芳杯、賽吩與二賽吩於金(111)上的自組裝研究
148
0
0
全文
(2) 芳杯、賽吩與二賽吩於金(111)上的自組裝研究. 學生 : 林昌諺. 指導教授 : 楊耀文 博士. 國立交通大學應用化學所. 中文摘要. 芳杯、賽吩與二賽吩在應用上都具有極大的潛力。芳杯在超分子 化學的主客化學範疇中被廣泛的應用作為主體,並且可應用於分子辨 識或是感測器,而賽吩與二賽吩因為在電子結構上具有去區域性的共 軛π電子雲,有助於電荷的傳導,所以其高分子在光電材料的應用上 相當的普遍,例如發光二極體、場效應電晶體與光伏特電池等。 本實驗利用真空吸附與溶液浸泡來製備芳杯、賽吩與二賽吩於金 (111)上的自組裝薄膜,並且利用 STM、EC-STM、TPD、XPS 與 NEXAFS 等表面分析技術來進行樣品的研究,希望能夠對於吸附分子 的電子結構、鍵結強度及分子排列結構有更深入的了解。 從芳杯的 STM 與 EC-STM 影像中並未能觀察到吸附分子的規則 排列結構,而 NEXAFS 光譜結果中,吸收譜線並不會隨 X 光入射角 的改變而有變化,表示芳杯分子並非以一特定傾角吸附於表面上。透 過 XPS 的量測,推測芳杯吸附於金(111)表面上時,分子中的兩個硫 醇基並非會同時與金進行強化學鍵結,因此導致吸附分子無法以一特. ii.
(3) 定傾角存在於表面上。 從真空吸附與溶液浸泡所得到的二賽吩自組裝分子在 XPS 與 TPD 實驗結果中有相當大的差異。利用真空吸附所得到的二賽吩分子 是以弱鍵結與金表面作用,而溶液浸泡所得到的二賽吩與表面間則是 以強化學鍵來進行吸附。STM 影像中並未能看到二賽吩吸附分子的 規則排列結構。雖然於 NEXAFS 光譜結果中譜線會隨著 X 光入射角 的改變而有變化,但是由於數據不足,所以無法準確的判斷二賽吩吸 附於表面上的傾角。於 STM 影像中並未能觀察到二賽吩分子的規則 排列結構。賽吩分子於各實驗中所得到的結果與二賽吩的結果相類 似。. iii.
(4) 誌謝. 兩年的光陰,說短不短,說長也不長。現在回頭看來,兩年似乎 轉眼間就過去了,但是其中各種喜怒哀樂、令人難以忘懷的回憶,又 讓人覺得兩年的歲月格外漫長。要畢業了,馬上就要離開這個待了兩 年的地方,說完全不會懷念、不會感傷,我想都是騙人的吧。對我來 說,在研究所期間所獲得的,絕不只是一張薄薄的畢業證書與碩士學 位而已,從各種實驗器材的準備、儀器的組裝與操作,到數據的處理 與分析,都讓我學習到了許多寶貴的知識與經驗。雖然只是短短兩 年,但是其中所得到的卻遠比大學四年多出許多。 在這裡首先要感謝我的指導教授楊耀文博士,感謝老師在這兩年 期間不論是研究或是生活上對我的指教與照顧。在楊老師的帶領之 下,我才有機會能夠在同步輻射研究中心內,利用各種高級的儀器來 進行表面科學的研究。在這裡的一切所見所聞,都大大開拓了我的視 野。再來感謝范良任學長在實驗方面所給予的所有教導與協助,在范 學長耐心的教導與幫助下,我的各項實驗才得以順利的進行下去。我 還要感謝實驗室裡的好夥伴楹璋與搞笑學弟文彦,因為有他們兩人的 陪伴,使我單調的研究生生活增色許多。新進學弟震東與仲翔的加 入,也替實驗室裡帶來不少的歡樂。感謝我最親愛的家人,你們永遠 的支持與無限的關愛,讓我知道我並不是孤獨的面對所有的困難,並 且能夠提起勇氣度過各種情緒上的低潮。最後要感謝各位關心我的朋 友們,在你們的鼓勵與幫助下,我才能順利完成學業,謝謝你們。. 昌諺 2005 年 8 月. iv.
(5) 目錄 頁碼 中文摘要…………………………………………………………………ii 致謝…………………………………………………………………...…iv 目錄………………………………………………………………………v 圖目錄……………………………………………………………….…viii 附圖目錄……………………………………………………………….xiv. 第一章 緒論……………………………………………………..………1 1.1 前言………………………………………………………...……1 1.2 自組裝單層膜…………………………………………..….……1 1.2.1 發展起源…………………………………………………1 1.2.2 原理………………………………………………………2 1.2.3 應用………………………………………………………4 1.3 芳杯化合物的介紹與應用……………………………...………6 1.3.1 起源及命名………………………………………………6 1.3.2 應用………………………………………………..……10 1.4 賽吩及其衍生物的介紹與應用……………………….………12 1.5 研究動機與目的……………………………………….………14. 第二章 實驗技術與數據分析處理……………………………………15 2.1 掃描穿隧顯微鏡(STM)…………………………………..……15 2.2 電化學掃描穿隧顯微鏡(EC-STM)……………………………16 2.3 程溫脫附法(TPD)………………………………………………17. v.
(6) 2.4 X 光光電子發射能譜(XPS)……………………………………17 2.5 XPS 能譜分析處理………………………………………….…20 2.6 近緣 X 光吸收細微結構光譜(NEXAFS)………………………22 2.7 NEXAFS 光譜分析處理…………………………….…………26 2.8 同步輻射光…………………………………………….………29. 第三章 實驗藥品、儀器設備清單與實驗步驟………………………33 3.1 藥品與儀器設備……………………………………….………33 3.2 超高真空系統…………………………………….……………35 3.3 STM 實驗方法與步驟…………………………………………39 3.4 EC-STM 實驗方法與步驟……………………..………………45 3.5 超高真空的準備…………………………….………….………47 3.6 TPD 實驗方法…………………………………………….……51 3.7 XPS 實驗方法……………………………………………….…52 3.8 NEXAFS 實驗方法……………………………….……………52. 第四章 實驗結果與討論………………………………………………58 4.1 芳杯分子自組裝薄膜實驗結果………………………….……58 4.1.1 STM 影像結果………………………………………..…58 4.1.2 EC-STM 影像結果………………………………………62 4.1.3 XPS 能譜結果……………………………………………67 4.1.4 NEXAFS 光譜結果………………………………...……72 4.1.5 結論………………………………………………………74 4.2 二賽吩自組裝分子實驗結果……………………………….…76 4.2.1 真空吸附製備的二賽吩 TPD 圖譜結果…………...……76 4.2.2 真空吸附製備的二賽吩 XPS 能譜結果…………………79 vi.
