國立交通大學
應用化學研究所
碩士論文
新穎藍光含蒽共軛高分子之合成及其在
高分子電激發光二極體之應用
Synthesis of New Blue Anthracene-based Conjugated
Polymers and Their Applications in Polymer
Light-emitting Diodes
研 究 生:林承叡 Cheng-Jui Lin
指導教授:許千樹 博士 Dr. Chain-Shu Hsu
新穎藍光含蒽共軛高分子之合成及其在 高分子電激發光二極體之應用
Synthesis of New Blue Anthracene-based Conjugated Polymers and Their Applications in Polymer Light-emitting Diodes
研究生:林承叡 Student : Cheng-Jui Lin 指導教授:許千樹 博士 Advisor : Dr. Chain-Shu Hsu
國立交通大學 應用化學研究所
碩士論文
A Thesis Submitted to Institute of Applied Chemistry National Chiao Tung University
in Partial Fulfillment of the Requirements for the Degree of Master
in
Applied Chemistry June 2009
Hsinchu, Taiwan, Republic of China
新穎藍光含蒽共軛高分子之合成及其在高分子電激發
光二極體之應用
研究生 : 林承叡 指導教授 : 許千樹 博士
國立交通大學
應用化學研究所碩士班
摘要
本研究的主要目的在於合成以Anthracene 為基礎的共軛高分子及 其 在 高 分 子 電 激 發 光 二 極 體 之 應 用 。 我 們 成 功 合 成 出 六 個 以 9,10-Bis(6-bromonaphthalen-2-yl)-2-tert-butylanthracene (TBADN) 與 Fluorene 衍生物混掺的藍光高分子。所有共聚物除了 TAZ-T50 是以 Yamamoto polymerization 做聚合外,其他的高分子如 F-T50、DPF-T5 ~ DPF-T50 及 TAZ5-DPF-T45 皆是以 Suzuki polymerization 作為合成 方法。第一部分將TBADN 與具有長碳鏈的 Fluorene(PFO)進行聚合反應 得到 F-T50,希望藉由長碳鏈的導入能有效改善 TBADN 同聚物 (homopolymer)溶解度不佳的問題。至於第二部分,為了解決一般高分 子材料於薄膜態易有共軛主鏈因π-πstacking 造成堆疊現象發生而產 生消光的情形,我們利用苯環衍生物的加入,企圖增加芴分子於九號 碳位上取代基的立體障礙,來減少高分子共軛主鏈的堆疊,故合成出 DPF-T 系列高分子,其中透過調整 TBADN 與 Fluorene 衍生物的比 例,希望高分子主鏈將所吸收到的能量以較有效的方式如 Host-dopant 的能量轉移機制來提升材料發光的效率。另外聚芴高分子本質上是以 傳輸電洞為主的材料,故在第三部分,藉由在高分子的側鏈導入具有 傳導電子能力的官能基如 1,2,3-三氮唑(1,2,3-triazole units),可以增加 正負電荷於發光層中再結合的比例,使其因電子電洞平衡而提升元件 效率,所以合成出TAZ-T50 與 TAZ5-DPF-T45。所有合成出來的高分 子熱穩定性佳,其熱裂解溫度高於 330 oC,而玻璃轉移溫度則高於 80 oC,並且對一般常用的溶劑,如 THF 及 Toluene 皆有不錯的溶解度, 有利於使用旋轉塗佈法在元件的製作。這些高分子在光學性質部分, 其溶液態與薄膜態之螢光光譜的主峰都在約 440 nm ~ 460 nm 的藍光 範圍,而且具有不錯的螢光量子效率 (Φsol=61~88 %,Φfilm=14~26
值約在-2.9 ~ -3 eV,大致上來說,我們所合成出來的高分子其 LUMO 值較低,有利於元件上電子從陰極端注入至發光層。 元件製作部分主要是以 ITO/PEDOT:PSS/Polymer/CsF/Al 的形式 做雙層的元件結構,其中最佳的材料為 DPF-T50。在驅動電壓為 13V 下可達最大亮度 1650 cd/m2,此時CIE 色度座標值為(0.22,0.33),光 色為藍綠光,而最大發光效率則為 0.39 cd/A。綜合上述雙層元件結果 來看,我們所合成出來的含蒽聚芴高分子,具有不錯的發光效率 (luminance efficiency,LE),而且在高操作電壓下也有不錯的穩定性。
S
ynthesis of New Blue Anthracene-based Conjugated
Polymers and Their Applications in Polymer
Light-emitting Diodes
Student : Cheng-Jui Lin Advisor : Dr. Chain-Shu Hsu
Institute of Applied Chemistry
National Chiao Tung University
Abstract
This study focused on the synthesis of anthracene-based copolymers and their applications in polymer light-emitting diodes (PLEDs). We had synthesized six copolymers, i.e., F-T50, DPF-T5, DPF-T25, DPF-T50, TAZ5-DPF-T45 and TAZ-T50, containing 9,10-Bis (6-bromonaph thalen-2-yl)-2-tert-butylanthracene (TBADN) and 2,7-disubstituted fluorene moieties. All of these copolymers were synthesized via palladium-catalyzed Suzuki polymerization except TAZ-T50 which was
synthesized by nickel-mediated Yamamoto polymerization.
The first polymer F-T50 were prepared by Suzuki coupling of TBADN with boronic ester of fluorene derivative which contains long alkyl chains in the C-9 position of fluorene units for improving the solubility of the obtained polymer. However, the PLED device performance of F-T50 was quite low. We introduced a bulky phenyl-ring in the C-9 position of fluorene units so as to reduce the aggregation caused by π-π interaction between polymer main chains in the other polymers DPF-T5~DPF-T50. We tuned the mole ratio between TBADN and fluorene derivatives among these polymers as to enhance the luminance efficiency through Förster energy transfer. Finally, we synthesized both TAZ5-DPF-T45 and TAZ-T50 polymers which contained electron transporting 1,2,3-triazole units on the C-9 position of a fluorene ring.
All the synthesized polymers showed good thermal stability ( Td >
330 oC ) and their glass transition temperature is higher than 80 oC. The synthesized polymers emit blue light in the region from 440 nm to 460 nm in both solution and thin film states. Their HOMO and LUMO energy
levels are in the range from -5.8 to -5.9 eV and -2.9 to -3.0 eV respectively. These results demonstrate that electrons could inject from cathode to polymer emitting layer easily. All of these polymers had good solubility in common organic solvent, such as toluene or THF. A double layer PLED device with the configuration of ITO/PEDOT:PSS/Polymer/ CsF/Al was fabricated. The EL spectra exhibited simalr emissions with those of PL spectra. The best EL device was achieved by using DPF-T50 as an emitting layer. Its maximum brightness was 1650 cd/m2 at 13 V, and maximum luminance efficiency was 0.39 cd/A. The CIE coordinates was (0.22, 0.33) at 13V. It means that this device emits a greenish-blue color.
謝誌
