國立臺灣大學醫學院微生物學研究所 碩士論文
Graduate Institute of Microbiology, College of Medicine
National Taiwan University Master Thesis
大鼠實驗性心內膜炎模式中轉糖鏈球菌形成生物模 之機制
Mechanism of Streptococcus mutans biofilm formation in the rat model of experimental endocarditis
鄭鴻偉
Hung-Wei Cheng
指導教授:賈景山 博士 Advisor: Jean-San Chia, Ph.D.
中華民國九十八年七月
July, 2009
中文摘要
轉糖鏈球菌(Streptococcus mutans)主要存在於人體口腔中,是造成齲齒的 主要病原菌。而當轉糖鏈球菌藉由口腔傷口進入血流,造成暫時性的菌血症,存 在於血流中的細菌黏附到受損的心臟瓣膜上,形成贅疣(vegetation)引發感染性 心內膜炎(infective endocarditis)。然而在以往的研究中,血小板在感染性心內膜 炎中扮演甚麼角色並不清楚。本實驗發現在缺乏血小板的血漿中,細菌生物膜的 形成會明顯被抑制,而當血小板存在時卻可以幫助細菌生物膜的形成。當外加入 不同的血小板活化抑制劑也可以明顯觀察到細菌生物膜形成的減少,此外,當給 與感染性心內膜炎大鼠抗凝血劑阿斯匹靈後,也可以有效干擾細菌體內生物膜的 形成。這些結果暗示了血小板的活化對於細菌在血漿中形成生物膜是重要的。而 進一步將血小板與細菌進行黏附,發現在人類免疫球蛋白存在下轉糖鏈球菌才能 有效與血小板結合,顯示轉糖鏈球菌可能利用人類免疫球蛋白與血小板黏附並活 化血小板有助其體內生物膜的形成。除了宿主因子與細菌的交互作用外,細菌本 身是利用怎樣的調控機制也是值得研究,在本實驗室奇玄學長研究中發現一個 response regulator 的缺失可以影響轉糖鏈球菌體內生物膜的形成,而在 microarray 中也發現 pspC 基因會明顯的被 response regulator 所調控。利用染色質免疫沉澱法 與膠體電泳阻滯實驗也發現此 response regulator 可能直接調控 pspC 基因。而在 pspC 基因有所缺失的菌株中也可以明顯觀察到體外與體內生物膜形成的缺失。然 而,測試多種細菌與宿主因子結合的實驗,發現野生株與PspC 突變株與宿主因子 結合並沒有明顯差異。顯示PspC 在轉糖鏈球菌體內生物膜形成扮演重要角色,卻 不影響轉糖鏈球菌與宿主因子的結合。
Abstract
Streptococcus mutans is one of the principal causative agents of caries and an opportunistic pathogen of infective endocarditis (IE). Bacteremia is caused by S. mutans through the oral trauma and the circulating bacteria can adhere to the damaged valve to form a compact structure, vegetation (fibrin-platelet bacterial biofilm). However, the role of platelet is controversial in the pathogenesis of IE. In this study, plasma without platelets could inhibit the bacterial biofilm formation but enhance biofilm formation in the addition of platelets, which suggested platelets play an important role in the in vivo biofilm formation. After the addition of platelet activation inhibitors, the bacterial biofilm was reduced. When one of the platelet activation inhibitor, Aspirin, was applied to the IE rat model, the bacterial biofilm could be disturbed and lower bacterial load was obtained from the vegetation. These results suggest the activation of platelets is important in the pathogenesis of IE. To further dissect the interaction between platelets, plasma factors and bacteria, purified platelets are incubated with different plasma ingredients. Human immunoglobulin is discovered to essentially involve in the adherence of bacteria on the platelets. Except for the interaction of bacteria and platelets, the regulatory mechanism in the pathogenesis of IE is also studied. In our lab previous study, the defect in response regulator 11 can lead to the defect in IE biofilm formation and pspC gene is supposedly regulated by response regulator 11 in the microarray data.
Chromatin immunoprecipitation and electrophoretic motility shift assay are performed in this study and further confirm response regulator 11 may directly regulate pspC gene.
The deletion in pspC gene has defect in the in vitro and in vivo biofilm formation and this defect can be restored in the pspC gene complementation strain. However, no significant differences are obtained in the interaction of bacteria and host factors between PspC mutant and wildtype GS5. These results suggest the role of PspC protein may involve in the in vivo bacterial biofilm formation but not the interaction with host factors.
目錄
中文摘要………...….……1
英文摘要………...2
目錄……….…...3
圖表目錄………....7
第一章 緒論……… ………...8
一、 轉糖鏈球菌(Streptococcus mutans)的介紹 ……….8
二、 感染性心內膜炎 ……….………...8
三、 Two component regulatory systems ………9
四、 細菌毒力因子與感染性心內膜炎的發生………10
(一) 細菌表面黏附因子…..………..10
(二) 細菌生物膜形成因子………..………..10
五、 血小板與細菌間的交互作用……….….…...11
六、 血小板與感染性心內膜炎的發生……….………12
七、 研究目的與實驗設計……….………13
第二章 實驗材料與方法……….………14
一、 Streptococcus mutans 的轉形作用……….………….14
二、 生物膜形成能力測定……….…….14
三、 人類血小板懸浮液(platelet suspension)的製備………….……14
四、 體外生物膜形成模式的建立……….…….15
五、 細菌黏附活化血小板試驗……….……….15
六、 模擬體內生物膜形成之體外模式……….…….16
七、 實驗性心內膜炎之老鼠模型的建立……….…...16
八、 細菌處理與感染老鼠之方法……….….17
九、 抗凝血藥物阿斯匹靈(Aspirin)給與感染性心內膜炎大鼠之方 法……….……….17
十、 以抗體 anti-CD42d 染感染性心內膜炎贅生物中血小板之方 法………...……….17
十一、 染色質免疫沉澱技術……….……….17
十二、 膠體電泳阻滯實驗……....………..19
十三、 全血殺菌能力試驗(whole blood killing assay) …...….….19
十四、 細菌與血漿中纖維蛋白原結合試驗……….….20
十五、 細菌與血小板的結合試驗……….…….20
第三章 結果……….…..22
第一部分:血小板參與轉糖鏈球菌所引發感染性心內膜炎中生物膜形 成………...22
一、 血小板對於血漿中轉糖鏈球菌形成生物膜的重要性..……...22
二、 血小板的活化對於轉糖鏈球菌生物膜的形成……….…22
三、 活化的血小板本身而非血小板活化後所釋放的因子對於轉糖 鏈球菌於血漿中形成生物膜是重要的………....….22 四、 血小板活化抑制劑 Aspirin 對於感染性心內膜炎大鼠中細菌生
物膜的形成……….…23 五、 血小板抑制劑影響細菌與活化血小板的黏附……….…23 六、 感染性心內膜炎體內細菌生物膜與血小板的交互作用….…24
第二部分:Response regulator 11 藉由調控 pspC 基因來影響感 染性心內膜炎中,轉糖鏈球菌生物膜的形成