(7) 4.2.3 真空吸附製備的二賽吩 NEXAFS 光譜結果…………81 4.2.4 溶液浸泡製備的二賽吩 STM 影像結果………...……85 4.2.5 溶液浸泡製備的二賽吩 TPD 圖譜結果…………....…91 4.2.6 溶液浸泡製備的二賽吩 XPS 能譜結果……….…...…94 4.2.7 溶液浸泡製備的二賽吩 NEXAFS 光譜結果…………96 4.2.8 結論……………………………………………….……98 4.3 賽吩自組裝分子實驗結果……………………………….…102 4.3.1 真空吸附的賽吩 TPD 圖譜結果…………………..…102 4.3.2 真空吸附的賽吩 XPS 能譜結果……………………...105 4.3.3 真空吸附的賽吩 NEXAFS 光譜結果……………..…109 4.3.4 溶液浸泡製備的賽吩分子 TPD 圖譜結果………..…109 4.3.5 溶液浸泡製備的賽吩分子 XPS 能譜結果………..…109 4.3.6 溶液浸泡製備的賽吩分子 NEXAFS 光譜結果….….113 4.3.7 結論………………………………………………....…116. 參考文獻…………………………………………………………...…119 附圖………………………………………………………………...…123 簡歷……………………………………………………………………133. vii.
(8) 圖目錄 頁碼 圖 1-1. 自組裝分子的結構與作用力示意圖。……………………3. 圖 1-2. 不同對位取代基之 calix[4]arene。……………………..…7. 圖 1-3. 4-三級丁酚與甲醛在鹼的催化條件下的產物。……….…8. 圖 1-4(a). 芳杯結構示意圖。……………………………………….…9. 圖 1-4(b). 芳杯化合物 calix[4]arene。………………………………..9. 圖 1-5. 芳杯化合物 8 以及與碳六十結合之示意圖。……….……11. 圖 1-6. 芳杯化合物 9、10。……………………………….…..…13. 圖 1-7. 不同官能基的芳杯化合物 11a 與 11b。…………………13. 圖 2-1. 測得的電子動能與電子動能分析儀功函數的關係圖。…18. 圖 2-2. 實際偵測縱深與電子起飛角的關係圖。…….……..……21. 圖 2-3. 原子與雙原子分子的電子能階圖及其相應的 NEXAFS 光譜。………………………………………..…23. 圖 2-4. 入射 X 光與分子未填滿軌域角度關係圖。………………25. 圖 2-5. X 光入射角與不同分子傾角下的 X 光吸收譜線強度 關係圖。…………………………………………………..27. 圖 2-6. 部分電子產率光譜的處理步驟說明圖。…………………30. 圖 3-1. 超高真空腔體配置俯視圖。……………….………….…36. 圖 3-2. 可進行樣品交換的樣品平台。……………….…….……37. 圖 3-3. 可進行樣品交換的樣品操作平台。…………….…….…37. 圖 3-4(a). 中空不銹鋼管的樣品平台。………………….……….…38. 圖 3-4(b). 樣品平台近照。…………………………………….…..…38. 圖 3-5(a). 利用電化學蝕刻法製作探針時所使用的裝置。……..…40. 圖 3-5(b). 探針製作裝置的近照。……………………………..……40 viii.
(9) 圖 3-6(a). 未進行回火步驟,直接進行 STM 掃描所得的影像。…43. 圖 3-6(b). 為經過回火步驟後再進行 STM 量測所得的影像。……43. 圖 3-7. 裝置於 STM 樣品平台上的金(111)晶面。………………44. 圖 3-8. 裝載於 STM 中的樣品。…………………………………44. 圖 3-9. EC-STM 專用金單晶電極。…………….…………….…46. 圖 3-10. 電化學樣品試槽。………………………..………………48. 圖 3-11. 裝置完成後的 EC-STM。………………….…………….48. 圖 3-12. 電子產率偵測器構造示意圖與實物。…………….…….53. 圖 3-13. 歐傑電子產生示意圖。…………………………….…….55. 圖 4-1(a). 乾淨金(111)晶面的 STM 大範圍掃描影像。……………59. 圖 4-1(b). 圖 4-1(a)的 STM 小範圍掃描影像。…………………….59. 圖 4-2(a). 金(111)表面浸泡 25 µM 芳杯溶液 1 分鐘後得到的 STM 大範圍掃描影像。……………………………..……60. 圖 4-2(b). 圖 4-2(a)的 STM 小範圍掃描影像。……………………..60. 圖 4-3(a). 金(111)表面浸泡 25 µM 芳杯溶液 5 分鐘後得到的 STM 大範圍掃描影像。……………………………….…61. 圖 4-3(b). 圖 4-3(a)的 STM 小範圍掃描影像。………………….…61. 圖 4-4(a). 金(111)表面浸泡 25 µM 芳杯溶液 15 分鐘後得到的 STM 大範圍掃描影像。…………………………….……63. 圖 4-4(b). 圖 4-4(a)的 STM 小範圍掃描影像。……………………..63. 圖 4-5(a). 金(111)表面浸泡 25 µM 芳杯溶液 30 分鐘後得到的 STM 大範圍掃描影像。……………………………….…64. 圖 4-5(b). 圖 4-5(a)的 STM 小範圍掃描影像。.…………………….64. 圖 4-6(a). 乾淨金(111)晶面的 EC-STM 大範圍掃描影像。…………65. 圖 4-6(b). 圖 4-6(a)的 EC-STM 小範圍掃描影像。…………………65. ix.
(10) 圖 4-7(a). 滴入 1 mM 芳杯溶液後進行掃描所得到的 EC-STM 影像。……………………………………………………..66. 圖 4-7(b). 圖 4-7(a)的 EC-STM 小範圍掃描影像。…………………..66. 圖 4-8(a). 滴入 1 mM 芳杯溶液後進行掃描所得到的 EC-STM 影像。……………………………………………………..68. 圖 4-8(b). 圖 4-8(a)的 EC-STM 小範圍掃描影像。…………………68. 圖 4-9(a). 以電子起飛角 30°進行量測所得的芳杯 S 2p XPS 能譜。….………………………………………………….69. 圖 4-9(b). 以電子起飛角 70°進行量測所得的芳杯 S 2p XPS 能譜。…………………………………………………..…69. 圖 4-10. 以靜置一段時間的芳杯溶液進行實驗所量測得到的 芳杯 S 2p XPS 能譜。…………………………………….71. 圖 4-11. 芳杯 C 1s XPS 能譜。……………………………………71. 圖 4-12. 各加熱溫度下所得到的芳杯 S 2p XPS 能譜變化。……73. 圖 4-13. 不同溫度下芳杯 S 2p XPS 能譜的分析。…………..…..73. 圖 4-14. 以 X 光入射角 20°、55°與 90°進行 NEXAFS 實驗所 得到的 TEY 訊號。……………………………………….75. 圖 4-15. 真空吸附二賽吩 2.5 L 的 TPD 圖譜。…………………..77. 圖 4-16. 減少物理吸附產生後的二賽吩 TPD 圖譜。……….…...77. 圖 4-17. 各低吸附量下質荷比 166 amu 的二賽吩 TPD 圖譜 變化。…………………………………………….……….78. 圖 4-18. 80 K 下真空吸附二賽吩 1.3 L 的 S 2p XPS 能譜分析。….80. 圖 4-19. 80 K 下真空吸附二賽吩 7.3 L 的 S 2p XPS 能譜分析。….80. 圖 4-20. 各加熱溫度下所得到的真空吸附二賽吩 S 2p XPS 能 譜變化。………………………………………….….……82. x.