在攻讀碩士學位的這兩年,所受到的幫助遠超過我所合出來的 compound,在這裡僅以寥寥幾句話實在難以表達心中的感謝。 首先感謝我的指導教授許千樹老師,提供了一個那麼舒適且資源 豐富的研究環境,使我們能夠不用為了借用儀器而到處奔波,至於您 平日的教誨及對學術的熱誠亦深深影響著我。同時感謝口試委員陳信 龍教授、何榮銘教授與鄭彥如助理教授,給我許多寶貴的建議,使這 本論文得以更加的充實完善。 感謝勝雄學長對實驗室無論是研究上還是管理上的付出,因為有 你細心的督導,我們才能以較有條理且有效率的方式完成老闆交付的 任務。感謝百哥,在我徬徨無助時伸出援手,你的好學上進與處理問 題的能力一直讓我很欽佩。晉彥學長的合成技巧真的很罩,多虧有你 我才能一次又一次度過難關。感謝小施在後期幫我製作元件,因為有 你,我合出來的 compound 才得以發光發熱,你的認真與努力總有一 天會得到回報的。感謝敏碩學長與群哥在我還是個菜鳥時,不厭其煩 的指導我做實驗。熱心的小明哥,謝謝你平日在生活上的照顧。幽默 風趣的小毛,當初就是因為你替我們介紹實驗室時的一席話,我才下 定決心進來 Hsu Lab.,果然不虛此行。謝謝大楠哥總在出奇不意時提供 compound 給我,讓我不用合得太辛苦。感謝 Martin 不時製造笑果, 讓我體會法式幽默! 秋翔看不出在你豪邁粗獷的外表下,還有如此聰明的頭腦與靈巧 的雙手,再搭配你勇於嘗試的實驗精神與細心,讓我不得不佩服,與 你一邊鬥嘴一邊做實驗真的蠻過癮的。再來是阿輝,我認識你最久, 感謝你一路走來對我的包容,你真的是一位很貼心的夥伴。善解人意 的天心,與妳聊天的過程總是很愉快,而妳悠閒的生活態度一直是我 所嚮往的。爆發力十足的大炮,你在尾牙上的一首〝衝衝衝〞,讓我 重新燃起了鬥志,希望你快點度過眼前的難關。憲哥你在認真做實驗 之餘,身體也要顧,有信心一點,學弟妹都要跟隨你的腳步。昌哥當 了你一年的室友,才發現你是一個這麼有趣的人,與你那嚴肅正經的 外表有頗大差距,以你的能力,遲早會升上去,跟憲哥一起輔佐老闆。 阿鴻你的努力我都看得到,繼續加油,polymer 一定會聚出來的。小 誠你辦事的能力比起你的外表還要可靠多了,再認真一點一定可以做 出一番成績。土榮你是一個非常熱心且相當有想法的人,我在你旁邊 學了不少東西。亭芝、佩蓉與逸芃,希望你們的實驗一帆風順。小 p、 冷翰與芳銘學長,多謝你們提供的實驗技巧與寶貴經驗,讓我獲益匪 淺。隔壁實驗室的球友,寶哥、狸貓、阿隆與阿國,感謝你們能在實 驗繁忙之餘偷跑出來陪我打球,有你們如此重情義的相挺,使我倍感
溫馨。另外特別感謝助理小燕姐、欣怡與似婷,因為有妳們在一旁協 助,我們的實驗才能進展的如此順利。最後,感謝父母長久以來的支 持與涵青的陪伴,在我沮喪時給予鼓勵,在歡樂時分享我的喜悅。
在這短短兩年的研究生涯中,說不辛苦、不覺得累是騙人的;慶 幸的是我擁有你們這群好夥伴,也因為有你們,才能成就現在的我。
目次
中文摘要………..I 英文摘要……….. .IV 謝誌... VII 目次... X 合成目錄...XIV 表目錄... XV 圖目錄...XVI 附圖目錄...XIX 第1 章 緒論...1 1.1 有機電激發光簡介...1 1.2 電激發光原理...4 1.3 電極的選擇...8 1.3.1 陽極 (Anode)...8 1.3.2 陰極 (Cathode)...81.4.1 主發光體材料 ( Host )... 11
1.4.2 客發光體材料 ( Guest )...12
1.4.3 電子傳導層 ( Electron transporting layer, ETL )...15
1.4.4 電洞傳導層 ( Hole transporting layer, HTL )...15
1.5 能階理論...16 1.5.1 雙層與多層之元件介紹...18 1.6 高分子發光二極體材料簡介...20 1.6.1 Fluorene(芴)衍生物的發展與性質介紹...21 1.6.2 聚芴藍光材料回顧...25 1.6.3 Anthracene(蒽)衍生物的發展與性質介紹...27 1.6.4 聚蒽藍光材料回顧...29 1.7 研究動機...31 第2 章 實驗部分...34 2.1 試藥...34 2.2 量測儀器...34
2.2.1 核磁共振光譜儀 ( Nuclear Magnetic Resonance,NMR )....34
2.2.2 微差掃描卡計 ( Differential Scanning Calorimeter, DSC )....35
2.2.3 熱重分析儀 ( Thermal Gravimetric Analyzer, TGA )...35
2.2.5 紫外線與可見光光譜儀 ( UV-Vis Spectrophotometer )...36 2.2.6 螢光光譜儀 ( Photoluminescence Spectrophotometer )...36 2.2.7 循環伏安計量儀 ( Cyclic Voltammetry, CV )...36 2.2.8 光譜掃描色度計 ( Spectroscan Colorimeter, PR-650 )...37 2.3 合成部分...37 2.3.1 單體 M1~M3的合成...41 2.3.2 高分子 F-T50, DPF-T5~DPF-T50, TAZ5-DPF-T45 與 TAZ-T50 的合成...48 第3 章 結果與討論...54 3.1 單體及含蒽聚芴高分子材料之合成與討論...54 3.2 高分子分子量鑑定...56 3.3 高分子熱性質分析...56 3.4 光學性質...59 3.4.1 溶液及薄膜態的 UV-vis 以及 PL 光譜 ...59 3.4.1.1 聚芴高分子材料的迴火實驗 (annealing)...59 3.4.2 螢光量子效率之量測...63 3.5 電化學性質...71 3.6 有機電激發光二極體元件製作與光電性質量測...75 3.6.1 ITO 圖形化製作 ...75
3.6.2 發光元件的結構...76
3.6.3 元件光電性質討論...78
第4 章 結論...89
第5 章 參考文獻...90
合成目錄
SCHEME 1. 單體M1~M3 的合成...38
SCHEME 2. 含蒽聚芴高分子F-T50~DPF-T25 的合成...39
SCHEME 3. 含蒽聚芴高分子DPF-T50~TAZ-T50 的合成 ...40
表目錄
表 1-1 陰極金屬及ITO 的功函數值 ...9 表 1-2 四種不同的金屬在PPV 元件中做為傳輸電子的發光效率值 ...10 表 1-3 ADN 衍生物的液態螢光光譜及其熱性質比較...28 表 2-1 含蒽聚芴高分子的組成...53 表 3-1 含蒽聚芴高分子的分子量分佈和熱性質...58 表 3-2 含蒽聚芴高分子的UV 吸收及 PL 放射光譜值...65 表 3-3 含蒽聚芴高分子的電化學性質...74 表 3-4 ITO 玻璃清洗程序 ...76 表 3-5 含蒽聚芴高分子在元件結構為ITO/PEDOT:PSS/POLYMER/ CSF/AL之電激發光表現...88圖目錄
圖 1-1 電激發光元件示意圖...3 圖 1-2 PVK 以及 PPV 結構示意圖...3 圖 1-3 能量轉移示意圖...6 圖 1-4 單層電激發光元件示意圖...7 圖 1-5 電子與電洞於發光層內再結合示意圖...7 圖 1-6 OLED 元件中常見主發光體化學結構 ...12 圖 1-7 OLED 元件中常見客發光體化學結構 ...12 圖 1-8 混摻系統的主體-客體之能量傳遞示意圖...13 圖 1-9 FÖRSTER 及 DEXTER能量轉移機制...14 圖 1-10 OLED 元件中常見電子傳輸材料化學結構...15 圖 1-11 OLED 元件中常見電洞傳輸材料化學結構...16 圖 1-12 雙層結構的 OLED 發光元件 ...19 圖 1-13 三層結構的 OLED 發光元件 ...20 圖 1-14 PPV 及其衍生物的化學結構以及光色範圍圖...21 圖 1-15 芴(FLUORENE)分子示意圖...22 圖 1-16 YAMAMOTO偶合法...24 圖 1-17 SUZUKI偶合法...24圖 1-18 聚芴高分子PF2-6AMX 之分子結構與元件表現...25 圖 1-19 聚芴高分子P4 之分子結構與元件表現...25 圖 1-20 聚芴高分子PF-OXD 之分子結構與元件表現...26 圖 1-21 聚芴高分子PFO+50%DFD 之分子結構與元件表現 ...26 圖 1-22 ADN 衍生物分子結構圖 ...28 圖 1-23 聚蒽高分子XPA之分子結構與元件表現...29 圖 1-24 聚蒽高分子P2 之分子結構與元件表現...29 圖 1-25 聚蒽高分子A 之分子結構與元件表現 ...30 圖 1-26 聚蒽高分子15 之分子結構與元件表現 ...30 圖 3-1 SUZUKI COUPLING的反應機制...55 圖 3-2 含蒽聚芴高分子在150OC 下加熱 2 小時後之 PL 光譜圖 ...62 圖 3-3 含蒽聚芴高分子的UV-PL 光譜 (A)溶液態 (B)薄膜態...68 圖 3-4 F-T50、DPF-T50 與 TAZ5-DPF-T45 之 UV-PL 光譜圖 (A)溶液 態 (B)薄膜態...69 圖 3-5 TAZ-T50、DPF-T5 與 DPF-T25 之 UV-PL 光譜圖 (A)溶液態 (B)薄膜態 ...70 圖 3-6 含蒽聚芴高分子之能帶關係圖...74 圖 3-7 PEDOT:PSS 分子結構示意圖...77 圖 3-8 元件結構示意圖...78
圖 3-9 F-T50 之 EL 光譜圖(16V)...79 圖 3-10 F-T50 之 J-V-B 關係圖...79 圖 3-11 DPF-T5 之 EL 光譜圖(10V)...80 圖 3-12 DPF-T5 之 J-V-B 關係圖...80 圖 3-13 DPF-T25 之 EL 光譜圖(11V)...81 圖 3-14 DPF-T25 之 J-V-B 關係圖...81 圖 3-15 DPF-T50 之 EL 光譜圖(13V)...82 圖 3-16 DPF-T50 之 J-V-B 關係圖...82 圖 3-17 TAZ5-DPF-T45 之 EL 光譜圖(12V)...83 圖 3-18 TAZ5-DPF-T45 之 J-V-B 關係圖...83 圖 3-19 TAZ-T50 之 EL 光譜圖(12V) ...84 圖 3-20 TAZ-T50 之 J-V-B 關係圖 ...84 圖 3-21 電流密度對操作電壓關係圖...87
附圖目錄
