國 立 交 通 大 學
材料科學與工程研究所
博 士 論 文
含共軛吡啶質子受體懸掛基氫鍵側鏈高分子之超分子
組裝於有機光電材料之應用
Supramolecular Assembly of H-Bonded Side-Chain
Polymers Containing Conjugated Pyridyl H-Acceptor
Pendants for Organic Electro-Optical Materials
Applications
研 究 生 : 梁宗琦
指導教授 : 林宏洲 博士
中華民國九十八年七月
含共軛吡啶質子受體懸掛基氫鍵側鏈高分子之超分子
組裝於有機光電材料之應用
Supramolecular Assembly of H-Bonded Side-Chain
Polymers Containing Conjugated Pyridyl H-Acceptor
Pendants for Organic Electro-Optical Materials
Applications
研 究 生 : 梁宗琦 Student: Tzung-Chi Liang
指導教授 : 林宏洲 Advisor: Prof. Hong-Cheu Lin
國立交通大學
材料科學與工程學系
博士論文
A Thesis
Submitted to Department of Materials Science and Engineering
College of Engineering
National Chiao Tung University
In Partial Fulfillment of the Requirement
For the Degree of Doctor of Philosophy of Science
In Materials Science and Engineering
July 2009
Hsinchu, Taiwan
中華民國九十八年七月
Supramolecular Assembly of H-Bonded Side-Chain
Polymers Containing Conjugated Pyridyl H-Acceptor
Pendants for Organic Electro-Optical Materials
Applications
Student: Tzung-Chi Liang Advisor: Prof. Hong-Cheu Lin
Department of Materials Science and Engineering National Chiao Tung
University
Abstract
First, two H-bonded acceptor polymers and were successfully prepared by polymerization of fluorescent pyridyl monomers, which were synthesized via Sonogashira coupling and Wittig-Horner reactions. Supramolecular side-chain and cross-linking polymers (i.e., H-bonded polymer complexes) were obtained by complexation of light-emitting H-acceptor polymers with various proton donor (H-donor) acids. Mesogenic and photoluminescent (PL) properties of light-emitting H-acceptor polymers can be adjusted not only by the central structures of the conjugated pyridyl cores but also by their surrounding non-fluorescent H-donor acids. Redder shifts of PL emissions in H-bonded polymer complexes occurred when the light-emitting H-acceptor polymers were complexed with H-donors having smaller pKa values.
Second, a series of PL and liquid crystalline (LC) self-H-bonded side-chain copolymers consisting of pyridyl H-acceptors and isomeric acid H-donors (i.e., para-,
meta-, and ortho-benzoic acids) were synthesized. Supramolecular H-bonded
complexes were also obtained by mixing the photoluminescent H-acceptor homopolymer with isomeric H-donor homopolymers. The formation of H-bonds was confirmed by FTIR, DSC, and XRD measurements. Moreover, PL and LC properties of the H-bonded copolymers and complexes were affected not only by the H-bonding effect of the supramolecular structures but also by the acid-substituted positions of isomeric H-donors. In combination with different functionalized gold nanoparticles (which bear acid or acid-free surfactants), the emission intensities of nanocomposites containing self-H-bonded copolymer (bearing both H-acceptor and H-donor moieties) and non-self-H-bonded copolymer (bearing acid-protected moieties), respectively, were quenched to different extents by varying the concentration of gold nanoparticles. The copolymeric H-acceptors and surface-modified gold nanoparticles demonstrated diverse morphological and PL quenching effects on the supramolecular architectures of nanocomposites, which result from competition between the H-donors from the acid pendants on copolymers and the acid surfactants on gold nanoparticles.
Third, this approach is exploring hydrogen-bonded (H-bonded) suparmolecular assembled behavior via both TEM and fluorescence quenching studies through organic solvent dissolving and evaporating processes. Different lateral methyl- and methoxy-substituted groups with pyridyl terminus of fluorescent side-chain polymers, it performed that the H-bonded interactions affect the fluorescence quenching effectively upon the addition of surface-modified gold nanoparticles bearing acid and acid-free surfactants in the fluorescence titrations experiments. We demonstrated that homopolymer PBOT1 has the highest Ksv constant in the compared fluorescent
side-chain polymers. In addition, we established the exponential equation to predict Stern-Volmer constant in various pyridyl units of polymers from the experimental information. TEM studies displayed that interesting H-bonded suparmolecular behavior of addition into the carboxylic acid units of surface-modified gold nanoparticles. It is clearly observed that homogeneously gold nanoparticles distributions are on the fluorescent side-chain polymers. Thus, the TEM morphologies of H-bonded architectures demonstrate the versatility of the self-assembled processes in supramolecular nanocomposites of H-acceptor polymers and surface-modified gold nanoparticles.
Finally, novel supramolecular side-chain polymers were constructed by complexation of H-acceptor polymers, i.e., side-chain conjugated polymers containing pyridyl pendants, with low-band-gap H-donor dyes (bearing terminal cyanoacrylic acids) in a proper molar ratio. H-bonded polymer complexes exhibited broad absorption peaks in the range of 440-462 nm with optical band-gaps of 2.11-2.25 eV. The PSC device containing H-bonded polymer complex blended with PCBM (1:1 w/w) gave the best preliminary result with an overall power conversion efficiency (PCE) of 0.50%, a short-circuit current of 3.17 mA/cm2, an open-circuit voltage of 0.47 V, and a fill factor of 34%.
含共軛吡啶質子受體懸掛基氫鍵側鏈高分子之超分子
組裝於有機光電材料之應用
學 生:梁宗琦 指 導 教 授:林宏洲 博士
國立交通大學材料科學與工程研究所
博士班
摘 要
本論文研究方向為探討一系列包含共軛吡啶質子受體氫鍵側鏈高分子之合 成,利用超分子組裝的特性,在三大方向在有機光電材料為研究主軸。第一個部份,成功的聚合出以Sonogashira coupling 和 Wittig-Horner 反應合 成出的兩種發光吡啶單體,進而將發光高分子與不同的質子予體錯合形成超分子 側鏈與交聯高分子錯合物,而此氫鍵高分子錯合物之液晶及發光性質可以被吡啶 共軛段及不同質子予體所調整。然而,含有較小 pKa 值的質子予體可以讓氫鍵 高分子錯合物得到紅位移的發光。 第二個部份,成功的將吡啶單體與苯酸同分異構物(即,para-、meta-、和 ortho-)共聚出液晶及發光氫鍵高分子。藉由 FTIR、DSC、XRD 鑑定其氫鍵的 形成。此外,此氫鍵高分子與其錯合物之液晶及發光性質不但與氫鍵效應有關, 也被共聚不同位置的苯酸同分異構物所影響。接著,結合不同改質的奈米金球(酸 或無酸改質),隨著加入的奈米金球濃度變化,自身氫鍵(含有吡啶與酸)與無
同程度的驟息。進一步,利用TEM 觀察自身氫鍵與無自身氫鍵高分子混合改質 奈米金球,而其微觀現象和PL 驟息程度相互呼應,解釋了自身氫鍵高分子的酸 與改質金球的酸相互競爭所造成。 第三個部份,描述兩種不同吡啶基比例系列的發光高分子超分子奈米複合材 料,即,側面甲基高分子和側面甲氧基高分子,進而與質子予體的奈米粒子發展 出可區分的聚集現象在TEM 影像裡。質子受體高分子不但顯示了高程度聚集, 而且可以使質子予體的奈米粒子均勻分散。並且,酸表面改質的奈米金球與側面 甲氧基的高分子之間的氫鍵作用力,比起無酸表面改質的奈米金球更有效的影響 了發光驟息的現象。此外,與側面甲基高分子比起來,側面甲氧基高分子更容易 的捕捉酸表面改質的奈米金球在超分子組裝的奈米複合材料裡。吡啶基質子受體 高分子與質子予體的奈米粒子的氫鍵作用力可以解釋相似的發光驟息效應。我們 以 實 驗 數 據 建 立 了 在 不 同 的 吡 啶 基 高 分 子 的 指 數 方 程 式 , 進 而 可 以 預 測 Stern-Volmer 常數。發展了多種奈米複合材料包含兩種發光高分子(甲氧基和甲基 高分子)與表面改質的奈米金球(酸和無酸改質),進而展示出差異性的聚集現象在 TEM 影像。未來我們可以基於奈米金球發光驟息的回復性,在化學感測與生醫 感測研究領域作進一步的發展。 最後一個部份,氫鍵側鏈高分子與染料型質子予體結合(含末端氰酸基)。 氫鍵高分子錯合物得到了從440 至 462 nm 寬廣的吸收能帶,且其光學能隙大約 在2.11-2.25 eV 之間。結果發現,在 AM 1.5G、100 mW/cm2的模擬太陽光下, 將合成出之氫鍵高分子錯合物與 PCBM([6,6]-phenyl C61-butyric acid methyl
ester)混合為主動層材料,成功地以 1:1 w/w 的混合比例得到一具有短路電流 3.17 mA/cm2、開路電壓0.47 V、填充因子 34%及最高之光電轉換效率 0.50%之有機 太陽能電池。
ACKNOWLEDGEMENTS
本論文首先感謝林宏洲老師這些年來對我的照顧及鼓勵,老師對
於研究上的辛苦用心及待人處世的教導,使我一路成長,如今順利完
成博士學業,老師指導的恩惠,學生將永記於心。感謝韋光華老師、
黃華宗老師、林建村老師、賴榮豊老師、韓建中老師於百忙之中審核
論文並給予寶貴的建議及指正。在研究的過程中,也非常感謝朱治偉
老師在實驗元件上大力的支持,使的本論文能更趨完善。
博士班近五年的時光使我獲益良多,很幸運也很快樂地在這實驗
室度過這些日子,在此特別感謝實驗室的學長們在實驗上的教導與幫
助,並感謝實驗室的衆多的同學及學弟妹在實驗上的協助,使我的實
驗得以順利完成,還有陳智實驗室、呂志鵬實驗室以及許許多多材料
所的學長、同學、學弟妹們在這些日子的陪伴,使我的交大生活更增
添的許多歡樂。
最後要特別由衷地感謝一直栽培我的父母親、姐姐、弟弟,謝謝
你們一路上的支持與鼓勵,讓我能在無後顧之憂下求學並完成博士學
位。
Table of Contents
Abstract... I Acknowledgements... VII Table of Contents ...VIII Table Lists... XII Figure Lists ...XIIII
Chapter 1 Introduction...1
1.1 Introduction of Supramolecular Polymers ...1
1.1.1 Hydrogen Bonding Enforced by Liquid Crystallinity ...3
1.2 Chemical Sensors Based on Amplifying Fluorescent Conjugated Polymers ..7
1.3 Assembly of Nanoparticles Based on Supramolecular Chemistry ...11
1.3.1 Miscellaneous Recognition and Sensors...13
1.4 Supramolecular in Organic Devices ...18
Chapter 2 Study of Supramolecular Side-Chain and Cross-Linking Polymers by Complexation of Various H-Donor Acids with H-Acceptor Copolymers Containing Pendent Carbazole and Fluorescent Pyridyl Units...22
2.1 Introduction...23
2.2 Experimental...30
2.2.1 Materials ...30
2.2.2 Preparation of Supramolecular Complexes ...38
2.2.3 Measurements and Characterization ...39