……….………...25一、 利用染色質免疫沉澱法(Chromatin immunoprecipitation)研究 response regulator 11 在細菌體內是否能直接調控 pspC 基因的 表現………..………...…...….…25 二、 膠體電泳阻滯實驗(electrophoretic motility shift assay) 研究
response regulator 11 是否可與 pspC 前可能的啟動子區位結 合……….………...….26 三、 利用共軛焦螢光顯微鏡觀察富含血小板血漿中 Wild-type GS5、rr11 mutant、pspC mutant 與 pspC complementation strain 生物膜形成的差異.……....………..…….26 四、 利用共軛焦螢光顯微鏡觀察感染性心內膜炎大鼠贅疣中野生
株 GS5、rr11 mutant、pspC mutant 與 pspC complementation
strain 生物膜形成的差異………..…………...……..27 五、 檢測野生株 GS5 和 pspC mutant 與宿主因子結合之差….…27 六、 檢測野生株 GS5 和 pspC mutant 在生物體內及全血中生存能
力……….28 第四章 討論………..……….………29
一、血小板參與轉糖鏈球菌引起之感染性心內膜炎中生物膜之形 成………...29 二、PspC 蛋白表現的缺失影響轉糖鏈球菌體內生物膜的形成….32
第五章 參考文獻……….…………..35
圖表目錄
Figure1. Platelets are important in the S. mutans biofilm formation in the
plasma. ……….………….41
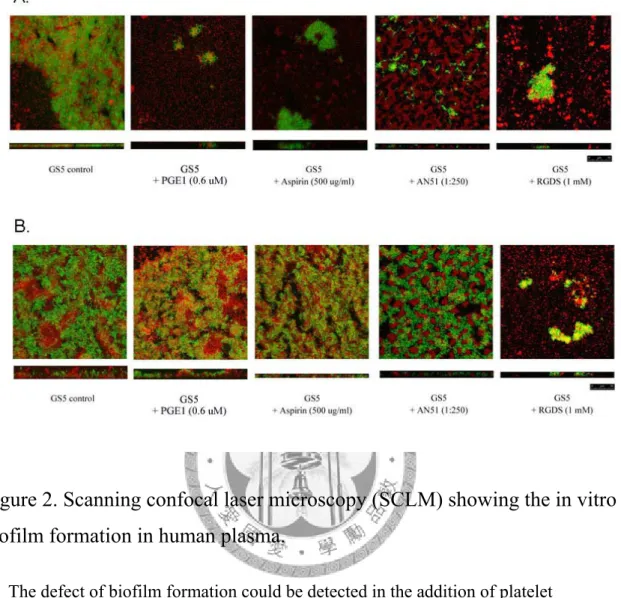
Figure 2. Scanning confocal laser microscopy (SCLM) showing the in vitro biofilm formation in human plasma. ……….………...43
Figure 3. Platelets are important host factors involving in the bacterial biofilm formation in infective endocarditis. ………44
Figure 4. In vitro model to mimic in vivo biofilm formation. ………….45
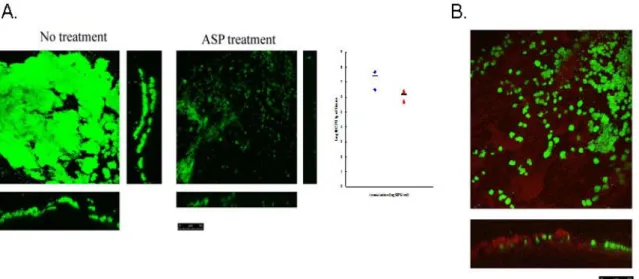
Figure 5. Platelet activation inhibitors have no effect in the BHI- incubated biofilm formation. ………...46
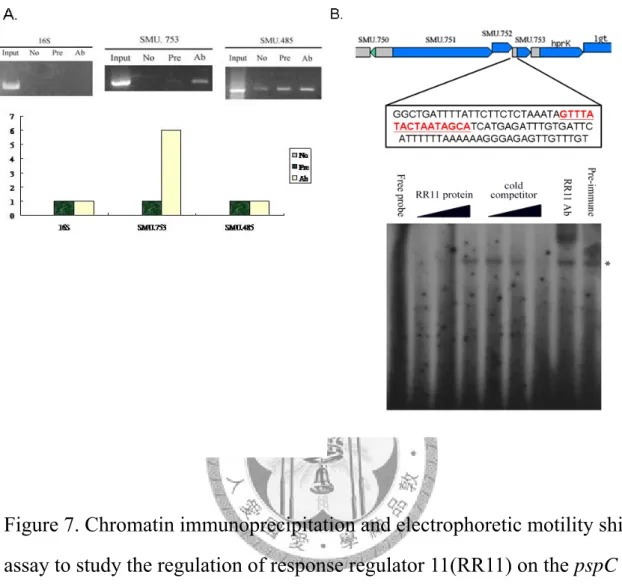
Figure 7. Chromatin immunoprecipitation and electrophoretic motility shift assay to study the regulation of response regulator 11(RR11) on the pspC gene. ………...……….………47
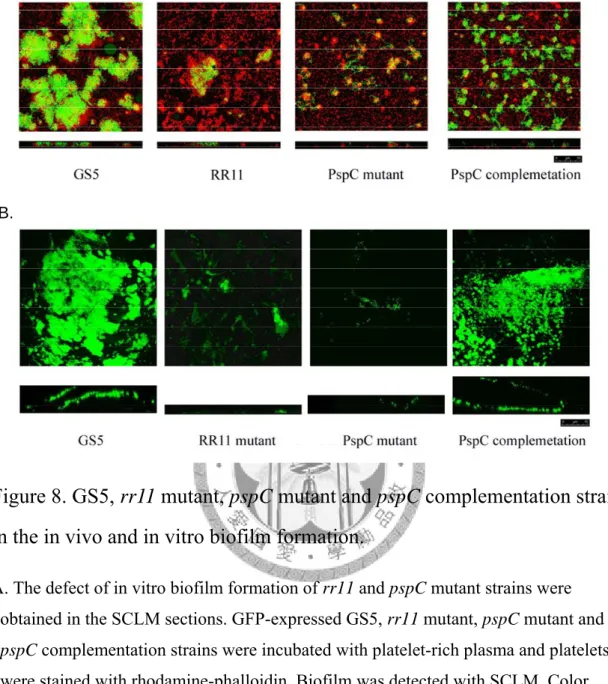
Figure 8. GS5, rr11 mutant, pspC mutant and pspC complementation strain in the in vivo and in vitro biofilm formation. ……….………48
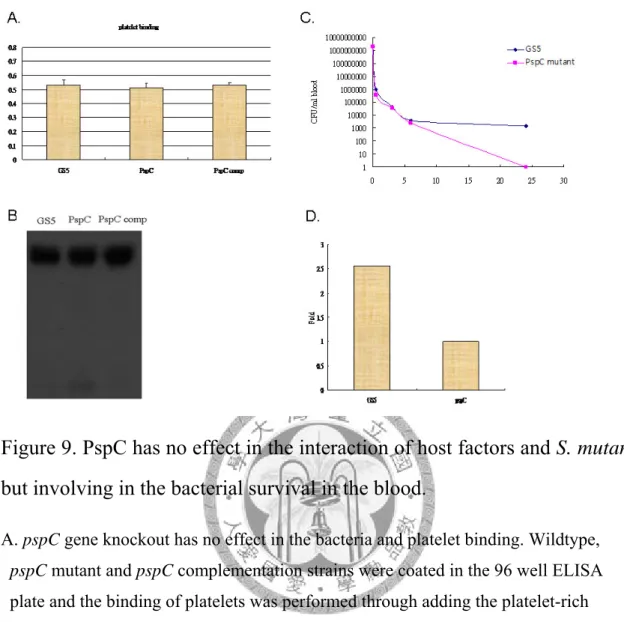
Figure 9. PspC has no effect in the interaction of host factors and S. mutans, but involving in the bacterial survival in the blood. ………...49
表 1、metamorph 軟體定量分析結果………..50
附圖 1、草綠色鏈球菌引起心內膜炎的初期致病機轉………..51
附圖 2、Two component regulatory system work model………..52
附圖 3、Phage-shock-protein response………...……..53
壹、緒論
一.轉糖鏈球菌(Streptococcus mutans)
轉糖鏈球菌存在於人體的口腔之中,為引起齲齒的主要致病菌。轉糖鏈球菌 最早在1924 年,由 Clarke 在人類齲齒窩洞內所分離出的革蘭氏陽性菌,培養於含 有綿羊血的培養基上會出現草綠色的不完全溶血(α-hemolysis)的菌落,屬於草 綠色鏈球菌屬(viridans streptococci)。但由於在顯微鏡下所觀察的形狀,並不同於 一般鏈球菌呈球狀,而為橢圓的短桿狀,因此推測可能為鏈球菌的突變株,故命 名為 Streptococcus mutans。
在2002 年時,血清型 c 型的 Streptococcus mutans 菌種 UA159 基因組已經被完整 定序(Honggui Jia et al., 2002)。S. mutans 具有一條環狀的染色體,整個基因組具 有2,030,936 個鹼基對,其中 GC 含量僅佔 36.82%,和其他低 GC 含量的菌種類似。
基因組中包含了1,963 個 ORFs(Open Reading Frame),其中 63%被認為是未知功 能,21% 和其他菌種有同源性,16%則是 S. mutans 特有的序列。經由和其它已知 基因組的革蘭氏陽性菌比對,發現 S. mutans 的 ORFs 和 Streptococcus pneumoniae 及 Streptococcus pyogenes 最為相似。
S. mutans 引起齲齒的第一個步驟為細菌利用表面蛋白附著於含有蔗糖的 牙齒表面,再利用本身具有的葡萄糖傳遞酵素(glucosyltransferses , GTFs)合成葡 聚糖(glucan)。隨著葡聚糖的產生,使得細菌附著於牙齒表面更加緊密,而透過 細菌表面的葡聚糖結合蛋白(glucan-binding protein)作用下,使得大量的轉糖鏈 球菌相互聚集而形成牙菌斑(dental plaque)。細菌所合成的葡聚糖之最終代謝產物 為乳酸則會導致酸化,使牙齒表面之牙釉質酸蝕而造成齲齒。
二.感染性心內膜炎
草綠色鏈球菌為口腔中正常菌叢,除了可能形成牙菌斑及攝取過多蔗糖導致 齲齒外,平時並不會對人體造成傷害。但當他們有機會進入血液中,形成暫時性
菌血症時,便可能經由血液循環進入心臟,進而黏附於先天有缺陷的心臟瓣膜或 是人工心瓣膜上,形成一種贅生物,稱為贅疣 (Vegetation) (Moreillon et al., 2004)。
而贅疣在心臟瓣膜上形成後,細菌除了黏附在贅疣上,也會被血小板與纖維蛋白 (Fibrin)所覆蓋,形成生物膜 (Biofilm)。藉由生物膜作為屏障,細菌得以在心臟瓣 膜上增殖,並且抵抗宿主免疫細胞的毒殺與抗生素的攻擊(附圖一)。
依照感染細菌的不同,感染性心內膜炎可分為兩型:(1)急性心內膜炎 (acute bacterial endocarditis),主要由金黃色葡萄球菌 (Staphylococcus aureus)所引起。為 爆發型的急性感染,病程發展迅速,瓣膜破壞快速,而在病變部位聚集大量纖維 母細胞 (fibroblast)與單核球 (mononuclear leukocyte),此型心內膜炎的贅疣通常較 大且易脫落,受傷部位也會聚集較多嗜中性球 (neutrophil)與病原體。(2)亞急性心 內膜炎(subacute bacterial endocarditis),感染症狀較輕微,但病程較長,可持續六 週至三個月。草綠色鏈球菌則常引起亞急性心內膜炎,當心瓣膜存在有無菌性贅 疣時,細菌才會附著上去(Moreillon and Que 2004)。
三.Two component regulatory systems
細菌經常藉由調控結構組成、生理狀態和行為模式以適應外界環境。在細菌 中最常見的一種訊息傳遞系統two component regulatory systems(TCS),使細菌面 對外界環境改變,例如溫度、滲透壓、養分來源等,可活化或抑制某些基因的表 現,調整細菌的結構組成或生理行為 ( Stock et al., 2000 )。這種系統一般由兩種蛋 白質所組成,其一為位於細胞膜上的histidine kinase,此蛋白可藉由感應外在環境 刺激而自體磷酸化histidine kinase 上的 histidine 殘基。最後再將高能的磷酸根傳遞 給response regulator N 端上的 aspartate 殘基,當 response regulator 被磷酸化,蛋白 質會發生結構性的改變,進而調控下游基因的表現 (West and Stock, 2001) (附圖 二)。
在以往的研究中發現轉糖鏈球菌中共有十四對two component regulatory systems,其中已經有許多被證實與生物膜形成、耐酸及 genetic competence 有關,
包括VicRK、CiaH、ComCD (Levesque et al., 2007;Qi et al., 2004;Li et al., 2002)。
但在轉糖鏈球菌中,two component regulatory systems 是否參與了心內膜炎的發
生,現今仍不是很清楚。
四.細菌毒力因子與感染性心內膜炎的發生 1.細菌表面黏附因子:
細菌的表面黏附因子,如:adhesin、fibrinogen-binding protein、
glucosyltransferase以及一些與胞外基質蛋白結合的分子,是決定細菌與宿主細胞外 基質結合的重要因素,這些因子統稱為Microbial surface components recognizing adhesive matrix molecules (MSCRAMMs)。在急性心內膜炎發生初期,金黃色葡萄 球菌可利用MSCRAMMs與內皮細胞進行結合,進而黏附到心臟瓣膜上。而現今已 發現,金黃色葡萄球菌中兩個重要的調控子,accessory gene regulator (agr)與 staphylococcal accessory regulator (sar),具有調控MSCRAMMs的表現的能力 (Cheung et al., 1994)。且在兩個調控子發生缺失的金黃色葡萄球菌突變株中,其導 致的感染性心內膜炎有明顯的減弱,顯示兩個調控子在金黃色葡萄球菌所引發的 心內膜炎中扮演著重要的角色 (Cheung et al., 1994)。而在亞急性心內膜炎的發生 的初期,草綠色鏈球菌必須先利用表面的黏附因子先與fibrin-platelet形成的無菌性 贅疣結合,而進一步黏附到心臟瓣膜後,才能造成心內膜炎的發生。相關草綠色 鏈球菌MSCRAMMs的研究,現在較為清楚的是Streptococcus parasanguis的FimA;
