國立臺灣大學生物資源暨農學院生物產業機電工程學系
碩士論文
Department of Bio-Industrial Mechatronics Engineering College of Bioresources and Agriculture
National Taiwan University Master Thesis
利用序列特徵提升使用染色質免疫沉澱定序平台 預測轉錄因子結合位點之準確度
Incorporating sequence motifs to improve accuracy of predicting transcription factor binding sites using ChIP-seq data
吳秉承 Ping-Cheng Wu
指導教授:陳倩瑜 博士 Advisor: Chien-Yu Chen, Ph.D
中華民國 105 年 6 月
June 2006
i
誌謝
碩士兩年,時光飛逝轉眼間結束了,如果說人生最重要的成長經歷,那一定 就是在碩士求學的這兩年。我從一個機械學科背景的大學生進入到了生物資訊領 域,學習以前不了解的資訊相關技能,且在老師、學長姐們的幫助與支持下,讓 原本懵懂無知的我,一點一點慢慢的進步與成長,最後終於成功躍過跨領域的大 門,在這艱辛的路程上,有太多的感謝說不完,而這些種種將會深深烙印在我的 回憶裡。首先十分感謝無時無刻都為我著想的指導教授─陳倩瑜教授,總是不厭 其煩的教導我這個領域的相關知識,而在研究的過程中,遇到了很多的未知與困 難,也經常面臨各種難關與煎熬,但倩瑜老師總是能用正面且積極的態度鼓勵我 們,一起面對這些各式各樣的問題,深深覺得能在這樣一個實驗室裡與夥伴們一 起努力,我真的很榮幸。
光陰荏苒,感謝實驗室的學長姐同學學弟充實了我碩班兩年的生活,首先感 謝哲栩,與我一起奮鬥碩班這兩年來一起修課的種種經過,在那無數個寧靜的夜 晚,我們一起打拼一起努力往前進,此外,要謝謝學長姐:玫如、祐榆、翊安、
育銓,謝謝兩年來的指導與照顧,從開始基本的程式學習,到後來生物知識上面 的指導,特別感謝玫如學姐&翊安學長的幫忙,與我討論碩論內容,並給我許多 的建議和加油打氣,我才能把最後的關頭撐過去。
最後,僅以此論文獻給我的家人,謝謝你們在我這段求學的路上,許多的呵 護與叮嚀,支持我度過種種的挫折與困難,我將繼續努力往人生下一步邁進,並 會盡我最大努力去實現自己的理想。
iii
中文摘要
染色質免疫沉澱定序技術,是用來尋找特定蛋白,例如轉錄因子,與其調控 的基因一種方法,藉由這種技術我們可以大略的知道轉錄因子在人體 DNA 片段 上的位置,然而這些被找到的轉錄因子結合位點的準確率尚未曾有研究做過系統 性的討論。因此,本論文裡用 TRANSFAC 資料庫提供之已知的轉錄因子結合位 點,針對染色質免疫沉澱技術鑑定之不同信心程度(FDR)下的轉錄因子結合位點 進行整體性的預測表現評估,並輔以序列特徵資訊,增進其預測準確度。本論文 使用了 ENCODE 資料庫的染色免疫沉澱資料來進行分析,且挑選了擁有不同細 胞株的轉錄因子來做比較,整體而言,各個細胞株的結果顯示,經由 ChIP-seq 鑑定出的峰值區中,約六成會包含至少一個該特定轉錄因子的轉錄因子結合位。
此外,本論文發現利用模序探勘所得之序列特徵結合 ChIP-seq 峰值區的資訊去 預測轉錄因子結合位,經觀察確實可增加預測轉錄因子結合位的準確率,然而,
使用不同 FDR 信心程度與不同的序列特徵,將會影響轉錄因子結合位的準確 率。本論文之研究結果點出單純使用染色質免疫沉澱技術預測轉錄因子結合位點 的缺陷,並提出序列特徵有助於改善預測結果,而可作為未來相關生物資訊預測 方法之重要基礎。
關鍵字:轉錄因子,轉錄因子結合位,模序探勘,染色質免疫沉澱,結合位特徵
ABSTRACT
Transcription factors (TF) regulate gene expression in living organisms and influence multiple biological processes. Chromatin immunoprecipitation sequencing (ChIP-seq) is a technology that have been widely used to find transcription factor binding sites (TFBSs) of a specific TF among the DNA sequences of a genome. However, the accuracy of the TFBSs identified by ChIP-seq has not been systematically evaluated.
In this regard, this thesis utilized TFBS information provided by the TRANSFAC database to validate the TFBSs identified by using ChIP-seq only with multiple false discovery rate (FDR). Moreover, in this thesis, a method incorporating de novo motif discovery was proposed to improve the performance of the predicted TFBSs.
ChIP-seq data sampled from different cell lines was collected from ENCODE database. In general, ∼60% of the peak regions identified by using the ChIP-seq only with a strict FDR cutoff (FDR = 0) contained at least one TFBS of the specific TF across multiple cell lines. In addition, by our proposed method, the prediction accuracy was improved and better than the results using ChIP-seq alone, though it was observed that the improved levels were affected by the used FDR cutoffs and discovered motifs. In conclusion, this thesis identified the accuracy problem of the ChIP-seq platform by observing from the data in a large scale, and address this issue by proposing a method incorporating de novo motif discovery. The observed results can serve as an important foundation for developing bioinformatics tools on TFBS prediction in future.
Keywords:
Transcription factor ; transcription factor binding site ; motif discovery ; Chromatin immunoprecipitation sequencing
v
目錄
論文口試委員審定書... i
誌謝... ii
中文摘要... iii
ABSTRACT ... iv
目錄... v
圖目錄... viii
表目錄... x
第一章 研究目的... 1
第二章 文獻探討... 3
2.1 分子生物學中心法則... 3
2.2 染色質免疫沉澱定序技術(ChIP-seq) ... 3
2.3.1 轉錄因子(Transcription factor) ... 4
2.3.2 轉錄因子結合位(Transcription Factor Binding Sites) ... 4
2.3.3 峰值(peak) ... 4
2.3.4 啟動子(Promoter) ... 5
2.4 細胞株與細胞系(Cell strain and cell line) ... 5
2.4.1 A549 細胞株 ... 7
2.4.2 HeLa-S3 細胞株 ... 8
2.4.3 GM12878 ... 8
2.4.4 K562 ... 9
2.4.5 Ishikawa ... 9
2.4.6 MCF-7 ... 10
2.5 使用的工具... 10
2.5.1 Bowtie2 ... 10
2.5.2 MACS ... 11
