行政院國家科學委員會補助專題研究計畫成果報告
台灣肺癌之基因體研究及臨床應用:著重於女性肺腺
癌-分項計畫一:台灣女性肺腺癌遺傳流行病學研究-女性肺腺癌基因定位之家族研究
計畫編號:91-3112-P-002-010
執行期間:91年05月01日至91年12月31日
計畫主持人:陳建仁
執行機構及單位名稱:國立台灣大學流行病學研究所
中華民國 90 年 12 月 31 日
Mater ials and Methods
Study subjects
The study subjects were recruited from National Taiwan University Hospital, Taipei,
Taiwan. National Taiwan University Hospital is the leading teaching hospital in
Taiwan, and is the most important referral center in Taiwan. Subjects were mostly
from great Taipei area; however, some of them were referred from other area from
other hospitals. Eligible cases were newly diagnosed and histologically (pathology
or cytology) confirmed primary lung adenocarcinoma by experienced pathologists
or chest specialists. A total of 301 eligible cases were recruited between July 1996
and March 2001. Among them, 30 subjects were proved to be not lung cancer,
including tuberculosis, ovary cancer, breast cancer, or other benign tumors; 4
subjects were of unknown diagnosis, and 263 subjects were proved to lung cancer.
Among 263 lung cancer patients, 200 subjects were adenocarcinoma, and 63 of them
were non-adenocarcinoma lung cancer, including squamous cell, small cell, large
cell, and adenosquamous cell carcinoma. About 76 % of lung cancer patients were
adenocarcinoma. Among adenocarcinoma, 16 subjects had no questionnaires due to
either too ill to response, discharged, or expired. The response rate was 92%. Among
the 184 subjects, 167 of them had blood sample, and 17 subjects had no blood
two stages. The first stage was between 1997 July and 1998 January. Hospital
controls were recruited from National Taiwan University Hospital health
examination department. A total of 279 controls were recruited. We did not perform
individual matching because of administrative consideration. However, the control
group was younger and receiving higher education than cases. In order to improve
the power and efficiency, we stared a secondary stage of control recruitment. This
time, controls were recruited from Taipei Municipal Chung-Hsiao hospital in 2001.
Female older than 65 years receiving free health examination provided by Bureau of
National Health Insurance, Taiwan was eligible controls. A total of 73 controls were
recruited. Among the total 352 controls, only 2 subjects refused interview, and 10
subjects refused to provide blood samples. Only 277 controls and 148 cases had
received genotyping for phase I ,2 xenobiotics-metabolizing enzymes, estrogen
metabolizing and receptor gene , and DNA repair genes polymorphism.
The catchments areas of the controls were from the great Taipei city. However, the
catchments areas of cases, though mostly were from great Taipei city, were from the
whole Taiwan in fact. We did not use proxy responder information in our study.
Data specification
Two trained interviews conducted personal interviews to collect risk factors data.
exposure status including the smoking status of the patient and her spouse, parents,
and co-workers, individual medical condition, incense smoke, dietary history,
alcohol consumption, occupational exposure, a family history of lung cancer and
cooking fume exposure were obtained from structured questionnaires. Cooking
habit before 40 was defined as cooking daily for at least 6 months before
40-year-old. Cooking fume exposure was defined as those who had cooking habit
before 40-year-old and no ventilator was used when she was cooking. Subjects who
had no cooking habits or used ventilator when cooking were defined as no cooking
fume exposure. Other cooking related items, such as Cooking fuels, including
electricity, natural gas, charcoal, wood, and coal, cooking oils, including lard oil or
vegetable oils (soybean, peanut, sunflower, and other vegetable oils), age at starting
cooking, total cooking years before 40, and fume extractor in kitchen (as
dichotomous variable) were also interviewed and analyzed. Ever smoker was
defined as having smoked daily for at least 6 months during her lifetime.
Nonsmoker was defined as never having smoked daily for at least 6 months during
her lifetime. Smoking duration (in years), and cumulative smoking amount (in
pack-year) were stratified to four levels to test the dose-response relationship. As to
the environmental tobacco smoke exposure, smoking status of father, mother, spouse,
smoking amount (in pack-years) were stratified into four levels to test the
dose-response relationship. Tobacco smoke exposure was defined as ever smokers
or spouse smoking nears her.
Hormone-related risk factors included age at menarche, age at menopause,
menstruation regularity, menstrual cycle length, length of menstrual period, number
of gestation, parity, spontaneous abortion, and total duration of breast-feeding.
External source of sex hormone included oral contraceptives and hormone
replacement therapy, and some Chinese herb drug for menstruation-regulation.
Per iod of hor mone exposure was defined as (age of recruitment – age of menarche)
× 12-10 × times of full term delivery- 5× times of abortion when she was not
menopausal, and was defined as (age of menopause – age of menarche) × 12-10 ×
times of full term delivery- 5× times of abortion when she was menopausal. Body
mass index was calculated by body weight six months before diagnosis in kilogram
divided by square of body height in meter.
Interviewer also asked about the history of pulmonary tuberculosis, chronic
obstructive airway disease (emphysema, chronic bronchitis), asthma, and history of
hysterectomy, oophorectomy, and family history of lung cancer.
venous blood for genotype analysis. Genomic DNA sample were extracted from
peripheral lymphocytes using a Puregene DNA isolation kit (Gentra System, Lnc.,
Minneapoils, MN, USA). After extraction, DNA was dissolved in a hydration
solution and stored at 4℃ until further analysis. Genotypes were detected using a
PCR-RFLP technique as the following condition:
Phase I xenobiotics-metabolizing enzymes genotyping CYP1B1 (Codon 432) Primer: 5’-GTG GTT TTT GTC AAC CAG TGG-3’ 5’-GCC TCT TGC TTC TTA TTG GCA-3’ Condition: 94℃ 4 mins à(94℃ 40” à 55℃ 30” à 72℃ 40”)à 72℃ 10 mins 35 cycles
Exon3, codon 432(ValàLeu)1294 GàC PCR product: 390 bp, create Eco57I site G/G: 390 bp, C/C: 330+60 bp
CYP1B1 (Codon 48)
Primer:
5’-TAC GGC GAC GTT TTC CAG AT-3’ 5’-CGT GAA GAA GTT GCG CAT CA-3’ Condition:
94℃ 4 mins à(94℃ 40” à 55℃ 30” à 72℃ 40”)à 72℃ 10 mins 35 cycles
PCR product: 230 bp; codon 48 AlaàSer (GàT) Create Ahd1 site, G/G bp: 230; T/T: 110+120 bp
CYP1A1-exon7 (Codon 462)
Primer: 5’-GAACTGCCACTTCAGCTGTCT-3’ 5’-GAAAGACCTCCCAGCGGTCA-3’ Condition: 94 °C 4 min → (94 °C 40” →60°C 25” →72 °C 30”) →72°C 10min 35 cycles
Codon 462 Ile→ Val (ATT→GTT)
PCR product 187 bp, A-G mutation create HincII site, Ile: 139+48, Val: 120+48+19 bp
CYP1A1-Msp1 (3’-flanking r egion)
C44: 5’-TAGGAGTCTTGTCTCATGCCT-3’ C47: 5’-CAGTGAAGAGGTGTAGCCGCT-3’
94 °C 4 min → (94 °C 40” →60°C 30” →72 °C 30”) →72°C 10min 35 cycles
PCR product 340 bp
3’-flanking T→C mutation create Msp1 site, wt/wt: 340, mt/mt 205+135
CYP2E1-Rsa1 Primer: 5’-CCAGTCGAGTCTACATTGTCA-3’ 5’-TTCATTCTGTCTTCTAACTGG-3’ Condition: PCR product 412 bp, Rsa1, wt/wt: 366+46, mt/mt: 412 CYP1A2 (-2964) Primer:
5’-GCT ACA CAT GAT CGA GCT ATA C -3’ 5’-CA GGT CTC TTC ACT GTA AAG TTA-3’
94℃ 4 mins à(94℃ 40” à 56℃ 30” à 72℃ 40”)à 72℃ 10 mins 35 cycles Gà A at position-2964, create BslI Size of PCR product: 596 bp G:/G: 343 +132+ 93+ 28 A/A: 475+ 93+ 28 CYP2C19 m1 Primer: 5’-AATTACAACCAGAGCTTGGC -3’ 5’-TATCACTTTCCATAAAAGCAAG-3’ Condition: 94 °C 4 min → (94 °C 40” →52°C 30” →72 °C 30”) →72°C 10min 35 cycles PCR product: 169 wt/wt: 120+49 mt/mt: 169
Phase II xenobiotics-metabolizing enzymes genotyping COMT (Val158Met)
5’-AGGTCTGAC AAC GGGTCAGGC-3’
94℃ 4 mins à(94℃ 40” à 55℃ 30” à 72℃ 30”)à 72℃ 10 mins 35 cycles
Size of PCR product: 217bp AàG loss of an NlaIII site
Met/Met: 40+96+81bp, Val/Val: 136+81bp.
