國立臺灣大學醫學院基因體暨蛋白體醫學研究所 碩士論文
Graduate Institute of Medical Genomics and Proteomics College of Medicine
National Taiwan University Master Thesis
亞洲特異性 ALDH2 突變造成肥胖與胰島素阻抗性的 分子機轉
Asian-specific ALDH2 mutation causes obesity and insulin resistance: molecular mechanism
劉采晴 Cai-Cing Liu
指導教授:張以承 博士
Advisor:Yi-Cheng Chang, M.D., Ph.D.
中華民國 108 年 7 月
July 2019
誌謝
兩年碩士生涯一晃即逝,回頭看看這一路上的歷程,心中實在有說不盡的感激。
首先感謝的是我的指導教授張以承老師,老師不管是在課業或是實驗方面都提供 了我很多的建議與幫助,另外也和我分享他自身的人生經歷與價值觀,也在我陷入 低潮的時候給予我極大的鼓勵與安慰,我非常幸運可以在老師指導下完成我的碩 士論文,這些日子真的很感謝老師的指導與教授。
另外,感謝莊立民教授在每次的實驗室會議中,對於我的進度報告提供了寶貴 的意見,並鼓勵學生去進一步思考,也感謝莊立民教授與黃祥博教授在擔任口委期 間針對我的論文給予建議,使論文得以更臻完善。
接著要感謝實驗室的所有同仁,謝謝蘇寧與曉薇學姊在我初進實驗室時教導 我許多相關的實驗,以及當我遇到低潮的時候傾聽我的煩惱,給我關心和溫暖。謝 謝孟倫學長與詩宜學姊對實驗室的付出。謝謝李博作為我論文的合作夥伴,與我一 起討論實驗的設計與實作,一起找出實驗有瓶頸的部分,你的意見往往提供我在實 驗上重要的線索,真的很感激能在你的協助之下完成這本論文。謝謝敬詠學姊與雅 鈴在動物實驗上幫了我很多的忙,讓我能夠順利的完成實驗,也感激你們平日的照 顧,總是從你們這邊收到很多關懷,讓我覺得很感動也窩心。另外也謝謝致中學弟 在這段期間的幫忙,希望你能夠順利畢業。
最後我要感謝我的家人,在這忙碌的兩年當中幾乎都沒有回家,但你們還是一 直默默的支持我,讓我能夠心無旁騖地專心做自己的事情,有你們的關心與打氣,
讓我更有動力能在繁忙的課業中順利撐過來,希望我的努力及成長能讓你們感到 欣慰及驕傲。。
最後,謹以此篇論文獻給我的師長、家人以及所有關愛我的朋友,謝謝你們。
采晴 2019.7
中文摘要
東亞地區,有過40%的東亞人群攜帶 ALDH2 特有的錯義點突變(Glu487Lys),
使其酵素在異合子酵素活性失去60-80%,同合子酵素活性失去約 90%,此基因突
變為全球盛行率最高之單一基因疾病(占~8%全球人口)。
ALDH2 (acetaldehyde dehydrogenase 2,mitochondria)是一粒線體內的酵素,代
謝乙醛為乙酸。除此之外,ALDH2 還會代謝多種有害醛類,包括人體的中間代謝、
腸道細菌發酵、環境當中煙霧、香菸、各種塑料以及氧化壓力引發細胞脂肪過氧化
而產生的有害醛類,如4-hydroxynonenal(4-HNE),而這些有害醛類會與蛋白質產
生共價鍵修飾而改變其功能。
ALDH2 特有的錯義性點突變(Glu487Lys)在東亞人全基因組掃描中被發現與肥
胖與相關表現型有關,我們以模擬人類突變的 ALDH2 *2/*2 基因嵌入鼠為模型,
發現 ALDH2 *2/*2 基因嵌入鼠在餵食高脂高糖的飲食下,相較於對照小鼠,體重
明顯上升,胰島素阻抗性增加並伴隨脂肪肝與脂肪細胞肥厚。
在這項研究中,我們探討 ALDH2 *2/*2 基因嵌入鼠產生肥胖的原因,發現此
小鼠能量消耗減少,起因推測可能與進食後產熱降低相關;另外觀察到嵌入鼠的棕 色脂肪組織明顯變小。這些現象顯示此嵌入鼠可能因棕色脂肪組織功能減少、產熱 降低,而產生肥胖。
我們進一步去探討分子機制,發現4-HNE 會降低棕色脂肪細胞的脂肪酸氧化
功能。利用LC-MS/MS,我們發現 4-HNE 會與脂肪酸氧化的酵素以及電子傳遞鏈
上的酵素結合,綜上所述,推測ALDH2 *2/*2 基因嵌入鼠會因為有害醛類累積,
影響脂肪酸氧化與粒線體功能而造成小鼠肥胖。
關鍵字:乙醛脫氫酶, 醛類, 4-羥基壬烯醛, 肥胖, 能量消耗
ABSTRACT
In East Asia, approximately 40% of the East Asian population carries an inactivating
missense Glu487Lys mutation of ALDH2 (SNP671) gene. The ALDH2 enzymatic
activity reduces to ~60-80 % in heterozygotes and ~90 % in homozygotes of mutants. It
is the most prevalent monogenetic disease in the world (~8% global population).
ALDH2 (acetaldehyde dehydrogenase 2, mitochondria) is the primary enzyme
responsible for metabolizing toxic acetaldehydes in mitochondria. ALDH2 also
metabolizes a variety of toxic aldehydes, including acetaldehyde from intermediate
metabolism, intestinal bacterial fermentation, environmental smog, cigarette smoke,
various plastics and toxic aldehydes generated from lipid peroxidation by oxidative stress,
such as 4-hydroxynonenal (4-HNE). These aldehydes modify proteins by forming
covalently bond crosslinks, thereby altering their biological functions.
Several recent large-scale meta-analysis of genome-wide association studies in East
Asian identified ALDH2 Glu487Lys polymorphism is significantly associated with
obesity and related metabolic phenotypes. In our study, we generated Aldh2 *2/*2 knock-
in mice mimicking the human mutation as a model. We found that Aldh2 *2/*2 knock-in
mice are prone to develop obesity, insulin resistance, fatty liver, and adipocyte
hypertrophy on high-fat high sucrose diet as compared to controls.