2.5.3 泊松分佈(Poisson distribution) ... 11
2.5.4 位置頻率矩陣(position frequency matrix,PFM)... 12
第三章 研究方法... 13
3.1 ENCODE 資料庫 ... 13
3.2 TRANSFAC 資料庫 ... 14
3.3 參考基因組資料(Reference Genome) ... 14
3.4 實驗流程... 15
3.4.1 序列回貼... 19
3.4.2 求出峰值... 19
3.4.3 篩選 FDR ... 21
3.4.4 結合位特徵(Consensus, annotated motifs) ... 23
3.4.5 TOP500 模序探勘 (De novo Motif Discovery) ... 25
第四章 結果與討論... 26
4.1 效果評估的參考數值... 26
4.2 探討只使用 ChIP-seq 平台鑑定轉錄因子結合位的效果 ... 27
4.3 透過整合各細胞株資料可增進預測之敏感度... 28
4.4 利用已知的序列特徵資訊確信染色質免疫沉澱定序技術與增加準確度 ... 32
4.5 利用模序探勘得到的序列特徵資訊可進一步增加準確度... 35
第五章 結論... 41
參考文獻... 42
附件(一) CEBPB ... 44
附件(二) NR3C1 ... 46
vii
附件(三) MAFK ... 47
圖目錄
圖 1.1 美國 2014 年癌症統計圖 Cancer Statistics, 2014,摘自文獻[2] .. 1
圖 2.1 ATCC 肺癌相關細胞株... 7
圖 3.1 ENCODE 資料庫,摘自文獻[12] ... 13
圖 3.2 實驗流程圖... 15
圖 3.3 ENCODE 資料庫下載資料頁面 ... 16
圖 3.4 為 MACS 輸出的 bed 檔格式 ... 20
圖 3.5 TRANSFAC 資料庫紀錄的位置與其對應的基因 ... 21
圖 3.6 CEBPB 在 TRANSFAC 資料庫被記載的位置 ... 22
圖 3.7 為 TRANSFAC 資料庫中紀錄的的 CEBPB 結合位特徵 ... 23
圖 3.8 峰值被抽取出來的序列... 24
圖 3.9 結合位特徵找到的峰值與其找到的特徵... 25
圖 4.1 CEBPB 轉錄因子的各細胞株不同 FDR 值的 Sensitivity 與 Precision 比較圖,單一條線上由左至右的點分別為 FDR0、FDR0.5、FDR1、 FDR5 與 No_filter。 ... 27
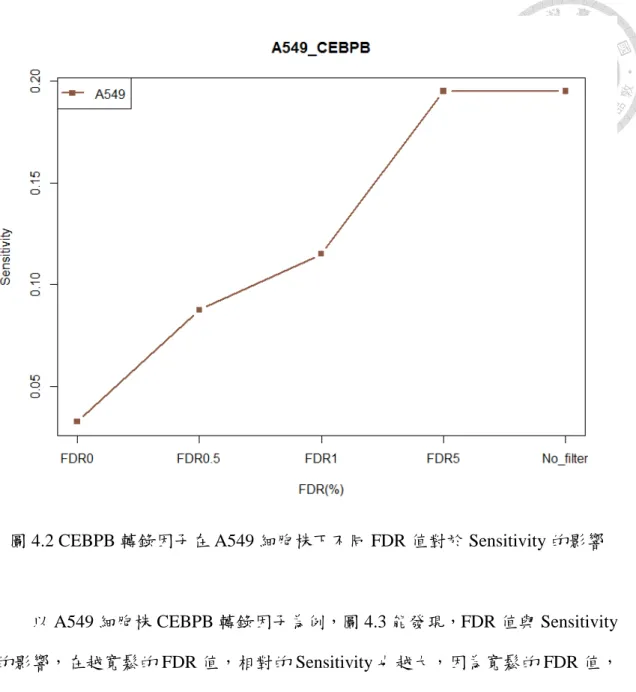
圖 4.2 CEBPB 轉錄因子在 A549 細胞株下不同 FDR 值對於 Sensitivity 的影響... 29
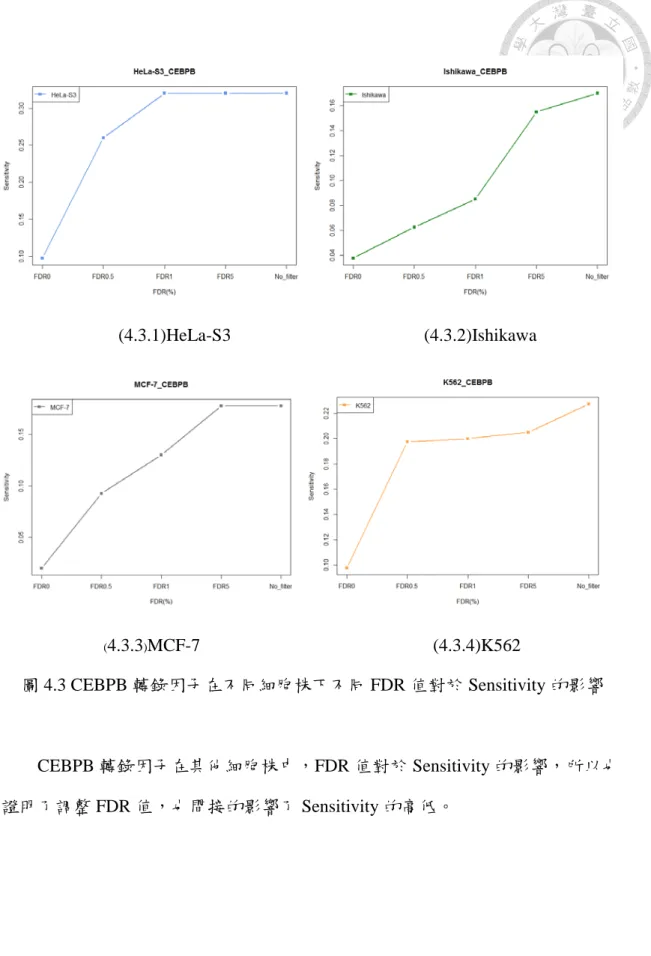
圖 4.3 CEBPB 轉錄因子在不同細胞株下不同 FDR 值對於 Sensitivity 的 影響... 30
圖 4.4 CEBPB 轉錄因子在所有的細胞株下不同 FDR 值對於 Sensitivity 影響的比較圖... 31
圖 4.5 A549 細胞株對到 TRANSFAC 的未致數目的 Precision 與存在著 TRANSFAC consensus 的 Precision 比較 ... 34 圖 4.6 CEBPB 在 Ishikawa 細胞株中,ChIP-seq 平台與 De novo method
ix
方法的 Precision 比較圖 ... 38
圖 4.7 CEBPB 在 MCF-7 細胞株中,ChIP-seq 平台與 De novo method 方 法的 Precision 比較圖 ... 38
圖 4.8 TRANSFAC 資料庫中 CEBPB 被紀錄的 motif ... 39
圖 4.9 為 Ishikawa 和 MCF-7 中 Precision 最高的 motif ... 39
圖 4.10 為 A549 細胞株與 K562 細胞株其中 eTFBS 找到的 motif ... 40
表目錄
表 2-1 A549 細胞株資訊 ... 7
表 2-2 HeLa-S3 細胞株資訊 ... 8
表 2-3 GM12878 細胞株資訊 ... 8
表 2-4 K562 細胞株資訊 ... 9
表 2-5 Ishikawa 細胞株資訊 ... 9
表 2-6 MCF-7 細胞株資訊 ... 10
表 3-1 轉錄因子與其對應的細胞株 ... 17
表 3-2 各個細胞株所代表的疾病 ... 18
表 3-3 為本實驗使用的細胞株和轉錄因子的免疫沉澱資料 ... 19
表 4-1 TRANSFAC 資料庫中 CEBPB 的位置在不同細胞株中被對到的位 置數目... 28
表 4-2 TRANSFAC 資料庫中 CEBPB 的位置在不同細胞株中被對到的峰 值數目... 32
表 4-3 TRANSFAC 資料庫中 CEBPB 的 consensus 對到不同細胞株的峰 值數目... 33
表 4-4 為 CEBPB 在 A549 細胞株中與 De novo method 的 Precision ... 35
表 4-5 為 CEBPB 在 HeLa-S3 細胞株中與 De novo method 的 Precision ... 36
表 4-6 為 CEBPB 在 Ishikawa 細胞株中與 De novo method 的 Precision ... 36
表 4-7 為 CEBPB 在 MCF-7 細胞株中與 De novo method 的 Precision 37 表 4-8 為 CEBPB 在 K562 細胞株中與 De novo method 的 Precision ... 37
1
第一章 研究目的
染色質免疫沉澱定序(ChIP-seq)技術,是用來尋找特定蛋白與其調控的基 因一種方法,藉由這種技術我們可以大略的知道這些特定蛋白在人體 DNA 片段 上的位置,進而可以更一步的去分析特定蛋白,這種技術是把染色質免疫沉澱定 序所得到的序列,拿去對回我們人類的參考基因組,然而這些對回到基因組上面 的片段,會形成一個一個的波峰,而這些波峰就可能是這些特定蛋白結合的位置。
圖 1.1 美國 2014 年癌症統計圖 Cancer Statistics, 2014,摘自文獻[2]
在 2014 年美國癌症統計中,圖 1.1 為美國 2014 年癌症統計,美國人在 2014 得到癌症機率男性最高前三名,為前列腺癌、肺癌和大腸癌,女性最高前三名為 乳癌、肺癌和大腸癌,致死率最高者都為肺癌,所以在致死率很高的情況下,眾
多科學家開始針對癌症進行了多項的研究,這些癌症的研究,跟轉錄因子蛋白質 有著密切的關係[3-6],因為轉錄因子與雙股 DNA 之間的相互作用對於基因表現 (gene expression)會產生活化或者抑制的影響,目前所知基因、轉錄因子結合位是 具有生物意義的片段,但他們僅占了 DNA 片段的一小部分,而轉錄因子如何辨 識其結合的位置進而達到調控基因表現量是目前非常重要的議題,且其結合位的 鏈結強弱可能會影響其調控基因的表現,所以我們想分析這些癌症與轉錄因子結 合位的關係,透過觀察發病的細胞株和不同轉錄因子的結合位是否與正常的基因 組有差異。
然而染色質免疫沉澱這個方法還存有很多問題,因為這些被尋找出來的波峰 並不完全是某一特定蛋白特有的位置,通常被找出來的位置,準確率是不高的。
所以我們想針對這樣的實驗,設計出一個能有效提升這些波峰準確率的方法,在 本實驗會從 ENCODE 資料庫中納入不同的細胞株,與其這些細胞株的特定蛋白 的染色質免疫沉澱資料,然後利用一些生物分析的工具來找出這些波峰,此實驗 使用 MACS 來作為尋找波峰的工具,透過這些不同的細胞株與特定蛋白去選擇 能幫助我們對於這些波峰的準確率與差異,進而提升尋找這些特定蛋白位置的準 確,且會使用 TRANSFAC 資料庫來作為這些位置的參考與標準,並利用位置頻 率矩陣的模序探勘去找尋峰值裡相似轉錄因子結合位的特徵,進而去比較分析結 果。
3
第二章 文獻探討
此章節將介紹本實驗所需要了解專有名詞的解釋、分析工具的介紹,以及之 前對於本實驗的相關研究內容的探討。
2.1 分子生物學中心法則
英國科學家 Francis Crick,於 1957 年針對遺傳訊息的傳遞提出了一個假設,
認為 DNA 會將訊息傳遞給 RNA,然後 RNA 再把訊息傳遞給蛋白質[7]。
可是在 1970 年,Francis Crick 做了補充及修正,生物遺傳訊息的傳遞是一 種 DNA 通過自我複製的過程,由親代 DNA 分子將訊息傳遞給子代 DNA 分子,
也就是 DNA 透過複製的過程而被保留下來,藉以保留遺傳物質。
另一方面,DNA 可以用一條鏈作為模板而進行合成 RNA,使得遺傳訊息可 以從 DNA 傳遞給 RNA 這就是轉錄,再經由 RNA 轉譯合成蛋白質[8]。
2.2 染色質免疫沉澱定序技術(ChIP-seq)