附圖 1 TGA OF F-T50...95 附圖 2 DSC OF F-T50 ...95 附圖 3 TGA OF DPF-T5 ...96 附圖 4 DSC OF DPF-T5...96 附圖 5 TGA OF DPF-T25 ...97 附圖 6 DSC OF DPF-T25...97 附圖 7 TGA OF DPF-T50 ...98 附圖 8 TGA OF TAZ5-DPF-T45...98 附圖 9 TGA OF TAZ-T50 ...99 附圖 10 DSC OF TAZ-T50...99 附圖 11 CV OF F-T50...100 附圖 12 CV OF DPF-T5 ...100 附圖 13 CV OF DPF-T25 ...101 附圖 14 CV OF DPF-T50 ...101 附圖 15 CV OF TAZ5-DPF-T45...102 附圖 16 CV OF TAZ-T50...102 附圖 17 1H-NMR SPECTRUM OF (1)...103附圖 18 13C-NMR SPECTRUM OF (1)...104 附圖 19 MASS SPECTRUM OF (1)...105 附圖 20 1H-NMR SPECTRUM OF (M1)...106 附圖 21 13C-NMR SPECTRUM OF (M1)...107 附圖 22 MASS SPECTRUM OF (M1) ...108 附圖 23 1H-NMR SPECTRUM OF (2)...109 附圖 24 13C-NMR SPECTRUM OF (2)...110 附圖 25 MASS SPECTRUM OF (2)... 111 附圖 26 1H-NMR SPECTRUM OF (M2)...112 附圖 27 13C-NMR SPECTRUM OF (M2)...113 附圖 28 MASS SPECTRUM OF (M2) ...114 附圖 29 1H-NMR SPECTRUM OF (M3)...115 附圖 30 13C-NMR SPECTRUM OF (M3)...116 附圖 31 MASS SPECTRUM OF (M3) ...117 附圖 32 1H-NMR SPECTRUM OF (ECP1)...118 附圖 33 13C-NMR SPECTRUM OF (ECP1) ...119 附圖 34 MASS SPECTRUM OF (ECP1)...120 附圖 35 1H-NMR SPECTRUM OF (ECP2)...121 附圖 36 13C-NMR SPECTRUM OF (ECP2) ...122
附圖 37 MASS SPECTRUM OF (ECP2)...123 附圖 38 1H-NMR SPECTRUM OF F-T50...124 附圖 39 1H-NMR SPECTRUM OF DPF-T5 ...125 附圖 40 1H-NMR SPECTRUM OF DPF-T25 ...126 附圖 41 1H-NMR SPECTRUM OF DPF-T50 ...127 附圖 42 1H-NMR SPECTRUM OF TAZ5-DPF-T45 ...128 附圖 43 1H-NMR SPECTRUM OF TAZ-T50 ...129
第
1章 緒論
1.1 有機電激發光簡介
有機電激發光(Organic Electroluminescence,OEL)的發現最早可回
溯到 1950 年代,Bernanose 等人於 1953 年將 acridine orange 與
quinacrine 薄膜加上直流高壓電,觀察到發光現象 [1-4],當時他們解釋
此發光原理為類似於傳統 III-V 族元素所組合的薄膜式電激發光板
(thin-film electroluminescence panel, TFEL),例如硫化鋅(ZnS)。
目 前 有 機 電 激 發 光 裝 置 的 雛 型 則 由 1963 年 Pope 等人在蒽
(anthracene)單晶兩端跨接 400 伏特以上的直流高壓電,並觀察到發光
現象[5]。1966 年,Helfrich 和 Schneideru 以含有 AlCl3-anthracene (陰
極) 和 Na-anthracene (陽極)的電解質溶液製備高亮度的 EL 元件[6],但 此元件的驅動電壓仍相當高。之後其它有機分子單晶也陸續被發現具 有電激發光現象。雖然有些有機單晶分子已可達到相當高的量子效 率,但因受限於單晶的厚度,一般單晶仍需要超過 100 伏特的電壓才 能驅動發光。一直到了1979 年左右,Roberts 等人以 Langmuir-Blodgett 技術製造 anthracene 衍生物的元件[7],利用多次重複的單層分子成膜 技術製造有機電激發光層,有效地降低了有機電激發光層的厚度,使
有機電激發光的驅動電壓大幅下降,有機電激發光才得以真正進入可 以實用化的階段。更進一步的改進則由 Vincett 等人在 1980 年以真空 蒸鍍的方式製造非晶相(amorphous)的 anthracene 薄膜[8],此方法可以 得到均勻的大面積的有機分子薄膜,也成為現今製造 OEL 元件的標 準方法之一。 真正商業化上的突破源自1987 年柯達 Kodak 公司 C. W. Tang 和 S. A. Vanslyke 等人利用真空蒸鍍非晶系(amorphous)有機薄膜的技術 以及創新的異質介面(hetero-junction)多層有機薄膜之元件結構製作出 高效率的 ITO/diamine/Alq3/Mg:Ag 雙層結構的電激發光元件[9] (如圖
1-1 所示)。其是以鎂銀合金為陰極,銦錫氧化物(indium-tin oxide, ITO)
做為陽極,8-hydroxyquinoline aluminium (Alq3) 作為電子傳輸層兼發
光層,芳香胺類(aromatic diamine) 化合物做為電洞傳輸層。此有機電 激發光元件的驅動電壓小於 10 伏特,發光效率大於 1 %,大幅改善 了 OLED 元件的性質。從此之後,OLED 發光材料開始受到廣泛的重 視。 在高分子電激發光(PLED)的發展方面,最早是由 Patridge 等人[10] 在 1982 年以 poly(N-vinyl carbazole) (PVK) 作為材料,利用溶液旋轉 塗佈(spin coating)的方式製作第一個高分子的電激發光元件。接著在
1990 年英國劍橋大學卡文迪西實驗室(Calvendish Lab.)的 Burroughes 等人所發表的有機高分子電激發光元件[11],其利用 poly(p-phenylene vinylene)(PPV)的前驅物高分子塗佈於導電玻璃的表面,再加熱此前驅 物,使之經由脫去反應得到 PPV 高分子共軛聚合物做為發光層,製 造出 ITO/PPV/Al 單層元件,得到綠光有機電激發光,這是第一個以 主鏈型共軛高分子作為發光材料的電激發光元件。PVK 與 PPV 的結 構見圖 1-2 所示。 圖 1-1 電激發光元件示意圖 圖1-2 PVK 以及 PPV 結構示意圖
接著在 1991 年 Heeger 等人[12]合成出對於一般有機溶劑溶解度相 當好的 MEH-PPV,利用其高分子側鏈的取代基可以有效的增加高分 子本身對於溶劑的溶解度,使高分子發光材料在製程上更加具有實用 性。之後在學術界及工業界有更多的人力投入有機電激發光研究,不 斷地開發出各種新的材料或元件構造,並在電激發光元件的壽命與發 光效率上都有豐碩的成果,使 LED 成為一個熱門的研究領域。對於 近年來蓬勃發展的平面顯示器產業而言,小分子的 OLED 顯示器具有 自發冷光、高亮度、廣視角、高應答速度、低驅動電壓、低耗電量、 製程簡易等優點,極具有潛力成為下一代平面顯示器的主流,尤其在 1992 年已有可撓曲的高分子 PLED 元件被研製出來[13],可望發展成 為可撓曲的平面顯示器,極具商業潛力。2005 年韓國三星電子成功研 發出全球最大的有機電激發光顯示器電視 (40 吋 OLED TV),使得 OLED 亦將正式迎接大尺寸化的時代。因此,有機電激發光堪稱二十 一世紀的明星產業,相當值得投入精力研究。
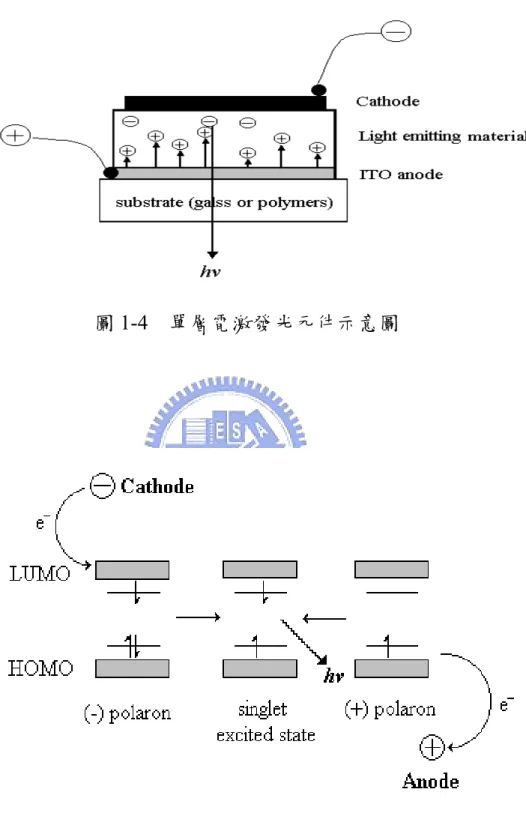
1.2 電激發光原理
化學分子在接受光能、電能或是化學能等能量後,其電子組態將 由基態(ground state)轉變為激發態(excited state),而分子處在此高能的 狀態是相當不穩定的,必須將能量釋放以回到基態才會形成穩定的電子組態(如圖 1-3 所示)。有機電激發光材料皆是具有共軛雙鍵的小分 子或高分子化合物,其特色為分子內單鍵雙鍵交互共存,且在化合物 的分子結構中存在許多非定域化(delocalized)的 π 電子,容易在共軛雙 鍵之間作共振與躍遷,因此分子軌域的價帶(valance band)和傳導帶 (conduction band)的位能差距較一般有機分子小,並具有半導體的性 質,故相當適合應用在電激發光元件上。 由於分子結構強烈的影響了分子軌域的能階,因此發光材料的分 子結構便決定了材料本身的發光光色與效率等特性。一般單層有機電 激發光元件的基本結構如圖 1-4 所示。將有機發光材料薄膜夾於上端 金屬陰極與下端 ITO 陽極之間,ITO 電極為透明電極。當元件加以順 向偏壓時,電子由上方的陰極注入發光層中,電洞則由下方的陽極注 入發光層中,在電場的作用下,電子、電洞克服各自的能障(energy barrier),在共軛分子鏈之間傳遞,向相對的方向靠近,之後再於發光 層中結合,放出可見光。其發光的原理如圖1-5 所示,通電之後電子 由陰極注入發光層的 LUMO 中,形成負的 polaron;電洞由陽極注入
發光層的 HOMO 中,形成正的 polaron。正、負的 polarons 於發光層
中 再 結 合 產 生 了 單 重 態 激 子(singlet exciton) 及 三 重 態 激 子 (triplet
exciton),激子緩解(relaxation)的過程亦如同圖 1-3 所示。單重態激子 經輻射衰退(radiative decay)回到基態而放出螢光,此過程即為電激發
光,而三重態激子則會以磷光或是非輻射方式衰退,並不會放出螢
光。然而,形成三重態激子的機率高達 75 % [14],因此OEL 的內部量
子效率(internal quantum efficiency)理論上最大只能達到 25 %。
圖 1-3 能量轉移示意圖 當在基態(S0)的電子之吸收光子 hν 後,由於電子之躍遷,使得分 子轉換成具較高位能之激發態(S1),此激發態之分子可經由與其它分 子或介質碰撞以非輻射性方式(nonradiative)釋放出能量(熱能)回歸到 基態(I);亦可經由釋出輻射能回歸到基態,以 S1到 S0之轉換釋出輻 射能(螢光)(II);若分子內含重原子(原子序>30)時,因 spin-orbital
coupling 作用力,使得分子之電子組態將由單重態(singlet state, S1)轉
圖 1-4 單層電激發光元件示意圖
1.3 電極的選擇
1.3.1 陽極 (Anode)
在有機電激發光元件中,載子注入的難易程度影響元件的驅動電 壓與發光效率甚巨,為了降低驅動電壓與增加發光效率,除了改變載 子注入層的材料之外,選擇合適的電極材料也非常重要。有機電激發 光元件一般使用的陽極為具有較高功函數(work function)的銦錫氧化 物(indium-tin oxide, ITO),因為它具備了穩定、透明及高導電度等特 性,而且它在一般標準的微影蝕刻製程中相當容易圖案化(pattern), 其功用在於有效的將電洞注入有機發光層中。1.3.2 陰極 (Cathode)