GST T1M1:
M1:G5 5’-GAAC TCCCTGAAAAGCTAAAGC-3’ G6 5’-GTTGGGCTCAAATATAC GGTGG-3’ T1: T1-R 5’-TCAC CGGATCATGGCCAGCA-3’ T1-F 5’-TTCCTTAC TGGTCCTCAC ATCTC-3’ B-globin: CAAC TTCATCCAC GTTCAC C
GAAGAGCCAAGGAC AGGTAC PCR condition: 94 °C 4 min → (94 °C 40” →55°C 30” →72 °C 40”) →72°C 10min 35 cycles 2.5 % agarose electrophoresis GSTP1 (Ile105Val)
P105F 5’-ACC CCA GGG CTC TAT GG-3’ P105R 5’-TGA GGG CAC AAG AAG CCC CT-3’
94 °C 4 min → (94 °C 40” →60°C 25” →72 °C 30”) →72°C 10min 35 cycles
PCR product: 176bp, Ile/Ile: 176 bp, Val/Val: 91+85 bp The Alw26I site created by the A→G mutation at codon105
NAT2
N4: 5’-TCT AGC ATG AAT CAC TCT GC-3’ N5: 5’-GGA AC A AAT TGG AC T TGG-3’ 94 °C 4 min → (94 °C 40” →52°C 30” →72 °C 90”) →72°C 10min 35 cycles PCR product: 1093 M1 (Kpn1) C/C: 660+433bp, T/T: 1093bp M2 (Taq1) G/G: 380+317+226+170, A/A: 396+380+317 M3 (BamH1) G/G: 811+282, A/A: 1093 NAT1 N1323 5’-TAAAAC AATCTTGTCTATTTG-3’ N1536NR 5’-ATAAC CAC AGGCCATCTTTAGAA-3’
94 °C 4 min → (94 °C 40” →52°C 30” →72 °C 40”) →72°C 10min 35 cycles
SNP site: *4/*4:1528T/T; 1535:C/C. *3/*3:1528T/T; 1535:A/A *10/*10:1528A/A; 1535:A/A
*11/*11:1520-1528 deleted; Run cycle-sequence to distinguish *3/4*/10/*11
EH (Tyr 113His)
5’-TGT CCT TCC CAT CCC TCT CAA CTT-3’
5’-CCT TCA ATC TTA GTC TTG AAG TGA CGG T-3’
94 °C 4 min → (94 °C 40” →55°C 30” →72 °C 40”) →72°C 10min 35 cycles
C→ T mutation loss of an Asp1 site.
PCR product 228bp, Tyr/Tyr: 201+ 27bp, His/His: 228bp.
EH (His139Ar g)
5’-AAC AC CGGGCCCAC CCTTGGC-3’ 5’-GGGGTAC CAGAGCCTGAC CGT-3’
94 °C 4 min → (94 °C 40” →60°C 25” →72 °C 30”) →72°C 10min 35 cycles
A→G mutation create a Rsa1 site
PCR product: 357 bp, His/His: 299+58 bp, Arg/Arg: 177+122+58 bp 2%agarose electrophoresis’
Estrogen metabolizing and receptor gene polymor phism genotying
COMT (Val158Met) 5’-TCGTGGACGCCGTGATTCAGG-3’ 5’-AGGTCTGACAACGGGTCAGGC-3’ 94 4 mins à(94 40” à 55 30” à 72 30”)à 72 10 mins 35 cycles Size of PCR product: 217bp AàG loss of an NlaIII site
Met/Met: 40+96+81bp, Val/Val: 136+81bp . CYP17 5’-CATTCGCACTCTGGAGTC-3’ 5’-AGGCTCTTGGGGTACTTG-3’ 94 °C 4 min → (94 °C 40” →57°C 30” →72 °C 40”) →72°C 10min
35 cycles
SNP located at 34 bp upstream from the initiation of translation T-C mutation create a MspA1I site,
PCR product: 419, wt/wt: 419, mt/mt: 295+124
CYP19 (TTTA)n in Intr on 5
5’-GTC TAT GAA TAT GCC TTT TT-3’ 5’-GTT TGA CTC CGT GTG TTT GA-3’ PCR product: 291-320 bp
94 4 mins à(94 40” à 55 30” à 72 30”)à 72 10 mins for 35 cycles
ESR (Codon 325)
5’-GCC CGC TCA TGA TCA AAC G-3’ 5’-GGA TCA TAC TCG GAA TAG AGA AT-3’
94 4 mins à(94 40” à 55 30” à 72 30”)à 72 10 mins for 35 cycles
Size of PCR product: 120 bp
Codon 325 CCCà CCG create Hinf1 CCC (120bp) àCCG (98+21 bp) CYP1A1 (exon 7) 5’-GAACTGCCACTTCAGCTGTCT-3’ 5’-GAAAGACCTCCCAGCGGTCA-3’ 94 °C 4 min → (94 °C 40” →60°C 25” →72 °C 30”) →72°C 10min 35 cycles
Codon 462 Ile→ Val (ATT→GTT)
PCR product 187 bp, A-G mutation create HincII site, Ile: 139+48, Val: 120+48+19 bp DNA repair enzyme genotying
Genotypes were examined using polymerase chain reaction-based restriction fragment length polymorphism (PCR-RFLP) assays. Primers for XRCC1 exon 6 were 5’-CGA GTC TAG GTC TCA ACC CTA CTC ACT-3’ and 5’-GTT CCG TGT GAA GGA GGA GGA-3’, which amplified a 138
bp DNA fragment. Primers for XRCC1 exons 9 and 10 were 5’-TTG ACC CCC AGT GGT GCT AA-3’ and 5’-GGC TGG GAC CAC CTG TGT T-3’, which amplified an 861 bp DNA fragment. Primers for XRCC3 exon 7 were 5’- TCG CCT GGT GGT CAT CGA CTC-3’ and 5’-GCA TCC TGG CTA AAA ATA CGA GC-3’, which amplified a 207 bp DNA fragment. Primers for hMLH1 5’-flanking region were 5’-AGT AGC CGC TTC AGG GA-3’ and 5’-CTC GTC CAG CCG CCG AAT AA-3’, which amplified a 259 bp DNA fragment. Primers for XPD exon 23 were 5’-AGG ATC AGC TGG GCC TGT CCC TGC-3’ and 5’-TGT GGA CGT GAC AGT GAG AAA T-3’, which amplified a 220 bp DNA fragment. All PCRs were under the same condition as follows: a 50µL reaction mixture containing 2µL of genomic DNA, 1µ L each dNTPS, 0.5 unit Tag (Promega, Madison, WI), and 1X PCR buffer. PCR program was consisted of an initial melting step of 94℃ for 4 minutes, followed by 35 cycles of 40 seconds at 94℃and 30 second at 55℃. The products were electrophoresed using 2% agarose gel and visualized by ethidium bromide.
The restriction enzyme Pvu II was used to distinguish the 26304 polymorphism of XRCC1 exon 6 in which the gain of a Pvu II restriction site occurred in the polymorphic allele. The Arg/Arg, Arg/Trp, and Trp/Trp genotypes for codon 194 resulted in 138 bp; 138 bp, 63 bp and 75 bp; and 63 bp and 75 bp digestion products, respectively. The restriction enzyme Rsa I was used to distinguish the 27466 polymorphism of XRCC1 exon 9. The Arg/Arg, Arg/His, and His/His genotypes for codon 280 resulted in 63 bp, 201 bp and 597 bp; 63 bp, 201 bp, 597 bp and 660 bp; and 660 bp and 201 bp digestion products, respectively. The restriction enzyme Msp I was used to distinguish the 28152 polymorphism of XRCC1 exon 10. The Arg/Arg, Arg/Gln, and Gln/Gln genotypes for codon 399 resulted in 115 bp, 285 bp and 461 bp; 115 bp, 285 bp, 461 bp and 576 bp; and 285 bp and 576 bp digestion products, respectively. The restriction enzyme Nla III was used to distinguish the 18067 polymorphism of XRCC3 exon 7. The Thr/Thr, Thr/Met and Met/Met genotypes for codon 241 resulted in 207 bp; 207 bp, 103 bp and 104 bp; and 103 bp and 104 bp, respectively. The restriction enzyme Mob II was used to distinguish the 35931 polymorphism of XPD exon 23. The Lys/Lys,
and 220 bp, respectively. The restriction enzyme Pvu II was used to distinguish the polymorphism at position -93nt of hMLH1 5’-flanking region. The G/G, G/A, and A/A alleles resulted in 125 bp and 134 bp; 125 bp, 134 bp and 259 bp; and 259 bp digestion products, respectively.
Statistical analysis
In the case-control study, odds ratios and their 95% confidence intervals were used
as estimates of relative risk. Univariate logistic regression model was applied to test
the potential risk factors mentioned above. The significant risk factors identified in
univariate logistic regression model were put into multiple logistic regressions. All
the models were adjusted for age and education levels. Stratified analysis was done
for evaluating the interactive effect of tobacco smoke exposure and cooking
exposure, tobacco smoke exposure and family history, and hormone exposure period
and BMI. All univariate and multivariate logistic regressions were adjusted for age,
education level. All non-smoking related risk factors analysis was adjusted for
smoking exposure. All p values were obtained by two-tail test.