We found that the Aldh2 *2/*2 knock-in mice had significantly lower energy
expenditure than controls. This reduction in energy expenditure may result from reduced
diet-induced thermogenesis. In particular, the brown adipose tissue was markedly smaller
in Aldh2 *2/*2 knock-in mice. This finding suggests the reduced thermogenesis of Aldh2
*2/*2 knock-in mice may result from impaired brown adipose tissue function. We further
explored the molecular mechanism by which the knock-in mice have reduced brown
adipose tissue function. We found 4-hydroxynonenal (4-HNE) inhibits fatty acid beta-
oxidation in brown adipose tissue. Using liquid chromatography-tandem mass
spectrometry (LC-MS/MS), we found that 4-HNE modifies enzymes involved in fatty
acid beta-oxidation and the electron transfer chain. In summary, we found that Aldh2
*2/*2 knock-in mice are prone to develop obesity, insulin resistance, and fatty liver due
to reduced thermogenesis, which may be related to impaired development of brown
adipose tissue and toxic aldehydes-mediated suppression of fatty acid oxidation and
mitochondrial function.
Key word: ALDH2, aldehyde, 4-hydroxynonenal, obesity, energy expenditure
CONTENTS
口試委員會審定書 ... #
誌謝 ... i
中文摘要 ... ii
ABSTRACT ... iii
CONTENTS ... v
Chapter I Introduction ... 1
1. Aldehyde dehydrogenase 2 (ALDH2) serves as a crucial enzyme that catalyzes aldehydes metabolism but the Glu487Lys mutation causes East- Asian specific ALDH2 deficiency ... 1
2. The pathogenic effects of aldehydes through form different adducts in vivo . 2 3. Lipid peroxidation-derived products association with obesity in adipose tissue ... 3
4. Brown adipose tissue thermogenesis and obesity ... 4
5. Rationale of this study ... 6
Chapter II Materials and Methods ... 7
1. Animal model ... 7
2. Glucose and insulin tolerance test ... 7
3. Energy expenditure, food intake and physical activity ... 8
4. Cold tolerance test and diet-induced thermogenesis test ... 8
5. RNA extraction and RTqPCR ... 9
6. Primary cell culture... 10
7. Western blot analysis ... 11
8. Fatty acid oxidation assay ... 12
9. Isolation of brown adipose tissue mitochondria ... 14
10. In-solution digestion ... 15
11. LC-MS/MS analysis ... 15
12. Database analysis ... 16
Chapter III Result ... 17
1. Mice with ALDH2 Glu487Lys mutation display much more obesity phenotype during High-Fat-Diet ... 17
2. Aldh2 KI mice exhibit insulin resistance and glucose intolerance ... 18
3. Aldh2 KI mice have lower energy expenditure in response to HFHSD feeding and loss the fatty acid oxidation capacity in BAT ... 18
4. Aldh2 KI mice express higher levels of modified proteins ... 21
Chapter IV Discussion... 24
FIGURE ... 27
TABLE ... 38
REFERENCE ... 39
Chapter I ― Introduction
1.Aldehyde dehydrogenase 2 (ALDH2) serves as a crucial enzyme that catalyzes
aldehydes metabolism but the Glu487Lys mutation causes East-Asian specific
ALDH2 deficiency
Aldehyde dehydrogenase 2 (ALDH2) belongs to a family of detoxifying enzymes,
aldehyde dehydrogenases, (ALDHs) involved in the detoxification of aldehydes. It is
located in the mitochondria and is responsible for ethanol metabolism. The pathway has
two steps: ethanol is first oxidized by the alcohol dehydrogenase (ADH) in the liver to
acetaldehyde. Then aldehyde dehydrogenase 2 (ALDH2) oxidizes acetaldehyde to
acetate[1]. Additionally, previous reports suggest that ALDH2 shows the lowest Km for ethanol[2, 3]. It also has the highest catalytic efficiency for acetaldehyde metabolism than
other isoforms. It is estimated that, ALDH2 catalyze ~95 % acetaldehyde after ethanol
intake[4, 5].
A portion of East Asian population carry a missense mutation in their ALDH2 gene
(SNPr671), which the guanine is replaced by adenine. This mutation, named
the ALDH2*2 allele, results in the substitution of lysine for glutamate in the protein at
residue 487 (E487K)[6, 7]. The heterozygotes (ALDH2 *1/*2) lost more than half the
wild-type activity, and homozygotes ( ALDH2 *2/*2) only have less than 1% enzymatic
activity of wild-type individuals (ALDH2 *1/*1) [8, 9]. Individuals carries the inactive
ALDH2 will develop “Asian flushing” syndrome, which is characterized by headache,
facial flushing, nausea, and tachycardia after ethanol consumption owing to, acetaldehyde
accumulation[10, 11]. This inactivating mutation is highly prevalent in East Asia,
particularly in Southeast China, Taiwan, Japan, and Korea which is considered the most
common monogenic disease across the globe (~8% global population)[12].
2. The pathogenic effects of aldehydes through form different adducts in vivo
ALDH2 mutation is involved in the pathogenesis of many diseases, including
cardiovascular diseases, diabetes mellitus, neurodegenerative diseases, cancer, Fanconi
anemia, Osteoporosis, pain, and aging[5]. These diseases are linked to aldehydes from
endogenous and exogenous from the environment. Aldehydes are generated during
metabolism that are intermediates or products from physiologic, biologic, and
pharmacologic processes. For example, microbes produce acetaldehyde in saliva,
stomach acid, and intestines. Fermented foods, alcoholic beverages, cigarette smoke,
exhaust gas from automobile, factory, and various chemicals and pollutants all contain
amounts of aldehydes [13, 14].
Biogenic aldehydes, primarily formed by oxidation of lipids, glucose, and primary
amines, are generated from tissues with high oxidative stress. They are highly reactive
electrophiles, which spontaneously react with macromolecules such as proteins, DNA,
and lipids through covalent modification[15-17]. Lipid peroxidation, especially oxidized
polyunsaturated fatty acids (PUFAs), leads to the generation of α,β-unsaturated aldehydes,
such as 4-hydroxynonenal (HNE) and 4-hydroxyhexenal (HHE), and malondialdehyde
(MDA), which are highly bioactive and are considered as “second toxic messengers”[18].