(11) 圖4-21. 210 K下吸附二賽吩4.3 L後進行NEXAFS實驗所得到的 PEY訊號。………………………………………………..83. 圖 4-22. 真空吸附法製備二賽吩 NEXAFS 光譜結果的的分子 吸附傾角分析。……………………………………….….84. 圖 4-23. 210 K 下吸附二賽吩 4.3 L 後進行 NEXAFS 實驗所得 到的 PEY 訊號。………………………………..…….….86. 圖 4-24. 真空吸附法製備二賽吩 NEXAFS 光譜結果的分子吸 附傾角分析。………………………………………..….…87. 圖 4-25(a) 金(111)表面浸泡 25 µM 二賽吩溶液 3 秒鐘後得到的 STM 大範圍掃描影像。…………………………….….…88 圖 4-25(b) 圖 4-23(a)的 STM 小範圍掃描影像。……………..….…88 圖 4-26(a) 金(111)表面浸泡 25 µM 二賽吩溶液 5 分鐘後得到的 STM 大範圍掃描影像。……………………………..……89 圖 4-26(b) 圖 4-24(a)的 STM 小範圍掃描影像。……………………89 圖 4-27(a) 金(111)表面浸泡 1 mM 二賽吩溶液 30 分鐘後得到的 STM 大範圍掃描影像。…………………………….…....90 圖 4-27(b) 圖 4-25(a)的 STM 小範圍掃描影像。…………….…..…90 圖 4-28. 樣品交換系統下乾淨的金(111)晶面 TPD 圖譜。……….92. 圖 4-29. 浸泡 1 mM 二賽吩溶液 8 小時的 TPD 圖譜。…..…..….93. 圖 4-30(a) 以電子起飛角 40°進行量測所得的二賽吩 S 2p XPS 能譜。…………………………………….…………….…95 圖 4-30(b) 以電子起飛角 70°進行量測所得的二賽吩 S 2p XPS 能譜。………………………………………….……….…95 圖 4-31. 各溫度下所得到的二賽吩 S 2p XPS 能譜變化。………..97. 圖 4-32. 於特定溫度下的二賽吩 S 2p XPS 能譜分析。…………..97. xi.
(12) 圖 4-33. 將浸泡 1 mM 二賽吩溶液 9.5 小時後進行 NEXAFS 實驗得到的 PEY 訊號。……………………………....…99. 圖 4-34. 溶液浸泡法製備二賽吩 NEXAFS 光譜結果的分子吸 附傾角分析。……………………………………………100. 圖 4-35. 真空吸附賽吩 1.7 L 的 TPD 圖譜。……………………103. 圖 4-36. 真空吸附賽吩 3.7 L 的 TPD 圖譜。………………….…..103. 圖 4-37. 各低吸附量下質荷比 84 amu 的賽吩 TPD 圖譜變化。..104. 圖 4-38. 80 K 下真空吸附賽吩 1 L 的 S 2p XPS 能譜分析。…..106. 圖 4-39. 80 K 下真空吸附賽吩 1.6 L 的 S 2p XPS 能譜分析。..….106. 圖 4-40. 受 X 光照射約 1.5 小時後的真空吸附賽吩 S 2p XPS 能譜。………………………………………………..…..108. 圖 4-41. 不同溫度下所得到的真空吸附賽吩 S 2p XPS 能譜 變化。……………………………………………………108. 圖4-42. 180 K下吸附賽吩1 L後進行NEXAFS實驗所得到的PEY 訊號。…………………………………………………….110. 圖 4-43. 真空吸附法製備賽吩 NEXAFS 光譜結果的分子吸附 傾角分析。………………………………………….....…111. 圖 4-44. 浸泡 1 mM 賽吩溶液 4.5 小時的 TPD 圖譜。……………112. 圖 4-25(a) 以電子起飛角 30°進行量測所得的賽吩 S 2p XPS 能譜。………………………………………………..…..114 圖 4-25(b) 以電子起飛角 70°進行量測所得的賽吩 S 2p XPS 能譜。……………………………………………..……..114 圖 4-46. 將浸泡 1 mM 賽吩溶液 32 小時後進行 NEXAFS 實驗 得到的 PEY 訊號。..……………………………………115. 圖 4-47. 溶液浸泡法製備賽吩 NEXAFS 光譜結果的分子吸附. xii.
(13) 傾角分析。……………………………………….…..…117. xiii.
(14) 附圖目錄 頁碼 附圖 1. 金(111)浸泡 1 mM 芳杯溶液中 5 小時後所量測到的 大範圍 XPS 能譜。……………………………….…..…123. 附圖 2. 各加熱溫度下所得到的芳杯 Au 4f XPS 能譜變化。….123. 附圖 3. 各加熱溫度下所得到的芳杯 C 1s XPS 能譜變化。……124. 附圖 4. 80 K 下吸附 1.3 L 二賽吩分子所得到的大範圍 XPS 能譜。……………………………………………………124. 附圖 5. 80 K 下真空吸附二賽吩 C 1s XPS 能譜。…………..…125. 附圖 6. 80 K 下吸附 7.3 L 二賽吩分子所得到的大範圍 XPS 能譜。………………………………………………….…125. 附圖 7. 80 K 下真空吸附二賽吩 C 1s XPS 能譜。…………..…126. 附圖 8. 各加熱溫度下所得到的真空吸附二賽吩 Au 4f XPS 能譜變化。……………………………………………….126. 附圖 9. 各加熱溫度下所得到的真空吸附二賽吩 C 1s XPS 能譜 變化。…………………………………………………..…127. 附圖 10. 金(111)浸泡 1 mM 二賽吩溶液中 15 小時後所量測到 的大範圍 XPS 能譜。……………………………………127. 附圖 11. 二賽吩 C 1s XPS 能譜。…………………………………128. 附圖 12. 各加熱溫度下所得到的二賽吩 Au 4f XPS 能譜變化。...128. 附圖 13. 各加熱溫度下所得到的二賽吩 C 1s XPS 能譜變化。….129. 附圖 14. 80 K 下吸附 1 L 賽吩分子所得到的大範圍 XPS 能譜。..129. 附圖 15. 80 K 下吸附 1.6 L 賽吩分子所得到的大範圍 XPS 能 譜。………………………………………………………130. xiv.