陰極材料的選用通常為低功函數的金屬或合金,如鎂、鈣或以鎂/ 銀 = 10/1 的比例,用共蒸鍍方式製成的鎂銀合金,其功用為將電子 有效的從陰極端注入有機發光層中。一般常使用電極材料的功函數見 表 1-1[15],其對單層導電高分子 PPV 的量子效率見表 1-2。柯達公司 發展出的鎂銀合金(約 10:1 比例,此適當的陰極材料,其好處是少 量的銀可以幫助鎂的沈積(deposition),同時延遲鎂的氧化,更有助於 降低元件的驅動電壓[9]。由表 1-1 與表 1-2 中,Ca、Mg,Al、Au 四種金屬的功函數大小依次為 Au > Al > Mg > Ca,其在 PPV 為發 光層的電激發光元件中做為陰極,其量子效率依次為 Ca > Mg > Al > Au。因此,選擇功函數較低的金屬做為元件的陰極可以大幅增 加電激發光的量子效率。 大多數的高分子有機發光二極體的螢光光譜(Photoluminescense, PL)與電激發光光譜(Electroluminescence, EL)非常相似,這是因為兩者 放光機制雷同,只是分別藉由光或是電來激發處在基態的電子,使其 形成激子後以輻射的方式由激發狀態衰退回基態而放出來。一個有機 共軛高分子發光元件,其最重要也最直接的評價在於它的亮度及發光 效率,這兩項因素又取決於激子的生成效率及電子和電洞是否能有效 的傳遞結合進而鬆弛放光。 表 1-1 陰極金屬及 ITO 的功函數值。
Material Work function (eV)
Au 5.1 ITO 4.7 Ag 4.5 Al 4.3 Mg 3.7 Ca 2.9
表 1-2 四種不同的金屬在 PPV 元件中做為傳輸電子的發光效率值。
Electron injection electrode
(low work function) Efficiency (%)
Ca 0.1 Mg 0.05 Al 0.002 Au 0.00005 以實用的觀點來看,低功函數的金屬雖可達較高量子效率,但因 其活性較高,在空氣中易氧化而導致元件具有可靠度不高的問題。因 此使用較高環境穩定度的金屬,如 Al,有其實際應用上的必要性。若 在 Al 陰極和有機層間蒸鍍一極薄的緩衝絕緣層如氟化鋰(lithium
fluoride, LiF)、氟化銫(cesium fluoride, CsF)、氟化鈉(sodium fluoride,
NaF) 或二氧化矽等,可以有效地增加使用 Al 當陰極的效率[16],主
要原因為 Al 與發光層在介面會產生化學反應,而產生較大能障以阻
礙電子注入,因此絕緣層的引入可防止介面的化學反應的發生。但絕 緣層的厚度不可太厚,否則電子的穿隧能力便會相對的受到影響。
1.4 發光層 (Emitting layer, EML)
許多的有機材料可以應用為發光層,發光顏色幾乎可包含整個可 見光的範圍。目前所用的有機發光材料分為兩大系統,一是以有機小 分子為主的元件,其所用材料的分子量通常來說小於兩千,其元件製 作方式通常採用真空蒸鍍而成;再者則是以高分子為主的元件,其分 子量約介於數萬至數百萬之間,主要是具螢光性的共軛高分子,其元 件製作方式通常採用旋轉塗佈的方式成膜。材料的發光特性會因材料 本性與成膜方法不同而異,但本質上並無不同。 不論是選用哪一種材料作為發光層,皆須符合以下兩點要素:(1) 適合加工製造以及可精準的控制其成膜性和厚度,而膜厚範圍在 5 ~ 200 nm。 (2) 具備相當程度的熱及化學穩定性 (對於高分子來說即表 示Tg及Td點要高),因為元件在操作下,很容易因為溫度的上升而嚴 重影響發光效率。1.4.1 主發光體材料 ( Host )
主發光體往往與傳電荷層一起使用,以期讓正負電荷再結合,並 將產生的激子被侷限在發光層上而發光。著名的例子有: BAlq、TAZ、 DPVBI、Zn(ODZ)2、PVK 等,如圖 1-6 所示:圖1-6 OLED 元件中常見主發光體化學結構
1.4.2 客發光體材料 ( Guest )
客發光體則常以共蒸鍍或分散方式與主發光體共同使用,並以能 量轉移或載子捕獲方式接受來自被激發的主發光體能量,導致不同光 色的產生並有效增強元件的發光效率。著名的例子有: Perylene、 Coumarin-545T、DCJTB、DCM-1、DCM-2 等,如圖 1-7 所示: 圖1-7 OLED 元件中常見客發光體化學結構主體與客體間的能量轉移
(Energy transfer)
[6-8]增加有機電激發光二極體效率的關鍵性技術,主要來自於主客體
摻雜發光(host-guest dopant emitter)系統之發展[7-8],如圖 1-8 所示,藉
由具備高電子傳輸特性的主體材料(host),結合具有高螢光或磷光效 率的發光客體材料(guest),可大幅提高元件整體的再結合與放光效 率。主體在經由電激發所產生之激子可轉移到高發光效率的混摻物 (dopant)中發光,此舉除了降低能量由非輻射衰減(non-radiative decay) 的機率之外,另一方面,低混摻濃度同時亦可減少發光客體發生自我 淬熄(self-quenching)的機率。 圖1-8 混摻系統的主體-客體之能量傳遞示意圖 主體與發光客體之間的能量轉移機制,包含有 Förster 及 Dexter 兩種型式的能量轉移(如圖 1-9)[6,9]。Förster 能量轉移是指藉由主體(施 體, donor) 和 發 光 客 體 ( 受 體 , acceptor) 分 子 之 間 的 偶 極 - 偶 極
(dipole-dipole)耦合來傳遞能量,是屬於長距離(約 30-100 Å)之非輻射 能量轉移。不論施體或是受體,其基態至激發態的躍遷須為自旋 允許。故只有單重態間的能量傳遞是屬於 Förster 型式。而電子交換 式的 Dexter 能量轉移則是藉由分子間軌域重疊,將激子由主體傳遞至 客發光體,屬於近距離(約 6-20 Å)之輻射能量傳遞。在 Wigner-Witmer 選擇律下,Dexter 能量轉移只需要遵守施體和受體之總自旋量子數守
恆(total spin conservation) 。 故 此 機 制 可 允 許 單 重 態 - 單 重 態
(singlet-singlet)及三重態-三重態(triplet-triplet)之能量轉移。
1.4.3 電子傳導層 ( Electron transporting layer, ETL )
一個好的電子傳輸材料可以容易的將電子從陰極導入,因為它的 電子親和力(electron affinity, EA)大於發光層(EML),除此之外,它甚 至還扮演電洞阻擋層(hole blocking layer, HBL)的角色,因為它的游離 能(ionization potential,IP)亦有可能大於 EML,此特點將可有效的將
電洞限制在 EML 及 ETL 的介面間。現今常用的材料為具有較高電子
親和性基團,如 PBD 等,如圖 1-10 所示。
圖1-10 OLED 元件中常見電子傳輸材料化學結構
1.4.4 電洞傳導層 ( Hole transporting layer, HTL )
電洞傳輸材料的特性為 IP 和 EA 皆小於 EML 層,使得電洞容易
注入,同時此層亦可將電子限制在 HTL 和 EML 的介面間,目前常見
圖1-11 OLED 元件中常見電洞傳輸材料化學結構 ETL 及 HTL 的功能主要為以下兩項: (1) 有效降低傳導電子或電洞時所需克服的能障。 (2) 將電子與電洞再結合的區域限制在 EML 層,避免電子或電 洞過於靠近兩極而產生淬息(quench)現象,降低量子產率。
1.5 能階理論
在發光材料的分子設計觀點中,若想讓一些基團的分子軌域重 疊,然後發生軌域互相影響以改變分子電子組態,進而影響此分子之 能量吸收與放光波長。首先可使分子骨架的 π-電子盡量位於共平面上,以改變其電子特性。第二種方法則是在這個重疊後的非定域化之 π-電子系統中引入不同屬性之官能基,官能基對於非定域化 π-電子系 統之影響模式基本上可分為兩種,一種是使非定域化 π-電子系統之電 子密度上升,即藉由共振效應(resonance effect,稱為 R 效應) 讓取代 基 p 軌域中的未成對電子進入分子骨架之 π-電子系統,例如硫、氧與 氮原子與其所形成之官能基可以增加原有之 π-電子系統密度。另外, 若取代基 d 軌域之未成對電子進入分子骨架之 π-電子系統者,則稱之
為金屬—配位基之間之電荷轉移(metal-to-ligand charge transfer,
MLCT),一般較常發生於 B 族的過渡金屬元素。 陰電性小於骨架分子之元素亦可透過誘導效應(inductive effect,稱 為 I 效應)而將本身之電子貢獻到分子骨架之 π-電子系統,而使其電子 密度增高,當骨架分子之電子密度上升時,將導致 HOMO 能階上升, 此軌域之提升表示價電帶電子移去將更為容易。前面所討論的可增加 骨架分子電子密度之共振效應與誘導效應稱之為 +R 效應與 +I 效 應。反之,如果外接原子或基團對於骨架分子產生的是 -R 效應或 -I 效應,則將導致骨架分子電子密度下降,而使(HOMO)之能階下降, 同時 LUMO 之能階也同時下降,由於分子的 LUMO 相對於有機發光 材料的傳導帶,此軌域之下降表示電子填入傳導帶更為容易。一般而 言,在 -R 與 -I 效應中,若 LUMO 能階下降幅度大於 HOMO,因
此其電子躍遷能階(energy gap)將縮小,可使原先骨架分子之螢光放射 往長波長移動而改變其光色,至於取代基(或原子)對於骨架分子究竟 是產生+R、+I 效應或-R、-I 效應,則取決於官能基屬性與其於骨架上 的連結位置。
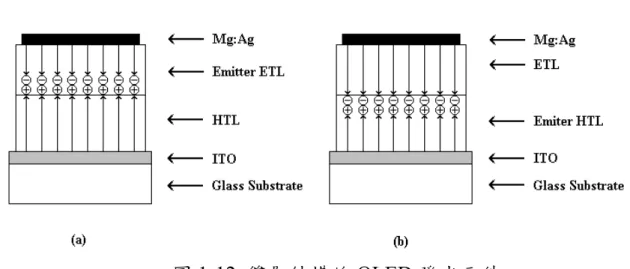
1.5.1 雙層與多層之元件介紹
不同的 OEL 材料傳遞電子、電洞的速率不同,在電激發光元件 中,若電子移動的速率較快,電子與電洞再結合的區域則會靠近陽 極;若電洞移動的速率較快,則其再結合的區域則會較靠近陰極。載 子靠近電極的再結合會發生發光淬息(quench)的現象,同時減少量子產率。為解決這個問題,柯達公司 Tang 和 Van Slyke 最早提出採用雙
層有機薄膜的組合[9],如圖1-12 (a)所示,電洞傳輸層為二苯基胺類材 料(diamine),發光層與電子傳輸層為 Alq3。 由於HTL 電子傳輸能力相當的弱,由陰極注入的電子將可被阻擋 在發光層與 HTL 的介面。另一方面,由陽極注入的電洞也因為不利 於在 ETL 層中傳輸而被阻擋在此介面。故再結合的現象將有可能發 生在 HTL 與 ETL 的介面附近,而遠離了會造成發光淬息的電極附近。 此創舉大幅降低OEL 的驅動電壓到小於 10 伏特,使外部量子效率達 到約 1 %,也為 OEL 的商業化帶來曙光。在此之後,日本九州大學