The genotype-genotype interaction effect was divided into four groups: individuals
with three, two, one, and no putative high-risk genotypes of phase I metabolizing
genes, phase II metabolizing genes, and estrogen metabolizing enzymes. The gene
dosage effect was evaluated by p value obtained from test for trend method. The
with three, two, one, and no putative high-risk factors among tobacco smoke
exposure, cooking fume exposure, and three putative high-risk genotypes. The
gene-environment dosage effect was evaluated by p value obtained from test for
Results
We showed demographic characteristics in Table 1-1. Among cases, the peak
incidence was among 60-69 years group (26.6%), which was consistent with
previous finding. And we found a small plateau over 40-60 years (comprised of
more than 40% cases). It seemed that more female adenocarcinoma occurring at
younger age than male. The mean age was 60.33 among case group, and was 56.27
among control group; the mean schooling year was 7.4 years among case group, and
9.72 years among control group. Overall, the control group was younger and
receiving higher education than cases. So, in the following analysis, we all adjusted
for age and education levels. As to the ethnic groups, we found no significant
difference among cases and controls. More than three fourth of the subjects were
Fukienese.
In Table 1-2, we showed the smoking-related risk factors in association with lung
adenocarcinoma. The ever-smoker possessed a 2.4-folds significant risk compared
with nonsmoker. Only 8.2% female adenocarcinoma were an ever smoker, which
was the lowest compared to previous study (4). The risk increased when the
smoking duration and cumulative smoking amount increased, showing a significant
trend. Those who smoked more than 25 pack-years possessed a 6.3-folds risk
factors. Spouse smoking exposure only carried a 1.2-folds non-significant risk;
however, if her husband smoked just besides her, the risk increased to 1.5-folds. We
stratified the cumulative amount of spouse smoking exposure into four levels to test
the dose-response relationship; however, there were no significant trend. We also
found that if more than 10 coworkers were ever smoker, the risk would be up to
2.8-folds. We categorized the subjects who were ever-smokers or her spouse smoked
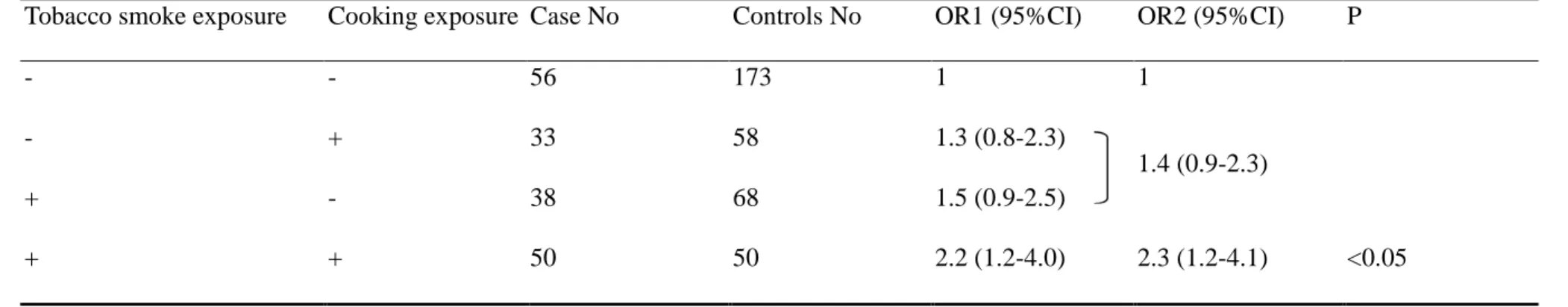
just besides her as having “tobacco smoke exposure”, and found that it carried a
1.7-fold (95%CI=1.1-2.5) significant risk.
Table 1-4 showed cooking-related risk factors before 40. Almost all subjects (>90 %
cases and controls) cooked daily before 40, which is consistent with traditional
Chinese women daily practice. The odds ratio for cooking habit did not show
significant association with lung adenocarcinoma (OR=0.6) because almost all
subjects had the exposure. However, if we compared those who had cooking habit
and did not have fume extractor in kitchen with those who did not cook or cooked
but had fume extractor in kitchen, we see a non-significant increased risk (OR=1.3,
95%CI= 0.8- 2.2). And then, we categorized those who did not cook or cooked but
had fume extractor in kitchen as no cooking fume exposure, and those who had
cooking habit and did not have fume extractor in kitchen as having cooking fume
development of lung adenocarcinoma. However, total duration of cooking before 40
showed a mild, but non-significant trend in association with the development of lung
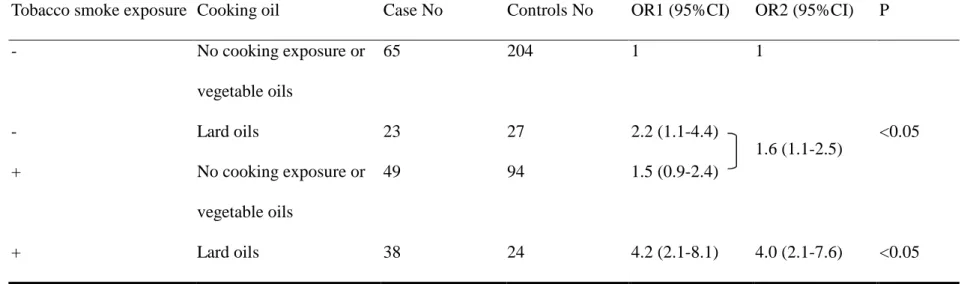
adenocarcinoma. As to the cooking oils, we found that lard oils possessed a
2.1-folds (95%CI=1.2-3.8) of significant risk compared with no cooking fume
exposure. Vegetable oils did not show significant risk (OR=0.7, 95% CI=0.4-1.3).
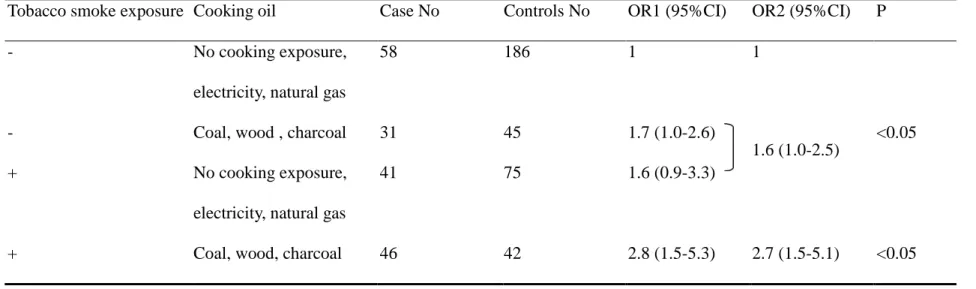
As to the cooking fuels, we found that coal, wood and charcoal possessed a 1.6-folds
(95%CI=1.0-2.8) of significant risk compared with no cooking fume exposure.
Electricity and natural gas did not show significant risk (OR=0.6, 95% CI=0.2-1.6).
We concluded that those who cooked, had no fume extractor, and used lard oils as
cooking oils or used coal, wood, and charcoal as cooking fuels possessed higher risk
for lung adenocarcinoma.
Table 1-5 showed hormone-related risk factor in association with lung
adenocarcinoma. Late onset of menarche (>=15 years) showed a borderline
significant risk for developing lung adenocarcinoma compared with early onset of
menarche. The earlier menopause the subjects were, the higher risk for lung
adenocarcinoma they would have, showing a significant trends. Those who were
menopausal carried a 6.6-folds risk (95%CI=2.9-14.7) compared with those who
were not yet menopausal, adjusting for age and education levels. Longer
(>=25 days) (OR=2.3, 95%CI=1.0-5.3) showed higher risk compared with shorter
menstruation period (<=6 days) and shorter menstruation cycle (<25 days). As the
numbers of gestation and parity increased, the risk for lung adenocarcinoma
increased simultaneously, and it showed a significant trend (p for trend <0.05).
Breast-feeding more than 18 months carried a 1.7-folds significant risk
(95%CI=1.0-2.8) compared with less than 18 months or no breast-feeding. As to the
external source of sex hormone, history of oral contraceptives and hormone
replacement therapy carried a borderline significantly protective effect (OR=0.6, 0.7,
respectively). And the protective effect increased as the duration of usage increased,
all showed a significant trend (p for trend <0.05). In those taking more than 1 year
compared with subjects never using, the ORs were 0.3, and 0.4 respectively for oral
contraceptives and HRT. Taking Chinese herb drug for menstruation-regulation
possessed a 1.2-folds significant risk (95%CI=1.0-1.5). BMI was inversely
associated with lung adenocarcinoma risk, and the more obese the women were, the
more unlikely she contracted lung cancer (p for trend =0.01). BMI >22.5 had a
0.6-fold significantly protective risk compared with BMI <=22.5. Hormone
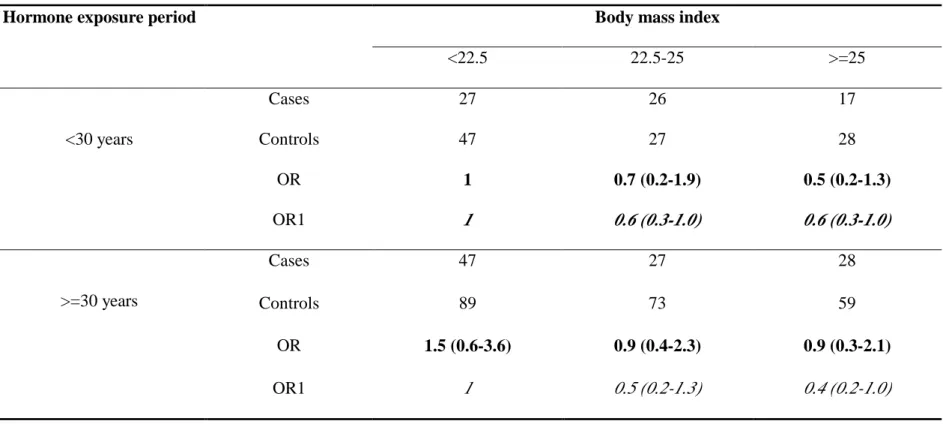
exposure period (>=30 years) had no association with lung adenocarcinoma.