2.3 Results and Discussion ...40
2.3.1 Synthesis and Characterization of Polymers...41
2.3.2 FT-IR Spectroscopy of H-Bonded Polymer Complexes ...42
2.3.3 Thermal Behavior ...44
2.3.5 Optical Properties...59
2.4 Conclusions...66
Chapter 3 Supramolecular Assembly of H-Bonded Copolymers/Complexes/Nanocomposites and Fluorescence Quenching Effects of Surface-Modified Gold Nanoparticles on Fluorescent Copolymers Containing Pyridyl H-Acceptors and Acid H-Donors ...68
3.1 Introduction...69
3.2 Experimental Section...74
3.2.1 Materials ...74
3.2.2 Preparation of H-Bonded Complexes (PBT1/P7-P9) ...78
3.2.3 Preparation of Nanocomposites Consisting of Gold Nanoparticles and Polymers ...78
3.2.4 Characterizations...79
3.3 Results and Discussion ...81
3.3.1 Synthesis and Characterization...81
3.3.2 FT-IR Spectroscopic Studies...82
3.3.3 Phase Characterization...85
3.3.4 Optical Properties...90
3.3.5 Fluorescence Quenching Effects of Copolymers by Surface-Modified Gold Nanoparticles ...94
3.3.6 TEM Analyses...100
3.4 Conclusions...103
Chapter 4 Supramolecular Fluorescence Quenching Effects of H-Donor Surface-Modified Gold Nanoparticles on Fluorescent H-Acceptor Polymers/Copolymers Containing Lateral Methyl- and Methoxy-Substituted Groups...105
4.2 Experimental Section...110
4.2.1 Materials ...110
4.2.2 Synthesis of Surface-Functionalized Gold Nanoparticles AuSC10 and AuSCOOH ... 110
4.2.3 Preparation of Nanocomposites Consisting of Gold Nanoparticles and Polymers ...112
4.2.4 Measurements and Characterization ...113
4.3 Results and Discussion ...113
4.3.1 FT-IR Spectroscopic Studies ...113
4.3.2 Fluorescence Quenching Effects of Copolymers by Surface-Modified Gold Nanoparticles ...115
4.3.3 TEM Analyses ...125
4.4 Conclusions...128
Chapter 5 Supramolecular Assembly of H-Bonded Side-Chain Polymers Containing Conjugated Pyridyl H-Acceptor Pendants and Various Low-Band-Gap H-Donor Dyes Bearing Cyanoacrylic Acid Groups for Organic Solar Cell Applications ...130
5.1 Introduction...131
5.2 Experimental...137
5.2.1 Materials ...137
5.2.2 Preparation of Supramolecular Polymer Complexes...137
5.2.3 Measurements and Characterization ...138
5.2.4 Device Fabrication and Characterization of Polymer Solar Cells (PSCs) ...140
5.3 Results and Discussion ...141
5.3.2 Phase Behavior...144
5.3.3 Optical Properties...150
5.3.4 Electrochemical Properties ...155
5.3.5 Photovoltaic Cell Properties ...160
5.4 Conclusions...168
Chapter 6 Conclusion ...170
Table Lists
Table 2.1 Molecular Weights and Thermal Properties of H-Acceptor Polymers ...51 Table 2.2 Thermal Properties of H-Bonded Polymer Complexes a,b...52 Table 2.3 The d-spacing and Tilt Angle Values of the Sc Phase in H-Bonded Polymer
Complexes...57
Table 2.4 Absorption and Photoluminescence Spectral Data of H-Acceptor Polymers
...61
Table 2.5 Photophysical Properties of H-Acceptor Polymers and H-Bonded Polymer
Complexes in Solid Films...65
Table 3.1 Characterization of Copolymers P1-P6 and H-Donor Homopolymers P7-P9
...77
Table 3.2 Thermal Properties of Polymers and H-Bonded Homopolymer Complexes
...88
Table 3.3 Absorption and PL Emission Spectral Data of Polymers and H-Bonded
Homopolymer Complexes in THF Solutions and Solid Films ...92
Table 3.4 Stern-Volmer Constants (Ksv) of Copolymers P1 and P4 Titrated with
Different Nanoparticle Quenchers (AuSCOOH and AuSC10) in THF Solutions ...98
Table 4.1 Stern-Volmer Constant (Ksv) for Polymers PBT1-PBOT3 Titrated with
Different Nanoparticle Quenchers (AuSCOOH and AuSC10) in THF Solutions ...121
Table 5.1 Thermal Properties of H-Acceptor Polymers (P1-P2), H-Donor Dyes
(S1-S4), Acid-Protected Dye S1P, H-Bonded Polymer Complexes (P1/S1-P1/S4 and
P2/S1-P2/S4), and Physical Blend P1/S1P ...148 Table 5.2 Absorption and Photoluminescence Spectral Data of H-Acceptor Polymers
(P1-P2), H-Donor Dyes (S1-S4), Acid-Protected Dye S1P, H-Bonded Polymer Complexes (P1/S1-P1/S4 and P2/S1-P2/S4), and Physical Blend P1/S1P ...152
Table 5.3 Electrochemical Potentials and Energy Levels of H-Acceptor Polymers
(P1-P2), H-Donor Dyes (S1-S4), and H-Bonded Polymer Complexes (P1/S1-P1/S4 and P2/S1-P2/S4)...158
Table 5.4 Photovoltaic Properties of PSC Devices Containing an Active Layer of
H-Bonded Polymer Complexes:PCBM = 1:1 (w/w) with a Device Configuration of ITO/PEDOT:PSS/H-Bonded Polymer Complexes:PCBM/Ca/Ala...165
Table 5.5 Photovoltaic Properties of PSC Devices Containing an Active Layer of
Dyes S1-S4:PCBM/Ca/Ala...165
Table 5.6 Photovoltaic Properties of PSC Devices Containing an Active Layer of
Mixture:PCBM = 1:1 (w/w) with a Device Configuration of ITO/PEDOT:PSS/ Mixture:PCBM/Ca/Ala...166
Table 5.7 Photovoltaic Parametersfor Bulk-Heterojunction PSC Devices Containing
Figure Lists
Figure 1.1 Molecular structures consisting of non-covalent bonds: such as metal
bonds, ionic forces, and H-bonds...3
Figure 1.2 Liquid crystalline supramolecular polymers developed by Lehn, based on
triple hydrogen bonds: from chiral, tartaric acid based monomers c and from rigid monomers d. ...6
Figure 1.3 Demonstration of amplified quenching in a CP. The determination of the
Stern-Volmer quenching reveals the constant for binding of paraquat to a single cyclophane receptor (top). In the polymer the larger (amplified) quenching constant reflects the fact that the quencher can be bound to any of the repeating units visited by the exciton (bottom)...10
Figure 1.4 (a) Scheme 1, a chemosensor is composed of two functional elements, a
receptor and a reporter group, which need not be separate in identity, (b) this account describes an approach to enhancing the sensitivity of chemosensors in effect by “wiring chemosensory molecules in series” (Scheme 2), (c) for the sake of clarity, some representative conducting polymers are shown in Scheme 3...11
Figure 1.5 Programmed pseudorotaxane assembly at the surface of AuNPs by
heterosupramolecular chemistry. ...13
Figure 1.6 Self-assembly based on selective control of non-covalent interactions
provides a powerful tool for the creation of structured systems at a molecular level. This paper present a polymer-mediated “bricks and mortar” strategy for the ordering of nanoparticles into structured assemblies. ...14
Figure 1.7 This paper has a system where recognition element-functionalized diblock
copolymers are used to self-assemblye complementary nanoparticles. The size of the aggregate both in solution and in thin films is controlled through block length. ...15
Figure 1.8 Amine-functionalized polymers have been used to simultaneously
assemble carboxylic acid functionalized gold and silica nanoparticles into extended aggregates, and nanoparticle segregation within the aggregate regulated through order of component addition. ...15
Figure 1.9 (A) Fluorescence resonance energy transfer (FRET) within a CdSe/ZnS
QD/Texas-Red-functionalized duplex DNA. (B) FRET process in the DNA 3/2 duplex structure. (C) FRET process in the DNA duplex structure consisting of the 1-functionalized Au NP and 2...17
Figure 1.10 Schematic presentation of reversion process from oxidized to reduced
glutathione based on the modulation in photoluminescent quenching efficiency between chromophore and AuNPs. (Bottom) Chemical structure of GSH and GSSG. ...17
Figure 1.11 Superstructure of self-assembly of [60]fullerene derivative 1 with
perylene bisimide 5 by H-bonding...20
Figure 1.12 The incorporation of a ruthenium complex into the donor-acceptor
conjugated polymers has the potential to facilitate the charge carrier generation...20
Figure 1.13 Pyrrolidinofullerenes bearing chelating pyridyl groups (PyFs) on
vacuum-evaporated films of zinc phthalocyanine (ZnPc) in donor-acceptor bilayer heterojunctions formed by deposition of solution-processed. It is shown that coordination complexes are formed at the interface between these donor and acceptor components; such association facilitates photoinduced charge separation and results in improved performance of the photovoltaic devices...21
