國立台東大學生命科學研究所 碩士論文
指導教授:胡焯淳 博士
毛細管電泳應用於藥品及農藥之分析
研 究 生: 蔡郁嫻 撰
- -
tt
1L it ~i\.~
~1!l.;~x..~~~ ~ - $~"
~ (iff Jiu . !t 4rff~hJt, PlT
~Ji
±
,1'!L ~~ ~D
t~±
~11r
~~ 5l:.~ x !f: 1l'l. ~ .~ ~ ~ i" : /ttJ
----"'----~---
$t~)
1b-.:>L
1ft;f~~
81:!!Jj: ,/15".if- -6 11 / ~
EJm
~:
1. ;J;..a
it .::.11] ~iR [l,,1ft ~ ~ -i- {Hi ' :i.! 3t ~ PIT ~ 0- &ttTAt m~: 'f!}~ -#'*
02. -*-.f<.4\ El1i.~ .;. _'fl " 1td A i l 5}~#:Nht~3!:fi}", ~1.t 0
~:arrf.j·~fJI~Pf[
*
tf-=J-f1fT~:ttZ~5[m_ _ _*ft .
·41~-AtE
~==..!-l.~-=--~-.:::=-: fiR
rJ '.£f!{:fi;.A..Jtij, !1IJ 1li1 'J!.t.
- , f/11l ~ ''I!$: ,m»~x.- ':~1iJ.~. '!f.IJJf'(Jlf c
----~~---_._---~~- --_._.-.__.--- ---....---~~-_.-
__ -+-_I_-_1f._~_/'_\ -: _+-I_if._·1i_0m_~ _
_ f . _ - - fMf:
, )
em
-JEt*),m&:'·lA~)
致謝辭
首先誠摯的感謝指導教授胡焯淳 博士及張煥宗 博士,兩位老師悉心的 教導使我得以一窺化學分析領域的深奧,不時的討論並指點我正確的方向,
使我在這兩年中獲益匪淺。感謝劉春櫻 教授審查並參與我的論文口試,老 師對學問的嚴謹更是我輩學習的典範。
本論文的完成另外亦得感謝馬偕紀念醫院台東分院的白明忠 醫師及台 東農改場的謝進來 博士大力協助,使得本研究的實驗條件可以應用於真實 樣品上,驗證其可行性。生科所助理 惠嵐對我的問題都能細心的一一答覆。
因為有你們的體諒及幫忙,使得本論文能夠更完整而嚴謹。
兩年裡的日子,實驗室裡共同的生活點滴,感謝各位學弟妹的共同砥 礪,你們的陪伴讓兩年的研究生活變得絢麗多彩。
感謝台大實驗室的學長姐、同學和學弟妹的照顧,柏齡學長指導我實驗 技巧,關心我什麼時候畢業,常常麻煩秋玲學姊帶我去吃好吃的東西。感恩 于鈊(學長/學弟)辛苦的管理實驗室的電腦,幫忙完成老師交辦的事情,且 總能在我電腦有問題時為我解決,也感謝玟伶、青芩的鼓勵及幫忙,恭喜我 們順利走過這兩年。一起討論人生大代誌的彩娥還有幫忙論文校稿及準備口 試用品的文璋和姿羽當然也不能忘記,從你們的身上,我獲益良多。還有很 多貴人,在生活上適時給我一個方向及幫助,多謝你們。
家人在背後的默默支持更是我前進的動力,沒有你們的體諒、包容,相 信這兩年的生活將是很不一樣的光景。也感謝奶奶千里迢迢從高雄送資料到 台東來,讓我能順利申請到獎學金,在生活上不虞匱乏。
最後,謹以此文獻給我摯愛的雙親。
毛細管電泳應用於藥品及農藥之分析
蔡郁嫻
國立台東大學生命科學研究所
摘要
本實驗以毛細管電泳連接螢光偵測器為分析方法,對藥品及農藥 進行分離及檢測。在藥品方面分離ofloxacin 異構物,使用 50 mM 的 磷酸緩衝溶液(pH 2.30)添加 60 mM 的 HP-β-CD 的條件下分離 ofloxacin,解析度大於 2,對 levofloxacin 最低之偵測極限為 10-8 M。
對levofloxacin 的標準品定量範圍可從 10-7 M~5×10-3 M,R2=0.9989。
對添加在尿液中的levofloxacin 定量線性範圍從 5×10-6 M 至 5×10-3 M 之間, R2=0.9943。應用在藥物的不純度判別上,一顆藥錠中約含有 93 毫克的 levofloxacin,R(+)-ofloxacin:levofloxacin 為 0.1:99.9。應 用在服用藥物病人的尿液檢測藥物含量上,可發現尿液中levofloxacin 含量約為7.9×10-4 M,且 levofloxacin:R(+)-ofloxacin 99.35:0.65。
此外,實驗中也提出在動相緩衝溶液中添加幾丁聚醣(chitosan)及 金屬離子的檢測方式,發現添加濃度為 5×10-7 (W/V)的幾丁聚醣及 5×10-8 M 的 Cu2+時,分離效果比單獨只有HP-β-CD 時略佳,將來可 朝向添加其他的金屬離子研究是否會有更佳的分離效果。
在農藥方面,開發以陽離子界面活性劑分離偵測五種農藥(加保 利、貝芬替、奈乙醯胺、奈乙酸和腐絕)的分析方法,最佳的分離條 件為15 mM 磷酸緩衝溶液(pH 6.47)添加 15 mM CTAB 和 10 % 正 丙醇,並以10 mM 和 5 mM HP-β-CD 分別添加在緩衝溶液和樣品中 作為螢光增強劑,使用-25 kV 進行電泳時,可在 13 分鐘內完成所有 的分離,偵測極限最低可達 0.06 ppm(加保利),最高為 0.191 ppm
(貝芬替)。應用在六種自然界的水質樣品上,無特殊前處理步驟,
可在無干擾的狀態下,偵測加保利、奈乙醯胺、腐絕及奈乙酸皆達到 10-6 M,貝芬替最低可偵測到 5×10-6 M,回收率從 93.5 %至 118.4 %。
應用在市面上的農藥(立倍利)偵測上,可成功的偵測及定量出加保 利,以甲醇為溶劑時所測得的加保利含量符合農藥上所標示的含量。
驗證此分析方法及條件可應用在水質及農藥成品上的偵測及定量。
關鍵詞: levofloxacin,毛細管電泳,螢光,農藥,陽離子界面活性劑
Analysis drug and pesticides with capillary electrophoresis
Tsai Yu-Hsien
Abstract
PART ⅠIn this study, we developed an analytical method for the enantioseperation of ofloxacin, using capillary electrophoresis with fluorescence detector. The optimum separation conditions were obtained with 60 mM hydroxylpropyl-β-cyclodextrin (HP-β-CD) dissolved in 50 mM phosphate buffer at pH 2.30. Under these conditions, the (+) and (-) forms could be completely separated and the detection limit of levofloxacin in distilled water was 1 × 10-8M. The linear ranges of levofloxacin were 1×10-7 to 5×10-3 M with R2=0.9989 and 5×10-6 to 5×10-3 M with R2=0.9943 in DI water and untreated urine, respectively.
We also applied this method to investigate the purity of the commercial drug. The results revealed that the ratio between R(+)-ofloxacin and S(-)-ofloxacin (levofloxacin) was 0.1: 99.9, and there is about 93mg levofloxacin per tablet (100mg). The concentration of levofloxacin in patient’s urine was founded to be 7.9×10-4M, and the ratio between the two optical isomers was 99.3: 0.7.
PART In this study, we developed an analytical method for five Ⅱ pesticides (CAR, NAD, TBZ, NAA and CAB) by using micellar electrokinetic capillary chromatography (MEKC). The optimum separation conditions for MEKC were 15 mM phosphate buffer (pH 6.47),
15 mM hexadecyltrimethylammonium bromide (CTAB) and excitation wavelength is 280 nm, emission wavelength is 336 nm by fluorescence detector. We also found that 10 mM HP-β-CD added in running buffer and 5 mM HP- -CD added in sample solution could increase fluorescence signal from 8 to 38 %. Under this condition, these pesticides were separated for less than 13 min and the detection limits in distilled water were 3×10-7 M(NAD, CAR, NAA),5×10-7 M (TBA) and 5×10-6 M (CAB) respectively. The linear ranges of five pesticides were 5×10-7 M to 1×10-4 M (NAD, CAR, NAA), 1×10-6 M to 1×10-4 M (TBZ) and 5×10-6 M to 7×10-5 M (CAB) with R2 all larger than 0.995. This method was applied to not only the analysis of the residual pesticides in nature water without pretreatment but also the analysis of commercial insecticide (carbaryl). The results revealed that the recovery ranged from 93.5 to 118.4 % for different nature water samples and the carbaryl contains in commercial insect ides was about 85.8 %.
Keywords: levofloxacin, capillary electrophoresis, fluorescence, pesticides, cationic surfactant.