These aldehydes would react with DNA and cause genotoxic DNA modification,
inhibit DNA and protein synthesis, modulate gene expression, and trigger cellular
apoptosis[19, 20]. The nucleophilic side chains of histidine, cysteine, and lysine residues
of protein are susceptible to the covalent attack of aldehydes. These covalent
modifications result in a free carbonyl attached to the protein, termed “carbonylation”.
Generation of carbonyl adducts lead to a range of pathologic damages[21-24].
3. Lipid peroxidation-derived products association with obesity in adipose tissue
In modern society, the obesity epidemic lead to a range of metabolic disorders,
including fatty liver, dyslipidemia, and type 2 diabetes mellitus (T2DM)[25]. Obesity and
these related metabolic disorders are associated with increased oxidative stress, lipid
peroxidation, and byproducts[26-28]. The general sequence of events is that obesity
induces oxidative stress level and then triggers lipid peroxidation and finally produces
aldehydes by-products. However, these aldehydes can further stimulate the production of
ROS that causes a vicious cycle between oxidative stress and carbonyl stress [28-30].
Oxidative stress induced by obesity is a major contributor to insulin resistance.
Adipose tissue is rich in polyunsaturated fatty acids (PUFAs) in cell membrane, which is
a target of lipid peroxidation. Peroxidation of polyunsaturated fatty acids (PUFAs) finally
gives rise to the emancipation of diffusible reactive lipid aldehydes. They can diffuse into
the cytoplasm and nucleus from membranes which formed in and react with a variety of
proteins and DNA[22].
Among the variety reactive aldehydes, 4-hydroxynonenal(4-HNE), derived from
oxidation of n-6 fatty acids, is the most reactive and cytotoxic, and most well studied [31].
For instance, mice lacking the gene mGSTA4-4 cannot catalyze HNE and are prone to
develop obesity and insulin resistance[32]. On the other hand, 4-HNE was found to
elevate the adipose tissue of obese humans and mice [33], and decrease insulin secretion
of pancreatic islets [34, 35]. Moreover, 4-HNE also has been reported to block the insulin-
signaling pathways [30, 36, 37].
4. Brown adipose tissue thermogenesis and obesity
The concept of energy homeostasis is the balance of body energy, including energy
intake, energy expenditure. Energy intake means the food intake and energy expenditure
consists of basal metabolic rate, the thermic effect of food (TEF), the physical activity,
and the thermogenesis corresponding to the environment. When the energy intake is equal
to the energy expenditure, the energy homeostasis approaches stable. However, obesity,
the well-known and significant drivers behind insulin resistance, often happens when
energy intake exceeds energy expenditure [38, 39].
Brown adipose tissue plays an essential role in modulating energy expenditure,
which is responsible for thermogenesis. Brown adipocytes have highly enriched
uncoupling protein‐1 (UCP1) located at mitochondria inner membranes, which makes
protons leak across the membrane to produce heat[40]. And fatty acid β-oxidation
provides fatty acid as the substrates for activating the process. The procedure of fatty acid
β-oxidation is transporting the long-chain fatty acid (LCFAs) into the mitochondria
matrix by the carnitine palmitoyltransferase (CPT) system, which includes CPT1, CPT2,
and carnitine/acylcarnitine translocase (CACT). And acetyl-CoA is generated eventually
and getting into the TCA cycle to produce ATP or uncoupling ATP production by UCP1
to produce heat. [41, 42].
Studies showed that brown adipose tissue participates in both cold-induced and diet-
induced thermogenesis[43]. Inhibition of fatty acid oxidation in mice has been
demonstrated severely cold intolerance[44, 45]. On the other hand, it is reported that BAT
mass and activity was reduced in obese and diabetic patients[46]. BAT-mediated
thermogenesis can promote energy expenditure and cope with the physiological functions
and may act as a potential organ to combat obesity.
Figure 1. Brown adipocyte possesses a high capacity for fatty acid β-oxidation[42]
5. Rationale of this study
Large-scale meta-analyses of genome-wide association studies in East Asian
identified ALDH2 Glu487Lys polymorphism are associated with blood pressure, lipids,
fasting plasma glucose, and body mass index[47, 48]. In our previous research also prove
that ALDH2 genetic polymorphism is associated with blood pressure and hypertension in
Han Chinese[49].
Here, we utilized Aldh2 *2/*2 knock-in mice which mimic the Glu487Lys (E487K)
mutation in human by homologous recombination with a C57BL/C background. We
sought to determine the effects of Aldh2 mutation on obesity and related metabolic
phenotypes and explore the underlying mechanism.
Chapter II― Materials and Methods
1.Animal model
Aldh2 *2/*2 knock-in mice were provided by the Dr. Che-Hong Chen from Dr. Daria
Mochly-Rosen’s lab, Stanford University. Aldh2 *2/*2 knock-in mice that on the
C57BL/C genetic background carry the Glu487Lys (E487K) were generated by
homologous recombination. All mice were housed in a temperature-controlled
environment at 23℃ under a 12 hr light/dark cycle. Mice were fed on high-fat-high-
sucrose diet (Cat. No. D12331, Research Diets, USA) or chow diet since the age of 4
weeks, respectively. Body weight was measured weekly. All animal experiments were
approved by the Institutional Animal Care and Use Committee of the Medical college of
National Taiwan University.
2.Glucose and insulin tolerance test
At 24 weeks, mice were evaluated by intraperitoneal glucose tolerance test (ipGTT)
after a 6-hour fasting at the age of 24 weeks. Tail blood glucose were measured at 0, 15,
30, 45, 60, 90, 120 min after intraperitoneal injection of glucose water (1mg/kg). The
insulin tolerance test (ITT) were conducted at the age of 25 weeks. Briefly, mice were
fasted 4 hours, and insulin (1.6IU/kg and 1.1IU/kg for HFHSD and chow diet respectively,
Humulin RTM, Eli Lily, USA) were injected intraperitoneally. Tail blood glucose were
measured at 0, 15, 30, 45, 60, 90, 120, 180 min. For oral glucose tolerance test (OGTT)
conducted at the age of 26 weeks, mice were fasted for 6 hours and glucose water (1mg/kg)
were given by oral gavage. Tail blood glucose was measured at 0, 15, 30, 60, 120 min
following glucose administration. Acute insulin response were measured using oral
glucose tolerance test (OGTT) at the age of 27 weeks. Mice blood was collected at 15 and
30 min during OGTT. Blood was centrifuged at 13000rpm, 10min, 4℃. Plasma was
collected for measurement insulin concentration.