(OR=1.1, 95%CI=0.7-1.7). Overall, many hormone-related items were associated
Table 1-6 showed personal medical history and family history of lung cancer in
association with lung adenocarcinoma. Pulmonary tuberculosis had a 2.3-folds
significant risk for lung adenocarcinoma. COPD, asthma, hysterectomy, and
oophorectomy were not associated with lung adenocarcinoma. As to the family
history, we found mother contracting lung cancer carried an 8.9-folds significant risk
for lung adenocarcinoma. Sibling contracting lung cancer also carried a 5.6-folds
significant risk for lung adenocarcinoma. Father contracting lung cancer was not
associated with lung adenocarcinoma. If we included all first-degree relatives, we
found 1.9-folds significant risk for lung adenocarcinoma. If we exclude father, all
first-degree relatives showed a 4.9-flods (95%CI=1.7-14.1). Table 1-7 showed the
results of hormone-related risk factors in multiple logistic regression models. The
analysis was adjusted for age, education levels, and smoking exposure. We found
that oral contraceptives (OR=0.6), hormone replacement therapy (OR=0.2), BMI
(OR=0.4), menopause (OR=9.0), and longer menstruation period (OR=1.7) showed
significant association with lung adenocarcinoma. However, “breast feeding longer
than 18 months” showed borderline significance (OR=1.8). Other items, including
shorter menstruation cycle length, menstruation regularity, age at menarche, and
Chinese herb drug did not show association with lung adenocarcinoma. In Table 1-8,
multiple logistic regression models. The analysis was adjusted for age and education
levels. We found that cooking oil with lard (OR=2.0), tobacco smoke exposure
(OR=2.0), oral contraceptives (OR=0.6), hormone replacement therapy (OR=0.2),
BMI (OR=0.5) and menopause (OR=9.8) showed significant association with lung
adenocarcinoma. However, longer menstruation period (OR=1.7) showed borderline
significance. Other items, including lung cancer history of first-degree relatives
(OR=3.0), and pulmonary tuberculosis (OR=0.9) did not show association with lung
adenocarcinoma.
Tables 1-9 to 1-13 showed the interactive effect of tobacco smoke exposure and
cooking fume exposure, tobacco smoke exposure and cooking oil, tobacco smoke
exposure and cooking fuels, tobacco smoke exposure and, and BMI and hormone
exposure period in relation to lung adenocarcinoma. We found multiplicative
patterns in “tobacco smoke exposure” and “cooking fume exposure”, “tobacco
smoke exposure” and “cooking oils”, “tobacco smoke exposure” and “cooking
fuels”, and “tobacco smoke exposure” and “family history of lung cancer”. We
investigated the modifier effect with regard to BMI in association with lung
adenocarcinoma. In shorter hormone exposure period, the ORs for lung
adenocarcinoma were 1, 0.6, 0.6 respectively for those BMI<=22.5, 22.5-25, and
adenocarcinoma were 1, 0.5, 0.4 respectively, for those BMI<=22.5, 22.5-25, and
BMI>25. It seemed no modifying among BMI and hormone exposure period.
Table 2-1 presents the overall distribution of cases and controls and adjusted ORs
and 95% CI s by genotypes of phase I genes. The CYP1A1 Ile/Ile genotype had
1.8-folds (95% CI=1.1-2.9) increased risk of developing lung adenocarcinoma
(compared with Ile/Val and Val/Val genotype as the referent group). The CYP1A2
G/G or G/A genotype had 3.9-folds (95% CI=1.4-11.3) increased risk of developing
lung adenocarcinoma (compared with A/A). Other phase I gene, i.e. CYP1A1 MspI
polymorphism (TT/TC vs. CC, OR= 1.4, 95% C.I.= 0.7-2.5), CYP2E1 RsaI
polymorphism (c1c1/ c1c2 vs. c2c2, OR= 1.4, 95% C.I= 0.4-4.3), CYP2E1 DraI
polymorphism (DD/ DC vs. CC, OR=1.5, 95% C.I= 0.5-3.9), CYP2C19 exon 5
(GG/GA vs. AA, OR= 1.3, 95% C.I.= 0.6-2.98), CYP1B1 codon 48 (Ala/Ala vs.
Ala/Ser, Ser/Ser, OR= 1.1, 95% C.I= 0.6-2.0), CYP1B1 codon 432 (Val/Val vs.
Val/Leu, Leu/Leu, OR= 1.4, 95% C.I= 0.7-2.5), did not show significant association
with lung adenocarcinoma.
To avoid gene-gene confounding effect, we put all phase I genes into multiple
logistic regression model. Because the CYP1A1 MspI and Ile/Val polymorphism,
and the CYP2E1 RsaI and DraI polymorphisms all showed strong linkage
polymorphism were included in the model. CYP1A1 Ile/Val polymorphism was
chosen due to their greater risk in the simple logistic regression. CYP2E1 RsaI
polymorphism was chosen because its phenotypic implication is more evident than
DraI polymorphisms in previous studies. As to CYP1B1, only codon 432
polymorphism was included in the model due to their greater risk in the simple
logistic regression than CYP1B1 codon 48. The results are showed in table 2-2: only CYP1A2 5’ flanking region polymorphism (GG/GA vs. AA) showed a 6.5-folds risk
of developing female lung adenocarcinoma (95% C.I 1.6-29.2), CYP2E1 RsaI
polymorphism (c1c2/c1c1 vs. c2c2) had a 1.3-folds risk; however, it did not reach
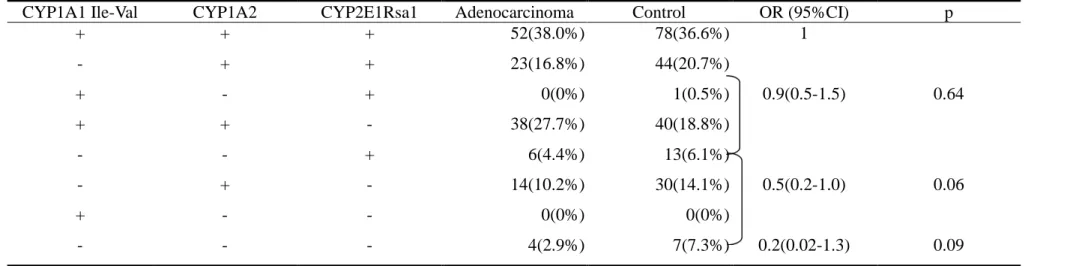
statistical significance. Table 2-3 presents the gene dosage effect. CYP1A1 Ile/Val, CYP1A2 5’ flanking region, and CYP2E1 RsaI polymorphisms were combined into a
four-level model of risk. A borderline significantly dose-response relationship was
noted between the numbers of putative high-risk genotype and the risk of lung
adenocarcinoma (p=0.06). OR=1 for those with three risk genotype (referent group),
adjusted OR=0.9 (95% CI=0.5-1.5) for those with two putative high-risk genotype,
adjusted OR=0.5 (95% CI=0.2-1.0) for those with one putative high-risk genotype,
and adjusted OR=0.2 (95% CI=0.02-1.3) for those with zero putative high-risk
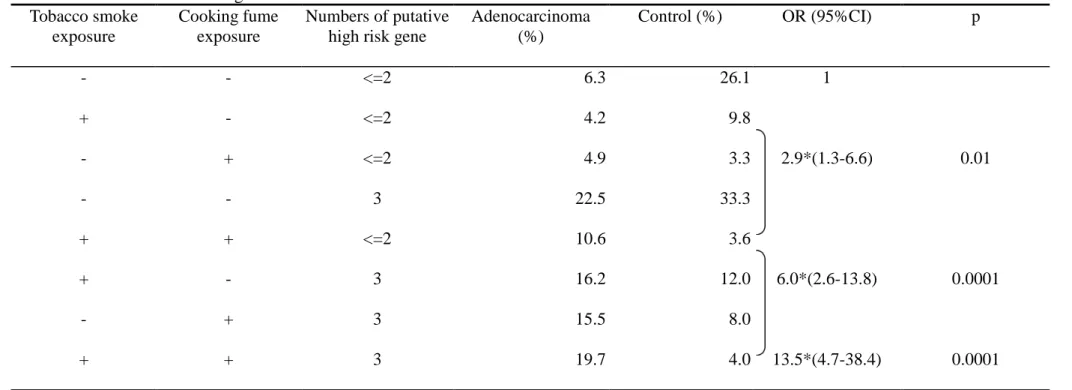
genotype. Table 2-4 presents the gene-environment dosage effect. Tobacco exposure,
risk. We categorized phase I gene into two groups: one group having less than three
putative high-risk genotypes, the other group having three putative high-risk
genotypes. We assigned those with neither risk factor as referent group. Having one
putative high-risk factor (including any one of tobacco smoke exposure, cooking
fume exposure, or three putative high-risk genotypes) is associated with a 1.4-folds
increased risk for developing lung adenocarcinoma. Having two putative high-risk
factors (including any two of tobacco exposure, cooking fume exposure, or three
putative high-risk genotypes) is associated with a significantly higher risk of lung
adenocarcinoma (OR=3.0; 95% CI=1.4-6.2). Women who had three putative
high-risk factors (those who exposed to tobacco, cooking fume, and having three
putative high risk genotypes) had a strong associated with lung adenocarcinoma
(OR=20.8; 95% CI =2.4-179.3). And it showed strong linear trend in our analysis
(p<0.0001).
