義 守 大 學
生物技術與化學工程研究所
碩 士 論 文
利用對甲苯磺醯基自由基與不對稱二烯化
合物進行自由基加成環化反應探討其位向
選擇性與立體選擇性
To Study the Free Radical Addition
Cyclization Reactions of the Tosyl Radical
with Asymmetric Diene Compound and to
Investigate the Regioselectivity and
Stereoselectivity
研
究
生 : 呂 嘉 瑜
指 導 教 授 : 吳 裕 文 博 士
中 華 民 國 一 百 零 四 年 七 月
To Study the Free Radical Addition Cyclization
Reactions of the Tosyl Radical with Asymmetric
Diene Compound and to Investigate the
Regioselectivity and Stereoselectivity
研
究 生
: 呂 嘉 瑜 Student:Chia-Yu Lu
指 導 教 授 : 吳 裕 文 Advisor:Yuh-Wern Wu
義守大學
生物技術與化學工程研究所
碩士論文
A ThesisSubmitted to Graduate Institute of Biotechnology and Chemical
Engineering
I-Shou University
in Partial Fulfillment of the Requirements
for the Master degree
in
Biotechnology and Chemical Engineering
July , 2015
Kaohsiung, Taiwan, Republic of China
i
中文摘要
近年,自由基在有機合成上的應用逐漸受到重視,尤其是自由基在 加成環化時所表現的位向選擇性與立體選擇性更是受到重視。 利用對甲苯磺醯基自由基,與不對稱二烯【(2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚、(3-丙烯醯基)-(4-溴-2-丁烯基)醚】,進行自由基加成環 化反應。利用取代基極性效應與立體效應的影響,探討自由基加成至不 同的雙鍵時的加成向選擇性,與自由基加成環化反應時其對位向選擇性 與立體選擇性之影響。 關鍵詞:自由基SH2'反應、自由基加成環化反應、對甲苯磺醯基自 由基、取代基立體效應、取代基極性效應。ii
英文摘要
In recent years, it is important that the application of free radical in organic synthesis, especially, the regioselectivity and the stereoselectivity of the free radical addition
cyclization reactions.
It were investigated that the regioselectivity and stereoselectivity of the reactions of the toluene sulfonyl radical with asymmetric dienes, (2-methyl
propenyl)-(4-bromo-2-butenyl) ether and E-4-bromobut-2-enyl acrylate by the substituent polar effect and steric effect and by the different polarity of free radicals.
Keyword : Free radical SH2' reaction、 Free radical addition cyclization reaction、
iii
謝誌
首先我要感謝指導教授吳裕文老師和楊志馮老師,在老師的教導下讓我 在課業與實驗上能有效且順利的進行,專業領域上可以越來越精進,也讓我 學習到很多做人處事的道理,在未來的道路上一定會記住老師的諄諄教誨。 感謝口試委員黃美利老師與郭曼娫博士,在你們的指導下可以讓我在論文上 內容更為紮實。 研究期間感謝孔堯璋學長、陳愛如學姊與陳欣鈴學姊,學長姊們無私的 教導讓我很快的進入實驗的運作與思考自己該如何進行研究,再來感謝同學 建峯、家駒、亭穎、文昌與學弟妹杰穎、德川、玉珮、建豪、士傑及許多的 朋友們,有你們在一起我的研究所生涯真的非常有趣,在此特別感謝建峯, 建峯的幫助與支持讓我可以在學業當中儀器使用可以順利使用,事情也都幫 忙處理完成。 最後感謝的是我的家人們,家人給我的支持讓我可以無後顧之憂的完成 研究所學業,最後再次感謝大家。iv
目錄
中文摘要 ... i 英文摘要 ... ii 謝誌 ... iii 目錄 ... iv 圖目錄 ... vii 表目錄 ... ix 附錄 圖譜目錄 ... x 第一章 前言 ... 1 1.1 自由基反應 ... 2 1.1.1 自由基加成反應 ... 2 1.1.2 自由基環化反應 ... 3 1.1.3 自由基加成環化反應 ... 6 1.2 自由基加成反應其取代基極性效應 ... 8 1.3 自由基加成反應其共振效應 ... 10 1.4 自由基加成環化反應其立體效應 ... 11 第二章 論文回顧 ... 13 第三章 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯溴進行自由基加 成環化反應 ... 24 3.1 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚合成 ... 28 3.2 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由基之加成 環化反應 ... 29v 3.3 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由基加成環 化反應之位向選擇性與立體效應 ... 32 3.4 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由基加成環 化反應之結構鑑定 ... 33 第四章 (3-丙烯醯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯溴進行自由基加成環 化反應 ... 42 4.1 (3-丙烯醯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由基之加成環化 反應 ... 46 4.2 (3-丙烯醯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由基之位向選擇 性與立體效應 ... 50 4.3 (3-丙烯醯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由基之加成環化 反應結構鑑定 ... 52 第五章 結論 ... 59 第六章 實驗部分 ... 61 6.1 藥品 ... 61 6.1.1 試藥級溶劑 ... 61 6.1.2 試劑 ... 61 6.2 儀器 ... 62 6.2.1 氣相層析質譜儀 (GCMS)... 62 6.2.2 氣相層析儀 (GC) ... 62 6.2.3 光化學反應器 (PR-2000) ... 63 6.2.4 核磁共振光譜儀 (NMR) ... 63 6.3 實驗步驟 ... 64 6.3.1 對甲苯磺醯溴製備(TsBr) ... 64
vi 6.3.2 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚製備(化合物 7) ... 64 6.3.3 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯溴進行自 由基加成環化反應 ... 65 6.3.4 (3-丙烯醯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯溴進行自由基加 成環化反應 ... 65 參考文獻 ... 66 附錄 圖譜目錄 ... 68
vii
圖目錄
圖 1 自由基分子內環化反應 ... 3 圖 2 己烯自由基環化路徑 ... 3 圖 3 自由基環化反應 ... 4 圖 4 自由基加成環化反應 ... 6 圖 5 化合物CIS與TRANS結構 ... 11 圖 6 苯磺醯基親電性自由基加成環化反應 ... 12 圖 7 化合物 7【(2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚】合成 ... 28 圖 8化合物 7 與對甲苯磺醯溴加成環化反應反應機構 ... 30 圖 9 化合物 9 之GC圖譜 ... 34 圖 10 化合物 9 之MS圖譜 ... 34 圖 11 化合物 9 之MS圖譜... 35 圖 12 化合物 9 之1 HNMR圖譜 ... 35 圖 13 化合物 9 之13 CNMR圖譜 ... 36 圖 14 化合物 9 之DEPT圖譜 ... 36 圖 15 化合物 91 HNMR化學位移 ... 38 圖 16 化合物 913 CNMR 化學位移 ... 38 圖 17 化合物 15 進行自由基加成環化反應之反應機構 ... 44 圖 18 化合物 12 進行自由基加成環化反應之反應機構 ... 49 圖 19 化合物 19 與化合物 20 之GC圖譜... 53 圖 20 化合物 19 與化合物 20 之MS圖譜 ... 53 圖 21 化合物 19 與化合物 20 之1 HNMR圖譜 ... 54viii 圖 22 化合物 19 與化合物 20 之13 CNMR圖譜 ... 54 圖 23 化合物 19 與化合物 20 之DEPT圖譜 ... 55 圖 24 化合物 191 HNMR與13CNMR化學位移 ... 56 圖 25 化合物 2013CNMR化學位移... 56 圖 26 化合物 201 HNMR化學位移 ... 56