(15) 附圖 16. 80 K 下真空吸附賽吩 C 1s XPS 能譜。……………..…130. 附圖 17. 各加熱溫度下所得到的真空吸附賽吩 Au 4f XPS 能譜 變化。………………………………………………….…131. 附圖 18. 各加熱溫度下所得到的真空吸附賽吩 C 1s XPS 能譜 變化。………………………………………………….…131. 附圖 19. 金(111)浸泡 1 mM 賽吩溶液中 32 小時後所量測到的 大範圍 XPS 能譜。………………………………..….…132. 附圖 20. 賽吩 C 1s XPS 能譜。……………………………………132. xv.
(16) 第一章 緒論. 1.1 前言. 由於科技的進步,各類電子產品的開發均以輕、薄、短、小為目 標,而如何將元件微小化成為了最重要的研究課題,自此開啟了對於 奈米領域的廣泛研究。其中,自組裝技術的突破,使科學家們得以開 始設計超分子(supramolecule)與其他各種奈米結構與材料。利用新的 製程技術所製造出的奈米電子元件,在元件密度、速度以及成本的效 益上,將遠遠超過現有的半導體產品,因此分子的自組裝技術對於奈 米材料的製造與控制,具有極大的發展潛力。. 1.2 自組裝單層膜 (self-assembled monolayer). 一般常見的有機單層薄膜依照形成方式的不同,可以區分為兩大 系統;一為 LB 薄膜(Langmuir-Blodgett film),另一為自組裝單層膜 (self-assembled monolayer)。自組裝單層膜研究的起步時間比 LB 薄膜 晚,但是因為其極大的發展潛力而至今仍被廣泛的研究與探討。. 1.2.1 發展起源. 1946 年 Zisman 將介面活性劑吸附於金屬表面上,製備出單層分 子薄膜 1,但是當時並沒有發現分子自組裝的特性。之後 Kuhn 利用 三氯矽烷的衍生物吸附於親水性的玻璃上,亦形成單層的薄膜。1983. 1.
(17) 2. 年,Nuzzo 和 Allara 將二硫化物(dialkyldisulfide)吸附於金表面上,結 果發現分子於表面上形成規則有序的單層膜結構,自此分子自組裝單 層膜開始被廣泛的研究。. 1.2.2 原理. 自組裝單層膜是分子藉由特定官能基以物理作用力或化學鍵結 吸附在固體表面上所形成的薄膜,製備方法通常是將特定基材表面浸 入一對該表面具有吸附性的化合物溶液中,或是將基材暴露於該化合 物蒸氣下,利用其自發性的化學吸附,加上吸附分子間非共價鍵作用 力的影響,分子會趨向熱力學最穩定的狀態,自然發生組裝排列,而 形成具有整齊排列的薄膜。 這種自組裝技術的特色,在於分子排列緊密、有秩序,並且製備 上又極為簡便,不需要外加太多的能量,所以在材料奈米製造及控制 技術上,極具發展潛力,常被用來取代 LB 薄膜。 以能量的觀點來看,自組裝分子的結構通常可分為三個部份,以 烷基硫醇為例,如圖 1-1 所示 3: 第一部份為頭基(head group),其與特定基材表面具有化學吸附作 用,並且於鍵結時產生放熱的過程,其能量為一百多個千焦耳/莫耳 (kJ/mol)。第二部份為長烷鏈的部份,利用鏈與鏈間的凡得瓦力(van der Waals force)形成有秩序性且緊密的組裝結構。此外,具有芳香環 結構的分子,除了分子間凡得瓦力的作用外,也要考慮π-π堆疊的 作用力。接下來的第三部份,則是具功能化的末端官能基部份(end group),此末端官能基將取代原本的基材表面,直接影響表面的能量 與性質,例如飽和的烷基末端,會形成疏水性的表面,而末端具磷酸. 2.
(18) 圖 1-1:自組裝分子的結構與作用力示意圖。3. 3.
(19) 根的分子,則造成親水性的表面。由於末端官能基部份的多樣性,使 得有機自組裝分子薄膜在應用上具有很大的潛力。 目前已發現多種以分子自組裝方式形成的單層有機薄膜,包括了 有機矽烷吸附於水合的表面(矽表面的氧化矽、鋁表面的氧化鋁、玻 璃等);烷基硫醇吸附於金、銀、銅表面;雙烷基硫化物吸附於金表 面;以及醇類及胺類吸附於鉑表面等。表 1-1 列出數種已知的分子自 組裝薄膜系統 3。. 1.2.3 應用. 自組裝薄膜的主要應用在於載體表面的改質,例如利用特定末端 官能基來增進表面潤滑、韌度、親水性、防腐蝕性等。除此之外,還 可應用於生化感應器、電路和半導體保護膜、人造生化薄膜、接著促 進劑、觸媒等,也可利用其特殊的光學現象,應用於非線性光學薄膜。. 1.3 芳杯化合物的介紹與應用. 主客化學(host-guest chemistry)是近年來相當熱門的研究主題。主體與 客體之間可藉由非共價鍵的靜電作用力而形成特殊的結構。芳杯為主 客化學中常見的主體之一,可與之作用的客體有各種陰、陽離子與分 子。藉由此特殊的主客化學特性,芳杯在辨識與感測應用上具有極大 的潛力。. 4.
(20) 表 1-1 各種形成分子自我組裝薄膜的系統 3 基材. 前驅物. 鍵結. Au. RSH, ArSH (thiol). RS-Au. Au. RSSR (disulfide). RS-Au. Au. RSR (sulfide). RS-Au. Au. RSO2H. RSO2-Au. Au. R3P. R3P-Au. Ag. RSH, ArSH. RS-Ag. Cu. RSH, ArSH. RS-Cu. Pd. RSH, ArSH. RS-Pd. Pt. RNC. RNC-Pt. GaAs. RSH. RS-GaAs. InP. RSH. RS-InP. SiO2, glass. RSiCl3, RSi (OR)3. Siloxane. Si/Si-H. (RCOO)2. R-Si. Si/Si-H. RCH=CH2. RCH2CH2Si. Si/Si-Cl. RLi, RMgX. R-Si. Metal oxides. RCOOH. RCOO …MOn. Metal oxides. RCONHOH. RCONHOH…MOn. ZrO2. RPO3H2. RPO32-…Zr4+. InO3/SnO2 (ITO). RPO3H2. RPO32-…Mn+. -. 5.