總目錄
中文摘要 i
英文摘要 iii 表目錄 viii 圖目錄 ix 縮寫全文對照 xiii 第一部分 以毛細管電泳分析尿液中的 ofloxacin 第一章 緒論 一、前言 1
二、文獻回顧 2
三、毛細管電泳簡介 10
(一) 電淌度 10
(二) 電滲流 11
(三) 毛細管區帶電泳 13
(四) 對掌選擇劑 14
第二章 材料與方法 一、儀器 16
二、藥品與溶液 17
三、樣品配製 17
四、毛細管電泳實驗流程 18
第三章 結果與討論 一、pH 值對螢光的影響 19
二、HP-β-CD 濃度對螢光的影響 20
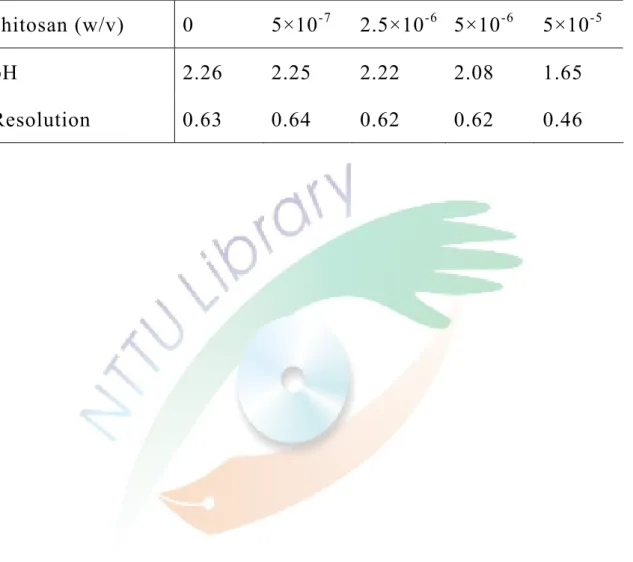
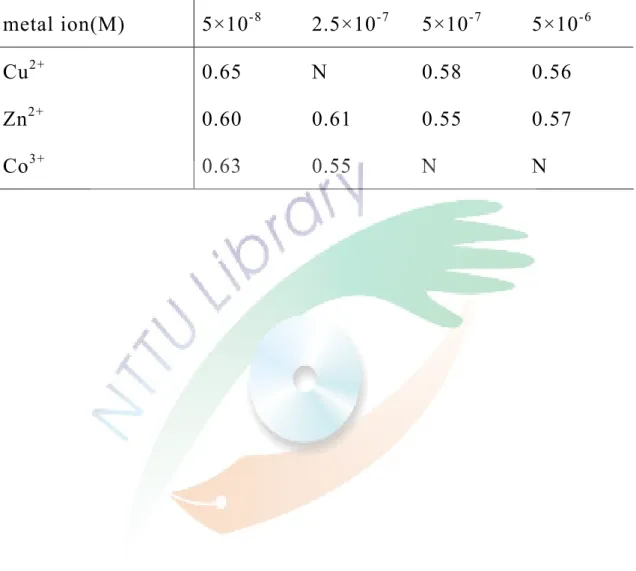
三、添加幾丁聚醣及金屬離子對分離異構物的影響 21
四、定量 23
五、應用 24
(一) 藥物不純度 24
(二) 病人尿液中藥品定量 25
第四章 結論 27
第五章 參考文獻 28
第二部分 以毛細管電泳分析疑似環境荷爾蒙之農藥 第一章 緒論 一、前言 44
(一) 加保利 45
(二) 貝芬替 48
(三) 腐絕 48
(四) 奈乙醯胺 52
(五) 奈乙酸 52
二、文獻回顧 53
三、微胞電動層析 65
(一) 微胞電動層析簡介 65
(二) 陽離子界面活性劑 65
第二章 材料與方法 一、儀器 68
二、藥品與溶液 69
三、實際樣品配製 70
四、毛細管電泳實驗流程 70
第三章 結果與討論 一、pH 值對螢光的影響 72
二、界面活性劑種類及濃度對分離度的影響 73
三、緩衝溶液的濃度對分離度的影響 75
四、有機溶劑的種類與濃度對螢光強度及分離的影響 76
五、電壓對分離的影響 77
六、添加HP-β-CD 對螢光增強的影響 77
七、定量 79
八、應用 80
(一)自然界水質的檢測 80
(二)商業農藥(立倍利)之偵測 81
第四章 結論 83
第五章 參考文獻 84
表目錄
第一部分 以毛細管電泳分析尿液中的 ofloxacin
Table 1. The analysis conditions collected from reference papers 6 Table 2. The pH value and the resolution of ofloxacin at different
concentrations of chitosan
31
Table 3. Effect of various metal ions at different concentrations on resolution of racemic ofloxacin
32
Table 4. Analytical parameters of the proposed method 33
第二部分 以毛細管電泳分析疑似環境荷爾蒙之農藥
Table 5. The analysis conditions collected from reference papers 60 Table 6. Optimum conditions for capillary electrophoresis experiment 88 Table 7. The linear range, LOD, LOQ and linear range of the proposed method 89 Table 8. Recovery values of pesticides in different water sample 90
圖目錄
第一部分 以毛細管電泳分析尿液中的 ofloxacin
Fig. 1. Structure of ofloxacin. 1
Fig. 2. Cravit that we used in this study is the pastille type. 2 Fig. 3. Development of the electroosmotic flow. 12 Fig. 4. Separation mechanism of capillary zone electrophoresis. 14 Fig. 5. (a) two-dimensional (b) three-dimensional structure of HP-β-CD 15
Fig. 6. Structure of chitosan 21
Fig. 7. Fluorescence intensity of ofloxacin at various pH value of the phosphate buffer.
34
Fig. 8. Enantioseparation of ofloxacin in various concentration of HP-β-CD and phosphate buffer(50 mM)at pH 2.30.
35
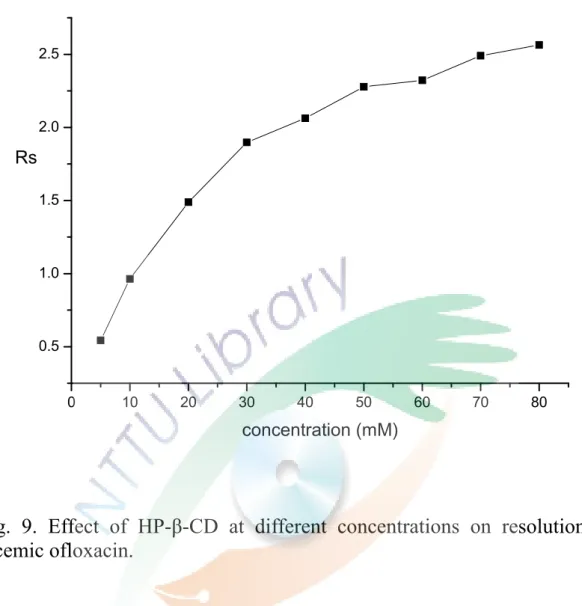
Fig. 9. Effect of HP-β-CD at different concentrations on resolution of racemic ofloxacin.
36
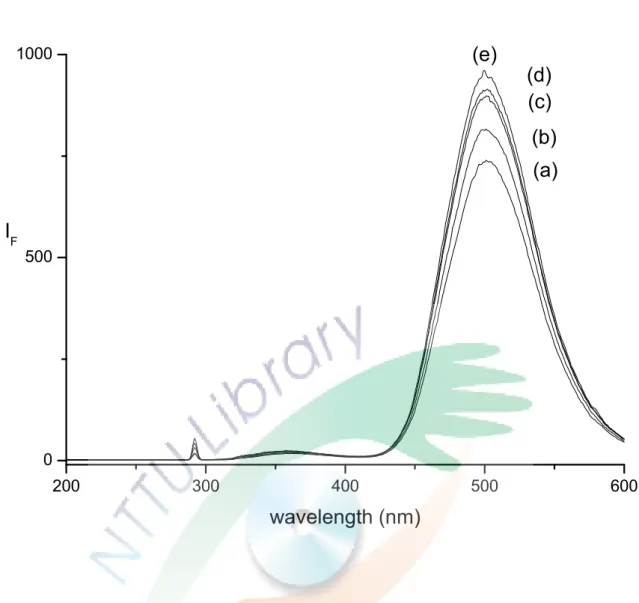
Fig. 10. Fluorescence intensity of ofloxacin at different concentration of HP-β-CD.
37
Fig. 11. Fluorescence intensity of ofloxacin at different concentration of HP-β-CD.
38
Fig. 12. Electropherograms of racemic ofloxacin in 50 mM phosphate with different concentration of chitosan.