Saito 教授實驗室提出另一種雙層元件組合,主要由電洞傳輸層發光 [17],如圖 1-12 (b)所示,電子電洞在靠近 HTL 與 ETL 介面的地方結 合,而在 HTL 層發光。 隨後Saito 又提出三層式元件的結構,在發光層的上下使用非晶相 (amorphous)的電洞傳輸層與電子傳輸層,HTL 與 ETL 之間夾著發光 層,元件構造如圖1-13(a)所示[18-19]。發光層的厚度只需相當薄的厚度 就可以控制激子在發光層內使之發出強烈的光[20]。爾後,Kido 再修 改這個結構,提出幽禁式(confinement)的結構,如圖 1-13(b)所示。使
用 一 層 激 發 態 能 階 高 的 激 子 幽 禁 層(excitation confinement layer,
ECL),其產生激子的能量高於 HTL 與 ETL,於是將能量轉移到 HTL
或 ETL,但保持本身不發光的特性。因此,調整 ECL 厚度可控制發
光層為 HTL 或 ETL,當厚度控制得當時則兩層可同時發光,並得到
混合的光色[21]。
圖1-13 三層結構的 OLED 發光元件
1.6 高分子發光二極體材料簡介
自 1990 年代以來已有許多不同的材料被應用在高分子電激發光 元件(PLED)上。為了達到全彩的顯示功能,顯示器需要有紅、綠、藍 三種光色來混合,而目前綠光材料在效率、壽命、色彩飽和度等方面 的表現皆較為成熟,但在紅光及藍光材料則還有相當大的空間有待改 進。PLED 的發光材料可經由結構的修飾,得以改變材料本身 HOMO 與 LUMO 之間的能隙,而發出不同波長的光,這種可藉由修飾局部 結構來達到調整發光光色的特性是有機電激發光材料的一項優點。例 如 1,4-仲苯基乙烯類衍生物(1,4-phenylene vinylene, PPV)是第一個被 應用在 LED 的高分子,隨後許多化學家針對 PPV 的結構進行修飾, 發展出各種具不同取代基的 PPV 衍生物,其光色可由原來 PPV 的黃 綠色變為紅色或綠色(圖 1-14) [9,22-30]。圖1-14 PPV 及其衍生物的化學結構以及光色範圍圖
1.6.1 Fluorene(芴)衍生物的發展與性質介紹
芴(fluorene)本身為一種多環的芳香族化合物,其英文名字起源於 其高規則度且共平面的共軛 π 電子系統,在吸收光子之後將可放射出 藍紫光。芴環的 2,7 位置,反應性相當的強(見圖 1-15),適合被親電 子基(electrophile)所攻擊,之後再配以適當的聚合方法即可合成出 堅硬的聚芴高分子(rigid-rod),其主鏈之間的共軛情形並不會因為聚合 過程而有所影響。聚芴高分子擁有高螢光量子效率、優良的熱及化學穩定性,對於一般有機溶劑(如甲苯、氯仿)溶解度佳等特點,相當適 合用來做為一藍光發光材料。以聚芴為發光層所製成的元件則擁有相 當低的驅動電壓,並於電壓操作之下經長久時間使用不會衰退等優 點。同時芴環的九號位置擁有兩個酸性氫,可透過鹼性環境來達到官 能化,以有效的提高聚合物對於有機溶劑的溶解度,或者達到避免高 分子鏈與鏈之間堆疊的情形發生。若再搭配不同功能性的單體(如傳 電子或電洞、樹枝狀側鏈等基團),即可得到各式各樣的聚芴高分子。 事實上,聚芴也是所有的共軛高分子中唯一能夠涵蓋紅綠藍三種光色 範圍,並同時擁有高螢光效率的發光材料。 圖1-15 芴(fluorene)分子示意圖 合成聚芴高分子的方法,撇開最早的電化學聚合法及以氯化鐵 (FeCl3)進行的氧化聚合法不提[33],目前最被廣泛使用的為 Yamamoto
聚合法以及 Suzuki 聚合法。Yamamoto 聚合法主要是利用鎳金屬(Ni)
做為媒介物(Nickel-mediated reaction),透過氧化還原機制變成二價的
14,000-60,000),但聚合的過程中單體結合較為雜亂而沒有規則性,適 合用來聚合同元聚合物(homopolymer)[31]。故進行共聚合反應時要小 心的控制反應條件,否則每次的結果可能都大不相同,另外金屬媒介 物的用量太大,反應結束後難以除去也是一大困擾之處。Miller 等人 於 1998 年,引入 Ni(cod)2/cyclooctadiene/2,2-bipyridyl 的配方,並將 聚合反應於甲苯及 N,N-二甲基甲醯胺(toluene-DMF)的溶劑中進行,可 將 poly(9,9-dihexylfluorene)的分子量進一步的提高到 Mn約 250,000, 將近有 500 個單體單元之多(見圖 1-16)[34]。從此之後,Uniax 與 Dow Chemical 兩家公司所販售的商品化聚芴高分子多是以此法合成,且有 相當多的專利問世[35-36]。 將 Suzuki 聚合法應用於聚芴最早由 Leclerc.等人所提出[37],其係 利用少量的 Pd(PPh3)4 做為催化劑,加入一介面活性劑於水及甲苯的 共溶劑中行聚合反應(見圖 1-17),此舉除了大大的減少上述 Yamamoto 聚合法中大量金屬媒介物的問題之外,單體之間的聚合也是遵行一對 一規則結合,即硼酯類單體與含有溴的單體的偶合,反應條件較為穩 定,但分子量通常較小,Mn 約 5,000 ~ 40,000。值得注意的是,在分 子量的控制上,除了聚合方法之外,最重要的是單體的純度究竟夠不 夠高,越純的單體所得高分子的分子量越大,當然芴環的側鏈取代基 大小及種類同時也決定了高分子的分子量。
圖 1-16 Yamamoto 偶合法 圖 1-17 Suzuki 偶合法 目前文獻上發表的聚芴高分子(polyfluorene)的數量平均分子量 (Mn)通常介於 10,000~200,000 之間,PDI 介於 1.5 ~ 3 之間。熱裂解 溫度(Td)介於 350 ~ 400 oC 之間,甚至某些較為堅硬的結構,Td亦有 可能超過 400 oC [32,38]。若芴環側鏈接有較長的取代基時[-dioctyl[39] 或-bis(2-ethylhexyl)[40]],則有可能會表現出液晶相,對於發展偏極化 的電激發光元件亦相當有幫助[38,41]。相較於其它的發光材料來說, 在光激發光量子效率方面,聚芴高分子的值通常較高,並介於 40~80% 之間。
1.6.2 聚芴藍光材料回顧
此處主要收錄2000 年以後效率較佳的聚芴藍光共軛高分子。
圖 1-18 PF2-6amX,雙層元件結構 ITO / PEDOT / 發光體 /
Ca,驅動電壓 3.5 V,CIE 座標 ( x = 0.150, y = 0.080 ),最大效率 1.1cd/A,最大亮度 1,600 cd/m2。多層元件結構,以三苯基胺類化 合物做為電洞傳輸層,效率可達 2.7 cd/A,最大亮度超過 5,000 cd/m2。[42] 圖1-19 P4,雙層元件結構 ITO/PEDOT/發光體/Ca/Ag,CIE 座標 ( x = 0.150, y = 0.160 ),於 100 cd/m2下可顯現效率 3.0 cd/A,驅動 電壓(Von) 4.6 V[43]。
圖1-20 PF-OXD,雙層元件結構 ITO/PEDOT/發光體/Ca/Ag,驅 動電壓(Von)為 5.3 V,在 10.8 V 下可達最大亮度 2770 cd/m2,最大 效率為0.25 cd/A[44]。 圖 1-21 PFO + 50% DFD,雙層元件結構 ITO/PEDOT/發光體 /Ca/Al,在驅動電壓為 10 V 下,效率為 0.20 cd/A,亮度為 100 cd/m2[45]。
1.6.3 Anthracene(蒽)衍生物的發展與性質介紹
[46] 有關 Anthracene 的發展已於 1.1.1 提及,此處主要是回顧蒽衍生 物 — 9,10-di(2-naphthyl)anthracene ( ADN )之相關文獻。 美國柯達的 OLED 研究團隊於美國專利中首次發表了以 ADN 為 主體的衍生物,ADN 在液態和固態均有相當好的螢光效率,目前已 成為 OLED 元件中被廣泛應用的藍光主發光體材料之ㄧ。2002 年石 建民及鄧青雲博士首度將柯達公司使用的藍光主發光體材料 ADN 發 表於期刊上,在此論文中將不同濃度的 tetra(t-butyl)perylene (TBP)摻雜於 ADN 中,在元件結構為 ITO (35 nm)/ CuPc (25 nm)/ NPB (50 nm)
/ADN:TBP(30 nm)/Alq3(40 nm)/Mg:Ag(200 nm)中,可得到藍光元件。
未摻雜 TBP 的元件 CIEx,y座標為(0.20, 0.26),摻雜 TBP 後元件 EL 圖
就呈現 TBP 的波形。顯見兩者間可以有很好的能量轉移,由於半波
寬變窄,元件光色變為CIEx,y(0.15, 0.23),發光效率更提升為 3.5 cd/A。
未摻雜元件壽命在起始亮度為 384 cd/m2下可達2000h,摻雜 TBP 後
元件壽命在起始亮度為636 cd/m2下可達 4000h,在當時公開發表的期
刊中可算是最穩定的藍光發光體。
在長時間電場操作下或升溫(95 oC)迴火(annealing)程序中,ADN
綠(CIEx,y=0.20, 0.26)。所以 Kodak 團隊在歐洲專利提出含 tert-butyl
取代基的衍生物,2-(t-butyl)-9,10-di(2-naphthyl)anthracene (TBADN)
來改善這些問題,在文獻中利用相同摻雜物及元件架構,TBADN 可
以發出深藍光,其CIEx,y為(0.13, 0.19)。不同取代基的 ADN 衍生物的
液態螢光光譜(在甲苯溶液下)及其熱性質比較列在表 1-3[46],ADN 衍 生物分子結構圖見圖 1-22[46]。 圖1-22 ADN 衍生物分子結構圖 表 1-3 ADN 衍生物的液態螢光光譜及其熱性質比較。 Compound Peak(nm) Td (oC) Tm (oC) Tg (oC) ADN 427 396 388 - TBADN 430 408 291 128 MADN 430 397 255 120
1.6.4 聚蒽藍光材料回顧
此處主要收錄2000 年以後效率較佳的含蒽藍光共軛高分子。 圖 1-23 XPa,雙層元件結構 ITO/PEDOT:PSS/發光體/LiF/Ca /Al,EL 光譜放射峰約在 468 nm,最大亮度為 587 cd/m2,當驅動 電壓為13 V 時,效率可達 0.26 cd/A[47]。 圖1-24 P2,雙層元件結構 ITO/PEDOT:PSS/發光體/Mg:Ag,EL 光譜放射峰約在468 nm,在電流密度為 20 mA/cm2下可達最大效 率0.4 cd/A[48]。圖 1-25 A,多層元件結構 ITO/PEDOT:PSS/polymer/BCP/Alq3 /Mg:Ag,EL 光譜放射峰為 538 nm,在電流密度為 0.1 A/cm2下可 達最大效率0.19 cd/A[49]。 圖 1-26 15 , 雙 層 元 件 結 構 ITO/PEDOT:PSS/polymer/ BaF2/Ca/Al,驅動電壓(Von)為 4.7 V,其最大亮度為 1000 cd/m2, 當驅動電壓為6.4 V 時有最大效率 0.7 cd/A[50]。