染色質免疫沉澱定序(ChIP)是用來研究細胞內蛋白質與DNA 的相互作用 關係,即確定特定蛋白(轉錄因子)是否會結合特定基因組區域(啟動子或其它 DNA 結合點位)[9]。
ChIP 實驗首先,附著在染色質上的蛋白和 DNA 以甲醛進行交叉結合反應,
然後再把細胞打破,以超音波將 DNA 切成每條長度約數百的 DNA 片段,再利 用辨識欲研究之特定蛋白的抗體,取出與該特定蛋白質結合的基因片段,沉澱步 驟完成,再把這些基因片段經由純化去除轉錄因子和所加之抗體,只留下 DNA 片段,將所得 DNA 片段定序完成,回貼至參考的基因組,完成染色質免疫沉澱
定序分析。
2.3.1 轉錄因子(Transcription factor)
轉錄因子是一種特定的蛋白質,轉錄是指將 DNA 序列轉換到 RNA 序列的 過程,過程中用一特定 DNA 片段作為模板,並利用 RNA聚合酶(polymerase)的 生物活性,進行合成 RNA 的過程。
因為轉錄因子是轉錄起始過程中 RNA 聚合酶所需要的輔助因子,真核生物 的基因在無轉錄因子時會處於不表現之狀態,因為 RNA 聚合酶自身無法啟動基 因轉綠,只有當轉錄因子結合在識別的 DNA 序列後,基因才會開始表現。
轉錄因子這一類蛋白質一般有不同的功能區域,如 DNA 的結合結構域和效 應結構域,DNA 結合結構域影響轉錄因子與 DNA 特異的結合特性,效應結構可 使轉錄因子與其他轉錄因子形成複合體來影響基因的轉錄的效率。
2.3.2 轉錄因子結合位(Transcription Factor Binding Sites)
轉錄因子結合位意指轉錄因子結合在基因組上的位置,而這些基因組可能是 轉錄的啟動或調控,找到轉錄因子結合位,也就可以知道轉錄因子結合在 DNA 序列上的位置,而研究指出轉錄因子結合位於啟動子區域中。
2.3.3 峰值(peak)
峰值就是我們把 ChIP-seq 貼回到參考基因組後,因為 ChIP-seq 是小片段的 序列,回貼時有些相同的片段會貼到一樣的基因組位置,而形成一個一個的小波 峰,這些小波峰我們就稱之為峰值(peak),峰值對我們找到轉錄因子結合位有極 大的關聯性,假如是單端定序(single-end)的 ChIP-seq 資料,貼回基因組所找到的 峰值,就可能是轉錄因子結合位之位置,假如是雙端定序(pair-end),則會貼回到 基因體的正反股,會依回貼序列的數量畫出常態分佈後,會得到正反股的峰值,
5
而峰值與峰值之間就是轉錄因子結合位之位置。
2.3.4 啟動子(Promoter)
啟動子為一段能促使基因開始進行轉錄的 DNA 序列,轉綠過程中,轉錄因 子會辨識認定的轉錄因子結合位的 DNA 序列,然後誘導特定的 RNA 聚合酶接 近啟動子與之結合並開始進行轉錄,轉錄因子結合位一般位於特定的啟動子區域 中,啟動子包含核心啟動子區域和調控區域,決定基因的活動進而控制細胞開始 產生蛋白質。
核心啟動子(core promoter)是引發轉錄的起始點,且為 RNA 聚合酶和一般轉 錄因子結合位點,近端啟動子(proximal promoter)為基因的近端序列上游,包含 基本的調控元件,且為特定的轉錄因子結合位點,核心啟動子大概位於轉錄啟始 位(transcription start site, TSS)的上下游 40 個鹼基,而近端啟動子位於 TSS 的上 下游 1000 鹼基中,為確保能找出所有的轉錄因子結合位,所以會取觀察 TSS 上 下游 10000 鹼基。
2.4 細胞株與細胞系(Cell strain and cell line)
細胞株是組織培養的一種形式,具有繼承能力且能隔代培養,通常是由細胞 生長成一個群落,而這樣的細胞通常有繼承且不變的特性,所以只要可以穩定生 長和不變的特性皆可稱為細胞株,培養細胞株的目的通常都是為了各種實驗研 究,且細胞培養的優點是可以比較容易的控制各種的可變因素,所以可以減少很 多人力成本、時間的消耗。
7
圖 2.1 ATCC 肺癌相關細胞株
ATCC 成立於 1925 年,為世界上最大生物資源中心,由美國 14 家生化、醫 學協會組成並負責管理,是一家全球性生物標準品資源中心。向全球發布其鑑 定、保存和開發的生物標準品,並推動科學研究的驗證、應用和進步,下面我們 會列出此研究有使用到的細胞株,和其細胞株的相關資訊。
2.4.1 A549 細胞株
Species Homo sapiens, human
Tissue lung
Disease Carcinoma
Age 58 years
Gender male
Ethnicity Caucasian
表 2-1 A549 細胞株資訊
表 2-1,為 A549 細胞株的資訊,可看到這個細胞株是有關於肺部組織,且 病因為癌症,年齡為 58 歲,且種族為白人男性。
2.4.2 HeLa-S3 細胞株
Species Homo sapiens, human
Tissue Cervix
Disease Adenocarcinoma
Age 31 years
Gender Female
Ethnicity Black
表 2-2 HeLa-S3 細胞株資訊
表 2-2,為 HeLa-S3 細胞株的資訊,可看到這個細胞株是有關於子宮頸,病 因則為腺癌,也是一種癌症,年齡為 31 歲,種族為黑人女性。
2.4.3 GM12878
Species Homo sapiens, human
Life stage Adult
Gender Female
Disease Epstein-Barr Virus
Ethnicity Caucasian
表 2-3 GM12878 細胞株資訊
表 2-3,為 GM12878 細胞株的資訊,這個細胞株的疾病是有關於人類皰疹 病毒第四型,種族為白人女性。
9
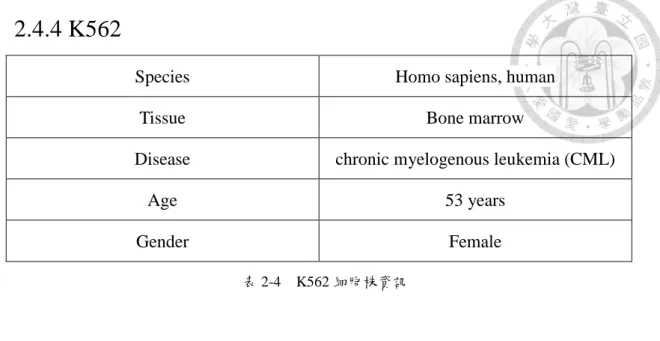
2.4.4 K562
Species Homo sapiens, human
Tissue Bone marrow
Disease chronic myelogenous leukemia (CML)
Age 53 years
Gender Female
表 2-4 K562 細胞株資訊
表 2-4,為 K562 細胞株的資訊,可看到這個細胞株是有關於人類骨髓的部 分,病因是慢性骨髓性白血病,是一種白血病,年齡為 53 歲的女性。
2.4.5 Ishikawa
Species Homo sapiens, human
Age 39 year
Gender Female
Disease Endometrial adenocarcinoma
Ethnicity Japanese
表 2-5 Ishikawa 細胞株資訊
表 2-5,為 Ishikawa 細胞株的資訊,這個細胞株是有關於子宮內膜腺癌,年 齡為 39 歲,日本人女性。
2.4.6 MCF-7
Organism Homo sapiens, human
Tissue mammary gland, breast; derived from metastatic site: pleural effusion
Disease adenocarcinoma
Age 69 years
Gender female
Ethnicity Caucasian
表 2-6 MCF-7 細胞株資訊
表 2-6,為 MCF-7 細胞株的資訊,可看到這個細胞株是有關於人類乳房組織 的部分,病因是乳癌,年齡為 69 歲的白人女性。
2.5 使用的工具
為了進行 ChIP-seq 實驗的分析,使用了 Bowtie2、MACS 和 Homer,來幫助實驗 分析的進行。
2.5.1 Bowtie2
Bowtie2[14]是一個速度很快且有效使用記憶體空間的序列回貼工具,且對於 較長(哺乳類動物)的基因組讀取有特別好的效果。所謂的回貼就是把我們所得到 的序列片段,貼至我們的參考基因組上,觀察其序列與參考基因組的差異。
11
2.5.2 MACS
染色質免疫沉澱定序技術,是用來探討全基因組蛋白與 DNA 相互作用的一 種策略,而 MACS[11]是一用來分析短序列的模型,類似基因組分析儀(如 Illumina 公司的 Solexa),MACS 藉由模型測序 ChIP 片段的長度,用來改善預測結合位的 辨識率,且 MACS 還採用了動態泊松分佈(Poisson distribution),能有效的捕捉基 因組序列的局部偏差,獲得更精準的預測結果[11]。
所以 MACS 是一個有效找出峰值(peak)的演算法,無論其是否有對照組的資 料,且 MACS 為一個對外公開的資源。
2.5.3 泊松分佈(Poisson distribution)
泊松分佈[15]是一種統計學與機率學裡常見到的離散機率分佈法,於 1838 年由法國數學家Siméon-Denis Poisson 發表。
泊松分佈適用於單位時間內隨機事件發生次數的機率分佈,如每小時服務台 的訪客人數、每天家中電話的通話次數、生產線上的瑕疵品個數、DNA 序列的 變異數等等,共同的特徵是在某時間區段內,平均發生若干次事件,但有時候很 少,有時候卻很多,因此事件發生是一個隨機變數。