Figure 2.1 Schematic illustration of singly/doubly H-bonded processes for H-bonded
side-chain/cross-linking polymers. ...27
Figure 2.2 Mono-acid (singly H-bonded) and bis-acid (doubly H-bonded) donors
used in supramolecular side-chain/cross-linking polymers, respectively...27
Figure 2.3 Infrared spectra for PBOT2 (18), THDA (22), and H-bonded polymer
complex PBOT2/THDA (18/22) at room temperature. ...43
Figure 2.4 DSC heating curves (second scans) of H-acceptor polymers 14-19. ...51 Figure 2.5 Phase diagrams of mesophases in (a) H-bonded polymer complexes of PBT and (b) H-bonded polymer complexes of PBOT upon heating...53 Figure 2.6 Liquid crystalline textures of H-bonded polymer complexes observed by
POM (a) the Sc phase in PBT1/ONA (14/21) at 160 °C (cooling) (b) the Sc phase in
PBT2/ONA (15/21) at 110 °C (cooling) (c) the nematic phase in PBT2/THDA (15/22) at 180 °C (cooling) (d) the Sc phase in PBOT1/THDA (17/22) at 160 °C
(cooling)...54
Figure 2.7 X-ray diffraction patterns of H-bonded polymer complexes (a) the Sc
phase in PBOT1/OBA (17/20) at 115 °C, PBOT1/ONA (17/21) at 120 °C, and
PBOT1/THDA (17/22) at 160 °C; (b) the nematic phase in PBT2/THDA (15/22) and PBOT2/THDA (18/22) at 130 °C. ...58 Figure 2.8 X-ray diffraction patterns for H-bonded polymer complex PBT1/ONA
Figure 2.9 (a) Absorption spectra and (b) PL spectra (excited at the maximum
absorption wavelengths) in THF solutions (c) normalized PL spectra (excited at the maximum absorption wavelengths) of H-acceptor polymers 14-19 in solid films...62
Figure 2.10 Normalized PL spectra (excited at the maximum absorption wavelengths)
of (a) H-acceptor polymer PBT2 (15) and its H-bonded polymer complexes
PBT2/OBA (15/20), PBT2/ONA (15/21), and PBT2/THDA (15/22) in solid films;
(b) H-acceptor polymer PBOT2 (18) and its H-bonded polymer complexes
PBOT2/OBA (18/20), PBOT2/ONA (18/21), and PBOT2/THDA (18/22) in solid
films. ...66
Figure 3.1 Schematic illustration of acid-functionalized gold nanoparticles
(AuSCOOH) blended with self-H-bonded copolymer P1 and non-self-H-bonded copolymer P4. ...73
Figure 3.2 Infrared spectra of (a) H-acceptor homopolymer PBT1, H-donor
homopolymer P7, and H-bonded homopolymer complexes PBT1/P7; (b) copolymers
P1 and P4 in solid films...84 Figure 3.3 (a) X-ray diffraction (XRD) patterns of copolymer P1 and H-bonded
complex PBT1/P7 in the Sc phase at 130°C. (b) XRD intensity against angle profiles obtained in the nematic phase of copolymer P2 at 70°C...89
Figure 3.4 (a) PL spectra of copolymers P1-P6 in THF solutions (b) normalized PL
spectra of copolymers P1-P6 and H-bonded homopolymer complexes PBT1/P7,
PBT1/P8, and PBT1/P9 in solid films. ...93 Figure 3.5 Fluorescence quenching spectra of copolymers P1 and P4 titrated by
surface-functionalized nanoparticles (AuSCOOH and AuSC10) in THF solutions: (a)
P1 and (b) P4 by varying the concentration of acid-donor-modified gold nanoparticles
(AuSCOOH); (c) P1 and (d) P4 by varying the concentration of non-acid-modified gold nanoparticles (AuSC10). ...99
Figure 3.6 Corresponding Stern-Volmer plots of copolymers P1 and P4 for increasing
concentrations of acid-modified gold nanoparticles (AuSCOOH) and non-acid-modified gold nanoparticles (AuSC10) in THF solutions. ...99
Figure 3.7 TEM images of acid-functionalized gold nanoparticles (AuSCOOH)
blended with (a) self-H-bonded copolymer P1, (b) acid-protected copolymer P4, and (c) alkyl-functionalized gold nanoparticles (AuSC10) blended with self-H-bonded copolymer P1. ...102
methoxy-substituted groups of H-bonded polymers blended with AuSCOOH. ...110
Figure 4.2 Molecular structures of different lateral methyl- and methoxy-substituted
groups of fluorescent side-chain polymers PBT1-PBT3 and PBOT1-PBOT3. ...112
Figure 4.3 Infrared spectra for AuSCOOH, PBOT1, and H-bonded nanocomposite PBOT1/AuSCOOH at room temperature... 115 Figure 4.4 Fluorescence quenching spectra of polymers in THF solutions (a) PBOT1
and (b) PBT1 by varying the concentration of acid-donor-modified gold nanoparticles
AuSCOOH (c) PBOT1 and (d) PBT1 by varying the concentration of
non-acid-modified gold nanoparticles AuSC10. ...122
Figure 4.5 Corresponding Stern-Volmer plots of polymers PBT1 (■), PBT2 (●), PBT3 ( ▲ ), PBOT1 ( ▼ ), PBOT2 ( ◆ ), and PBOT3 ( ★ ) by increasing the
concentration of acid-modified gold nanoparticles AuSCOOH and polymers PBT1 (□), PBT2 (○), PBT3 (△), PBOT1 (▽), PBOT2 (◇), and PBOT3 (☆) by increasing the concentration of non-acid-modified gold nanoparticles AuSC10 in THF solutions, where polymers PBT1-PBOT1 by increasing the concentration of non-acid-modified gold nanoparticles AuSC10 are replotted from inset of Figure..123
Figure 4.6 Schematic curves showing the dependence of Stern-Volmer constant (Ksv)
on pyridyl units’ X mol% of polymers, which follow the experimental equation of Ksv = A[1-exp(-BX)], where A and B are constants for the exponential curve fittings. ..124
Figure 4.7 UV-visible spectra of polymers (a) PBOT1 titrated by varying the
concentration of acid-donor-modified gold nanoparticles (AuSCOOH) and (b) PBT1 by varying the concentration of non-acid-modified gold nanoparticles (AuSC10) in THF solutions...125
Figure 4.8 TEM images of (a) acid-modified gold nanoparticles AuSCOOH and
blended with polymers (b) PBOT1, (c) PBT1, and (d) alkyl-functionalized gold nanoparticles (AuSC10) blended with polymer PBT1. ...128
Figure 5.1 Schematic illustration of complexation processes for H-bonded side-chain
polymers...136
Figure 5.2 H-acceptor polymers (P1 and P2) and H-donor dyes (S1-S4) used in the
H-bonded polymer complexes (P1/S1-P1/S4 and P2/S1-P2/S4)...136
Figure 5.3 Acid-protected dye S1P without carboxylic end functionality...137 Figure 5.4 FTIR spectra of (a) H-acceptor polymer P2, H-donor dye S1, and
acid-protected dye S1P, H-bonded polymer complex P1/S1, and physical blend
P1/S1P. ...143 Figure 5.5 (a) Optical texture of the nematic phase in H-bonded polymer complex P2/S4 observed by POM at 130 °C (cooling) and (b) XRD intensity against angle
profiles obtained from H-bonded polymer complexes P1/S1 and P2/S4 at 130 °C (in the nematic phase)...149
Figure 5.6 UV-visible absorption spectra of H-acceptor polymers P1-P2 and H-donor
dyes S1-S4 (a) in THF solutions and (b) in solid films. ...153
Figure 5.7 UV-visible absorption spectra of (a) H-bonded polymer complexes P1/S1-P1/S4 and (b) H-bonded polymer complexes P2/S1-P2/S4 in solid films...154 Figure 5.8 Cyclic voltammograms of H-bonded polymer complexes (a) P1/S1-P1/S4
and (b) P2/S1-P2/S4. ...159
Figure 5.9 Energy band diagram with HOMO/LUMO levels of H-bonded polymer
complexes P1/S1-P1/S4 (as electron donors) and PCBM (as an electron acceptor) in relation to the work functions of ITO, PEDOT:PSS, and Al (HOMO value of PCBM was obtained from Reference 112). ...160
Figure 5.10 I-V curves (under simulated AM 1.5 solar irradiation) dependencies of
PSC devices with an active layer of blended (a) H-bonded polymer complexes
P1/S1-P1/S4:PCBM (1:1 w/w) and (b) H-bonded polymer complexes P2/S1-P2/S4:PCBM (1:1 w/w)...167 Figure 5.11 I-V curves of PSC devices containing an active layer of H-bonded
polymer complex P1/S3:PCBM (w/w) with different weight ratios under simulated AM 1.5 solar irradiation...168
Chapter 1
Introduction
1.1 Introduction of Supramolecular Polymers
With the introduction of supramolecular polymers, which are polymers based on monomeric units held together with directional and reversible secondary interactions, the playground for polymer scientists has broadened and is not restricted to macromolecular species, in which the repetition of monomeric units is mainly governed by covalent bonding. The importance of supramolecular interactions within polymer science is beyond discussion and dates back to the first synthesis of synthetic polymers; the materials properties of, e.g., nylons, are mainly the result of cooperative hydrogen bonding. More recently, many exciting examples of programmed structure formation of polymeric architectures based on the combination of a variety of secondary supramolecular interactions have been disclosed.
