1110 2.75 100 nm 4.27 x 106 9.40 x 109 0.275 1μm 4.27 x 105 9.40 x 108 0.0275 10μm 4.27 x 104 9.40 x 107 0.00275 100μm 4.27 x 103 9.40 x 106 0.000275
(f) 熔點與燒結溫度之降低 因晶體表面原子振幅約為晶體內部原子振幅之兩倍,因此可預 期,隨顆粒粒徑變小與表面原子所佔總原子數比例增加,晶體之熔點 將不斷降低。例如,一般鎢粉燒結溫度為3000℃,然加入0.1%~0.5 %奈米級鎢粉後,燒結溫度可有效降低至1300℃。表2-3 為一些奈米 粒子與其塊材之熔點比較表3。因此奈米材料除可於較低溫度燒結而 得之外,若將此些奈米材料摻雜於塊材中亦可降低整體之燒結溫度。 表2-3 金屬奈米粒子之熔點變化【3】。 單位:℃ 材料種類 Fe Ag Ni W Au Pt Cu Nanopowder (50 nm) 300-400 60-80 ~200 1100 900 800 200 Bulk 1536 960.8 1453 3380 1063 1769 1083 (g)表層吸附性 表層原子本身具有非結合(non-bonding)的電子,除表現於催化性 外,亦表現於強烈之導電、順磁與傳感作用上。當材料尺度降至奈米 範圍,將使吸附方式大異於傳統化學所能預測之結果,此可由奈米材 料表面張力等因素探討吸附現象。當材料尺寸愈小,其表面張力愈 小,則表面位能愈高,如圖2-2 所示。為平衡與環境之界面位能差,
此些粒子將吸附環境中其它原子或分子以平衡界面位能,因此奈米材 料之吸附能力遠高於一般塊材。因奈米粒子傳感器,表面活性位置將 增加,除加強敏感度之外,因粒徑小已致孔隙度亦有效縮小,使得訊 號之傳遞迅速而不受干擾。故信號與雜音比也能獲改善。 圖2-2 界面張力與粒子相對大小之關係【2】。 (h)結合力 受強烈凡得瓦作用力與大量表層原子之作用影響,造成奈米陶瓷 複合體具有良好之硬度、耐壓力與耐滑能力。 (i) 化學鍵之性質變化 一般,當粒徑大小逐漸趨近奈米顆粒範圍時,原屬於離子型晶體 將呈現出更強之共價鍵性質,原屬於共價鍵型晶體會逐漸呈現離子鍵 或共價鍵之特性。 奈米材料具同材料於塊材狀態截然不同之化學性質與物理性
延展性、高催化性、高硬度、低熔點等,將隨著粒徑大小不同而有所 變化,此些特性與傳統塊材材料截然不同,此些有趣之特性,應用性 十分廣泛,其囊括物理、化學、化工、電子與材料科學等各領域,造 成各界對奈米材料之研究與重視。
2.2 一維奈米磁性金屬線
當金屬線之寬度接近電子之平均自由徑(mean free path,電子運 動碰撞另一個電子前所能移動之距離)時,電子於金屬線內之傳導現 象將由散射(diffusive)轉為彈射(ballistic),如圖2-3 所示,其電阻將因 電子碰撞機率減少而減至相當低。當此金屬線之直徑進一步縮短至奈 米尺寸,亦即接近電子之費米波長(fermi wavelength),其導電性將與 長度無關且變為量子化。 G = 2Ne2/h = N(2e2/h) (2-1) 其中G 為導電度,h 為浦朗克常數,e 為電子電量,而N 則為導線 中通道之數目,亦即能階之數量。此時導電性將取決於導線直徑且為 非線性關係,當導線直徑減小時,其導電性則以階梯狀方式減小。 除電性外,於奈米尺度下電子之自旋亦有相當大之改變,例如不同直 徑之鎳金屬奈米線,由其磁滯曲線圖2-4 中可知, 隨著直徑由55 nm 逐漸減小至30 nm,鎳奈米線中平行線材方向之矯頑磁力(Hc)由600 Oe、1000Oe 而增加至1200 Oe,其角形比[squareness, 外加磁場為零
時樣品之殘留磁化量(Mr)/飽和磁化量(Ms)]亦由30 %增加至98 %。 然而若將外加磁場施於垂直線材之方向,其矯頑磁力僅為100 Oe,且 角形比亦較磁場施於平行線材方向時降低許多。為固定鎳奈米線間之 磁交互影響,圖2-4中不同直徑之奈米線之間距均控制於100 nm,而 長度則固定為1 μm。由上述可知,磁性金屬奈米線其於不同方向之矯 頑磁力存在相當大之差異,當奈米線之長寬比(aspect ratio)增加時(即 圖2-4中奈米線直徑由55 nm減小至30 nm),其與長端平行之矯頑磁力 將隨之增大,此將有利其於磁性紀錄媒體之應用。此外,若將鐵磁性 金屬(如鐵、鈷與鎳等)與非鐵磁性金屬(如銅與銀等)交替接合成層狀 奈米線材,其導線電阻將隨外加磁場之方向與大小而有極大之變化, 即磁電阻(magnetoresistance; MR)特性,此特性可應用於磁感測元件。
圖2-3 電子於一維導線中之散射(diffusive)與彈射(ballistic)傳導
圖2-4 不同直徑之鎳金屬奈米線於垂直(●)或平行(□)線材方向之 磁滯曲線【5】
。
2.3 一維奈米材料之合成方式
奈米線製程部分,由於目前黃光技術之瓶頸限制由薄膜蝕刻成奈 米結構之線寬,無法以top down 方法任意製造不同線徑之奈米結 構;因此bottomup 之方法為目前被積極發展之一維奈米結構材料製 程技術。而bottom up方式成長一維奈米結構材料的方法可分為 template 輔助成長法、氣相-液相-固相成長法(vapor-liquid-solid; VLS)法、氧化物輔助成長法、奈米晶粒輔助成長法、超臨界流體溶 液(supercritical fluid solution-phase)、雷射剝除法與電弧放電法等,以 下即個別簡介各合成方法。(1) 模板輔助成長法:
所謂模板輔助成長法又可分為AAM(anodic alumina membranes) 輔助成長法與界面活性劑輔助合成法兩類:
