行政院國家科學委員會專題研究計畫 成果報告
生物晶片之專利保護、授權、侵權及上市前程序之研究(III)
計畫類別: 個別型計畫 計畫編號: NSC93-3112-H-009-001- 執行期間: 93 年 05 月 01 日至 94 年 04 月 30 日 執行單位: 國立交通大學科技法律研究所 計畫主持人: 劉尚志 共同主持人: 倪貴榮 報告類型: 完整報告 處理方式: 本計畫可公開查詢中 華 民 國 94 年 8 月 1 日
生物晶片之專利保護、授權、侵權
及上市前程序之研究(III)
第一章、前言
第二章、生物晶片的應用與產業現況
第三章、美國醫藥器材管制法規介紹
第四章、歐盟對醫療器材的管制規範模式
第五章、我國現行相關管制法規
第六章、從管制理論出發看生物晶片上市前審查之制度
第七章、結論
生物晶片之專利保護、授權、侵權
及上市前程序之研究(III)
第一章、前言 本計劃的第一階段(第一年)以研究基因晶片專利為主要目標,以美國專利 資料庫QPAT-US 為檢索資料來源,經軟體分析及人工解讀方式共篩選出 533 案 與微陣列基因晶片製作和應用有關的專利案,並進行其專利管理地圖與專利技術地圖的分析。重要競爭公司以Affymetrix 和 Nanogen 生技公司為首,而 Agilent
Technologies、Incyte Pharmaceuticals、Affymax 與 MicroFab Technologies 等公司
亦佔有一席之地,而前十大重要專利案皆為 Affymetrix 和 Nanogen 公司所有。
在專利技術分析方面,Affymetrix 公司在光罩蝕刻法、Nanogen 公司在電磁式晶 片、MicroFab Technologies 與 PE Corporation 在印刷法各擁有重要關鍵技術。在 其他各項重要關鍵技術上,Affymetrix 公司仍居重要角色,除此之外 Affymax 與 Agilent Technologies 於晶片的表面處理、Amersham Pharmacia Biotech 於核酸標 定物質、Molecular Dynamics 於影像訊號處理、Incyte Pharmaceuticals 於核酸探 針性質中皆有其重要的影響力。 第二年則以LexisNexis 及 Westlaw 二資料庫,搜尋相關公司所涉之專利侵權 訴訟新聞、判決等。以有關微陣列晶片製造及微流體生物晶片相關專利侵權訴訟 為重心。Affymetrix 擁有許多的微陣列晶片製造相關專利,亦是相關專利侵權訴 訟之中心。不過,自2001/1/22,北加州地院對判決其 5,800,992 號專利無效、Incyte 不侵害第 5,445,934 號專利任何一項專利權利範圍等等敗訴、及資金壓力等。 Affymetrix 於是先後與多家公司達成和解,並進行專利交互授權,尋求未來商業 合作的機會。Caliper Technologies Corp.與 Aclara Biosciences, Inc.則是有關微流體 生物晶片專利之侵權訴訟值得關注的另一議題。目前尚不知雙方是否上訴或已達 成和解。 本年度目標為上市前程序之研究。應用於臨床檢驗的生物晶片係屬醫療檢驗 器材,而醫療器材的安全及效能攸關人命與健康,各國政府無不定立嚴謹的法規 加以管理。雖然生物晶片的發展已有十年,然(於本計畫開始時)世界各國尚無專 門法規加以管理。因此,本計畫先比較美國、歐盟與我國對於醫療器材的管制法 規(第三章 - 第五章),並對各國醫療器材的相關上市前審查管制法規進行探 討,並各作簡單的評論與討論;其後並針對率先在歐美通過上市前審查的 AmpliChip 晶片進行個案研究。第六章由經濟分析理論層面出發,試圖逐段理出 最適當的管制脈絡,使生物晶片的科技能持續發展應用,並能保障公共健康,同 時讓政府的管制資源更有效率。
第二章、生物晶片的應用與產業現況分析 第一節、概述 生物晶片工業是一個極爲年輕的産業,從第一個産品問世到現在,還不到 10 年的光景1 。十幾年前,美國史丹福大學在美國國家衛生研究院的資助下,著 手研究將不同的 DNA 點在一小塊濾紙上,利用濾紙上已知序列或已知功能的 DNA,來偵測檢體中未知的 DNA。這種技術啓發了日後生物晶片研發的風潮, 配合著微電子工業中微小化技術,而産生了今天日益蓬勃的生物晶片工業。 依其產品應用的市場區隔,生物晶片可分為「研究用晶片」及「臨床檢驗 用晶片」。研究用晶片主要供應研究單位或新藥研發公司,目前生物晶片多供應 此一市場。據統計,2000 年全球生物晶片的市場規模大約為 1.9 億美元,若加上 掃瞄器、微流體晶片、及晶片生產設備,則達3.97 億美元的規模,產出 20.6 萬 片,在 2001 年有 34 萬片需求,產值近 2.5 億美元,預計將以每年約 18.9%的 速率快速成長,今年(2005)將達 5.36 億美元。整體市場成長的關鍵點在於生物晶 片本身的價格能否大幅降低。如高雄醫學大學於於2004 年發展出 K-ras oncogene 診斷晶片2,此類晶片要價頗高,每片要2 萬 4 千元3。 據 1999 年我國工研院生醫中心之預測,至 2008 年,醫療檢驗用晶片之 平均價格將降至 10 美元,銷售量將至 8,200 萬片,遠超過研究用晶片,銷售額 亦超越研究用晶片之水準。一旦晶片能取代目前的檢驗試劑被醫院或診所大量使 用於篩檢和臨床檢驗,成為可丟棄式的健康檢查或疾病檢測儀器,甚至當生物晶 片被應用於消費市場時,將如同電腦普及至一般家庭,市場潛力無可限量4。 然經歷了自 2001 年以來的全球經濟衰退及數件生技公司併購案,再加上 Affymetrix 等大公司的訴訟纏身,生技產業無可避免地亦正處於非常困難及挑戰 的時期5 。所幸近年來,在醫療技術持續進步與人口結構高齡化的驅動下,儘管 經濟環境波動不已,美國醫療器材產業卻是充滿生氣與進步6 。另一方面,我國 1 請參閱,<21 世紀生物晶片將獨領風騷 成為生物產業明星 >,資料來源: http://www.bioweb.com.tw/feature_content.asp?ISSID=634&chkey1=生物晶片&chkey2=基因晶片 &chkey3=微陣列晶片&chkey4=&chkey5=,2004/12/6 拜訪。 2 請參閱,林糸秀茹、陳怡芳,<以微矩陣列分析技術探討 K-ras 致癌基因在腎上腺腫瘤之作用機轉 >。資料來源:http://www.genome-kmu.com/genomeweb/diagram_12.htm,2005/7/22 拜訪。 3 請參閱,楊欣怡,<癌症晶片靈敏度 93% 抓得住 0.2 公分腫瘤>,2004.07.14,中時晚報。資 料來源: http://news.chinatimes.com/Chinatimes/newslist/newslist-content/0,3546,130503+132004071400701,0 0.html,2005/7/14 拜訪。 4 請參閱,<基因時代產業新主流──生物晶片簡介 (下) >,資料來源: http://www.sinphar.com/medical/no30/product_01.html,2004/12/04 拜訪。 5 請參閱,林珮慈,<生物資訊及生物晶片產業發展現況>,資料來源: http://www.bionet.org.tw/innovation/new_21.html,2004/12/04 拜訪。 6 根據 Espicom 的估計,1994 年至 2004 年間,美國醫療器材市場規模由 411.4 億美元,上升為
的醫療器材產業也在成長中7 ,未來生物晶片市場乘此之勢,其市場未來應可期 待有更多發展空間。 第二節、美國生物晶片產業 自2003 年至今,美國多出 122 家新上市或上櫃的生技公司8。以下茲就美國 五家與基因或生物晶片相關的大型生物晶片廠商的利基與困境,作一簡單的介紹 9: (一)、Affymetrix 公司:
位於加州Santa Clara,是全美第一家發展 DNA 晶片技術的公司,起源於 1980
年代以Stephen P.A. Foodor 為首的科學家團隊,並於 1991 年成立 Affymetrix 公
司,為Affymax N.V.的分支10。擁有多項DNA 晶片製造技術的專利權,而在 DNA
晶片製造和銷售的市場上佔有不容忽視的競爭潛力。其全力推行多種行銷策略, 期使其DNA 晶片 Gene Chip oligonucleotide arrays 能領先其他公司,成為顧客使
用DNA 晶片的最佳選擇。其中的“Easy Access”行銷策略,襲自”Gillette”可換式
安全刮鬍刀的策略,故其部分營利也是來自出售大量的可丟棄式的DNA 晶片。
Affymetrix 公司與美國國家癌症中心(National Cancer Institute)簽約,提供其較優 惠的價格享用服務和設備,亦為該公司行銷策略之一,除可吸引學術界專業研究 人員的使用,並可建立該公司權威形象。除前述產品及技術之外,該公司擁有其
他DNA 晶片的專利權,包括具有超過 400 種不同的 DNA 探針的”spotted cDNA