Table 3-1 presents the overall distribution of cases and controls and adjusted ORs
and 95% CI s by genotypes of phase II genes. The GSTM1 null genotype has
1.5-folds (95% CI=0.9-2.5) borderline significantly increased risk for developing
lung adenocarcinoma (compared with non-null genotype). The COMT Val/Met,
Met/Met genotype has 1.7-folds (95% CI=1.1-2.8) increased risk for developing
(null vs. non-null, OR= 0.9, 95% CI=0.5-1.4), GSTP1Ile105Val polymorphism (Ile/Ile vs. Ile/Val and Val/Val, OR= 1.3, 95% CI=0.8-2.1), NAT1 (slow acetylator vs. rapid
acetylator, OR=1.2, 95% CI=0.7-2.4), NAT2 (slow acetylator vs. rapid acetylator,
OR=1.2, 95% CI= 0.6-2.3), Epoxide hydrolase Tyr113His (His/His, Tyr/His vs. Tyr/Tyr, OR=1.4, 95% CI=0.9-2.3), Epoxide hydrolase His139Arg (Arg/Arg, Arg/His vs. His/His, OR=1.2, 95% CI =0.7-2.3), do not show significant association with
female lung adenocarcinoma.
To avoid gene-gene confounding effect, we put all phase II genes into multiple
logistic regression models. The results are showed in Table 3-2. COMT Met158Val (Met/Met, Met/Val vs. Val/Val) shows a 2.2-folds increased risk for developing
female lung adenocarcinoma (95% C.I 1.2-4.0). Epoxide hydrolaseTyr113His(Tyr/His, His/His vs. Tyr/Tyr) shows a 2.0-folds increased risk (95% CI =1.1-3.7), and
GSTM1 null genotype shows borderline significantly association with female lung
adenocarcinoma compared with GSTM1 non-null genotype (OR=1.7, 95% CI
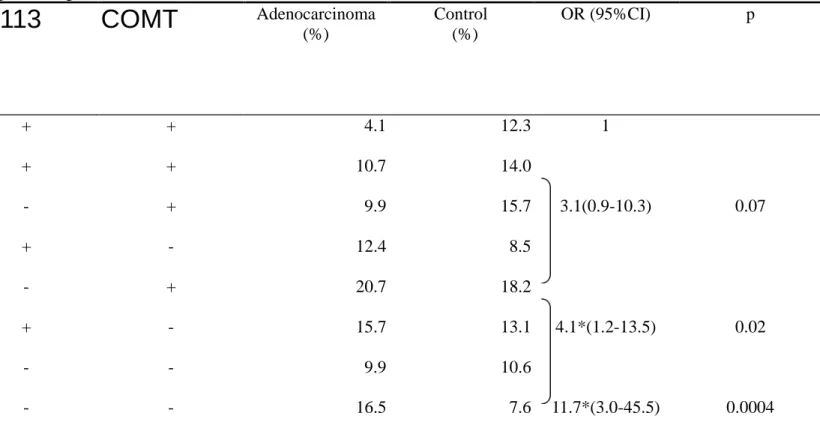
=0.9-3.1). In order to see the gene dosage effect, GSTM1, EH Tyr113His, and COMT
Met
158Val polymorphisms are combined into a model of four-level risk. The results
are shown in Table 3-3: OR=1 for those with zero putative high-risk genotype
(referent group), adjusted OR=3.1 (95% CI =0.9-10.3) for those with one putative
high-risk genotype, and adjusted OR=11.7 (95% CI =3.0-45.5) for those with three
putative high-risk genotype. A significantly dose-response relationship is noted
between the numbers of putative high-risk genotype and the risk of lung
adenocarcinoma (test for trend p<0.001). Table 3-4 presents the gene-environment
dosage effect. Tobacco exposure, cooking fume exposure, and phase II gene are
combined into a model of four-level risk. We categorize phase II gene into two
groups: one group has less than three putative high-risk genotypes; the other group
has three putative high-risk genotypes. OR=1 is for those with neither risk factor
(referent group), adjusted OR=2.9 (95% CI=1.3-6.6) is for those with one putative
high-risk factor (including any one of tobacco exposure, cooking fume exposure, or
three putative high-risk genotypes). Having two putative high-risk factors (including
any two of tobacco exposure, cooking fume exposure, or three putative high-risk
genotypes) is associated with a significantly higher risk for lung adenocarcinoma
(OR=6.0; 95% CI=2.6-13.8). Having two putative high-risk factors (those who
exposed to tobacco, cooking fume, and having three putative high risk genotypes) is
strongly associated with lung adenocarcinoma (OR=13.5, 95% CI =4.7-38.4). And a
significantly dose-response relationship is noted between the numbers of putative
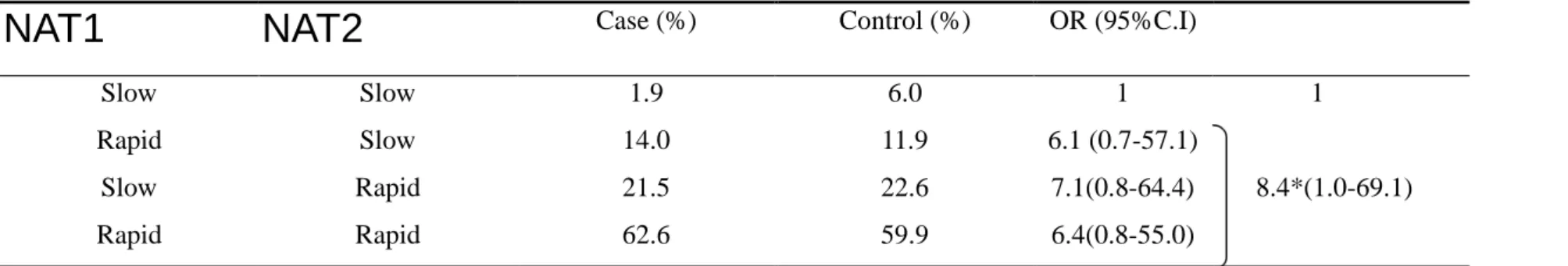
high-risk factors and the risk of lung adenocarcinoma (test for trend p<0.001). Table
subject possessing one or two rapid acetylator shows an 8.4-folds significantly
increased risk for developing lung adenocarcinoma, compared with the slow/slow
acetylator combination. Table 3-6 shows the combined effect of GSTM1, GSTT1,
and GSTP1. We found that the subjects possessing three putative high-risk
genotypes have a 2.2-folds increased risk for developing lung adenocarcinoma,
compared with those who have none putative high-risk genotype. And the subjects
possessing one or more than one putative high-risk genotype have a 1.9-folds of
increased risk compared with those who have none putative high-risk genotype, but
all do not reach statistical significance.
Table 4-1 presents the overall distribution of cases and controls and adjusted ORs
and 95%CIs by genotypes of hormone-related genes. The CYP17 A2A2 genotype
has 2.2-folds (95%C.I.=1.1-4.5) significantly increased risk for developing lung
adenocarcinoma (compared with A1A1 genotype). The COMT Val/Met, Met/Met
genotype has 1.7-folds (95% CI=1.1-2.8) increased risk for developing lung
adenocarcinoma (compared with Val/Val). Other hormone-related gene, such as
CYP19 microsatellite number and ESR codon 325 polymorphisms do not show
statistically significant association with lung adenocarcinoma.
To avoid gene-gene confounding effect, we put all hormone-related genes into
we put all the hormone related genes into the model. In model 2, only CYP17,
CYP19, and COMT are put into the model. In model 2, COMT (Met/Met, Met/Val
vs. Val/Val) shows a 1.7-folds increased risk for developing female lung
adenocarcinoma (95% C.I=1.0-2.9), and CYP17 A2A2 shows a 1.7-folds increased
risk for developing female lung adenocarcinoma (95% C.I=1.0-3.0).CYP19 and ESR codon 325 do not show statistical significance with lung adenocarcinoma. In
order to see the synthesis gene and metabolizing gene interactive effects stratified
analysis of CYP17 and COMT in relation to lung adenocarcinoma is shown in table
4-3. Those who possess CYP 17 A2/A2 and COMT Met carrier have a 4.2-folds risk
compared with those who possess CYP17 A1/A1 and COMT Val/Val. In table 4-4,
we show that the synthesis gene and metabolizing gene interactive effect is modified
by BMI of the subjects. Those who possess CYP17 A2/A2 and COMT Met carrier
have a 6.7-folds significantly increased risk compared with those who possess
CYP17 A1/A1 and COMT Val/Val among thinner subjects (BMI<=23), but only
2.5-folds non-significant risk among fatter subjects (BMI>23). In table 4-5, we show
that the synthesis gene and metabolizing gene interactive effect is modified by
hormone exposure period of the subjects. Those who possess CYP17 A2/A2 and COMT Met carrier have a 10.4-folds significantly increased risk compared with
exposure period group (<=363 months), but only 1.1-folds non-significant risk
among longer hormone exposure period group (>363 months). In order to see the
three genes gene-dosage effect, CYP17, CYP19, and COMT polymorphisms are
combined into a model of four-level risk. The results are shown in table 4-6: OR=1
for those with three putative high-risk genotypes (referent group), adjusted OR=0.5
(95%CI=0.3-0.9) for those with two putative high-risk genotypes, adjusted OR=0.4
(95%CI=0.2-0.8) for those with one putative high-risk genotype, and adjusted
OR=0.2 (95%CI=0.01-2.3) for those with zero putative high-risk genotype. A
significantly dose-response relationship is noted between the numbers of putative
high-risk genotype and the risk of lung adenocarcinoma (test for trend p<0.002).