(a) AAM(anodic alumina membranes)輔助成長法:
乃 以 陽 極 氧 化 法 形 成 具 有 奈 米 尺 度 孔 洞 之 多 孔 性 氧 化 鋁 為 template,分別利用各種化學方法,如CVD、溶液化學法、凝膠(sol-gel) 法、電鍍法等,於奈米尺度孔洞中沉積奈米線結構之材料。此外,亦 以碳微管或多孔性高分子基材為template沉積奈米線之研究結果發 表,以聚碳酸酯模為例,首先準備一面無孔洞之聚碳酸酯模,其厚度
約為6-20 mm,爾後利用高能離子轟擊表面,經轟擊之表面將出現 許多凹洞,再藉由化學方式蝕刻(配方為6.25 M 之NaOH)即可得均一 孔徑分布之多孔膜,其孔徑大小可小至10 nm【6】,圖2-5即為以聚 碳酸酯製備之多孔模板。然由於離子轟擊之凹洞並非規則排列,故此 些孔洞之排列亦無次序,此外由於部分凹洞並非完全垂直於膜表面, 因此所形成之孔洞其成長方向相對於表面並非為90°角,根據文獻報 導其傾斜可達34°。8雖孔洞之排列與方向均不規則,所合成之奈米線 其直徑仍不受影響,故此法仍常用於奈米線之製備。若須奈米線於模 板中規則排列,則模板之材料則須改用陽極氧化鋁。故本研究以前述 之多孔性陽極氧化鋁優勢為template 藉由無電鍍沉積法配製一維奈 米材料。 圖2-5 以聚碳酸酯製備之多孔模板,孔徑約為1μm【7】。
(b)Soft template 界面活性劑輔助合成法: 以微胞或逆微胞合成奈米金屬及半導體晶粒已發展多時,利用此 方法可合成尺寸分布非常均勻之奈米晶粒,對於金屬、半導體晶粒之 各種物理性質與奈米晶粒尺寸之關係建立有極大貢獻。近來,界面活 性劑(被稱之為soft template)輔助合成一維奈米線材料之研究亦漸漸 被重視,如何提高其aspect ratio 乃為一重要課題【8-11】。
(2) 氣相-液相-固相成長法(vapor liquid solid; VLS)法:
VLS方法其早已廣泛應用於鬚晶之成長【12】,目前也被利用於 奈米碳管及半導體奈米線之成長,寬能距材料如GaN等的奈米線也可 以利用VLS法有效成長【13】。利用此機制成長奈米線之優點為,可 藉由觸媒顆粒之大小來控制奈米線之直徑分布【14】;此外藉由觸媒 薄膜或顆粒之選擇性沉積,可於基板上選擇性成長奈米管或奈米線。 (3)氧化物輔助成長法:
Lee 等研究者發現利用SiO2 與Si 固態混和物為靶材,以laser
ablation將之氣化,於無觸媒之成長條件下,可於適當溫度下成長Si 奈
米線。Ge奈米線亦可以此氧化物輔助成長法,利用GeO2 與Ge 之混
(4)奈米晶粒輔助成長法:
此法於無觸媒之成長條件下,先於基板上成長一奈米晶粒薄膜作 為緩衝層,而後於其上成長相同成分的一維奈米結構材料。目前利用 此法成長的一維奈米結構材料有SiCN 奈米柱【16】與GaN 奈米線等 【17】。
(5)雷射剝除法(laser ablation method)
雷射剝除法最早乃由Smalley 等人應用於單壁奈米碳管之大量 製備,其合成裝置包含五部分:雷射源、石英或氧化鋁管、含有反應 物及催化劑之靶材、水平管狀爐以及真空系統。首先將靶材置入石英 管中,放入管狀爐內,爾後將雷射光照射靶材使其氣化,此些蒸氣原 子於適當之氣氛及溫度控制下將以VLS 之方式形成奈米線【18,19】 。
(4)電弧放電法(arc discharge method)
將樣品粉末塞入一石墨陽極內,另取一石墨棒作為陰極,兩電極
間之距離為數mm,在腔體內充入500 torr 之Ar / O2 (1 / 4)混合氣,並
於兩電極間施加約55-65 A 之直流電流,電壓為13-15 V,電弧放 電約5-6 秒後,樣品粉末將形成奈米線吸附於陰極與腔體表面,並 將兩電極間形成網絡狀之構造【18】。
2.4 一維磁性奈米金屬線文獻回顧
以下將介紹利用多孔性陽極氧化鋁膜作為金屬沉積模板之研究 於近十年來受至各界之高度重視,其中涵蓋各類材料之製備與其特性 分析。其相關文獻如下所述: Whitney 等人(1993)【20】利用以track-etch 方式製得具垂直奈米 孔洞之聚碳酸酯作為基版,以濺鍍(sputter)方式於其上濺鍍一層金屬 銅作為背電極,並藉由三電極法於孔洞內電化學沉積鎳與鈷之奈米 線,其直徑隨聚碳酸酯基版孔洞大小之不同而改變,其值可為30 nm 至200 nm。Whitney 等人於此篇文獻中闡述電化學沉積過程中電流於 奈米線不同成長階段之變化,並探討不同長寬比之鈷與鎳奈米線其矯 頑磁力(coercivity ; Hc)與角形比(squareness)之變化。作者發現此二種 磁性奈米線其易磁化之軸向方向均與基板垂直,亦即平行於線之方 向,而矯頑磁力之數值則隨奈米線直徑之增加而減小,如圖2-6 所 示,鎳奈米線之矯頑磁力由直徑50 nm 時之500 Oe 減少為200 nm 時 之300Oe,鈷奈米線之變化亦相似,此變化乃因奈米線內磁區之數量 隨線之直徑增大而增加所致。此磁區增加之現象將造成奈米線內各原 子磁矩之排列方向不一致,使其較易受外加磁場之影響而翻轉方向, 亦即其矯頑磁力降低。 除矯頑磁力之外,作者尚探討角形比之變化趨勢。由圖中可知直徑50 nm之鎳或鈷磁性奈米線其垂直基板之角形比較平行基板者為 高,此形狀異向性(shape anisotropy)乃因長軸方向(易軸方向)磁矩間交 互作用產生之反鐵磁偶合現象較弱,故於移除外加磁場後之殘存磁化 量較高所致。 圖2-6 (A)鎳與(B)鈷奈米線陣列其於垂直(實心圓點)與平行(空心圓點) 奈米線外加磁場下之矯頑磁力對直徑之關係圖【20】。 Piraux 等人(1994)【21】以多孔聚碳酸酯作為基版,於一面蒸鍍銅作 為背電極,藉由三電極法沉積鈷/銅多層奈米線。其中鈷與銅之金屬