1.7 研究動機
有機電激發光二極體近年來廣為學術界、工業界所廣泛研究。電 激發光元件的壽命、亮度、效率等性質為評定一發光元件好壞以及是 否適合商業化的首要指標。目前小分子有機電激發光元件在穩定性、 色純度及效率上都優於高分子元件,但以高分子材料為主的有機電激 發光元件仍受重視,主要原因即為高分子材料可採用如旋轉塗佈 (spin coating) 或噴墨印刷 (inkjet printing) 等成膜技術,這些薄膜製 作程序具有快速、簡易和低成本的優點。而高分子發光二極體由於在 大面積的平面顯示器應用方面亦有不錯的潛力而日益受到重視。為達 到全彩化顯示的目的,必須擁有同時兼具高效率、色澤穩定且高色純 度的紅、綠、藍發光材料;而目前在這三種主要發光材料中,最急需 突破的就是研發出高效率的藍光材料。擁有一個好的藍光材料,其本 身除了可以發藍光外,還能在材料本身摻雜其他較低能隙的材料,如 綠光或是紅光材料,利用快速且有效率的能量轉換(Energy conversion) 與調控摻雜物(dopant)的比例等方式,發出其他顏色甚至是白色的 光。至於身為一個好的藍光材料首要條件,就是材料本身的能隙 (Energy gap)要夠大;而當能隙夠大時,其 HOMO 與 LUMO 能階也必 要須能夠配合其他像是電洞或電子傳輸層的相對應之能階,才能達到高發光效率元件的門檻。 我們選用Anthracene(蒽)的衍生物 – TBADN,做為藍光高分子材 料的基礎,其原因包括 Anthacene 衍生物無論在液態或是固態都具有 不錯的螢光量子效率,並且在Anthracene 分子上有 2,6 與 9,10 四個位 置可以作官能基的修飾,而在這四個位置上若接有立體障礙較大的取 代基,如苯環衍生物,會與環繞在Anthracene 分子上的氫原子產生排 斥力,造成分子結構的扭曲,破壞共軛高分子的平面性,利用這個特 點,我們可以有效的控制共軛長度;另外還選用 Fluorene(芴)的衍生 物做為建構此藍光高分子的另一部份,其理由不外乎是Fluorene 衍生 物容易藉由不同取代基的導入達到改善溶解度、增加電子或電洞傳輸 的能力,而在作為光電材料的表現上亦擁有不錯的光激發光與電激發 光效率,並且具有極佳的熱穩定性。在本研究中,利用巨大的苯環衍 生物導入在芴分子的九號位置,另外還導入具有傳輸電子能力的 1,2,3-三氮唑(1,2,3-triazole units)官能基,希望藉由這些官能基在高分 子側鏈的修飾,一方面可以抑制因高分子鏈堆疊或是芴分子九號位置 受熱及電壓操作下氧化產生的 Keto defect,另一方面透過較為平衡的 正負電荷傳輸以增進發光材料中電子電洞再結合的能力。最後我們結 合這兩種極具潛力的小分子,透過聚合的方式得到較大分子量的高分 子以獲得成膜性,利用旋轉塗佈等製程方式應用於元件上,並對這些
第
2章 實驗部分
2.1 試藥
實驗中所使用之藥品均分別採購自Aldrich、Merck、Acros、TCI、
Alfa Aesar 與聯工公司。所有溶劑皆購自 Merck 及 Fischer 公司。無水
四氫呋喃( tetrahydrofuran THF )以鈉金屬除水,並加入二苯甲酮 ( benzophenone )為指示劑,在氮氣條件下迴流二日後蒸餾出使用。無 水甲苯( toluene )以氫化鈣除水,在氮氣條件下迴流二日後蒸餾出使 用。
2.2 量測儀器
為了鑑定中間產物、前驅物單體或聚合物之化學結構及物理特 性,採用下列測試儀器:2.2.1 核磁共振光譜儀
( Nuclear Magnetic Resonance,NMR )使用Varian-300 MHz 核磁共振光譜儀。其中以 d-chloroform 作為
溶劑,化學位移單位為 ppm,氫譜分別以 δ = 0.00 (TMS) or 7.26
為內部基準。光譜資料中:符號 s 表示單峰(singlet),d 表示二重峰 (doublet),t 表示三重峰(triplet),q 表示四重峰(quartet),m 則表示多 重峰 (multiplet)。
2.2.2 微差掃描卡計
( Differential Scanning Calorimeter, DSC )使用TA Instruments Unpacking the Q Series DSC 及 RCS 冷卻系統
提供低溫環境。實驗所需秤取樣品 2 ~ 5 mg,加熱及冷卻的掃描速率
分別為 10 oC/min,以溫度對熱流作圖,取圖形的最大反曲點(inflection
point)為玻璃轉移溫度(glass transition temperature, Tg)。
2.2.3 熱重分析儀
( Thermal Gravimetric Analyzer, TGA )使用Perkin Elmer Pyris 熱重分析儀。實驗所需樣品 2 ~ 5 mg,樣
品之加熱速率為 10 oC/min,範圍從 50 oC ~ 750 oC,並在氮氣流量為
100 mL/min 下測量其熱裂解情形,將樣品重量損失 5 wt %處定義為熱 裂解溫度(thermal decomposition temperature, Td)。
2.2.4 凝膠滲透層析儀
( Gel Permeation Chromatography,GPC )使用Viscotek VE2001GPC 高壓幫浦系統,偵測器為 Viscotek T50A
American Polymer Column,所填充之 Gel 尺寸大小各為 105、104 和 103Å,並使用 polystyrene 標準樣品製作分子量校正曲線。測試時以 THF 為沖提液,並保持於 35 oC 的恆溫槽中。樣品溶液之配製方式為 將秤取好的 4.0 mg 聚合物溶於 2 mL THF 中,將配置溶液超音波震盪 15 分鐘後,以 0.2 μm 的 PTFE filter 過濾後使用。
2.2.5 紫外線與可見光光譜儀
( UV-Vis Spectrophotometer ) 使用HP 8453 型 UV-Visible 光譜儀。用以偵測樣品之吸收光譜, 量測時樣品以溶劑溶解後置於石英槽內,或將溶液滴在ITO 玻璃上直 接旋轉塗佈成膜後量測。光譜單位為 nm。2.2.6 螢光光譜儀
( Photoluminescence Spectrophotometer ) 使用ARC SpectraPro-150 型螢光光譜儀。用以偵測樣品之放射光 譜,儀器使用之激發光源為 450W 之 Xenon 燈,量測時激發波長根據 個 別 樣 品 之 吸 收 光 譜 而 有 所 不 同 , 所 得 數 據 即 為 光 激 發 光 (photoluminescence, PL)光譜。光譜單位為 nm。2.2.7 循環伏安計量儀
( Cyclic Voltammetry, CV ) 使用Autolab ADC 164 型電位儀來記錄氧化-還原電位,以 0.1 M之(n-Bu4)NBF4 (tetra-n-butylammonium tetrafluoroborate)的 CH2Cl2溶
液為電解液,將待測物溶在電解液內,以 Ag/AgCl 為參考電極
(reference electrode),ferrocene/ferrocenium (Fc/Fc+)為內參考電位,白
金 片 作 為 工 作 電 極(work electrode) , 白 金 絲 為 對 應 電 極 (counter
electrode)。量測時以 50 mV/sec 的速率掃描記錄其氧化還原曲線。
2.2.8 光譜掃描色度計
( Spectroscan Colorimeter, PR-650 ) PR-650 型。將元件施以電壓趨動發光之後,再以此光譜色度計量 測發光強度與光色。2.3 合成部分
關於單體M1~M3、高分子F-T50~TAZ-T50 及末端終止試劑 1 與 2 的合成示意圖,見 Scheme 1 ~ Scheme 3,實驗步驟則於 2.3.1 與 2.3.2 中詳述,至於各化合物相關的光譜鑑定見附圖 17 ~ 43。Scheme 4. 末端終止試劑 1 與 2 的合成
2.3.1 單體 M1~M3
的合成
9,10-Bis(6-bromonaphthalen-2-yl)-2-tert-butyl-9,10-dihydroanthra- cene-9,10-diol (1) 之合成 將2,6-dibromonaphthalene (6.49 g,22.7 mmol)放入 250 ml 三頸 甁,2-tert-butylanthraquinone (3 g,11.4 mmol)放入固體加料甁,在真 空下以火焰除水後,於氮氣環境下用針筒注入 dry THF (約 150 mL) 將 2,6-dibromonaphthalene 溶解,在-78 oC 下用針筒緩慢注入 1.6 Mn-BuLi (10.64 mL,17.1 mmol)並維持在低溫攪拌 1 小時後,於-78 oC
下加入 2-tert-butylanthraquinone,使反應逐漸回溫並繼續攪拌 12 hr。
加入適量 H2O 除掉尚未反應掉的 n-BuLi,上 rota vapor 除去 THF 後,
再用 ethyl acetate(EA)與 H2O 萃取,收集有機層,以無水 MgSO4除水
後濃縮,最後用管柱層析(EA : Hexane = 1 : 4)純化,得白色固體約 5.94
g,產率: 77%。 MS (FAB-MS) m/z︰678。 1H-NMR (300 MHz, CDCl3)
δ 1.32 (s, 9H, CH3), 2.93 (s, 1H, -OH), 2.98 (s, 1H, -OH), 6.98 (s, 1H,
aromatic protons), 7.02 (d, J = 5.1 Hz, 3H, aromatic protons), 7.23 (dd, J = 1.5 Hz, J = 8.7 Hz, 2H, aromatic protons), 7.29 (d, J = 2.1 Hz, 1H, aromatic protons), 7.31 (d, J = 1.8 Hz, 1H, aromatic protons), 7.35 (d, J = 8.7 Hz, 2H, aromatic protons), 7.41~7.48 (m, 2H, aromatic protons), 7.66 (s, 2H, aromatic protons), 7.71 (d, J = 8.1 Hz, 2H, aromatic protons), 7.75~7.83 (m, 3H, aromatic protons)。 13C-NMR (75 MHz, CDCl3) δ 31.3, 34.8, 75.1, 75.4, 120.1, 123.6, 125.4, 126.1, 126.3, 126.4, 126.4, 126.5, 126.5, 126.7, 126.8, 128.2, 128.9, 129.4, 130.5, 133.0, 138.1, 140.3, 141.1, 141.2, 141.6, 141.7, 151.4。 9,10-Bis(6-bromonaphthalen-2-yl)-2-tert-butylanthracene (M1)之合成 將1 (7.08 g,10.4 mmol)、KI (6.24 g,37.6 mmol)、NaH2PO2 (7.52