ix
表目錄
表 1 自由基環化速率表 ... 5 表 2 第三丁基自由基加成至 2-取代基丙烯氯之相對速率 ... 8 表 3 化合物 1 與第三丁基氯化汞照光反應 ... 24 表 4 化合物 4A/4B與第三丁基氯化汞照光反應 ... 26 表 5 化合物 7 與對甲苯磺醯溴照光反應 ... 31 表 6 化合物 9 之13 CNMR化學位移 ... 39 表 7 化合物 9 之1 HNMR化學位移 ... 40 表 8 化合物 12 與第三丁基氯化汞進行照光反應結果 ... 43 表 9 化合物 15 與第三丁基氯化汞進行照光反應結果 ... 44 表 10 化合物 12 與對甲苯磺醯溴照光反應 ... 48 表 11 化合物 19 與 20 之13 CNMR化學位移 ... 57 表 12 化合物 19 之1 HNMR化學位移 ... 57 表 13 化合物 20 之1 HNMR化學位移 ... 58x
附錄 圖譜目錄
圖譜 1TSBR之1HNMR光譜 ... 69 圖譜 2TSBR之13CNMR光譜 ... 70 圖譜 3TSBR之DEPT光譜 ... 71 圖譜 4 化合物 7 之1 HNMR光譜 ... 72 圖譜 5 化合物 7 之1 HNMR光譜(放大圖) ... 73 圖譜 6 化合物 7 之1HNMR光譜(放大圖) ... 74 圖譜 7 化合物 7 之13 CNMR光譜 ... 75 圖譜 8 化合物 7 之DEPT光譜 ... 76 圖譜 9 化合物 9 之1HNMR光譜 ... 77 圖譜 10 化合物 9 之1HNMR光譜(放大圖) ... 78 圖譜 11 化合物 9 之1HNMR光譜(放大圖) ... 79 圖譜 12 化合物 9 之13CNMR光譜 ... 80 圖譜 13 化合物 9 之13CNMR光譜(放大圖) ... 81 圖譜 14 化合物 9 之DEPT光譜 ... 82 圖譜 15 化合物 19 與化合物 20 之1 HNMR光譜 ... 83 圖譜 16 化合物 19 與化合物 20 之1 HNMR光譜(放大圖) ... 84 圖譜 17 化合物 19 與化合物 20 之1 HNMR光譜(放大圖) ... 85 圖譜 18 化合物 19 與化合物 20 之13 CNMR光譜 ... 86 圖譜 19 化合物 19 與化合物 20 之13 CNMR光譜(放大圖) ... 87 圖譜 20 化合物 19 與化合物 20 之13 CNMR光譜(放大圖) ... 88 圖譜 21 化合物 19 與化合物 20 之DEPT光譜 ... 891
第一章 前言
近年來越來越多研究團隊發展自由基環化反應的研究,目前國內也有多 位學者從事這方面的研究,然而自由基加成環化反應的研究,這方面的論文 文獻相對比較少,自由基加成反應及自由基加成環化反應所表現的位向選擇 性與立體選擇性,深受研究團隊的重視。 自由基具有未配對的電子,是一種活性很高的物質。一般的自由基是不 帶電荷的中性物質,如果自由基上的取代基不同,自由基會表現出不一樣的 性質,因此導致不同的反應性。當自由基上有拉電子的取代基時因誘導效應 導致自由基具有親電子性,此自由基被視為親電子性自由基。自由基在加成 反應上喜歡加成至不飽和雙鍵上,並且會因為雙鍵上不同的立體效應、極性 效應或共振效應而產生不同的影響,本次實驗我們所要討論的重點則著重在 於親電子性的自由基進行加成環化反應其極性效應與立體效應的影響。 在自由基加成環化反應上,化合物本身先需要有兩組以上的不飽和鍵, 自由基會先加成至其中一組雙鍵後所產生的中間體化合物再去進行分子內自 身環化反應,此反應會受到自由基中間體的順反結構或立體障礙影響,也可 能受到環化反應後所產生的鍵角不穩定所影響,這些都是值得探討的原因。 本實驗室對於自由基加成環化反應有長期的研究,本實驗選擇對甲苯磺 醯基自由基,此自由基為親電子性的自由基,其進行加成環化反應、立體效 應與離去基效應則是我們想要探討的。2
1.1 自由基反應
1.1.1 自由基加成反應 自由基與不飽和鍵進行加成是自由基的主要反應。自由基會受到本身性 質所影響,分為親電子性自由基與親核性自由基,親核性自由基會喜好加成 至電子密度低的不飽和鍵(方程式 1)1。 t-BuHgCl X Cl hv X t-Bu X=H, CH3, Cl, CH2OPh, CH2SiMe3 (1) 當自由基加成的化合物上其雙鍵上有兩個甲基取代基增加立體障礙,此 時自由基不易加成至雙鍵上,反而會加成至立體障礙較小的參鍵上(方程式 2)2。 自由基與不飽和鍵進行加成反應,其影響自由基加成主要以不飽和鍵的極性 效應與立體效應為最主要影響因素,而又以立體效應影響較大。因此在自由 基的加成反應中,在欲加成的不飽和鍵上立體效應越大,自由基越不容易加 成到此不飽和鍵。 O PhSO2Br , hv CH3CN , r.t. (2) O Br SO2Ph O O3 1.1.2 自由基環化反應 碳自由基進行分子內環化反應時,主要分為兩種路徑,一種是自由基進 行雙鍵內側加成環化反應,稱exo cyclization;另一路徑是自由基往雙鍵外側 加成環化反應,稱endo cyclization,如圖 13、圖 24。 endo exo endo cyclization exo cyclization 圖 1 自由基分子內環化反應 X 6-endo 5-exo 圖 2 己烯自由基環化路徑
4 以熱力學觀點,碳自由基穩定度是三級 > 二級 > 一級,但從反應看來 並非如此,大部份結果都以五環結構為主並且形成一級自由基,顯然這結果 並不符合熱力學原理,代表可能為其他因素所為;以動力學來說,自由基的 活性非常高,在環化反應中形成五環的速率比形成六環的速率來的快許多, 因此在自由基環化反應當中皆已五環為主。因此自由基環化反應大部份都是 以動力學控制而非熱力學控制,但若自由基在環化反應中,被加成的雙鍵上 有取代基的影響,則會因為取代基立體效應的影響,導致反應偏向六環使六 員環產物增加,如圖 35。 98% 2% 99% 1% 33% 66% 圖 3 自由基環化反應
5 分子內自由基進行exo或endo環化反應時,其化合物的結構與反應速率有 很大的影響,如表 13。當化合物要進行環化反應時,此時進行exo cyclization 的速率是比endo cyclization快,當碳鏈越長或加成的雙鍵上有立體效應時,此 時反應是以endo cyclization為主,反應是受到環化反應後所形成的環形結構之 鍵角所影響。 表 1 自由基環化速率表 kexo kendo 2.3×105 4.1×103 5.4×103 7.5×102 <0.7 1.2×102 5.3×103 9.0×103 3.5×105 6.0×103 3.6×106 1.0×105 5.1×106 1.0×105 3.2×106 1.0×105 O 8.5×106 1.0×105 O 5.0×104 -
6 1.1.3 自由基加成環化反應 自由基加成環化反應是將自由基加成反應和自由基環化反應結合在一起, 而且反應物必須要有兩個或兩個以上的不飽和鍵才可以進行此反應,首先碳 自由基與不飽和鍵進行分子間的加成反應,形成自由基中間產物後再進行分 子內環化反應,反應途中化合物立體結構會有很大的影響,自由基中間產物 應與另一組不飽和鍵在同一個側邊,才能有效的進行環化反應,如圖 46。 t-Bu O DMSO hv O t-Bu DMSO hv O t-Bu O t-Bu O t-Bu t-BuHgI DMSO hv O t-Bu HgI t-Bu 圖 4 自由基加成環化反應
7 在加成環化反應中,可以利用立體效應來控制自由基加成至需要的位置, 如方程式 32,在帶有碳氧雙鍵取代基的不飽和鍵上增加兩個甲基取代基,苯 磺醯基自由基在加成時會因為立體效應的影響有很高的選擇性加成,其自由 基加成後的中間產物則不會受到立體效應影響進行環化反應,達到加成環化 反應的效果。 O O PhSO2Br , hv CH3CN , r.t. O O SO2Ph (3)
8
1.2 自由基加成反應其取代基極性效應
自由基是一種活性很高的物質,因為活性很高則會產很多不一樣的反應 性,這些反應與自由基所連接的取代基極性有所關聯。當自由基要加成至雙 鍵時,在雙鍵上放置不同的取代基因誘導效應改變雙鍵上的極性,進而影響 自由基加成反應。 本實驗室曾經報告過SH2'加成反應如方程式 11有很明顯的取代基效應, 第三丁烷基自由基與 2-取代基丙烯氯進行SH2'加成反應,表 2 當不飽和鍵上 取代基為拉電子基時,第三丁烷基自由基為親核性自由基會因為取代基誘導 效應使親核性自由基更容易加成至雙鍵上,導致加成加反應速率變快。 t-BuHgCl X Cl hv X t-Bu X=H, CH3, Cl, CH2OPh, CH2SiMe3 (1) 表 2 第三丁基自由基加成至 2-取代基丙烯氯之相對速率Substituents X σm Relative rate