泊松分佈的機率函數為:
參數 為單位時間或單位面積內隨機事件平均發生率
2.5.4 位置頻率矩陣(position frequency matrix,PFM)
位置頻率矩陣,是將特定轉錄因子的結合位,去計算每一個位置鹼基 A、T、
C、G 出現的次數,然後統計起來,所以將會是一個四乘以 M 的矩陣,M 代表 該 轉 錄因 子的 結合 位 特徵 的 長 度 。而位 置 權重矩陣 (position weight matrix, PWM),這個會依不同的實驗設定而有所不一樣,中心準則是將位置頻率矩陣的 每一欄視為互相獨立,並且將每一個值正規化以後取對數,然而每一個位置權重 矩陣通常不會剛好是整數,所以每一個都代表在轉錄因子結合位上出現的比例,
藉由這些數字,我們可以清楚的了解轉錄因子結合位特徵。美國基因學家 Gary Stormo 於 1982 年將位置權重矩陣的方法概念應用在找尋 DNA 的結合位[9],奠 定了尋找結合位特徵的基礎。
13
第三章 研究方法
此章節將概述整個實驗的分析方法和實驗的過程,以及所使用的 ENCODE 資料庫、TRANSFAC 資料庫和參考基因組的介紹。
3.1 ENCODE 資料庫
圖 3.1 ENCODE 資料庫,摘自文獻[12]
ENCODE 是由美國國家人類基因研究所(NHGRI)發起的聯合研究項目,旨 在找出人類基因組中所有功能組件,且該項目產生的研究數據都會被公開,是一 個公共的資料庫。
ENCODE 上所收錄的 ChIP-seq 都是有疾病的細胞株資料,且每個細胞株都 有詳細的來源,因為資料完全公開和容易取得,所以本實驗將使用 ENCODE 資 料來做為實驗的分析,圖 3.1 為 ENCODE 資料庫圖示。
3.2 TRANSFAC 資料庫
TRANSFAC 資料庫中收納了真核生物的轉錄因子與 miRNA,並且記錄了這 些轉錄因子的結合位置的資訊和所調控的基因,而這些被收納在資料庫裡的資 料,都已經從生物實驗中得到證實,而在 TRANSFAC 資料庫中有提供人類有關 的轉錄因子結合位置與其參考基因比對後得到的結合位數量為 17441,其記錄的 資訊包含結合位置的起點與終點,和其所對應得基因。
3.3 參考基因組資料(Reference Genome)
本 研 究使 用 的 參考 基 因組 是 從聖 塔克魯 茲 加州大學 基 因 資料 庫 (USCS Genome Browser)下載,基因組為 GRCh37 在聖塔克魯茲加州大學資料庫上的名 稱為 HG19。
ChIP-seq 分析時,我們需要用到人類的參考基因組(hg19),把我們所下載的 ChIP-seq 資料對我們的人類基因組進行回貼,再將所得到的 SAM 檔繼續下面的 分析及研究。
15
3.4 實驗流程
圖 3.2 實驗流程圖
實驗的流程如圖 3.2 所示,首先我們將從 ENCODE 的資料庫中,下載我們 欲分析的 ChIP-seq 資料,選擇了 2014 年後且為成年人的 ChIP-seq 資料如下圖 3.3 所示。
圖 3.3 ENCODE 資料庫下載資料頁面
可以發現到 ENCODE 的 ChIP-seq 資料,都有詳細的標註哪一個細胞株且用 這個細胞株所要探討的轉錄因子名稱,但因為 2014 年後的 ChIP-seq 資料有包含 很多的細胞株和轉錄因子,因此需要寫程式來區找出是有哪幾種細胞株和其對應 要找的轉錄因子是那些。
轉錄因子 細胞株
ARID3A
ChIP-seq of GM12878 (Homo sapiens, adult)
ATF7
ChIP-seq of K562 (Homo sapiens, adult 53 year)
BACH1
ChIP-seq of A549 (Homo sapiens, adult 58 year)ChIP-seq of GM12878 (Homo sapiens, adult)
BHLHE40 ChIP-seq of GM12878 (Homo sapiens,
adult)
BMI1
ChIP-seq of K562 (Homo sapiens, adult 53 year)ChIP-seq of K562 (Homo sapiens, adult 53 year)
BRCA1
ChIP-seq of A549 (Homo sapiens, adult17
58 year)
CBX5
ChIP-seq of K562 (Homo sapiens, adult 53 year)CEBPB
ChIP-seq of GM12878 (Homo sapiens, adult)CHD1
ChIP-seq of A549 (Homo sapiens, adult 58 year)ChIP-seq of HeLa-S3 (Homo sapiens, adult 31 year)
CREB3L1 ChIP-seq of K562 (Homo sapiens, adult
53 year)
CREM
ChIP-seq of GM12878 (Homo sapiens, adult)ChIP-seq of K562 (Homo sapiens, adult 53 year)
表 3-1 轉錄因子與其對應的細胞株
結果如表 3-1 所示,我們可以看到不同細胞株探討的轉錄因子,統計後得到 2014 年 ENCODE 資料庫成人 ChIP-seq 資料總共有 68 個轉錄因子和 20 個細胞 株,且各個細胞株所探討的疾病分別為下表 3-2。
細胞株 相關疾病
ChIP-seq of A549 (Homo sapiens, adult 58 year)
肺癌ChIP-seq of GM12878
皰疹病毒ChIP-seq of T47D (Homo sapiens, adult 54 year)
乳癌ChIP-seq of NB4 (Homo sapiens, adult 23 year)
白血病ChIP-seq of MCF-7 (Homo sapiens, adult 69 year)
乳腺癌ChIP-seq of HeLa-S3 (Homo sapiens, adult 31 year) 子宮頸癌
ChIP-seq of Oci-Ly-3 (Homo sapiens, adult 52 year) 淋巴癌
ChIP-seq of GM12878 (Homo sapiens, adult)
皰疹病毒ChIP-seq of Ishikawa (Homo sapiens, adult)
子宮內膜腺癌ChIP-seq of LNCaP clone FGC (Homo sapiens,
前列腺癌adult 50 year)
ChIP-seq of DOHH2 (Homo sapiens, adult 60 year)
淋巴癌ChIP-seq of Caco-2 (Homo sapiens, adult 72 year)
大腸腺癌ChIP-seq of Oci-Ly-7 (Homo sapiens, adult 48 year) 淋巴癌 ChIP-seq of SUDHL6 (Homo sapiens, adult 43
year)
淋巴癌
ChIP-seq of H54 (Homo sapiens, adult 36 year)
多形性膠質母 細胞瘤ChIP-seq of Karpas-422 (Homo sapiens, adult 73
year)
淋巴癌
ChIP-seq of K562 (Homo sapiens, adult 53 year)
髓細胞性白血 病ChIP-seq of HL-60 (Homo sapiens, adult 36 year)
骨髓細胞白血 病ChIP-seq of U-87 MG (Homo sapiens, adult 44 year)
多形性膠質母 細胞瘤
ChIP-seq of Ishikawa (Homo sapiens, adult 39
year)
子宮內膜癌
表 3-2 各個細胞株所代表的疾病
因此我們能看到這些細胞株和其對應的轉錄因子,所以我挑選了幾個細胞株 (A549、GM12878、K562、HeLa-S3、Ishikawa、MCF-7)以及至少兩個細胞株擁 有 的 相 同 轉 錄 因 子 , 如 CEBPB(CCAAT/enhancer-binding protein beta) 、 NR3C1(Glucocorticoid receptor)、SP(Transcription factor Sp1)、MAFK(Transcription factor MafK),分別對於這些轉錄因子進行分析。
19
轉錄因子 細胞株
CEBPB
A549 Ishikawa HeLa-S3 MCF-7 K562MAFK
A549 MCF-7NR3C1
A549 Ishikawa GM12878SP
A549 K562表 3-3 為本實驗使用的細胞株和轉錄因子的免疫沉澱資料
3.4.1 序列回貼