換不同電位而還原沉積不同之金屬。此文獻中製得之鈷/銅奈米線其 直徑為40 nm,單一鈷層之厚度為10nm,銅層亦為10 nm,而鈷/銅之 層數則達至500 層,顏色較淡者為鈷層,較淺者為銅層。作者量測其 磁特性,發現其具GMR 效應,當外加磁場於平行基板方向時,此些 奈米線於4.2 K 之MR%為19 %,而於290 K 時則為15 %,如圖2-7 所 示。 (a) (b) (c)
(d) 圖2-7(a)約500 層之鈷(10 nm)/ 銅(10 nm)奈米線之明視野穿透電子顯 微鏡相片,(b)為放大圖,圖中顏色較淡者為鈷層,顏色較深者為銅 層,(c)為溫度4.2 K 下之MR%為19 %,(d)於290 K 之MR%為15 % 【21】 。 Blondel(1994)【22】以多孔聚碳酸酯作為基版,利用與Piraux 等 人所發表類似之方式沉積鈷/銅與(鐵鎳合金)/ 銅奈米線【21】,其中 鐵磁性層之厚度為5nm,非鐵磁性層之厚度亦為5 nm,線之直徑則為 80 nm。作者將磁場分別施於平行與垂直奈米線之方向,量測其磁滯 曲線,發現於此二種不同方向之外加磁場作用下其磁滯曲線十分相 似,亦即消磁場類似,此外於此兩種模式下所測得之MR%亦類似, 為14%(溫度300 K),圖2-8 所示。
圖2-8 鈷(5 nm)/ 銅(5 nm)之磁電阻與磁滯曲線【22】。 Blondel 等人(1996)【23】利用與前一篇相同之方式沉積鈷/銅奈 米線【22】,但作者於此篇文獻則分別嘗試利用單一反應槽與雙反應 槽之方式進行實驗,其中單一反應槽之實驗與前篇相同,而雙反應槽 之方式乃為將鈷與銅之金屬鹽分別置入個別之反應槽中,沉積不同金 屬時則須將基板來回浸入兩反應槽中進行反應。利用單反應槽所合成 之鈷銅奈米線因銅較鈷易還原,故於製備鈷層時必然有部分之銅被還 原而形成合金,藉由元素分析可知其中銅之百分比為15%,而利用雙 反應槽所製得之奈米線則幾無合金產生。作者將利用此兩種方式合成 之奈米線進行磁電阻效應之量測。結果證明以雙反應槽合成之奈米線 由於鈷層中無銅之雜質存在,其電阻值較以單反應槽合成者為小,理 論之MR%應較高,但實際測得之MR%卻較低,僅10%以下,而以單
反應槽所合成者則可達20%以上,由此可知鈷層中雜質之電阻並非影 響MR%之主要因素。此外,作者另嘗試同時改變鈷與銅層之厚度, 量測其MR%之變化,發現其隨鈷與銅層之厚度增加而呈現降低之趨 勢。圖2-9 為鈷/ 銅之磁電阻曲線。 圖2-9 為鈷/銅之磁電阻曲線【23】。 B. Doudin 等人(1998)【24】以孔徑80 nm 徑深6μm 之多孔聚碳酸酯 作為基版,以電化學沉積鎳奈米金屬線,並測其MR ratio。於300 K 可 得1%MR ratio,如圖2-10 所示。 圖2-10 為80 nm 鎳奈米金屬線之磁電阻曲線【24】。
Piraux 等人(1999)【25】以多孔聚碳酸酯作為基版,於一面蒸鍍 銅作為背電極,藉由三電極法沉積鈷(10 nm)/銅(5 nm)及鎳鐵/銅多層 奈米線。於77K 可獲得鎳鐵/銅MR值67%及鈷(10 nm)/銅(5 nm) MR 值約30%。於4.2K 可獲得鎳鐵/銅MR 值71%,如圖2-11。 圖2-11 於77K 所量測MR 分別實線為鎳鐵/銅奈米線及虛線鈷(10 nm)/銅(5nm)奈米線,縮圖為4.2K 鎳鐵/銅奈米線之MR【25】。 Zheng 等人(2000)【26】利用硫酸溶液陽極氧化鋁片製備孔徑約 8 nm 之氧化鋁膜,並以磷酸進行擴孔改變其孔徑,大小可由8 nm 擴 大至22 nm。爾後作者利用此些氧化鋁膜以脈衝法電化學沉積鎳奈米 線於孔洞內,並觀察其矯頑磁力之變化,圖2-12為其矯頑磁力對直徑 變化之關係圖。由圖中可清楚得知矯頑磁力起初隨直徑上升而增加, 爾後於直徑大於18 nm 以上則急遽下降,此種變化乃因奈米線之磁區
數量變化所致。磁性奈米線於直徑較小時其內部之原子磁矩傾向以同 一方向排列而形成單一磁區,當直徑逐漸增加至某一極限時,其原子 磁矩則因原先排列模式能量過高無法繼續維持而產生部分翻轉現 象,翻轉與未翻轉之磁矩個別形成不同之磁區,而此些磁區間磁矩之 交互作用力則有助於降低整體之能量,使排列穩定,此由單一磁區奈 米線轉變為多重磁區奈米線其半徑之分界值稱之為臨界半徑。上述磁 區增加之現象亦將使其較易受外加磁場之影響而翻轉方向,亦即其矯 頑磁力降低。由以上敘述可知,作者所合成之鎳奈米線其臨界半徑約 為9 nm,當半徑大於此值則矯頑磁力下降,而半徑小於臨界半徑依理 論則應保持不變,然則卻出現降低之趨勢,作者認為此乃因奈米線中 之晶體缺陷所導致。 圖2-12 鎳奈米線陣列其矯頑磁力對直徑(擴孔時間)之關係圖,圖中之
磁滯曲線由左至右分別為直徑8 nm、18 nm 以及21 nm 之奈米線量測 結果【26】。 Chu 等人(2002)【27】以氧化銦錫(ITO)作為基材,於其上利用 濺鍍方式成長鋁層,並將鋁層利用陽極氧化製成具孔洞之氧化鋁層, 爾後藉由此作為基板成沉積鎳奈米金屬線。作者分別利用硫酸、草酸 及磷酸等不同之酸液進行陽極氧化,可得具不同直徑孔洞之氧化鋁 膜,進而改變合成鎳奈米線之直徑。圖2-13為其製備不同長寬比奈米 線之磁滯曲線圖,由圖中可知隨其長寬比增加,垂直基板方向之角形 比亦增加,此乃因形狀異向性所導致。