Cl 0.37 22.08
CH2OPh 0.06 3.04
H 0 1.00
Me -0.069 0.82
9 所以在不飽和鍵上放置適當的取代基,則可以利用取代基的極性效應, 來控制自由基加成的選擇性,利用此原理可望解決自由基加成環化反應位置 選擇性的問題。 本實驗室使曾使用第三丁烷基自由基來進行自由基加成環化反應來討論 極性效應的影響,不飽和鍵旁碳氧雙鍵為拉電子基團,此親核性自由基容易 加成至電子密度低的不飽和鍵上再來進行環化反應如方程式 47,本次研究是 使用對甲苯磺醯基自由基去探討其加成環化反應,此自由基為親電子性自由 基,性質應與第三丁烷基自由基不同,預期與第三丁烷基自由基應有不同的 效應。 O O Br t-BuHgCl hv O t-Bu O 77% O O t-Bu 19% (4) 當起始物不飽和鍵上有拉電子性的取代基,對親電子基自由基而言其活 性不高,理論上親電子自由基應該接在電子密度較大的雙鍵,此反應卻相反 卻加成到電子密度較低的α、β不飽和的雙鍵如方程式 58,而且選擇性相當 高,可能是自由基加成到有碳氧雙鍵取代基的不飽和鍵上,中間產物所產生 的未配位電子與碳氧雙鍵產生共振效應導致穩定性變大,其作用大於碳氧雙 鍵所產生的極性效應所造成的不利因素,所以提高加成環化反應的位置選擇 性。 O N R TosX N R O Tos X (5) 96%
10
1.3 自由基加成反應其共振效應
除了可以利用不飽和鍵上的取代基極性效應來提高位置選擇性以外,以 共振效應來提高穩定性進而增加位置選擇性則是另一個選擇的方式。 O O PhSO2Br , hv CH3CN , r.t. O PhO2S Br O (6) 當化合物上雙鍵與叄鍵沒有立體障礙時,苯磺醯基自由基此種親電子性 自由基欲加成至不飽和鍵上,雙鍵旁的碳氧為一種拉電子基團,叄鍵旁兩組 甲基取代基為推電子基團,相較之下雙鍵上的電子密度應小於三鍵上的電子 密度,苯磺醯基自由基應該會往電子密度較高的叄鍵加成,但實驗結果卻顯 示是往雙鍵加成,結果與預期不相符合。因此原本苯磺醯基自由基不應該加 成至雙鍵上,但此自由基加成至雙鍵上時所產生的自由基中間產物,其自由 基電子與碳氧雙鍵會有共振效應的產生,因而穩定這個中間產物使自由基加 成至雙鍵上,如方程式 62。11
1.4 自由基加成環化反應其立體效應
自由基在反應中,最容易受到的影響是立體效應,立體效應在自由基加 成環化反應中有兩種,一種是自由基加成時不飽和鍵上取代基所產生的立體 障礙,另一種為自由基加成至不飽和鍵上形成自由基中間體後,其本身的立 體效應對環化反應所產生的的影響。 第一種情形如方程式 72,化合物的雙鍵上若增加兩個甲基取代基,苯磺 醯基自由基則會選擇加成至叁鍵上。因此在自由基的加成反應中,在欲加成 的不飽和鍵上的立體效應越大,自由基越不容易加成到此不飽和鍵,因此立 體效應的影響會大於共振效應與極性效應的影響。 O O PhSO2Br , hv CH3CN , r.t. O O Br SO2Ph 50% O O SO2Ph 16% (7) 自由基加成環化成為中間體之後,環化反應時結構所產生的s-cis與s-trans 的影響,此為空間上鍵角所旋轉方式不同而產生立體效應,會產生化合物只 有加成反應而沒有環化的現象,如圖 52。 O O R2 R1 O O R1 R2 s-cis s-trans 圖 5 化合物 cis 與 trans 結構12 第二種情形如圖 6,化合物本身因為 s-cis 與 s-trans 的影響,使自由基在 加成至雙鍵後因化合物結構順反的原因而無法進行環化反應,而只進行加成 反應,對於化合物本身所產生的立體結構,也是影響自由基環化反應是否能 順利環化的一個很重要的影響,如圖 6。 O O R2 R1 PhSO2Br , hv CH3CN , r.t. R1=H , R2=H R1=CH3 , R2=H R1=CH3 , R2=CH3 O O PhO2S O O PhO2S O O Br PhO2S 圖 6 苯磺醯基親電性自由基加成環化反應
13
第二章 論文回顧
1989 年Chuang與Ngoi9利用對甲苯磺醯基自由基與 1,6-對稱的二烯類進行 自由基加成環化反應。探討磺醯基親電性自由基的立體效應與加成選擇性的 不同,發現自由基的加成反應會因取代基產生立體效應而有不同的結果,當 在雙鍵上有兩個甲基取代基時自由基會受到立體效應的影響而不易加成,此 加成環化反應得到cis 和trans二種產物,可是選擇性不高(cis/trans) ratio在 1.4~6.0 之間其反應機構如下圖。 E E E E Ts Cl TsCl Ts E E Ts E E14 1990 年Serra10等人利用照光產生自由基,討論自由基加成至雙丙烯基化 合物的環化反應。發現在環化反應時需要雙丙烯結構的雙鍵在同一側邊才會 進行環化反應,以自由基與二丙烯基硫化物的加成環化反應中,環化過後的 化合物全部都為cis的結構,推測兩邊的雙鍵會因為軌域對稱性,影響自由基 的加成方向,環化時會因此產生很高的選擇性。 X R X R X R RY RY X R Y X R Y X Ts Cis X Ts Trans
15 1991年Correa11等人利用對甲苯磺醯溴與對甲苯磺醯碘對丙烯酸丙酯進 行自由基加成環化反應,討論加成速率與選擇性的關係,發現對甲苯磺醯溴 與對甲苯磺醯碘在形成自由基速率方面差很大,對甲苯磺醯碘可以在很短的 時間內產生自由基並加成至丙烯酸丙酯,但因為速率很快的關係則選擇性大 幅下降,但這兩種化合物所產生的自由基加成到丙烯基的雙鍵或是丙酯基的 雙鍵兩種加成速率是相同的,在選擇性上對甲苯磺醯基自由基則喜好加成至 丙酯類雙鍵上,因反應中自由基加成至丙酯的雙鍵上會產生共振效應而使的 能階較低,因此選擇性較高,加成過後的環化反應則會因為空間上的立體效 應所影響,而形成有環化與沒環化的兩種結構方式。
16 1992年Bertrand8等人發現對甲苯磺醯基自由基在加成反應上,對甲苯磺 醯基自由基加成至N-烷基-N-丙烯基丙烯醯胺其1,6-不對稱二烯在立體結構方 面必須呈現s-cis的狀態才能進行分子內環化反應,維持這樣的s-cis立體結構需 要N-烷基-N-丙烯基丙烯酰胺的烷基取代基能夠產生較大立體效應,因烷基取 代基如果有較大的立體障礙,則化合物在s-cis與s-trans的兩種結構中會因為 s-trans有較大的立體效應而選擇轉變成s-cis的結構,此結構才能成功的進行加 成環化反應。 N R O O N R cis trans R cis trans CH2Ph 40 60 CH2CHCH2 50 50 t-Bu 100 0 Ph 0 100
17 1995年Bertrand12等人利用對甲苯磺醯基自由基與1,6-非對稱二烯類進行 自由基環化加成反應,此反應受到1,2-和1,5-取代基所影響,反應後有很好的 位向選擇性,在1,2-取代基加成選擇性上皆為1,2-trans,在1,5-的立體選擇性上 則cis/trans比為75:25。1,2-取代基加成時取代基可能位於axial或equatorial兩 種不同的位置,在equatorial的位置其立體結構上是比較穩定,因此會有比較 好的選擇性,在1,5-取代基上因有chair與boat兩種立體結構,加成後則會產生 兩種不同的立體異構物,因chair相對穩定因此選擇性相對較高。 O TsCl O O CH2Ts Cl 75% O CH2Ts Cl 25% Ts
18 1999 年Russell與Wang2利用磺醯基自由基與丙炔基丙烯酸酯進行加成反 應,發現親電子性磺醯基自由基喜好加成在有拉電子基的丙烯酸雙鍵上,且 在丙炔基R1R2上為烷基取代基時,會有良好的環化效果,自由基在加成後環 化至三鍵時的加成則不影響反應的進行,影響自由基加成環化反應的只有在 加成後的立體效應,反應物s-cis與s-trans的不同,如果不飽和鍵不能在同一側 邊,則無法順利進行環化而只能進行加成反應,而R基為烷基取代基時,能有 效的使化合物呈現s-cis的結構,因此容易進行加成環化反應。 O O R2 R1 PhSO2Br hv O O PhO2S Br R2 R1 O O R R R=H R=Me O SO2Ph O Br 94% O O Br SO2Ph 50% O SO2Ph O 16%
19 同一年Russell與Wang13也發表了用磺醯基自由基與N-烷基-N-(丙烯基)丙 烯醯胺進行自由基加成環化反應合成內醯胺結構,探討取代基立體效應所造 成的影響,當結構上有立體障礙較大的第三丁基取代基時進行環化反應的產 率就會提高,也發現到自由基加成後的中間產物,會因為共振效應穩定性的 不同而在環化速率方面有不同的變化,加成至有碳氧雙鍵的取代基會因為自 由基與碳氧雙鍵的共振因穩定而容易進行環化反應,加成至沒有取代基的雙 鍵上則會因較不穩定而會有其他反應的影響。 X O X
fast intermediate slow
X=O, NSO2Ar X=O, NR
N O hv.CH3CN PhSO2Br N PhO2S Br O 93%,cis/trans=1:5
20 1999 年Dolbier14等人討論自由基在自身環化反應時,自由基進行exo cyclization與endo cyclization的動力學控制,在自由基中間體裡,因動力學控 制關係碳鏈越小進行exo的速率相對較快,碳鏈越長的會越來越趨向熱力學控 制因而反應相對較慢。作者也使用一些電負度很高的取代基,利用自由基加 成的速率減緩,促進endo cyclization的環化,但結果還是不如預期,表示自由 基還化反應路徑主要還是以動力學控制來進行。 (CH2)n kc (CH2)n (CH2)n kexo/s-1 kendo/s-1 n=1 2.7×105 5×103 n=2 5.4×103 7.5×102 n=3 1.2×102 C F2 CF2 CF2 C F2 CF2 F2 C 89% C F2 CF2 CF2 11% k5-exo 4.5×107 S-1 k6-endo 5.6×106 S-1