將此基因剔除後,細菌結合fibrin的能力大幅降低,導致老鼠心內膜炎的發生率下 降(Burnette-Curley et al.,1995)。此外,研究發現將FimA打入動物體內,使其產生 抗體後可預防心內膜炎的產生(Viscount et al.,1997)。但是Caldelari在1999年發表 的文獻中指出,將S. gordonii的SecA(FimA homologue)剔除後,卻無法降低心內 膜炎的感染率。所以不同細菌其致病的機轉與毒力因子也有所不同。
2.細菌生物膜形成因子:
在醫學上,大約65%的細菌感染症有生物膜的參與,在慢性感染症發生過程中,細 菌會適應環境並形成生物膜幫助細菌抵抗許多抗生素藥物的攻擊。在細菌感染的 過程中,細菌進入人體到達感染的目標組織後,會先利用表面黏附分子與宿主細 胞結合,進行黏附。隨後,細菌會形成微聚落(microcolony),並且釋放出一些胞外
多醣(exopolysaccharide)包覆這樣的細菌微聚落,完成生物膜的構成(Hall-Stoodley et al., 2004)。在Gram negative細菌中,Pseudomonas aeruginosa 已經被發現可以在 囊狀症纖維化(cystic fibrosis)的病人中,形成完整的生物膜,造成P. aeruginosa抵抗 抗生素的能力增加,導致治療上的困難 (Murray et al., 2007)。而感染性心內膜炎的 發生一般也認為與生物膜的形成有關。然而,在Streptococcus godornii中卻發現,
體外生物膜形成有所缺失的突變株並不會去影響其造成感染性心內膜炎發生的能 力 (Bizzini et al., 2006),顯示體外生物膜的建構方法並不能完全模擬體內生物膜發 生的狀態。
五.血小板與細菌間的交互作用
血小板為直徑2-4 μm,圓形或橢圓型的碟型物,主要是由骨髓中的
megakaryocyte 所產生。一般人一毫升的血漿中約有 3X108至15X108個血小板,血 小板沒有核但保留了內質網、高基氏體、粒線體等胞器,並且在細胞質內有許多 濃稠體(dense granule)以及 α 顆粒(α-granule)存在。α-granule 中儲存許多蛋白質,
如fibronectin、fibrinogen、von Willebrand factor (vWF)等;dense granule 則儲存一 些小分子,如ADP、ATP、Ca2+、Mg2+等。在以往的研究發現,有些細菌感染宿 主後,可以藉由表面的一些分子直接活化血小板或透過誘發大量tissue factor 間接 活化凝血機轉,而其中研究較多的是 Staphylococcus aureus 與 viridans group of Streptococci。在 S. aureus 中已經發現一個蛋白 clumping factor A(ClfA),可以藉由 與血漿中的纖維蛋白原(Fibrinogen)結合後,進一步與血小板黏附,然而這樣的黏 附並不足以誘發血小板的活化。在ClfA 與纖維蛋白原結合後,還需要 anti-ClfA 的 人類免疫球蛋白與ClfA 結合,細菌可以藉由免疫球蛋白(human immunoglobulin) 的Fc 區位與血小板的 FcγRIIa 結合,進一步誘發血小板的活化與凝集(Fitzgerald et al., 2006)。此外,S. aureus 也具有另一個蛋白 Fibronectin-binding protein A(FnBPA),
也具有黏附與活化血小板功能。FnBPA 的作用機制類似於 ClfA,需要與纖維蛋白 原結合後幫助 S. aureus 先與血小板 GPIIb/IIIa 受體黏附後,更進一步藉由與 anti-FnBPA 的免疫球蛋白的 Fc 區位與血小板的 FcγRIIa 結合,誘發血小板的活化 (Fitzgerald et al., 2006)。然而,FnBPA 也被發現具有一段與 fibronectin 結合的區位 BCD domain 可以直接誘發血小板的凝集。而在先前的研究中也發現 S. aureus 表現
這兩種蛋白的時機並不相同,在細菌生長對數期(exponential phase)時,FnBPA 蛋 白會大量的表現,而ClfA 表現較為低落(Saravia-Otten et al., 1997),直到進入生長 靜止期(stationary phase)ClfA 蛋白才會被大量表現(Que et al.,2005)。相關的研究在 viridans group of Streptococci 中也有許多的發現,在 Streptococcus pyrogenes 中,M protein 被發現具有活化並誘發血小板凝集的能力,且 M protein 的作用機制也被發 現需要透過人類免疫球蛋白與補體(Complement)的作用(Beachey et al., 1971)。
Streptococcus sanguis 也被發現可以促使血小板的凝集,而且也需要免疫球蛋白參 與(Sullam et al., 1988),而 S. sanguis 表現有 Platelet aggregation-associated protein (PAAP)與 serine-rich protein A (SrpA) (Erickson et al., 1990;Plummer et al., 2005),
PAAP 已經被發現可以促使血小板的凝集,而 SrpA 蛋白則被發現可能與血小板的 GPIb 受體結合而與血小板黏附。在 Streptococcus gordonii 中也發現類似的蛋白 GspB 與 Hsa,兩個蛋白都被發現可與血小板的 GPIb 結合並刺激血小板活化 (Bensing et al., 2004),且 Hsa 依照序列結構也被猜測很有可能與血小板另一個受體 GPIIb/IIIa 結合(Yajima et al., 2005)。而在 Streptococcus agalactiae 中,FbsA 蛋白呈 現與S. aureus ClfA 相似的作用機制,可藉由和纖維蛋白原結合並借用宿主免疫球 蛋白達到黏附與活化血小板的能力(Pietrocola et al., 2005)。
六. 血小板與感染性心內膜炎的發生。
在感染性心內膜炎中,受損的瓣膜會有血小板的沉積,而致病菌可能藉由吸 附於這些血小板沉積物或吸附在贅疣上,間接引起局部血小板活化與fibrin 的堆 積,導致贅疣的增大。在 S. sanguis 中,Platelet aggregation -associated protein (PAAP) 具有凝集血小板的能力(Erickson et al., 1990),而在心內膜炎動物模式中也證實,
在血小板凝集能力缺失的 S. sanguis 中,其造成心內膜炎的程度會有所減輕,而加 入抗PAAP 的抗體也可以減輕心內膜炎發炎程度 (Herzberg et al.,1992)。而在其他 細菌中也發現與血小板凝集相關的蛋白,如 S. aureus ClfA、Group A streptococci M protein、S. agalactiae FbsA 蛋白等 (Rivera et al., 2007),其中在 S. aureus 已經有動 物模式證明,clfA 發生缺失的突變株,其造成心內膜發炎的程度也有明顯減輕 (Sullam et al., 1996 ),顯示血小板的凝集參與了細菌引發感染性心內膜炎的發生。
七.研究目的與實驗設計
在本實驗室奇玄學長的研究中發現,轉糖鏈球菌中第十一對two component regulatory systems的response regulator (RR11)被剔除後,會導致心內膜炎形成的贅 疣中菌數明顯的減少,且喪失在受損心臟瓣膜上形成的生物膜的能力,這似乎顯 示RR11調控與感染性心內膜炎相關的細菌毒力因子。而奇玄學長也利用富含血小 板的血漿部分(PRP)模擬細菌進入血液受到刺激後,生物膜形成的狀況,他發現兩 者生物膜形成的結構和在心臟瓣膜上形成的生物膜相當類似。而在本實驗室俊達 學長的研究中,他利用microarray的方式檢測野生株GS5與RR11 mutant基因表現的 差異,發現一個基因SMU. 753在RR11 mutant中表現量會有明顯的下降,利用生物 資訊校對發現SMU.753蛋白結構上與 Escherichia coli和Yersinia enterocolitica 的 phage-shock protein C (PspC)有相似性,因此我們將此基因名為PspC。根據以上結 果,我更進一步想要研究RR11是否藉由調控PspC,幫助轉糖鏈球菌附著在受損的 心臟瓣膜上。另一方面,我利用生物膜形成能力測試的方法發現,將轉糖鏈球菌 養在血小板貧乏的血漿中(Platelet-Poor Plasma),並無法形成良好的生物膜,這似 乎也顯示血小板對於轉糖鏈球菌生物膜的形成是重要的。因此我也想要探討血小 板在轉糖鏈球菌的生物膜形成過程中扮演怎樣的角色。
貳、實驗材料與方法
1. Streptococcus mutans 的轉形作用
將 BHI (購自 BD )隔夜培養的 S. mutans,以百分之一的比例種入含有 5%的 horse serum 的 BHI 中,養至 OD550約0.2 至 0.3 之間,以 3000 rpm (Eppendorf centrifuge 5810R)離心後,去上清液後,以 300 μl 的 BHI 將菌體懸浮起來並加入 1 μl 帶有綠螢光蛋白 (GFP)的 plasmid DNA (pDL278),混合培養 30 分鐘後,以 BHI 將體積補至1 ml,培養 90 分鐘。最後以 3000 rpm 離心 5 分鐘,去上清液,以 100 μl 的 BHI 懸浮菌體後,將菌液塗於含有 500 μg Spectinomycin 的 BHI 培養基中。
培養兩天後,以UV 照射測試菌體是否可發綠色螢光,以進行進一步實驗。
2.生物膜形成能力測定