線圖,線之直徑分別為180 nm、50 nm 及18 nm【27】。 Takeshi Ohgai等人(2003)【28】利用經陽極氧化之多孔氧化鋁膜 與聚碳酸酯作為基板,沉積直徑為60 nm 之鎳、銅及鈷/銅多層奈米 線。於300K 量其MR,結果如圖2-14所示,鎳與鈷於此兩模板之磁電 阻差異,顯示出典型之異向性磁電阻(AMR)大約為1.0% 及藉由調配 沉積時間使銅與鈷之厚度被控制為5 nm 至15nm。當調配沉積時間使 銅與鈷之厚度被控制10 nm,此Co/Cu 多層磁性奈米金屬線於陽極氧 化鋁膜基板可獲得MR 20% 。 (a) (b) 圖2-14 (a)為以60 nm 陽極氧化鋁膜與聚酯膜之鎳與鈷磁電阻值,(b) 為以60 nm 陽極氧化鋁膜與聚酯膜之鈷/銅磁電阻值【28】 。
Leszek M. Malkinski等人(2005)【29】以陽極氧化鋁膜沉積磁性 多 層 之 模 板 , 孔 徑 為60 nm,孔洞間距為 100 nm。 以 Magnetron sputtering system 沉積鈷/銅磁性多層膜,量測其多層膜沉積於陽極氧 化鋁膜模板與改變其中非鐵磁性層(銅)厚度,並與沉積於矽晶圓比較 其MR%差異。由圖2-15(a)可明顯獲知鈷/銅之GMR 磁滯迴圈,其以 鐵為seed layer,各層厚度為鐵8 nm(鈷1.5 nm/銅4 nm/鈷2.5nm)。圖 2-15(b)為於陽極氧化鋁膜與矽晶圓上分別改變其沉積非鐵磁性層銅 厚度對MR %之影響。可觀察以陽極氧化鋁膜為模板能獲較高之 MR% ,且於非鐵磁性銅層為4 nm 時具最大之MR% 約12 % ,量測 溫度均為10K 。 (a) (b) 圖2-15 (a)為以60 nm陽極氧化鋁膜合成鐵8 nm(鈷1.5 nm/銅4 nm/鈷 2.5 nm)之磁電阻值,與(b)改變銅層對磁電阻之影響【29】。
2.5 多孔性陽極氧化鋁膜
陽極處理技術於1920 年起就已被研究探討並應用於鋁合金之抗 氧化層與著色之處理上,為歷史悠久之防蝕方法【30-32】。近年, 因奈米科技之蓬勃發展,再次受到各界關注。陽極處理鋁之處理技 術,其為將傳統之陽極處理技術經由改善,使得此具多孔性陣列奈米 結構之氧化鋁膜,提昇應用領域進入奈米技術。多孔性陽極氧化鋁膜(anodic aluminum oxide;AAO)之結構具奈 米尺寸之結構,於電磁(magnetic)【33-35】、電子(electronic)與光電 子(optoelectronic)【36】元件領域上有廣泛之應用, 其ㄧ 原因為陽 極氧化鋁膜有自我組織(self-assembly)之性質。其配製為將鋁箔利用 電化學反應原理與裝置,控制特定之條件下於鋁箔表面產生具多孔性 陣列結構,奈米級之氧化鋁膜乃藉由酸性溶液,如硫酸、草酸、磷酸 作為電解液,獲得六角型蜂巢式(honeycomb)結構形成之陣列【37】。 其特性為孔洞大小尺寸一致、孔洞均一分布,且孔洞均一分布,且孔 洞分布密度與大小可藉由調整製備參數而加以控制。其中鋁箔表面之 粗糙程度影響電場之一致性,電解液溫度影響氧化鋁反應之速率,電 解液之濃度並且直接影響氧化鋁之品質。孔洞直徑與外加電壓有線性 之關係並且與使用不同之電解液有關【37】,一般電解液使用之硫酸 所獲得之孔洞直徑比草酸、磷酸小。為使氧化鋁膜孔洞有一致性與規
則之排列,適當控制外加電壓、陽極氧化時間、電極距離、鋁箔表面 之粗糙程度、電解液溫度與電解液濃度均為重要之條件。 近年國內外發表關於陽極氧化鋁製造技術之期刊皆用磷酸添加 鉻酸(chromic acid)移除第一次陽極處理產生之不規則氧化鋁,留下底 部規則之凹陷缺口,進行第二次或多次之陽極處理,產生規則性之氧 化鋁孔洞【38-43】,然鉻酸具有重金屬特性對生物而言為致癌物, 如能避免使用較佳,本研究能夠完全不用鉻酸達到與其相同目的。有 鑑於多孔性氧化鋁膜具備孔洞大小尺寸一致、孔洞均一分布,且孔洞 均一分布與高度規則性,故可作為優良之奈米結構成長模板,此外多 孔性氧化鋁膜更具高強度、優良化學安定性、絕緣性與耐高溫等特 性,故可應用於多種製程與處理方式。 以純度為99.0%之鋁箔為基材,於低溫下以草酸或硫酸等電解 液,藉由定電壓或定電流方式進行陽極處理,即可獲得多孔性氧化鋁 膜圖2-16(a),然因此時所得之氧化鋁膜其孔洞大小與排列並非理想, 後續可於將此氧化鋁膜溶解後2-16(b),再重複進行陽極氧化處理2-16 (c),而可得具較佳孔洞排列之氧化鋁膜2-16(d) ,圖2-16為多孔性氧 化鋁膜之製備概略圖,其中(a)為一次陽極處理、(b)為氧化鋁膜溶除、 (c)二次陽極氧化處理與(d)為氧化鋁膜剝落。典型草酸溶液所產生之 氧化鋁層如圖2-17所示。為多孔性氧化鋁構成之蜂巢式結構,形成六
角形支單胞(cell)與其孔洞(pore)垂直於鋁基材,底部之半圓形障壁 (barrier layer)接鄰著鋁基材。氧化鋁單胞有兩個主要區域,第一為鋁 純度較高之區域(relatively pure aluminum) , 第二為非晶相低純度鋁 之區域(amorphous contaminated alumina)其性質介於純鋁與氧化鋁之 間【44】。
孔洞之大小主要受外加電壓與不同種類之電解液所影響,孔洞之大小 可以從幾奈米到幾百奈米,並與外加電壓呈線性關係,如圖2-18。
圖2-17 多孔性氧化鋁膜之示意圖【44】。
2.5.1 多孔性氧化鋁之生長機制
氧化鋁其生長機制根據G.E. Thompson【45】與O. Jessensky【46】