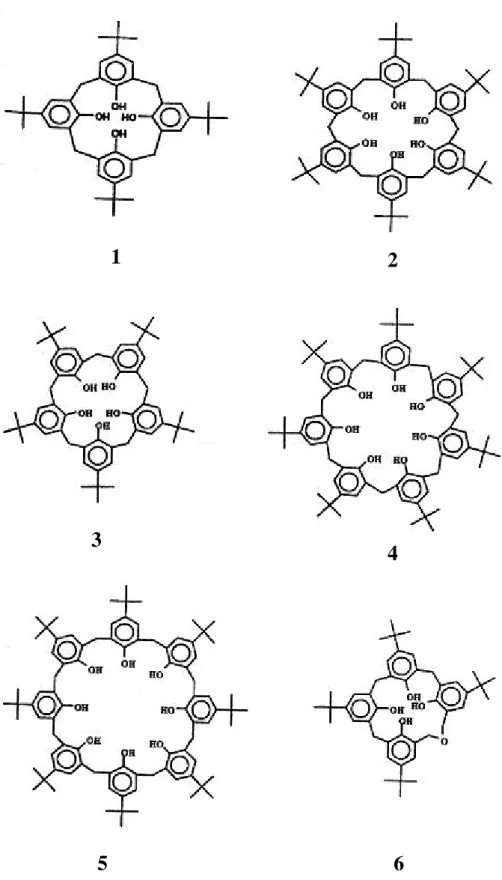
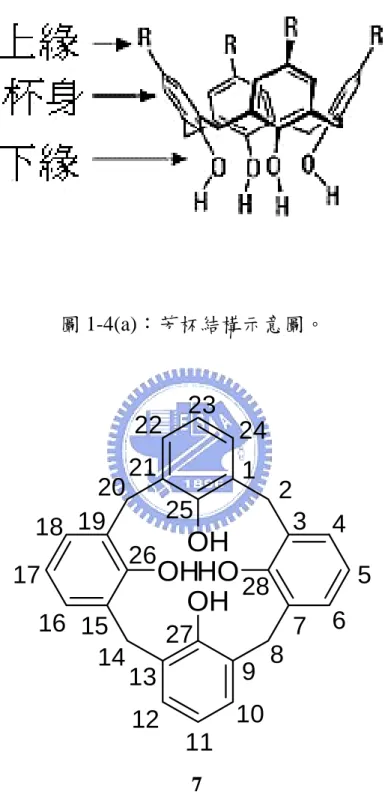
(21) 1.3.1 起源及命名. 在 1940 到 1950 年間,Zinke4 等人將對位上含不同取代基的酚和 37%甲醛水溶液,在氫氧化鈉催化下進行反應,處理後得到一系列由 亞甲基連結苯環的環狀四合物,如圖 1-2 所示。 1955 年,Cornforth5 利用與 Zinke 相同的方法,卻分離出不同熔 點的兩種化合物,經過 Kammerer6 與 Munch7 對環狀四合物 1H-NMR 的動力學研究後,發現此組異構物在室溫下能迅速翻轉,所以為構形 異構物,而非 Cornforth 所認為的一組環狀四合物的結構異構物。1978 年,Gutche8 發現 4-三級丁酚和甲醛在鹼的催化下,可生成數種不同 大小的環狀聚合物,包括環狀四合物 1、環狀六合物 2、少量的環狀 五合物 3、環狀七合物 4、環狀八合物 5 與 p-tert-butyldihomooxa 環氧 化物 6,如圖 1-3 所示。 此類的化合物外形似杯狀,可分成上緣、杯身及下緣部分,如圖 1-4(a)所示,故 Gutche 將酚和甲醛的環狀聚合物命名為 calixarene (在 希臘文中,calix 為酒杯之意,而 arene 則表示含芳香環排列成的巨環 結構),並且在 calix 和 arene 之間插入數字以表苯環的數目,用此簡 化這類化合物的命名方式。以圖 1-4(b)中化合物 7 為例,在 IUPAC 系統中正式命名為 pentacyclo[19.3.1.13,719,13115,19]octacosa-1(25),3,5,7(28), 9,11,13(27),15,17,19(26),21,23-dodecaene-25,26,27,28-tetraol,利用此命 名規則,可稱之為 25,26,27,28-tetrahydroxycalix[4]arene,或更簡單稱 之 calix[4]arene,以下以芳杯稱呼之。. 6.
(22) R. R. OH OHHO OH. R. R = methyl tert-butyl tert-amyl 1,1,3,3-tetramethylbutylcyclohexyl benzyl. R. 圖 1-2:不同對位取代基之 calix[4]arene。4. 7.
(23) 1. 2. 3. 4. 6. 5. 圖 1-3:4-三級丁酚與甲醛在鹼的催化條件下的產物。8. 8.
(24) 圖 1-4(a):芳杯結構示意圖。. 22 20 18 19 17 16 15 14. 23 24. 21. 1 25. 2 3. 26 OH OHHO 28 OH 27 13. 9. 12. 10. 4 5. 7. 6. 8. 11 7. 圖 1-4(b):芳杯化合物 calix[4]arene。. 9.
(25) 1.3.2 應用. 於辨識功能上的應用. 芳杯具有能與分子或離子形成可逆錯合型態的特性,而且將芳杯 做不同的修飾,對分子或離子的選擇性也會有所不同。 可與芳杯進行錯合的金屬離子包括有鹼金族 9、鹼土族. 10. 、過渡. 金屬離子 11,甚至連鑭系 12、錒系 13 金屬離子都曾被報導過。 文獻中,可與陰離子進行錯合作用的芳杯衍生物大多含有醯胺基 團(amide)14,醯胺基團與陰離子之間可形成氫鍵,而使陰離子與芳杯 錯合。其他例如含有尿素或硫脲(thiourea)的芳杯也可與陰離子作用 15. 。 芳杯除了能與離子形成錯合外,也能與中性分子作用。於 2000. 年,Kutateladze 利用光不穩定的 spiro-bis-dithiane 基團連接兩個芳杯 而得到化合物 8,而化合物 8 可以容納碳六十 ([60]fullerene)16,如圖 1-5。. 於感測器上的應用. 在芳杯的上緣或下緣接上發色團或螢光基團,則當芳杯與離子或 分子形成錯合時,可由顏色、紫外-可見光吸收光譜或螢光放射光譜 來觀察其變化。於應用上,可發展為鈣 17、鋰 18、鈉 19 等離子的感測 器。. 10.
(26) 8 圖 1-5:芳杯化合物 8 以及與碳六十結合之示意圖。16. 11.
(27) 於掌性分子辨識上的應用. 利用芳杯來辨識或分離鏡像異構物(enantiomers),是近年來廣受 注意的研究方向。一般此類的芳杯可分為兩大類型 : 一為本身構形 具有掌性(chiral)20 (如圖 1-6 中化合物 9), 二為在其上緣或下緣接上 掌性基團 21 (如圖 1-6 中化合物 10)。 除此之外,1996 年 Kubo 合成出含發色團之芳杯 (如圖 1-7 中 11a、11b),藉由錯合前後顏色變化來辨識鏡像分子 22,例如化合物 12、13、14。. 1.4 賽吩及其衍生物的介紹與應用. 賽吩為一雜環有機化合物,存在於原油中,並且具有毒化催化表 面的特性。除了早期關於賽吩在石化工業應用方面的研究之外,由於 分子電子學的崛起,使得賽吩至今仍不斷的被研究與討論。 分子電子學(molecular electronics)於近十餘年蓬勃的發展,其主 要觀念為,電子電路的個別元件可以利用物質的分子來構成。這將大 大增加晶片上的電路密度並使它們得以更快的速度來運作。目前該領 域最重要的課題之一為,利用有機分子來取代傳統的半導體材料,特 別是在平面顯示器方面。其優點在於低生產成本、發光材料的多變性 以及環境上的考量。雖然於應用上的研究已經快速的發展,但是對於 很多基本物理機制上的了解卻很缺乏,需要更多對於理論系統的詳細 研究。 目前π-共軛高分子在半導體工業上正被廣泛採用,因為這些有機 高分子在電子結構上具有去區域性(delocalization)的共軛π電子雲,有 12.