When the covalent bonds that hold together the monomeric units in a macromolecule are replaced by highly directional noncovalent interactions (Figure 1.1), supramolecular polymers are obtained. In recent years, a large number of concepts have been disclosed that make use of these noncovalent interactions. Although most of the structures disclosed keep their polymeric properties in solution,
it was only after the careful design of multiple-hydrogen-bonded supramolecular polymers that systems were obtained that show true polymer materials properties, both in solution and in the solid state. Polymers based on this concept hold promise as a unique class of novel materials because they combine many of the attractive features of conventional polymers with properties that result from the reversibility of the bonds between monomeric units. Architectural and dynamic parameters that determine polymer properties, such as degree of polymerization, lifetime of the chain, and its conformation, are a function of the strength of the noncovalent interaction, which can reversibly be adjusted. This results in materials that are able to respond to external stimuli in a way that is not possible for traditional macromolecules. These aspects of supramolecular polymers have led to a recent surge in attention for this promising class of compounds1-3 and have stimulated us to bring together materials
science and supramolecular chemistry.4 On the other hand, it is obvious that a large
number of important properties of polymers require the covalent and irreversible bonding of the repeating units in the main chain. For applications in which all of these properties are important, supramolecular polymers are not the perfect choice. However, the opportunity to combine macromolecules and concepts derived from supramolecular polymers also has an enormous potential to alter the properties of polymers in a controlled way.
Figure 1.1 Molecular structures consisting of non-covalent bonds: such as metal
bonds, ionic forces, and H-bonds.
1.1.1 Hydrogen Bonding Enforced by Liquid Crystallinity
The first hydrogen-bonded supramolecular polymers all showed liquid crystallinity, although the separate components making up these polymers displayed a narrow liquid crystalline regime or no liquid crystallinity at all. The liquid crystalline phase in the supramolecular polymer is stabilized by the increased aspect ratio of aggregates compared to the constituent molecules. There is a strong cooperativity between association and the induction of the liquid crystalline phase, because anisotropy in the liquid crystal strongly enhances the degree of polymerization of the hydrogen-bonded molecules.5 Liquid crystalline supramolecular polymers are unique
in the respect that they combine the potential to exhibit the electrooptical properties associated with lowmolar-mass liquid crystals with the benefit of the good mechanical properties of conventional polymers.6 Odijk,7,8 van der Schoot,9 and Ciferri10
supramolecular liquid crystals.
The group of Lehn is recognized to be the first to develop a supramolecular main-chain polymer. By triple hydrogen bonding between difunctional diaminopyridines b and difunctional uracil c derivatives (Figure 1.2) supramolecular polymer chains were formed c.11,12 The 1:1 mixture of a and b exhibits liquid crystallinity over a broad temperature window, whereas, in contrast, the pure compounds are solids which melt in an isotropic liquid without displaying a liquid crystalline phase. Because of the chirality of the tartaric acid spacer used, the fibers observed by electron microscopy showed biased helicity.13 Lehn and co-workers
expanded the scope of supramolecular polymers by the development of rigid rod polymers (d, Figure 1.2).14,15 In these polymers, a rigid 9,10-dialkoxyanthracene core connects the hydrogenbonded groups via an imide group. Because of the increased molecular rigidity, the system is not thermotropic liquid crystalline, but a lyotropic liquid crystalline phase is observed in apolar solvents, that is birefringent and highly viscous.
A number of supramolecular liquid crystalline polymers based on a single hydrogen bond have been reported. Incorporation of single hydrogen-bonding units is synthetically more straightforward than those with triple hydrogen bonds, and, particularly, the single hydrogen bond between a pyridyl unit and carboxylic acids has
been utilized frequently in supramolecular liquid crystalline polymers (LCP’s).16-20
The complexation between a pyridyl unit and a carboxylic acid is stronger than carboxylic acid dimerization; a Ka value of approximately 500 M-1 was estimated for
the pyridyl/carboxylic acid complex.17 Kato and Fréchet have described a variety of
self-assembled side-chain liquid crystalline polymers (SLCPs), with various backbones.18,19 Polyacrylates and polysiloxanes functionalized with pendant benzoic
acids display stable mesophases upon selfassembly with stilbazoles. The reverse principle has been employed for the formation of supramolecular liquid crystalline polyurethanes.20 Furthermore, the stability of the induced mesophase has been
enlarged by employing the double hydrogen bond between benzoic acids in the polymer main-chain and 2-(acylamino) pyridines.21,22
Figure 1.2 Liquid crystalline supramolecular polymers developed by Lehn, based on
triple hydrogen bonds: from chiral, tartaric acid based monomers c and from rigid monomers d.
Utilization of the single hydrogen bond between pyridine and benzoic acids in SLCP’s has been a source of inspiration for other groups in the development of main-chain supramolecular polymers based on diacids and dipyridines.23-26
Supramolecular rodcoil polymers have been developed by assembly of 4,4’-bipyridines and telechelic polypropylene oxide with benzoic acid end-groups, which show highly ordered liquid crystalline phases.27 The use of tartaric acid
derivatives in combination with bipyridine units resulted in the formation of hydrogen-bonded, chiral main-chain LCP’s, as has been shown by circular dichroism measurements, optical microscopy, and X-ray data.28,29
1.2 Chemical Sensors Based on Amplifying Fluorescent Conjugated
Polymers
The field of chemical sensing is becoming ever more dependent upon novel materials. Polymers, crystals, glasses, particles, and nanostructures have made a profound impact and have endowed modern sensory systems with superior performance. Electronic polymers have emerged as one of the most important classes of transduction materials; they readily transform a chemical signal into an easily measured electrical or optical event. The dominant attribute that has driven interest in fluorescent conjugated polymers (CPs) sensory materials is their ability to produce signal gain in response to interactions with analytes. This has led to them being referred to as amplifying fluorescent polymers (AFPs), while some researchers have referred to this gain in terms of superquenching. In analogy to microelectronic devices, the increased sensitivity (amplification) is derived from the ability of a conjugated polymer to serve as a highly efficient transport medium. However, unlike a silicon circuit, which transports electrons or holes, AFPs transport electronic excited states. The geometric relaxation of molecular structure around an excited state gives it a finite size. As a result, it is typical to refer to these excited states as quasiparticles called excitons. Excitons in AFPs are highly mobile and can diffuse throughout an isolated polymer chain or within an AFP solid by mechanisms that involve both
through space dipolar couplings and strong mixing of electronic states.