3.Energy expenditure, food intake and physical activity
Indirect calorimetry measurements were performed using the Promethion Metabolic
Cage System (Promethion® , Sable Systems, Las Vegas, NV) at National Health Research
Institutes Laboratory Animal Center. VO2 and VCO2 of individual mouse were measured
for calculation of energy expenditure and the respiratory exchange ratio (RER). Wheels
in the chamber was used for measurement of the distance that mice run within 24 hours.
The food intake was measured for each mouse.
4. Cold tolerance test and diet-induced thermogenesis test
For a cold tolerance test, 10-week-old mice fasted for 4 hours were placed at 4℃
chamber and then the rectal temperature of mice was measured at 0, 1, 2, 3, 4 hour
respectively. Before the diet-induced thermogenesis test, 10-week-old mice fasted
overnight for 18 hours. The rectal temperature after HFHSD re-feeding were measured
at 0, 15, 30, 60, 120, 180, 240 min.
5.RNA extraction and RTqPCR
Brown adipose tissue from Aldh2 WT and KI mice was minced and harvested in 1ml
of REzol C&T (Cat. No. PT-KP200CT, Protech, Taiwan). 200μl of chloroform was added
to 1 ml of sample in Rezol, and samples were mixed vigorously by shaking for 30 seconds
and then were incubated at room temperature for 5 min. Then, samples were centrifuged
at 12000xg, 15min, 4℃, and 400 μl of the upper aqueous phase were transferred to a new
1.5 ml Eppendorf tube. An equal volume of isopropanol was added and samples were
inverted several times to mix and centrifuged at 12000xg, 10 min, 4℃. The RNA
precipitate, which formed a pellet at the bottom of 1.5 ml Eppendorf tube, was washed
with 75% ethanol for three times and are dried for 15 min. Pellets were dissolved the in
RNase-free water. The concentration of RNA was measured by Nanodrop. cDNA were
synthesized with reverse transcription kit (Cat. No. K1622, Thermo Scientific) by using
the oligo(dT)18 random primer. Real-time quantitative PCR (RT-qPCR) was performed in a 10 μl reaction with 50 ng cDNA and 0.4 μM primer using SYBR green reagent (Cat.
No. 1123ES08, YEASEN, China). Mouse Cyclophilin A mRNA was used as the internal
control. RT-qPCR reactions were performed by using ABI7900HT FAST (Applied
Biosystems, USA) and Sequence Detection Systems (SDS v2.3, Applied Biosystems).
All qPCR reactions were run in duplicates for each sample.
6.Primary cell culture
Brown adipose tissue from 4-6 weeks Aldh2 WT and KI mice were minced and
digested in 0.5% type I collagenase (Thermo Scientific) in 5 μM HEPES buffer for 40
min at 37℃. The digests were centrifuged at 600g for 5 min and the supernatant was
removed. The stromal vascular fraction (SVF) cell pellet was resuspended with PBS then
centrifuged at 600g for 5 min. After removal of the supernatant, pellet was resuspended
and cultured in 5μM Dulbecco’s modified Eagle medium: Nutrient Mixture F-12 (DMEM/F-12) (Cat. No. 12500062, Hyclone, USA) supplemented with 10% FBS (Cat.
No. 04-001-1A, Biological Industries, USA) and 1% antibiotic/antimycotic solution (Cat.
No. SV30079.01, Hyclone). For differentiation, primary brown preadipocyte were
exposed to induction medium containing 10% FBS, 0.5mM isobutyl-methylxanthine (Cat.
no. sc-201188A, Santa Cruz Biotechnology, USA), 1μg/ml insulin (Humulin RTM, Eli
Lilly), 5μM dexamethasone (Cat. no. D4902, Sigma-Aldrich, USA), 1nM T3(cat.no.
T5516, Sigma-Aldrich, USA), 125μM indomethacin (Cat. no. I7378, Sigma-Aldrich,
USA) as indicated. After 2 days, the medium was changed into DMEM/F12 containing
10% FBS, 1μg/ml insulin, and 1nM T3. Then we changed medium every 2 days until assay.
7.Western blot analysis
BAT homogenates from 30-week HFHSD Aldh2 WT and KI mice were prepared
using Radioimmunoprecipitation assay buffer (RIPA buffer) with protease inhibitor
cocktails (cOmplete™ ULTRA Tablets, Mini, EDTA-free, EASYpack Protease Inhibitor
Cocktail, Roche). The protein concentration was measured using the Bradford assay. We used 50 μg protein on the SDS-PAGE gel and then transferred it to the polyvinylidene
fluoride microporous membrane (GE Healthcare Life Science). The membranes were
blocked with 10% milk in PBS containing 0,1% Tween-20, and were probed with
antibodies UCP1 (Cat. no. GTX10983, GeneTex), 4HNE[HNEJ-2] (Cat. no. ab48506,
Abcam), Hsp70 (Cat. no. ab45133, Abcam), GAPDH (Cat. no. MAB374, Merck).
Corresponding secondary antibodies conjugated to horseradish peroxidase were used the
chemiluminescence. Signal were detected using TOPBIO Chemiluminescence image
system (MultiGel-21, MGIS-21-C2-6M).
The protein using for detecting with biotin hydrazide and 4HNE antibody was
incubated in 10mM NaBH4 for 30min. For carbonylated proteins, samples chemically
reduced by NaBH4 were then incubated with 5mM EZ-link biotin hydrazide (Cat. no.