將細菌加入於 BHI 培養液中培養 16 至 20 小時,使其處於代謝旺盛階段。接 著以 1:100 的比例將菌液加入新鮮的 BHI 培養液或是富血小板血漿(platelet-rich plasma, PRP)中,以 200 μl/well 的體積加入 96 孔平底盤,放置於厭氧箱中培養 24 小時。定量前,更換平底盤上的 BHI 培養基,並用清水緩慢沖洗兩次。等待平底 盤風乾後,以50 μl/well 的體積加入 0.01 %的 crystal violet,在室溫下進行染色 15 分鐘,接者再以清水緩慢沖洗兩次。最後加入 ethanol-acetone 混合液(8:2)進行 退染(200 μl/well),測定 OD550nm的吸光值。
3.人類血小板懸浮液(platelet suspension)的製備
利用真空採血管(購自 BD)將健康捐贈者的血與抗凝血劑(3.8% sodium citrate) 以9:1 比例混合,以 200xg,25℃離心 10 分鐘,收集上層富含血小板血漿
(Platelet-Rich Plasma),加入 heparin (6.4U/ml)以及 PGE1 (1μM)(Sigma Aldrich),再 以790xg 離心 10 分鐘,除去上清液。將離心後的血小板以 1 ml Tyrode’s solution 打散後,再加入3 ml 的 tyrode’s solution,隨後加入 heparin (6.4U/ml)以及 PGE1
(1μM),以 790xg 離心 10 分鐘,除去上清液,最後將血小板懸浮於 Tyrode’s solution 中,並逐步稀釋後,以1:1 的比例與 trypan blue 混合,以血球計數器進行計數。
4.體外生物膜形成模式的建立
將12 mm 的圓型玻片先放入細胞培養用 24 well 培養盤中,以 UV 燈照射 30 分鐘以上,隨後將健康捐贈者所取出的血液以1200 rpm 離心後,取出富含血小板 的血漿部分 (PRP),將 PRP:細菌為 99:1 的比例,種入 BHI 隔夜培養可表現綠螢光 蛋白 (GFP)之轉糖鏈球菌,16 至 24 小時後,去除上清液並利用 1XPBS 清洗一次 後,以4 % paraformaldehyde 處理 15 分鐘。去除 paraformaldehyde 後,加入含有 1% TritonX-100 的 PBS 處理 15 分鐘。之後以 1XPBS 清洗後,加入濃度為百分之 一的Rhodamine-phalloidin (invitrogen)染色 1 小時。最後,以 1XPBS 清洗一次後,
挑出圓形玻片置於載玻片上,蓋上蓋玻片後,以共軛焦顯螢光微鏡觀察生物膜之 形成。在某些樣品處理中,外加入血小板活化抑制劑PGE1(0.6 μM) (Sigma
Aldrich)、aspirin (500 μg/ml)(南光製藥)、RGDS (1 mM) (Sigma Aldrich)或單株抗體 AN51(1:250) (Dakocytomation),來探討血小板活化對於生物膜形成之影響。
另一部分,將健康捐贈者所取出的血液,以3000 rpm 離心後,取出上清液血 小板貧乏的血漿部分(platelet-poor plasma, PPP),加入純化後的血小板懸浮液(約 107個細胞)。以百分之一的比例,將隔夜培養可表現綠螢光蛋白 (GFP)之轉糖鏈 球菌分別種入外加血小板與未加血小板的血漿中,培養16 至 24 小時後,以共軛 焦螢光顯微鏡觀察。
5.細菌黏附活化血小板試驗
利用先前的分離方式取得富含血小板的血漿部分(PRP),隨後取 500μl 富含血 小板的血漿置入含有12 mm 圓形玻片的 24 孔盤,並外加入終濃度 10μM 的 ADP,
於室溫中震盪處理30 分鐘,以促使血小板完全活化。接著以 3000 rpm 離心 10 分 鐘,將活化的血小板固定在圓形玻片上,並去除上清液。最後,將帶有綠螢光蛋 白的轉糖鏈球菌以百分之一的比例種入血小板缺乏的血漿(PPP)中,培養 16-20 小 時。之後與體外生物膜模式試驗的後續處理相同,最後以共軛焦顯微鏡觀察。在 某些樣品處理中,外加入血小板活化抑制劑PGE1(0.6 μM)、aspirin (500 μg/ml)、
RGDS (1 mM)或單株抗體 AN51(1:250),來探討這些血小板抑制劑是否會去干擾活 化血小板與細菌間的黏附。
6.模擬體內生物膜形成之體外模式
利用先前的分離方式取得富含血小板的血漿部分(PRP),隨後取 500μl 富含血小 板的血漿置入含有12 mm 圓形玻片的 24 孔盤,並外加入終濃度 10 μM 的 ADP,
於室溫中震盪處理30 分鐘,以促使血小板完全活化。接著以 3000 rpm 離心 10 分 鐘,將活化的血小板固定在圓形玻片上,並去除上清液。最後,將帶有綠螢光蛋 白的轉糖鏈球菌以百分之一的比例種入富含血小板的血漿(PRP)中,培養 16-20 小 時。之後與體外生物膜模式試驗的後續處理相同,最後以共軛焦顯微鏡觀察。在 某些樣品處理中,外加入血小板活化抑制劑PGE1(0.6 μM)、aspirin (500 μg/ml)、
RGDS (1 mM)或單株抗體 AN51(1:250),來探討這些血小板抑制劑是否會去干擾細 菌生物膜的形成。
7.實驗性心內膜炎之老鼠模型的建立
本實驗方法係參考 1978 年由 Santoro 及 Matthew 之實驗設計並加以修改。進 行插管手術之前,準備26.5 號針筒吸取每毫升 50mg 的 Ketamin 麻醉劑(購自 Pfizer) 約0.6~1.0 毫升 (老鼠體重每 100 公克施打約 0.2 毫升),以腹腔注射Wistar 品系 大鼠(體重約300 至 500 克),待確定老鼠麻醉後即剔除頸部毛髮並以碘酒和 70
%酒精消毒該處。接著以麻繩固定老鼠四肢與頭部於手術平台上,以手術刀縱切 老鼠左側頸部約2 至 3 公分大小的切口,利用剪刀將頸部肌肉分開後,以鑷子挑 出左側頸動脈,再用動脈夾將頸動脈兩端夾緊,並以絲線綁死靠近頭部的一端。
使用動脈剪將頸動脈剪出一個缺口,將含有細鐵絲的導管(PE10),插入頸動脈的 缺口中,待插入頸動脈的導管抵住下端的動脈夾後,以絲線將導管與頸動脈打結 固定,接著用鑷子夾住已插入導管之頸動脈,並取走兩端的動脈夾。將導管推入 頸動脈珠,直至鑷子夾住導管可感覺心臟跳動,或導管碰到阻礙物為止。此外,
將鐵絲抽出確定血流之順暢以確保導管沒有刺穿心臟,或由倒流出之血液是否有 上下震動確保血流之無礙。最後以絲線綁死導管與頸動脈,將露在頸動脈外面的 導管剪短至頸動脈插入處並縫合頸部切口。完成插管手術之老鼠須觀察24 小時並 給予進食餵水。
8.細菌處理與感染老鼠之方法
將細菌加入於 BHI 培養液中培養 16 至 20 小時,使其處於代謝旺盛階段,離 心並以PBS(phosphate buffer saline)反覆沖洗二次,接著利用超音波震盪(40W)
於冰上間斷作用5 分鐘,打散凝集菌塊。離心並以 PBS 將菌液調至 O.D550為1.5,
以26.5 號針筒吸取菌液,由尾巴靜脈注射至老鼠體內,當針頭插到血管至針筒出 現回血時,即可將菌液慢慢打入,注射過程可看見靜脈由淺藍色轉為白色,表示 已成功將菌液打入老鼠血液中。
9.抗凝血藥物阿斯匹靈(Aspirin)給與感染性心內膜炎大鼠之方法
在打菌前30 分鐘以腹腔注射方式先給予一次 Aspirin (25 mg/kg),打菌後 30 分鐘再次給予Aspirin (25 mg/kg),最後在打菌後六小時給予最後一次 Aspirin (25 mg/kg),隔天犧牲大鼠並取出心臟瓣膜上的贅疣部分以共軛焦顯微鏡觀察。另外一 方面,也將取出的贅生物部分利用超音波震盪(40W)於冰上間斷作用 5 分鐘打 散贅疣,隨後以1XPBS 稀釋到適當比例後,塗在洋菜培養基上,於厭氧箱中培養 2 天後,計數菌量。
10.以抗體 anti-CD42d 染感染性心內膜炎贅生物中血小板之方法
在感染性心內膜炎大鼠中取出贅生物後,置於玻片上烘乾,隨後加入 100μl 的 1:50 rabbit anti-CD42d (Santa cruz)抗體染 2 小時後,以 1XPBS 清洗兩次,烘乾玻 片後,加入100μl 的 1:100 goat anti-rabbit Texas-red (Jackson ImmunoResearch)二抗 染 1 小時,以1XPBS 清洗兩次後烘乾,以共軛焦顯微鏡觀察。
11.染色質免疫沉澱技術(chromatin immunoprecipitation)
將隔夜培養的菌繼代到40 ml 的 BHI 中,培養至 OD550為0.4 後,加入 1.1 ml 37%
formaldehyde,室溫振盪 15 分鐘後,加入 2 ml 2.5M glycine 震盪培養 5 分鐘,3000 rpm、4℃離心 10 分鐘。以 40 ml 1XPBS 清洗沉澱物兩次,再以 3000 rpm、4℃離 心10 分鐘。取 800 μl lysis buffer (50mM Tris,500mM sodium chloride,1mM EDTA,
0.1% TritonX-100,0.1% SDS,0.1% sodium deoxycholate,100 mM PMSF) 將沉澱 物重新懸浮置溶液中,並將溶液等量分置兩個裝有1.5 ml 破菌珠(biospec)的
eppendorf 中,以震盪破菌的方式打破細菌後,以 10000 rpm、4℃離心 10 分鐘。取 出上清液的部分,利用超音波震盪(40W)於冰上間斷作用 5 分鐘,隨後取 5 μl 跑0.8% DNA 膠確定 DNA 片段已被打斷約 500~1000 bp 後,以 12000 rpm、4℃離 心5 分鐘。取出上清液部分並外加入 200μl 的 protein A bead (invitrogen),以迴轉式 混和器旋轉處理2 小時,去除會與 protein A bead 非專一性結合的蛋白與 DNA 片 段。取1/10 的量到新的試管並標示為 Input 儲存在-20℃,剩餘的部分等分置於三 個不同試管中,其中一個為不加抗體的控制組,另外兩個分別加入5μl 的 rabbit anti-RR11 antibody 與 rabbit pre-immune serum,以迴轉式混和器旋轉混合,隔天取 100μl 的 protein A bead 加入三個試管中,以迴轉式混和器 4℃旋轉處理 1 小時。隨 後,spin down 並去除上清液,以 lysis buffer 重新懸浮沉澱物後,以迴轉式混和器 4℃旋轉處理 5 分鐘後,離心並去除上清液,重複步驟兩次後,外加 1ml DOC buffer(10 mM Tris,0.25 M LiCl,0.5% NP40,0.5% sodium deoxycholate,1mM EDTA) 以迴轉式混和器4℃旋轉處理 5 分鐘,離心並去除上清液。之後再以 1ml TE buffer (10mM Tris,1mM EDTA)清洗並以迴轉式混和器 4℃旋轉處理 5 分鐘,離心並去除 上清液。取0.5 ml TE buffer 重新懸浮 bead 並換至新的試管中,隨後加 0.5ml TE buffer 到原本的試管中將剩餘的 bead 懸浮後加至新的試管中,在室溫以 3000 rpm 離心5 分鐘。去除上清液後,加入 50 μl 的 TES buffer (50mM Tris,10mM EDTA,