39
Fig. 13. Electropherograms of racemic ofloaxacin 50 mM phosphate buffer with 40
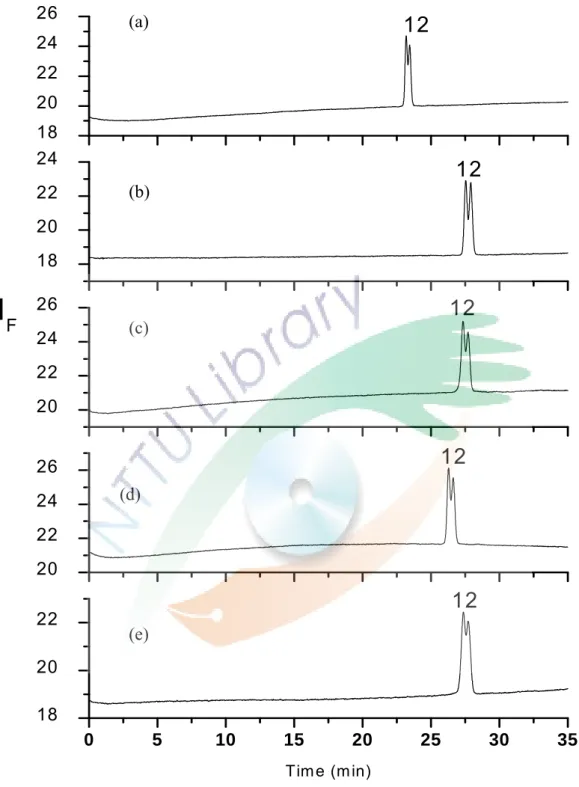
5 mM HP-β-CD , 5×10-7 (w/v) chitosan and various metal ions.
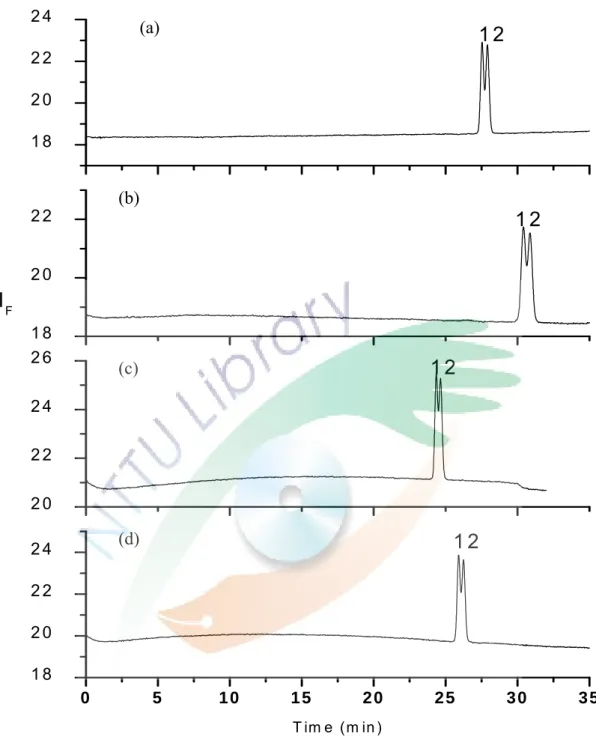
Fig. 14. Electropherograms of the commercial drug. 41 Fig. 15. Electrograms of (a) urine from a healthy person; (b) urine from a patient
diluted 10 times; (c) urine from a patient diluted 10 times and spiked with 5×10-5 M ofloxacin .
42
Fig. 16. The Standard addition method used to determinate the concentration of levofloxacin in patient’s urine.
43
第二部分 以毛細管電泳分析疑似環境荷爾蒙之農藥
Fig. 17. Structure of carbaryl. 45
Fig. 18. The metabolism pathway of carbaryl. 46
Fig. 19. Structure of carbendazim. 48
Fig. 20. Structure of thiabendazole. 49
Fig. 21. The metabolism pathway of thiabendazole. 50
Fig. 22. Structure of 1-naphthylacetamide 52
Fig. 23. Structure of 1-naphthylacetic acid 52
Fig. 24. The EOF of MEKC 66
Fig. 25. Structure of cationic surfactants 67
Fig. 26. Electropherograms of the four pesticides in 15 mM phosphate buffer with 15 mM TTAB and 10 % n-propanol at various pH value.
91
Fig. 27. The fluorescence intensity of pesticides in 15 mM phosphate buffer (pH 6.47) .
92
Fig. 28. Electropherograms of the four pesticides in 15 mM phosphate buffer 93
(pH 6.47) with different surfactant and 10 % n-propanol.
Fig. 29. Electropherograms of the four pesticides in 15 mM phosphate buffer (pH 6.47) with different concentrations of CTAB and 10 %
n-propanol.
94
Fig. 30. Electropherograms of the four pesticides in (a)5 mM; (b)15 mM; (c)20 mM; (d)30 mM; (e)45 mM phosphate buffer (pH 6.47) containing 15 mM CTAB and 10 % n-propanol.
95
Fig. 31. Electropherograms of the four pesticides in 15mM phosphate buffer (pH 6.47) with 15 mM CTAB and 10 % (a) methanol ; (b) ethanol ; (c) n-propanol ; (d) iso-propanol ; (e) ACN.
96
Fig. 32. Electropherograms of the four pesticides in 15 mM phosphate buffer (pH 6.47) with 15 mM CTAB and (a) 0 %; (b) 2 % ; (c) 6 % ; (d) 10
% ; (e)12 % n-propanol.
97
Fig. 33. Electropherograms of the four pesticides in 15 mM phosphate buffer (pH 6.47) with 15 mM CTAB and 10 % n-propanol.
98
Fig. 34. Electropherograms of the five pesticides in 15 mM phosphate buffer (pH 6.47) with 15 mM CTAB, 10 % n-propanol, and (a) no HP-β-CD in sample and buffer; (b) 5 mM HP-β-CD in sample and no HP-β-CD in buffer; (c) 10 mM HP-β-CD in buffer and no HP-β-CD in sample;
(d) 5 mM HP-β-CD in sample and 10 mM HP-β-CD in buffer.
99
Fig. 35. Electropherograms of the five pesticides in 15 mM phosphate buffer (pH 6.47) with 15 mM CTAB and 10 % n-propanol, addition (a) 0 mM; (b) 5 mM; (c) 10 mM; (d) 15 mM; (e) 20 mM HP-β-CD.
100
Fig. 36. Electropherograms of the five pesticides in 15 mM phosphate buffer 101
(pH 6.47) with 15 mM CTAB, 10 mM HP-β-CD and 10 % n-propanol, (a) 0 mM; (b)5 mM; (c)10 mM; (d) 15mM HP-β-CD in sample.
Fig. 37. Electropherograms of the (a) blank; (b) five pesticides spiked in tap water.
102
Fig. 38. Electropherograms of the (a) blank; (b) five pesticides spiked in water sample 1.
103
Fig. 39. Electropherograms of the (a) blank; (b) five pesticides spiked in water sample 2.
104
Fig. 40. Electropherograms of the (a) blank; (b) five pesticides spiked in water sample 3.
105
Fig. 41. Electropherograms of the (a) blank; (b) five pesticides spiked in water sample 4.
106
Fig. 42. Electropherograms of the (a) blank; (b) five pesticides spiked in water sample 5.
107
Fig. 43. The commercial sample of carbaryl. 108
Fig. 44. Electropherograms of the commercial smple dissolved in (a) distilled water (b) methanol.
109
縮寫全文對照
ACN acetonitrile
BAL bronchoalveolar lavage
CAB carbendazim CAR carbaryl CD cyclodextrin
CE capillary electrophoresis
CEKC capillary electrokinetic chromatography
CTAB hexadecyltrimethylammonium bromide
CZE capillary zone electrophoresis
DAD diode array detector
DNA deoxyribonucleic acid
DTAB dodecyltrimethylammonium bromide
ELISA enzyme-linked immunosorbent assays
EOF electroosmotic flow
ESI electronspray ionization
FI flow injection
FL fluorescence
GC gas chromatography
HP-β-CD hydroxylpropyl-β-cyclodextrin
HPLC high performance liquid chromatography HPTLC high performance thin layer chromatography
IT ion trap
IUPAC International Union of Pure and Applied Chemistry LD50 lethal dose, 50 percent kill
LLE liquid-liquid extraction
LOD limited of detection LOQ limited of quantification
MAE microwave-assisted extraction
MEKC micellar electrokinetic chromatography
MS mass spectroscopy
MS/MS tandem MS
NAA 1-naphthylacetic acid
NAD 1-naphthylacetamide
PAD photodiode array detector
Ref. reference SFE supercritical fluid extraction SDS sodium dodecyl sulfate
SPE solid phase extraction
TBAA tetrabutylammonium acetate
TBZ thiabendazole TEA triethylamine
TTAB trimethyl(tetradecyl)ammonium bromide UV ultraviolet
第一部份
偵測藥物
-以毛細管電泳分析尿液中的
ofloxacin
第一章 緒論
一、 前言
Ofloxacin 是 氟 化 奎 林 酮 類
( fluoroquinolones ) 的 一 種 , IUPAC(The International Union of Pure and Applied Chemistry)命名 為
9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7
H-pyrido[1,2,3-de]-1,4-benzoxazine-6-carboxylic acid hemihydrate,結構如Fig. 1,有光學異構物存在,”*”處為其
對掌光學中心。氟化奎林酮類是人工合成的抗生素,主要抗菌機 制是抑制細菌的DNA gyrase及type IV topoisomerase,使細菌無法 進行DNA的轉錄及轉譯,抗菌範圍包括革蘭氏陽性菌及陰性菌。
ofloxacin常用於治療呼吸道、尿道、皮膚及全身性感染。研究顯 示ofloxacin的左旋異構物levofloxacin具有較高的抗菌活性,是右 旋的8-128倍[1],目前已有成藥”可樂必妥”在市面上販賣,見Fig.