(28) 9. 10 圖 1-6:芳杯化合物 9、10。. 圖 1-7:不同官能基的芳杯化合物 11a 與 11b,可用來辨識化合物 12、 13、14。. 13.
(29) 助於電荷的傳導。而聚賽吩(polythiophene)與寡聚賽吩(oligothiophene) 為被廣泛研究的有機高分子之一。例如發光二極體(light-emitting 23. 24. diode) , 場 效 應 電 晶 體 (field-effect transistor) , 光 伏 特 電 池 25. (photovoltaic cell) 等,均為寡聚賽吩在光電材料的應用。由於這些分 子具有良好的化學穩定度,並且可以透過取代基的改變來進行光電性 質方面的改良與控制,而極具應用價值。 為了開發有機高分子在光電性質方面的應用潛力,所以必須要加 強對於有機薄膜的電子與幾何結構的控制。除此之外,有機分子材料 與金屬接觸界面的熱力學與機械性的穩定,對於電子元件的表現十分 重要,所以對於有機材料與金屬之間的作用需要更深入的了解與探 討。2,2'-bithiophene (以下簡稱為 bithiophene)為最短的α-寡聚賽吩, 由於長鏈分子的低溶解度,以及短鏈自由基離子所具有的良好聚合反 應性,所以常被拿來進行研究。. 1.5 研究動機與目的. 利用各種表面分析技術來進行芳杯、賽吩與二賽吩於金(111)表面 上吸附行為的研究,希望能夠藉此對於吸附分子的電子結構、鍵結強 度及分子排列結構有綜合的了解,並且對於其介面表面性質能有更佳 的掌握,而有助於以上化合物在應用方面的發展。. 14.
(30) 第二章、實驗技術與數據分析處理. 2.1 掃描穿隧顯微鏡 (scanning tunneling microscope, STM). 在介紹 STM 的操作原理之前,必須先介紹何為「穿隧效應」 (tunneling effect)。所謂的「穿隧效應」就是指粒子可穿過比本身總能 高的能量障礙,在古典力學中,這是不可能發生的,不過以量子物理 的觀點來看,卻有此可能性。穿隧的機率與距離有關;距離愈近,穿 隧的機率愈大。在金屬探針與導體樣品表面間加一偏壓,並使之相隔 距離為數個原子大小範圍時,即可造成穿隧電流的產生,而電子穿隧 的機率是和發生穿隧效應兩端的間距成指數反比的關係,如以下公式 所示:. ﹣Cd. It = V‧e. 其中 It 為穿隧電流強度,V 為加於探針與表面之間的偏壓,C 為一常 數,而 d 為探針上最尖端處的原子與樣品表面原子間的距離。由上式 可知,探針與表面之間距離的些微改變,即可造成穿隧電流顯著的變 化,所以於量測上具有相當高的解析度。 在掃描穿隧顯微鏡中,樣品被安置在一壓電材料的平台上,透過 電壓對壓電材料的控制,可使樣品在三度空間中作小範圍的移動。量 測時,先將探針慢慢接近樣品表面,隨著探針與表面之間距離的縮 小,所產生的穿隧電流呈指數增加。當穿隧電流達預定值大小時,壓 電平台則開始 XY 水平方向的移動,使探針進行影像掃描。在影像的 取得有兩種方法,分別為等電流取像(constant current imaging)與等高 15.
(31) 度取像(constant height imaging)。本實驗中是使用等電流取像法,即 以設定的穿隧電流作為回饋訊號。由於探針與樣品表面的間距和穿隧 電流有十分靈敏的關係,所以設定穿隧電流值即鎖定探針和樣品表面 之間距。當探針在樣品表面掃描時,探針必須隨表面之起伏調整其高 度,以保持探針與表面的距離,此時可由回饋電路控制壓電平台於 Z 方向的移動,使兩者間的距離為一恆定值。因此,以平台的高度變化 來呈像,就反映出樣品表面的形貌。等電流取像法的優點在於可容忍 較大的表面高低變化,但是由於必須以回饋信號來調制,掃描速度較 慢,容易受低頻雜訊的干擾。由於 STM 量測的是樣品表面與探針之 間的穿隧電流,所以樣品必須為導體或半導體,對於不導電的材料, STM 無法進行量測,此為 STM 較美中不足之處。. 2.2 電 化 學 掃 描 穿 隧 顯 微 鏡 (electrochemical scanning tunneling microscope, EC-STM) EC-STM 可視為是電化學與 STM 的結合,於本實驗中將工作電 極控制於特定電位下,再以 STM 掃描樣品表面,觀測樣品於基材上 的吸附狀況。EC-STM 的電化學系統是由四個電極所組成,分別為工 作電極(working electrode)、參考電極(reference electrode)與相對電極 (counter electrode),以及在電位控制下進行掃描的探針。傳統之電化 學方法,在量測上為了避免電流直接流經參考電極,產生 IR 電位降 之不良影響,而多採用三電極組態方式,利用相對電極來提供電流的 流通。 除了電化學控制之外,EC-STM 的工作原理與 STM 相同。比較 特別的是,EC-STM 所使用的探針上需要塗附一層絕緣層,而 STM 則否,塗附上絕緣層的目的是為了降低法拉第電流的產生,關於絕緣 16.
(32) 層的塗附方法,請參見 3.4 小節中 EC-STM 探針前處理的部分。由於 EC-STM 探針在實驗進行時必須浸泡於溶液中,除了來自於探針尖端 與樣品表面之間的穿隧電流外,透過溶液的傳導,整個金屬探針表面 與樣品表面之間也會產生電流,而此電流可能會影響穿隧電流的監 測,所以才利用絕緣層的塗附來避免穿隧電流以外的電流產生。 2.3 程溫脫附法 (Temperature-Programmed Desorption, TPD). 當一試片被加熱達到特定溫度時,吸附物與表面之間的鍵結會被 熱能所破壞,造成吸附物的脫附,而從表面上脫附的物質則可由四極 質譜儀所偵測,並且得到脫附物質譜訊號與試片溫度的關係曲線。 TPD 所偵測的是從表面上脫離的物質,與「XPS 能譜所偵測的是表 面上物質的化學態」是恰好相反的;因此,這兩種技術常同時被運用 來研究物質在表面上的性質。 2.4 X 光光電子發射能譜 (X-ray Photoemission Spectroscopy, XPS). 當 X 光光束照射樣品表面時,可使內核層(core level)電子被激發 並且脫離材料表面,最後被電子動能分析儀所偵測。經由電子動能分 析儀測得之電子動能,推算出電子之束縛能(binding energy)。 圖 2-1 中樣品與電子動能分析儀經由接地,將兩者的費米能階(EF) 拉齊至同一能量高度。在量測脫離樣品表面的電子動能時,量測到的 電子動能是來自於入射光能量克服電子束縛能(Eb)與樣品表面的功函 數(eφ)之後,基於能量守恆定律,所遺留的能量以電子動能方式展現 的結果;但在進入電子動能分析儀之後,因為還需要克服電子動能分 析儀內偵測器(detector)的功函數(eφ sp),因此測得的電子動能實際上 17.