Although more recently Swager and co-workers used exciton mobility within conjugated polymers to develop other mechanisms for amplified fluorescent detection, this study serves as a general introduction to the prototypical mechanism of chemical sensing by amplified fluorescence quenching with conjugated polymers.30
Amplification in a poly(phenylene ethynylene) (PPE) possessing well-defined cyclophane receptors integrated directly into the polymer backbone, effectively connecting the receptors in series. Cyclophane receptors were chosen because they form complexes with paraquat, a powerful electron acceptor and well-known electron-transfer quenching agent. To determine the ability of exciton transport to amplify binding events, a model compound containing a single receptor with the same local environment was synthesized. The large binding constant of the receptors resulted in paraquat quenching processes that were static in nature. In other words, the quencher is bound to the receptor and, once generated, the excited state is immediately and quantitatively quenched. Quenching due to diffusion of the quencher to an excited state was insignificant in this case. The quenching processes in AFPs can be analyzed by Stern-Volmer relationships (eqs 1 and 2),
F0/F = (1 + KSV[Q]) (1)
where F is the fluorescence intensity as a function of quencher concentration [Q], τ0 is
the lifetime without added quencher, and τ is the lifetime at [Q]. The slope of the plots gives the Stern-Volmer quenching constant (KSV) or the diffusional quenching
constant (kq).
A linear relationship of [Q] with F0/F implies a purely static (bound to the
polymer) or dynamic (diffusion limited) quenching. Moderate to large binding constants give rise to Stern-Volmer quenching constants (KSV) that exceed the rate
achievable at the diffusion limit, and hence, static quenching can be inferred. Another method to determine if quenching in an AFP is static or dynamic is to determine the dependence of the lifetime on the quencher concentration (eq 2).
The apparent binding constant of the receptor-containing AFP measured by the Stern-Volmer method is the individual repeating unit binding constant multiplied by the amplification factor. Comparison of the apparent binding constant (KSV) of
receptor PPEs revealed a linear increase with molecular weight up to a critical molecular weight of ~ 1.3 x 102 phenylene ethynylene repeating units (Figure 1.3).31
Higher chain lengths did not increase the apparent binding constant. These results reveal that the exciton was not able to visit the entire length of the higher molecular weight polymers because of its limited mobility and its finite lifetime (there is always competitive relaxation to the ground state). The amplification is due to exciton
mobility and should be properly separated from the binding constant. However, this analysis is seldom performed because of the need for nontrivial synthesis and other complexities inherent to polymer aggregates and thin films (Vide infra). This method also can be used to determine the exciton diffusion length of a CP in solution (Figure 1.4).32
Figure 1.3 Demonstration of amplified quenching in a CP. The determination of the
Stern-Volmer quenching reveals the constant for binding of paraquat to a single cyclophane receptor (top). In the polymer the larger (amplified) quenching constant reflects the fact that the quencher can be bound to any of the repeating units visited by the exciton (bottom).
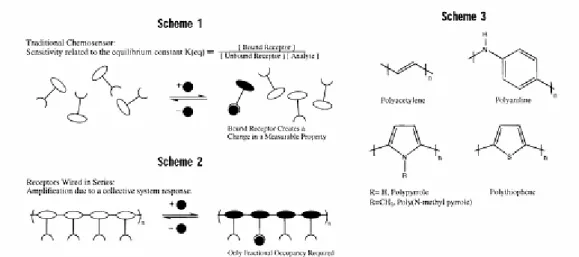
Figure 1.4 (a) Scheme 1, a chemosensor is composed of two functional elements, a
receptor and a reporter group, which need not be separate in identity, (b) this account describes an approach to enhancing the sensitivity of chemosensors in effect by “wiring chemosensory molecules in series” (Scheme 2), (c) for the sake of clarity, some representative conducting polymers are shown in Scheme 3.
1.3 Assembly of Nanoparticles Based on Supramolecular Chemistry
Acid-base chemistry has been involved in molecular recognition and has been studied using various techniques.33-35 For instance, the structures formed by
adsorption of carboxyalkylphosphonic acids on metal oxides were compared to those formed with ω-mercaptocarboxylic acids and gold nanoparticles (AuNPs) cores using
1H fast magic angle spinning (MAS), heteronuclear correlation (HETCOR), and 1H
doublequantum (DQ) MAS solid-state NMR.36 Infrared reflection spectroscopic data
were used to characterize the interfacial structures derived from the interfacial reactivity at the interconnecting linkages of core-shell nanoparticle networks and pH-tunable networks consisting of head-to-head hydrogen-bonded carboxylic acid terminals.37,38 Nanoparticle arrays obtained by mixing anionic bilayer membranes and
cationic, quaternary ammonium-stabilized AuNPs were immobilized densely into the hydrophilic interlayers of dispersed lamellar structures to form a quasi-1D structure.39
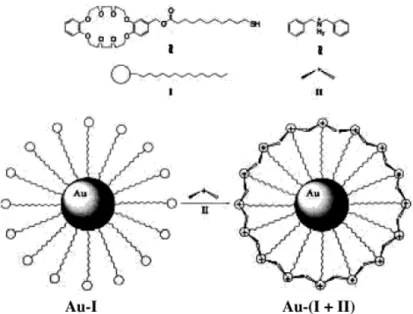
A pseudorotaxane assembly was achieved at the surface of AuNPs, pointing to similarities with the binding of a drug molecule by the receptor sites on the surface of a cell (Figure 1.5).40 Evidence for film formation was also found in the case of AuNPs
terminated by carboxylic acid groups.41-43
For amide-functionalized AuNPs, IR and 1H NMR studies revealed that
intramolecular H-bonding was highly dependent on the distance of the amide from the core.44 Polyhedral oligomeric silsesquioxanes functionalized by diaminopyridines
self-assembled with complementary thymines into spherical aggregates.45 IR
spectroscopy and cyanide-mediated decomposition of the gold cores of amide-stabilized AuNPs showed a strong correlation between the strength of the intramolecular H-bonding and the rate of decomposition.46 Interaction of nitroxyl
radicals with AuNPs, monitored by paramagnetic probes, resulted in loss of the EPR signal, due to exchange interaction of the unpaired electron with conduction band electrons of the AuNPs. Catalytic oxidation of a probe was also found when dioxygen was present.47
Figure 1.5 Programmed pseudorotaxane assembly at the surface of AuNPs by
heterosupramolecular chemistry.
1.3.1 Miscellaneous Recognition and Sensors
The controlled assembly of nanoparticles in solution based on supramolecular chemistry, i.e., noncovalent bonding,48 is a general strategy leading to well-organized
AuNP materials. Thus, approaches have been reported using hydrogen-bonding,48b
π-π,48c host-guest,48d van der Waals,48e electrostatic,48f charge-transfer,48g and
antigen-antibody48h interactions.
Amide-functionalized AuNPs were also used as optical sensors for anions.49
AuNPs functionalized with 15-crown-5 recognize K+ in water,50a and Li+50b and heavy
metal ions51,52 were recognized using AuNP-based sensors. Rotello and co-workers
reported the synthesis and self-assembly of gold nanoparticles with inherent optical properties (Figure 1.6). The recognition properties based on H-bonding were used to
assemble AuNPs into micelles using polymers (Figure 1.7).53 Furthermore,
amine-functionalized polymers have been used to simultaneously assemble carboxylic acid functionalized gold and silica nanoparticles into extended aggregates, and nanoparticle segregation within the aggregate regulated through order of component addition (Figure 1.8).54
Figure 1.6 Self-assembly based on selective control of non-covalent interactions
provides a powerful tool for the creation of structured systems at a molecular level. This paper present a polymer-mediated “bricks and mortar” strategy for the ordering of nanoparticles into structured assemblies.
Figure 1.7 This paper has a system where recognition element-functionalized diblock
copolymers are used to self-assembly complementary nanoparticles. The size of the aggregate both in solution and in thin films is controlled through block length.
Figure 1.8 Amine-functionalized polymers have been used to simultaneously
assemble carboxylic acid functionalized gold and silica nanoparticles into extended aggregates, and nanoparticle segregation within the aggregate regulated through order of component addition.