g,70.9 mmol)置入 250 mL 雙頸甁中,加入適量醋酸(約 100 mL),在 110 oC 下加熱迴流 2 hr,反應回溫後,會有淡黃色固體析出,將整鍋 溶液滴入約 1 L 的水中劇烈攪拌 6 hr 後,抽氣過濾,並用大量清水清 洗固體後上真空抽乾,不再作進一步的純化,可得淡黃色固體6.05 g, 產率: 90 %。 MS (FAB-MS) m/z︰644。 1H-NMR (300 MHz, CDCl3) δ 1.22 (s, 9H, CH3), 7.28~7.33(m, 2H, aromatic protons), 7.44 (dd, J = 2.1
Hz, J = 9.3 Hz, 1H, aromatic protons), 7.62~7.70 (m, 8H, aromatic protons), 7.78~7.83 (m, 2H, aromatic protons), 7.97~8.03 (m, 4H, aromatic protons), 8.20~8.21 (m, 2H, aromatic protons)。 13C-NMR (75 MHz, CDCl3) δ 30.7, 34.9, 120.2, 121, 124.8, 124.9, 125.1, 126.1, 126.8,
127.1, 128.6, 129.8, 129.9, 130.2, 130.6, 131.8, 133.8, 136.2, 136.4, 137.2, 147.7。 Anal. Calcd. for C38H30Br2: C, 70.60 ; H, 4.68. Found: C, 70.47 ;
H, 4.78。
2,7-Dibromo-9,9’-bis-(4-hydroxyphenyl)fluorene (2) 之合成
取一 250mL 雙頸甁,在氮氣下置入 2,7-dibromo-9-fluorenone (4 g,
11.8 mmol)和 phenol (8 g, 85 mmol),在 150 oC 下慢慢用加液漏斗滴
入 Eaton’s reagent (24 mL)。滴完後再反應 20 min,待反應回溫,將
水 MgSO4除水後濃縮,最後用管柱層析(EA : Hexane = 1 : 4)純化,可
得一白色固體 4.21 g,產率: 70 %。 MS (EI-MS) m/z︰508。 1H-NMR
(300 MHz, DMSO-d6) δ 6.66 (d, J = 9 Hz, 4H, aromatic protons), 6.90 (d,
J = 8.7 Hz, 4H, aromatic protons), 7.49 (d, J = 1.8 Hz, 2H, aromatic protons), 7.57 (dd, J = 1.8 Hz,J = 8.4 Hz, 2H, aromatic protons), 7.89 (d, J = 8.1 Hz, 2H, aromatic protons), 9.41 (s, 2H, phenol-OH)。
13C-NMR (75 MHz, DMSO-d 6) δ 116.2, 122.0, 123.7, 129.5, 129.6, 131.6, 135.4, 138.5, 154.8, 157.3。 2,7-Dibromo-9,9’bis-[(4-hexyloxy)phenyl]-9H-fluorene (M2) 之合成 將 2 (6 g,11.8 mmol)、K2CO3(4.90 g,35.4 mmol)、KI(0.39 g, 2.36 mmol)與適量 Acetone(約 60 mL)置入 100mL 雙頸甁內,在氮氣 環境下攪拌 30 min,再緩慢打入 1-bromohexane,於 80 oC 下加熱迴
流 12 hr。反應回溫後,上 rota vapor 將 solvent 抽乾,用稀鹽酸水溶液
與 EA 萃取,收集有機層,以無水 MgSO4除水後濃縮,最後用管柱層
析(Hexane)純化,得白色固體 4.05 g,產率: 52 % 。 MS (FAB-MS) m/z︰676。 1H-NMR (300 MHz, CDCl3) δ 0.92 (t, J = 6.6 Hz, 6H,-
OCH2(CH2)4-CH3), 1.34~1.47 (m, 12H, -OCH2-(CH2)4-CH3), 1.72~1.79
6.79 (d, J = 9 Hz, 4H, aromatic protons), 7.08 (d, J = 8.1 Hz, 4H, aromatic protons), 7.46 (d, J = 1.5 Hz, 2H, aromatic protons), 7.49 (d, J = 3.6 Hz, 2H, aromatic protons), 7.57 (d, J = 8.1 Hz, 2H, aromatic protons)。
13C-NMR (75 MHz, CDCl
3) δ 14.0, 22.6, 25.7, 29.2, 31.5, 64.3, 67.9,
114.3, 121.5, 121.8, 129.0, 129.2, 130.7, 136.2, 137.8, 153.7, 158.2。 Anal. Calcd. for C37H40Br2O2: C, 65.69 ; H, 5.96. Found: C, 65.59 ; H,
5.83。 2,2’-{9,9’-[Bis(4-hexyloxy)phenyl]-9H-fluorene-2,7-diyl}bis(4,4,5,5-tet ramethyl-1,3,2-dioxaborolane) (M3) 之合成 將 M2 (4.05 g,5.98 mmol)置入 100 mL 的雙頸甁中,在真空下以 火焰除水後,在氮氣環境下打入 dry THF 將其溶解,於 -78 oC 下用 針筒緩慢注入 2.5 M n-BuLi ( 5.98 mL,15 mmol ) 並維持在低溫 攪拌 1 小時後,於-78 oC 下用針筒緩慢打入 2-isopropoxy-4,4,5,5- tetramethyl-1,3,2-dioxaborolan (3.66 mL, 17.9 mmol),使反應逐漸回溫 並繼續攪拌 12 hr。加入適量 H2O 除掉尚未反應掉的 n-BuLi,上 rota vapor 除去 THF 後,再用 EA 與 H2O 萃取,收集有機層,以無水 MgSO4 除水後濃縮,用EA 作再結晶,可得白色固體約 3.10 g,產率: 59 %。 MS (FAB-MS) m/z︰770。 1H-NMR (300 MHz, CDCl3) δ 0.88 (t, J = 6 Hz, 6H, -OCH2(CH2)4-CH3), 1.25~1.44 (m, 36H, -OCH2-(CH2)4-CH3,
-CH3), 1.69~1.78 (m, 4H, -OCH2-(CH2)4-CH3), 3.89 (t, J = 6.6 Hz, 4H,
-OCH2-(CH2)4CH3), 6.73 (d, J = 9 Hz, 4H, aromatic protons), 7.12 (d, J =
8.7 Hz, 4H, aromatic protons), 7.75~7.82 (m, 6H, aromatic protons)。
13C-NMR (75 MHz, CDCl
3) δ 14.0, 22.6, 24.9, 25.7, 29.3, 31.6, 64.1, 67.8,
83.7,114.0, 119.8, 129.4, 132.2, 134.0, 137.6, 142.6, 151.8, 157.7。Anal. Calcd. for C49H64B2O6: C, 76.37 ; H, 8.37. Found: C, 76.03 ; H, 8.32。
N,N-Bis(4-methylphenyl)-N-(4-bromophenyl)amine (End-capping
reagent 1) 之合成
將 4-bromo aniline (5 g,29 mmol)、1-iodotoluene (15.84,72.6
mmol)、CuCl (0.143g,1.44 mmol)、1,10-phenanthroline (0.262,1.44 mmol)、KOH (13.044g,232 mmol)及 Toluene (約 120 ml)加入 250 mL
雙頸甁中,在氮氣環境下約 130 oC 加熱迴流 24 hr 後,反應回溫,加
入稀鹽酸中和成中性,再用 EA 與 H2O 萃取,收集有機層並以無水
MgSO4除水後濃縮,最後用管柱層析(Hexane)純化,可得白色固體 4.63
g,產率: 45 %。 MS (EI-MS) m/z︰352。 1H-NMR (300 MHz, CDCl3)
δ 2.31 (s, 6H, CH3), 6.88 (d, J = 9 Hz, 2H, aromatic protons), 6.96 (d, J =
8.4 Hz, 4H, aromatic protons), 7.06 (d, J = 8.4 Hz, 4H , aromatic protons), 7.28 (d, J = 2.1 Hz, 2H, aromatic protons)。13C-NMR (75 MHz, CDCl3)
δ 113.6, 123.9, 124.6, 130.0, 131.9, 132.9, 144.9, 147.4。 Anal. Calcd. for C20H18BrN: C, 68.19 ; H, 5.15. Found: C, 68.31 ; H, 5.28。
N,N-Di(4-methylphenyl)-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan
-2-yl)phenyl] amine (End-capping reagent 2) 之合成
將 N,N-Bis(4-methylphenyl)-N-(4-bromophenyl)amine (5 g , 14.2 mmol)放入 100mL 雙頸甁中,在真空下以火焰除水後,於氮氣環境下 用 針 筒 注 入 dry THF ( 約 30 mL) , 將 N,N-Bis(4-methylphenyl)- N-(4-bromophenyl)amine 溶解,在 -78 oC 下用針筒緩慢注入 2.5 M n-BuLi ( 7.38 mL,18.5 mmol ) 並維持在低溫攪拌 1 小時後,於-78 oC 下用針筒緩慢打入 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolan (3.77 mL,18.5 mmol),使反應逐漸回溫並繼續攪拌 12 hr。加入適量