21339, Pierce) for 1 hour. After coupling, the samples are separated by SDS-PAGE gel
and transferred to the polyvinylidene fluoride microporous membrane (GE Healthcare
Life Science). The membrane was blocked with 10% milk in PBS containing 0,1%
Tween-20 overnight at 4℃. Rinsed with PBST and incubated with Streptavidin-HRPA (Cat. no. 890803, BD Biosciences) for 1hour. Signal were detected using TOPBIO Chemiluminescence image system (MultiGel-21, MGIS-21-C2-6M).
8.Fatty acid oxidation assay
Differentiated primary brown adipocytes from Aldh2 WT and KI mice cultured in
24-well-plate were washed with PBS three times, and incubated with 250μl
palmitate/BSA buffer including 0.5 μCi [3H]-palmitate (Cat. no. PK-NET043001MC,
3H-Palmitic acid,1mCi, PerkinElmer) for 2 hours. At the end of the incubation period,
the reaction mixture was transferred to the 2ml Eppendorf tube. We then add 750μl chloroform, 250μl chloroform/methanol (1:2 volume mixture), and 250μl 2M KCl/HCl to previous 2ml Eppendorf tube in sequence. The samples were vortexed for 5 seconds
and centrifuged at 3500rpm, 10min, 4℃. 600μl upper aqueous phase was transferred to
new 2 ml Eppendorf tubes. 400 μl chloroform was then added and vortexed for 5 seconds, followed by addition of 400 μl methanol. Then 360 μl 2M KCl/HCl was added and vortexed for 5 seconds. The samples were centrifuged at 3500rpm, 10min, 4℃. 1ml upper
aqueous phase was transferred to scintillation vial which contains 4 ml of scintillation
fluid and was measured the average counts per minute (CPM) by a liquid scintillation
counter.
Regarding fatty acid oxidation of tissues, Aldh2 KI and WT mice were fed with
HFHSD for 5 weeks. Mouse BAT was isolated, weighed, and added STE buffer (0.25M
Sucrose,10mM Tris-HCl, 1mM EDTA, pH7.4), which the volume we added was
100mg/ml of tissue. BAT was homogenized using a glass Dounce homogenizer with the
“Loose” pestle down and up 10 strokes each. The homogenate was poured to 1.5 ml
Eppendorf tube and centrifuged at 500g for 10min at 4℃. The supernatant was decanted
to new 1.5ml Eppendorf tube. We transferred 30 μl of tissue homogenate to 24-well-plate containing 370μl reaction mixture buffer which had prepared previously. Each sample
was running in triplicate. The reaction mixture buffer was prepared with 100mM sucrose,
10mM Tris-HCl, 5mM KH2PO4, 0.2mM EDTA, 80mM KCl, 1mM MgCl2, 2mM L-
carnitine, 0.1mM malate, 0.05mM Coenzyme A, 2mM ATP, 1mM DTT, 7% BSA/5mM palmitate/ 0.01 μCi/μl [3H]-palmitate. The samples were incubated for 60min at 37℃. At
the end of the incubation, the total volume of each sample was transferred to 2ml Eppendorf tube. We then add 750μl chloroform, 250μl chloroform/methanol (1:2 volume
mixture), and 250μl 2M KCl/HCl in sequence. The samples were vortexed for 5 seconds and centrifuged at 3500rpm, 10min, 4℃. 600μl upper aqueous phase was transferred to
new 2 ml Eppendorf tubes. 400 μl chloroform was then added and vortexed for 5 seconds, followed by addition of 400 μl methanol. Then 360 μl 2M KCl/HCl was added and vortexed for 5 seconds. The samples were centrifuged at 3500rpm, 10min, 4℃. 1ml upper
aqueous phase was transferred to scintillation vial which contains 4 ml of scintillation
fluid and was measured the average counts per minute (CPM) by a liquid scintillation
counter.
9.Isolation of brown adipose tissue mitochondria
The BAT from 30-week-old Aldh2 KI and WT mice were homogenized in the
Dounce homogenizer with ice-cold mitochondrial isolation buffer (210mM Mannitol,
70mM sucrose, 1mM EGTA, 5mM HEPES pH7.5, 0.5%BSA), which the volume we
added was 100mg/ml of tissue. The homogenate was centrifuged 800g for 10 min at 4℃
and the supernatant was decanted and filtered through two-layer cheesecloth to remove
residual particulates. The filtered homogenate was centrifuged by 8000g for 10 min at
4℃ and carefully removed the supernatant. The pellet was resolved with 1ml
mitochondrial isolation buffer and centrifuged again. The mitochondrial pellet was re- suspended in 30 μl of STE buffer (0.25M Sucrose,10mM Tris-HCl, 1mM EDTA, pH7.4)
and protein concentration was quantitated using the Bradford assay.
10.In-solution digestion
The proteins samples in 6 M urea were reduced by 20 mM dithiothreitol for 1 h at
60°C, and alkylated by 55mM iodoacetamide for 45 min in the dark at room temperate.
Then, the samples were diluted 6-fold with 50 mM triethyl ammonium bicarbonate buffer
and digested with trypsin or chymotrypsin at an enzyme:protein ratio of 1:50 w/w for 16-
18 h at 37 °C and 25 °C individually. Digestion was quenched by an addition of
trifluoroacetic acid to a final concentration of 1%. Peptides were desalted with C18 ziptip
(Millipore Corp., Billerica, MA, USA) according to the instructions of the manufacturer.
Desalted and dried peptides were resuspended in 10μL of 0.1% formic acid (FA), and
ready for LC-MS/MS analysis.
11.LC-MS/MS analysis
LC-MS/MS analysis was performed on a NanoACQUITY UPLC System (Waters,
USA) coupled to a high-resolution mass spectrometer (QE HF-X, Thermo Fisher Scientific, USA). The peptides were injected into a trap column (2 cm × 75 μm i.d.,
Symmetry C18), then separated in a 25 cm × 75 μm i.d. BEH130 C18 column (Waters,
USA) by a gradient from 0% to 85% buffer B (buffer A, 0.1% formic acid in H2O; buffer
B, 0.1% formic acid in acetonitrile). The mass spectrometer is operated in data-dependent
mode with the following acquisition cycle: a MS scan (m/z 350–1600) recorded at
resolution R = 60,000; MS/MS scans recorded at resolution R = 15,000, which are
acquired by HCD fragmentation with collision energy of 28.