(33) 圖 2-1:測得的電子動能與電子動能分析儀功函數的關係圖。26. 18.
(34) 應描述為下式: Ekin(sp) = hν - Eb - eφ sp. 而我們在光譜所能獲得的資訊中,一般都將脫離樣品表面具有最小動 能的電子當作基準,其電子動能經由電子動能分析儀所測得的結果 為: Ekin(sp)min = eφ - eφ sp 而測得的電子動能與基準點的差值(ΔE),便是上二式相減: ∆E = Ekin(sp) - Ekin(sp)min = hν - Eb - eφ. 藉由基準點,儀器本身的功函數便可以被忽略,簡化了我們用來計算 束縛能的算式。 目前已經了解 XPS 束縛能的偏移源自於(1)分子中各元素之間的 電負度差異(2)分子與金屬表面的作用力差異,所造成的不同電荷轉 移在化合物形成過程中,原子間藉由價電子形成鍵結,產生電子轉移 的現象,使電負度(electronegativity)較大的元素帶有負電荷,而電負 度較小的原子則帶有正電荷。因為價電子轉移的緣故,使內層電子能 階因靜電位差(electrostatic potential)而產生變化。若元素具有正電 荷,則光電子束縛能會較原子態的電子束縛能高;反之,具有負電荷 的元素所量測到的光電子束縛能會往低束縛能偏移。化合物之間的作 用也會造成相同的結果。利用推算出的束縛能可得知元素種類以及其. 19.
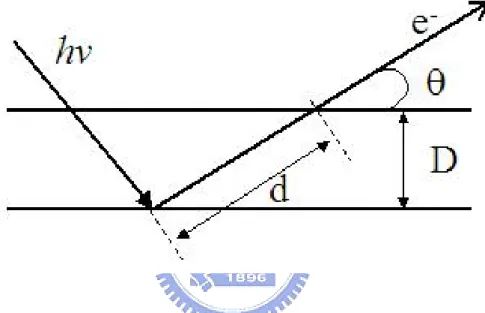
(35) 電子組態,而由束縛能的偏移量則可推測出元素周圍的化學環境,例 如其鍵結與化學態等。 此外,利用改變電子動能分析儀與表面的相對角度來進行角解析 (angular resolved)量測。首先定義電子出射方向(電子動能分析儀電子 透鏡中心軸)與表面的夾角稱為電子起飛角(take-off angle)。實際偵測 縱深與電子起飛角之間的關係如圖 2-2 所示。圖中 d 為光電子於樣品 中運動的距離,D 為偵測縱深,θ為電子起飛角。而從圖 2-2 中可得 到下列關係式:. D = d sinθ. 當起飛角θ變小時,來自固定深度 D 的光電子在試片中運動的距 離 d 變長,因而導致電子產生非彈性散射的機率增加,於是來自深層 的光電子會因為更多能量的減損,而最後轉變成微弱的背景訊號。因 此當起飛角小時,來自於表層原子的訊號相較於深層原子訊號會變 大;反之,起飛角變大時,表層原子的訊號相較於深層原子訊號會變 小。 2.5 XPS 能譜分析處理. 由於儲存環的電子束強度會隨時間而減弱,同步輻射光強度也會 因此而遞減,所以 XPS 光譜資料必須經過歸一化(normalization)處 理,去除時間對光強度的影響。可由實驗時 X 光穿透過置於樣品前 的金薄網所產生的光電流變化,得到 X 光強度相對於時間的改變量。 關於 XPS 譜圖能量的校正,所有的光電子能譜圖皆以基材 Au 4f = 7/2. 84.0 eV 作為 X 光能量校正的基準。之後再經過歸一化處理,最後利 20.
(36) 圖 2-2:實際偵測縱深與電子起飛角的關係圖。. 21.
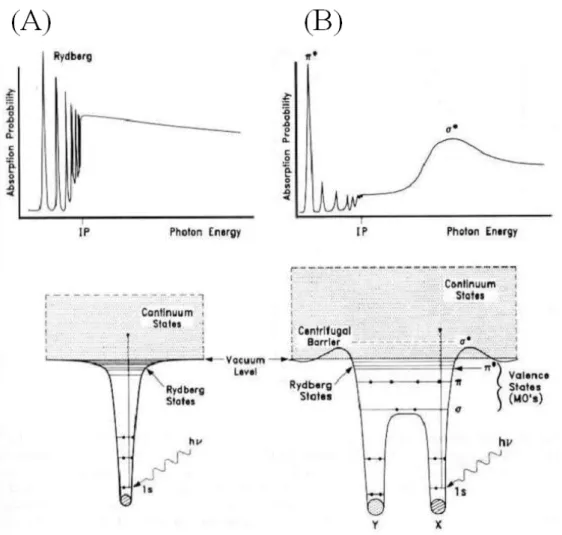
(37) 用 Unifit2002 進行能譜波峰的配湊(fitting)分析。其中參數的設定,S 2p 與 S 2p 譜線強度比為 1:2,所以設定兩波峰強度比例參數時 1/2. 3/2. 則定為 1:2。根據資料所提供的 spin-orbit splitting 數據,S 2p 與 S 1/2. 2p 譜線束縛能能量差值定為 1.18 eV,XPS 能譜中自旋分裂值 3/2. (spin-orbit splitting)通常與化合物之鍵結狀態無關。經過程式運算之 後,再根據運算出的配湊波形結果來改變參數。其中高斯分布考慮的 是來自於光源及儀器解析度誤差所造成的波形特性的變化,而勞倫茲 分布考慮的是光電子本身生命期(life time)所造成的自然線寬。. 2.6 近緣 X 光吸收細微結構光譜 (Near Edge X-ray Absorption Fine Structure, NEXAFS) 由吸收截面與光子能量的關係圖中,可看出來在 X 光能量區內 出現了數個不連續處,在此數個光子能量的範圍之內即為吸收邊緣 (absorption edge),在這些範圍內 X 光吸收係數有劇烈的變化。X 光 吸收光譜(X-ray Absorption Spectroscopy, XAS)可進一步以 X 光能量 與吸收邊緣的能量差值來區分成近緣(Near-Edge)X 光吸收光譜區和 延伸(Extended-Edge)X 光吸收光譜區兩部份,此兩部份的物理現象來 源不一致,所得到的資訊也不相同,在我們的實驗中,只討論 NEXAFS 的部分。 當以 X 光來照射物質,其中內核層(core level)電子可被 X 光激 發到一系列的未填滿軌域,以圖 2-3(A)的氣體原子為例,這些未填滿 軌域,包含電子尚未游離前之一系列雷德堡能態(Rydberg states),以 及電子游離後之連續能態(continuum states)。這些能態的存在使得. 22.