Table 4-7 shows four genes gene-dosage effect. CYP1A1, CYP17, CYP19, and COMT polymorphisms are combined into a model of five-level risk. OR=1 for those
with four putative high-risk genotypes (referent group), adjusted OR=0.4
(95%CI=0.4-0.7) for those with three putative high-risk genotypes, adjusted OR=0.2
(95%CI=0.1-0.5) for those with two putative high-risk genotypes, and adjusted
OR=0.3 (95%CI=0.1-0.9) for those with one putative high-risk genotype. No any
cases possess zero high-risk genotype. A significantly dose-response relationship is
noted between the numbers of putative high-risk genotype and the risk of lung
Table 5-1 compares the genetic polymorphisms of four DNA-repair enzymes
between cases and controls. Cases had higher percentages of Arg/Arg and Arg/Trp
genotypes of XRCC1 codon 194, Gln/Gln genotype of XRCC1 codon 399, Thr/Met
genotype of XRCC3 codon 241, Lys/Gln and Gln/Gln genotypes of XPD codon 751,
and GA and AA genotypes of hMLH1 at -95 nucleotide. Cases and controls had
similat genotype frequency of XRCC1 codon 280. The age-adjusted OR (95% CI) of
developing lung adenocarcinoma was 5.6 (1.2-26.2) for Arg/Arg and Arg/Trp
genotypes of XRCC1 codon 194 compared with Trp/Trp genotype as the referent;
2.2 (1.1-4.6) for Gln/Gln genotype of XRCC1 codon 399 compared with Arg/Arg
and Arg/Gln genotypes; 2.8 (1.0-7.8) for Thr/Met genotype of XRCC3 codon 241
compared with Thr/Thr genotype; 2.7 (1.5-4.8) for Lys/Gln and Gln/Gln genotypes
of XPD codon 751 compared with Lys/Lys genotype; and 2.9 (1.2-7.1) for GA and
AA genotypes of hMLH1 compared with GG genotype.
There was significant correlation with genotype of XRCC1 codon 399 for genotypes
of XRCC1 codon 194 and codon 280. The percentages of Trp/Trp genotype of
XRCC1 codon 194 and His/His genotype of XRCC1 codon 280 were less than 2%.
Accordingly, only the genotype of XRCC1 codon 399 was included in the further
multiple regression analysis as shown in Table 5-2. Genetic polymorphisms of all
adenocarcinoma after adjustment for age, exposures to tobacco smoke and cooking
fume, and genotypes of other DNA-repair enzymes. The multivariate-adjusted ORs
for high-risk genotype of these DNA-repair enzymes ranged from 2.5 to 3.1. Table
5-3 presents the association with lung adenocarcinoma for the combination of
high-risk genotypes of four DNA repair enzymes. Neither cases nor controls had
high-risk genotypes of all four enzymes. There were more cases had a higher
number of high-risk genotypes than controls. A significant dose-response
relationship was observed between the risk of lung adenocarcinoma and the number
of high-risk genotypes of DNA-repair enzymes (p<0.0001 for trend). Compared
with those who had no high-risk genotype as the referent group, the
multivariate-adjusted ORs (95% CI) were 4.3 (1.0-19.67), 11.8 (2.5-54.8) and 18.9
(3.1-115.8) for those who had one, two and three high-risk genotypes, respectively.
The dose-response relationship remained statistically significant in the stratification
analyses by exposures to tobacco smoke and cooking fume. Table 5-4 shows the
effects of combination of genetic and environmental factors on the development of
lung adenocarcinoma. We categorized DNA repair gene into two groups. A
significantly increased risk of lung adenocarcinoma was observed with the number
of both environmental and genetic risk factors showing a dose-response relationship
Discussion
The cause of female lung adenocarcinoma in Taiwan remained unknown. Our
case-control study was conducted to elucidate the possible risk factors, including
active smoking, passive smoking, cooking fume exposure, hormone-related risk
factors, and personal and family history. We focused on female lung
adenocarcinoma in Taiwan, where had the lowest sex ratio of lung cancer incidence,
relatively low smoking prevalence among female lung cancer, higher proportion of
adenocarcinoma among lung cancer, and the most rapid increased rate of lung
cancer during past fifty years in the world. To the best of our knowledge, this was
the first study focusing on lung adenocarcinoma conducted in Taiwanese women. In
controls were recruited. The response rate in cases was 92%; only 8% of the cases
did not received interview due to too ill, death, and discharge. The response rate in
controls was 99.4% (350/352); only 2 eligible controls refuse to be interviewed. The
causes for not participating the study were not associated with risk factors we
intended to investigate in our study; so, it did not influence the accuracy of our
results. In our study, we did not use proxy responders in both cases and controls to
avoid information bias. The catchments area of cases was slightly different from the
catchments area of controls. However, we compared the ethnicity for cases and
controls, we found no significant difference. Our control group was selected from
health examinees. The risk factors we intended to investigate (such as smoking,
cooking, hormone-related factors) were not associated with the characteristics of the
health examinees. So, the selection bias may be limited. As to recall bias, it is
common problem in case-control study. However, the cooking habits and smoking
habits of herself or her coworker and co-inhabitants were so consistent and
unchangeable in her life, so the effects of recall bias were also limited. As to the
sample size, our study is adequate for OR=1.5, under the assumption of α
level=0.05,β level =0.8, exposure p=0.5, 1: 2 match. In most of risk factors we
investigated, the sample size was adequate.
lung adenocarcinoma occurred at younger age than male lung cancer. A small
plateau was noted between 40-60 years, and most of them were nonsmokers.
Chinese cooking style was considered as important risk factors in previous studies (2,
3-6). The cooking related items, such as cooking frequency, cooking oils, cooking
fuels, fume extractor and ventilation device were considered as important factors for
lung cancer (3-6). There were some debates as to cooking fume exposure being the
major determinants for female lung adenocarcinoma. Firstly, the use of fume
extractor in Taiwan now is very popular, but the incidence of female lung
adenocarcinoma remains steadily increased; secondly, Chinese women had cooked
for thousands of years; however, the incidence rate of lung cancer increased for
about 8-folds during past thirty years, and 50-folds during past fifty years (1). Ko (5)
had proposed several reasons: fume extractors are not positioned properly, modern
housing is small, vegetable oils were increasingly used, cohort effect and longer life
expectancy, and other risk factors (such as passive smoking, air pollution) interacted
with the mutagenicity of cooking oil fume. However, we did not think that the
reasons could fully explain the discrepancy. In Ko study (5), using fume extractor
had a more than 2-folds significant protective effect compared with no using fume
extractor before age 40. In our study, we found a non-significant risk for those who
might not be positioned properly; however, it was better than no using fume
extractor. Till now, there is no consistent evidence showing vegetable oils more
hazardous than lard oils. Cooking fume analysis showed mutagenicity and
genotoxicity in both lard oils and vegetable oils (7-8). PAHs and other carcinogens
were also found in both (7-8). And in our study, we showed much higher risk for
using lard as cooking oils (OR=2.1), compared with using vegetable oils (OR=0.7).
So, the increased usage of vegetable oils could not explain the increase of lung
adenocarcinoma. As the life expectancy prolonged, other competing cause for lung
cancer, such as other malignancy, cardiovascular disease, and cerebrovascular
diseases did not show the same magnitude of increase as lung cancer. The only
plausible reason was that modern housing is smaller than before due to the effect of
urbanization and industrialization. So, more cooking fume was exposed during past
thirty years. In our study, we found that cooking habit was not associated with lung
adenocarcinoma, because more than 90% cases and controls had cooking habits
before 40. So, the cooking habit was not the main determinant for cooking hazards.
However, we categorized the subjects having no cooking habit or cooking but using
fume extractor as “no cooking fume exposure”. Those who cooked and did not use
fume extractor were considered as having “cooking fume exposure”. We found that
adenocarcinoma. Then, we further stratified the subjects into three groups: no
cooking exposure, using vegetable oils, and using lard oils. We found lard oils had a
2.1-folds risk compared to no cooking fume exposure. We also found that using coal
or charcoal as cooking fuels possessed a 1.6-folds significant risk for developing
lung adenocarcinoma. Age at starting cooking and total cooking duration before 40
did not show any significant trends for developing lung adenocarcinoma. Our results
were slightly difference from previous studies. It seemed that cooking-related risk
factors not so important in our study. The style of cooking (stir-frying, deep frying,
and frying) and cooking frequency were also not associated with lung
adenocarcinoma (not shown in our analysis). In Chinese studies, especially
conducted in Shanghai, rapeseed oils seemed to be hazardous, and the component
analysis and mutagenicity and genotoxicity assay all showed compatible results (9).