12.Database analysis
MS/MS spectra were searched with the Mascot engine (v2.6, Matrix Science, UK)
against the UniProtKB mouse protein database using the following parameters: the mass
tolerance of precursor peptide was set as 20 ppm, and the tolerance for MS/MS fragments
was 0.02 Da with maximum two missed cleavage. The modifications of peptides were set
as follows: static carbamidomethylation on cysteine, variable oxidation on methionine,
variable deamidation of asparagine or glutamine, and variable 4-HNE modification on
cysteine, histidine and lysine. The cut-off threshold of significant peptide-to-spectrum
matches is p < 0.05.
Mitochondrial modified proteins biological function were confirmed by using the Uniport database. Then we used g:Profiler to get the “Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathways” and “Gene ontology biological process” information.
Chapter III― Results
1. Mice with ALDH2 Glu487Lys mutation display much more obesity phenotype
during High-Fat-Diet
To assess the effect of ALDH2 Glu487Lys mutation on obesity, we placed wild-type
mice and Aldh2 *2/*2 knock-in mice (herein Aldh2 WT mice and Aldh2 KI mice) on a
regular chow diet or high-fat high-sucrose diet (HFHSD) for 16 weeks. Aldh2 KI mice
exhibited a significant weight gain compared to Aldh2 WT mice (Figure1A). Body
composition, analyses showed that Aldh2 KI mice have significantly higher fat mass, lean
mass and total water (Figure1B). However, Aldh2 KI mice fed with a chow diet for 16
weeks did not exhibit differences in body weight and body composition (Figure 2A and
2B). Aldh2 KI mice had a significant increase in each depot of white adipose tissue,
including inguinal and perigonadal fat. Conversely, the brown adipose tissue is
significantly smaller in Aldh2 KI mice (Figure 2C). Compared with Aldh2 WT mice,
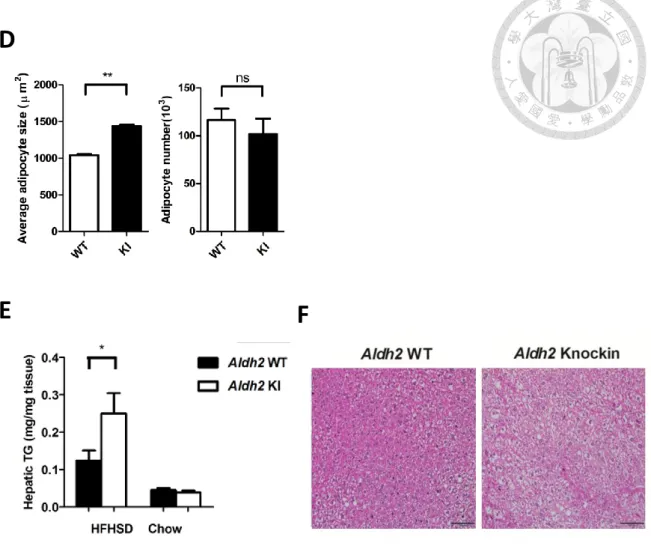
Aldh2 KI mice had significantly larger adipocyte size but similar adipocyte number in
perigonadal white adipose tissue (Figure 2D). Besides, HFHSD-fed Aldh2 KI mice had
higher mass and hepatic triglycerides content of the liver and developed severe hepatic
steatosis (Figure 2E and 2F).
2. Aldh2 *2/*2 knock-in mice exhibit insulin resistance and glucose intolerance
Aldh2 KI mice have worsened glucose tolerance than Aldh2 WT mice during
intraperitoneal glucose tolerance test (ipGTT) (Figure 3A). Insulin tolerance test (ITT)
showed impaired insulin sensitivity in HFHSD-fed Aldh2 KI mice (Figure 3B).
Consistently, the oral glucose tolerance test (OGTT) demonstrated the same trend. Aldh2
KI mice also exhibited compensatory increase in acute insulin secretion (Figure 3C and
3D). However, there was no difference between Aldh2 KI mice and WT mice on the chow
diet (Figure 3).
These data show East-Asian-specific Aldh2 mutation causes obesity, hepatic
steatosis, insulin resistance, and glucose intolerance in mice on HFHSD.
3. Aldh2 *2/*2 knock-in mice have lower energy expenditure in response to HFHSD
feeding and loss the fatty acid oxidation capacity in BAT
To explore the mechanism by which Aldh2 KI mice are prone to develop obesity
and related metabolic phenotypes, we measured their energy balance including energy
intake and expenditure. Indirect calorimetry showed that Aldh2 KI mice exhibit lower
oxygen consumption and energy expenditure at dark phase (Figure 4A). The basal
metabolic rate is similar between Aldh2 KI mice and WT mice. There is also no change
in food intake and physical activity between two strains (Figure 4B). We next examine
thermogenesis function including cold-induced thermogenesis and diet-induced
thermogenesis. When mice were placed at a 4°C cold environment, there is no difference
of temperature change between Aldh2 KI mice and Aldh2 WT mice (Figure 4C). However,
Aldh2 KI mice exhibited significantly lower diet-induced thermogenesis (DIT), defined
as the thermogenesis food ingestion (Figure 4D). These data suggest that reduced diet-
induced thermogenesis in Aldh2 KI mice might be a major cause of decreased energy
expenditure.
We next investigated the causes of reduced thermogenesis in Aldh2 KI mice. Brown
adipose tissue plays a vital role in adaptive thermogenesis and its unique protein,
uncoupling protein-1 (UCP1), generates heat by uncoupling oxidative phosphorylation.
[40]. For this process, fatty acid β-oxidation is essential and provides substrates for
uncoupling in BAT[50, 51]. And it has been showed that thermogenic brown adipocyte
possesses a high capacity for fatty acid β-oxidation. Chondronikola et al. also reported
that BAT mitochondrial thermogenesis is 45-fold greater than that of white adipose tissue.
[52]. Thus, we hypothesized the reduced thermogenesis and energy expenditure of Aldh2
KI mice are probably due to reduced brown adipose tissue mass.