arrays”。其他 DNA 晶片的生技公司若要採用類似的技術,必須付專利授權費用 給。 Affymetrix 公司一直是以高密度基因晶片專利及微影光罩(Light-directed in situ synthesis )之特殊晶片製程,維持基因晶片市場領導者的地位。該公司的晶片 812.0 億美元,期間年複合成長率為 7.04%,呈現穩定成長的態勢。請參閱,李維欽,<持續看 好的美國醫療器材市場>,工研院經資中心生醫組。資料來源: http://mdm.bionet.org.tw/Usr/DBDetail.asp?rid={1BCE7CE7-B130-4C9B-AA81-42B782DEC71A}, 2005/7/29 拜訪。 7 請參閱,馮晉嘉,<我國醫療器材產業 2004 年回顧與展望>,工研院經資中心生醫組。資料 來源: http://mdm.bionet.org.tw/Usr/DBDetail.asp?rid={835FB56A-2165-4F6E-8721-6EDAD9A770F8}, 2005/7/29 拜訪。 8 請參閱,李慧瑜,<只要一滴血–談生物晶片產業>,華南金控,16 期,2004 年 4 月,頁 38-43。 9 請參閱,<生物晶片是台灣生物科技的明星產業,然而台灣的發展契機究竟在哪裡呢﹖>。資 料來源:http://www.ncku.edu.tw/~cbst/biochip.htm,2004/12/04 拜訪。 10 請參考,Affymetrix 公司網站。資料來源: http://www.affymetrix.com/corporate/history/index.affx,2005/7/15 拜訪。
售價曾高達 US$2,000,但隨著各項專利訴訟紛紛落幕,Affymetrix 並未取得明
顯勝利,甚至部分訴訟如與Oxford Gene Technology 之訴訟,還屈居下風。再加
上其產品問題連連,製程成本居高不下,導致晶片售價居高不下,其應用面也無 法隨時間擴大。所以該公司雖然長期佔有領導產業發展的戰略地位,卻因為製程 技術無法更上層樓,造成整體產業停滯不前。 Affymetrix 從 1990 年提出第一項專利申請開始計算,技術研發已超過十 年,若改以 1993 年公司從 Affymax 衍生成立開始計算,研發投入也馬上要滿 十週年,所以投資人對於公司的期望,已不是偶而來點「取得某項專利」、「技 術突破」等消息而已,而是更希望看到銷售額成長,公司財務達到損益平衡及出 現盈餘。但是,如前所述,該公司投入十餘年研發的製程技術,無法達到預期目 標,若是放棄微影光罩的製程,另外發展其他技術,不但該公司先前之投入回收 無望,同時也喪失技術領先的利基。 在此背景之下,Affymetrix 改變營運方針,首先於 2000 年 10 月宣布轉投 資成立Perlegen Sciences 公司,篩選 50 個選定個體在基因上的差異,並將以產 生的資料庫為基礎,探討基因變異與疾病之間的關係,作為開發新藥的基礎11。 隨後又在2001 年 10 月底宣布與纏訟多年的對手 Hyseq 和解,並共同投資成立 一家名為N-MER 的公司,負責基因晶片後續技術研發。至今已有 230 個美國專 利,更有420 個專利正在申請中12,個人醫療檢測應用為該公司今後主要發展重 點所在。 (二)、Nanogen 公司:
位於加州San Diego,自 1993 年設廠以來致力於發展電子 DNA 晶片13的技
術。該公司產品最大的特色在於其自由度大,顧客可自由選擇他們想要放在晶片 上的 DNA 片段,只要幾個小時就可以設計一片自己想要的 DNA 晶片。此外, 其可放25 萬個探針在面積也還比 Affymetrix 公司的產品小、只有一平方公分的 晶片上,且利用電子吸引力的原理,其 DNA 晶片只需要 15 秒即可完成雜交 (Hybridization)。由於其可快速又大量的檢測檢體的優點,Nanogen 公司大力推廣 將其產品應用在法醫學的犯罪證據檢定上。 (三)、Sequenom 公司: 為一跨國公司,工廠位於美國,而辦公室在德國。其主要產品MassARRAY® 11 請參閱,同前註5 。 12 請參考,Affymetrix 公司網站。資料來源: http://www.affymetrix.com/corporate/history/factsheet.affx,2005/1/16 拜訪。
13 即該公司的主力產品,the NanoChip® Electronic Microarray,每一晶片上有一百個測試點(test
sites),每一個測試點由電腦控制。請參考,Nanogen 公司網站,資料來源: http://www.nanogen.com/products/nanochip_micro.htm,2005/1/16 拜訪。
system 的特殊偵測技術能夠檢定一個核甘酸的差別,可應用到作大規模的篩選人 類族群中單一核醣核酸的個體差異(single nucleotide polymorphisms, SNPs),且此
系列檢測一個檢品只需3.5 秒,相當快速。但其產品面積、體積仍然過大,有待
改進。
(四)、Illumina 公司:
亦位於加州。該公司的DNA 晶片為目前面積最小,密度最高者。由於其面
積小、製造簡單,價格勢必比Affymetrix 公司的產品便宜得多,此為它市場競爭
的利器。此外,Illumina 公司聲稱其[DNA 片段—beads]技術可應用在各種不同的
分析技術上,因此將會有更廣泛的市場。 (五)、Nuvelo 公司:
其前身為 Hyseq 公司,曾經在生物晶片領域中佔有一席之地,但由於與
Affymetrix 之專利侵權訴訟、營運策略、資金等問題,於 2001/10/25 與 Affymetrix
達成和解,雙方同意針對微陣列專利部分進行雙向商業授權,並共組 Callida 及
其子公 司 N-Mer, Inc.,由 N-Mer 利用 Hyseq 所有之 SBH (sequencing by hybridization)技術及基因晶片技術發展高速晶片。Affymetrix 為 N-Mer 之專屬晶 片 、 系 統 供 應 者 , 並 擁 有 其 產 品 獨 家 銷 售 權 。Hyseq, Inc. 改 名 為 Hyseq Pharmaceuticals, Inc.,全力利用其收集之特殊基因開發藥物。之後,Hyseq 又與 Variagenics, Inc.合併,改名為 Nuvelo, Inc.,專注人體藥物之研發。
第三節、臺灣生物晶片產業 第一項、產業現況概述 政府視生物科技為本世紀的明星產業,至今已投入超過3000 億元的相關研 發經費14。不過,相較於新加坡或中國,台灣生物科技發展籌資不易、優惠條件 不佳等因素居於劣勢,難以吸收外資與生技人才的返台。然而,台灣有足夠優秀 的生技研發人才,而且結合台灣既有的電子、電機、化工、資訊產業利基與半導 體與電子科技產業所累積的基礎,為我國發展生物晶片產業的優勢。若政府能擬 定良好的決策,增加研發經費與輔助設廠的優惠條件,使台灣產、官、學、研的 力量能夠分工整合,選定發展策略與目標,整合資源,突破現階段的窘況,台灣 生技產業的未來仍可期待頗有發展。 相較於大型國際企業耗費巨資投入生物晶片產業,國內的中小型生物晶片公 司要具有競爭力,可以切入一些特定的晶片項目上,特別是部份研究機構所需要 的基因晶片,或投入歐美研究比較少的亞洲型疾病的技術或資訊,或鎖定中草藥 科學化的分析平台建立,作為未來與國際基因晶片大廠進行交互授權的基礎,都 14 請參閱,同前註9。
是將來可能的發展方向15。 第二項、現有廠商簡介 國內的生物晶片產業,自1998 年起陸續有國內廠商投入研發與製造,形成 新竹、台南二個生物技術產業聚落,以下作簡要介紹16: 1. 晶宇生物科技實業: 正式於1998 年 10 月底成立的晶宇生技(DR.CHIP),是臺灣第一個以 研發製造為導向的生物晶片公司,主要研發核醣核酸檢驗晶片,包括 DNA 及 RNA 等生物晶片及相關生物科技技術儀器與設備。投資 1.8 億 元在竹科竹南基地興建國內第一座生物晶片GMP 標準廠房,單日最大 產能可達五萬片生物晶片。 主要產品有腸病毒檢驗晶片、乳房炎晶片、食品病源菌晶片。目前 在開發 lab-on-a-chip 式「高通量生物晶片自動操作系統」。在國際上, 晶宇在歐、澳、義大利、日本與中國均有所發展,目前與英國Microzone 公司合作開發樣本前處理試劑,另與德、法國及義大利等公司合作,著 重於開發量產設備及動物臨床檢斷晶片。在日本,合作的伙伴為關東化 學。晶宇採取集中發展低密度生物晶片的經營策略,使其與國際生物晶 片大廠做出了市場區隔,並不斷在技術研發或產品行銷上尋求合作夥 伴,期使晶宇的技術水準與產業實力能在最短的時間內發揮最大的效益 17。 2. 達灣生化科技公司: 2001 年 12 月獲核准進駐竹科,投資金額約 2.5 億。其技轉中研院 生醫所所開發的生物晶片,產品以基因晶片、蛋白質晶片與檢驗試劑為 主。目前與晶宇進行產銷聯盟18。 15 請參閱,同前註4。 16 請參閱,陳婉如,<萌芽中的我國生物晶片>,光連雙月刊,45 期,2003 年 5 月,頁 31-33。 17 請參閱,<晶宇生技計劃於明年第一季掛牌上市 王獻煌:生物晶片產業唯一不變就是「變」 >,資料來源:http://www.2300.com.tw/investment/ShowContent.asp?contentid=12707,2004/12/4 拜訪。 18 請參閱,<達灣「羊乳快速檢驗試劑」應市,摻假佯乳無所遁形 >,資料來源:2004/12/6 拜 訪。