觀點,陽極氧化之成長可分為兩個階段:1.孔洞之形成,2.穩態成長。 (1)孔洞之形成於陽極處理之初期,首先電場將O2-/OH-離子從電解液 中推入金屬-電解液介面,即將Al3+從金屬溶出至電解液介面中而形成 緻密之障壁層,如圖2-19 (a)均勻之電位分布;穿透路徑(penetration path)將從表面之裂縫開始形成,圖2-19 (b),很快於路徑下方,電力 線開始集中,圖2-19(c)顯示電場集中之現象。隨時間增長,穿透路徑 數亦增加,電場集中之現象也越趨明顯,集中之電場有效將Al-O鍵極 化並產生局部之焦耳熱效應加速反應,使高電場區相較於低電場區有 更多Al3+離子被溶解,而導致電場集中區之膜面產生局部溶解之現 象。最後穿透路徑尖端下方之電場其側向分量發生橫向擴張,於膜面 產生最初始之孔洞。於接下來之孔洞成長裡,電場及電流大多集中於 孔洞下方之障壁層中,強大電場持續將O2-/OH-離子從溶液中推向障 壁層,而Al3+從金屬溶出與氧離子結合成為氧化鋁,因此於電場集 中區域,隨著反應之進行,金屬-障壁層介面開始呈現半圓形凹槽, 此些凹槽逐漸之擴張成長直至凹槽與凹槽互相接觸在一起,而進入穩 態成長多孔氧化鋁膜,圖2-20。
(a)初始電場分布均勻(b)穿透路徑形成(c)局部電場集中變強 圖2-19 氧化鋁表面形成局部電場集中之過程示意圖【45】。 圖2-20 陽極氧化鋁孔洞形成之機制【45,46】。 (2)穩態成長 穩態成長之多孔氧化鋁膜,膜面上電場分布決定氧化鋁膜之生長 情形。孔洞底部之電解液-氧化鋁膜界面處具較集中電場使此處之氧 化鋁膜快速被溶解,而與底部形成之障壁層生長速度相同而達至動態 平衡。因此於孔洞底部之氧化膜一直維持固定厚度,而氧化膜之其他 部分則持續之向下生長,結果便為形成多孔之氧化鋁膜。 於穩態成長下,僅電壓與溫度維持恆定,氧化膜之結構參數即固 定,若陽極處理時間越久,底部之單胞互相擠壓而形成六角形並愈趨
焦離子束(focus ion beam)【47,48】或用電子槍微影技術(electron beam lithographic technology)製作模具【49】,將設計之有序形狀刻於SiC 模具上以約1600 kg/cm2 油壓方式將SiC模具打印在鋁材表面印下SiC 上的形狀,再經後續之陽極處理鋁程序,即可得有序之孔洞與形狀, 達到全面性之規則,然缺點為複雜且耗時,其流程與實驗結果如圖 2-21、圖2-22、圖2-23與圖2-24與所示。故本研究藉由簡單、費用便 宜、具有規則自我組織之氧化鋁。其規則性可藉由適當條件來控制, 高純度之鋁箔藉由長時間陽極處理將部分氧化鋁剝除,所留下具有規 則性之孔洞,再一次之陽極處理。 圖2-21 藉 由 聚 焦 離 子 束 得 到 有 序 之 孔 洞 與 形 狀 之 製 作 流 程 圖 【47,48】。
圖2-22藉由聚焦離子束得到有序之孔洞與形狀之SEM 上視圖【47,48】 。
圖2-23 藉由SiC 模版於鋁箔上壓模得到有序之孔洞之製作流程圖 【49】。
圖2-24 藉由SiC模版於鋁箔上壓模得到有序之孔洞之SEM側視圖 【49】。
2.5.2 多孔性陽極氧化鋁膜之應用
近年因奈米材料之蓬勃發展,使得此類具奈米結構之模板材料受 到極度重視,一般常用之模板材料包括氧化鋁模版、高分子模板與金 屬模板,然因氧化鋁膜具高強度、優良化學安定性、絕緣性與耐高溫 等特性,故為最常用之模板技術材料。以下將介紹之氧化鋁膜即其應 用。 (1)無電極電鍍-奈米金屬線與金屬管 無電極電鍍為藉由化學還原方式使溶液中之金屬離子於還原後 沉積於模板內,與電化學沉積之差異於其基材可為非導體。此方法與 電化學沉積法相較之最大缺點於無法有效控制奈米金屬管之長度,僅 可藉由調控沉積時間以控制奈米管之內徑大小【50,51】。 (2)電化學沉積-奈米金屬線與金屬管 藉由電化學沉積法配製奈米金屬線與金屬管,一般將先蒸鍍一層 導電金屬於模板某一面,以利電鍍之進行。電鍍開始時,金屬將由底 部向上生長使鍍出之金屬順這模板規則成長,形成規則之奈米金屬線 陣列。因氧化鋁膜之孔洞大小一致且方向固定,故藉由氧化鋁膜配製 之奈米金屬線,其尺寸大小、形狀、長寬比與方向性皆能精確控制 【52-55】。(3)化學氣相沉積法-半導體 化學氣相沉積法(CVD)為藉由單一或數種氣體產生化學反應,並 經由反應物透過擴散、吸附至模板之孔洞表面進行沉積【56】。 (4)溶膠-凝膠法-金屬氧化物 溶膠-凝膠法於金屬氧化物中,經溶液溶膠、凝膠而固化,再經 低溫熱處理而形成奈米粒子,將此技術應用至奈米模板中,可將奈米 模板浸泡於金屬溶膠液中使奈米孔動中充滿金屬溶膠液,經乾燥與樂
處理後即可得奈米金屬氧化物,如TiO2、ZnO 與WO3…..等【57】。
(5)化學聚合法-奈米碳管 化學聚合法分為化學或電化學方法使單體於模板孔洞中進行聚 合應。藉由化學配製奈米結構為將奈米模板預先浸泡於含單體與起始 濟之溶液中,再經照光或加熱方式使單體發生聚合反應,於孔洞內表 面形成奈米管或線。此外電化學聚合法則必須採用可導電模板,否則 必須先將模板某一面鍍上金屬以作為電極,再藉由通電使模版孔洞中 之高分子單體產生聚合。該法特點為可配製導電性高分子與絕緣性高 分子等聚合物【58】。
2.6 無電鍍(electroless plating)
2.6.1 無電鍍原理
無電鍍是一種不外加電流,而能在材料比面經由催化作用下,控制化學還原作用,將鍍液中之金屬離子還原,而在基材表面析出之過 程。前處理對鍍膜品質有很大的影響,當表面催化後,無電鍍反應進 行而附著第一層金屬後,此層金屬又繼續催化下一層金屬的還原沉 積,使得鍍層可以連續成長,故無電鍍又稱為自催化析鍍(autocatalytic plating)。 無電鍍法可鍍在玻璃、陶瓷...等非金屬基材及各種金屬基材上,無論 基材幾何形狀如何都可以,且鍍膜經過適當熱處理後,更可以製造硬 度甚大之皮膜,因此在工業界上利用範圍相當廣泛。具代表性之無電 鍍有鍍鎳及鍍鉻。在製備磁性及導電性之材料,因鍍液所使用之還原 劑含量不同,而使鍍膜特性不同【59】。