To understand the function of fatty acid β-oxidation, we measured the fatty acid β-
oxidation related genes and thermogenic gene in brown adipose tissue form Aldh2 KI and
WT mice fed HFHSD. Aldh2 KI BAT showed a significantly reduced expression of fatty
acid β-oxidation-related genes, including Cpt1b, Cpt2, and Aco1. On the other hand, there
is no difference in the expression of UCP1 (Figure 5A and 5B). Since previous studies
showed that aldehydes accumulate in Aldh2 KI mice, we evaluated the effect aldehydes
on the function of brown adipose tissue. Primary brown preadipocytes were isolated from
4-week-old mice and induced into mature brown adipocytes. We found that 4-HNE
inhibited fatty acid oxidation in a concentration-dependent manner (Figure 5C).
Furthermore, primary brown adipocytes isolated from Aldh2 KI mice showed a
significant decrease of fatty acid oxidation as compared to Aldh2 WT mice (Figure 5D).
Following this finding, we further investigated the fatty acid oxidation capacity of
whole brown adipose tissue. Aldh2 KI mice and WT mice were fed HFHSD for five weeks,
and fresh BAT was isolated for fatty acid oxidation assay. There is a non-significant
reduction in fatty acid oxidation capacity of whole brown adipose tissue of Aldh2 KI mice.
These data show that the decrease fatty acid β-oxidation of BAT in Aldh2 KI mice may
explain the decreased energy expenditure observed in Aldh2 KI mice.
4. Aldh2 *2/*2 mice express higher levels of modified proteins
ALDH2 mutation is characterized by the inactive ALDH2 enzyme activity, which
fails to metabolize toxic aldehydes. As a result, these bio-reactive aldehydes, such as
4HNE, will attack proteins and alter their functions. To examine the hypothesis, we
explored the extent of 4HNE-adduction and protein carbonylation in the Aldh2 KI and
WT mice.
Based on previous research, 4HNE modified proteins has been extensively studied
and involved in the development of a variety of metabolic diseases[53-55]. However, the
protein modification by 4HNE in brown adipose tissue is still unclear. Chronic dietary
nutrient overload accelerated obesity and is associated with enhanced mitochondria
oxidative stress, increasing lipid peroxidation in adipose tissue[56]. The brown adipose
tissue has a large capacity for mitochondria fatty acid β-oxidation, which is critical for
regulating energy balance. Loss of the brown adipose tissue mitochondria fatty acid
oxidation function due to modification by aldehyde adduction may result in energy
imbalance in Aldh2 KI mice.
We first compared the pattern of 4-HNE modified proteins and carbonylation
proteins of BAT from Aldh2 KI and WT mice using 4HNE antibody and biotin hydrazide-
tagged aldehyde reactive probe, respectively. Western blot showed increased levels of
carbonylation proteins (left panel) and 4-HNE adducted protein (right panel) in Aldh2 KI
mice as compared to controls (Figure 6A).
The workflow presented an analytical scheme for identifying the 4-HNE-modified
proteins in the Aldh2 KI and WT mice at the age of 30 weeks. Aldh2 KI and WT mice
were fed HFHSD and the brown adipose tissue mitochondrial proteins were isolated.
Proteins were digested with trypsin and subjected to liquid chromatography-tandem mass
spectrometry (LC-MS/MS). Then the MS data were queried by the Mascot software
(Figure 6B).
We used 2 Aldh2 KI and 2 WT mice in the study. After removing all the proteins that
were detected without 4HNE modified, a list of soluble proteins considered as targets of
4HNE modification in BAT were generated (Table1). A total of 20 proteins were
identified, and ten mutual proteins were showed between Aldh2 KI and WT mice. And it
revealed that Aldh2 KI mice have more modified proteins than Aldh2 WT mice (Figure
6C and 6D).
We used online bioinformatics resources g:Profiler to obtain the Kyoto Gene
Encyclopedia and genomic pathway (KEGG pathway) and Gene ontology biological
process to assess the association of these proteins. Protein functional information were
provided from the Uniprot database. Some of proteins are associated with mainly
metabolic processes, including fatty acid, amino acid, and nucleotide metabolism and
TCA cycle and others are related to oxidative phosphorylation, respiratory electron
transport chain, NADH and ATP metabolic process (Table 1). Acetyl-CoA acyltransferase
2 (Acaa2), one mutual protein in Aldh2 WT and KI mice, is involved in fatty acid
metabolism. And it is also the last step of the mitochondrial fatty acid β-oxidation
pathway by breaking down fatty acid into acetyl-CoA. On the other hand, proteins are
involved in energy metabolism, such as NADH dehydrogenase [ubiquinone] 1 subunit
C2 (Ndufc2), cytochrome b-c1 complex subunit 7 (Uqcrb) and so on.
Chapter IV― Discussion
In this study, we found that Aldh2 knock-in mice mimicking the East-Asian-specific
ALDH2 Glu487Lys mutation are prone to develop obesity, fatty liver, insulin resistance
and glucose intolerance. The obesity phenotype is probably caused by reduced energy
expenditure and thermogenesis. Unexpectedly, we observe that the brown adipose tissue
is remarkably smaller in Aldh2 KI mice compared to wild-type mice, either on chow diet
or HFHSD. We further found the fatty acid oxidation capacity is reduced in BAT of Aldh2
KI mice, which may lead to the observed reduced thermogenesis.
Brown adipose tissue generates heat primarily within the mitochondria through
fatty acid oxidation and uncoupling of oxidative phosphorylation mediated by UCP1. The
fatty acid can be activators of the UCP1 for thermogenesis or be suppliers in the electron
transport chain for generating ATP. The oxidation of fatty acid in brown adipocyte
represents a major source of thermogenesis[57]. Suppression of fatty acid oxidation
compromise thermogenesis. For instance, adipose-specific knockout CPT2 mice
presented the hypothermic when they were exposed in a cold environment. And the
thermogenic genes in CPT2 mice brown adipose tissue didn't up-regulate under the
agonist-induced stimulation[45, 58]. When ACSL1, an enzyme catalyzing the formation
of acyl-CoA that are used for β-oxidation, are impaired in adipose tissue. The Acsl1A−/−
mice are also showed cold intolerance due to impaired fatty acid oxidation[44]. It
indicates that mitochondrial fatty acid β-oxidation is critical for the thermogenesis.