3. 聿新生物科技公司: 聿新生物科技股份有限公司創立於1999 年,總公司設於新竹科學園 區,擁有檢驗試劑處、生物醫材處、生物晶片處三個研發生產部門,以 及新竹及台北兩個營運中心。針對肺癌早期檢驗用生物晶片作研發19 。 4. 國朕生物科技公司: 該公司自 1993 年起投入生物科技產業,除了研發有關基因工程、 病理、生化等技術外,並針對水產養殖業進行技術再造的工程,整合 魚蝦養殖的上、中、下游技術,導入免疫技術及病理檢驗技術,並運 用基因工程中相關技術篩檢病毒毒性疾病,以去除在育成各階段可能 造成的感染,並輔以環境工程控制及營養調配等相關技術20。 於2002 年 6 月獲核准進駐竹科,投資金額約 4.95 億。其開發目標 為水產養殖科技化解決方案,目前有蝦生物晶片。 5. 聯華生物科技公司: 聯華生技股份有限公司為一家專精於製造體外免疫診斷試劑系列 的公司,所生產製造的產品線包括傳染性疾病測試劑及成癮性藥物濫 用測試劑,產品普及於一般使用者及專業人員使用。之後擴大領域至 基因晶片、生物晶片之研發與製造,為工研院「生物晶片研發計畫」 之技轉公司,以高密度基因晶片、生物晶片讀取機、微型電泳分析儀 為開發重點21。 6. 台岳生物科技公司: 於2002 年 6 月於台南科學園區設立。其主要營業項目區隔為兩大 事業群,第一事業群為以微陣列生物晶片平台開發免疫相關疾病之快速 檢測晶片及中草藥現代化平台;第二事業群為以轉基因動物技術平台開 發藥用蛋白質。投資金額約為 4 億元。其主為產品為自體免疫檢測晶 片、過敏原檢測晶片。 7. 聯華國際企業公司: 2002 年 3 月獲核准進駐南科。投資金額約為 5 億元。生物基因、 蛋白質晶片之診斷試劑為主要產品。 8. 榮睿生物科技公司: 19 請參閱,聿新生物科技企業網站,資料來源:http://www.bioptik.com.tw/about/,2004/12/6 拜訪。 20 資料來源: http://www.104.com.tw/cfdocs/2000/job2000/introduce.cfm?invoice=70457608000&jobnum=967347。 21 請參閱,聯華生技股份有限公司網站,資料來源:http://www.aspire.com.tw/cindex.html。
成立於 1998 年,已自行開發出多項快速檢測試劑,如腸病毒 71 型快速檢測試劑、性病系列快速檢測試劑、食物中毒系列快速檢測試 劑、驗孕快速檢測試劑、排卵快速檢測試劑及各類濫用藥物系列快速 檢測試劑等,其中多項產品已進行專利申請中22。 於2001 年 11 月獲核准進駐南科。投資金額約為 5 億元。以蛋白 質晶片、快速檢驗試劑為主要產製目標。 另外,高雄市政府成立生物科技園區推動委員金,期以BOT 之方式, 在大坪林推動生物園區之設置。在高雄地區有科景生物科技、華肝基因等 生物因片廠商。 國內生物晶片市場還很小,且以代工為主。然而生物晶片的研發階段需要 投入大量的資源,且目前國內生技公司營收偏低,需要改變傳統的經營模式 以提昇競爭力,包括: 1) 產業聯盟: 各公司彼此之間的聯合經營模式。如晶宇藉由與國內生技公司建立的策 略聯盟,積極地在臺灣、大陸及歐洲等地建立行銷的通路與管道。在國際上, 與 數 家 歐 洲 公 司 與 機 構 , 亦 有 相 當 程 度 的 技 術 合 作 。 現 階 段 正 和 英 國 Microzone 公司合作樣本前處理試劑的開發。此外,更與德國及法國的公司合 作,著重於量產設備及動物臨床檢斷晶片的開發23 。 2) 產業育成: 仰賴科學園區周邊的大學、醫學中心、研究機構所提供的研究資源。如 成大於2002 年整合校內生醫、微積電、材料工程三大領域學者群,著手成立 微電泳生物晶片衍生公司。公司初期資金募集額度為新台幣二億元。該衍生 公司以微流體晶片製作技術、電泳晶片、PCR 晶片、微流體相關技術、光學 (UV、螢光)吸收偵測技術、構裝及整合技術為主要的研發技術。進一步產 品化且同時開發整合之技術,近期發展出多樣微生物晶片儀器系統產品,包 括Single channel CE 晶片、Multichannel CE 晶片、PCR 晶片、前處理晶片、
整合 PCR/CE 晶片、及晶片儀器系統。中長期產品開發包括新藥篩選晶片、 生物資訊晶片和免疫檢測晶片為主的蛋白質晶片,以及以細胞計數和分類晶 22請參閱,榮睿生物科技有限公司網站,資料來源: http://www.oncoprobe.com.tw/company/index.html,2004/12/6 拜訪。 23 請參照,晶宇生物科技實業的網站。資料來源:http://www.bio-drchip.com.tw/partner.asp, 2005/1/16 拜訪。
片為主的細胞晶片24。 3) 多元經營模式: 結合生物資訊、代理外國醫療器材進口、諮詢等服務。例如晶宇除生物 晶片與相關產品之生產外,亦成立了以生物晶片實驗為基礎之檢驗中心,提 供多種分子生物學的檢驗服務25 。 第三項、臺灣產業發展優勢與劣勢分析 目前臺灣發展生物晶片的利基與劣勢分析如下26: 劣勢: i. 生物晶片相關產業的研發費用比起生產所需成本高出甚多,降 低製造成本之重要性相對微小,所以鮮少看見生技業的代工廠。 ii. 現階段生物晶片的市場還很小,無法與半導體產業相提並論。 iii. 生物晶片製造上之突破,如何利用科技合成各種探針、如何將 探針植於晶片上、晶片表面處理如何做出細小而又佈滿管道的 晶片等挑戰,此不僅為台灣的,亦為世界各國生物晶片產業發 展的重要課題。 利基: i 我國生技及半導體、光學上人才屬亞洲佼佼者之一。 ii 生物晶片之製造亦需整合性,如晶片設計開發公司、晶片元件製 造、系統整合、應用軟體、試劑藥品及電子化學材料。在生技 及半導體、光學領域有眾多人才的我國,發展生物晶片產業的 潛力無窮,應有機會進入比代工更高層次的領域。 24 請參閱,<成功大學衍生公司營運計畫簡介 –案件二:微電泳生物晶片技術營運計劃簡介, 資料來源:http://www.tlo.ncku.edu.tw/Html/SubFolder/1news/com_2.htm。 25 請參照,晶宇生物科技實業的網站。資料來源:http://www.bio-drchip.com.tw/PRODUCT3.asp, 2005/1/16 拜訪。 26 請參閱,<台灣策略性技術領域「生物電子學」之技術地圖>。資料來源: 科技政策智庫網 站,http://www.stic.gov.tw/policy/intelligence_1_c_5rs.htm,2004/12/4 拜訪。
展望未來國內生物晶片產業的發展,主要取決於下述三大關鍵點,即晶 片價格能否低價化、能否突破美國Affymetrix 公司的專利、以及能否從眾多 不同的競爭技術中脫穎而出。 目前國內以經濟部技術處支持之工研院生醫中心擁有較多之各種資 源,其藉由整合41 項的專利技術,推動廠商成立研發技術聯盟一次承接, 希望能藉著專利交互授權與策略結盟的方式,與國際大廠平起平坐。其中包 括可降低成本至十分之一的基因探針快速佈放技術。在業界方面,如晶碁生 技,以半導體雷射光製程所獨創的星雲晶片(Galaxy chip),將一平方公分大 小之生物晶片,切割成數萬點。寬度為頭髮十分之一的微細晶片,不但可望 突破上述Affymetrix 之專利限制,更晉升國際上正激烈競爭中的優勢晶片技 術之林27。 27 請參閱,同前註錯誤! 尚未定義書籤。。
第三章、美國醫療器材管制法規介紹
依照美國聯邦食品、藥物、化妝品法案(Federal Food, Drug, and Cosmetics Act,簡稱 FD&CA)的規定,所有的藥品、生物製劑(biologics)與醫療器材(含試 劑),無論是在研發、試驗、生產、銷售與使用等所有階段,均屬於聯邦食品藥 物管理局(Food and Drug Administration,簡稱 FDA)的管轄範圍,以訂定聯邦法
規管理之。目前FDA 下設兩大分支機構,一是藥品評估研究中心(Center for Drug
Evaluation and Research,簡稱為 CDER),負責藥品與生物製劑的管理,另一為 器材與放射性健康中心(Center for Devices and Radiological Health,簡稱為 CDRH),負責各種醫療器材的管理與查驗登記,其管理的範圍不僅種類繁多, 且器材間之性質差異亦甚。一般而言,臨床上所需之儀器及套件,在上市前,製 造商或是進口商必須提供足夠的資料,以證明該產品有足夠的安全與有效,經過 FDA 的核准後才能在市場上販售。FDA 在診斷試劑及醫療器材方面,與藥品的 上市前審核採用不同的管理模式,其中最大的差異在於前者在一開始就會被 FDA 依照產品的不同性質加以分類;對於不同類別的診斷試劑與器材,FDA 審 核的規則與嚴格度自然也有差異。我國對於醫療器材之管理模式原則上參考美 國,例如美國關於醫療器材等相關產品須於上市前經過FDA 核准,始得於市面 上銷售等程序規範,以及醫療器材之分級管理制度等。 為因應生物晶片類產品之發展,FDA 於 2003 年公佈了關於此類醫療器材的 管理草案,經過公聽會及聆聽各方意見後,終於於今(2005)年 3 月 10 日正式公 告”Class II Special Control Guidance Document: Instrumentation for Clinical Multiplex Test System”28。期間經歷Roche 公司(與 Affymetrix 合作開發)AmpliChip
的申請、許可及要求由Class III 改為 Class II 等。如今應是塵埃落定,其他各家
公司也可依循提出申請。 第一節、歷史沿革
美國聯邦政府於1906 年實施的食品藥品法案(Food and Drugs Act)並未將
醫療器材納入管理的範圍內,一直到1938 年的食品藥物化妝品法案(Food Drug
and Cosmetics Act) 才將 FDA 的職權範圍擴展至醫療器材與化妝品上,並要求
所有上述的產品在上市前必須充分證明其安全性29。此法對於醫療器材的定義 為:“用於診斷、治療、或是預防人類與動物疾病的任何器械、儀器、設備與 裝置包括其零件與組成物,均為醫療器材”。當時如果產品上市後,經檢測發 現含有有害物質會是會變質,或是製造過程不合乎衛生者,FDA 有權令產品下 架,甚至於對製造者與供應商進行刑事追訴,但是當時 FDA 並無權對產品實 施上市前的試驗與審核。直到1962 年的 Kefauver-Harris 藥物修正案的實施, FDA 才有權要求藥品在上市前,必須經過 FDA 的核准,而且藥品除了安全性 28 www.fda.gov/cdrh/oivd/guidance/1546.pdf 29 21 U.S.C. 301 et seq.