1% SDS),震盪混勻後,於 65℃靜置 10 分鐘。隨後在室溫以 10000 rpm 離心 5 分 鐘,將上清液取到新的試管中。之後再外加150 μl TES buffer 到含有 bead 的試管 中,震盪混勻後,在室溫以10000 rpm 離心 5 分鐘,之後將上清液取至先前的試管 中,靜置於65℃處理隔夜。同時也加 150 μl TES buffer 到 Input 的試管中,一樣 65℃
處理隔夜。之後,將試管中的液體換至新的試管中並外加入12.5 μl 的 proteinase K (20mg/ml) (invitrogen)及 200 μl TE buffer,37℃水浴 2 小時。加入 400 μl 的
phenol/chloroform 並震盪混勻 30 秒,於室溫 13000 rpm 離心 10 分鐘。之後取出上 層液的部分置新的試管中並加入400 μl 的 chloroform 震盪混勻 30 秒,於室溫 13000 rpm 離心 10 分鐘。取出上層液的部分加入 45 μl 的 3M sodium acetate 及 1 ml 的 100%
冰的ethanol,混勻後靜置於-80℃ 1 小時,隨後於 4℃以 10000 rpm 離心 30 分鐘,
去掉上清液後再加入500 μl 的 70% ethanol 混勻後,於 4℃以 10000 rpm 離心 10 分 鐘,將上清液去除後,風乾沉澱物。最後加入50 μl TE buffer 將沉澱物回溶後,以 PCR 的方式將 DNA 片斷擴增,跑膠。
12.膠體電泳阻滯實驗(electrophoretic motility shift assay) A. DNA 探針製作。
訂製 5’-GTTTATACTAATAGCA 及反向序列 5’-TGCTATTAGTATAAAC (Stock:20 mM),各取 5 μl 加入 35 μl 10X EMSA annealing buffer 及 5 的 nuclease-free water,以 PCR program 設定 95℃(10 分鐘)-70℃(4 分鐘)-20℃(10 分鐘)- 4℃,隨後 將濃度稀釋至2 mM。
B. DNA 探針之末端標定(End labeling)
取製作好之DNA 探針 5 μl,外加入 2μl 10X PNK buffer(New England Biolabs)、1μl T4 PNK enzyme (New England Biolabs)、7μl ddH2O 及 5μl ATP-γP32,
置於37℃ 30 分鐘後,換至 70℃處理 10 分鐘,最後加入 140 μl 的 ddH2O 將 DNA 探針濃度稀釋至62.5 nM,置於-20℃儲存。
C.膠體電泳阻滯。
取一定量純化的RR11 蛋白,外加入 15μl 2X binding buffer (40mM Tris,
100mM KCl,10mM MgCl2,1mM EDTA,20% glycerol,5μg BSA,1mM DTT)、
1μl polydI-dC (Sigma Aldrich)及 2μl 的 isotope-labeled DNA 探針,最後加 ddH2O 將 總體積補至30μl,4℃培養 30 分鐘。在某些樣品中外加入未 label isotope 的 1X、
10X 及 100X 的 cold competitor 2μl,另有些處理則先將 4 μg 的 RR11 polyclonal antibody 或 rabbit pre-immune serum 與 RR11 蛋白先行混合,於 4℃培養 30 分鐘後,
在外加入上述buffer。最後將與各樣品跑 6% non-denaturing TBE gel 1~2 小時後,
乾膠 1 小時,最後置入片匣中並置入底片,將片匣置於-80℃中靜置隔夜後洗片。
13.全血殺菌能力試驗(whole blood killing assay)
將隔夜培養的轉糖鏈球菌以3000 rpm 離心 10 分鐘後,以 1×PBS 重新懸浮,再 次以3000 rpm 離心 10 分鐘,之後再以 1×PBS 將菌液重新懸浮,並利用超音波震 盪(40W)於冰上間斷作用 3 分鐘,最後將菌液調整至 OD550為1.5 (約 2×109
cfu/ml)。將菌液更進一步稀釋至 105 (約 2×104 cfu/ml),取 450 μl 的稀釋後的菌液,
加入50 μl human serum,以迴轉式混和器於 37℃培養 30 分鐘進行調理作用 (opsonization)。隨後取 30 μl 菌液加入 270 μl 的 1×PBS,取 100 μl 菌液塗培養基定 量菌數。此外,取30 μl 菌液與 270 μl 的全血(heparin 管抽取之血液)混合後,置於 迴轉式混和器上培養60 分鐘與 120 分鐘後,塗在培養基上定量全血的殺菌能力。
14.細菌與血漿中纖維蛋白原結合試驗
將隔夜培養的轉糖鏈球菌以3000 rpm 離心 10 分鐘後,以 1×PBS 重新懸浮,再 次以3000 rpm 離心 10 分鐘,之後再以 1×PBS 將菌液重新懸浮,並利用超音波震 盪(40W)於冰上間斷作用 3 分鐘,最後將菌液調整至 OD550為1.5 (約 2×109 cfu/ml)。取 1 ml 的菌液以 10000 rpm 離心,去上清液後外加入 1 ml 血小板貧乏血 漿(platelet-poor plasma)置於迴轉式混和器上 37℃混合培養 60 分鐘,以 10000 rpm 離心將菌體離下,去上清液後,加入20 μl 的 sample treat buffer 於室溫下靜置 30 分鐘,再以10000 rpm 離心,將上清液取出放置於另一個 eppendorf 中,並外加入 20 μl 的 protein dye,以沸水煮 10 分鐘後,跑 10% SDS PAGE。隨後將蛋白轉漬到 PVDF 膜上,取 10 ml blocking buffer(2 mM Tris pH=7.4、150 mM NaCl、0.4 % non-fat milk)加入 PVDF 膜 blocking 30 分鐘,隨後加入 1:5000 goat anti-human Fg antibody (Sigma Aldrich)室溫處理 2 小時,倒去含一抗的 buffer 後,以 TBST 清洗三次,每 次10 分鐘。隨後再加入 10 ml 含有 1:2500 anti-goat HRP (Jackson ImmunoResearch) 的blocking buffer(2 mM Tris pH=7.4、150 mM NaCl、0.4 % non-fat milk)室溫處理 2 小時,倒去含二抗的buffer 後,以 TBST 清洗三次,每次 10 分鐘。將 PVDF 膜稍 微瀝乾後,外加化學冷光顯色劑後,壓片進行觀察。
15.細菌與血小板的結合試驗
將隔夜培養的轉糖鏈球菌以3000 rpm 離心 10 分鐘後,以 1 ml ELISA coating buffer 重新懸浮,再次以 3000 rpm 離心 10 分鐘,之後再以 1 ml ELISA coating buffer 將菌液重新懸浮,並利用超音波震盪(40W)於冰上間斷作用 3 分鐘,最後將菌 液調整至OD550為1.5 (約 2×109 cfu/ml)。取 50 μl 菌液置於 96 孔的 ELISA plate 中,
放置於4℃處理隔夜。控制組部份則是加入 50 μl 含有 1%BSA 的 ELISA coating
buffer。隨後倒去上清液,並以 1×PBST (1×PBS+1% Tween-20)清洗三次。每個 well 外加入50 μl 富含血小板的血漿(platelet-rich plasma),於 37℃處理 2 小時後倒去液 體,並以1×PBST (1×PBS+1% Tween-20)清洗三次。之後每個 well 加入 50 μl 含有 1%BSA 與 1:2000 一抗 AN51 (mouse anti-human platelet GPΙb clone) 1×PBS。於 4℃
處理隔夜後,去除上清液並再以1×PBST (1×PBS+1% Tween-20)清洗三次,之後每 個well 加入 50 μl 含有 1%BSA 與 1:2000 二抗 anti-mouse alkaline phosphatase (Jackson ImmunoResearch)的 1×PBS,於 37℃處理 2 小時。去除上清液並再以 1×PBST (1×PBS+1% Tween-20)清洗三次後,以 alkaline phosphatase substrate (Sigma Aldrich)進行呈色,之後以 405 nm 進行偵測。
參、結果
第一部分:血小板參與轉糖鏈球菌所引發感染性心內膜炎中生 物膜形成。
1.血小板對於血漿中轉糖鏈球菌形成生物膜的重要性。
利用生物膜形成測試方法,我發現血小板貧乏的血漿部分並不能促使轉糖鏈 球菌形成生物膜 (圖一 A),這似乎顯示血小板在轉糖鏈球菌形成生物膜的過程中 扮演著重要的角色。我更進一步利用共軛焦螢光顯微鏡觀察在缺乏血小板與外加 血小板的實驗組中,兩者生物膜形成的狀況。我發現利用血小板貧乏的血漿培養 的轉糖鏈球菌並不能形成完整的生物膜,而外加入血小板後便可以促使生物膜的 形成 (圖一 B),這也代表血小板參與了轉糖鏈球菌在血漿中生物膜的形成。
2.血小板的活化對於轉糖鏈球菌生物膜的形成。
在先前的實驗已經發現,血小板對於轉糖鏈球菌於血漿中形成生物膜是重要 的。接著我想探討血小板的活化是否與轉糖鏈球菌生物膜的形成有關。因此我在 實驗中外加不同的血小板活化抑制劑來觀察血小板的活化被抑制後是否仍可以促 使轉糖鏈球菌形成生物膜,而在本實驗室筱菁學姊先前的研究曾發現Aspirin、