2。
Fig. 2. Cravit that we used in this study is the pastille type. The diameter is about 0.81cm and each tablet contains 100 mg levofloxacin.
ofloxacin經由口服能快速的被人體吸收,人體中只能代謝很 少量的ofloxacin,大約有87 %服入的藥物在48小時內會恢復為原 來的形式從尿液中排泄,少於4 %的藥物會在72小時內從糞便中 排出,只有少於5 %的藥物會代謝成desmethyl和 N-oxide代謝物 從尿液中排出[2]。
二、 文獻回顧
文 獻 中 偵 測 levofloxacin 以 高 效 能 液 相 層 析 方 法 ( high performance liquid chromatography, HPLC)居多。Wong 等人提 出以單一步驟液相萃取(liquid-liquid extraction, LLE)人類尿液 及血漿中的levofloxacin,再以Inertsil C18管柱採用逆相HPLC-UV 偵測及定量levofloxacin,線性範圍(linear range)分別為0.08~5.18 μg/mL(血漿),23~1464 μg/mL(尿液)。並可使用ligand-exchange
操 作 模 式 將 細 胞 液 中ofloxacin的 異 構 物 分 離[3]。Wright等 人 使 用 逆 相HPLC-UV 可 在 生 長 基 質 ( growth media ) 中 偵 測 到 包 含 ofloxacin 的 三 種 奎 林 酮 類 抗 生 素 , 線 性 範 圍 為 0.0625~20 μg/mL[4]。Böttcher實驗團隊則是利用逆相HPLC-FL分析偵測軟組 織、骨頭和血清中的levofloxacin,線性範圍為0.1~40 μg/mL,偵 測極限(limited of detection, LOD)為1 ng/mL(血清與膽汁)[5]。 Tsai 等 人 使 用 微 透 析 和 HPLC 偵 測 老 鼠 血 液 和 膽 汁 中 的 levofloxacin,線性範圍為0.1~5 μg/mL ,偵測極限為50 ng/mL[6]。 Stewart實驗團隊提出使用固相萃取(solid phase extraction, SPE)
及HPLC 的 方 法 分 離 人 類 血 漿 中 包 含 levofloxacin 的 氟 化 奎 林 酮 類,levofloxacin的線性範圍為 51.2~5069 ng/mL,偵測極限為25 ng/mL , 定 量 極 限 ( limited of quantification, LOQ ) 為 51.2 ng/mL[7]。Sowinski等使用HPLC連接UV和螢光偵測器分離血漿中 的包含levofloxacin的六種氟化奎林酮類,levofloxacin在血漿中的 線性範圍分別為50~10000 ng/mL(UV),20~5000 ng/mL(FL)[8]。 Mayer實 驗 室 提 出 以 逆 相 HPLC連 接 螢 光 偵 測 器 偵 測 血 漿 中 的 氟 化奎林酮類,並與微透析萃取方法比較。levofloxacin的線性範圍 分別為0.0156~5 μg/mL (微透析),0.02~12.5 μg/mL(血漿),偵測 極限分別為0.01 μg/mL (微透析)和0.0125 μg/mL(血漿)[9]。Breilh 等 則 提 出 偵 測 血 漿 、 脊 髓 液 和 骨 頭 組 織 中 的levofloxacin 的 HPLC-UV方 法 , 線 性 範 圍 分 別 為 0.25~25 μg/mL ( 血 漿 )、 1~6
μg/mL (脊髓液)和0.5~10 μg/g(骨頭組織),偵測極限分別為 0.050 μg/mL(血漿)、0.10 μg/mL(脊髓液)和0.20 μg/g(骨頭組 織)[10]。Greelet實驗團隊使用column-switching HPLC-螢光偵測 血 清 中 三 種 氟 化 奎 林 酮 類 ,levofloxacin 線 性 範 圍 為 125~4000 ng/mL , 偵 測 極 限 為 60 ng/mL[11]。 Santoro 實 驗 團 隊 則 使 用 HPLC-UV定 量 藥 錠 中 的 四 種 氟 化 奎 林 酮 類 , levofloxacin線 性 範 圍 為4.0~24.0 μg/mL, 偵 測 極 限 為 0.15 μg/mL, 定 量 極 限 為 0.46 μg/mL[12]。
Mochón提出另一種偵測藥錠、尿液及血清中levofloxacin的 螢光發光法,在pH 4的醋酸緩衝液時,可偵測到20~3000 ng/mL 的levofloxacin,在pH 5的醋酸緩衝溶液中添加 6 mM SDS可增強 螢光,偵測極限分別為5 μg/mL(血清)與420 μg/mL(尿液)[13]。 Du 實 驗 室 提 出 利 用 電 子 轉 移 形 成 錯 合 物 而 產 生 的 螢 光 偵 測 levofloxacin、ofloxacin等四種奎林酮類,線性範圍分別是0.02~1.0 μg/mL(ofloxacin)、0.04~1.2 μg/mL (levofloxacin),偵測極限分別 為0.006 μg/mL (ofloxacin)、0.012 μg/mL (levofloxacin),可應用在 藥錠、膠囊、尿液和血漿中[14]。El-Sherif等提出使用玻璃碳電極 以方波伏安法偵測levofloxacin,線性範圍為6×10-9 M~5×10-7 M,
偵 測 極 限 為10-9 M,可應用在稀釋後的病人尿液偵測上[2]。Radi 等人利用DNA與levofloxacin的反應,以玻璃碳電極使用循環伏安 法偵測,線性範圍為5×10-7 M~5×10-6 M,偵測極限為1×10-7 M,
應 用 在 尿 液 偵 測 中 , 偵 測 極 限 可 達25 μg/mL[15]。Sursh實驗團隊 提 出 利 用 高 效 能 薄 層 分 析 ( high-performance thin layer chromatography, HPTLC),線性範圍為0.8~3.0 μg/mL,偵測極限 為0.08 μg,定量極限為 0.3 μg[16]。
在毛細管電泳(capillary electrophoresis, CE)方面,有三篇 關 於 分 離ofloxacin 異 構 物 的 文 獻 。 Horimai[17]的 團 隊 提 出 使 用 γ-CD-Zn(II)-D-phenyl -alanine 錯 合 物 分 離 數 種 氟 化 奎 林 酮 類 藥 物 的 異 構 物 , 利 用Zn(Ⅱ) 和D-phenyl 與 氟 化 奎 林 酮 類 作 用 產 生 ligand-exchange , γ-CD 與 氟 化 奎 林 酮 類 產 生 host-guest interaction,使其異構物分離。Boer[1]的實驗團隊則是採用毛細管 電動層析(capillary electrokinetic chromatography, CEKC)系統 連接UV偵測器在50 mM phosphate buffer(pH 2.5)中添加了0.35 mM sulfated β-cyclodextrin,可在五分鐘內分離ofloxacin異構物,
在 未 經 前 處 理 的 尿 液 中 對R-(+)-ofloxacin 最 低 偵 測 極 限 為 2 μg/mL ( 5.5×10-6 M)。 Ouyang和 Baeyens[18]的 團 隊 提 出 毛 細 管 電 泳 連 接UV 偵 測 器 使 用 70 mM phosphate buffer 添 加 40 mM HP-β-CD 使 用 不 同 的 pH 值 分 離 lomefloxacin 、 gatifloxacin 、 pazufloxacin 及 ofloxacin , 對 所 有 的 異 構 物 線 性 範 圍 都 為 7~105 μg/mL,最低偵測極限皆為7 μg/mL(約1.94×10-5 M),以上文獻 實驗條件及結果整理在Table 1。
Table 1. The analysis conditions collected from reference papers
1. High performance liquid chromatography (HPLC)
Sample Matrix Preparat-
ion
Condition Detector Linear range LOD Ref.
ofloxacin plasma, urine
Single- step LLE
Ligand-exchange mode
Mobile phase: 5 mM copper (Ⅱ ) sulfate pentahydrate
containing L-isoleucine (10 mM)/methanol (87.5:12.5,v/v) Flow rate: 1.0 mL/min
UV (330 nm)
plasma:
0.08~5.18 µg/mL urine:
6.36×10-5 M ~4×10-3 M
- [3]
Ciprofloxacin, ofloxacin, sparfloxacin
Muelle- Hinton broth
- Mobile phase: pH 3.0, phosphate buffer with SDS and
TEA –ACN (65 %: 35 %) Flow rate: 1.75 mL/min
UV (280 nm)
0.0625~20 μg/mL - [4]
levofloxacin serum, soft tissue, bone, bile
- Mobile phase: pH 3.0, phosphate buffer/methanol/triethylamine
(750:250:4, v/v/v)
Flow rate: 1.5 mL/min
FL (295 nm/
490 nm)
0.1~40 μg/mL 1 ng/mL( serum and bile)
[5]
levofloxacin serum, - Mobile phase: ACN/1 mM FL 0.1~5 μg/mL 50 ng/mL [6]
bile 1-octanesulfonic acid( 40:60)
Flow rate: 1 mL/min
(292 nm/
494 nm) zidovudine,
levofloxacin
plasma SPE Mobile phase: 25 mM phosphate buffer, 0.1% trifluoroacetic acid (pH 2.4)/ACN (86:14,v/v) Flow rate: 1.5 mL/min
UV (266 nm)
51.2~5069 ng/mL 25 ng/mL [7]
levofloxacin, ciprofloxacin, gatifloxacin, moxifloxacin, trovafloxacin, cinoxacin
plasma - Mobile phase: 10 mM SDS, 10 mM TBAA, 25 mM citric acid and 43% ACN at pH 3.5.