AuNPs have also been used for vapor sensing.55a,b The sensitivity of the plasmon
response (Surface Plasmon Band) has been modelized.56 Electrochemical genosensors
for the detection of the Factor V Leiden mutation from polymerase chain reaction amplicons using the oxidation signal of AuNPs at +1.20 V were described.55c
DNA-AuNP assemblies have also been used as colorimetric lead biosensors.55d
N-Methylimidazole-functionalized AuNPs were reported to recognize bis- and tris-Zn
porphyrins.55e Conjugates of AuNPs-oligonucleotides are of great current interest
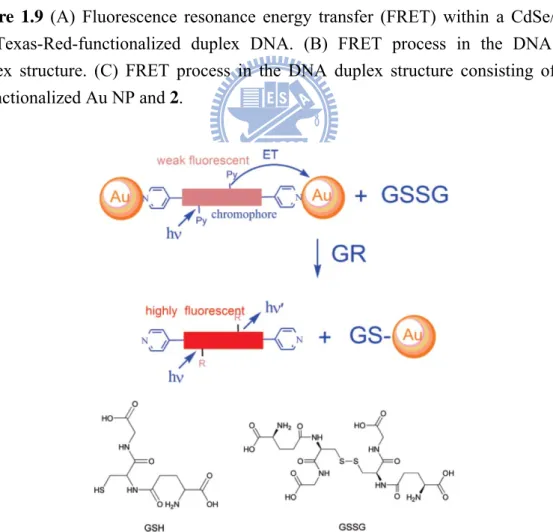
because of the potential use of the programmability of DNA base-pairing to organize nanocrystals in space and the multiple ways of providing a signature for the detection of precise DNA sequences (Figure 1.9).57 Furthermore, in the presence of other
metal ions, such as Cu2+, Co2+, Fe3+, Ni2+, Zn2+, Pb2+ and Ag+, the gold nanoparticles
of the quenched nanocomposites (containing fluorophores) can be replaced with the metal ions to different extents due to the stronger re-coordination or re-complexation of the metal ions with fluorophores, and therefore recover the fluorescence of the chromophores to behave as chemosensors. In addition, the gold nanoparticles of the quenched nanocomposites can also be reacted with the reduced glutathione in the presence of glutathione reductase enzyme, and then recover the fluorescence of the chromophores to behave as biosensors (Figure 1.10).58 The sensor applications of this
work can be further developed to detect the metal ions and biomolecules based on the modulation of fluorescence quenching and recovery.
Figure 1.9 (A) Fluorescence resonance energy transfer (FRET) within a CdSe/ZnS
QD/Texas-Red-functionalized duplex DNA. (B) FRET process in the DNA 3/2 duplex structure. (C) FRET process in the DNA duplex structure consisting of the 1-functionalized Au NP and 2.
Figure 1.10 Schematic presentation of reversion process from oxidized to reduced
glutathione based on the modulation in photoluminescent quenching efficiency between chromophore and AuNPs. (Bottom) Chemical structure of GSH and GSSG.
1.4 Supramolecular in Organic Devices
One of the major challenges in the field of electronics based on organic molecules is the design of functional, multicomponent architectures possessing long-range ordering. Having an electron-donating as well as an electron-accepting chromophore is a prerequisite to obtain organic photovoltaics. It has also been demonstrated that incorporation of energy- or electron-accepting chromophores into
π-conjugated polymer backbones can rigorously alter the electrooptical properties of
the resulting copolymers. The copolymers can be characterized by either almost exclusive emission from the incorporated acceptor moiety or efficient charge transfer. Recently, an end was put to a long discussion about undesired green emission in polyfluorenes, which appeared to arise from efficient energy transfer to small amounts of oxidized monomer,59 indicating the necessity for high-purity polymers.
The viability of this functional approach has been shown by the successful tuning of electroluminescence in a light-emitting diode (LED) consisting of PPV with dangling porphyrins.60 Also, a photovoltaic device based on a blend of PPV and
polyfluorene with dangling perylene proved to yield high external quantum efficiencies in both the perylene and the polyfluorene absorption regions, due to energy transfer from the polyfluorene to the perylene with near unit efficiency.61
established through the supramolecular interactions, e.g. H-bonds, in organic, dendritic, and polymeric H-bonded complex systems. This was illustrated by a recent report on a triple hydrogen-bonded triad consisting of a central perylene that was connected to two C60 chromophores (Figure 1.11).62 Novel monomer and PPV-donor
and PPV-acceptor bearing a terminal terpyridine chelating unit were reported (Figure 1.12).63 Three new photoactive supramolecular dyads have been prepared by
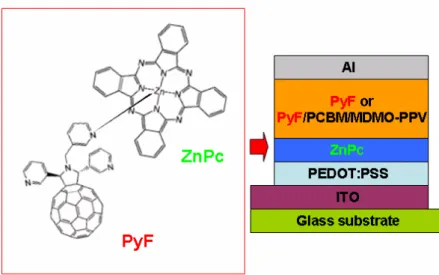
complexing the ruthenium with the PPV-terpyridine ligands. Studies on the incorporation of such ligands and supramolecular building blocks into polymers were performed through photophysical properties. An efficient energy transfer process from the conjugated polymer block to the metal complex was shown. Moreover, the preparation of several different fullerene-based materials bearing chelating pyridyl groups (PyFs, Figure 1.13)64 was reported, with a comparative investigation of their
interactions with ZnPc in solid thin films and evaluation of the performance of these materials in bilayer organic solar cells. To achieve efficient photocurrent generation in a wide spectral region, they merged bulk heterojunction MDMO-PPV/PCBM cells and bilayer ZnPc/PyF cells in a multicomponent device structure. The combination of bilayer fullerene/ZnPc and bulk heterojunction fullerene/polymer photovoltaic devices can be considered a promising approach to achieve a wide spectral response in organic solar cells while retaining the good charge-transfer and transport properties
of established materials.
Figure 1.11 Superstructure of self-assembly of [60]fullerene derivative 1 with
perylene bisimide 5 by H-bonding.
Figure 1.12 The incorporation of a ruthenium complex into the donor-acceptor
Figure 1.13 Pyrrolidinofullerenes bearing chelating pyridyl groups (PyFs) on
vacuum-evaporated films of zinc phthalocyanine (ZnPc) in donor-acceptor bilayer heterojunctions formed by deposition of solution-processed. It is shown that coordination complexes are formed at the interface between these donor and acceptor components; such association facilitates photoinduced charge separation and results in improved performance of the photovoltaic devices.
Chapter 2
Study of Supramolecular Side-Chain and
Cross-Linking Polymers by Complexation of Various
H-Donor Acids with H-Acceptor Copolymers
Containing Pendent Carbazole and Fluorescent
Pyridyl Units
Two H-bonded acceptor (H-acceptor) homopolymers 14 and 17 were successfully prepared by polymerization of fluorescent pyridyl monomers PBT and
PBOT (12 and 13), which were synthesized via Sonogashira coupling and
Wittig-Horner reactions. In order to increase the glass transition temperatures as well as reduce the π-π stacking of the photoluminescent (PL) H-acceptor copolymers and their H-bonded polymer complexes, fluorescent monomers 12 and 13 were copolymerized with N-vinylcarbazole monomer CAZ (23) to produce H-acceptor copolymers 15-16 and 18-19. Supramolecular side-chain and cross-linking polymers (i.e., H-bonded polymer complexes) obtained by complexation of light-emitting H-acceptor polymers 14-19 with various proton donor (H-donor) acids 20-22 were further characterized by DSC, POM, FTIR, XRD, and PL measurements. The mesomorphic properties can be tuned from the nematic phase in H-acceptor
homopolymers (14 and 17) to the tilted smectic C phase in their H-bonded polymer complexes (14/20-21 and 17/20-22) by the introduction of H-donor acids (20-22). Moreover, the PL properties of light-emitting H-acceptor polymers can be adjusted not only by the central structures of the conjugated pyridyl cores but also by their surrounding non-fluorescent H-donor acids. In general, redder shifts of PL emissions in H-bonded polymer complexes occurred when the light-emitting H-acceptor polymers were complexed with H-donors having smaller pKa values.