prostaglandin E1、RGDS 及單株抗體 AN51 可有效抑制轉糖鏈球菌所誘發的血小板 的凝集。因此我也選用這幾種血小板凝集的抑制劑來進行我的實驗。我發現血小 板抑制劑的加入對於轉糖鏈球菌於富含血小板的血漿中生物膜的形成有明顯的抑 制現象 (圖二 A)。而將這些血小板抑制劑加入培養基中,測試培養基培養生物膜 之形成。由結果發現,這些抑制劑並不會對培養基培養的生物膜形成有所影響 (圖 五)。顯示這些抑制劑的作用是影響血小板的活化,而非藥物本身有抑菌的作用造 成轉糖鏈球菌於血漿中形成生物膜有明顯抑制。
3.活化的血小板本身而非血小板活化後所釋放的因子對於轉糖鏈球菌 於血漿中形成生物膜是重要的。
由上述實驗我發現血小板的活化對於轉糖鏈球菌於血漿中形成生物膜是重要 的,然而血小板活化後會釋放出許多分子,我更進一步想要探討究竟是活化血小 板本身還是血小板活化後所釋放出的因子有助於細菌在血漿中形成生物膜。我將 10 μM 的 ADP 加入富含血小板的血漿中先將血小板活化。隨後離心,區分成活化 的血小板及含有活化血小板所釋放因子的血小板貧乏的血漿部分,將兩者與細菌 一起培養後,觀察細菌在何種狀況下能形成完整的生物膜。此外,我也利用2%
paraformaldehyde 先將血小板殺死後離心,取得死亡的血小板,在與細菌一同培養,
觀察死亡的血小板是否仍舊可以幫助細菌形成生物膜。由結果我發現,在缺乏活 化血小板或是血小板被殺死的樣品中,我都無法觀察到明顯細菌生物膜的形成(圖 一C),顯示活化的血小板本身對於轉糖鏈球菌於血漿中形成生物膜是重要的。
4.血小板活化抑制劑 Aspirin 對於感染性心內膜炎大鼠中細菌生物膜的 形成。
更進一步,我們想探討在感染性心內膜炎致病過程中若給予血小板抑制劑 Aspirin 是否可以去影響轉糖鏈球菌體內細菌生物膜的形成。當我們分批給與感染 性心內膜炎大鼠Aspirin 後,以共軛焦顯微鏡觀察其細菌生物膜的形成,由結果發 現其細菌生物膜的形成狀態有明顯的缺失(圖三 A)。此外,定量贅疣中的細菌量,
也一樣發現細菌的量會有明顯的減少,顯示血小板抑制劑Aspirin 可以有效干擾感 染性心內膜炎中細菌生物膜的形成(圖三 A)。
5.血小板抑制劑影響細菌與活化血小板的黏附。
由感染性心內膜炎大鼠模式中我們發現給予Aspirin 可以有效干擾轉糖鏈球 菌於受傷瓣膜上形成生物膜,而由生物膜的三級結構中我們也觀察到給予Aspirin 後的體內生物膜並無法形成多層次的結構。我們推測在人工造成心臟瓣膜缺損 時,便會誘發血小板活化凝集在受傷的組織上,當我們將菌打入大鼠體內時,細 菌便能黏附到活化的血小板上。而給予抗凝血劑Aspirin 後,會抑制未活化的血小 板受細菌刺激而活化,卻無法回復已活化的血小板,因此我們雖然不能觀察到明 顯的多層結構生物膜形成,但仍可見到明顯的生物膜形成。為了證明細菌能與已 活化的血小板黏附,我們將血小板活化後再與轉糖鏈球菌一起培養,觀察細菌黏
附的狀態。我發現當血小板先被ADP 活化後,外加入血小板抑制劑 PGE1、Aspirin 或單株抗體AN51 都無法有效抑制細菌與血小板的黏附(圖六)。暗示在感染性心內 膜炎動物模式中可能因為心臟瓣膜附近早有活化的血小板凝集,所以當我們給與 Aspirin 之後並不能抑制細菌與活化血小板的結合。然而,當我加入纖維蛋白原的 一段胜肽RGDS 後,卻可以觀察到 RGDS 可以干擾細菌與血小板的黏附(圖二 B),
這也暗示纖維蛋白原可能在細菌與血小板之間交互作用扮演重要的角色。
6.感染性心內膜炎體內細菌生物膜與血小板的交互作用。
在以往的研究中,學者們推測在體內環境中,除了活化的血小板之外,還會 有許多未活化的血小板循環在人體中,一旦細菌進入人體,黏附到受傷的心臟瓣 膜後,可以進一步活化血小板的凝集,完成致病的過程(Moreillon et al., 2004)。然 而迄今並沒有相關研究證實在感染性心內膜炎中,細菌生物膜的形成與血小板的 活化堆疊有關,因此我將感染性心內膜炎大鼠上的贅生物取出後,利用anti-rat CD42d 的一抗,再加入帶有 Texas-red 二抗去染贅生物中的血小板。由結果我們發 現在生物膜形成的附近都可以看到明顯的血小板堆疊(圖三 B),顯示血小板對於細 菌生物膜的形成是相當重要的。
7.模擬體內生物膜形成體外模式觀察血小板抑制劑對於細菌生物膜形 成的影響。
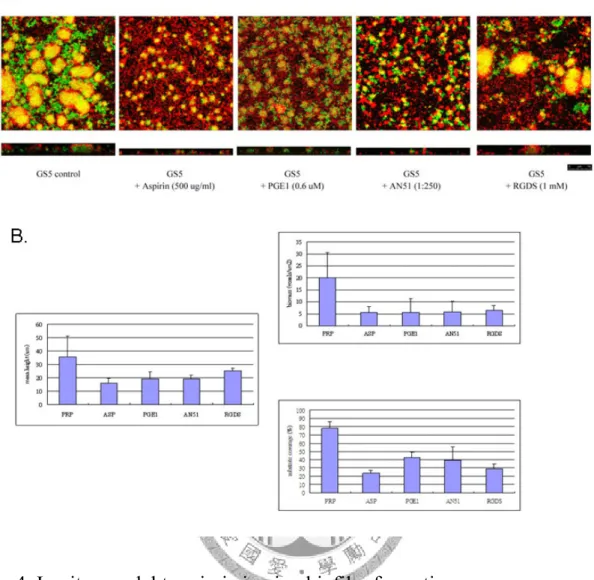
為了更進一步研究血小板對於生物膜形成之影響,我們修改先前建立的體外 模式,我們先利用ADP將血小板活化並固定在玻片上,以模擬受傷的心臟瓣膜,
更進一步再利用富含血小板的血漿與細菌培養,模擬細菌與血液中未活化的血小 板作用的情況,並同時加入不同的血小板抑制劑,觀察細菌生物膜的形成。由結 果我們發現利用此方法可以觀察到明顯細菌生物膜的形成,而其形成的結構與體 內生物膜形成結構相似。而在外加入不同的血小板抑制劑後,我們利用共軛焦顯 微鏡觀察生物膜形成的三級結構,發現這些血小板抑制劑無法干擾細菌與底層活 化血小板的黏附,但可以觀察到在細菌上層僅有少數血小板被活化(圖四),顯示血
小板活化抑制劑可以抑制未活化血小板受細菌刺激而被活化,進而抑制細菌生物 膜的形成。為了確定血小板抑制劑確實可以干擾細菌生物膜的形成,我進一步利 用軟體metamorph定量分析生物膜形成後,生物膜形成厚度、全部細菌的生物質量 (biomass)與表面基質覆蓋率(substrate coverage)。根據Beloin等人的研究,他們利用 另一個定量軟體COMSTAT可以有效訂定E.coli 形成的生物膜狀態(Beloin et al., 2004)。搜尋原始的公式計算後,發現COMSTAT軟體藉由計算出3D像素的多寡來 模擬可發光的菌量,隨後除以表面積便可以得到所謂的生物質量(biomass),單位 為μm3/μm2。此外COMSTAT軟體也可以計算出在固定表面積下,細菌黏附的比例,
稱為表面基質覆蓋率(substrate coverage)(Heydorn et al., 2000)。我利用metamorph軟體 執行相似的功能,一樣訂定生物質量與表面基質覆蓋率。由統計結果發現外加入 血小板抑制劑的樣本中,可以明顯觀察到生物質量與表面基質覆蓋率的減少 (P<0.05)(圖四B、C,表一)。這樣的結果也顯示血小板抑制劑可以有效干擾細菌生 物膜的形成。然而,觀察生物膜形成的平均厚度(Mean height)則發現在外加入血小 板抑制劑的樣品中,生物膜形成厚度有減少,然而在統計上卻沒有顯著差異 (P>0.05)(圖四A,表一)。
第二部分:Response regulator 11 藉由調控 PspC 基因來影響感 染性心內膜炎中,轉糖鏈球菌生物膜的形成。
1.利用染色質免疫沉澱法(Chromatin immunoprecipitation)研究 response regulator 11 在細菌體內是否能直接調控 PspC 基因的表現。
在microarray 與 RT-PCR 確定 response regulator 會調控 PspC 的表現後,我更 進一步想探討response regulator 11 是否直接調控 PspC 基因的表現來影響細菌生物 膜的形成。首先,利用純化的RR11 蛋白免疫兔子得到 RR11 polyclonal antibody,
之後以染色質免疫沉澱法來研究response regulator 11 是否可以直接調控 PspC 基因 的表現。由實驗結果發現response regulator 11 可以直接調控 PspC 基因的表現(圖
七A)。然而在先前筱菁學姊的研究中發現 PspC 會與前段的另外兩的基因 SMU.751 與SMU.752 形成一個 operon,而利用生物資訊的方式我們也發現 PspC 前有一段 小段的intergenic region,我們推測 PspC 基因除了可以 operon 的方式被表現,另外 可能也會受response regulator 11 的調控。所以我另外也以染色質免疫沉澱法來觀 察response regulator 11 是否可以調控 SMU.751 基因,我發現 response regulator 11 在細菌體內似乎無法直接調控SMU.751 的表現。在以往的研究曾經發現 Two component regulatory systems 有時可藉由自我調控(autoregulation)的方式來調控 two component system sensor 與 regulator 的表現,所以我也利用染色質免疫沉澱法來觀 察response regulator 11 (SMU.485)是否可以有自我調控的狀況。由結果發現
response regulator 11 似乎不會自我調控基因的表現(圖七 A)。
2.膠體電泳阻滯實驗(electrophoretic motility shift assay) 研究 response regulator 11 是否可與 PspC 前可能的啟動子區位結合。
更進一步,我利用膠體電泳阻滯實驗來驗證 response regulator 11 是否確實可 與PspC 前可能的啟動子區位結合,進而調控 PspC 的表現。利用 microarray 的結 果,一一比對在可能受response regulator 11 所調控基因前的 intergenic region 與 PspC 之間是否有相類似的區位。由結果發現有一段約 16 mers
( GTTTATACTAATAGCA )的 DNA 序列,在多個基因中有相當高的相似性。因此 我選用這段DNA 序列來進行膠體電泳阻滯實驗。由結果我發現 response regulator 11 確實可與這段序列進行結合(圖七 B),顯示 response regulator 11 可能是與此段 DNA 序列結合,進一步調控 PspC 的表現。
3.利用共軛焦螢光顯微鏡觀察富含血小板血漿中 Wild-type GS5、RR11 mutant、PspC mutant 與 PspC complementation strain 生物膜形成的差異。
利用共軛焦顯微鏡的觀察可以發現GS-5、RR11 mutant 與 PSPC mutant 在富 含血小的血漿中形成生物膜的狀態有所不同。在野生株 GS-5 中,生物膜的形成是 較為緊實而且其生物膜形成範圍也較大。但在RR11 mutant 中,生物膜的狀態可見 到明顯的微聚落 (microcolonies)形成,且其結構也較為鬆散,而在 PspC 基因有所
缺失的菌株,則可以發現其生物膜幾乎無法形成,然而在將PspC 送回的
complementation strain 中,我也發現其生物膜形成能力有明顯被恢復,但其生物膜 形態卻不似野生株 GS5,反而更相近於 RR11 mutant 所形成的微聚落(圖六 A)。