從 ENCODE 資料庫,下載上述所說的細胞株與其轉錄因子的資料,這些從 ENCODE 資料庫下載的資料檔案,都為 fasta 格式,如圖,我們可以看到 Chip-seq 資料,是很多的 reads 合成的檔案,接下來就把這些資料,回貼到人類參考基因 組 hg19,這邊使用 Bowtie2 作為回貼的工具軟體,回貼完後觀察回貼的狀況並 進行下一步的分析。
3.4.2 求出峰值
序列回貼完後,開始要尋找峰值(peak calling),找出的峰值就可以假設為特 定蛋白可能結合之位置,序列回貼的時,很多的序列會被貼到重複的 DNA 片段,
而 MACS 可以把這些回貼的片段,做一個常態分布,然後經由工具本身的試算 會得到 MACS 的輸出檔。
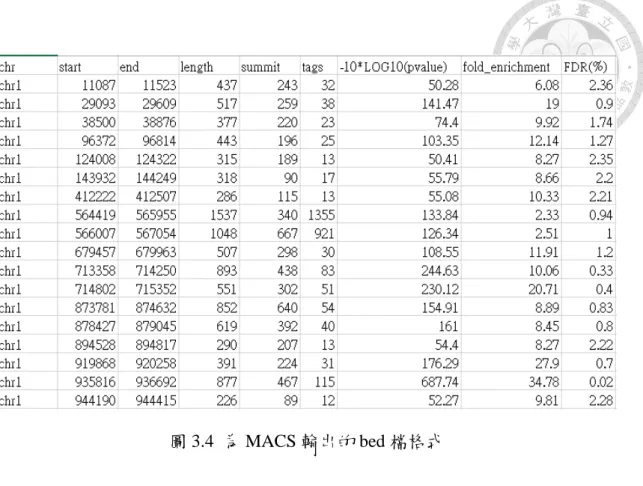
圖 3.4 為 MACS 輸出的 bed 檔格式
圖 3.4,為一 MACS 輸出檔案,可以看到輸出檔的資訊,其格式每一列都是一個 峰值,第一欄是這個峰值出現在參考基因組的第幾條染色體,二三欄分別是這個 峰值出現的起始位和終點,第四欄是這個峰值的總長度是多少,當然長度越長的 峰值比較有可能包含到特定蛋白的結合位,五六欄分別為峰值的高度和多少 reads 被貼到這個片段,後面就是一些分數的檢定,也因為 MACS 所釋出的峰值 數目很多,因此我們要在這些輸出檔裡做一些篩選,在此有興趣的是最後一行的 FDR 值,因為 FDR 值為找尋波峰時,對於這個波峰的真實性為何,FDR 實際就 是 false discovery rate 的縮寫,所以我們設定一些 FDR 值,並且觀察這些 FDR 值下的波峰數目,以及準確性的關係。
21
3.4.3 篩選 FDR
為了要選擇不同 FDR 值下的峰值的數目以及其準確率,我們拿了
TRANSFAC 資料庫的資訊,來幫助整個流程的進行,先以 CEBPB 這個轉錄因子 的分析為例,TRANSFAC 資料庫有的資訊是,已經被實驗驗證過的轉錄因子結 合位點位,和其轉錄因子為何,目前被收錄在 TRANSFAC 資料庫中的位置,總 共有 17441,所以我們把這些資訊,先把他們配對起來,把轉錄因子他自己的 ID 與名字,對到屬於他自己的位置上,如下圖 3.5。
圖 3.5 TRANSFAC 資料庫紀錄的位置與其對應的基因
我們可以比較容易看出 TRANSFAC 資料庫的資訊,第二行是代表這個位置 的資訊是出現在哪一條染色體上,二三行分別是該點位在其染色體出現的起點和 終點,後面是代表在這個位置上,有什麼基因出現在這個位置。
接下來我們要統計在 CEBPB 轉錄因子在 TRANSFAC 資料庫所收入的位置 總共有多少,整理完後得知 CEBPB 轉錄因子在 TRANSFAC 資料庫裡總共有 400 個位置,整理完的資訊如圖 3.6,
圖 3.6 CEBPB 在 TRANSFAC 資料庫被記載的位置
接下來我們把這 400 個位置與我們做出的 MACS 輸出檔進行比較,我們先 取了 5 個 FDR 值(%),分別為 FDR(0%)、FDR(0.5%)、FDR(1%)、FDR(5%)和 FDR(無 篩選),且在 ENCODE 得到的 ChIP-seq 資料中,擁有 CEBPB 這個轉錄因子的細 胞株有 ChIP-seq of A549 (Homo sapiens, adult 58 year)、ChIP-seq of K562 (Homo sapiens, adult 53 year)、ChIP-seq of HeLa-S3 (Homo sapiens, adult 31 year)、
ChIP-seq of MCF-7 (Homo sapiens, adult 69 year)和 ChIP-seq of Ishikawa (Homo sapiens, adult 39 year)這五個細胞株,所以這些 ChIP-seq 資料,分別都篩選了 FDR 值且與 TRANSFAC 資料庫做比較,我們要看這些 ChIP-seq 資料所得到的峰值,
是否出現在 TRANSFAC 所記錄的位置之中。
23
3.4.4 結合位特徵(Consensus, annotated motifs)
想要了解 ChIP-seq 峰值的可信程度,我們把 TRANSFAC 資料庫所記載的轉 錄因子的 consensus 拿出來,CEBPB 在 TRANSFAC 資料庫中紀載的 consensus ID 分別是 V$CEBPB_01、V$CEBPB_02、V$CEBPB_03、V$CEBPB_04、V$CEBPB_06 和 V$CEBPB_07,這些 consensus 都是由對應到的位置,透過這些位置的資訊建 立的 PWM 所得到。
圖 3.7,是 TRANSFAC 資料庫收入的 CEBPB 的 consensus,以 V$CEBPB_01 這個 ID 為例,他的 consensus 為 RNRTKDNGMAAKNN,意思是轉錄因子 CEBPB 結合到 DNA 上的序列為[AG][ATCG][AG]T[GT][AGT][ATCG]G[AC]AA[GT][ATCG][ATCG]
,這是經由統計結位置建立的 PFM 得到的 consensus,在這個 V$CEBPB_01ID 的 consensus 大小為 14 鹼基對(bp),所以我們就把這樣的 consensus 從 MACS 輸 出的峰值中,對回去找這些峰值是否存在著這樣的 consensus,進一步來確定 ChIP-seq 資料的可信度,因此把 MACS 輸出的 bed 檔找到的峰值,寫程式去把 整個峰值的序列把它抽取出來。
圖 3.7 為 TRANSFAC 資料庫中紀錄的的 CEBPB 結合位特徵
圖 3.8 峰值被抽取出來的序列
這樣抽取出來的序列,是根據峰值在哪一條染色體的位置,從參考基因體 hg19 取出,所以圖 3.8,代表在參考基因體 hg19 的第一條染色體(chr1)的
11203(start)到 11620(end)的位置,然後我們將 TRANSFAC 資料庫記載的 CEBPB 轉錄因子的 consensus,寫程式從這些峰值的序列中,去尋找有沒有符合的峰值 和 pattern。
25
圖 3.9 結合位特徵找到的峰值與其找到的特徵
從圖 3.9,我們可以看到,在這些峰值的序列中,確實有發現符合 TRANSFAC 資料庫所記載的 consensus,我們找到的這些 motif 在正股和反股端都有出現,在 反股端出現的 motif 我們加了 rc 作為註解,因此我們把所有細胞株的 MACS 輸 出檔,和所篩選的 FDR 五個值都做了以上的步驟,然後使用 R 語言[16]繪製出 圖表,將在本論文的第四章進行討論。
3.4.5 TOP500 模序探勘 (De novo Motif Discovery)
這邊我們使用了 eTFBS[14]來做 De novo motif discovery,我們取了所有峰值 的前 500 名和後 500 名(用 P-value 排列),來當正相關(positive)序列和負相關 (negative)序列,我們主要是想要在前 500 名的序列中找出共同存在的模序特徵,
後 500 名是來當作濾掉一些可能被找到不是正確的模序特徵,所以求出了 CEBPB 中五個細胞株(A549、HeLa-S3、Ishikawa、MCF-7、K562)的模序特徵和位置頻 率矩陣,所以我們將找到前三名的模序特徵對回輸出的總數峰值,重複之前的動 作來比較。
第四章 結果與討論
本論文透過 TRANSFAC 資料庫提供的已知的轉錄因子結合位點,整體性評 估免疫沉澱技術平台鑑定的準確與否,並輔以序列特徵增進其鑑定轉錄因子結合 位點的預測準確度。各章節內容摘要如下:章節 4.1 效果評估的參考數值(敏感 度、準確度);章節 4.2 探討只使用 ChIP-seq 平台鑑定轉錄因子結合位的效果;
章節 4.3 透過整合各細胞株資料可增進預測之敏感度;章節 4.4 利用已知的序列 特徵資訊確信染色質免疫沉澱定序技術與增加準確度;章節 4.5 利用模序探勘得 到的序列特徵資訊可進一步增加準確度。
4.1 效果評估的參考數值
各方法的效果評估以 Sensitivity (敏感度)、Precision (準確度)與 F-score 進行 比較。詳述如下:Sensitivity 的定義為在 TRANSFAC 資料庫的轉錄因子 site 被峰 值對到的 site 數目除以轉錄因子總 site 數目,Precision 的定義是 MACS 輸出檔的 峰值對到 TRANSFAC 資料庫轉錄因子位置的峰值數目,除以輸出的峰值總數目。
意即為;
Sensitivity = Precision =
以 CEBPB 轉錄因子為例,因為在下載的染色共免疫沉澱資料中,CEBPB 轉錄因 子擁有最多共同的細胞株,所以使用 CEBPB 轉錄因子,去比較不同細胞株間是 否 Sensitivity 與 Precision 也會有所不同。
27
4.2 探討只使用 ChIP-seq 平台鑑定轉錄因子結合位的效果