21
2002 年Chatgilialoglu15等人研究在自由基環化反應中進行 5-endo與 4-exo
其中的反應機制,在進行 4-exo環化反應中,五員環過渡態比四員環的更穩定, 在熱焓的變化上 5-endo也比 4-exo能量低了許多,原因可能是出在四環的鍵角 比較不穩定,在過渡態當中容易還原成中間體。環化時不飽和鍵上的立體效 應是可以影響 4-exo或 5-endo的形成,卻不會影響在過渡狀態時的穩定度。 R 4-exo R 5-endo R
radical cyclization Ea, kcal/mol Hr, kcal/mol
N
H O
5-endo 16.1 -20.8
22 2004 年Landais與James16使用對甲苯磺醯基自由基與 1,6-非對稱二烯 進行SH2'加成環化反應,產生一個五員環的結構,最高產率可以到達 85%, 也可以維持一個很高的位向選擇性加成。 PhMe2Si R SO2Tol TolSO2SePh,hv R PhMe2Si Tos R PhMe2Si Tos R=H (85%,cis/trans=98:2) R=CO2Me (72%,cis/trans=95:5)
23 2004 年Ernest與Sean 17利用催化的方式,使化合物本身變成一個自由基中 間體,討論自由基環化反應時所進行的路徑,結果磺醯基自由基自身加成環 化時,原本有 5-exo與 6-endo兩種選擇性,但因為磺醯基自由基在動力學上穩 定性不好,因此在環化時影響加成的選擇性,此時全部都是 5-exo的選擇性, 而自由基加成時雙鍵的選擇性,顯示雙鍵所產生的立體效應影響大於極性效 應,因此成為自由基環化的一個重要因素。 SO2Ph Bu3SnH SO2Ph 100% S TsBr S Ts S Ts S Ts S Br Ts S Br Ts 100% 0% SO2 Bu3SnH SO2 2.5% 97.5% SO2
24
第三章 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚與對甲苯
磺醯溴進行自由基加成環化反應
本實驗室曾經5利用化合物 1 與第三丁基氯化汞溶於二甲基亞碸中進行照 光反應,得到化合物 2 與化合物 3,而化合物 2 與化合物 3 的比例大約是 1: 2,如方程式 8,表 3。 C2 C1 Cl O C6 C7 Cl t-BuHgCl hv O C6 C7 C2 C1 t-Bu Cl C2 C1 Cl O C6 C7 t-Bu 1 2 3 (8) 表 3 化合物 1 與第三丁基氯化汞照光反應 化合物 1 (mmol) t-BuHgCl (mmol) 反應時間 (hr) 產物 2a (%) 產物 3a (%) 0.1 0.1 2 19 39 0.1 0.1 4 23 44 0.1 0.1 6 21 44 a 此面積比是以固定內標準品 10% mmol 聯苯的面積而換算出 化合物 1 中,兩組不飽和鍵旁為氯取代基與甲基氯取代基,其中氯的拉 電子效應大於甲基氯的拉電子效應,所以親核性自由基第三丁基自由基應該 會加成至氯取代基的雙鍵上,進而進行加成環化反應,然而結果與預期的不 相符,所以認為影響自由基加成至不同雙鍵的選擇性,取代基的極性效應並 非影響的唯一因素。25 自由基在加成至雙鍵時的選擇性有兩種可能:一為C1C2雙鍵、二為C6C7 雙鍵,當自由基加成至任何一個雙鍵,其活化能取決於自由基加成時打斷π鍵 的能量。而加成至C6C7雙鍵時雖然因為取代基的極性效應的關係使得其活化 能較高,但取代基甲基氯含有氯離去基,因此在自由基加成至C6C7雙鍵時進 行同步式的反應,因此自由基的加成與離去基的離去會同步發生,亦即自由 基加成會打斷π鍵,但是會同步產生另一組新的π鍵,此為SH2'同步式反應, 導致自由基加成至C6C7雙鍵時活化能相對較低,所以選擇加成至C6C7雙鍵上, 所以離去基效應則變成影響反應一個不能忽略的因素。 為了要增加自由基的加成至C1C2雙鍵上的選擇性,我們將化合物 1 改成 化合物 4,把化合物 1 中裸露的C6C7雙鍵將其改成一種隱藏式的雙鍵,因而 增加其立體效應,使自由基加成至C6C7雙鍵因立體效應增加而不容易加成至 此雙鍵。化合物 4a與第三丁基氯化汞進行照光反應,形成了化合物 5 和化合 物 6(方程式 9),結果如表 4;化合物 5 與化合物 6 的產率比例約 4:1,結果 和預期的相符。 C1 C2 Cl O C6 C7 X hv t-BuHgCl O t-Bu Cl O t-Bu Cl 4a X=Br 4b X=OPh 5 6 (9)
26 表 4 化合物 4a/4b 與第三丁基氯化汞照光反應 化合物 4a (mmol) t-BuHgCl (mmol) 反應時間 (hr) 產物 5a (%) 產物 6a (%) 0.1 0.1 2 21.16 5.62 0.1 0.1 4 41.49 10.57 0.1 0.1 6 48.69 12.08 化合物 4b (mmol) t-BuHgCl (mmol) 反應時間 (hr) 產物 5a (%) 產物 6a (%) 0.1 0.1 2 2.10 0.32 0.1 0.1 4 2.51 0.39 0.1 0.1 6 3.19 0.46 a 此面積比是以固定內標準品 10% mmol 聯苯的面積而換算出 從表 4 化合物 6 的產率相對變少,但還是有少量的產生,可能原因是自 由基加成至C6C7雙鍵後取代基的離去效應所產生,因此為了降低離去基的離 去效應,我們改變離去基的種類,因離去基相對離去速率為I>Br>SPh>Cl >>OPh,SiMe318,所以將溴的離去基換成苯氧基的化合物 4b,希望將化合物 6 的產率能夠減少,而實驗結果發現化合物 5 與化合物 6 的產率比例從 4:1 提升至 6.5:1,但是產率降低,如表 4。從實驗結果可以得知,取代基離去效 應對於自由基SH2'的加成環化反應是有所影響的,離去基效應會影響到加成 選擇性與產率,當選擇離去效應較差的離去基時自由基在加成方面選擇性會 較高,但產率會相對減少;而離去基如果有較好的離去效應,則自由基在加 成時選擇性會較低,但也會產生較高的產率,因此可以在自由基SH2'反應中 調整離去基的種類,達到所需要的要求。
27 本次實驗所使用的自由基為對甲苯磺醯自由基,對甲苯磺醯基自由基為 一種親電子性的自由基,與親核性自由基第三丁基自由基有很大的不同,為 了瞭解不同性質的自由基在SH2'加成環化反應的結果有何不同,取代基極性 效應與離去基效應是是否會影響結果,因此我們設計了化合物 7,因對甲苯磺 醯自由基是親電子性自由基,所以在C1C2雙鍵放上一個推電子性的甲基取代 基,利用誘導效應使對甲苯磺醯基自由基容易加成至C1C2雙鍵,C6C7雙鍵上 也利用隱藏式的雙鍵來增加其立體效應使自由基不易加成至此雙鍵,進而達 到所需要的自由基SH2'加成環化反應。 C2 C1 O C6 C7 Br 7
28
3.1 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚合成
首先將 trans-1,4-dibromo-2-butene 與 sodium hydride 置於反應瓶中與溶劑 正己烷攪拌,加熱回流 30 分鐘,此反應需在氮封下進行。待降至室溫後,再 取 2-methyl-2-propen-1-ol 緩慢加入其中,反應 6 個小時。反應完畢後使用乙 醚進行萃取,以氣相層析質譜儀檢測產物,有預期產物(2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚(化合物 7)產生(產率 60%)與少量化合物 8(產率 10%)生成,如 圖 7。 OH NaH O O Br Br O Br O O 7 8 圖 7 化合物 7【(2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚】合成
29
3.2 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由
基之加成環化反應
利用對甲苯磺醯溴在照光系統下,產生對甲苯磺醯基自由基來進行自由 基加成環化反應,此自由基是一種親電子性的自由基。我們將使用化合物 7 與甲苯磺醯溴溶於乙腈中進行照光反應,預期可以得到化合物 9(方程式 10)。 C2 C1 O C6 C7 C8 Br TsBr hv C6 C2 O C1 Ts C7 C 8 7 9 (10) 化合物 7 進行自由基加成環化反應時,我們推測可能有兩種反應路徑, 如圖 8,當對甲苯磺醯溴在照光下產生對甲苯磺醯基自由基,對甲苯磺醯基自 由基會加成至雙鍵上,在化合物 7 的結構中有C1C2與C6C7兩組雙鍵,對甲苯 磺醯基自由基加成有兩種路徑,一種是自由基加成在C1C2雙鍵上,形成自由 基中間體 11,此自由基進一步與C6C7雙鍵進行SH2'加成環化反應,而形成化 合物 9;另一種則是自由基加成在C6C7雙鍵上,直接進行SH2'同步式反應形成 化合物 10。30 C2 C1 O C6 C7 C8 Br TsBr hv C6 C2 O C1 C7 C2 C1 O C6 C7 C8 Ts 7 9 10 C2 C1 O C6 C7 C8 Br Ts 11 5-exo Ts C8 C2 C1 O C6 C7 C8 Br Ts 12 SH2' 圖 8 化合物 7 與對甲苯磺醯溴加成環化反應反應機構 化合物 7 與對甲苯磺醯溴的照光加成環化反應其結果如表 5,此反應得到 的主要產物為化合物 9,並沒有觀察到任何產物 10,因為對甲苯磺醯基自由 基為一種親電子性的自由基,利用C1C2雙鍵上的甲基取代基產生誘導效應並 且在C6C7雙鍵上產生立體效應,使自由基加成位向選擇性提升,加成後形成 自由基中間體 11 再經由 5-exo cyclization反應,進行SH2'同步式反應形成化合 物 9,達到預期產物。 改變不同的溶劑對於產物的位向選擇性沒有太大的影響,用乙腈溶劑有 最高產率 69%,使用正己烷或 THF 當溶劑時會產生很多的副產物而影響導致反應不 好,DMSO 溶劑則會與對甲苯磺醯溴直接產生作用而無法進行自由基加成環化反應, 而最後選擇使用乙腈當作這次反應溶劑如表 5。