這些結果顯示RR11 mutant 在富含血小板的血漿中會喪失部分生物膜形成的能 力,而PspC mutant 則是喪失大部分在富含血小板的血漿中形成生物膜的能力,將 PspC 基因送回的互補株中,也可以觀察到在富含血小板血漿中形成的生物膜有明 顯的回復。這樣的結果暗示RR11 可能藉由調控 PspC 基因來影響轉糖鏈球菌在富 含血小板的血漿中形成生物膜的能力。
4.利用共軛焦螢光顯微鏡觀察感染性心內膜炎大鼠贅疣中野生株 GS5、RR11 mutant、PspC mutant 與 PspC complementation strain 生物膜 形成的差異。
在利用體外模式觀察生物膜形成後,我們更進一步利用感染性心內膜炎大鼠 模式來探討野生株GS5、RR11 mutant、PspC mutant 與 PspC complementation strain 體內生物膜形成的差異。由結果我們發現,在PspC 基因有所缺失的菌株中,其形 成體內生物膜的能力會有明顯的缺失(圖六B)。而在 PspC complementation strain 中則可以發現其體內生物膜形成有明顯被恢復。顯示PspC 基因在感染性心內膜炎 中,細菌生物膜的形成是相當重要的。
5.檢測野生株 GS5 和 PspC mutant 與宿主因子結合之差異。
在第一部分研究中顯示轉糖鏈球菌與宿主因子的作用對於轉糖鏈球菌於生物 體內形成生物膜是重要的。因此我檢測野生株GS5 和 PspC mutant 與宿主因子結合 是否有所差異。利用ELISA 的方法將野生株 GS5、PspC mutant 與 PspC
complementation strain 先 coating 在 ELISA plate 後,外加富含血小板的血漿部分與 之結合,觀察PspC 的缺失是否與血小板的黏附有關。由結果發現 PspC 的缺失並 不影響轉糖鏈球菌與血漿中血小板的結合。而我將野生株GS5 和 PspC mutant 與血 小板貧乏血漿培養後,利用西方墨點法(western blot)研究 PspC 的缺失是否影響轉 糖鏈球菌與血漿中的纖維蛋白原的結合。由結果發現PspC 的缺失並不影響轉糖鏈
球菌與纖維蛋白原的結合。
6.檢測野生株 GS5 和 PspC mutant 在生物體內及全血中生存能力。
我進一步將GS5 與 PspC mutant 打到老鼠體內觀察生物體內細菌被清除的速 率是否有所不同,我發現PspC mutant 比起 GS5 會更容易在體被被清除。而利用人 類全血殺菌試驗也發現PspC mutant 在全血中的存活率也是較 GS5 低。這些結果顯 示PspC 蛋白可能參與了細菌對抗宿主免疫系統的功能。
四、討論
1.血小板參與轉糖鏈球菌引起之感染性心內膜炎中生物膜之形成。
轉糖鏈球菌為造成人類齲齒的主要病原菌。在引發齲齒的過程中,轉糖鏈球 菌已被發現可利用葡萄糖傳遞酶(glucosyltransferase)將蔗糖分解成葡萄糖聚合物後 (glucan),進一步穩定黏附於牙齒表面。而這樣細菌群聚的緊密結構一般被視為一 種細菌形成的生物膜。而在先前的研究中也發現在心臟瓣膜有所缺損的病人身 上,當轉糖鏈球菌進入血液形成暫時性菌血症時,便有機會黏附於有缺損的心臟 瓣膜上,形成一種贅生物,稱為贅疣。而在Durack 1975 年的研究中,利用電子顯 微鏡觀察贅疣的結構發現,贅疣中是由細菌、血小板與纖維蛋白所構成的緊密包 裹的結構,而這樣的結構符合Parsek 等人於 2003 年針對細菌形成生物膜所造成疾 病的臨床特徵。因此,以往也認為在贅疣中轉糖鏈球菌是以生物膜的形式存在。
然而,細菌在體外生物膜的形成機制與體內生物膜形成機制似乎有著相當大的差 異。在 Streptococcus gordornii 的研究中曾發現體外生物膜有明顯缺失的菌株,在 動物實驗中,並不會影響其造成感染性心內膜炎的致病力 (Bizzini et al ., 2006)。
而有學者利用signature-tagged library selection 方式,篩選出多個 Streptococcus sanguinis 的突變株,在體外生物膜培養中會發生明顯缺失,然而,這樣的突變卻 不會影響他們造成感染性心內膜炎的能力(Ge et al., 2008)。因此我們嘗試建構一套 新的體外篩選模式可模擬細菌進入血液,受到血漿的刺激後,在體內形成的生物 膜。然而,在我初步利用BHI 培養基加上 1%葡萄糖培養轉糖鏈球菌生物膜形成時,
外加入不同比例的血小板貧乏的血漿部份(platelet-poor plasma),發現隨著血漿比例 的增加,卻會抑制轉糖鏈球菌生物膜的形成。然而,當我以富含血小板的血漿部 份(platelet-rich plasma)進行培養時,卻可以觀察到明顯細菌生物膜的形成。而我更 進一步利用帶有綠螢光蛋白的轉糖鏈球菌與血小板貧乏的血漿和富含血小板的血 漿進行培養後,以共軛焦顯微鏡觀察細菌生物膜的形成,我也發現只有在富含血 小板的血漿中才能觀察到細菌生物膜的形成。顯示血小板對於轉糖鏈球菌在血漿 中形成生物膜是重要的。
然而,血漿中的什麼物質有可能抑制培養基培養的細菌生物膜形成,卻不干
擾細菌與血小板的結合而形成生物膜?在以往的研究中,免疫球蛋白
(Immunoglobulin)已經被發現可以黏附於 S. aureus 的表面,並進一步抑制 S. aureus 培養基培養生物膜的形成(Merino et al., 2009)。然而,有學者發現在 S. Sanguis 中,
S. Sanguis 可能藉由免疫球蛋白與血小板的 FcγRIIA 結合,進一步誘發血小板的活 化(Ford et al., 1997)。而在 S. pyrogenes 中也有報導指出,S. pyrogenes 可以藉由細 菌的表面蛋白M1 protein 先與血漿中的纖維蛋白原結合,並黏附到未活化的血小 板上,當血液中對抗M1 protein 的免疫球蛋白與 M1 protein 結合後,S. pyrogenes 可以藉由免疫球蛋白的Fc 與血小板的 Fc receptor 結合,最後誘發血小板的活化 (Shannon et al., 2007)。此外,在 Kahn 等人的研究中也發現,S. pyrogenes 可以利用 M1 protein 與纖維蛋白原及免疫球蛋白結合後,進一步活化血液中的嗜中性球 (Neutrophil)促使 heparin-binding protein 的釋放,引發血管的受損,最後造成鏈球 菌毒性休克症候群(streptococcal toxic shock syndrome)的發生(Kahn et al., 2008)。因 此我們猜測在缺乏血小板的狀況下,血漿中的免疫球蛋白很有可能去抑制細菌形 成生物膜。而在培養基中外加入serotype C 的抗體後,也發現細菌生物膜形成有明 顯被抑制,顯示免疫球蛋白在培養基培養細菌生物膜形成有抑制的作用。之後可 以嘗試利用流式細胞儀來研究細菌與血小板之間的作用是否需要免疫球蛋白的參 與。
更進一步我也想探討血小板活化與否對於轉糖鏈球菌在血漿中形成生物膜是 重要的,藉由加入四種不同的血小板活化抑制劑,我發現血小板的活化對於轉糖 鏈球菌形成生物膜是重要的。而由感染性心內膜炎動物模式中,我們給予感染性 心內膜炎大鼠抗凝血劑阿斯匹靈後,可以發現其細菌生物膜的形成有明顯的缺 失,而贅疣中菌量也有些微的減少,然而卻無法有效清除贅疣中的細菌。在臨床 的研究中,也試圖利用阿斯匹靈以及其他抗血小板凝集藥物來作為治療感染性心 內膜炎的佐劑,然而臨床試驗的結果差異性卻相當大。在2003 年 Chan 等人的研 究中發現給與感染性心內膜炎病患阿斯匹靈並不會增加抗生素治療的效力,也無 法改善心內膜炎病患常發生的血栓情形(Chan et al., 2003)。而在近期的研究中卻發 現給予病患抗血小板治療(antiplatelet therapy)可以有效減少病患血栓(Embolism)的 形成,但不影響六個月內病患的死亡率(Anevakar et al.,2007)。然而在 2009 年 Pepin 等人的研究中卻又有不同的發現,他們篩檢出有感染性心內膜炎早期症狀之病
人,並同時給予長期的抗血小板治療(Chronic antiplatelet therapy)後。他們發現這樣 長期的治療可以有效減低感染性心內膜炎造成的致死率,但並不會影響病患血栓 之形成(Pepsin et al., 2009)。此外,也有醫療案件指出阿斯匹靈的給予曾有效減輕 一新生兒的感染性心內膜炎中贅疣的增生(Adler et al., 2004)。根據這些研究結果,
迄今對於抗血小板藥物是否適用於感染性心內膜炎患者仍舊留有爭論。然而為何 這些抗血小板藥物(尤其是 salicylate 系列藥物)所造成之結果有如斯的差異,現在 主要有兩種想法:第一是個體間的差異性造成的藥物抗性不同。在臨床研究中發現 大約有5%至 45%的急性冠狀動脈症候群患者(Acute coronary syndrome patients)會 對阿斯匹靈產生抗藥性,而其中又有約8%到 18%的患者服用阿斯匹靈後,兩年中 會發生週期重複性的心血管問題(Mason et al., 2005)。此外,有學者也發現女性比 男性似乎更容易有阿斯匹靈抗藥性的狀況(Dorsch et al., 2007),且低藥量的阿斯匹 靈(75 mg)可有效改善男性所發生的心肌梗塞(Myocardial infarction)情形,但對於女 性則無顯著差異(Kjeldsen et al., 2000)。第二是藥物劑量的多寡。在 Levison 等人 1977 年的研究中發現當他們給予感染性心內膜炎兔子單次高劑量的阿斯匹靈(600 mg)或批次性給與阿斯匹靈(75 mg)八次後,他們發現高劑量阿斯匹靈並無法減少贅 疣的增生與贅疣中的菌量(Levison et al., 1977)。然而,卻有另外的研究指出 24 小 時給予一次感染性心內膜炎兔子低劑量的阿斯匹靈(5 mg/kg),總共五次後,可以 發現贅疣的重量與菌量有明顯的減少(Nicolau et al., 1993)。而在 Kupferwasser 等人 的研究中又更一步指出阿斯匹靈的治療效果並不會隨著濃度增加而增加,而是有 其治療的劑量範圍(Kupferwasser et al., 1999)。根據這些結果,現今的治療不斷嘗 試找出阿斯匹靈治療感染性心內膜炎的Goldilocks effect(恰到好處的效果),但迄 今對於阿斯匹靈的劑量給與仍未有定論(Eison et al., 2008)。而在我的研究中先利用 ADP 將血小板先行活化後,我發現外加的血小板抑制劑便無法有效抑制細菌形成 生物膜,這也暗示血小板活化後細菌便可以黏附上去,形成生物膜。而在亞急性 感染性心內膜炎的發生過程中,第一步是心臟瓣膜要先有所缺損,造成血小板與 纖維蛋白的凝集後,草綠色鏈球菌才能有效黏附於受傷的瓣膜上(Moreillon et al., 2004)。就此我們推測在人工造成心臟瓣膜缺損時,便會誘發血小板活化凝集在受 傷的組織上,當我們將菌打入大鼠體內時,細菌便能黏附到活化的血小板上。而 給予抗凝血劑阿斯匹靈後,會抑制未活化的血小板受細菌刺激而活化,卻無法回 復已活化的血小板,因此我們雖然不能觀察到明顯的多層結構生物膜形成,但仍