2.6.2 無電鍍鎳 Ni 反應機制
無電鍍鎳的基本反應藉由鍍液中之化學反應物(還原劑)釋出電 子供給鎳離子,使鎳離子還原沉積在具有催化活性(如VB 族至 VIIIB 族之過渡元素)或是經過活化之基材表面上,並引發連鎖析鍍反應。 無電鍍鎳的反應機構如下: (1) 脫氫(Dehydrogenation): ads catalyst H HPO PO H2 2 ⎯⎯ →⎯ 2 + Hads:吸附的氫原子 (2) 氧化(Oxidation): ads H H HPO O H HPO2 + 2 ⎯⎯→ 32− + + +(3) 游離(Liberation): − + + ⎯→ ⎯ H e Hads (4) 再結合(Recombination): 2 H H Hads + ads ⎯⎯→ (5)還原(Reduction): Ni e Ni2++2 − ⎯⎯→ (6)磷的生成(Formation of phosphorous): O H P HPO H PO H2 2 32 2 3 2 3 −+ + ⎯⎯→ −+ + 其全反應式如下所示: 2 2 3 2 2 2 2 2 2 6 4 6H PO − + Ni + ⎯catalyst⎯ →⎯ P+ Ni+ H+ + HPO −+H 由反應式中可知,無電鍍鎳鍍層之合金成份與其所使用之還原劑有 關,即以次磷酸鈉作為還原劑之無電鍍鎳鍍層為鎳磷合金;而以硼氫 化鈉或胺基硼烷為還原劑,其得到的無電鍍鎳鍍層為鎳硼合金;或以 聯胺為還原劑者,其無電鍍鎳之鍍層為高純度之鎳鍍層。
2.7 原子層化學氣相沉積(Atomic Layer Deposition,
ALD)
ALD 屬 CVD 反應,藉由交替通入前驅物與低反應氣體,在試 片表面吸附並與試片表面產生化學反應,除此之外其前驅物彼此之間
管路及反應腔體的溫度不能太高。對於雙元素AB 化合物的成長,依 序通入前驅物 A、清潔氣體(purging gas)、前驅物 B、清潔氣體這樣 即完成一次反應循環。每次通入前驅物時皆使表面達成飽和狀態,前 驅物均勻鍍覆並且化學吸附在表面上或者與表面原子反應形成單一 層鍵結緊密的原子層,接下來清潔氣體沖掉多餘的反應分子,當另一 種前驅物鍍覆上時只有單一種原子層於其反應,如此得到想要的固態 產 物 及 氣 態 副 產 物 。 其 上 所 提 的 成 長 機 制 為 自 我 侷 限 機 制 (self-limitation mechanism),因為每一個循環的固態沉積量取決於在飽 和情況下能行成單一原子層之前驅物分子的數量。ALD 的成長特 性:1.自我侷限機制,薄膜厚度只與成長循環次數有關,且不需要相當 均質的反流場,可以控制精確的材料成份;2.獨立控制反應氣體,沒 有氣相的反應,足夠的時間去完成每一個反應循環;3.製程溫度的容 許範圍(process window)相當寬,不同的材料可以在相似的製程條件 下。ALD 的實際優點:1.準確的控制薄膜厚度;2.可大面積成長,可 ㄧ次成長相當多數量,具有相當好的一致性;3.對於不穩定固態前驅 物的蒸發速率也是可以容許的;4.好的再現性;5.對於高深寬比的凹 槽或尖銳的表面可以成長的相當均勻;6.可在低溫下成長高品質的薄 膜;7.可以成長多層材料或超晶格。ALD 最大的缺點在於低成長速 率,但對於某些IC 產業而言,薄膜厚度只需要幾奈米厚這相對是個
優點。理論上單一次循環成長之厚度應為單一原子層厚度,因此可以 精確的控制膜厚且可以大面積鍍覆平坦薄膜。當ALD 成長出的薄膜 為多晶材料時其表面通常較粗糙,因為成長過成長過程中包含成核階 段類似於一般CVD 的成長模式,即成長過程不符合 ALD 機制,因 每ㄧ個循環成長的厚度不足一個原子層。而當成長出的薄膜為磊晶或 非晶質時,其表面通常為光滑平整【60】。
2.8 ㄧ維奈米線之異質接面
近年來各種不同種類的奈米線被合成出來後,有關奈米線的研究逐漸 朝向光學與電學方面的基礎科學研究,希望可瞭解各種奈米線的特 性。例如,在與實際應用相關的研究方面,Lieber 的研究群嘗試將奈 米線作成積體電路元件,並且成功地研發出以P 型與N 型矽奈米線 所組合的二極體與場效電晶體,提升了半導體奈米線整合在電子與電 機元件裝置上面的能力【61】。此外,為了使得半導體奈米線能夠具 有更廣泛的應用性,許多人更合成出各種複雜的結構,如超晶格奈米 線(nanowire superlattices)與核層(core-shell)奈米線。Yang 的研究群與 Lieber 的研究群分別合成出具有軸向異質接面結構的超晶格奈米 線,使得不同成分的奈米線可以互相銜接起來,例如一個PN junction 就僅靠一條奈米線就可以完成【62,63】。他們認為利用這個方法,這可以大大縮小以奈米線做成的電子元件容積。此外,Lieber 的研究 群也利用矽與鍺合成具有徑向異質接面結構(lateral heterostructure)的 core-shell奈米線【64】。不同於之前提到的是,他們發現,這種core-shell 奈米線的異質接面的介面面積相當大,但是其接面擴散寬度卻在1 nm 左右。藉由能夠順利整合這種結構的奈米線到場效電晶體等電子元件 上面,他們認為在未來,無論是研究電子的量子效應或是實際的電子 元件應用,這種異質接面材料都可能提供許多發展的潛力。
2.9 參考文獻
1.H. Froes and C. Suryanarayana, JOM, June 1989.