之外,還必須證明其有效性才能獲得核准,不過該修正案僅適用於藥品,對於 醫療器材與化妝品並無同等的規範。
隨著科技的進展,醫療器材越來越複雜,而且開始有侵襲性(invasive)的產 品出現,對於病人的健康與生命有重大的影響,並導致諸多的醫療糾紛。因此,
為 因 應 各 方 的 要 求 ,1976 年終於通過了醫療器材修正案(Medical Device
Amendments,簡稱 MDA)30,此修正案賦予FDA 對於醫療器材從研發、臨床試
驗(含人體試驗)、生產、銷售與使用等各階段,都有管理的權力。而 1990
年的安全醫療器材法(Safe Medical Devices Act,簡稱 SMDA)31更使FDA 對於 醫療器材的管理權擴大到設計有效性、瑕疵產品的回收、產品使用後的追蹤監 控,並增加對於違規廠商的罰金處分等。依據此法,醫療器材的定義為:「符 合以下條件之裝置、儀器、用具、機器、器械、植入物、體外試劑、或是其他 類似及相關的物件(包括組件、零件與附件):
(1) 明列於美國 國家處方輯(National Formulary) 或美國藥典 (US Pharmacopeia)中的器材。 (2) 使用於動物或人類疾病或其他身體狀況的診斷、治癒、緩解、 治療或預防者。 (3) 影響動物或人體身體功能或結構,但並非依賴人體內的化學反 應與代謝作用來達成其目的者」32。 最後一段的定義是要區分藥品與醫療器材,而此定義幾乎將所有除了藥品 之外的醫療相關產品都涵蓋在內。依此定義,臨床遺傳檢測的儀器與試劑,即 使是預測性的檢驗,可能會屬於定義上所謂預防疾病的器材,或是診斷疾病以 外的其他狀況(other conditions)的器材,當然可被歸類為醫療器材。
1997 年的 FDA 現代化法案(FDA Modernization Act,簡稱 FDAMA),為
美國有關醫療器材法規的最近修正。該法案藉著規範 FDA 的審核運作程序,
間接影響到藥品與醫療器材的上市;其主要內容是加速部份特定新藥的核准, 並且藉著免除上市前通知(premarket notification)程序來加速部分低風險的新醫
療器材之上市。此外本法案規定在藥物查驗的過程中,允許 FDA 使用外部專
家小組(outside consulting board)來協助審查。其實美國國會在 1990 年醫療器材
30 Public Law 94-295 31
Public Law 101-629
32 The term “device” means:
an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is
(A) recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them,
(B) intended to use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
(C) intended to affect the structure or any function of the body of man or other animals, and
which does not achieve its primary intended purpose through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes.
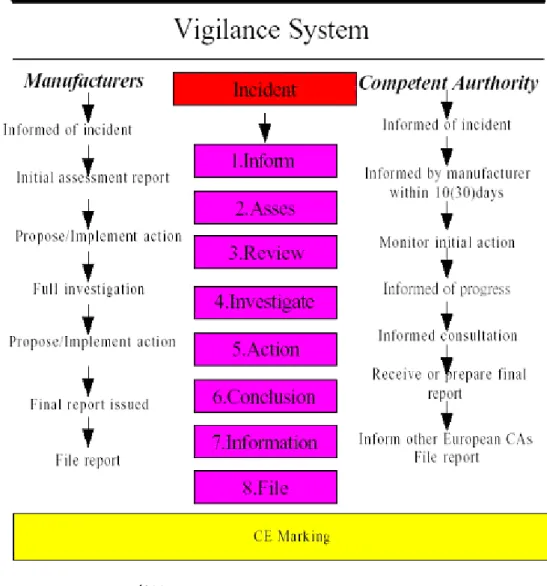
安全法案中已制訂醫療器材上市後監督辦法,然而FDAMA 仍賦予 FDA 在醫 療器材上市後監督的工作上更多的處理權。新的醫療器材上市後監督辦法於 2002 年 6 月 5 日公告,並於 2002 年 6 月 8 日正式生效,主要目的在要求醫療 器材廠商蒐集醫療器材功效性上的有用資料,用以揭露不可預期的產品副作用 及其他相關資訊,以期保護大眾的健康。 有些醫療器材伴隨的問題與風險,即使在有臨床試驗資料輔助的狀態之 下,也無法在尚未進入市場使用前進行預測。上市後監督的規定將幫助 FDA 和醫療器材製造商鑑定很少發生、但卻是潛在的嚴重問題。這些問題在產品開 發時並未被明顯的揭露,或所鑑定的問題並未嚴重到不允許該產品上市。上市 後監督辦法將提供早期評估醫療器材潛在風險的方法,並透過任何可能的工作 來減低並病患可能遭受的風險,例如加強治療者教育訓練、加強產品標示或改 變產品設計。 第二節、醫療器材分類及其管理模式 從醫療器材的定義來看,美國的立法試圖將所藥品以外的所有醫療產 品,全部歸類為醫療器材,以致於醫療器材的種類繁多,各類品項的性質也迴 異;而從法規的延革趨勢來看,美國國會賦予 FDA 對於醫療器材的管理權限 也越來越大,從上市後查核進展到上市前審查,最後又加上上市後的調查與監 控。但是,如果所有的醫療器材都使用同一規格的審核方式,勢必造成醫療器 材成本的增加,而且也增加 FDA 的行政負擔,此為醫療器材分級管理制度
(Midical Device Classification)所由設。
依照最新的聯邦食品藥物化妝品法33的規定,用於人的醫療器材依其個別
的安全性與有效性分為三個等級34。以下介紹醫療器材的分級原則與管理方式:
第一級醫療器材:低度危險性(Class I ---Low Risk)
依照聯邦食品藥物化妝品法第 513 條的定義,凡是只受一般管制 (General Controls)的管理就足以被認為具有可靠的安全性與有效性者,為 第一級醫療器材。或是,雖然目前有關此醫療器材的資訊不足,以至於無 法判斷是否一般管制的控管方式能夠確保該器材的安全性及有效性,但 是,(1)該器材並非用來支持(supporting)與維持(sustaining)人類生命,或是 對於人類疾病的預防無實質的重要性,而且(2)對人體無產生疾病與傷害 的潛在風險者,亦屬於第一級醫療器材。 所謂的一般管制,則規定在同法的第501 條「攙雜」、第 502 條「錯 誤標示」, 第 510 條「登錄程序」,第 516 條「禁用」, 第 518 條「通知 與其他救濟」,第519 條「紀錄與報告」以及第 520 條「人用醫療器材的 一般管理條款」等各條文中。簡言之,即所有的醫療器材需符合一般管制
33 Federal Food Drug and Cosmetics Act, Section 513, Classification of Devices Intended For Human
Use.