However, in Taiwan, rapeseed oil was not used. Lard oils, soybeans oils, peanut oils,
and sunflower oils were the most often used cooking oils. In the early decades, lard
oils were not refined, and were frequently repeated used due to economic
consideration. Chinese cooking style, including deep-frying, frying, stir-frying,
might reach high temperature (250-300℃) while cooking. The unrefined lard oils
repeated used in high temperature could produce large amount carcinogens. As to
contains genotoxic PAHs (10). In China, Xuan Wei County had the highest lung
cancer incidence in both male and female. However, the smoking rate was low in
Xuan Wei. It was believed that the high incidence of lung cancer might be due to
indoor air pollution from coal combustion (11). However, there were no reports for
natural gas and electricity. So, our study showing that cooking fuels with coal, wood,
or charcoal possessing higher risk for lung adenocarcinoma was biological plausible.
Passive smoking was a proved risk factor to lung cancer. Meta-analysis for 13
studies reported in NRC (12) showed a 1.34 (95%CI=1.18-1.53) significant risk for
lung cancer. Hackshaw reported a meta-analysis recently conducted for 37 studies
(13), and showed a 1.23-folds (1.13-1.34) significant risk. As we know, the major
histological type of lung cancer related to cigarette smoking is SCC, and
adenocarcinoma is weakly associated with cigarette smoking. In our study, we found
that eve-smoker possessed only 2.4-folds of risk for lung adenocarcinoma. Some
carcinogens, such as 4-aminobiphenyl, have higher concentration in side-stream
smoke than in the mainstream smoke (up to 30 folds). One important limitation of
studies investigating the relationship between passive smoking and lung cancer was
that the true exposed amount of smoke is difficult to be measured, depending on
number of ever-smoker exposed, duration of exposure per day, and whether the
may be a useful markers, it is not yet widely used in researches. In our study, spouse
smoking exposure carried a 1.2-folds non-significant risk; however, if the spouse
smoked just besides her, the risk was increased to 1.5-folds (p<0.05). The hazard
was slightly greater than previous studies (OR=1.23) (13). In Taiwan, the husbands
often smoke just besides her wives, and the average living space is smaller than that
in American. So, the higher risk for spouse smoking exposure in Taiwan is
reasonable. In our study, we couldn’t find significant trends in spouse smoking
duration and cumulative smoking amounts, because the duration and cumulative
smoking amount couldn’t represent the true amount of smoke the subjects exposed.
For more precisely estimating the total sources of passive smoking, we must
evaluate the childhood passive cigarette smoke exposure (including father and
mother smoking history) and workplace passive cigarette smoke exposure. The
childhood passive cigarette smoke exposure is important because the early event in
life may play an important role on cancer initiation. The workplace passive cigarette
smoke exposure is important because one may spend more than 8-10 hours per day
in workplace during her adulthood. In our study, we found maternal smoking status
possessed 1.8-folds significant risk. Paternal smoking status did not possess higher
risk. We also found “more than 10 coworkers were ever-smoker” carrying a
hazard of spouse smoking. What role did the passive cigarette smoke play in the
growing epidemics of female lung adenocarcinoma in Taiwan? In 1972, the smoking
prevalence is about 30% in male, and 2-3% in female; the smoking prevalence
changed to 55-60% in male, and 3-4% in female in 1996. However, the sex ratio was
still about 2.0-2.3. The increased incidence rate of lung cancer in male might be due
to smoking epidemics and urbanization in Taiwan since 1950. How to explain the
simultaneous increase of female incidence rate? Could it be explained by
simultaneous increase in passive smoking prevalence (increase smoking rate of her
father, husband, and male co-workers during the thirty years) and the same
urbanization effect? Risch had mentioned that females are more susceptible to
smoking induced lung cancer (14). The most possible explanation was different
genetic susceptibility between sexes. Taioli had mentioned that hormone-related
factors were associated with lung cancer. He also found that smoking could interact
with hormone to contract lung cancer (15). The interaction of passive smoking with
hormone-related factors might be the possible contributor for the growing epidemics
for lung adenocarcinoma in Taiwanese women.
The most amazing finding in our study was that hormone-related factors were
associated with female lung adenocarcinoma. As we previously mentioned,
sex hormone was considered as promoter effect; however, recently, estrogen was
considered a complete carcinogen due to accumulated evidence for estrogen action
in the breast cancer and endometrial cancer (16). However, sex hormone possessed
bi-directional effect, in other words, pro-oxidant or anti-oxidant. It had been
proposed that in lower concentration of catechol estrogen, its lipid peroxidation
effect predominates and shows carcinogenic effect. In higher level, its free radical
scavenging effect predominates, and shows protective effect (17). Many papers
reported that estrogen or progesterone receptors expression in lung tumor tissues
(18-23). In our study, we found that early menopause, late menarche, more gestation
and parity, longer duration of breast-feeding all carried significant higher risk for
lung adenocarcinoma. Oral contraceptives, hormone replacement therapy, and larger
BMI all showed significant protective effect. Longer hormone exposure periods
showed non-significant. Length of menstrual period (>6 days), longer menstrual
cycle all showed significant risk for lung adenocarcinoma. What did the above
results mean? We propose that: longer and higher estrogen exposure seemed to be
protective in lung adenocarcinoma in Taiwan, so we obtained just opposite results to
breast cancer. After menopause, estrogen was mainly from peripheral fat tissue
conversion. The more obese the subjects are, the higher serum level of estrogen the
level, thus showing protective effect for lung adenocarcinoma. In our study, the
number of cases who were not menopause was so small that we could not further
stratify it to test the interaction between BMI and menopause status. It had been
reported (24) that mean serum estrogen level in Chinese women is lower than that in
American. The results may be due to dietary factors (low fat and cholesterol), less
obesity, and genetic components. So, among nonsmokers, the incidence rate of lung
cancer in female is higher in Chinese women than in Caucasian women; however,
the breast cancer incidence is much higher in Caucasian than in Chinese women.
Taioli showed that smoking might interact with hormone to lung cancer (15). So, the
baseline higher incidence of lung adenocarcinoma in Chinese women might be due
to lower mean level of estrogen compared with Caucasian, and the growing
epidemics of female lung adenocarcinoma in Taiwan might be due to interaction of
passive smoking, cooking fumes with sex hormone. What is the precise mechanism
of interaction? We know that some CYP enzymes, such as CYP1A1, CYP1A2, and CYP1B1 were all responsible for metabolizing catechol estrogen. And the CYPs
enzymes expression was modulated by Ah receptor, which is in induced by many
inducers, such as dioxin, or some substances from tobacco smoke. In other word,
tobacco smoke and dioxin may induce CYPs enzymes expression, thus influencing
know that Taiwan is an area of heavy industrial pollution, and dioxin is especially
notorious. Another study will be needed to elucidate the impact of environmental
hormone.
If the sex hormone acted opposite roles among lung adenocarcinoma and breast
cancer; as the western life style becoming more popular, the breast cancer incidence
will be increased, and we can infer that: the incidence of lung adenocarcinoma will
be decreased in the future. In Taiwan, the breast cancer incidence increased rapidly
in recent decades; however, the incidence of lung adenocarcinoma did not decrease
recently. However, some trend was still noted: the sex ratio increased gradually
(1962-66, sex ratio= 1.6; 1987-1991, sex ratio=2.3) (25). In other words, the female
adenocarcinoma increased more slowly than male in past thirty years. We proposed
that the incidence rate of female lung adenocarcinoma in Taiwan might be decrease
in the future.
Personal medical history was considered as risk factor for lung cancer in previous
studies. Among them, pulmonary tuberculosis was most important. Cohort study (26)
and case-control studies (27) all showed pulmonary tuberculosis associated with
lung cancer, especially adenocarcinoma. Pulmonary tuberculosis is associated with
lung cancer in several aspects: pulmonary tuberculosis is the risk factor for lung
tuberculosis is the competing cause of death for lung cancer; pulmonary tuberculosis
may be misdiagnosed as lung cancer; and lung cancer may be misdiagnosed as
tuberculosis. In Taiwan, the age-adjusted mortality rate of tuberculosis per 100,000
person was 88.6 for male, 46.6 for female in 1960, and 18.7 for male, 5.0 for female
in 1991, decreased about 5-folds in male and 9-folds in female during past thirty
decades (25), largely contributed to nutrition status improvement and widely use of
anti-TB drug. However, in Taiwan, drug compliance was so poor that the
tuberculosis often was not completely treated and relapsed frequently. Persistent
inflammatory lung condition may provide the adequate environment for tumor
formation. And according to the competing cause of death theory: if two diseases
had the same etiology, as the one mortality decreased, the other one mortality
increased simultaneously. This phenomenon might explain why female lung
adenocarcinoma increased rapidly during the thirty years. Indirect evidence can
support the hypothesis: in Taiwan, Aborigine had the highest smoking rate among
different ethnicity; however, the lung cancer rate was lowest. And the pulmonary
tuberculosis mortality rate was highest among different ethnic group. In our study,
we found a 2.4-folds of risk for tuberculosis to develop lung adenocarcinoma.