2.1 Introduction
Supramolecular chemistry is a new and exciting branch of chemistry encompassing systems held together by non-covalent bonds,and such complexes have considerable application potentials in the rapidly developing fields of molecular electronics and optoelectronics.48a,65,66 More recently, the concept of supramolecular
chemistry has been applied to the design of liquid crystalline (LC) polymers in the expectation that molecular interactions may be amplified into macroscopically observable phenomena of self-assembled phases, i.e., liquid crystallinity.67
Supramolecular liquid crystals are molecular complexes generated from complexation of molecular species through non-covalent interactions, e.g., hydrogen bonding. Kato and Fréchet first exploited two different and independent components to generate
liquid crystals through intermolecular hetero-hydrogen-bonding interaction, and this concept in turn resulted in numerous findings of such supramolecular liquid crystals.18,19 The mesogenic properties can be easily modified by miscellaneous
proton donors and acceptors, and new liquid crystalline properties, which are different from those of their original moieties, can be easily obtained by the introduction of supramolecular structures. Many kinds of H-bonds and building elements have been explored in the H-bonded structures to stabilize liquid crystalline phases.68 Therefore,
side-chain liquid crystalline polymers consisting of polymer backbones, flexible spacers, and mesogenic pendants have great potentials in various utilizations as novel technological materials, such as optical switching elements, optical storage devices, and information displays. Among these approaches, intermolecular H-bonding is simply acquired by complexation of H-bonded donor (H-donor) carboxylic (or benzoic) acid groups with H-bonded acceptor (H-acceptor) pyridyl moieties. Several series of H-bonded polymer complexes and side-chain liquid crystalline polymers through intermolecular H-bonding (interaction between benzoic acid and pyridine) have been reported lately.69,70
The advantages of using organic materials to manufacture electroluminescent (EL) devices are their excellent film-forming properties, processing feasibilities of flexible devices, highly efficient EL properties, and low costs of fabrication.71 As we
know, poly(N-vinylcarbazole) (PVK) has attracted attention due to its applications related to polymer light-emitting diodes (PLEDs) in which the hole transporting layer is formed by PVK or it can be blended with other light-emitting materials. Such PLED devices have shown remarkably high luminescence efficiencies and relatively facile color tunabilities.72-74 In contrast to PVK, Romero et al.75 observed an increase
in the external quantum efficiency of PLED devices based on the copolymerization of carbazole units with short thiophene segments, so carbazole units were also used to copolymerize with fluorescent pyridyl moieties in our study. Moreover, tuning emission colors in organic light-emitting materials have been established through the supramolecular interactions, e.g. H-bonds, in organic, dendritic, and polymeric H-bonded complex systems.76-78
In this report, fluorescent pyridyl H-acceptors as pendent groups were incorporated into the side-chain polymeric structures rather than as small molecules in our previous studies.76-78 The purpose of the present study for side-chain conjugated
pyridyl polymers is to explore the self-assembled utilization of singly and doubly H-bonded structures (as shown in the schematic illustration of Figure 2.1) in preparing for supramolecular side-chain and cross-linking polymers, respectively. As shown in Schemes 2.1 and 2.2, fluorescent H-acceptor monomers PBT and PBOT (12 and 13) and their corresponding H-acceptor homopolymers (14 and 17)
containing three-conjugated aromatic rings (including two lateral substituted methyl and methoxy groups with one pyridyl terminus) were prepared, and both pyridyl H-acceptor monomers 12 and 13 were further reacted with different molar ratios of carbazole monomer CAZ (23) to produce copolymers 15-16 and 18-19, respectively. Thus, the glass transition temperatures of the H-acceptor polymers can be controlled by the contents of pendent carbazole monomer CAZ (23) in H-acceptor polymers (14-16 and 17-19). In addition to the syntheses of such fluorescent H-acceptor monomers and polymers, two series of different H-acceptor polymers PBT1-PBT3
(14-16) and PBOT1-PBOT3 (17-19) were complexed with asymmetric
mono-functional H-donors OBA (20) and ONA (21) as well as symmetric bi-functional H-donor THDA (22), respectively (as shown in Figure 2.2). By incorporating of H-acceptor polymers to H-donor acids with different pKa values, the light-emitting properties of the supramolecular polymer complexes can be easily adjusted. Singly/doubly H-bonded processes of side-chain/cross-linking H-bonded polymers were confirmed and investigated by means of their liquid crystalline properties, X-ray diffraction (XRD) patterns, and photoluminescent (PL) properties.
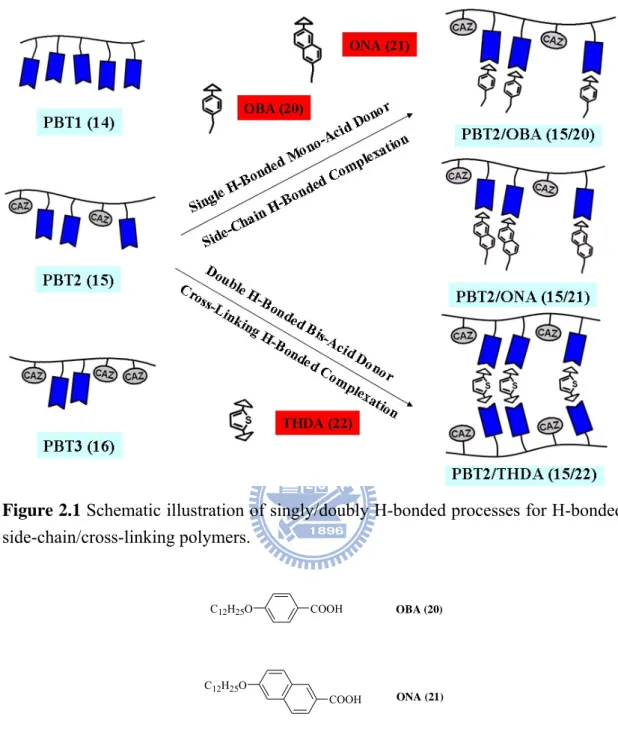
Figure 2.1 Schematic illustration of singly/doubly H-bonded processes for H-bonded
side-chain/cross-linking polymers.
Figure 2.2 Mono-acid (singly H-bonded) and bis-acid (doubly H-bonded) donors
used in supramolecular side-chain/cross-linking polymers, respectively.
COOH C12H25O C12H25O COOH S COOH HOOC OBA (20) ONA (21) THDA (22)
Scheme 2.1 Synthetic Routes of Monomer PBT (12). Br Br Br P(OEt)3 Br HO Br HO(CH2)10O Br Br(CH2)10OH/K2CO3/KI Acetone, reflux OH PO(OEt)2 Cl HO(CH2)10O OH HO(CH2)10O N CHO LDA THF, -78oC Br N Pd(PPh3)2Cl2/CuI/PPh3 Et3N/THF, 70oC N HO(CH2)10O N O(CH2)10O O Br Br THF, -78oC n-BuLi/DMF NaBH 4 MeOH/THF, r. t. HCl 1,4-Dioxane, reflux CHO HC CC(CH3)2OH Pd(PPh3)2Cl2/PPh3/CuI/Et3N, 70oC KOH 1,4-Dioxane, reflux CCH3COOCH H2C reflux 1 2 3 4 5 6 8 9 10 6 + 10 11 PBT (12) 7 CH2 Cl2Bu4Sn2O/THF, 50oC
Scheme 2.2 Synthetic Routes of H-Acceptor Polymers. O (CH2)10 O N PBT2 : X=60 Y=40 (15) PBT3 : X=34 Y=66 (16) PBT1 : X=100 Y=0 (14) C O H2 C HC2HC N X Y O (CH2)10 O O O N PBOT2 : X=62 Y=38 (18) PBOT1 : X=100 Y=0 (17) C O H2 C HC2HC N X Y PBOT3 : X=30 Y=70 (19) N O (CH2)10 O O O N O + AIBN THF 60°C PBOT (13) CAZ (23) N O (CH2)10 O N O + AIBN THF 60°C PBT (12) CAZ (23)
2.2 Experimental
2.2.1 Materials
N-vinylcarbazole CAZ (23) was purchased from Aldrich Chemical Co. and used
without further purification. Azobisisobutyronitrile (AIBN) was purchased from Kanto Chemical Co. and recrystallized from ethanol at 40 oC followed by drying in a
vacuum oven. Proton donors OBA (20) and ONA (21) were identified as the required materials by 1H and 13C NMR spectroscopy and elementary analyses, which were
reported in our previous results,78 and proton donors thiophene-2,5-dicarboxylic acid
THDA (22) was purchased from Aldrich Chemical Co. Chemicals and solvents were
reagent grades and purchased from Aldrich, Acros, TCI, and Lancaster Chemical Co. Dichloromathane and THF were distilled to keep anhydrous before use. The other chemicals were used without further purification.
Syntheses of H-Acceptor Monomers PBT (12) and PBOT (13)
The synthetic route of monomer PBT (12) is shown in Scheme 2.1, and its synthetic procedures are described as follows:
4-Bromo-2,5-dimethylbenzaldehyde (2). 2,5-Dibromo-p-xylene 1 (6.9 g, 26.3
mmol) was dissolved in 60 mL of dry THF purged with nitrogen. A solution of n-BuLi (13.7 mL, 34.2 mmol, 2.5 M in hexane) was added dropwise to a rapidly stirred THF at -78 °C. The rate of addition was adjusted to keep the temperature
below -78 °C. After the solution was stirred to react for 2 h at -78 °C, a solution of DMF (4.1 mL, 52.6 mmol) was added dropwise to keep at the same temperature. After 2 h, the reaction was quenched with water and extracted with ethyl acetate. The organic extracts were dried over Na2SO4 and then evaporated. The crude product was
purified and recrystallized from n-hexane to give a white crystal. Yield: 5.0 g (90%).
1H-NMR (ppm, CDCl
3): δ 10.19 (s, 1H), 7.63 (s, 1H), 7.47 (s, 1H), 2.60 (s, 3H), 2.43
(s, 3H).