的規定,其規範內容包括註冊、列名、GMP35、禁止不當標示、遵守銷售 及廣告規定、傷害報告管理程序等。
其中最重要的是第 520 條,規定人用醫療器材的製造必須符合 GMP
的規範,以及FDA 可限制部分醫療器材使用、銷售等,但是對於個人用
器 材(custom devices) 、 試 驗 性 用 途 (investigational use)36與 人 道 性 質
(Humanitarian)的醫療器材則有一些例外性的規定。 第510 條 k 項則規定,任何想要跨州鋪貨或是販售人用醫療器材者, 必須在開始販售或是鋪貨前九十天,向FDA 秘書處提出報告,即所謂上 市前通知(premarket notification),業界將之簡稱為 510(k)通知37。而通知的 內容則授權由秘書處訂定,大致的項目包括:基本資料、目錄、器材名稱、 註冊號碼、分類、性能標準、標示、實質相等性比較、摘要、產品描述、 生物相容性、以及色素添加物等。其中有部份毫無醫療風險可言的第一級 醫療器材,FDA 可免除其 510(k)通知的義務,允許廠商直接在市場上販 售。
第二級醫療器材:中度危險性(Class II ---Medium Risk)
部分醫療器材無法以一般管制確保其安全性與有效性,但仍可藉由特 殊管制(special controls)來提供確保醫療器材的安全與有效的資料者,則被 歸類在第二級醫療器材。而所謂的特殊管制的內容則包括公告的性能標準 (performance standard)、上市後的調查、病人登錄、對器材的實質建議、 以及FDA 秘書處認為其他能確保醫療器材安全有效的必要措施等。 同法還提到,如果是用來支持與維持人類生命的醫療器材,秘書處必 須認真檢討,並確認特殊管制是否能確保該醫療器材是有效且安全的、了 解該特殊管制是如何確認的。換句話說,FDA 對於用來支持與維持人類 生命的醫療器材。必須特別注意,本類醫療器材有可能被歸類至第三級醫 療器材。除了受到特別管制的規範外,第二級醫療器材仍然要遵守一般管 制的規定。 第三級醫療器材(Class III): 若因為資訊不足而無法斷定一般管制足以確保該醫療器材為安全、有 效,而同樣也因資訊不足而無法斷定特殊管制足以確保該醫療器材為安全 有效,因而無法被歸類為第一級與第二級醫療器材,且同時該醫療器材是
35 ISO9000 及 ISO13485 已將醫療器材 GMP 修改為 Quality System Regulation (QSR)。
36 新的醫療器材在進行人體試驗前,就必須先申請試驗性醫療器材例外(Investigational Device
Exemption Application,簡稱為 IDE),以免除部分一般管制的規定。
37 Federal Food Drug and Cosmetics Act, Section 510: (k) Each person who is required to register
under this section and who proposes to begin the introduction or delivery for introduction into interstate commerce for commercial distribution of a device intended for human use shall, at least ninety days before making such introduction or delivery, report to the Secretary (in such form and manner as the Secretary shall by regulation prescribe).
用來支持與維持人類生命、或對於人類疾病的預防有實質的重要性、或是 對人體有產生疾病與傷害的潛在風險者,皆屬於第三級醫療器材
第三級醫療器材亦須經第515 條上市前核准(premarket approval, PMA)
的程序,以合理確認其安全與有效性,方得在市場上販售38。而上市前核
准最重要的條件,就是要有臨床試驗的結果,而且在產品開發時,也受到 FDA 的管理。
以上是美國醫療器材分級管理的原則,然而實務上依照第 513 條 b 項的規
定,FDA 秘書處應組織分類工作小組(classification panels)。至於分類工作小組的 分級標準是什麼?此涉及風險評估研究,並將原有的醫療器材加以分類。而對於 新的醫療器材的初次分類,則必須按同法第513 條 f 項「特定醫療器材的初次分 類與再分類」的規定辦理。 聯邦食品藥物化妝品法第513 條 f 項規定: 「在本法實施後才上市的任何人用的醫療器材,除非有下列的情況,否則 應先歸類為第三級醫療器材39: (1)該醫療器材之類型(type)已被歸類為第一級或是第二級醫療器 材、或是與已被歸類為第一級或是第二級醫療器材的類型雖不 相同但是實質上是相等的(substantially equivalent)。或是 (2)秘書處已接受申請而將該醫療器材歸類為第一級或是第二級。」 所謂 510(k)的上市前通知,就是給廠商一個說明的機會,提供證據以闡明 新的醫療器材與原先已分類為第一級或第二級的醫療器材是同一類型或是實質 上相等的類型,而不需要經過臨床試驗才能上市,以期能快速地獲得FDA 的許 可而上市。而即使已經分類的醫療器材,FDA 也會接受廠商的申請而與以再分 類(reclassification)。不過,因為第二級與第三級醫療器材管理方式並不同,通常 由第三級的產品要重新分類為第二級前,FDA 必須先完成建立該器材的性能標 準,才能重新分類。 而所謂的實質相等,係指新的醫療器材與已分類確定的醫療器材同樣安 全、有效;依照FDA 的說明,一個新的醫療器材,與已分類確定的醫療器材比 較起來有相同的用途,而且隸屬於相同的技術領、或是雖然技術領域不同,但是 並無增加安全與有效性方面的疑慮40。因此,「實質相等」並非「完全相同」這 麼狹隘,尚須考量其用途、材質、性能、安全、有效、標示、生物相容性及其他 能相比較的性質等,經過整體的比較,而廠商在510(k)通知中的說明愈詳細,被 判定為實質相等的機會愈大。 38 此程序如同新藥上市之程序。 39 http://www.fda.gov/cdrh/devadvice/313.html#determine
然無論是申請510(k)或是上市前核准(PMA),醫療器材的安全性與有效性是 FDA 考量的最重要因素。例如,在申請 PMA 文件中,廠商需證明新的醫療器材 對人體有合理的安全性與有效性;而510(k)中廠商則需要證明新的醫療器材與已 分類確定的醫療器材是同樣安全有效的。然而,何謂安全與有效?其實法規並未 明確定義,實質的內涵則留待FDA 的裁量。事實上,世界上並沒有絕對安全的 醫療器材,「安全」僅只是醫療器材使用的一種概念,任何一種相當安全的醫療 器材,若使用者操作不當,仍可產生某種程度的危險性。因此,充其量,醫療器 材臨床試驗僅只能夠規範其功效與安全概念的內在產品條件而已,所以安全可解 釋為可接受的風險(acceptable risk),而這種風險必須視個案情況而定。原本 FDA 認為「有效」是指醫療器材具備高度可靠的定量或定性條件,也就是具有分析的 有效性(analytic effectiveness),但是並不意味該器材的使用必定能達成期望臨床 目的所具備的診斷或治療能力與性能,只要對醫師有參考價值即可,不一定能得 到臨床上的有效性(clinical effectiveness)。然此一觀點引起高度的爭議,輿論傾向 要求FDA 對於器材的臨床有效性需加以審核。 美國的法規規定新的醫療器材或是檢驗試劑必須從第三級開始歸類,之後 再依「從新分類」方式來降級。我國並未明定新的醫療器材應如何分級之程序。 我國學者多將生物晶片歸類於第三等級之體外檢驗器材,並依現行醫療器材管理 辦法裡的「第三等級產品」來管理,審查要點將著重在產品的安全、性能與功能, 關於我國管理法規部分詳如後述。 第三節、Affymetrix 申請案例簡介 第一項、 產品介紹
GeneChip 主要利用 Northern blotting 的概念,將原本轉漬在膜上的 target RNA 轉變成以 biotin 標示的 cRNA(由使用者提供 RNA 樣品而合成)。至於引子 (probe)則是由 Affymetrix 公司事先直接在晶片上合成好,再經由 hybridization 的
方式,將標示好的cRNA 雜合到晶片上頭的引子上41
。
第二項、 申請過程及依據42
依21 U.S.C. 360 section 513 (f)(1),若一設備未於 1976 年 5 月 28 日–即 the Medical Device Amendments of 1976 通過之日–之前跨州上市,稱作「修法 後設備」(postamendments devices),應自動被歸於 class III,而須為上市前審 查;除非之後被FDA 分類、再重新分類(reclassify)至 Class I 或 Class II,或
41 請參閱,Affymetrix GeneChip 中文快速攻略本。資料來源:http://www.affymetrix.com,2005/4/02
拜訪。
42
相關資料請參照,〈Rules and Regulations〉,Federal Register: March 10, 2005,Vol.70, No.46, 11867-11869。資料來源:http://www.fda.gov/ohrms/dockets/98fr/05-4760.htm,2005/4/28 訪。
FDA 依同法 section 513(i)認為該設備與一已判定的設備實質相似。必需該 class 能提供足夠的管理控制,以合理確保該 device 在其目的用途上的安全性 與有效性,FDA 才會作出上述動作。 同法§ 513(f)(2)規定任何人依§ 510(k)為某一未曾分類的設備的申請上 市前通知,應在收到一依§ 513(f)(1)將該設備歸類於 class III 的命令後三十日 內向FDA 要求依§ 513(a)(1)設定的標準,將該設備分類。FDA 應於收到此項 要求的六十日內 以書面命令將該設備分類。此一分類應是該設備的起始分 類。在對該設備分類的命令發布後的三十日內,FDA 應於聯邦公報(Federal Register)上發佈此一分類的公告。
FDA 在 2004 年 10 月 29 日發佈 the Affymetrix GENECHIP Microarray Instrumentation System 歸於 Class III 的公告。2004 年 11 月 3 日,Affymetrix 依 § 513 (f)(2)向 FDA 提出申請書(petition),希望將係爭設備歸類至 Class II。