31 表 5 化合物 7 與對甲苯磺醯溴照光反應 溶劑 化合物 7 (mmol) TsBr (mmol) 反應時間 (hr) 化合物 9a (%) 乙腈 0.5 0.5 1 55.6 0.5 0.5 2 52.4 0.5 0.5 3 69.2 甲苯 0.5 0.5 1 31.2 0.5 0.5 2 43.2 0.5 0.5 3 69 二氯甲烷 0.5 0.5 1 59.2 0.5 0.5 2 55.4 0.5 0.5 3 54.8 乙酸乙酯 0.5 0.5 1 27.5 0.5 0.5 2 41.8 0.5 0.5 3 52.3 乙醚 0.5 0.5 1 18.4 0.5 0.5 2 21.4 0.5 0.5 3 33 THF 0.5 0.5 1~3 無預期產物 DMSO 0.5 0.5 1~3 無預期產物 正己烷 0.5 0.5 1~3 無預期產物 a 此面積比是以固定內標準品 20% mmol 聯苯的面積而換算出
32
3.3 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由
基加成環化反應之位向選擇性與立體效應
影響自由基在加成反應時的因素有取代基極性效應與立體效應和離去基 效應,以對甲苯磺醯基自由基與化合物 7 的自由基加成環化反應中,化合物 7 有兩組雙鍵加成時最少就有四個位置可以進行加成,因此要控制自由基加成 至所想要的位置,則變成一個重要的問題。 以極性效應來說我們在化合物 7 的C1C2雙鍵與C6C7雙鍵上放置不同的取 代基,試著影響親電子性的自由基加成時的位向選擇性,在C6C7雙鍵上取代 基為溴甲基為一種拉電子基,而在C1C2雙鍵上放置一個甲基為一種推電子基, 因自由基是種親電子性的自由基,容易加成至有推電子基的甲基取代基之雙 鍵,因此對甲苯磺醯基自由基加成至C1C2雙鍵的速率會比加成至C6C7雙鍵速 率來的快。立體效應方面,C1C2雙鍵上C2碳上有一個甲基取代基,甲基取代 基在β−碳上,所以對自由基加成反應其立體效應該非常小,而在C6C7雙鍵上 的基取代基在α−碳(C6)上,所以對自由基加成反應產生較高的立體效應,因 此自由基不容易加成至C6C7雙鍵,相對的自由基容易加成至C1C2雙鍵上。自 由基加成至C1C2雙鍵後會在C2上形成一個自由基,然後與C6C7雙鍵進行加成 環化反應,因溴為一種很好的離去基,反應容易進行SH2'同步式反應,最終 形成化合物 9。33
3.4 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由
基加成環化反應之結構鑑定
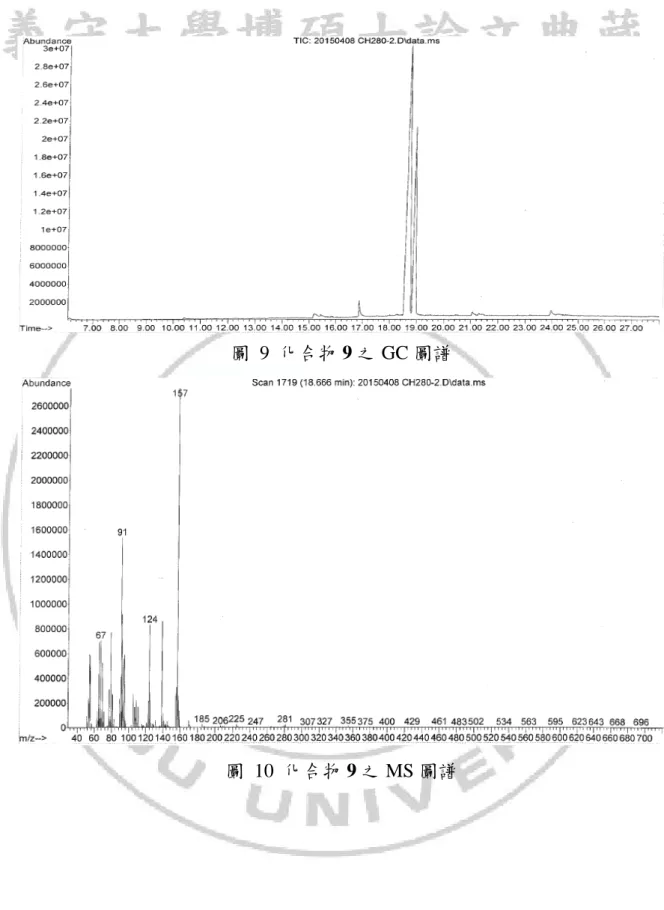
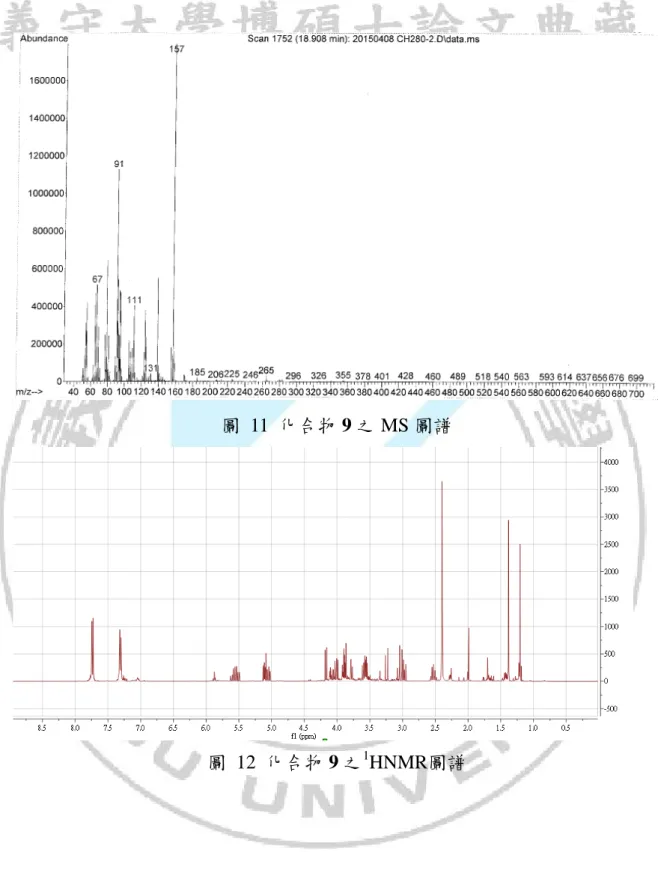
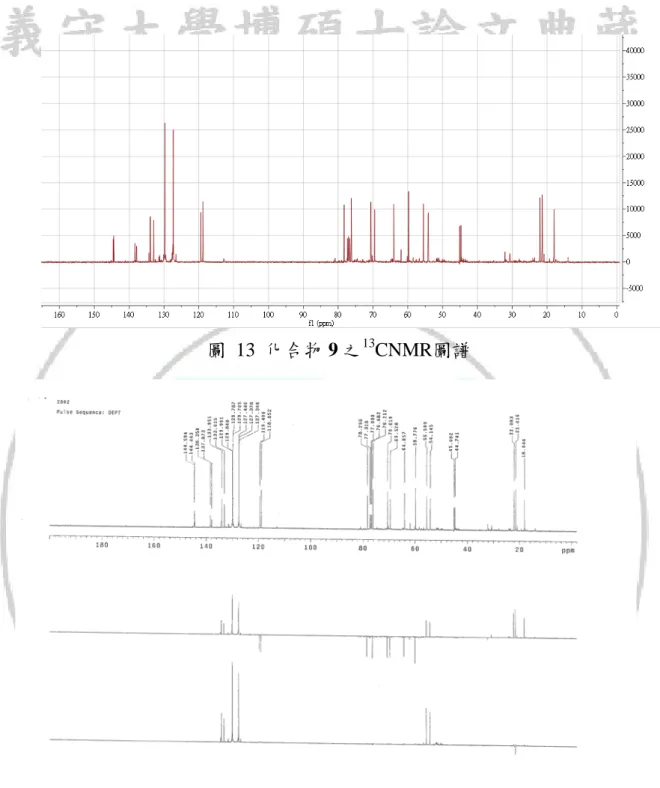
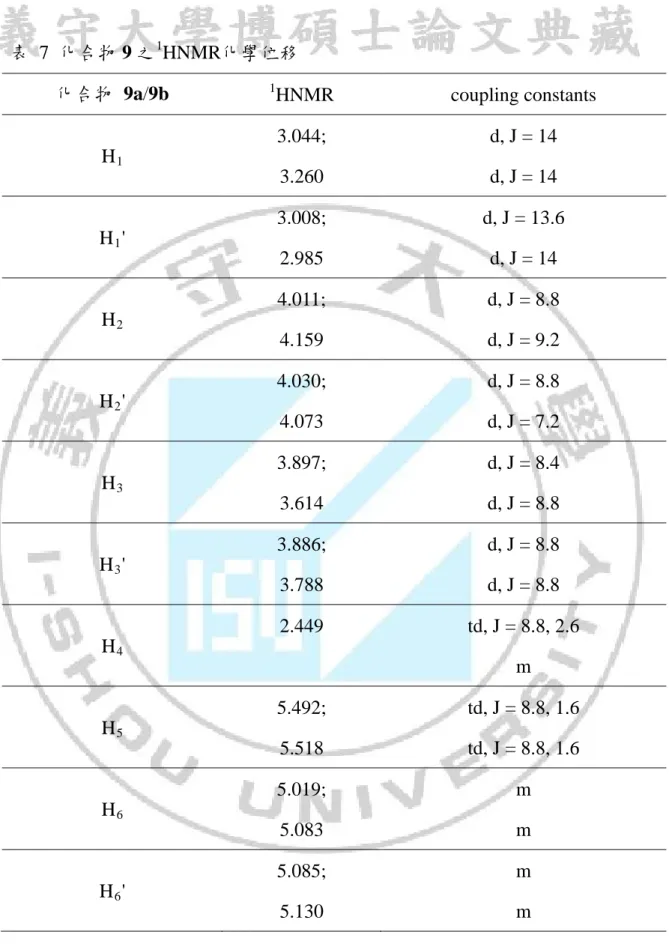
從化合物 7 與對甲苯磺醯溴照光反應結果可以得知,加成時有良好的位 向選擇性(如方程式 11),主要產物為兩種立體異構物。 C2 C1 O C6 C7 C8 Br TsBr hv C6 C2 O 7 9a (11) C1 Ts H3C H C6 C2 O C1 Ts H3C H 9b 由於兩種異構物在GCMS圖譜中滯留時間相當接近(如圖 9),因此在利用 管柱層析純化時的過程無法將其分離,從質譜中無法確定其分子量,不過從 兩者的分子斷裂的片段都相同推測兩者可能為同分異構物(如圖 10 與圖 11), 此混合物的NMR圖譜中發現13 CNMR圖譜中每一個碳似乎有兩種吸收峰, 在1 HNMR也有類似的情況(如圖 12 與圖 13),在DEPT的圖譜上這每組碳的氫 數也都相同(如圖 14) ,利用1 HNMR與13CNMR與DEPT推測產物的分子量與 預期產物 9 相符合,在結構中也發現在五員環上C2、C6碳是兩個不對稱碳, 如果此兩種結構為enantiomers這兩種結構的物理性質與化學性質應該會相同, 則不會有13 CNMR圖譜中的現象產生,而推測這兩種產物可能為diastereomer。34
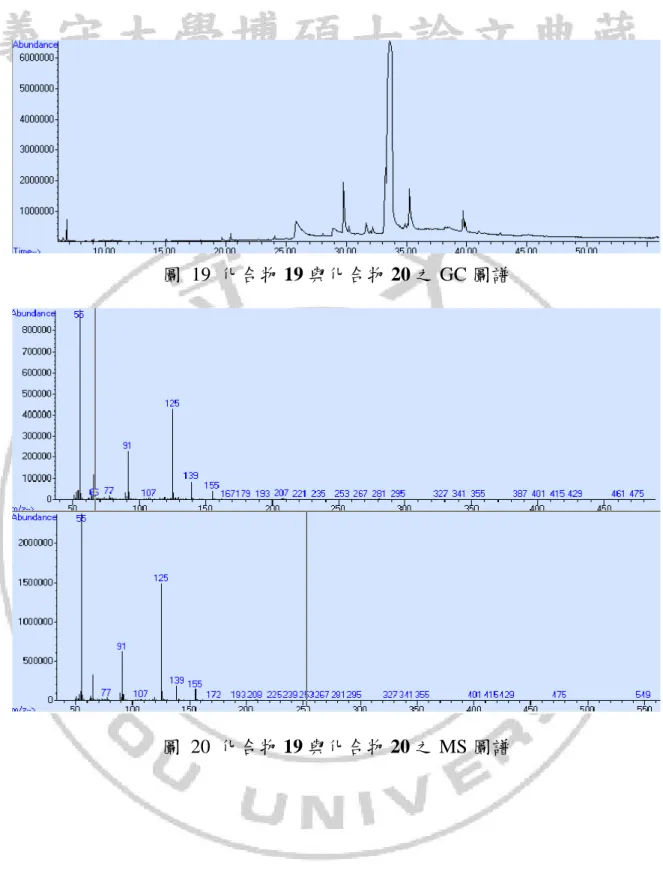
圖 9 化合物 9 之 GC 圖譜
35
圖 11 化合物 9 之 MS 圖譜
圖 12 化合物 9 之1
36
圖 13 化合物 9 之13
CNMR圖譜
37 質譜圖無法顯現M+,所以分子量無法確定,利用13 CNMR與DEPT圖譜來 推斷可能的分子量,如果預期產物為化合物 9,應該會有 15 個碳原子、1 個 硫原子、3 個氧原子和 20 個氫原子,分子量推測為 280。 化合物 9 的二種異構物其13 CNMR與DEPT圖譜(如表 6),從此二種圖譜可 以先行歸納出有二組碳,每一組各有 13 種不同的碳,化合物 9 的二種異構物 也有 13 種碳,13 CNMR圖譜中 120~150 ppm之間的訊號為不飽和鍵上的碳, 在化合物 9 結構中不飽和鍵的碳有 8 個,其中苯因為對位取代所以有兩組相 同的碳,所以在13 CNMR圖譜上應該要有 6 組符合我們所推測,C3、C4、C5、 C6為苯環上的碳所以可以明顯判斷為 127~129 ppm區域上的吸收峰,C2、C 7因分別有拉電子與推電子的取代基,所以可以判斷各別位置在 144 ppm與 138 ppm,C14、C15從DEPT也可以判斷位置為 133 ppm與 119 ppm,剩下的再 從DEPT圖譜中推測氫的數目判定各別位置的碳,60~80 ppm中有 3 組吸收峰, 氫的數目都是兩個推測為C8、C11、C12,因在環上C11、C12因為接氧吸收峰會 可能為 70~80 ppm,C11旁有拉電子對甲苯磺醯基取代基所以其化學位移較大, C8 、C13吸收峰大約為 60 ppm, C9從DEPT可以推測出吸收峰大約在 45 ppm, 最後吸收峰 20 ppm附近的則為C1、C10,分析光譜後發現此兩異構物其C1的 吸收峰可能為重疊。 1 HNMR圖譜中(如表 7),此結構比較明顯的是在 7~8 ppm左右有苯環的吸 收峰出現,此化合物 7 結構原本沒有苯環,此出現可以證明對甲苯磺醯基自 由基是有確實加成至化合物 7 中,而原本在化合物 7 當中C1C2與C6C7雙鍵的 吸收峰都消失,產生另一組新形成的雙鍵(C14C15)上的H5H6,但因為無法清楚 判斷圖譜中H6的分裂情況,所以無法準確的判定這個雙鍵的順反結構,在環 化結構中從模擬圖譜推測各別氫的吸收峰,最後判斷結構為化合物 9。
38 O H3C H Ts O H3C H Ts 9a 9b (18.4) (144.44; 144.59) (129.76; 129.78) (127.34; 127.44) (137.87; 138.35) (57.77;64.05) C13 C9 C11 O C12 C8 C10 C14 C 15 (21.41;22.08) (133.01;133.95) (118.85;119.40) (54.14;55.50) (69.52;70.61) (76.21;78.209) (44.74;45.08) 9 S C7 O O C6 C4 C2 C3 C5 C1 (129.76; 129.78) (127.34; 127.44) 圖 15 化合物 9 1 HNMR化學位移 O CH3 H1 H1' H2 H2' H3' H3 H4 H5 H6' H6 (2.386) (3.044; 3.260) (3.008;2.985) (4.011; 4.159) (4.030; 4.073) (3.897;3.614) (3.886; 3.788) (2.449) (5.085;5.130) (5.019;5.083) (5.492;5.518) (1.205; 1.381) 9 S O O H3C 圖 16 化合物 9 13 CNMR 化學位移
39 表 6 化合物 9 之13 CNMR化學位移 化合物 9a/9b 13 CNMR 化學位移 H 的數目 模擬值a C1 18.04;18.04 3;3 24.3 C2 144.44;144.59 0;0 143.4 C3,C4 129.76;129.78 1;1 130.1 C5,C6 127.34;127.44 1;1 128.2 C7 137.87;138.35 1;1 135.9 C8 59.77;64.05 2;2 58.9 C9 44.74;45.08 0;0 30.5 C10 21.41;22.08 3;3 17.6 C11 76.21;78.29 2;2 86.1 C12 69.52;70.61 2;2 75.2 C13 54.14;55.50 1;1 48.7 C14 133.01;133.95 1;1 139.0 C15 118.85;119.40 2;2 112.6 a模擬值由軟體ChemDraw Ultra 8.0 模擬而成