2.李傳宏、黃佩珍、盧成基、彭國光與徐文泰,材料奈米技術專刊, 60期 (2001)。
3.林景正與賴宏仁,工業材料,153 期,95 (1999)。
4.K. Takayanagi, Y. Kondo and H. Ohnishi, JSAP Int. 3, 3 (2001). 5. K. Nielsch, R. B. Wehrspohn, J. Barthel, J. Kirschner, U. Gösele, S. F. Fischer and H. Kronmüller, J. Appl. Phys. 79, 1360 (2001).
6. Y. Li, G. W. Meng and L. D. Zhang, Appl. Phys. Lett. 76, 2011 (2000). 7. J. C. Hulteen and C. R. Martin, J. Mater. Chem. 7, 1075 (1997).
8. Poretics Corporation, Product Guide; Livermore, CA (1995).
9. C. C. Chen, C. Y. Chao and Z. H. Lang, Chem. Mater 12, 1516 (2000). 10. L. Manna, E. C. Scher and A. P. Alivisatos, J. Am. Chem. Soc. 122,
12700 (2000).
11. X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature 404, 59 (2000).
12. A. M. Alper ed., Academic Press. (1995).
13.X. Duan and C.M. Lieber, J. Am. Chem. Soc. 122, 188 (2000).
14. M. S. Gudiksen and C. M. Lieber, J. Am. Chem. Soc. 122, 8801 (2000).
15. S. T. Lee, Y. F. Zhang, N. Wang, Y. H. Tang, I. Bello and C. S. Lee, J. Mater. Res. 14, 4503 (1999).
16. L. C. Chen, S. W. Chang, C. S. Chahg, C. Y. Wen, J-J. Wu, Y. F. Chen, Y. S. Huang and K. H. Chen, J. Phys. Chem. Solids. 62, 1567 (2001).
17. M. He, I. Minus, P. Zhou, S. N. Mohammed and J. B. Halpern, Appl. Phys. Lett. 77, 3731 (2000).
18. S. Bandyopadhyah and H. S. Nalwa, Quantum Dots and Nanowires; American Scientific Publishers, 2003.
19. A. Thess, R. Lee, P. Nikolaev, H. J. Dai, P. Petit, J. Robert, C. H. Xu, Y. H. Lee, S. G. Kim, A. G. Rinzler, D. T. Colbert, G. Scuseria, D. Tomek, J. E. Fischer and R. E. Smalley, Science 273, 483 (1996). 20. T. M. Whitney, J. S. Jiang, P. C. Searson and C. L. Chien, Science
261,1316 (1993).
21. L. Piraux, J. M. George, J. F. Despres, C. Leroy, E. Ferain, R. Legras, K. Ounadjela and A. Fert, Appl. Phys. Lett. 65, 2484 (1994).
22. A. Blondel, J. P Meier, B. Doudin and J.-Ph. Ansermet, Appl. Phys. Lett. 65, 3019 (1994).
23. A. Blondel, B. Doudin and J.-Ph. Ansermet, J. Appl. Phys. 79, 6090 (1996).
24. J. E. Wegrowe, S. E. Gibert, D. Kelly, B. Doudin and J.-Ph. Ansermet, IEEE Trans. Magn. 34, 903 (1998).
25. A. Fert and L. Piraux, J. Magn. Mag. Mater. 200, 338 (1999).
26. M. Zheng, L. Menon, H. Zeng, Y. Liu, S. Bandyopadhyay, R. D. Kirby and D. J. Sellmyer, Phys. Rev. B 62, 12282 (2000).
27. S.-Z. Chu, K. Wada, S. Inoue, S.-I. Todoroki, Y. K. Takahashi and K. Hono, Chem. Mater. 14, 4595 (2002).
28. Takeshi Ohgai, Xavier Hoffer, Andrea Fabian, Laurent Gravier and J.-Ph113 Ansermet, J. Mater. Chem. 13, 2530 (2003).
29. Leszek M. Malkinski, Athanasios Chalastaras, Andriy Vovka, Jin-Seung Jung, E.-M. Kimb, J.-H. Jun and Carl A. Ventrice Jr, J. Magn. Mag Mater. 286, 108 (2005).
30. 王正全、周淑金、李秉彰與王正和,工業材料,185 期,165 (2002)。
31. J. S. Goode, R. C. Furneaux, G. C. Wood, J. A. Richadson and G. E. Thompson, Nature 272, 433 (1978).
32. R. V. Parthasarathy and C. R. Martin, Nature 369, 298 (1994). 33. K. Nielsch, F. Muller, A. P. Li and U. Gosele, Adv. Mater. 12, 582
(2000).
34. K. Nielsch, R. Hertel and R. B. Wehrspohn, IEEE. 38, 2571 (2002). 35. R. M. Metzger, V. V. Konovalov and M. Sun, IEEE. 36, 30 (2000). 36. Y. Li, Appl. Phy. Lett. 76, 2011 (2000).
37. A. P. Li, F. Muller, A. Birner, K. Nielsh and U. Gosele, J. Appl. Phys. 86, 6023 (1998).
38. Y. Kanamori, K. Hane, H. Sai and H. Yugami, Appl. Phys. Lett. 78, 142 (2001).
39. Y. Kanamori and K. Hane, Appl. Phys.Lett.78, 142 (2001).
40. G. Yoon and J. S. Suh, Bull. Korean Chem. Soc. 23, 1519 (2002). 41. Z. Wang, Y. K. Su and H. L. Li, Appl. Phys. A 74, 563 (2002). 42. J. S. Suha and J. S. Lee, Appl. Phys.Lett. 75, 2047 (1999).