在審理該申請書後,FDA 作出判定,認為 Class II special controls 能提供 該device 安全性與有效性的合理保障。FDA 分配予此設備「Instrumentation for Clinical Multiplex Test Systems」的屬名(21 CFR §862.2570 - Instrumentation for Clinical Multiplex Test Systems)。Instrumentation for Clinical Multiplex Test Systems 被定義為一種用來測試及分類自一個臨床測試分析報告的多樣訊息 的設備。此測試設備與一特定的分析報告一起使用,可衡量多樣相似的分析, 建立單一指標以協助診斷。FDA 認為用「Instrumentation for Clinical Multiplex Test Systems」加以規制對健康無直接風險。然而不正確的結果,可能會不正 確的診斷或不當的病人管理。此外,若此裝配失誤未產生任何結果,可能會 拒絕或延遲有益的、適當的治療。FDA 所建議可緩和上述風險的方法載於 “Class II Special Controls Guidance Document: Instrumentation for Clinical Multiplex Test Systems”此份指導文件(the guidance document)中,包括成果 確認(performance validation)與標示(labeling)。
因此,本設備除了21 U.S.C. 360 的一般管制外,尚須符合「Class II special controls Guidance Document: Instrumentation for Clinical Multiplex Test Systems」此一特殊管制指南。
另外,FDA 未豁免本設備的上市前通知義務。依§ 510(m),若 FDA 認 為依§ 510(k)作上市前通知(pre-market notification)非提供屬於 class II 的設備 安全性與有效性之合理保證所必須,可豁免該設備上市前通知的義務。 FDA 判定上市前通知是提供本設備安全性與有效性之合理保證所必 須,因而未豁免免該設備上市前通知的義務。是以,在此分類生效後,任何 意欲在市場上銷售本設備之人,必須於其上市前按此特殊管制指南,向FDA 提出含有其欲上市設備資訊的上市前通知申請書。 第三項、 環境影響 FDA 依 21 CFR 25.34(b)認定該設施對人類環境無個別地或累加地顯著影
響。 因此,不需要作環境評估(an environmental assessment)或環境影響報告 書(an environmental impact statement)。
第四項、 影響分析
FDA 已依 Executive Order 12866、the Regulatory Flexibility Act (5 U.S.C. 601-612)及 the Unfunded Mandates Reform Act of 1995 (Public Law 104-4)檢視此 決定的影響。
Executive Order 12866 指示政府部門應評估可能的管制替代方法的所有成 本與利益。而且,當管制是必需時,應選擇能極大化淨利益的管制方式。FDA 認為本決定非該行政命令所指涉之管制行為。
The Regulatory Flexibility Act 要求政府部門分析管制選擇以極小化該決定對 小事業體(small entities)的影響。因將該設備歸於 class II 的分類,將免除該設備 的製造商依§ 515 of the act (21 U.S.C. 360e)的要求進行上市前批准的成本,並可
能因降低小型潛在競爭者的成本而讓其得以進入市場,FDA 擔保(certifies)43此終
局決定不會對大多數小事業體造成顯著影響。
The Unfunded Mandates Reform Act of 1995 的§ 202(a)要求政府部門在提出 「所有包括任何可能致使州、地方與部落政府的支出總額或私營部門的支出達一 年$100,000,000 以上(隨通貨膨脹作調整後)之聯邦命令的決定」前,須備妥一 份包含預期成本與利益的書面報告書。依最新(2003)的 Implicit Price Deflator for the Gross Domestic Product,調整後的門檻為 $115 million。FDA 不認為此終局決 定將導致任何等於或高於此數額的年支出。
第五項、 聯邦主義(Federalism)
FDA 依 Executive Order 13132 分析此終局決定。FDA 認定此決定不含有對 各州、國家政府與各州關係、各級政府間權責分配有實質直接影響的政策。據此, FDA 斷定此決定不含有該執行命令所定義的聯邦暗示(federalism implications), 因而不需作聯邦主義總括影響報告。
第六項、 1995 年文書減量法(the Paperwork Reduction Act of 1995)
此終局決定不含大量資訊。因此,不需要由管理與預算局(the Office of Management and Budget)依 1995 年文書減量法進行清除工作。
第七項、 附件:Affymetrix 申請案的審查資料
510(k) Premarket Notification Database
Device Classification Name instrumentation for clinical multiplex test systems
510(k) Number K042279
Device Name AFFYMETRIX GENECHIP MICROARRAY
INSTRUMENTATION SY
Applicant
AFFYMETRIX, INC. 3380 central expressway santa clara, CA 95051
Contact Robert Lipshutz
Product Code NSU
Date Received 08/23/2004
Decision Date 12/23/2004
Decision approved evaluation of automatic class iii designa (AN) Classification Advisory
Committee Clinical Chemistry
Review Advisory
Committee Toxicology
FOI ITEM LETTER
Type Evaluation of Automatic Class3 Designation
Reviewed by Third Party No
Expedited Review No
資料來源:
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm?ID=16003, 2005/4/28 拜訪。
第四節、「Class II special controls Guidance Document: Instrumentation for Clinical Multiplex Test Systems」之簡介
FDA 在 2003 年就公佈了關於生物晶片類器材的管制文件草稿,經過了公開 的說明,聆聽各方提出意見與報告之後,經過兩年的政策研商,在今(2005)年 3 月 10 日,公佈了 Class II Special Control Guidance Document: Instrumentation for Clinical multiplex Test Systems44文件。
這分文件特別針對臨床上使用單一器材針對多種不同信號進行測試的醫療 器材作特別的管制45 。而本文件中所謂的醫療器材,包含了整套測試的儀器,從 44 全文見 www.fda.gov/cdrh/oivd/guidance/1546.pdf 45 屬於 21 CFR 862.2570 規範範疇中的醫療器材。
測試用的晶片,乃至於整套分析的電腦軟硬體都屬其中。文件中也仔細的將其適 用的範圍進行限制,原則上此類的醫療器材仍應隸屬於Class III 的管制範疇,但 若為符合本文件敘述,以檢測為主要功能的此類醫療器材(其中很可能包括生物 晶片及其分析儀器之組合),則可以例外採用Class II 的審查標準,提出報告,並 證明相等性,就可以上市。FDA 也特別強調,這樣的作法之目的,就在於平衡 審查效率與安全性。 基於這幾年來的經驗,FDA 評估此類醫療器材可能帶來健康上的風險,主 要集中在對診斷結果可能出現的不準確的判讀。這個不正確的判讀結果,可能會 導致不適當的診斷,甚而可能會使病患之管理不適當,而帶來危險的後果。這些 錯誤的判讀,在整套醫療檢測器材使用的流程中:例如檢體採集、數據收集、乃 至電腦統計分析,各階段中都有可能出現。因此,FDA 特別要求廠商,必須針 對此部份,進行完善的風險評估報告,並詳加說明風險評估的方法,才能據此文 件提出申請案。 第五節、FDA 審查程序之討論 作為美國的藥品、食品(包括膳食補充劑-美國之保健食品)、醫療器材、化妝 品等等相關人體健康之各類商品的主要管制機構,FDA 所掌握的是極為龐大的 經濟利益與市場— 超過每年十兆美元的收益,相關市場佔了美國民眾超過四分 之一的消費量46。牽涉如此龐大的商業利益,加上維護大眾健康之公共利益的要 求,FDA 的管制決策程序,牽涉了複雜的利益團體糾葛,當然也成了高度政治 性的討論議題。 隨著生物科技的日新月異,各大藥廠研發能力提昇,以及大量的資金不斷投 入新的研發與生產,相關醫藥產品的數量快速的提昇,FDA 面臨了非常大的挑 戰。如何能夠有足夠的人力和物力,有效率的因應龐大的申請案件,又能夠善盡 把關者的責任,做好風險之控管,使通過上市前審查程序的商品,不至於對大眾 的健康造成威脅? 這個議題在美國的政壇、醫學界、法律學界、產業界已經被討論許久。本文 嘗試針對幾個比較常被提及的觀點,作簡單的討論。他山之石,可以攻錯。或許 美國所面臨的管制難題,也會出現在我國。且美國的應對及立法,亦常影響我國 的立法與實務走向。如何記取其缺失的教訓,如何擷取其優點,才能真正訂出合 乎國人最佳利益的法律與政策。 第一項、安全第一?CDRH 作為民眾健康安全的守門人 基於FDA 一貫的理念,其進行上市前審查最重要的工作就是,針對產品的 風險程度進行分級、審查,以保障公眾的健康安全。CDRH 在進行醫療器材的上 市前程序審查時,希望能針對所有的申請文件作完整的行政審查及紀錄。
46 President Bill Clinton & Vice President Al Gore, National Performance Review: Reinventing Drug
& Medical Device Regulations 2 (1995) [hereinafter Nat’l Performance Review]; see also <http:// www.fda.gov/bbs/topics/NEWS/NEW00414.html >(FDA home page).