40 表 7 化合物 9 之1 HNMR化學位移 化合物 9a/9b 1 HNMR coupling constants H1 3.044; 3.260 d, J = 14 d, J = 14 H1' 3.008; 2.985 d, J = 13.6 d, J = 14 H2 4.011; 4.159 d, J = 8.8 d, J = 9.2 H2' 4.030; 4.073 d, J = 8.8 d, J = 7.2 H3 3.897; 3.614 d, J = 8.4 d, J = 8.8 H3' 3.886; 3.788 d, J = 8.8 d, J = 8.8 H4 2.449 td, J = 8.8, 2.6 m H5 5.492; 5.518 td, J = 8.8, 1.6 td, J = 8.8, 1.6 H6 5.019; 5.083 m m H6' 5.085; 5.130 m m
41 從13 CNMR圖譜中推測這個化合物 9 是有兩種立體異構物產生,所以 在1 HNMR圖譜中,可以找到同樣對稱性吸收峰,除了判斷化學位移我們利用 J值與吸收峰的分裂來判定這兩個立體異構物的個別氫原子吸收峰位置,因目 前所得到的資料中,只能分出各個對稱的吸收峰,還沒辦法完全鑑定這些吸 收峰所代表的各別的化合物,但所推測的化合物 9 應該是相符的,在13 CNMR 圖譜中是沒有問題的,1 HNMR圖譜中也都能找到相應符合的吸收峰,但 在1 HNMR還有些沒有辦法解釋的吸收峰,把同分異構物分離過後才有辦法解 釋清楚,而我們也正嘗試的再利用HPLC想要分離此兩種異構物,但可以確定 的是對甲苯磺醯基自由基是加成在化合物 7 的C1C2雙鍵上,且化合物進行 5-exo環化反應。因此使用親電子性的自由基再把原本雙鍵上的氯取代基換成 甲基取代基,而取代基極性效應還是確實的有影響,加成選擇性因C6C7上的 取代基立體效應,而有不錯的選擇性,立體選擇性方面化合物 9 的cis與trans 比為 3:1。
42
第四章 (3-丙烯醯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯
溴進行自由基加成環化反應
在自由基加成反應裡除了取代基極性效應、立體效應或是離去基效應會 影響自由基的加成選擇性外,Tedder和Walton19曾經報告自由基加成反應如果 雙鍵上帶有含π軌域的取代基會加速自由基的加成速率,本實驗室曾經使用親 核性自由基第三丁基自由基與(3-丙烯醯基)-(4-溴-2-丁烯基)醚(化合物 12)反 應(如方程式 12),此化合物結構有一組碳氧雙鍵的取代基,反應時親核性自 由基會加成至碳氧雙鍵取代基旁的C1C2雙鍵上產生自由基中間體,自由基中 間體的未成對電子會與雙鍵上氧的孤對電子產生共振效應,此舉有利於親核 性自由基的加成時位向選擇性增加,反應結果化合物 13 與化合物 14 比例大 約 6:1,如表 8。 C2 C1 O O C5 C6 Br C5 C2 O C1 t-Bu O C6 C2 C1 O O C5 t-Bu C6 (12) 12 13 14 t-BuHgCl hv43 表 8 化合物 12 與第三丁基氯化汞進行照光反應結果 化合物 12 (mmol) t-BuHgCl (mmol) 反應時間 (hr) 產物 13 (%)a 產物 14 (%)a 0.2 0.2 0.2 0.2 1 2 69 72 12 15 0.2 0.2 0.4 0.4 1 2 74 76 15 17 a 此面積比是以固定內標準品 10% mmol 聯苯的面積而換算出 本實驗室也設計了化合物 15 與第三丁基氯化汞在DMSO下進行照光反應, 預期產物為化合物 16 與化合物 17(如方程式 13),但實驗結果卻發現完全沒有 這兩種產物的產生,是另外一種未知化合物的產生,從資料推測結果猜測可 能為化合物 18,如圖 17。但我們可以確定的是從化合物 15 與第三丁基自由 基進行加成環化反應時,第三丁基自由基是確實加成在C1C2這組雙鍵上,在 加成方面則有很高的位向選擇性,如表 9。 C2 C1 N O Bn C5 C7 C6 15 t-BuHgCl hv Cl C6 C2 N C5 O Bn C1 t-Bu C7 16 C2 C1 O N Bn C5 C6 C7 t-Bu 17 (13)
44
表 9 化合物 15 與第三丁基氯化汞進行照光反應結果
化合物 15 (mmol) t-BuHgCl (mmol) 反應時間 (hr) 化合物 18 (%)a
0.2 0.2 0.4 0.4 1 2 84 88 0.2 0.2 0.6 0.6 1 2 83 87 a 此面積比是以固定內標準品 10% mmol 聯苯的面積而換算出 C2 C1 N O Bn C5 C7 C6 15 t-BuHgCl hv Cl C6 C2 N C5 O Bn C1 t-Bu C7 16 C2 C1 O N Bn C5 C7 17 C6 t-Bu 6-endo 5-exo C5 O Bn t-Bu C6 C7 Cl SH2 C5 O Bn t-Bu C6 C7 18 圖 17 化合物 15 進行自由基加成環化反應之反應機構
45 當第三丁基自由基加成至C1C2雙鍵上時,產生自由基中間體,自由基形 成在C2上,會與碳氧雙鍵上的孤對電子產生共振效應,表示在取代基含π軌域 的情況下,確實是有提高對於親核性 自由基加成至雙鍵上時的選擇性,結果 可以證明對於加成時,有取代基碳氧雙鍵除了可以提供極性效應也產生了共 振效應的影響。 本實驗使用親電子性的對甲苯磺醯基自由基與化合物 12 進行反應,想利 用極性效應與共振效應來探討不同性質的自由基在自由基SH2'加成環化反應 會有什麼不同結果。
46
4.1 (3-丙烯醯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由基之
加成環化反應
對甲苯磺醯溴在照光系統下產生對甲苯磺醯基自由基與化合物 12 進行加 成環化反應。我們使用對甲苯磺醯溴與化合物 12 在二氯甲烷中進行照光反應, 得到產物 19 與產物 20(如方程式 14)。 C2 C1 O O C6 C7 Br 12 TsBr hv C2 C1 O O C6 C7 Br Ts 19 C2 C1 O O C6 C7 Br Ts 20 (14) 化合物 12 在進行自由基加成環化反應時,原本推測自由基會加成在C1C2 雙鍵上形成一個自由基中間體,再進行 5-exo環化反應但結果不如預期,此反 應有兩種不同的路徑讓自由基加成,第一種對甲苯磺醯基自由基直接加成在 C1C2雙鍵上,形成自由基中間體而進行抓氫反應,沒有與C6C7雙鍵形成環化 反應;第二種路徑則是對甲苯磺醯基自由基加成至C6C7雙鍵上,然後也是直 接進行抓氫反應而沒有接下去的環化反應或自由基SH2'反應,自由基的加成 選擇性化合物 19 與化合物 20 從1 HNMR圖譜中積分面積判斷大約為 2︰3。47 化合物 12 與對甲苯磺醯溴在不同的溶劑進行照光反應其結果如表 10,產 率普遍都不高,最好的產率也只有溶劑為甲苯的 20%,在溶劑效應上影響不 大,普遍副產物為自由基耦合所形成的產物,表示對甲苯磺醯基自由基對於 化合物 12 的加成選擇性不好,所以才產生許多自由基耦合的產物,當溶劑為 THF 時會產生很多的副產物而影響導致反應不好,而 DMSO 溶劑則會與對甲苯磺醯 溴直接產生作用而無法進行自由基加成環化反應,最後在此選擇甲苯為本次實驗所 使用的溶劑。 從表 10 得知,化合物 12 與對甲苯磺醯溴在照光系統下,得到的產物並 非一開始我們所預期的產物 21,實驗結果發現對甲苯磺醯基自由基加成至 C6C7雙鍵的速率大於C1C2雙鍵,從立體效應來看自由基加成至C1C2雙鍵應該 比加成C6C7雙鍵來得快,可是事實不然,其原因可能是對甲苯磺醯基自由基 加成至C6C7雙鍵是進行同步式的SH2'反應,得到化合物 22,然後與溴自由基 進行加成反應得到產物 20,因為同步式的SH2'反應,其活化能小於加成至C1C2 雙鍵的活化能,所以反應速率較快,也因為化合物 12 進行SH2'反應,所以會 先形成化合物 22,然後再進行溴自由基的加成反應最後形成化合物 20,如圖 18。
48 表 10 化合物 12 與對甲苯磺醯溴照光反應 溶劑 化合物 12 (mmol) TsBr (mmol) 時間 (hr) 產率%a (19 與 20) 乙腈 0.5 0.5 2 2 0.5 0.5 4 6 0.5 0.5 6 13 0.5 0.5 8 13 苯 0.5 0.5 2 1 0.5 0.5 4 1 0.5 0.5 6 3 0.5 0.5 8 5 甲苯 0.5 0.5 2 7 0.5 0.5 4 15 0.5 0.5 6 19 0.5 0.5 8 20 二氯甲烷 0.5 0.5 2 3 0.5 0.5 4 10 0.5 0.5 6 13 0.5 0.5 8 16 THF 0.5 0.5 2~8 無預期產物 DMSO 0.5 0.5 2~8 無預期產物 a 此面積比是以固定內標準品 10% mmol 聯苯的面積而換算出
49 C2 C1 O O C6 C7 Br TsBr hv C2 C1 O O C6 C7 Br Ts C2 C1 O O C6 Ts 5-exo 12 C2 C1 Ts O O C6 C7 Br 21 19 C2 C1 O O C6 C7 Ts Br 20 hydrogen abstraction C2 C1 O O C6 C7 Ts Br 22 SH2' 圖 18 化合物 12 進行自由基加成環化反應之反應機構