可見到明顯的生物膜形成。而在人體中很有可能也是相似的原因造成阿斯匹靈無 法有效治療感染性心內膜炎病患。
2.PspC 蛋白表現的缺失影響轉糖鏈球菌體內生物膜的形成。
在本實驗室奇玄學長的研究中曾經發現在轉糖鏈球菌第十一對雙分子調控系 統(two component regulatory system)的 response regulator (RR11)發生缺失的菌株 中,可以觀察到其體內生物膜的形成有明顯的缺失(林奇玄,2008)。而在俊達學長 的研究中,利用microarray 的方式檢測野生株 GS5 與 RR11 mutant 基因表現的差 異,發現 pspC 基因在 RR11 mutant 中表現量會有明顯的下降。而我更進一步利用 染色質免疫沉澱法(Chromatin immunoprecipitation)證明 pspC 基因在體內很有可能 是直接受RR11 所調控。而利用生物資訊的方法找到 pspC 基因前段的 intergenic region 有一段約 16 mers ( GTTTATACTAATAGCA )的 DNA 序列,在多個可能受 RR11 調控的基因前段可能啟動子區位中有相當高的相似性。利用膠體電泳阻滯實 驗(electrophoretic motility shift assay)也更一步確定這段序列很有可能是 RR11 與 DNA 結合的區位。
在先前的研究中,Psp 蛋白首先是在 Escherichia coli 中被發現,當 E. coli 受到 絲狀嗜菌體(Filamentous phage)感染時,Psp 蛋白會大量被表現,因而稱為
phage-shock-protein (Psp) (Brissette et al., 1990)。此外,學者們發現 Psp 蛋白是一種 具高度保留性的蛋白,在大多數的革蘭氏陽陰性菌,如:Escherichia coli、Yersinia enterocolitica、Salmonella enterica 中,都有發現高度相似性的蛋白存在(Darwin ., 2005)。此外,在革蘭氏陰性菌中,Psp 蛋白的調控的生理機制多由數個 Psp 蛋白 所構成,一般也稱為phage-shock-protein system (Psp system)。其中以 E. coli 的 Psp system 研究較多也對其調控機制較為瞭解。在 E. coli 中,Psp system 主要被發現有 PspF、PspA、PspB、PspC、PspD、PspE 與 PspG 七個蛋白所構成,而 PspBCDG 被發現為具有穿膜結構(transmenbrane domain)的膜蛋白,而 PspF 蛋白則是被發現 具有轉錄因子(transcription factor)的功能,在一般情況下 PspF 蛋白會與 PspA 蛋白 結合而抑制PspF 轉錄因子的活性。然而當外在有誘導訊號(Inducing signal)刺激菌
體後,這樣的訊號會被PspB 與 PspC 蛋白所接收,PspC 會更進一步延攬 PspA 過 來,將PspA 蛋白固定在內膜區位上並干擾 PspA 與 PspF 蛋白的結合,驅使 PspF 蛋白的轉錄因子活性恢復,調控下游基因的表現(如附圖三)。而 PspG 與 PspD 在 整個Psp system 中的調控機制為何,則迄今仍不是很清楚(Darwin ., 2005)。而現今 對於是怎樣的誘導訊號會誘發Psp 蛋白的表現也有些許研究。在先前的研究裡,
絲狀嗜菌體的感染被發現可以促使Psp system 的表現(Brissette et al., 1990)。此外,
質子內驅力(Proton motive force)的減少被發現也會誘導 Psp system 的表現(van der Lann et al., 2003),然而質子內驅力的減少究竟是直接或間接促使 Psp sytem 的表 現,現今仍不清楚。而也有研究發現secretin 蛋白的錯誤定位也可能參與誘發 Psp system 的表現(Model et al., 1997),secretin 蛋白被發現可能參與了許多細菌的 type II 與type III secretion system,因此也被認為可能影響細菌的致病力,而在 Y.
enterocolitica 中發現在 Ysc type III secretion system 合成過程中,Psp response 對於 細菌的致病能力是重要的(Darwin et al., 2001)。雖然對於 Psp system 的調控機制與 誘發訊號都有許多的發現,然而,在細菌體內Psp system 到底扮演了怎樣的生理 功能仍舊不清楚。
在我們構築的體外模式中,PspC 蛋白有所缺失的突變株並不能有效形成完整 的生物膜。而在感染性心內膜炎大鼠中,若感染PspC 有所缺失的突變株,其贅疣 上也無法觀察到明顯的生物膜生成。在將PspC 送回的 complementation strain 則又 可以觀察到明顯的生物膜形成,這樣的結果也顯示PspC 蛋白對於細菌體內生物膜 的形成是重要的。而在我第一部分的研究中顯示在感染性心內膜炎發生過程中,
血小板與細菌之間的交互作用扮演著重要的角色。因此我也進一步探討在PspC 蛋 白有所缺失的菌株中,是否會去影響其與宿主因子(Host factors)的結合,最後影響 了體內生物膜的形成。然而,我測試了細菌與血小板的黏附,發現PspC 蛋白的缺 失並不影響其血小板黏附的能力。此外,PspC 蛋白的缺失也並不影響細菌在血漿 中與纖維蛋白原(Fibrinogen)的結合。然而在體內病菌清除試驗(In vivo clearance) 中卻意外發現在PspC 蛋白發生缺失的狀況下,會使 PspC 突變株更容易在體內被 清除。而在全血殺菌試驗(Whole blood killing)中也發現,PspC 突變株比野生株更 容易被全血殺死。在先前的研究中也發現,當 Shigella flexneri 被多型核白血球 (polymorphonuclear leukocyte)吞噬後,可以觀察到 Psp system 會被大量表現
(Lucchini et al., 2005)。這些結果似乎暗喻著 PspC 蛋白對於細菌於在生物體內生存 是重要的。然而,由這些結果我們無法釐清究竟是因為PspC 突變株容易在體內被 清除所以不容易在體內形成生物膜,抑或是PspC 蛋白的缺失影響了體內生物膜的 形成。這些問題需要更進一步的實驗證明。
五、參考文獻
Adler A, Litmanovitz I, Bauer S, Dolfin T. Aspirin treatment for neonatal infectious endocarditis. Pediatr Cardiol. 2004 Sep- Oct;25(5):562-4.
Ajdić D, McShan WM, McLaughlin RE, Savić G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc Natl Acad Sci U S A. 2002 Oct 29;99(22):14434-9.
Anavekar NS, Tleyjeh IM, Anavekar NS, Mirzoyev Z, Steckelberg JM, Haddad C, Khandaker MH, Wilson WR, Chandrasekaran K, Baddour LM. Impact of prior antiplatelet therapy on risk of embolism in infective endocarditis. Clin Infect Dis. 2007 May 1;44(9):1180-6.
Beachey EH, Stollerman GH. Toxic effects of streptococcal M protein on platelets and polymorphonuclear leukocytes in human blood. J Exp Med. 1971 Aug 1;134(2):351-65.
Beloin C, Valle J, Latour-Lambert P, Faure P, Kzreminski M, Balestrino D,
Haagensen JA, Molin S, Prensier G, Arbeille B, Ghigo JM. Global impact of mature biofilm lifestyle on Escherichia coli K-12 gene expression. Mol Microbiol. 2004
Feb;51(3):659-74.
Bensing BA, López JA, Sullam PM. The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibalpha. Infect Immun. 2004 Nov;72(11):6528-37.
Bizzini A, Beggah-Moller S, Moreillon P, Entenza J M. Lack of in vitro biofilm formation does not attenuate the virulence of Streptococcus gordonii in experimental endocarditis. FEMS Immunol Med Microbiol. 2006;48:419-423.
Burnette-Curley D, Wells V, Viscount H, Munro CL, Fenno JC, Fives-Taylor P, Macrina FL. FimA, a major virulence factor associated with Streptococcus parasanguis endocarditis. Infect Immun. 1995 Dec;63(12):4669-74.
Brissette JL, Russel M. Secretion and membrane integration of a filamentous