資源的分配是CDRH 首先面臨的課題,過去的「先到先服務」(first come, first serve)作法在面臨日漸提昇的申請件數量,如何加強效率就成了重點47。也因此, 對於醫療器材申請上市產品的歸類就顯得很重要,被歸類為高度風險的第三級醫 療器材,FDA 將以較多的人力作完整的測試;而相對的,FDA 就分配給較中低 度風險器材較少的資源及較簡便的審查程序。 提到人力資源,CDRH 內部有許多的科學研究實驗室,有論者也建議應該將 機構編制內的所有科學專家和實驗室,都用來進行上市前審查的工作,以增加效 率48。不過,原則上,CDRH 認為仍然有必要持續研究工作,因為保護公共健康 並不僅限於產品的審查。 然而,安全與效率之間的權衡,其間的利益衝突逐漸浮上檯面,而許多激進 的改革 FDA 的建議也紛紛被提出,例如將審查程序外放至私人機構等提議,都 曾引發熱烈討論。 再者,許多申請的文件在許多重要的資訊上有所闕漏。FDA 希望加強文件 形式上的審查,如果在早期就發現有所闕漏,即應予以退件,才不會浪費了 FDA 檢驗科學家團隊的人力資源。 第二項、效率至上?法人化上市前審查程序的提議 如前段所提及,在大量且快速的產品問世且殷切等待上市許可的情況之下, 無可避免的造成FDA 審查效率之不彰。這種效率不張所帶來的影響,對產業界 來說,每拖遲一天核准的程序,所造成的是無比可觀的商業損失,這種情況大大 提昇了美國製藥產業、醫療用品製造業的內在成本,對於其競爭力造成很大的影 響49。美國國內的產業甚至擔心,長久以來美國在生物醫藥產業的領導地位,將 因此而搖搖欲墜。對消費者來說,FDA 審查的機制在於降低生物醫藥品之風險, 保護公眾的健康安全;但相對來說,許多急需新藥上市的重症患者,卻可能在 FDA 為了降低風險的過程中遭致不幸,慢速的審查程序,可能使許多創新的藥 品或醫療用品無法臨床使用。報導指出,在1965 到 1995 年間,有兩萬名以上的 美國民眾因此來不及挽救其生命50。1975 年,兩位藥理學家 William Wardell 和 Louis Lasagna 指出,「因為未能審查出新藥物的風險而貿然將藥品上市,固然會 導致災難;但我們也常常忽略未能即時將藥品上市,而使病患來不及使用新藥所 造成的損失51」。效率的提昇,對消費者權益的提昇的確具有相當重要的意義。從 FDA 的獨占權力來看,更會讓人擔心手握巨大商業利益的機構,加上外界無法 透明得知的審查方法,也會讓人擔心在公權力的披風背後,是否會出現不當的利
47 J. Schwartz, Promising Medical Devices to be Speeded to Market , Wash. Post , June 25, 1993, A2
48 D. Bruce Burlington, An Overview From The Director of The Center For Devices And Radiological Health, 49 Food & Drug L.J. 175 (1994).
49 David A. Kessler, Remarks by the Commissioner of Food and Drugs, 51 FOOD & DRUG L.J. 207, 208 (1996).
50 Daniel Green, Obstacle Course for Drug Producers: Pressure for Reform of the U.S. Food & Drug
Administration is Growing, Fin. Times, Aug. 21, 1995, 12.
益交換52。調查訪視相關廠商也發現,廠商即便得知某些不公正的情形,多半也
擔心遭到FDA 後續的報復,而不敢勇於擔當 whistleblower 的角色。
基於以上種種原因,私人化審查機制的呼聲開始出現。1995 年民主黨眾議 員 Ron Wyden 提出相關改革法案。他有三點主張:(一)開放本國法人審查機構 (institutional review boards, IRBs)能針對藥品、生物製劑、及醫療用品進行上市前 審查。但 FDA 仍然保留在上述機構做出核准後,三十天內得否決(veto)該許可
的權力。(二)賦予FDA 和私人機構簽訂進行毒物測試、統計資料分析的委任何
約的權力。(三)對於治療嚴重的、對生命有重大威脅的疾病之新研發藥品,可由
Department of Health and Human Service 部長所任命之專家團隊先行進行測試,若
通過該測試則視為取得FDA 之核准53。
同年年底,民主黨參議員 Nancy Kassebaum 也提議主張:(一)若 FDA 無法
在某一法定期限內完成百分之九十五的測試,則應該放第三人進行審查。(二) 若已經在歐盟或英國審查通過者,則視為已經取得核准上市的許可54。 面對強大的壓力,FDA 本身也有一些因應55。例如,推動第一級和第二級的 醫療器材委由國內其他單位進行審查。但是,此一改變普遍被批評範圍太小,甚 至醫療器材產業,也因為委由私人單位審查必須另外繳交審查費用,而不願支持 這樣的計畫。 法人化審查的立法並沒有通過,但針對其中內容的討論則包括56: (a).法人機構審查之資格 相對於公立政府機關,私立性的法人審查機構或許能夠較具有效率,但是那 些單位有能力進行審查?並沒有政府公權力背書的審查結果是否值得民眾相 信?會不會因為太過講求「效率」,反而忽略了審查機制真正重要的功能---「風 險控管」?這一連串的問號都會是反對者重要的考量。 因此,選擇具有審查能力的私人機構將十分重要,而這個重要的任務仍然應 該由政府的主管機構來進行,必須要能明定基準,選擇適當的機構。由消費者、 專業醫師、醫療院所,一起來協助政府主管機關來監督。市場的反應,其實也考 驗著私人機構的公信力,若某私人機構的審查出了問題,將大大降低其在醫藥界 的公信力。 (b).外國審查的承認 這在向來不採用他國標準而以領導者自居的美國來說是不常見的,當然也遭 致了一些批評57 。不過,這個情況對台灣來說,卻滿值得考慮。所謂「先進國家」 的審查結果如何來採用?每個國家都建立出不同的風險控管標準,基於國家文化
52 Peter Brimelow & Leslie Spencer, Food & Drugs & Politics, FORBES, Nov. 22, 1993, at 115. 53 H.R. 1742, 104th Cong., 1st Sess. (1995).
54 Christina Kent, Controversial FDA Overhaul Advances in Congress: Several Bills in Preparation,
Am. Med. News, Apr. 22, 1996, at 3.
55 60 Fed. Reg. 28,618 (June 1, 1995).
56 Elizabeth Price , Teaching The Elephant To Dance: Privatizing The FDA Review Process. 51 Food
& Drug L.J. 651.
57 諷刺的是,這個構想在私人化 FDA 的討論中被提出來,可是其實外國的衛生單位根本就不是
的不同,能有一體適用?或必須經過那些程序來簡化其適用程序?都值得我們後 續的討論,本段暫不處理,留待後文討論。 第三項、利益的妥協產物---FDAMA 1997 經歷了十多年的辯論,無數利益團體的遊說,FDAMA 可以說是一個多方妥 協之下的產物58。這個法案適度的賦予 FDA 外放審查權利至外部單位,但是並 不若前段所提的徹底法人化審查程序般激進,新的法案仍然保留了FDA 的管制 結構,但是賦予(也要求)FDA 在考量自身能力後,適度的開放其權利,將部 分審查權責外放至法人機構,而由FDA 主管。本法案也要求修正審查之標準, 要求修改複雜的審查程序,希望能使上市前審查的程序更加順暢,同時寄望能加 強產業界和管制者的溝通。 國會的這個立法,仍然無法根本的解決關於醫藥用品管制程序的兩個對立矛 盾—公眾對於安全的信心,以及市場對新藥上市審查程序效率的要求。但是在長 久以來以科學家為主組成的這個管制單位,對於安全與風險的把關一向要求嚴 格,以守門人的角色自居。不過在國會的立法介入之後,對於 FDA 的許多實質 審查標準都有了重大影響。其目的在於增進審查的效率59。 FDAMA 大多數的條款都在 1998 年開始生效,FDA 也開始針對許多審查的 基準,準則和政策文件進行修正60 。FDA 主席也將此現代化過程當作機關的主要 工作目標。當然,降低審查的標準及複雜的程序,無可避免的會引起部份保守傾 向消費者團體的疑慮。不過本法案賦予FDA 更多的上市後監督機制。只要能通 過初步的上市審查,進到市場後,並不代表能通行無阻,如果有任何安全上的疑 慮,仍然要靠著更完善的通報機制及上市後的監督程序,進行損害及風險的管理。 任何的政策選擇,都是價值之間的權衡,FDA 朝著更有效率的審查過程前 進,一方面也是因應龐大的工作量及市場需求不得不的走向,但是如何在加速之 餘,仍然做好公共安全的把關工作,就有賴FDA 的努力,建立起人民及產業界 的信心。 第四項、科學風險、管制機構與社會大眾 近幾十年來,科學的不確定性(scientific uncertainty)一直是熱門的議題;而關 於科學不確定性的討論,在新興科技的議題之中,又格外令人關注。從核能安全 的議題,一直到基因科技的進展,科技的日新月異帶給人們生活上很大方便,但 由於伴隨著科技的不確定性所產生的風險,相對的也要由人們來承受。在生技醫 藥產業也是如此,作為政府的管制機關,面臨了風險管理的責任,面對來自產業