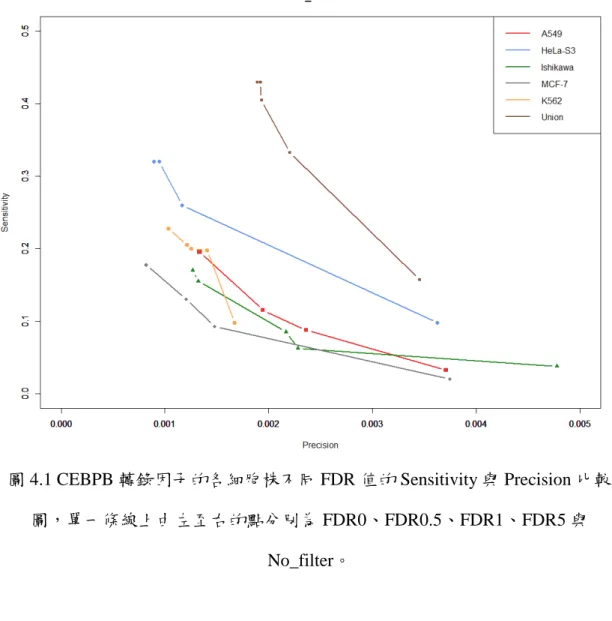
由圖 4.1,可看出在不同的 FDR 值的 Sensitivity 與 Precision 的關係,可以發 現,在越嚴謹的 FDR 值下,峰值的 Sensitivity 較低但 Precision 的準確率比較高。
這代表了在比較嚴謹的 FDR 值下,有存在著真的轉錄因子的結合位。
圖 4.1 CEBPB 轉錄因子的各細胞株不同 FDR 值的 Sensitivity 與 Precision 比較 圖,單一條線上由左至右的點分別為 FDR0、FDR0.5、FDR1、FDR5 與
No_filter。
4.3 透過整合各細胞株資料可增進預測之敏感度
表 4-1,為這五個細胞株的峰值出現在 TRANFAC 資料庫中的個數和並把它 們聯集之後所得到的數目,我們可以發現在嚴謹的 FDR 值下,峰值對到的位置 數目明顯是比較少的,隨著 FDR 的條件越來越寬鬆,所對到的數目也慢慢的增 加。
site FDR0 FDR0.5 FDR1 FDR5 No_filter
A549
13/400 35/400 46/400 78/400 78/400HeLa-S3
39/400 104/400 128/400 128/400 128/400Ishikawa
15/400 25/400 34/400 62/400 68/400MCF-7
8/400 37/400 52/400 71/400 71/400K562
39/400 79/400 80/400 82/400 91/400Union
63/400 133/400 162/400 172/400 172/400表 4-1 TRANSFAC 資料庫中 CEBPB 的位置在不同細胞株中被對到的位置數目
(對到的位置數目/總位置數目)
而且在聯集的部分可以明顯看出,透過比較多的細胞株找到的峰值,對到的結合 位位置數目是有增加的趨勢,這代表了不同的細胞株之間,有存在著屬於自己獨 有的轉錄因子結合位。
29
圖 4.2 CEBPB 轉錄因子在 A549 細胞株下不同 FDR 值對於 Sensitivity 的影響
以 A549 細胞株 CEBPB 轉錄因子為例,圖 4.3 能發現,FDR 值與 Sensitivity 的影響,在越寬鬆的 FDR 值,相對的 Sensitivity 也越大,因為寬鬆的 FDR 值,
被保留下來的峰值數目比較多,所以對到的 TRANSFAC 資料庫的轉錄因子位置 數目,也因此對到比較多,這樣的情形在其他細胞株也能見到。
(4.3.1)HeLa-S3 (4.3.2)Ishikawa
(4.3.3)MCF-7 (4.3.4)K562
圖 4.3 CEBPB 轉錄因子在不同細胞株下不同 FDR 值對於 Sensitivity 的影響
CEBPB 轉錄因子在其他細胞株中,FDR 值對於 Sensitivity 的影響,所以也 證明了調整 FDR 值,也間接的影響了 Sensitivity 的高低。
31
圖 4.4 CEBPB 轉錄因子在所有的細胞株下不同 FDR 值對於 Sensitivity 影響的比 較圖
圖 4.4,可以看到在同一個轉錄因子在不同的細胞株中,聯集(union)後我們 可以發現,Sensitivity 是有上升的,這也暗示了轉錄因子結合位,存在著細胞株 獨有的結合位位置,
4.4 利用已知的序列特徵資訊確信染色質免疫沉澱定序技術 與增加準確度
表 4-2,為這五個細胞株的 MACS 輸出的 bed 檔,所得到的峰值總數目,與 這些峰值有對應到 TRANSFAC 資料庫的峰值數目,我們可以看出雖然在 FDR 值嚴謹的情況下,對應到的峰值在總峰值數目的比例有變高,但事實上比例都是 極低的,讓我們懷疑 ChIP-seq 資料的可信度不高,還是其實還有很多位置,在 目前的資料庫中還沒被驗證,所以沒有收入進來,所以需要繼續往下的分析。
所以為了解釋這樣的情形,我們把 TRANSFAC 資料庫中,這些轉錄因子 PFM 矩陣的 Consensus 一起加進來探討,在 CEBPB 這個轉錄因子,我們使用了六個 TRANSFAC 資料庫的 Consensus,其在 TRANSFAC 資料庫的 ID 分別為
V$CEBPB_01、V$CEBPB_02、V$CEBPB_03、V$CEBPB_04、V$CEBPB_06、
V$CEBPB_07,來幫助我們解決問題。
peak FDR0 FDR0.5 FDR1 FDR5 No_filter
A549
18/4,854 41/17,352 54/27,767 86/64,270 86/64,822HeLa-S3
37/10,200 115/98,572 140/148,212 140/156,644 140/156,644Ishikawa
8/1,674 22/9,641 31/14,303 56/42,344 73/57,649MCF-7
7/1,868 35/23,674 53/44,021 81/98,863 81/98,863K562
40/23,932 87/61,850 89/70,995 91/74,995 94/90,754union
110/31,841 300/136,204 367/190,327 454/236,440 474/251,190表 4-2 TRANSFAC 資料庫中 CEBPB 的位置在不同細胞株中被對到的峰值數目
(對應到 TRANSFAC 資料庫的峰值數目/峰值總數目)
33
為了確信 ChIP-seq 資料,使用 TRANSFAC 資料庫的 consensus 去尋找輸出 的峰值中,是否有存在著一樣的 consensus,因為我們相信如果峰值中存在著這 些 TRANSFAC 資料庫中收集的 consensus,就代表其實 ChIP-seq 是有一定的可 信程度。
利用 TRANSFAC 資料庫的 consensus 去掃描後,我們發現峰值中確實有這 些相似的 consensus,,所以我們可以推測,雖然在 TRANSFAC 資料庫中,MACS 的輸出檔對應到 site 的峰值不多,但其實他們確實都有存在類似的 consensus,
因此 MACS 的輸出波峰的可信度是有的,所以我們相信存在著這些 consensus 的 峰值是有轉錄因子結合位的。
peak FDR0 FDR0.5 FDR1 FDR5 No_filter
A549 3,044/4,854 4,514/17,352 6,724/27,767 13,558/64,270 13,635/64,822 HeLa-S3 6,353/10,200 21,183/98,572 28,972/148,212 29,811/156,644 29,811/156,644 Ishikawa 1,128/1,674 2,461/9,641 3,360/14,303 7,215/42,344 9,635/57,649
MCF-7 1,315/1,868 6,257/23,674 10,342/44,021 19,465/98,863 19,465/98,863 K562 11,517/23,932 12,863/61,850 14,232/70,995 14,951/74,995 17258/90,754
表 4-3 TRANSFAC 資料庫中 CEBPB 的 consensus 對到不同細胞株的峰值數目
(存在 TRANSFAC 的 consensus 峰值數目/總峰值數目)
圖 4.5 A549 細胞株對到 TRANSFAC 的未致數目的 Precision 與存在著 TRANSFAC consensus 的 Precision 比較
圖 4.5 是由表 4-2 與 4-3 而來,可以看出在這些峰值中(數值取 log10),發現 這些有存在 TRANSFAC 的 consensus 的峰值數目是蠻多的,代表 ChIP-seq 資料 是可以被信賴的,也因此覺得目前 TRANSFAC 資料庫收錄實驗驗證過的轉錄因 子位置數目還不夠齊全。
35
4.5 利用模序探勘得到的序列特徵資訊可進一步增加準確度
解決 Precision 偏低的問題,使用了 TRANSFAC 資料庫的 consensus 來增加 準確度,再來使用 Top 500 的 motif discovery (De novo method)來做比較,目的是 為了從 De novo method 方法找到相似的 motif,來過濾原本的峰值數目,這樣就 可以再進一步提升準確率,這邊使用了 eTFBS 工具來尋找這些 motif,而且把前 三名的 motif 都拿回去過濾峰值,並且再作比較,De novo methodz 方法分別用 eTFBS_1(使用 eTFBS rank1 的 motif)、eTFBS_2(使用 eTFBS rank2 的 motif)、
eTFBS_3(使用 eTFBS rank3 的 motif)、eTFBS_12(使用 eTFBS rank 前二的 motif)、
eTFBS_123(使用 eTFBS rank 前三的 motif)代表。