However, in multiple logistic regression analysis, the association became
variables. The possibility of inaccurate recall of tuberculosis history existed.
However, tuberculosis is a major event of life, and needs to take drug for a long
duration. So, the possibility of recall bias was in limited range. Several studies had
been conducted to evaluate the association of chronic obstructive pulmonary disease
(chronic bronchitis and emphysema) with lung cancer (28-29), and the results
mostly supported the association between lung cancer and COPD. However, in our
study, we did not find any association with lung adenocarcinoma. COPD is highly
correlated with cigarette smoking; however, in Taiwanese cohort study (30), the
association of cigarette smoking with COPD was not so strong as that in western
population. In Taiwan, COPD was frequently misdiagnosed as asthma or congestive
heart failure. Thus, no association of COPD with lung adenocarcinoma might be the
result of misclassification bias. In this situation, a medical record was more reliable
than questionnaire interview. Other personal history, such as asthma, hysterectomy,
and oophorectomy were also not associated with lung adenocarcinoma. Wu (31) had
found the association of hysterectomy with lung cancer, and he proposed that pelvic
thrombus during hysterectomy which might produce multiple showers of small
emboli in the lungs, resulting in localized proliferative changes in the bronchial
epithelium, thus causing lung cancer. We think that the hypothesis has too many
association of hysterectomy with lung adenocarcinoma, because hysterectomy did
not influence the hormone status. As to oophorectomy, we did not classify the age
(pre-menopausal or post-menopausal), the causes (incidental or for cancer treatment),
and the methods (unilateral or bilateral). So we could not accurately estimate the
effect of oophorectomy on lung adenocarcinoma.
As to family history, previous studies (32-33) had shown that first-degree relatives
carried a two to five folds risk for developing lung cancer. In our study, we found
that the risk of family history differs between sexes. Among parents, mother
contracting lung cancer carried an 8.9-folds significant risk, but father contracting
lung cancer did not have any increased risk (OR=1.1). Sibling contracting lung
cancer had a 5.6-folds significant risk for lung adenocarcinoma, however, the sisters
contracting lung cancer possessing higher risk compared with brother (not shown
here). According to our results, we found that: all first-degree relatives excluding
father had a 4.9-folds for developing lung adenocarcinoma. Combined with the
finding that hormone-related factors were associated with lung adenocarcinoma, we
propose that: the hormonal factors related to lung adenocarcinoma were genetic
inherited, and were transmitted among female relatives. So, hormone-related gene
and X chromosome may be the target for linkage analysis in the future family study.
smoking were still important risk factors for lung adenocarcinoma in Taiwan. Higher
prevalence of passive smoking among Taiwanese women may be the contributor to
female lung adenocarcinoma epidemics during the fifty years. Cooking fume
exposure was also the contributors for the lung adenocarcinoma; however, its
importance was limited. Hormone-related risk factors were important determinants
for lung adenocarcinoma. Higher and longer estrogen exposure had lower risk for
lung adenocarcinoma, just opposite to the results found in breast cancer. The
interaction between sex hormone and tobacco smoke may be the major contributors
to lung adenocarcinoma epidemics. Family history carried a high risk for lung
adenocarcinoma, especially female first-degree relatives. The decline of tuberculosis
mortality during the thirty years might be another contributor to lung
adenocarcinoma. Further studies for evaluating the genetic contribution for lung
adenocarcinoma were needed. Family study and genome-wide scan for major genes
or large-scale genetic association study for candidate genes approaches might be the
Refer ences
1. Department of Health, Executive Yuan, Republic of China: Health Statitics. Vol II, Vital Statistics, 1981-1997, Taipei, Department of Health, 1982-1997.
2. MacLennan R, Da Costa J, Day NE, et al. Risk factors for lung cancer in Singapore Chinese, a population with high female with high female incidence rate. Int J Cancer, 20: 854-860, 1977.
3. Law CH, Day NE, Shanmugaratnam K. Incidence rates of specific histological types of lung cancer in Singapore Chinese dialect group and their etiological significance. Int J Cancer, 17: 304-309, 1976.
4. Liu ZY, He XZ, Chapman RS. Smoking and other risk factors for lung cancer in Xuan Wei, China. Int J Epidemiol, 20: 26-31, 1991.
5. Ko YC, Lee CH, Chen MJ, et al. Risk factors for primary lung cancer among non-smoking women in Taiwan. Int J Epidemiol, 26: 24-31, 1997.
6. Gao YT, Blot WJ, Zheng W, et al. Lung cancer among Chinese women. Int J Cancer, 40: 604-609, 1987.
7. Wu PF, Chiang TA, Ko YC, Lee H. Genotoxicity of fumes from heated cooking oils produced in Taiwan. Environ Res Section A, 80: 122-126, 1999.
8. Wu PF, Chiang TA, Wang LF, Chang CS, Ko YC. Nitro-polycyclic aromatic hydrocarbon contents of fumes from heated cooking oils and prevention of mutagenicity by cetechin. Mutat Res, 403: 29-34, 1998.
9. Zhong LJ, Goldberg MS, Gao YT, Jin F. Lung cancer and indoor air pollution arising from Chinese-style cooking among nonsmoking women living in Shanghai, China. Epidemiol, 10: 488-494, 1999.
10. Oanh NTK, Reutergårdh LB, Dung NT, Emission of polycyclic aromatic hydrocarbons and particulate matter from domestic combustion of selected fuels. Environ Sci Technol, 33: 2703-2709, 1999.
11. Chuang JC, Wise SA, Cao S, Mumford J. Chemical characterization of
mutagenic fractions of particles from Indoor Coal Combustion: A study of lung cancer in Xuan Wei, China. Environ Sci Technol, 26 (5): 999-1004, 1992. 12. National Research Council Committee On Passive Smoking Environmental
Tobacco Smoke: Measuring Exposures and Assessing Health Effects, Washington, DC, National Academy Press, 1986.
13. Hackshaw AK, Law MR, Wald NJ. The accumulated evidence on lung cancer and environmental tobacco smoke. Brit Med J, 315: 980-988, 1997.
14. Risch HA, Howe GR, Jain M, et al. Are female smokers at higher risk for lung cancer male smokers? Am J Epidemiol, 138: 281-293,1993.
women. J Natl Cancer Inst, 86: 869-870, 1994.
16. Huang CS, Chern HD, Chang KJ, Cheng CW, Hsu SM, Shen CY. Breast cancer risk associated with genotype polymorphism of the estrogen-metabolizing genes
CYP17, CYP1A1, and COMT: A multigenic study on cancer susceptibility.
Cancer Res, 59: 4870-4875, 1999.
17. Dabrosin C, Hammer M, Olinger K. Impact of oestradiol and progesterone on antioxidant activity in normal human breast epithelial cells in culture. Free Radic Res, 28: 241-249, 1998.
18. Beattle CW, Hansen NW, Thomas PA. Steroid receptors in human lung cancer. Cancer Res, 45: 4206-4214, 1985.
19. Cagle PT, Mody DR, Schwartz MR. Estrogen and progesterone receptors in bronchogenic carcinoma. Cancer Res, 50: 6632-6635, 1990.
20. Ollayos CW, Riodan GP, Rubin JM. Estrogen receptor detection in paraffin sections of adenocarcinomas of colon, pancreas, and lung. Arch Pathol Lab Med, 118: 630-632, 1994.
21. Canver CC, Memoli VA, Vanderveer PL, et al. Sex hormone receptors in non-small cell lung cancer in human beings. J Thorac Cardiovas Surg, 108: 153-157, 1994.
22. Su JM, Hsu HK, Chang H, et al. Expression of estrogen and progesterone receptors in non-small cell lung cancer: immunohistochemical study. Anticancer Res, 16: 3803-3806, 1996.
23. Kaiser U, Hofmann J, Schilli B, et al. Steroid-hormone receptors in cell lines and tumor biopsies of human lung cancer. Int J Cancer, 67: 357-364, 1996. 24. Bernstein L, Yuan JM, Rose RK, et al. Serum hormone levels in
pre-menopausal Chinese women in Shanghai and white women in Los Angels: results from two breast cancer case- control studies. Cancer Causes control, 1: 51-58, 1990.
25. Department of Health, Executive Yuan, Republic of China: Health Statitics. Vol II, Vital Statistics, 1960-1991, Taipei, Department of Health, 1961-1991.
26. Hinds MW, Cohen HI, Kolonel LN. Tuberculosis and lung cancer risk in nonsmoking women. Am Rev Respir Dis, 125: 776-778, 1982.
27. Zheng W, Blot WJ, Liao ML, et al. Lung cancer and prior tuberculosis infection in Shanghai. Br J Cancer, 56: 501-504, 1987.
28. Wu AH, Fontham ETH, Reynolds P, et al. Previous lung disease and risk of lung cancer among lifetime nonsmoking women in the United States. Am J Epidemiol, 141: 1023-1032, 1995.
29. Alavanja MCR, Brownson RC, Boice JD, Hock E. Preexisting lung disease and lung cancer among nonsmoking women. Am J Epidemiol, 136: 623-632, 1992.