50
4.2 (3-丙烯醯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由基之
位向選擇性與立體效應
以對甲苯磺醯基自由基與化合物 12 的自由基加成環化反應裡,自由基加 成至化合物 12 過程中,自由基的加成選擇性有C1C2雙鍵與C6C7雙鍵這兩組 雙鍵,各別有碳氧雙鍵拉電子基與溴甲基拉電子基,以對甲苯磺醯基自由基 這種親電子性自由基來說,C6C7雙鍵接溴甲基取代基的極性效應會小於C1C2 雙鍵接碳氧雙鍵取代基的極性效應,所以在自由基會喜歡加成至C6C7雙鍵, 且加成至C6C7雙鍵上後會進行同步式SH2'反應,從產物 19 與產物 20 的比例 為 2︰3,可以應證極性效應與自由基SH2'反應二者有利於自由基加成至C6C7 雙鍵。但還是不少的自由基加成至C1C2雙鍵,從立體效應的影響看來C6C7 雙鍵上其α−碳(C6)有取代基造成自由基加成時其立體效應較大,C1C2雙鍵其 α−碳(C1)上沒有取代基所以自由基加成時其立體效應較小,因此自由基容易 加成至C1C2雙鍵,而在自由基加成後自由基中間體會與碳氧雙鍵上氧的孤對 電子產生共振效應,加成時能量降低使也會讓自由基容易加成在C1C2雙鍵。 雖然不管加成在C1C2雙鍵或C6C7雙鍵上,都各別有共振效應與SH2'反應, 但從最後產物的比例來看共振效應的影響會小於SH2'反應的影響,在一般使 用對甲苯磺醯溴所產生的自由基加成環化反應中,對甲苯磺醯基自由基在加 成至雙鍵後,溴自由基會與加成過後的自由基中間體進行加成反應,可是此 種現象並未在此反應發生,對甲苯磺醯基自由基在兩種加成路徑上,加成至 C1C2雙鍵的自由基中間體直接進行抓氫反應,推測除了對甲苯磺醯基自由基 本身在動力學上就不穩定以外19,自由基中間體也是一個二級的自由基,比起 三級自由基來的活潑,而加成至C6C7雙鍵上的自由基中間體進行了同步式51
52
4.3 (3-丙烯醯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯基自由基之
加成環化反應結構鑑定
由化合物 12 與對甲苯磺醯溴進行照光反應,主要產物為化合物 19 與化 合物 20,如方程式 14。 C2 C1 O O C6 C7 Br 12 TsBr hv C2 C1 O O C6 C7 Br Ts 19 C2 C1 O O C6 C7 Br Ts 20 (14) 在1 HNMR與13CNMR圖譜中,無法直接輕易分辨此兩種化合物結構,在 GCMS圖譜中兩種化合物的滯留時間相當接近無法準確的分離開來,如圖 19, 使用TLC板檢測後用管柱層析法也無法準確將兩種產物分離,從兩種化合物 的質譜看來推測兩者可能為同分異構物(如圖 20),模擬各種可能性之後我們 推測這兩種產物為化合物 19 與化合物 20,我們可以從NMR圖譜中去找出這 兩個化合物的吸收峰各別分離出來,利用13 CNMR、1HNMR與DEPT圖譜(如圖 21、圖 22、圖 23)驗證推測的兩種化合物結構,在圖譜中吸收峰與碳上氫的 數目都有幾乎相符的模擬位置,可以推測在此兩種化合物的結構有可能是相 符的。53
圖 19 化合物 19 與化合物 20 之 GC 圖譜
54
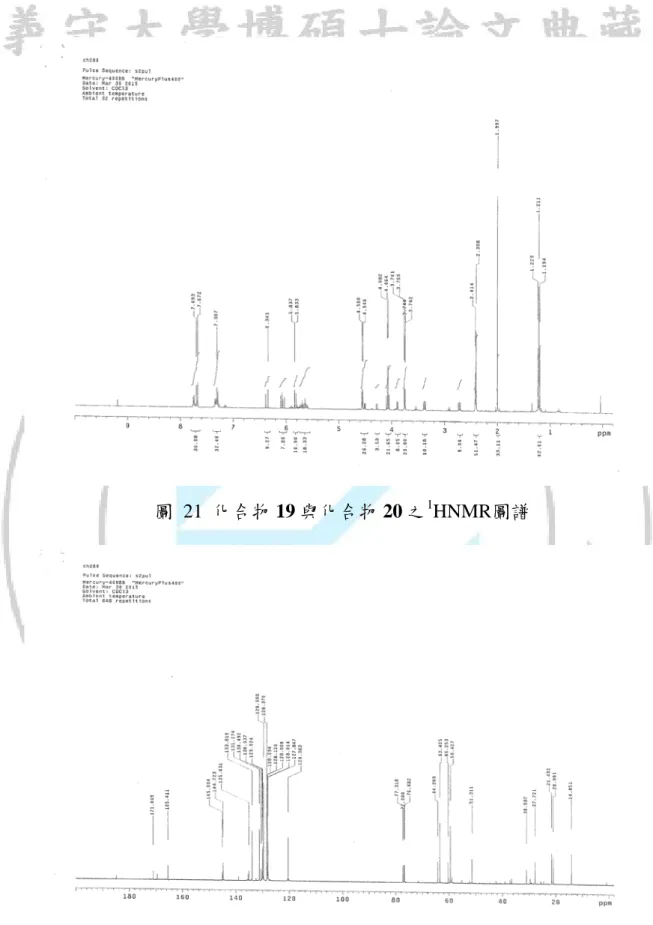
圖 21 化合物 19 與化合物 20 之1
HNMR圖譜
圖 22 化合物 19 與化合物 20 之13
55 圖 23 化合物 19 與化合物 20 之 DEPT 圖譜 先從13 CNMR與DEPT圖譜中(如表 11)過濾出化合物 19 與化合物 20 各別 的碳,13 CNMR圖譜上 120~180 ppm的吸收峰為不飽和鍵上的碳,從整個苯環 來看可以找出兩種化合物Cc、Cd與C3、C4的訊號位置大約在 128~130 ppm, Cb、Ce與C2、C5則在 144 ppm、135 ppm與 145 ppm、135 ppm,碳氧雙鍵上 的Cj和C8則各別為 165 ppm與 171 ppm,而最後剩下的不飽和鍵碳碳雙鍵就要 利用DEPT碳上氫的數目來判斷,推斷化合物 20 的Ck、Cl的吸收峰為 120 ppm、 131 ppm,化合物 19 上的雙鍵C10、C11則吸收峰為 130 ppm、127 ppm,其它 飽和的碳當中Ca、C1可以從DEPT圖譜中容易判斷位置為 21 ppm、20.9 ppm, DEPT圖譜中飽和鍵上還有一個碳上是只有一個氫,我們推測可能為Cf碳吸收 峰在 14 ppm上,但吸收峰位置不應該如此up field這數據則有待討論,最後 DEPT圖譜上剩下的氫數都為 2 的飽和鍵,我們依照模擬值去推論出最後的Cg、
56 Ch、Ci、C6、C7、C9、C12的吸收各別位置在 30 ppm、59 ppm、63 ppm、60 ppm、 27 ppm、64 ppm、51 ppm。 C7 C8 C6 O O S C5 C4 C3 C2 C3 C4 C1 C9 C10 C11 C12 Br O O S Br H1 H2 H3 H5 H4 H6 19 19 (20.901) (145.004) (130.037) (128.060) (135.327) (60.253) (27.721) (171.049) (64.209) (130.492) (127.847) (51.311) (3.88) (3.76) (4.40) (5.63) O O O O (5.67) (4.10) 圖 24 化合物 19 1 HNMR與13CNMR化學位移 C k C j l C O O C i f C C g h C Br S O O e C d C C c C b C c d C C a 20 (131.174) (120.383) (165.411) (63.405) (30.987) (59.427) (14.051) (128.370) (129.598) (144.723) (21.492) (135.031) 圖 25 化合物 20 13 CNMR化學位移 O O Br S O O H l' l H H k H i H i' f H h H H g 20 (6.34) (5.90) (3.57) (2.75) (6.10) (3.37) (4.53) (4.58) 圖 26 化合物 20 1 HNMR化學位移
57 表 11 化合物 19 與 20 之13 CNMR化學位移 化合物 19/20 13 CNMR化學位移 H 的數目 模擬值a C1;Ca 20.901;21.492 3;3 24.3;24.3 C2;Cb 145.004;144.723 0;0 143.4;143.4 C3;Cc 130.037;129.598 1;1 130.1;130.1 C4;Cd 128.060;128.370 1;1 128.2;128.2 C5;Ce 135.327;135.031 0;0 135.9;135.9 C6;Cf 60.253;14.051 2;1 66.3;57.7 C7;Cg 27.721;30.987 2;2 24.9;26.2 C8;Ch 171.049;59.427 0;2 173.1;29.8 C9;Ci 64.209;63.405 2;2 66.3;61.7 C10;Cj 130.492;165.411 1;0 133.5;166.5 C11;Ck 127.847;120.383 1;1 128.2;128.3 C12;Cl 51.311;131.174 2;2 34.6;130.2 a 模擬值由軟體ChemDraw Ultra 8.0 模擬而成 表 12 化合物 19 之1 HNMR化學位移 化合物 19 1 HNMR coupling constants H1 3.88 dt, J = 1.6, 0.8 H2 3.76 dt, J = 1.6, 0.8 H3 4.40 dd, J = 5.2, 1.6 H4 5.63 m H5 5.67 m H6 4.10 q, J = 7.2
58 表 13 化合物 20 之1 HNMR化學位移 化合物 20 1 HNMR coupling constants Hf 3.37 m Hg 2.75 td, J = 7.6, 0.8 Hh 3.57 m Hi 4.58 dd, J = 6, 0.8 Hi' 4.53 dd, J = 5.6, 0.8 Hk 6.10 dd, J = 17.2, 10.4 Hl 5.90 dd, J = 17.2, 1.2 Hl' 6.34 dd, J = 10.4 , 1.6 從1 HNMR、13CNMR圖譜資料中(表 11、表 12、表 13),我們基本上能從 裡面分離出化合物 19 與化合物 20 的吸收峰訊號位置,可以確定的是對甲苯 磺醯基自由基在與化合物 12 加成時有各別加成在C1C2雙鍵上與C6C7雙鍵上 的兩種產物,利用1 HNMR判斷積分面積產物比為 2:3,對於此反應親電子性 自由基與化合物 12 的加成上,因為極性效應與立體效應的影響,不但大大的 減少了自由基加成時的位向選擇性,產率方面也沒有很好的結果,表示取代 基極性效應方面對於不同極性的自由基是有很大的影響。
59
第五章 結論