A549 FDR0 FDR05 FDR1 FDR5 No_filter Chip-seq 0.627112 0.260143 0.242149 0.210937 0.210329 eTFBS_1 0.629404 0.261228 0.245034 0.220305 0.219713 eTFBS_2 0.626131 0.260895 0.245754 0.221874 0.221286 eTFBS_3 0.629408 0.261135 0.244098 0.218321 0.2176 eTFBS_12 0.627052 0.260605 0.243461 0.217457 0.216888 eTFBS_123 0.627475 0.260159 0.242409 0.214446 0.21387
表 4-4 為 CEBPB 在 A549 細胞株中與 De novo method 的 Precision
HeLa-S3 FDR0 FDR05 FDR1 FDR5 No_filter Chip-seq 0.622843 0.21489 0.195461 0.190295 0.190295 eTFBS_1 0.636256 0.223057 0.206318 0.202076 0.202076 eTFBS_2 0.633074 0.220524 0.203537 0.199222 0.199222 eTFBS_3 0.637653 0.222264 0.20622 0.202418 0.202418 eTFBS_12 0.627558 0.220041 0.202909 0.198469 0.198469 eTFBS_123 0.62625 0.219008 0.201579 0.197061 0.197061
表 4-5 為 CEBPB 在 HeLa-S3 細胞株中與 De novo method 的 Precision
Ishikawa FDR0 FDR05 FDR1 FDR5 No_filter Chip-seq 0.675045 0.255264 0.234916 0.17039 0.167129 eTFBS_1 0.74346 0.301021 0.28645 0.235544 0.236268 eTFBS_2 0.802646 0.357715 0.349602 0.325935 0.323826 eTFBS_3 0.691104 0.270317 0.253141 0.19348 0.19052 eTFBS_12 0.74346 0.301021 0.28645 0.235544 0.236268 eTFBS_123 0.690141 0.268124 0.250995 0.190797 0.187496
表 4-6 為 CEBPB 在 Ishikawa 細胞株中與 De novo method 的 Precision
37
MCF-7 FDR0 FDR05 FDR1 FDR5 No_filter Chip-seq 0.703961 0.264298 0.234933 0.196889 0.196889 eTFBS_1 0.788859 0.328926 0.309077 0.286763 0.286763 eTFBS_2 0.738504 0.292795 0.267046 0.234945 0.234945 eTFBS_3 0.722513 0.301015 0.279739 0.255902 0.255902 eTFBS_12 0.732108 0.289521 0.262723 0.230989 0.230989 eTFBS_123 0.718343 0.28368 0.257819 0.227715 0.227715
表 4-7 為 CEBPB 在 MCF-7 細胞株中與 De novo method 的 Precision
K562 FDR0 FDR05 FDR1 FDR5 No_filter Chip-seq 0.481239 0.207961 0.200456 0.199352 0.190156 eTFBS_1 0.507213 0.215464 0.208308 0.207031 0.196988 eTFBS_2 0.470578 0.196287 0.189458 0.188789 0.182501 eTFBS_3 0.503007 0.215907 0.20842 0.206964 0.196882 eTFBS_12 0.489598 0.208434 0.201129 0.200035 0.190866 eTFBS_123 0.486118 0.208967 0.201451 0.200268 0.190947
表 4-8 為 CEBPB 在 K562 細胞株中與 De novo method 的 Precision
表 4-4 到表 4-8 是 CEBPB 在不同細胞株中,ChIP-seq 平台與 De novo method 的 precision 比較,可以看出在細胞株 Ishikawa 和 MCF-7 的 precision 提升的比較 多,而其他三個細胞株(A549、HeLa-S3、K562)雖然有提升,但整體上提升不明 顯。
圖 4.6 CEBPB 在 Ishikawa 細胞株中,ChIP-seq 平台與 De novo method 方法的 Precision 比較圖
圖 4.7 CEBPB 在 MCF-7 細胞株中,ChIP-seq 平台與 De novo method 方法的 Precision 比較圖
39
再把表 4-6(Ishikawa)和表 4-7(MCF-7)拿出來畫圖,如 4.7 與圖 4.8,我們可 以明顯的發現到,在 De novo method 的方法 precision 的效果是來的比 ChIP-seq 平台優秀的,所以因此我們推斷應該是這兩個細胞株有找到與 TRANSFAC 資料 庫 consensus 相似度較高的 motif,所以過濾掉比較多的雜訊峰值,因此 precision 相對表現就比較優秀,但為了證實有找到相似 consensus 的 motif,這邊我們拿了 他們的位置頻率矩陣,利用 JASPAR[18]的網站,來看看這些 consensus 與 motif 是否相似。(其餘細胞株的 precision 比較圖請參考附件(一))
(4.8.1)V$CEBPB_01 (4.8.2)V$CEBPB_02 (4.8.3)V$CEBPB_03
(4.8.4)V$CEBPB_04 (4.8.5)V$CEBPB_06 (4.8.6)V$CEBPB_07 圖 4.8 TRANSFAC 資料庫中 CEBPB 被紀錄的 motif
(4.9.1)Ishikawa_eTFBS_2 (4.9.2)MCF-7_eTFBS_1 圖 4.9 為 Ishikawa 和 MCF-7 中 Precision 最高的 motif
由圖 4.8 和 4.9 可看見,在 Ishikawa 和 MCF-7 兩個細胞株中,eTFBS 找到 了跟 TRANSFAC 資料庫 consensus 相似度很高的 motif,precision 進一步的提升,
所以使用 De novo method 的方法,去增進 precision 時,取決於找到的 motif 是不
是原本轉錄因子本身的 consensus,而其他的細胞株為什麼沒有發現相似的 consensus,因為從表格看起來被 TRANSFAC 資料庫 consensus 留下來的峰值有 六成多,可是為什麼沒找到相似的 motif,一種可能是因為我們只用了前 500 條 序列去做 motif discovery,可能是這樣的 motif 在前 500 條裡沒有被發現到,受 限於工具的關係,這部分可以在未來的研究上加以探討,另一種可能是或許轉錄 因子與轉錄因子之間有交互作用的關係,所以在剛開始染色質免疫沉澱定序技術 時,被留下來的序列其實是交互作用的轉錄因子的 motif,這部分如附件(一),可 以看出 A549 細胞株與 K562 細胞株找出的 motif 有些相似,可能是代表了另一 個轉錄因子的 consensus,不過這部分也可以在未來的研究上在更加的去分析。
(4.10.1)A549_eTFBS_1 (4.10.2)K562_eTFBS_1 圖 4.10 為 A549 細胞株與 K562 細胞株其中 eTFBS 找到的 motif
41
第五章 結論
在鑑定轉錄因子及合位準確度時,我們發現在不同的細胞株中可以看到屬於 自己獨有的結合位置,且在過低的峰值準確度下,利用 TRANSFAC 資料庫的結 合位特徵來驗證染色質免疫沉澱定序技術的可信程度。
染色質免疫沉澱定序找出的峰值在 FDR 等於 0 下,峰值的可信度非常高,
就是能準確的預測轉錄因子結合位,再加上模序探勘(De novo method)的方法,
如果能找到與參考資料庫相似度高的模序特徵,能大大的增加峰值的準確率,所 以使用模序探勘,更過濾我們的峰值雜訊,達到增加準確率的效果。