Flow rate: 1 mL/min
UV (293 nm) FL (280 nm/
450 nm)
UV: 50~10000 ng/mL FL: 20~10000 ng/mL
- [8]
levofloxacin, ciprofloxacin
plasma - Mobile phase: 0.11%
orthophosphoric acid and 1.5%
ACN (pH 3.0) Flow rate: 0.4 mL/min
FL (310 nm/
467 nm)
microdialysates:
0.0156 ~5 μg/mL plasma:
0.02~12.5 μg/mL
microdialysates:
0.01 μg/mL plasma:
0.0125 μg/mL
[9]
levofloxacin plasma, bronchoal- veolar lavage,
liquid- solid extraction
Mobile phase: 0.4 %
triethylamine (pH 3.0) /ACN (83:17, v/v).
Flow rate: 1.2 mL/min
UV (299 nm)
plasma:
0.25~25 μg/mL BAL:
1~6 μg/mL
plasma:
0.050 μg/mL BAL:
0.10 μg/mL
[10]
bone tissues
bone tissues:
0.5~10 μg/g
bone tissues:
0.20 μg/g levofloxacin,
gatifloxacin, moxifloxacin
serum column- switching
Mobile phase: 2 mM
tetrabutylammonium bromide, 10 mM phosphate buffer (pH 2.5), 12 % ACN
Flow rate: 1 mL/min
FL (296 nm/
504 nm)
125~4000 ng/mL 60 ng/mL [11]
gatifloxacin, levofloxacin, lomefloxacin, pefloxacin
tablets - Mobile phase: water/ ACN (80:20, v/v) with 0.3 % of triethylamine (pH 3.3) Flow rate: 1 mL/min
UV (279 to 295 nm)
4~24 μg/mL LOD:
0.15 μg/mL LOQ:
0.46 μg/mL
[12]
2. Capillary electrophoresis(CE)
Sample Matrix Condition Detector Linear range LOD Ref.
quinolone drug - 10 mM γ-CD, 10 mM ZnSO4, 10 mM
D-phenylalanine, 10 mM ammonium acetate (pH 6.5)
UV (295 nm, 300 nm)
- - [17]
ofloxacin urine 50 mM phosphate buffer (pH 2.5), 0.35 mM sulfated β-cyclodextrin
UV (291 nm)
2~12 μg/mL 2 μg/mL [1]
lomefloxacin, gatifloxacin, urine 70 mM phosphate buffer (pH 2.16), 40 UV 7~105 μg/mL 7 μg/mL [18]
pazufloxacin, ofloxacin mM HP-β-cyclodextrin (254 nm)
3. Other methods
Sample Matrix Condition Linear range LOD Ref.
ofloxacin, levofloxacin, lomefloxacin, pipemidic acid
tablet, plasma, urine
charge transfer complexes formation 7,7,8,8-tetracynaoquinodimethane
ofloxacin:
0.02~1.0 μg/mL levofloxacin:
0.04~1.2 μg/mL
ofloxacin:
0.006 μg/mL Levofloxacin:
0.012 μg/mL
[14]
levofloxacin patient’s urine
cyclic and square-wave voltammetry at glassy carbon electrode
6×10-9 M~5×10-7 M 10-9 M [2]
DNA and levofloxacin
urine cyclic voltammetry at glassy carbon electrode 5×10-7 M~5×10-6 M 1×10-7 M urine:
25 μg/mL
[15]
levofloxacin - high-performance thin layer chromatography Mobile phase: water/methanol/n-butanol/ammonia
solution (25 %) (5:5:5:0.4, v/v)
0.8~3.0 μg/mL 0.08 μg LOQ:
0.3 μg
[16]
三、毛細管電泳簡介
電泳的定義是帶電離子受到電場作用而產生的不同速率運動,最 早是用來分離蛋白質等大分子,主要的分離機制是分子質量的差異。
使用高電壓進行電泳會產生熱擴散和對流的問題,所以需要在膠體
(gel)這類的物質中進行實驗,解決對流的問題,也不能使用過高的 電壓,避免焦耳熱過大,但因此導致分離時間長,效率低。直到1980 年,使用內徑為75 μm 的毛細管電泳實驗首次被提出,毛細管因為大 表面積/體積比及細小的管柱內徑克服了熱擴散和對流的問題,也因此 可以使用更高的電壓,使得分離速率變快、分離效率更高,且可在毛 細管中使用水相進行電泳實驗,提供了多樣化的操作模式,可以分離 更多種類的樣品,而不再只是侷限在大分子或帶電物質的分離上,開 啟了毛細管電泳的時代。以下簡單介紹毛細管電泳分離的理論。
(一)電淌度
電泳是以帶電物質在電場中的不同速率作為分離基礎,帶 電離子的遷移速率可以下式來表示:
v=μeE (1)
v=離子遷移速度,μe=電泳淌度,E=電場強度。μe為該離子的 物理常數,從標準表中可以査到物質的電泳淌度,這是假設物質 在帶最大電荷時所得之常數,但在不同 pH 值的緩衝溶液中,還 是會有不同的淌度,稱為有效淌度。電泳淌度會與離子在毛細管
內受到的電場力 F 成正比和摩擦力 f 成反比,電場力又可寫成:
F =qE (2) q 為帶電量,摩擦力 f 也可以下式計算:
f =-6πηrv (3) η 為溶液黏度,r 為離子半徑,v 為速度。電泳的過程中是電場 力與摩擦力兩個力互相平衡,此時兩力的方向相反。
qE=-6πηrv
rv 6 - πη
v= qE (4) 將(4)帶入(1),可得:
rv 6 - πη μe = q
由上式可知,帶電量大的離子,電泳淌度較大,帶電量小的 離子,電泳淌度較小。
(二)電滲流(electroosmotic flow, EOF)
除了物質自己本身的電泳淌度之外,管柱內的溶液受到電場 作用而形成的電滲流,也是另一個分離的重要因素。毛細管壁 是由矽醇基所組成,使用毛細管進行實驗前會先以NaOH 活化 管壁,使管壁帶負電,如下Fig. 3(a),注入緩衝溶液後,會吸 引溶液中的正離子聚集在管壁,形成緻密層。電荷吸引力與距 離的平方成反比,所以離管壁越遠吸引力越弱,會有一層較為 稀疏的正離子組成的擴散層,此兩層稱為電雙層,如下Fig. 3
O‐ O‐ O‐ O- O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐
‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O
O‐ O‐ O‐ O- O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐
‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O
++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++
++ ++ +
++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++
++ ++ +
-
+
++ + ++ + +- ++ + ++ + ++ +- ++ + ++ -+ ++
++ +-
+ + + + + + + - + + + + + + + + + - + + + + + - + + +
+ + + -
- -
-
-
-
- +
+
+
+
+
+
+ +
+
O‐ O‐ O‐ O- O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐
‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O
++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++
++ ++ +
++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++
++ ++ +
(b)。因為電雙層與靠近的溶液之間的電荷差異形成一個電位 差,稱為zeta 電位,施加電壓後組成擴散層的離子會由正極往 負極移動,帶動所有的體相溶液,形成電滲流,如Fig. 3(c)。
Fig. 3. Development of the electroosmotic flow:
(a) negatively charged fused silica surfance.
(b) hydrated cations accumulating near surfance.
(c) bulk flow towards the cathode upon application of electric field.