58 Pub.L. No. 105-115, 111 Stat. 2296 (codified in scattered sections of 21 U.S.C. (1994)).
59 Jennifer Kulynych, Will FDA Relinquish The "Gold Standard" For New Drug Approval? Redefining
"Substantial Evidence" in The FDA Modernization Act of 1997, 54 Food & Drug L.J. 127, 128
60 Food & Drug Admin., FDA's FDAMA Accomplishments One Year After Enactment (Nov. 23, 1998)
界、消費者團體、醫療團體、衛生團體的壓力,FDA 的處境其實相當艱難。 作為管制機構,首先要關注的應該是社會大眾的感知。研究顯示,一般民眾 對於新科技(在本文指新藥、新醫療器材,如生物晶片)本身的疑慮及不信任, 其實並不是最主要的,民眾所更不信任的可能是管制機構本身。而這就是 FDA 以及我國(及其他國家)的管制機關所面對的課題。 任何形式的上市審查程序,不論是效率取向或是安全取向,都是社會價值的 權衡。管制機構應該要是著建立自己的公信力,接受公共論壇的多元意見,不要 使社會大眾感覺到這個機構被利益團體所把持,才能夠建立初一套有說服力,而 能為普遍接受的上市前審查機制,而達到對新興科技之風險控管的社會目標。
第四章、歐盟對醫療器材的管制規範模式 第一節、前言 在全球經濟增長過程中,生物技術產業展示出高速、持續的發展態勢。全球 生物技術產業的研發力度加大,生物技術產業化能力加快。美國、歐盟和日本在 世界生物技術產業發展中發揮著主導性作用。其中,美國是現代生物技術的發源 地,目前美國生物技術產業不僅已經形成了相當的規模,而且發展勢頭強勁。歐 盟是居於世界生物技術第二位的集團,不過其目前的生物產業規模只相當於美國 的三分之一61。 雖然歐盟至今尚未公佈生物晶片相管管理法規,不過卻是第一個給予Roche
與Affymetrix 合作開發的 AmpliChip 產品 IVD 許可的單位62。 簡而言之,歐盟對於生技藥品管制規範議題有三:
(一) 在過去二、三十年來,歐盟不斷針對不同藥物法規進行整合,整合對 象除了歐洲各國之外,包括國際醫藥法規協會(International Conference for Harmonization, ICH)、世界衛生組織(World Health Organization, WHO),生 技藥品應如何規範,將尊重這兩單位要求,以達歐盟會員國之間規範的一 致性(to ensure consistency)63。 同時為統一國際標準,美國與歐盟並簽署相 互認可協定,詳後述之。
(二) 歐盟已於二○○三年底及二○○四年上半年公布相似生技醫藥的臨床試 驗準則,原則上針對不同的生技藥品,可以允許各別公司和歐洲藥品檢驗 局(European Medicines Agency, EMEA)討論如何進行臨床試驗64;
(三) 生技藥品依不同分類等級(classification)來決定臨床試驗的審核作業。 對於早期的干擾素及胰島素等生技藥品,臨床試驗程度可能不需要太複 雜,但未來針對基因治療的生技藥物,產品在上市前,可想而知臨床試驗 (clinical investigations)過程也會相當複雜65。 第二節、歐盟醫療器材分類及其管理模式 對於醫療器材上市前審查之規範制度,其挑戰在於如何建立一個有效的管制 架構以保障病人66。歐盟對於醫療器材的管制規範分類相似似於前述美國的分級 標準,依產品風險、保護層級的高低而區分。在進入市場前須符合一定的審查標 準要求67,如一定的檢驗測試(testing),並有 Notified Body (could be private or
61 http://www.blogchina.com/new/display/64331.html
62 EU approves Roche’s AmpliChip as in vitro device; competitors consider next moves,11/6/04, PGx
Reporter, www.pgxreporter.com
63 Council Directive 93/42/EEC, Art. 13. 64 http://www.emea.eu.int/
65 Council Directive 93/42/EEC, Art. 15, annex VIII. Special requirements for custom-made devices
are also prescribed in the Directive.
66 涉及管制規範理論,詳後述。
67 Anselmann, EC Legislation, Council Directive 84/529 on the approximation of the laws of the
public)介入審查68。 此外,歐盟對此管制規範架構之建立,尚須考量歐盟會員國間規範的一致性 69,以免造成不同規範適用後產生的貿易壁壘70,因此,對於醫療器材規範的相 互承認在歐盟會員國間亦具有一定重要性71。以下,將就歐盟對於醫療器材的分 類標準作一簡介。 基本上歐盟對於醫療器材的分類標準,亦是基於該器材的風險高低而為以下 的分類:
@Adopted from Nealda Leila M Yusof, Ph.D. Regulatory Scientist, Centre for Medical Device Regulation Health Sciences Authority
adopting CENELEC HD 395-1), The syringe is an example of the EU legal inconsistencies at the time work began on the medical device directives. In some Member States syringes were regulated as drugs, in another a medical device law governed them if sold as sterile, and in others they were subject to no regulations at all. Remarks by Norbert Anselmann, European Comm'n, The Forthcoming EC Legislation on Medical Devices: Provisional Results and Outlook 5-6, at the Reg. Aff. Professionals Soc'y Conference on International Harmonization of Medical Device Regulation (June 22-23, 1992), at 5-6.
68 Council Decision 90/683: "The essential objective of a conformity assessment procedure is to enable
the public authorities to ensure that products placed on the market conform to the requirements as expressed in the provisions of the directives, in particular with regard to the health and safety of users and consumers."
69 Council Directive 93/42/EEC, Art. 2.
70 E. Jongen, The Creation of an Internal Market For Industrial Goods in Europe Through Technical
Harmonization, Standardization, Certification And Mutual Recognition (Jan. 1991).
71 Statement by Jacques MacMillan, European Comm'n, at U.S.-EU negotiations on Mutual
Recognition Agreements (Apr. 1995).
Higher
Lower
Device
Class
I
IIa
IIb
III
General List B &
self-testing
歐盟對於醫療器材的現行規範有72:
(1) Active Implantable Medical Device Directive 90/385/EEC
(AIMMD)73 :主動植入式醫療器材指令。
(2) The Medical Device Directive 93/42/EEC (MDD)74 :一般醫療器材 指令。
(3) In Diagnostic Device Directive 98/79/EC (IVDD)75 :體外診斷器材 指令。
其中涉及歐盟醫療器材的主要規範又集中於MDD,其管制範圍涵蓋歐盟主
要的醫療器材規範;同時EU 亦根據 AIMMD、MDD、IVDD 訂定許多參考標準
76。
歐盟將醫療器材分為三大類:
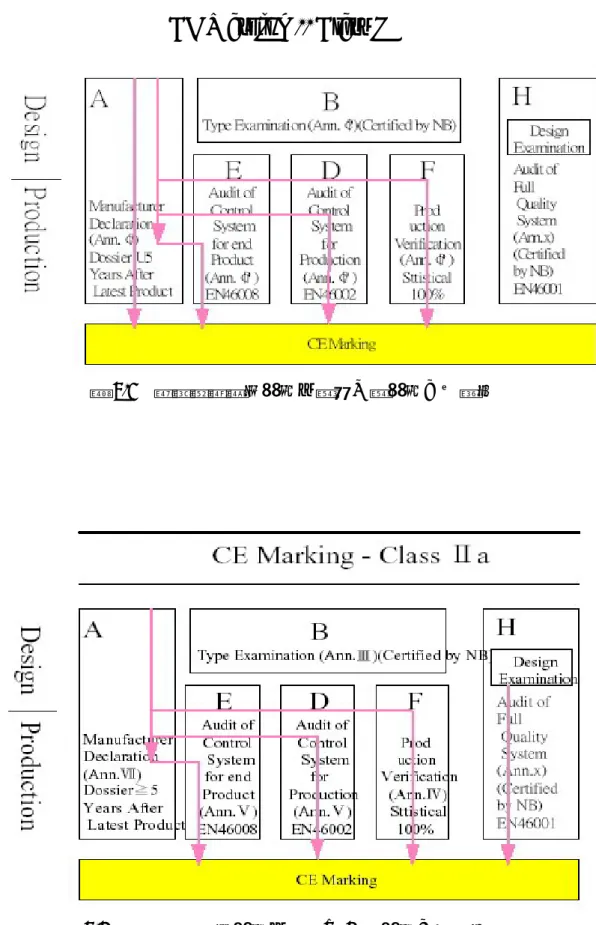
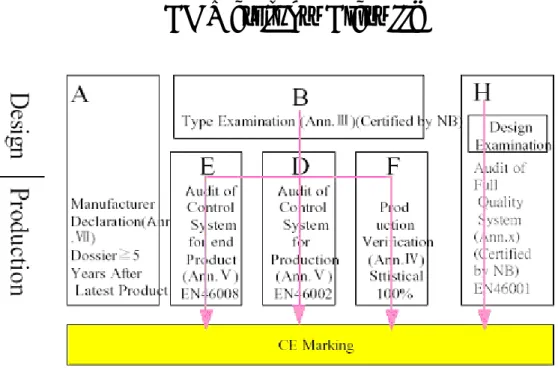
(1) 主動植入式醫療器材(active implantable medical device):經由手術 或醫療方法,全部或部份植入人體或插入並留置於人體孔道之主 動裝置,所謂主動裝置是指藉電力或其他能源而達成其功能者。 這類器材是歐盟最早公佈管理的醫療器材(90/385/EEC)。 (2) 一般醫療器材(medical device): 供人類使用於以下用途之儀器、 設備、用品、材料或其他除主動植入式醫療器材以外之物品:診 斷、預防、追蹤、治療或減輕疾病;診斷、追蹤、治療或修整傷 處或殘障部位;解剖或生理過程之檢查、置換或修正;生育控制。 但不可藉藥性、免疫力或新陳代謝的方法在人體內達到其主要目 的,但輔助前述功能之物品仍屬醫療器材。這項指令稱為Medical Device Directive(93/42/EEC),是最重要的醫療器材管理規定。 (3) 體外檢驗器材(in vitro diagnostic device):任何自成單位或由其他
物件組成之任何試劑、試劑品、校正器、套組、控制物質、儀器、 設備或系統形式之裝置,製造商設計用以檢查人體組織標本(含血 液、組織切片等)之裝置,或用於以上目的之容器亦屬之。體外檢 驗試劑的指令原本預定於1996 年公佈,但因各國尚未達成共識, 所以未能公佈,目前的指令編號是COM(95)130。 歐盟的醫療器材指令包含了絕大多數的醫療器材,以下簡單地說明其分 72 EU guidelines: http://www.mdc-ce.de/stand_02.htm 73 See http://www.mdss.com/AIMDD/aimdd_directive.htm 74 http://europa.eu.int/smartapi/cgi/sga_doc?smartapi!celexapi!prod!CELEXnumdoc&lg=en&numdo =31993L0042&model=guichett 75 http://www.mdc-ce.de/downloads/Ivd_d_en.pdf 76 http://www.mdc-ce.de/downloads/harm_st1.pdf