本研究指出,染色質免疫沉澱定序技術透過 TRANSFAC 資料庫的模序特 徵,證明其是可信賴的,也點出了目前收錄在 TRANSFAC 資料庫的轉錄因子整 體位置資訊還不夠完全,再者峰值的 FDR 值確實有一定的程度能解釋峰值的準 確率且是否含有轉錄因子結合位,FDR 值越小,峰值的準確率就越高,代表了 存在轉錄因子真的結合位,最後本文提到,如果能利用模序探勘找到最佳的模序 特徵時,將可以濾掉其餘雜訊的峰值,並將準確度在更進一步提升。
參考文獻
1. American Society for the Control of Cancer, A. American Cancer Society | Information and
Resources. 12 October 2015. Available at: http://www.cancer.org/.
2. Siegel, R., et al. 2014. Cancer statistics, 2014. CA: a cancer journal for clinicians. 64(1).
9-29.
3. Basseres, D.S., et al. 2012. Frequent downregulation of the transcription factor Foxa2 in lung
cancer through epigenetic silencing. Lung cancer. 77(1). 31-37.
4. Cheung, W.K., et al. 2013. Control of alveolar differentiation by the lineage transcription
factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer cell. 23(6).
725-738.
5. Fujita, J., et al. 2003. Expression of thyroid transcription factor-1 in 16 human lung cancer cell
lines. Lung Cancer. 39(1). 31-36.
6. Ishii, J., et al. 2014. Class III/IV POU transcription factors expressed in small cell lung cancer
cells are involved in proneural/neuroendocrine differentiation. Pathology international. 64(9).
415-422.
7. Crick, F.H., J.S. Griffith, and L.E. Orgel. 1957. Codes without commas. Proceedings of the
National Academy of Sciences of the United States of America. 43(5). 416.
8. Crick, F. 1970. Central dogma of molecular biology. Nature. 227(5258). 561-563.
9. Valouev, A., et al. 2008. Genome-wide analysis of transcription factor binding sites based on
ChIP-Seq data. Nature methods. 5(9). 829-834.
10. Jkwchui. 2012. ChIP-sequencing workflow. 13 October 2015. Available at:
https://en.wikipedia.org/wiki/ChIP-sequencing#/media/File:Chromatin_immunoprecipitation_
sequencing.svg.
43
11. Zhang, Y., et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome biology. 9(9).
R137.
12. Institute, N.H.G.R. 2011. ENCODE: Encyclopedia of DNA Elements – ENCODE. 12
October 2015. Available at: https://www.encodeproject.org/.
13. Chien-Yu Chen , Huai-Kuang Tsai , Chen-Ming Hsu , Mei-Ju May Chen, Hao-Geng
Hung ,Grace Tzu-Wei Huang , and Wen-Hsiung Li .2007 Discovering gapped binding sites
of yeast transcription factors
14 Ben Langmead,Steven L Salzberg .2012 Fast gapped-read alignment with Bowtie 2
15 Ronald J. Evans, J. Boersma, N. M. Blachman, A. A. Jagers. The Entropy of a Poisson
Distribution: Problem 87-6. SIAM Review. 1988
16 Andy Bunn,Mikko Korpela, Processed with dplR 1.6.4 in R version 3.2.4 (2016-03-10) on
March 15, 2016Time Series Analysis in dplR
17 Teemu D Laajala, Sunil Raghav, Soile Tuomela, Riitta Lahesmaa, Tero Aittokallio and Laura
L Elo BMC Genomics2009. A practical comparison of methods for detecting transcription
factor binding sites in ChIP-seq experiments
18 Team, J.W.2013. The JASPAR database. 12 October 2015. Available at:
http://jaspar.binf.ku.dk/.
附件(一) CEBPB
CEBPB 轉錄因子在不同細胞株中 ChIP-seq 平台與 De novo method 方法 Sensitivity FDR 值的比較關係
45
左到右分別為 CEBPB A549 細胞株 A549_eTFBS_1、A549_eTFBS_2、A549_eTFBS_3 的 motif
左到右分別為 CEBPB HeLa-S3 細胞株 HeLa-S3_eTFBS_1、HeLa-S3_eTFBS_2、HeLa-S3_eTFBS_3 的 motif
左到右分別為 CEBPB Ishikawa 細胞株 Ishikawa_eTFBS_1、Ishikawa_eTFBS_2、
Ishikawa_eTFBS_3 的 motif
左到右分別為 CEBPB MCF-7 細胞株 MCF-7_eTFBS_1、MCF-7_eTFBS_2、MCF-7_eTFBS_3 的 motif
左到右分別為 CEBPB K562 細胞株 K562_eTFBS_1、K562_eTFBS_2、K562_eTFBS_3 的 motif
附件(二) NR3C1
NR3C1 轉錄因子在不同細胞株中 ChIP-seq 平台 與 De novo method 方法 Sensitivity FDR 值的比較 關係
左到右分別為 NR3C1 A549 細胞株 A549_eTFBS_1、A549_eTFBS_2、A549_eTFBS_3 的 motif
左到右分別為 NR3C1 GM12878 細胞株 GM12878_eTFBS_1、GM12878_eTFBS_2、GM12878_eTFBS_3 的 motif
左到右分別為 NR3C1 Ishikawa 細胞株 Ishikawa_eTFBS_1、Ishikawa_eTFBS_2、Ishikawa_eTFBS_3 的 motif
47
附件(三) MAFK
MAFK 轉錄因子在不同細胞株中 ChIP-seq 平台與 De novo method 方法 Sensitivity FDR 值的比較關係
左到右分別為 MAFK A549 細胞株 A549_eTFBS_1、A549_eTFBS_2、A549_eTFBS_3 的 motif
左到右分別為 MAFK MCF-7 細胞株 MCF-7_eTFBS_1、MCF-7_eTFBS_2、MCF-7_eTFBS_3 的 motif