EOF (a)
(b)
(c)
-
+
電滲流可以用下面這個式子表示:
E vEOF
η
= εξ 或
η μEOF = εξ
vEOF為速度,μEOF為電滲淌度,ξ 為 zeta 電位,ε 為介電常數。
電滲流會因為溶液的pH 值而影響其大小。
一般由壓力進行分離的分析技術,靠近管壁的液體會有較大 的摩擦力,導致管柱內的流速產生差異,形成層流,這就會使得 樣品的波峰變寬。而電滲流因為是由靠近管壁的液體帶動流動,
就不會產生流速的差異導致樣品峰的擴散。
電滲流的另一個優點是可以使所有的離子往同一方向移 動,不論其所帶的電荷為何。在帶負電荷的毛細管壁狀態下,電 滲流是由正極往負極移動,帶正電荷的離子因與電滲流同方向,
其電泳速度會是電泳淌度加上電滲流,所以會最快。不帶電的物 質會隨著電滲流出來,帶負電的離子會因為與電滲流反向,所以 最慢被流出。
(三) 毛細管區帶電泳(capillary zone electrophoresis, CZE)
本研究使用的操作模式為毛細管區帶電泳,只在管柱中注入 緩衝溶液,讓樣品因為其帶電量及質量的不同而形成不同的區塊 分離,而中性離子就隨著電滲流被分離出來。見Fig. 4 [19]。
+
++
Fig. 4. Separation mechanism of capillary zone electrophoresis.
(四) 對掌選擇劑(chiral selectors)
對於電泳遷移速率相近之對掌異構物(enantiomers)毛細管 區帶電泳無法有效的將其分離,常須藉由添加對掌選擇劑於緩衝 溶液中分離對掌異構物。在1997年就有毛細管電泳中應用環糊精
(cyclodextrin, CD)分離異構物的文獻回顧,可知環糊精是最常 使用且有效的對掌選擇劑。環糊精的結構為D form的葡萄糖分子 用C-1, C-4 鍵結而成的環狀聚合物,可因鍵結的分子數目不同而 分為α、β、γ三種,性質也有所差異,比較如下[20]。
α-CD β-CD γ-CD
Number of glucose unit 6 7 8
Molecular mass 972 1135 1297
Internal cavity diameter (nm) 0.47~0.52 0.60~0.64 0.75~0.83 External cavity diameter (nm) 1.46±0.05 1.54±0.04 1.75±0.04 Cravity height (nm) 0.79~0.80 0.79~0.80 0.79~0.80 Solubility in water at 25 ℃
(g/100 mL) 14.5 1.85 23.20
+ -
O‐ O‐ O‐ O- O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐ O‐
‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O ‐O
++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++
++ ++ +
++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++ ++
++ ++ +
+
++
- +
-
-
-
-
-
-
- EOF
O
HO O HO
O OH
O
HO
HO O OH
O HO
O HO
O
OH O
OH O
OH O
O HO
O
OH
O OH O O OH O OH HO
O
OH O
OH O HO
O HO
因應不同的異構物可以使用不同類型的環糊精。此外,環糊 精還可以經由衍生化形成許多陽性、陰性和中性的衍生物,不但 解決了環糊精對水溶解度不佳的問題,並且可混合使用於電泳中 分離異構物,這就是環糊精可以廣泛的應用在對掌異構藥物選擇
上的原因[21,22]。
本 次 研 究 中 所 採 用 的 是 β-CD 的 衍 生 物 , hydroxylpropyl-β-cyclodextrin(HP-β-CD),比 β-CD 有較好的水 溶性。分子結構式如Fig. 5 (a),立體結構如 Fig. 5 (b),為內部疏 水,外部親水的結構,可以幫助難溶於水的物質溶解。一級氫是 位在小的開口,二級氫是位在大的開口。分離原理主要是以環糊 精本身的空腔孔洞為主體(host),分析物為客體(guest),利 用環糊精與異構物於空腔孔洞中形成主-客錯合物(host-guest complexes)或內包錯合物(inclusion complexes)的程度差異,進 而影響內包錯合物的電泳遷移速率,達到分離對掌異構物的效果。
Fig. 5. (a) two-dimensional (b) three-dimensional structure of HP-β-CD (a)
(b)
第二章 材料與方法
毛細管電泳因為具備了操作簡單、分離快速、低消耗量而在 近幾年被廣泛使用。螢光偵測器具有高靈敏度、低干擾和非破壞 性的優點。所以本實驗採用毛細管電泳連結螢光偵測器發展分離 ofloxacin 的分析方法。在緩衝溶液中添加 HP-β-CD 作為對掌選 擇劑,測試不同 pH 值、濃度的緩衝溶液、不同濃度的 HP-β-CD 對 分 離 的 影 響 , 並 嘗 試 添 加 低 黏 度 的 幾 丁 聚 醣 (chitosan) 和 金 屬離子於緩衝溶液中,測試其對分離效果的影響,最後使用最佳 化的實驗條件對 levofloxacin 作定量的線性範圍及應用在測試藥 物的不純度與服藥病人尿液中 levofloxacin 的含量。以期對監控 病人的服藥量、藥物的代謝路徑提供一種便利、快速的分析方法。
一、 儀器:
測試樣品激發與放射波長的螢光儀為 Hitachi F-4500,毛細 管電泳儀器連接的螢光偵測器型號為 Jasco FP-1520,設定激發波 長(excitation) 292 nm,放射波長(emission) 493 nm。 電泳 實 驗 是 以 商 業 化 的 儀 器 進 行 , 型 號 為 Model prince 4-tray CE System 使用 WPrince 32 bits 軟體從電腦控制,SISC 32 2.0 中文 版 軟 體 系 統 擷 取 數 據 , 計 算 面 積 及 分 離 度 。pH 計 使 用 Horiba
pH/ion meter D-23。所有的水溶液皆以 Barnstead 製造的純水配 製,電阻值皆大於 18 MΩ/cm。所使用的空毛細管內徑為 75 μm,
外徑 375 μm,總長度為 80 公分,有效長度為 54 公分。
二、 藥品及溶液
動相緩衝溶液(running buffer)的配製是混合不同比例的磷 酸、磷酸二氫鈉及磷酸氫二鈉,並測定其 pH 值。hydroxypropyl β-cyclodextrin (Aldrich) 母液(stock solution)的濃度為 200 mM,
溶在純水中。levofloxacin (Fluka)、ofloxacin (Sigma)都溶在甲醇 中,母液濃度分別為 10-2 M 及 10-3 M。存放在冰箱中,可保存約 二個星期,實驗時以純水稀釋至需要的濃度。低黏度的幾丁聚醣
(low-viscosity chitosan, Fluka)在酸性的條件下溶解度較好,以 重量百分比為 0.01 % 溶解在 0.05 M HCl,CuCl2 (Aldrich) 、 ZnCl2 (Riedel-de Haën)及 CoCl3 (J.T.Baker)的母液為 10-3 M 溶在水 溶液中,以上所使用的溶劑皆為 LC 級。
三、 樣品配製