43. D. Almawlawi, K. A. Bosnick, A. Osika and M. Moskovits, Adv. Mater. 12, 1252 (2000).
44. D. Routkevitch, A. N. Govyadinov and P. P. Mardilovich, MEMS. 2, 39 (2000).
45. G. E. Thompson, Thin solid films. 297, 192 (1997).
46. O. Jessensky, F. Muller, U. Gosele, Appl. Phys. Lett. 72, 1173 (1998).
47. C. Y. Liu, A. Datta and Y. L. Wang, Appl. Phys. Lett. 78, 120 (2001). 48. N. W. Liu, A. Datta, C. Y. Liu and Y. L. Wang, Appl. Phys. Lett. 82,
1281 (2003).
49. H. Masuda, H. Yamada, Masahiro Satoh and H. Asoh, Appl. Phys. Lett. 71, 2770 (1997).
50. V. P. Menon and C. R. Martin, Anal. Chem. 67, 1920 (1995).
51. M. Nishizawa, V. P. Menon and C. R. Martin, Scence 268, 700 (1995).
52. C. A. Foss, G. L. Hornyak and J. A. Stockert, J. Phys. Chem. B 98, 2963 (1994).
53. A. Despic and V. P. Parkhutik, Moder Aspects of Electrochemistry; Plenum Press: New York, 1989.
54. C. J. Brumlik, V. P. Menon and C. R. Martin, J. Mater. Res. 9, 1174 (1994).
55. H. Masuda and K. Fukuda, Science 268, 1446 (1995).
56. G. S. Chen, L. D. Zhang and Y. Zu, Appl. Phys. Lett. 75, 2455(1999). 57. B. B. Laskshmi, P. K. Dorhont and C. R. Martin, Chem. Mater. 9, 857
(1997).
58. R. V. Parthasarathy and C. R. Martin, Adv. Mater. 7, 896 (1995). 59.化學鍍理論及實踐 姜曉霞,沈傳 國防工業出版社.
60.科儀新知第29卷第一期 國家實驗研究院 儀器科技研究中心出版. 61.X. F. Duan, C. M. Lieber, Nature 409, 66 (2001).
62. Y. Wu, R. Fan, P. D. Yang, Nano. Lett. 2, 83 (2002).
63. M. S. Gudiksen, L. J. Lauhon, J. F. Wang, D. C. Smith, C. M. Lieber, Nature 415, 617 (2002).
64. L. J. Lauhon, M. S. Gudiksen, D. Wang, C. M. Lieber, Nature 420, 57 (2002).
第三章 實驗流程與方法
本研究包含三種試片結構:
1.一維鎳奈米柱陣列(1-D Ni nanorod arrasy)
2.一維鎳-氧化鎳之核-殼奈米柱陣列(1-D Ni-NiO core-shell nanorod arrays) 3. 一 維 鎳 - 二 氧 化 鈦 之 核 - 殼 奈 米 柱 陣 列 ( 1-D Ni-TiO2 core-shell nanorod arrays) 以下將依序介紹各種試片之製作流程與方法
3.1 一維鎳奈米柱陣列 (1-D Ni nanorod arrays)
本研究利用AAO 在矽基材上做為模板,搭配無電鍍鎳製程,在 矽基板上製作出鎳奈米陣列。圖 3-1 為一維鎳奈米柱陣列之實驗流程 圖。圖3-1 為一維鎳奈米柱陣列之實驗流程圖。
Si基板上製作AAO模板
無電鍍鎳沉積
RTA退火30 sec/1 min/2 min
移除AAO
SEM 分析 TEM 分析 SQUID分析
Si基板上製作AAO模板
3.1.1 Si 基板上製作 AAO 模板:
在 AAO 模板製程中,本研究所使用試片的基材為 p-type (100)
面的矽基材,熱阻絲蒸鍍系統(thermal evaporator coater)在高真空環境 下(< 4×10-6 Torr)蒸鍍 1 μm 的純鋁(99.999%)。完成上述步驟後,把試 片切割為2 cm×2.5 cm 大小,便開始正式進行陽極氧化鋁處理,本研 究所使用製具為抗酸鹼的PVC 材質電解槽,並在一端開出直徑 1.2 cm 的圓孔做為反應區域,將試片接上陽極,使用白金片當作陰極如圖 3.2 所示。 本研究使用兩階段的陽極氧化處理法,如圖3.3 所示,先進行第 一階段的陽極氧化鋁反應,反應電壓設定為 40 V,只留下所需厚度
的純鋁,再利用1.8 wt%鉻酸(chromic acid, H2Cr2O4)加上 6 wt%磷酸
(phosphoric acid, H3PO4),在 60 oC 下浸泡 40 分鐘移除第一階段所生
成的AAO。由於在 AAO 孔洞底部會有規則的阻障層結構在氧化鋁及
純鋁界面生成,因此將第一階段生成的AAO 移除後,便會留下表面
凹痕結構的純鋁,第二階段陽極氧化鋁反應(second anodization)所使 用條件與第一階段相同,電解液便會依照凹痕的位置向下反應,形成
規則的 AAO 奈米孔洞,最後再利用 5 wt%的磷酸(phosphoric acid,
圖 3-2 AAO 製具與試片示意圖。
Substrate
Al Substrate
a) 1st-step Anodization c) 2nd-step Anodization
b) 1st-step AAO Removal
Substrate Substrate
d) Pore Widening Treatment
Substrate Substrate Substrate Al Substrate Al Al Substrate
a) 1st-step Anodization c) 2nd-step Anodization
b) 1st-step AAO Removal Substrate
Substrate SubstrateSubstrate
d) Pore Widening Treatment
Substrate Substrate
3.1.2 無電鍍鎳沉積: 製備好 AAO 模板後,便可進行無電鍍鎳沉積,由於 AAO 基板 不具催化無電鍍反應之能力,所以基板必須進行前處理後才可進行無 電鍍鎳沉積。 前處理的可分為兩個部份: 清潔: 將試片利用丙酮沖洗過後,利用氮氣槍吹乾試片表面。 敏化及活化處理: (1) 敏化:將試片放入敏化溶液(成分如表 3-1 )中,浸泡 2 分鐘 使Sn 析鍍在基板表面上。 (2) 活化:將試片放入活化(成分如表 3-1 )溶液中 30 秒,使原本 在基板表面的Sn 置換成 Pd,反應式為: Sn2++Pd2+→Pd+Sn4+ 試片經敏化和活化處理後,便可進行無電鍍鎳沉積,其沉積條件鍍浴 配方如表3-1 所示,沉積時間約為 1 分鐘。