In this study, we did not observe altered UCP1 expression in Aldh2 knock-in mice.
However, either primary brown adipocytes or whole brown adipose tissue isolated from Aldh2 KI mice exhibited reduced fatty acid oxidation capacity. We hypothesize it may link to the higher aldehyde production in the BAT mitochondria of Aldh2 KI mice. Indeed, we observed that either 4-HNE-modified proteins or carbonylated proteins are enriched in the BAT isolated from Aldh2 KI mice.
Using LC-MS/MS, we further demonstrated that 4-HNE conjugated to the proteins
involved in mitochondrial electron transfer chain and fatty acid oxidation. And these
proteins demonstrated higher mount in Aldh2 KI mice. The conjugation of 4-HNE to these
proteins may interfere fatty acid oxidation. Consistent with our finding, mitochondrial
dysfunction caused by oxidative stress and the ROS-initiated lipid peroxidation
byproducts has been reported to be involved a range of disease in recent years[59, 60].
And it also indicates about 30% of 4-HNE-modified proteins formed in cell existing in
mitochondria and is considered as the major candidate to cause mitochondrial
dysfunction[61, 62]. Previous studies have demonstrated the 4-HNE modified proteins
plays important role in the pathogenesis of Alzheimer's disease[63, 64], rheumatological
diseases and other autoimmune disease[65], gastrointestinal diseases[66], myocardial
diseases[67], and cancer[68, 69].
Our study supports that 4-HNE or other toxic aldehydes may damper the function of
BAT, leading to obesity and related metabolic phenotypes. In addition to the reduced fatty
acid oxidation, which is probably caused by the adduction of aldehydes to protein
involved in fatty acid oxidation and mitochondrial electron transfer chain, we also observe
that the BAT of Aldh2 KI mice is much smaller in volume than wild-type controls. The
underlying mechanism is currently unknown. It may be related to the impaired embryonic
development of BAT. Therefore, we are currently investigating the embryonic
development of BAT in Aldh2 KI and WT mice.
FIGURE
A
B
C
Figure 2. Phenotype of Aldh2 *2/*2 knock-in mice and wild-type mice
(A) Body weight on HFHSD (n=21:24) and chow diet (n=19:15)
(B) Body composition on HFHSD (n=20:12) and chow diet(n=19:15)
(C) Distribution tissue weight of inguinal fat (subcutaneous), perigonadal fat
(intraabdominal fat), liver, and brown adipose (BAT) of Aldh2 *2/*2 knock-in mice (KI) and wild-type (WT) mice on HFHSD(n=78:67) or chow diet (n=12:10)
(D) Perigonadal fat adipocyte size and number from Aldh2 *2/*2 knock-in mice (KI)
and wild-type (WT) mice fed HFHSD (n=17:24)
D
E F
(E) Hepatic triglyceride content of HFHSD (n=23:20) and chow diet (n=8:10)
(F) Representative H&E stain of liver
* P < 0.05, ** P < 0.01, ***P<0.001. Data presented as mean ± s.e.m.
A
B
C
D
Figure3. Glucose and insulin tolerance test of Aldh2 *2/*2 knock-in mice and wild-
type mice
(A) Intraperitoneal glucose tolerance test on HFHSD(n=44:49) or chow diet(n=19:15)
(B) Insulin sensitivity test on HFHSD(n=44:49) or chow diet(n=19:15)
(C) oral glucose tolerance on HFHSD(n=44:49) or chow diet(n=19:15)
(D) insulin response during OGTT on HFHSD(n=41:47)
* P < 0.05, ** P < 0.01, ***P<0.001. Data presented as mean ± s.e.m.
ipGTT: Intraperitoneal glucose tolerance ;OGTT: oral glucose tolerance; ITT: insulin
tolerance test
C D
A B
Figure4. Aldh2 *2/*2 knock-in mice decrease energy expenditure in response to
HFHSD
(A) Relative oxygen consumption (VO2) and energy expenditure over a 24-hr period
(n=12:12)
(B) Metabolic parameters of food intake and wheel meters (n=12:12)
(C) The temperature of cold-induced thermogenesis (n=7:8)
(D) The temperature of diet-induced thermogenesis (n=8:8)
* P < 0.05, ** P < 0.01, ***P<0.001. Data presented as mean ± s.e.m.
C D
E
B
A
Figure 5. The effect of impaired ALDH2 activity inhibits fatty acid oxidation in
brown adipose tissue
(A) Quantitative real-time polymerase chain reaction(qPCR) analysis of mRNA for fatty
acid oxidation and thermogenic genes in BAT (n=22:20)
(B) Western blot of UCP1 in BAT of Aldh2 *2/*2 knock-in mice and wild-type mice fed
HFHSD (n=4:4)
(C) Fatty acid oxidation was measured in primary brown adipocytes which treated
different 4HNE concentration
(D) Fatty acid oxidation was measured in primary brown adipocytes from Aldh2 *2/*2
knock-in and wild-type mice (n=15:15)
(E) Fatty acid oxidation was measured with BAT from Aldh2 *2/*2 knock-in and wild-
type mice fed 5week HFHSD. The body weight and BAT mass were measured(n=14:10)
* P < 0.05, ** P < 0.01, ***P<0.001. Data presented as mean ± s.e.m.
A
B
C
Figure 6. LC-MS/MS Analysis the BAT mitochondrial modified protein by 4HNE
(A) Detection of carbonylated proteins and 4-HNE modified proteins in BAT from Aldh2
*2/*2 knock-in mice and wild-type mice fed HFHSD (n=4:4)
(B) Experimental design and workflows used in the study
(C) Comparison of identified proteins from Aldh2 *2/*2 knock-in mice and wild-type
mice at the age of 30 weeks
(D) Venn diagram of identified proteins from Aldh2 *2/*2 knock-in mice and wild-type
mice
![Figure 1. Brown adipocyte possesses a high capacity for fatty acid β-oxidation[42]](https://thumb-ap.123doks.com/thumbv2/9libinfo/9608813.634299/15.892.235.789.115.463/figure-brown-adipocyte-possesses-high-capacity-fatty-oxidation.webp)