目前販售的 levofloxacin 成藥分為藥錠及藥劑,本次實驗所 使用的是藥錠,外觀看起來為白色、圓凸形,上面有紅色的記號 D 723,一粒藥錠重約 200 毫克,包裝上標明內含 100 毫克的
levofloxacin。藥錠樣品配製依以下步驟進行:將藥錠秤重紀錄,
以研缽磨碎後再次紀錄取用的重量,用 10 mL 的甲醇溶解,以超 音波器震盪 15 分鐘,以 6000 rpm 離心後取上層澄清液,將不溶 物去除,再以 0.22 µm 的尼龍過濾膜過濾後,稀釋到 25 mL,置 於冰箱作為母液,實驗時依需要以純水稀釋成不同濃度使用。
依患者感染的部位不同,levofloxacin 的投藥量也不盡相同,
服藥後 1 至 2 小時內會達到血中最高濃度,以原來的形式從尿液 中 排 出 , 故 可 以 在 服 藥 的 病 人 尿 液 中 直 接 檢 測 到 levofloxacin。
此次分析的尿液樣品來源是每天服藥量約 400 毫克,連續服藥四 天的病人,收集第四天服藥後 3~4 小時的尿液,保存於冰箱中,
實驗時依需要的濃度以純水稀釋偵測。
四、 毛細管電泳實驗流程
新的空毛細管先以 0.5 N NaOH 充滿,隔夜後以純水沖洗 10 分鐘後,再以緩衝溶液沖洗 15 分鐘,就可開始使用。樣品以 100 psi 施壓 0.1 分鐘注入毛細管中,以 15 kV 進行電泳,電泳完後 用緩衝溶液以 1000 psi 推出剩餘物質,接著以緩衝溶液沖洗三分 鐘,便可開始下一次的實驗。實驗結束後,以 0.5 N NaOH 沖洗 15 分鐘,再以純水沖洗 10 分鐘即可關機。
第三章 結果與討論
一、 pH 值對螢光的影響
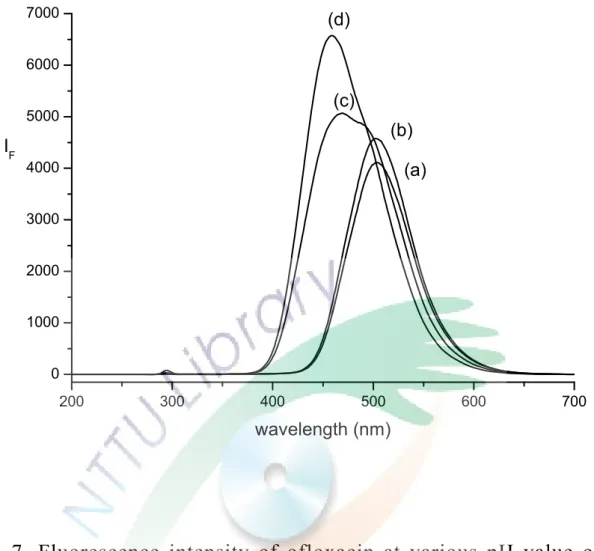
在 文 獻[1,18]中 大 都 以 酸 性 的 條 件 作 為 最 佳 的 分 離 酸 鹼 度 , 在 本實驗中先混合不同比例的磷酸、磷酸二氫鈉及磷酸氫二鈉,測 試不同的 pH 值對 ofloxacin 的螢光強度及分離的影響。如 Fig. 7,
使用波長 292 nm 激發,在高 pH 值時,ofloxacin 的最大放射波 長約位於 450 nm 左右,隨著 pH 值降低,放射波長產生了紅位移
(red shift),最大放射波長移到了約 507 nm,但螢光強度變得較 弱,因為 ofloxacin 的 pKa 為 6.0 和 8.0,Fig. 7 (c)的 pH 為 6.47 恰好介於這兩個 pH 值之間,導致(c)的圖形為兩個最大吸收波長 的波形重疊。本實驗是為了分離 ofloxacin 異構物,故希望能使 EOF 值較小,讓樣品有足夠的時間與 HP-β-CD 反應。pH 值越高,
分離時間越快,停留在管柱的時間就越短,也使得 ofloxacin 與 對掌選擇劑反應的時間不夠長,導致分離效果變差,所以為了降 低 EOF,延長停留在管柱裡的時間,故選擇酸性的 pH 值,為了 使緩衝溶液有較好的緩衝性,故本實驗採用以 1:1 的磷酸和磷 酸二氫鈉混合而成的緩衝溶液,緩衝溶液最佳化的條件為 50 mM 的磷酸緩衝溶液(pH 2.30),此時的 EOF 值幾近於 0,螢光偵測
器的激發波長設定為 292 nm,放射波長設定為 493 nm 作為最佳 實驗條件。
二、 HP-β-CD 濃度對分離度的影響
對掌異構物的差異只在光學活性的不同,其分子量和帶電量 都相同,使用單純的 CZE 無法分離,故需添加對掌選擇劑,本 研究選擇 HP-β-CD 作為對掌選擇劑,在此討論 HP-β-CD 的濃度 對分離度以及對 ofloxacin 螢光強度的影響,在 50 mM 磷酸緩衝 溶液(pH 2.30)中添加不同濃度的 HP-β-CD 的實驗結果為 Fig. 8,
由圖中可知添加的 HP-β-CD 濃度越高,分離效果越好,但遷移時 間也會隨之增長,HP-β-CD 濃度為 40 mM 時,分離度就可以達 到 2.0,濃度對分離度的結果呈現於 Fig. 9。且由 Fig. 8 發現添加 不同濃度的 HP-β-CD 後,ofloxacin 的峰高及面積有變大的趨勢。
為了驗證 HP-β-CD 是否可以增強 ofloxacin 的螢光強度,本 實驗固定 ofloxacin 的濃度添加不同濃度的 HP-β-CD 測試其螢光 強度的變化及增強的百分率,結果呈現於 Fig. 10。發現 HP-β-CD 的濃度從 0 mM 增加到 40 mM 時,螢光強度可以增加約 20 %,
但到了 40 mM 以後,螢光增強的幅度就漸趨緩慢,見 Fig. 11,
HP-β-CD 濃度為 60 mM 和 80 mM 時,螢光分別增強 22 %和 27
%,證實了添加 HP-β-CD 在緩衝溶液中不但可以作為對掌選擇劑 分離 ofloxacin,也可作為螢光增強劑,文獻[23]中也有以 HP-β-CD
增強其他樣品的探討,但對使用毛細管電泳分離 ofloxacin 這方 面 尚 未 看 到 類 似 的 文 獻 。 推 測 可 能 是 因 為 ofloxacin 進 入 了 HP-β-CD 後 , 形 成 了 一 個 較 為 剛 性 的 結 構 , 導 致 其 螢 光 強 度 增 加。但過了 40 mM 後,HP-β-CD 和 ofloxacin 的結合呈現飽和狀 態,導致螢光強度增強的幅度變緩。雖然高濃度的 HP-β-CD 可以 提供高分離度及較強的螢光強度,但在高濃度 HP-β-CD 下 S/N 會降低,而使偵測極限提高,可能是因為高濃度的 HP-β-CD 對螢 光 強 度 會 有 些 微 干 擾 , 影 響 基 線 (baseline) 的 穩 定 度 。 但 本 次 研 究 中 的 藥 錠 樣 品 需 要 在 高 濃 度 的 左 旋 異 構 物 中 看 到 右 旋 異 構 物,需要高分離度才可以作出區別。考慮以上幾點因素,選擇 60 mM HP-β-CD 作為最適合的實驗條件,可以提供高分離度並增強 27 %的螢光。使用 50 mM 磷酸緩衝溶液(pH 2.30)添加 60 mM HP-β-CD 的最佳條件對溶解在純水中的 levofloxacin 最低偵測極 限可達 10-8 M。
三、 添加幾丁聚醣及金屬離子對分離異構 物的影響
幾 丁 聚 醣 與 HP-β-CD 都 為 葡萄糖分子聚合而成,但幾丁聚 醣的 C-2 置換成一個胺基,結構
O HO
OH
NH2 O
O HO
OH
NH2 O
HO OH
NH2 O
OH
n<15
式如 Fig. 6,本身也如 HP-β-CD 一樣是個多分子聚合物,但為直 線型聚合。因為幾丁聚醣與 HP-β-CD 在單體結構上相似,故想測 試直線型幾丁聚醣對於對掌異構物是否同樣具有分離的效果,以 此開發新的對掌選擇劑。故嘗試將幾丁聚醣添加在緩衝溶液中,
測試對分離 ofloxacin 是否有與 HP-β-CD 相同的結果。因為幾丁 聚醣需在酸性的條件下才有較好的溶解度,添加大量的幾丁聚醣 會使得 pH 值變化過大,所以在 50 mM 緩衝溶液(pH 2.30)中 固定 5 mM HP-β-CD 濃度,添加少量的幾丁聚醣,測試對分離度 的影響。電泳結果呈現於 Fig. 12。可以發現添加了幾丁聚醣後,
遷移時間變長,但是沒有一個很好的規律性,這也可能是因為實 驗誤差或因為幾丁聚醣添加在緩衝溶液後改變了 pH 值,由圖中 可發現分離度皆無較佳的改變,反而添加的濃度越大,分離度越 差。各分離度及 pH 變化整理在 Table 2。
可能是因為其為線形聚合物,無法與 ofloxacin 形成完整的 錯合物,因此想利用加入金屬離子與幾丁聚醣結合使其變得較為 剛性而有利於與 ofloxacin 結合。所以,固定 5 mM 的 HP-β-CD 及幾丁聚醣為 5×10-7 (w/v)測試添加不同濃度與不同的金屬離子 於緩衝溶液中,測試的結果見 Fig. 13,分離度整理在 Table 3。