4-Bromo-2,5-dimethylbenzyl alcohol (3). To a stirred solution of compound 2
(5.0 g, 23.7 mmol) in 100 mL of THF/MeOH (1:1), NaBH4 (0.9 g, 23.7 mmol) was
added very slowly and reacted at room temperature. After 1 h, the solution was cooled to 0 °C by ice bath, acidified with dilute HCl solution, and extracted with ethyl acetate. The resulting extracts in organic phase were combined and washed with water. Then, the organic extracts were dried over Na2SO4 and evaporated. The crude product was
purified and recrystallized from dichloromethane/2-propanol to give a colorless crystal. Yield: 4.1 g (80%). 1H-NMR (ppm, CDCl
3): δ 7.33 (s, 1H), 7.21 (s, 1H), 4.61
(s, 2H), 2.35 (s, 3H), 2.27 (s, 3H).
1-Bromo-4-chloromethyl-2,5-dimethoxybenzene (4). A stirred solution of
compound 3 (4.1 g, 19 mmol) in 1,4-dioxane (150 mL) was added with concentrated HCl (20 mL, 3N), and then the mixture was refluxed for 10 h. After the reaction was
completed, the crude mixture was added with water. The organic layer was extracted with ethyl acetate, dried over Na2SO4 and evaporated. The crude product was purified
by flash column chromatography (silica gel, n-hexane/ethyl acetate 40:1) to give a white solid. Yield: 4.0 g (89%). 1H-NMR (ppm, CDCl
3): δ 7.36 (s, 1H), 7.15 (s, 1H),
4.51 (s, 2H), 2.36 (s, 6H).
(4-Bromo-2,5-dimethylbenzyl)diethylphosphonate (5). Compound 4 (4.0 g,
17.1 mmol) was mixed with an excess of triethylphosphite (20 mL) and heated to reflux for 12 h under reduced pressure. The excess of triethylphosphite was removed after reaction. The crude product was purified and washed with hot hexane to give a white solid. Yield: 5.1 g (90%). 1H-NMR (ppm, CDCl
3): δ 7.28 (s, 1H), 7.07 (s, 1H),
4.08-3.95 (m, 10H), 3.06 (s, 1H), 2.99 (s, 1H), 2.28 (s, 3H), 2.26 (s, 3H).
1-Bromo-2,5-dimethyl-4-[2-(4-pyridyl)ethenyl]benzene (6). Compound 5 (5.1
g, 15.1 mmol) was dissolved in 60 mL of dry THF purged with nitrogen. A solution of lithium diisopropylamide (22.7 mL, 45.3 mmol, 2.5 M in hexane) was added dropwise to a rapidly stirred solution at -78 °C. The rate of addition was adjusted to maintain the temperature below -78 °C. After the solution was stirred to react for 30 min at -78 °C, a solution of pyridine-4-carboxaldehyde (2 mL, 21.1 mmol) was added dropwise and stirred for 30 min to come back to room temperature. After that, the mixture was stirred to react for 18 h at room temperature. The reaction was quenched
with water and extracted with dichloromethane. Subsequently, the organic layer was dried over Na2SO4 and evaporated. The crude product was purified by column
chromatography (silica gel, dichloromethane/acetone 20:1) to give a yellow solid. Yield: 3.7 g (85%). 1H-NMR (ppm, CDCl
3): δ 8.56 (d, J = 4.8 Hz, 2H), 7.42 (s, 1H),
7.40 (d, J = 16.2 Hz, 1H), 7.36 (s, 1H), 7.35 (d, J = 4.8 Hz, 2H), 6.88 (d, J = 16.2 Hz, 1H), 2.38 (s, 3H), 2.35 (s, 3H).
10-(4-Bromophenoxy)-decan-1-ol (8). A mixture of 4-bromophenol 7 (4.9 g,
28.5 mmol), potassium carbonate (8.7 g, 62.7 mmol), 10-bromodecanol (7.4 g, 31.4 mmol), and a few amount of potassium iodide in acetone (200 mL) was heated to reflux and stirred under nitrogen for 48 h. After cooling to room temperature, the solvent was removed under reduced pressure. The residue was taken up in water and extracted with ethyl acetate. Then, the organic layer was dried over Na2SO4 and
evaporated. The crude product was purified by column chromatography (silica gel, n-hexane/ethyl acetate 3:1) to give a white solid. Yield: 8.3 g (88%). 1H-NMR (ppm,
CDCl3): δ 7.33 (d, J = 9.0 Hz, 2H), 6.75 (d, J = 9.0 Hz, 2H), 3.89 (t, J = 6.3 Hz, 2H),
3.62 (t, J = 6.6 Hz, 2H), 1.78-1.69 (m, 2H), 1.59-1.50 (m, 2H), 1.41-1.29 (m, 12H).
4-[4-(10-Hydroxy-decyloxy)-phenyl]-2-methyl-3-butyn-2-ol (9). A solution of
compound 8 (8.3 g, 25.3 mmol), PPh3 (13.1 mg, 0.51 mmol), and CuI (73 mg, 0.38
2-Methyl-3-butyn-2-ol (3.7 mL, 38 mmol) and Pd(PPh3)2Cl2 (180 mg, 0.25 mmol)
were added to the solution at room temperature and the mixture was stirred to react at 70 oC for 12 h. The mixture was filtered and the solvent was removed in vacuum.
Afterward, the crude mixture was extracted using dichloromethane. The organic solution was washed with water, and then dried over Na2SO4 and evaporated. The
crude product was followed by purifying with column chromatography (silica gel, n-hexane/ethyl acetate 2:1) to give a light yellow solid. Yield: 4.7 g (56%). 1H-NMR
(ppm, CDCl3): δ 7.31 (d, J = 9.0 Hz, 2H), 6.79 (d, J = 9.0 Hz, 2H), 3.92 (t, J = 6.6 Hz,
2H), 3.62 (t, J = 6.6 Hz, 2H), 1.77-1.70 (m, 2H), 1.60 (s, 6H), 1.58-1.50 (m, 2H), 1.42-1.29 (m, 12H).
4-Ethynyl-1-(10-hydroxydecan-1-yloxy)benzene (10). A solution of compound
9 (4.7 g, 14.2 mmol) and finely powdered KOH (2.39 g, 42.6 mmol) in 1,4-dioxane
(80 mL) was refluxed under nitrogen for 3 h. After cooling to room temperature, the solvent was removed under reduced pressure. The residue was taken up in water and extracted with ethyl acetate, and then acidified with 150 mL of HCl (3 N). The organic solution was washed with water, and then dried over Na2SO4 and evaporated.
The crude product was purified by column chromatography (silica gel, n-hexane/ethyl acetate 4:1) to give a light yellow solid. Yield: 3.6 g (92%). 1H-NMR (ppm, CDCl
3): δ
6.6 Hz, 2H), 2.97 (s, 1H), 1.80-1.70 (m, 2H), 1.57-1.50 (m, 2H), 1.42-1.29 (m, 12H).
10-{4-[2,5-Dimethyl-4-(2-pyridin-4-yl-vinyl)-phenylethynyl]-phenoxy}-decan
-1-ol (11). A mixture of compound 6 (3.7 g, 12.7 mmol), PPh3 (170 mg, 0.64 mmol),
and CuI (120 mg, 0.64 mmol) in dry triethylamine (80 mL) was degassed with nitrogen for 5 min. Compound 10 (3.6 mL, 13.3 mmol) and Pd(PPh3)2Cl2 (90 mg,
0.13 mmol) were added to the solution at room temperature, and afterward the reaction mixture was stirred to react at 70 oC for 12 h. The mixture was filtered and
the solvent was removed in vacuum. Next, the crude mixture was extracted using dichloromethane. The organic solution was washed with water, and then dried over Na2SO4 and evaporated. The crude product was purified by column chromatography
(silica gel, dichloromethane) to give a light yellow solid. Yield: 4.4 g (72%). 1H-NMR
(ppm, CDCl3): δ 8.61 (d, J = 6.0 Hz, 2H), 7.56 (d, J = 15.9 Hz, 1H), 7.51 (s, 1H), 7.49 (d, J = 8.7 Hz, 2H), 7.44 (d, J = 6.0 Hz, 2H), 7.36 (s, 1H), 6.98 (d, J = 15.9 Hz, 1H), 6.90 (d, J = 8.7 Hz, 2H), 4.00 (t, J = 6.6 Hz, 2H), 3.68 (t, J = 6.6 Hz, 2H), 2.53 (s, 3H), 2.44 (s, 3H), 1.82 (m, 2H), 1.60 (m, 2H), 1.49-1.16 (m, 12H). 2-Methyl-acrylic acid 10-{4-[2,5-dimethyl-4-(2-pyridin-4-yl-vinyl)-phenylethynyl]-phenoxy}-decyl ester
PBT (12). Compound 11 (1.0 g, 2.1 mmol), vinyl methacrylate (1.24 ml, 0.01 mmol),
![Figure 1.11 Superstructure of self-assembly of [60]fullerene derivative 1 with perylene bisimide 5 by H-bonding](https://thumb-ap.123doks.com/thumbv2/9libinfo/8362112.176905/39.892.151.737.221.419/figure-superstructure-assembly-fullerene-derivative-perylene-bisimide-bonding.webp)


![HPSH [ 分子間作用力 - 氫鍵 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)