H2O 除掉尚未反應掉的 n-BuLi,上 rota vapor 除去 THF 後,再用 EA
與 H2O 萃取,收集有機層,以無水 MgSO4除水後濃縮,先用管柱層
析( Hexane )純化,最後再用 Hexane 作再結晶,可得白色固體約 2.56
g,產率: 45 % 。 MS (EI-MS) m/z︰399。 1H-NMR (300 MHz, CDCl3)
δ 1.33 (s, 12H, CH3), 2.32 (s, 6H, CH3), 6.97 (d, J = 12.3 Hz, 2H,
aromatic protons), 6.99 (d, J = 12.3 Hz, 4H, aromatic protons), 7.07 (d, J = 8.1 Hz, 4H, aromatic protons), 7.62 (d, J = 8.4 Hz, 2H, aromatic
protons)。 13C-NMR (75 MHz, CDCl3) δ 20.8, 24.8, 83.4, 120.4, 120.5,
125.2, 129.9, 133.1, 135.7, 144.8, 150.9。 Anal. Calcd. for C26H30BNO2 :
C, 78.2 ; H, 7.57. Found: C, 78.45 ; H, 7.24。
2.3.2 高分子 F-T50, DPF-T5~DPF-T50, TAZ5-DPF-T45 與
TAZ-T50 的合成
高分子 TAZ-T50 的合成 將2,2-bipyridine (0.19 g ,1.24 mmol)、1,5-cyclooctadiene (0.15 mL , 1.24 mmol) 先 置 入 50 mL 雙 頸 甁 中 , 再 到 手 套 箱 秤 取 bis(1,5-cyclooctadiene) Nickel(0) (0.34 g,1.24 mmol)後,於氮氣環境下打入dry DMF 與 dry Toluene 各 5 mL,並於 80 oC 下加熱 1 hr。然後
將 M1 (0.2 g,0.31 mmol)與 M5 (0.30 g,0.31 mmol)溶在適量 dry
Toluene(約 8 mL)中再打入反應甁,繼續反應 5 天。於第 6 天加入 End-capping reagent 1(0.1g,0.28 mmol)再反應一天,待反應回溫後,
將其滴入約 500 mL 的 Methanol 中做再沉澱,重力過濾收集固體,再
次用適量 THF 將固體溶解,滴入 Methanol 中做再沉澱,一直重覆上
述再沉澱步驟至所收集到的固體顏色由灰黑變黃綠色,再用 Acetone
作為溶劑,連續萃取3 天後,收集固體上真空抽乾,可得ㄧ金黃色固
-O(CH2)5CH3), 1.21~1.53(m, 21H, -OCH2CH2(CH2)3CH3, C(CH3)3), 1.78(m, 4H, -OCH2CH2(CH2)3CH3), 3.95~3.99(m, 4H, -OCH2(CH2)4CH3), 6.91~8.21(m, 43H, aromatic protons)。 高分子 F-T50 的合成 將M1 (0.3 g,0.46 mmol)、M4 (0.30 g,0.46 mmol)、K2CO3(0.49 g, 3.51 mmol)、Aliquat 336(0.06 g,0.14 mmol)先置入 50 mL 雙頸甁中, 再到手套箱秤取 Pd(PPh3)4 (0.01 g,0.0093 mmol),裝置迴流管與血清 塞,雙頸甁外以鋁箔紙包覆,在氮氣環境下打入 dry Toluene(約 15 mL) 與除氣後的 H2O(約 3 mL),於 85 oC 下加熱 5 天。於第 6 天加入
End-capping reagent 1 (0.1g,0.28 mmol)反應一天後,於第 7 天加入 End-capping reagent 2 (0.1g,0.25 mmol)再反應一天。待反應回溫後,
將其滴入約 200 mL 的 Methanol 中做再沉澱,重力過濾後收集固體, 再用 Acetone 作為溶劑,連續萃取 3 天後,將收集到的固體上真空抽 乾,可得黃綠色固體約 0.23 g,產率: 58 % 。 1H-NMR (300 MHz, CDCl3) δ 0.80~0.89(m, 10H, -(CH2)6CH2CH3), 1.17~1.48(m, 29H, -CH2(CH2)5CH2CH3,-C(CH3)3), 1.98~2.22(m, 4H, -CH2(CH2)6CH3), 7.34~8,34(m, 25H, aromatic protons)。 高分子 DPF-T5 的合成
將M1 (0.02 g,0.03 mmol)、M2 (0.20 g,0.29 mmol)、M3 (0.25 g,
0.32 mmol)、K2CO3(0.33 g,2.42 mmol)、Aliquat 336(0.06 g,0.14 mmol)
先置入 50 mL 雙頸甁中,再到手套箱秤取 Pd(PPh3)4 (0.007 g,0.0064
mmol),裝置迴流管與血清塞,雙頸甁外以鋁箔紙包覆,在氮氣環境
下打入 dry Toluene(約 10 mL)與除氣後的 H2O(約 2 mL),於 85 oC 下
加熱 5 天。於第 6 天加入 End-capping reagent 1 (0.1g,0.28 mmol)反應
一天後,於第7 天加入 End-capping reagent 2 (0.1g,0.25 mmol)再反應
一天。待反應回溫後,將其滴入約200 mL 的 Methanol 中做再沉澱, 重力過濾後收集固體,再用Acetone 作為溶劑,連續萃取 3 天後,將 收集到的固體上真空抽乾,可得棕色固體約 0.17 g,產率: 52 %。 1H-NMR (300 MHz, CDCl 3) δ 0.86~0.90(m, 6H, -O(CH2)5CH3), 1.22~1.43(m, 12H, -OCH2CH2(CH2)3CH3), 1.55~1.75 (m, 4H, -OCH2CH2 (CH2)3CH3), 3.86~3.91(m, 4H, -OCH2(CH2)4CH3), 6.74~6.76(m, 4H,
aromatic protons), 7.14~7.17(m, 4H, aromatic protons), 7.50~7.55(m, 4H aromatic protons), 7.73~7.76(m, 2H, aromatic protons)。
高分子 DPF-T25 的合成
將M1 (0.13 g,0.19 mmol)、M2 (0.13 g,0.19 mmol)、M3 (0.3 g,
先置入 50 mL 雙頸甁中,再到手套箱秤取 Pd(PPh3)4 (0.009 g,0.0078
mmol),裝置迴流管與血清塞,雙頸甁外以鋁箔紙包覆,在氮氣環境
下打入 dry Toluene(約 10 mL)與除氣後的 H2O(約 2 mL),於 85 oC 下
加熱 5 天。於第 6 天加入 End-capping reagent 1 (0.1g,0.28 mmol)反應
一天後,於第7 天加入 End-capping reagent 2 (0.1g,0.25 mmol)再反應
一天。待反應回溫後,將其滴入約200 mL 的 Methanol 中做再沉澱, 重力過濾後收集固體,再用Acetone 作為溶劑,連續萃取 3 天後,將 收集到的固體上真空抽乾,可得深綠色固體約0.27 g,產率: 70 %。 1H-NMR (300 MHz, CDCl 3) δ 0.71(m, 6H, -O(CH2)5CH3), 1.10~1.50(m, 21H, -OCH2CH2(CH2)3CH3, C(CH3)3), 1.64~1.74 (m, 4H, -OCH2CH2 (CH2)3CH3), 3.89~3.91(m, 4H, -OCH2(CH2)4CH3), 6.74~8.23(m, 33H, aromatic protons)。 高分子 DPF-T50 的合成 將M1 (0.3 g,0.39 mmol)、M3 (0.25 g,0.39 mmol)、K2CO3(0.41 g, 2.94 mmol)、Aliquat 336(0.07 g,0.17 mmol)先置入 50 mL 雙頸甁中, 再到手套箱秤取 Pd(PPh3)4 (0.009 g,0.0078 mmol),裝置迴流管與血 清塞,雙頸甁外以鋁箔紙包覆,在氮氣環境下打入 dry Toluene(約 10 mL)與除氣後的 H2O(約 2 mL),於 85 oC 下加熱 5 天。於第 6 天加入
End-capping reagent 1 (0.1g,0.28 mmol)反應一天後,於第 7 天加入 End-capping reagent 2 (0.1g,0.25 mmol)再反應一天。待反應回溫後,
將其滴入約 200 mL 的 Methanol 中做再沉澱,重力過濾後收集固體, 再用 Acetone 作為溶劑,連續萃取 3 天後,將收集到的固體上真空抽 乾,可得黃綠色固體約 0.29 g,產率: 75 % 。 1H-NMR (300 MHz, CDCl3) δ 0.87~0.89(m, 6H, -O(CH2)5CH3 ), 1.17~1.49(m, 21H, -OCH2CH2(CH2)3CH3, C(CH3)3), 1.74~1.77(m, 4H, -OCH2CH2(CH2)3- CH3), 3.94(m, 4H, -OCH2(CH2)4CH3), 6.85~8.23(m, 33H, aromatic protons)。 高分子 TAZ5-DPF-T45 的合成 將M1 (0.20 g,0.32 mmol)、M3 (0.27 g,0.35 mmol)、M5 (0.03 g,
0.03 mmol)、K2CO3(0.37 g,2.66 mmol)、Aliquat 336(0.06 g,0.15 mmol)
先置入 50 mL 雙頸甁中,再到手套箱秤取 Pd(PPh3)4 (0.008 g,0.0071
mmol),裝置迴流管與血清塞,雙頸甁外以鋁箔紙包覆,在氮氣環境
下打入 dry Toluene(約 10 mL)與除氣後的 H2O(約 2 mL),於 85 oC 下
加熱 5 天。於第 6 天加入 End-capping reagent 1 (0.1g,0.28 mmol)反應
一天後,於第7 天加入 End-capping reagent 2 (0.1g,0.25 mmol)再反應








![HPSH [ 分子間作用力 - 凡得瓦力 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)