除了 Cu2+以外,添加 Zn2+及 Co3+對分離度沒有正面的幫助,反而 使得分離度變差。添加微量的 Cu2+對分離有正面的幫助,但改變 的幅度也不大。因為幾丁聚醣與金屬離子結合的最佳 pH 值為 6,
推測可能是因為 pH 值導致幾丁聚醣未與金屬離子發生錯合反應 或是錯合反應未完全,因此無法作為有效的光學選擇劑。也可能 是 ofloxacin 對幾丁聚醣與金屬離子形成的錯合物不會結合,導 致對分離度沒有大幅度的改善。由此實驗結果可知,幾丁聚醣對 分離如 ofloxacin 之類的異構物沒有良好的助益。
四、 定量
由以上的實驗結果得知最佳的實驗條件為 50 mM 磷酸緩衝 溶 液 (pH 2.30 ) 添 加 60 mM HP-β-CD 。 以 此 實 驗 條 件 對 levofloxacin 定量。對溶在純水中的 levofloxacin 標準樣品,線性 範圍可從 1×10-7 M~5×10-3 M,R2=0.9989。levofloxacin 添加在尿 液中的定量範圍可從 5×10-6 M ~ 5×10-3 M,R2=0.9943。最低偵測 極限在 S/N 比大於 3 的狀態下分別達到 1×10-8 M 和 1×10-7 M,
最低定量極限(在線性定量範圍的最低濃度)分別為 1×10-7 M 和 5×10-6 M。總整理見 Table 4。
相較於Ouyang 和 Baeyens[18]的團隊提出毛細管電泳連接UV 偵測器的分析方法,使用 70 mM phosphate buffer(pH 2.16)添 加 40 mM HP-β-CD 為實驗條件分離 ofloxacin 及其他有對掌異構 物的藥物,對 ofloxacin 的線性範圍為 7~105 μg/mL(1.94×10-5 M~2.91×10-4 M),最低偵測極限為 7 μg/mL(約 1.94×10-5 M)。
因為本研究使用螢光偵測器,因此所測得的偵測極限值遠比此文
獻中的偵測極限值要低 2000 倍,同時線性範圍也較廣。文獻中 只檢驗溶在純水中的標準品,而本實驗除了檢測在水中及尿液中 的標準品外,還測試真實藥物及服用藥物病人的尿液,所以文獻 中 HP-β-CD 的濃度只需添加 40 mM 就足夠,但本研究卻因此需 要添加至 60 mM,才能在極大量的左旋異構物中分離出右旋異構 物。
五、 應用
(一)
藥物的不純度
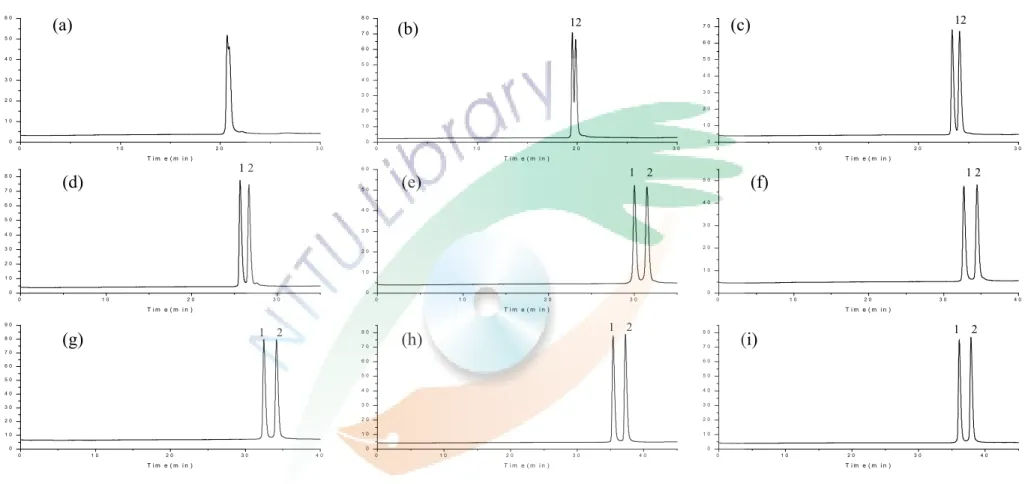
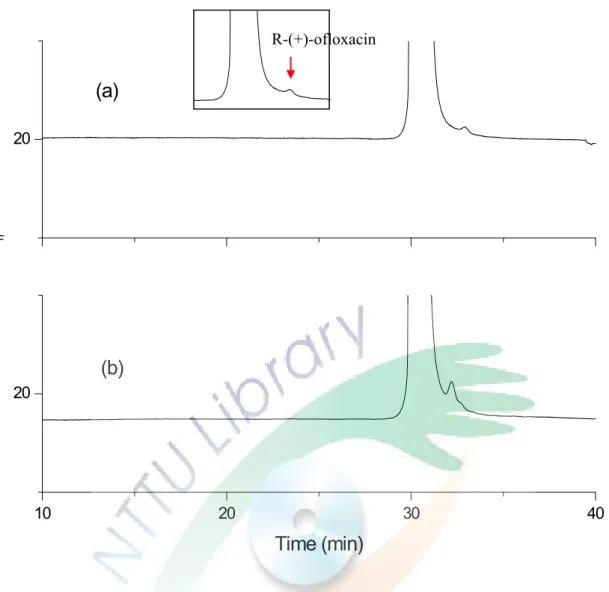
levofloxacin 目 前 已 是 應 用 在 醫 療 上 的 成 藥 -可 樂 必 妥 (cravit) 的主成分,將應用本研究所得之分析條件檢測該藥 物 的 純 度 , 是 否 真 的 只 含 有 levofloxacin 及 一 顆 藥 錠 中 levofloxacin 的真實含量為多少,實驗結果為 Fig. 14。Fig. 14
(a)為將樣品加純水稀釋 10 倍直接進行實驗所得之結果,
可以發現在高濃度的 levofloxacin 旁邊有一個小小的峰形。
Fig. 14(b)為添加了 5×10-5 M ofloxacin 於樣品中,兩電泳 圖比較後可以發現在高濃度的 levofloxacin 旁邊存在著微量 的右旋異構物。經由面積的比例得到藥物中 R(+)-ofloxacin 對 S(-)-ofloxacin (levofloxacin)的比率約為 0.1:99.9。藥物 包裝上寫內含 100 mg/錠的 levofloxacin,經由實驗測得的 levofloxacin 含量約為 93 mg。
(二)
病人尿液中藥品定量
Fig. 15(a)的電泳圖是應用此分析方法的條件檢測添 加 levofloxacin 於未服用藥物,25 歲健康女性的尿液,可從 圖中發現此分析方法應用在尿液的偵測上,基線平穩不會產 生基質干擾效應,因此尿液樣品不需經過複雜的前處理,可 稀釋後直接應用此方法作檢測。
Fig. 15(b)為未經過前處理,只以純水稀釋 10 倍的病 人尿液直接進行實驗得到的電泳圖,可知此條件應用在病人 的 尿 液 分 析 中 不 會 因 為 病 人 服 藥 或 生 理 狀 況 的 不 同 而 有 其 他的干擾發生。從圖中可以看到在 levofloxacin 旁邊依舊有 一個小小的峰形存在,經由 Fig. 15(c)添加了 5×10-5 M ofloxacin 比較後,可以確認此峰為右旋異構物,因此在病 人 尿 液 中 也 存 在 著 微 量 R(+)-ofloxacin, 且 對 levofloxacin 的比率約為 99.35:0.65。左右旋藥物的比率和藥物的比率 略有不同,推測可能是在經過人體的代謝後,有些左旋的藥 物被轉換成了右旋或左右旋藥物的代謝速率不同,導致尿液 中的比率有所差異。使用標準添加法,在稀釋 100 倍的病人 尿液中分別加入不同體積的 levofloxacin 標準品,使得最後 添 加 的 濃 度 為 0 、 1×10-5 M 、 2×10-5 M 和 5×10-5 M 的 levofloxacin,以面積對添加的 levofloxacin 濃度作圖,見 Fig.
16,得到 y=257226x+147579,R2=0.9996,測定出尿液中的
levofloxacin 含量約為 5.7×10-4 M,使用校正曲線得到的數 據是 8.6×10-4 M。由校正曲線得到的數值略高於標準添加法 所得之數值,可能是因為每個人的飲食及生活習慣的差異,
尿液基質中還是有一些成份的差異存在,導致校正曲線法所 得的數據略高於標準添加法之數據,因為在標準添加法中每 個樣品都有一樣的基質干擾,因此所得的數據較具可信度。
一般成人每日的排尿量約 1~1.5 L,依照其代謝的數據,假 設此病人服用的藥物在一天內以尿液排出 80 %,平均含量 約為 4.40×10-4 M~2.96×10-4 M,實驗的樣品是在服藥後的 3~4 小時內收集,故此樣品的濃度略高於平均濃度應該是屬 於 可 接 受 的 , 證 明 此 方 法 對 於 測 定 病 人 尿 液 中 的 levofloxacin 的含量是有效且合理的,對監控病人的服藥量 及尋找藥物的代謝途徑提供一個簡便的方法。
第四章 結論
本實驗在 50 mM 磷酸緩衝溶液(pH 2.30)中添加 60 mM HP-β-CD 可 得 到 最 佳 的 分 離 度 及 螢 光 強 度 增 強 效 果 , 對 levofloxacin 最低偵測極限可達 1×10-8 M ,溶在水中的標準品定 量範圍從 1×10-7 M~5×10-3 M 之間有良好的線性,添加在尿液中 的 線 性 範 圍 從 5×10-6 M~5×10-3 M, R2 可 分 別 達 到 0.9989 和 0.9943 。 應 用 在 藥 物 的 不 純 度 判 別 時 , 可 得 知 藥 物 中 R(+)-ofloxacin 對 S(-)-ofloxacin (levofloxacin)的比率約為 0.1:
99.9,一粒藥錠約含有 93 mg 的 levofloxacin。應用在服用藥物病 人的尿液檢測藥物含量,可發現 R(+)-ofloxacin 對 S(-)-ofloxacin (levofloxacin) 的 比 率 約 為 0.65 : 99.35 。 使 用 標 準 添 加 法 測 得 levofloxacin 的含量約為 5.7×10-4 M。此外,實驗中也提出在 buffer 中添加幾丁聚醣及金屬離子的檢測方式,發現添加濃度為 5×10-7 (w/v)的幾丁聚醣及 5×10-8 M 的 Cu2+時 , 分 離 效 果 比 單 獨 只 有 HP-β-CD 時略佳,將來可朝向添加不同的金屬離子或改變緩衝溶 液的 pH 研究是否會有更佳的分離效果。