第三丁基自由基與對甲苯磺醯基自由基在特性上有很大的差異,本實驗 室利用(2-氯丙烯基) (2-氯甲基丙烯基)醚與第三丁基自由基進行自由基加成環 化反應,因極性效應成功的使親核性第三丁基自由基加成到氯取代基的雙鍵 位置上增加位向選擇性,但在另一組取代基為氯甲基的雙鍵,因SH2'同步式 反應的影響使的自由基更容易加成至此,自由基加成至各邊的產物比為 1:2。 嘗試以(2-氯丙烯基) (4-溴-2-丁烯基)醚與第三丁基自由基進行自由基加成環 化反應,把甲基氯取代基換成甲基溴取代基,把原本甲基氯取代基的雙鍵位 置的改變,改變取代基位置可以增加雙鍵上的立體效應,因而自由基加成位 向選擇性明顯上升,自由基加成至氯取代基上的雙鍵與溴甲基取代基上的雙 鍵的產物比提升至 4:1,可見極性效應與立體效應對於自由基的加成影響是 很大的,可以有效的影響自由基加成在至雙鍵時的位向選擇性。 使用化合物 7 與親電子性的對甲苯磺醯基自由基進行反應,有甲基取代 基在β−碳上,反應結果發現甲基取代基,可以很有效的誘導對甲苯磺醯基自 由基加成至取代基旁的C1C2雙鍵上,使加成位向選擇性提高,在C6C7雙鍵上 的基取代基在α−碳(C6)上,所以對自由基加成反應產生較高的立體效應,有 立體效應的影響使自由基不容易加成,當自由基加成後形成自由基中間體, 因甲基為推電子基,形成中間體時的三級自由基相對較穩定,再加成至C6C7 雙鍵,自由基進行SH2'同步式反應。可惜在立體結構方面,還沒辦法確實分 離出兩種立體異構物來鑑定出結構,無法辦定最後自由基加成時結構的順反 立體異構物。60 化合物 13 與對甲苯磺醯溴的自由基加成環化反應,自由基有各別加成至 C1C2與C5C6雙鍵上,加成至C1C2雙鍵時形成自由基中間體然後再進行抓氫反 應,而自由基加成至C5C6雙鍵上後,自由基進行SH2'同步式反應,溴自由基 離去後形成π鍵,溴自由基加成至π鍵形成自由基中間體然後再進行抓氫反應, 產物比為 2:3。本實驗室曾研究(3-丙烯醯基)-(4-溴-2-丁烯基)醚(化合物 13) 與第三丁基自由基進行加成環化反應,加成至C1C2與C5C6雙鍵上的產物比為 4:1,C1C2雙鍵旁的碳氧雙鍵拉電子基對於這個親核性自由基,有很大的誘 導效應,加成後的中間體也會因共振效應更加穩定,最後進行環化反應。 不同極性的自由基和同一種化合物與進行自由基SH2'加成環化反應,卻 有很大不同的結果,親核子性自由基第三丁基自由基加成至化合物 13 時,因 極性效應與共振效應的影響讓自由基可以有效的加成在C1C2雙鍵上,並且進 行自由基SH2'加成環化反應,親電子性自由基對甲苯磺醯基自由基與化合物 13 進行反應時,因極性效應與SH2'同步式反應的影響,使的自由基加成C1C2 與C5C6雙鍵上的產物比為 2:3,文獻中對甲苯磺醯溴照光後會產生對甲苯磺 醯基自由基與溴自由基,兩種自由基都會加成至雙鍵,在此反應中發現,對 甲苯磺醯基自由基在加成至C1C2雙鍵上後形成自由基中間體是進行抓氫反應, 而不是與溴自由基反應,這與文獻觀察到的現象有所不同。
61
第六章 實驗部分
6.1 藥品
6.1.1 試藥級溶劑: 正己烷 ( n-C6H14, Macron ) 四氫呋喃 ( (CH2)4O, Macron ) 乙醚 ( (C2H5)2O, UinRegion ) 甲苯 ( C7H8, J. T. Baker ) 二氯甲烷 ( CH2Cl2, Duksan ) 乙腈 ( CH3CN, Macron ) 6.1.2 試劑:Silical gel (Silicycle) Acrylic acid (Acros)
β-Methallyl Alcohol (TCI)
Trans-1,4-Dibromo-2-butene (Acros) Triethylamine (Sigma)
Sodium hydride (Alfa Aesar) N-Bromosuccinimide (Alfa Aesar) p-Toluenesulfinic acid (Alfa Aesar) Sodium (日本試藥)
62
6.2 儀器
6.2.1 氣相層析質譜儀 (GCMS)
氣相層析儀型號:Agilent Technologies 6890N Network GC System 質譜儀型號:Agilent Technologies 5975 inert Mass selective Detector
管柱:SUPELCO 的 EquityTM-5 管柱,長度 60m,內徑 0.25mm,coating 厚
度0.25μm
操作條件:載流氣體為高純度氦氣,壓力 21.57 psi,注射入口溫度為 280℃, 偵測器溫度為 280℃;條件:oven 100℃以每分鐘 10℃的速率上升至 280℃維 持 10 分鐘,總共所需時間為 28 分鐘。
6.2.2 氣相層析儀 (GC)
廠牌:PERKIN ELMER AutoSystem
管柱:J&W SCIENTIFIC 的 DB-5 管柱,長度 60m,內徑 0.25mm,coating 厚
度0.25μm
操作條件:載流氣體為高純度氮氣,壓力 18.0 psi,注射入口溫度為 220℃, 偵測器溫度為 270℃;oven 100℃以每分鐘 10℃的速率上升至 280℃維持 10 分鐘,總共所需時間為 28 分鐘。
63
6.2.3 光化學反應器 (PR-2000)
廠牌:PANCHUM PR-2000 盤面轉動系統 燈管總數:16 pcs
燈管控制:8 pcs lamps per switch 光頻率:60 Hz
6.2.4 核磁共振光譜儀 (NMR)
規格:Nuclear Magnetic Resonance 400 MHz Varian Unity plus&Nercury Plus
檢測學校:高雄醫學大學
測定項目:1
64
6.3 實驗步驟
6.3.1 對甲苯磺醯溴製備(TsBr)20
取兩個 1000mL的燒杯,各別將Sodium p-toluenesulfinate 8.9 g(50 mmol) 溶入 500mL水中與NBS 8.9 g(50 mmol)溶入 700mL水中,各別攪拌至溶解。溶 解後把兩液體緩慢混和在 2000mL的燒杯中後會產生白色粉末固體,攪拌 20 分鐘後使用抽氣過濾過濾產物,利用二氯甲烷萃取過後,利用減壓濃縮機將 二氯甲烷去除乾燥並保存在 4℃環境中,產率 60%。GCMS(EI) m/z(relative intensity):M+ 234(1),155(85),91(100),65(40)。1H NMR (400 MHz, CDCl3) δ 2.48(s, 3H), 7.38(d, J = 8.4 Hz, 2H), 7.87(d, J = 8.4, 2H). 13 C NMR (101 MHz, CDCl3) δ 21.82, 126.50, 130.08, 144.59, 146.71. 6.3.2 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚製備(化合物 7) 取一個雙口 250ml的圓底瓶,放入trans-1,4-dibromo-2-butene 10.6g(50 mmol)與sodium hydride 2 g(50 mmol)架回流管後進行氮封(此反應必須無水無 氧,在氮氣封存下所進行),再加入 200mL的正己烷,先加熱迴流 30 分鐘, 待冷卻至室溫時再用注射針筒吸取 2-methyl-2-propen-1-ol 4.23 ml(50 mmol)加 入(因反應劇烈加入時會產生放熱反應,需緩慢滴加,加入時間約為 15 分鐘), 並加熱迴流 3 小時。反應完降至室溫後,利用減壓濃縮機去除正己烷,加入 適量乙醚萃取,並水洗三次,使用無水硫酸鎂脫水後過濾,最後以氣相層析 質譜儀檢測產物。以管柱層析做最後的純化分離,所使用的沖提液為正己烷,
分離過後產率為 50%。GCMS(EI) m/z(relative intensity):[M-C4H7]+ 149(10),
65 3.86(d, J = 0.4 Hz, 2H), 3.94(m, 4H), 4.88(m, 1H), 4.94(m, 1H), 5.85(m, 1H), 5.95(dt, J = 15.2, 7.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 19.38, 31.95, 68.99, 74.15, 112.23, 128.34, 131.68, 141.86. 6.3.3 (2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯溴進行自由基 加成環化反應 取(2-甲基-2-丙烯基)-(4-溴-2-丁烯基)醚 0.102 g(0.5 mmol)與對甲苯磺醯 溴 0.117 g(0.5 mmol),溶於 5mL 的乙腈,待完全溶解後,取乾燥過後的 NMR 試管將其溶液加入其中,置於光化學反應器裡反應 3 小時。反應完後使用減 壓濃縮機去除乙腈,再加入適量乙酸乙酯萃取後,並水洗三次,使用無水硫 酸鎂脫水後過濾,最後以氣相層析質譜儀檢測產物。以管柱層析做最後的純 化分離,所使用的沖提液為正己烷與乙酸乙酯,比例為 3:1。。 6.3.4 (3-丙烯醯基)-(4-溴-2-丁烯基)醚與對甲苯磺醯溴進行自由基加成 環化反應 取(3-丙烯醯基)-(4-溴-2-丁烯基)醚 0.102g(0.5 mmol)與對甲苯磺醯溴 0.117 g(50 mmol),溶解在 20mL 的甲苯中攪拌至完全溶解,並將此溶液加入 乾燥過後的 NMR 試管,置於光化學反應器裡反應 8 小時。反應完後使用減 壓濃縮機去除甲苯,再加入適量乙酸乙酯萃取後,並水洗三次,使用無水硫 酸鎂脫水後過濾,最後以氣相層析質譜儀檢測產物。以管柱層析做最後的純 化分離,所使用的沖提液為正己烷與乙酸乙酯,比例為 3:1。