(38) 圖 2-3:(A)原子與(B)雙原子分子的電子能階圖及其相應的 NEXAFS 光譜。27. 23.
(39) 跨過 K-edge(即 1s 電子被游離)的 X 光吸收係數對 X 光能量的關係, 不再是一平滑的函數,而相應具有許多吸收峰(peak)以及階梯(step)。 習稱的近緣 X 光吸收細微結構光譜區,即指從 X 光吸收邊緣 (absorption edge),如 K-edge,往高能量方向約 50 eV 區域內的光譜。 再以圖 2-3(B) X 光激發氣態雙原子分子所得到的 K-edge NEXAFS 光 譜為例。除了前述的雷德堡能態所造成的 X 光吸收峰之外,分子還 存在有具π或σ特性的價電子軌域以及未填滿的π*和σ*軌域。內核層 1s 電子躍遷到π*和σ*空軌域所造成的 X 光吸收峰有著截然不同的形 狀。1s → π*吸收峰非常窄,且因為電子緩解(electronic relaxation)效 應,導致此吸收峰的能量甚至可低於電子的游離位能(ionization potential),相形之下 1s → σ*的躍遷能量較高,而且吸收峰非常寬, 且其形狀非常不對稱,較不適合做進一步分析。 光譜的成因主要是因為光子激發原子內層電子,使其躍遷至能量 較高的空軌域而產生。若以 1s 電子受 X 光激發為例,X 光吸收係數 與躍遷機率的矩陣乘積的平方值成正比,如下列所示:. G I i , f ∝ eˆ ⋅ f r i. 2. ∝ cos 2 χ. 其中 i 為具球對稱之起始 1s 電子狀態, eˆ 為入射 X 光電場的單位向 G. 量,而向量形式的矩陣元素(vector matrix element) f r i 的方向,與被 激發原子的末電子狀態的 p 分量一致。因此當 X 光的電場方向指向 末電子狀態最大電子密度的方向時,吸收係數值最大;而偏離此方向 時,X 光吸收係數則依 cos2χ的形式遞減,其中χ為兩向量的夾角。 圖 2-4 中 X 光入射角為θ,因此與入射光方向垂直的入射電場方. 24.
(40) 圖 2-4:入射 X 光與分子未填滿軌域角度關係圖。. 25.
(41) 向( E & ),與樣品表面法向量(n)的夾角同樣也是θ;分子排列向量(O)與 樣品表面法向量夾角為α。基材的方位角(azimuthal)角度為φ,在本實 驗中由於基材具有三重對稱(threefold)以上的對稱性,cos2φ被平均至 1/2,使得樣品分子軌域與 X 光電場的共振強度關係式簡化成:. Iν// = cos2θ cos2α + 1/2 (sin2θ sin2α) = 1/3[ 1 + 1/2 (3cos2θ-1) (3cos2α-1)] 當α = 54.7 °(magic angle)時 3cos2α-1 = 0 ,此時不論如何改變θ角, 皆不對會軌域與電場的共振強度造成變化。而實驗所使用的 24A 寬 頻光束線的 X 光極化率為 86%(入射光電場有 86%為 E &,14%為 E ⊥ ), 如果以 85%對上式作圖可得圖 2-5,下文中將採用來對照各吸收峰強 度,用來算出分子傾角α。 本研究主要是利用上述原理來分析吸附分子於表面上的吸附結 構 。 由 於 同 步 輻 射 光 源 所 產 生 的 X 光 主 要 為 線 偏 振 (linearly polarized),而且偏振向量平行於水平面,因此只需要沿水平面方向轉 G. 動樣品,即可變化 X 光電場向量( E & )與分子軌域向量( O )的夾角(δ, 上述原理為將其拆解為θ和α來分別分析)。最後利用樣品轉動的角度 與光譜上吸收峰的強弱變化關係,則判斷出吸附分子與表面之夾角角 度。 2.7 NEXAFS 光譜分析處理. 由於儲存環電子束位置會有些微的變動,造成分光後的入射光能 量有所變化,因此必須要先校正光譜結果的能量,才能繼續之後的能 26.
(42) 27. 圖2-5:X光入射角與不同分子傾角下的X光吸收譜線強度關係圖。27.
(43) 譜分析。能量位置的校正是先以 HOPG 為樣品來進行掃描,之後再 將光譜中 285 eV 附近 1s→π*的特徵譜線確實對準在文獻中所報導的 285.38 eV 位置之後,各樣品的光譜結果再以校正後的 HOPG 光譜結 果作為依據,進行能量位置的校正。完成能量的校正後,即可開始總 電子產率(total electron yield, TEY)與部分電子產率(partial electron yield, PEY)的數據處理。總電子產率與部分電子產率的數據處理公式 如下:. sam I Ssam I 0sam ITEY _ Au TEY = subs = subs subs ITEY I S I 0 _ Au sam sam S MCP I 0sam S PEY _ Au PEY = subs = subs subs S PEY S MCP I 0 _ Au. 其中上標 sam 代表是來自於樣品(sample)的訊號,上標 subs 則代表來 自於乾淨基材(substrate)的訊號。利用樣品訊號除以乾淨基材的訊 號,來進行背景訊號的扣除。 一般總電子產率與部分電子產率訊號均有除以金網電流(I0_Au), 以除去時間對於同步輻射光強度的影響。但是由於本實驗中金網電流 會在能量 260 至 270 eV 之間的位置引進氧的訊號,所以於本實驗數 據處理中不除以金網電流,而改以下列公式處理:. sam I Ssam I 0sam ITEY I Ssam _ Au TEY = subs = subs subs ≈ subs ITEY I S I 0 _ Au I S sam sam sam S MCP I 0sam S PEY S MCP _ Au PEY = subs = subs subs ≈ subs S PEY S MCP I 0 _ Au S MCP. 28.
(44) 關於時間對於光源強度的影響,則盡量在相同的儲存環電流下進 行樣品與乾淨基材訊號的量測,藉此降低光源強度變化所造成的影 響。 以部分電子產率光譜的處理步驟為例(圖 2-6),比較樣品 MCP 訊 sam subs 以及除以乾淨基材 MCP 訊號 S MCP 後所得到的訊號,可以發現 號 S MCP. 原本出現於 pre-edge 處(265 至 270 eV)的特徵吸收峰,在處理過後的 光譜中並沒有出現,表示雜質訊號的干擾已經除去。 除此之外,於不同測量角度下樣品受到 X 光照射的面積不一樣, 因此碳訊號對光譜的貢獻量也不同,所以當進行掠角光譜的掃描時, 樣品的暴露面積最大,其光譜會有最大的 edge jump。理論上 edge jump 強度不會因為吸附分子排列位向的變化而改變,而與所監測的元素數 量成正比。為了觀察各譜峰強度隨 X 光入射角的變化,則必須將各 量測角度的光譜對 edge jump 進行歸一化(normalized)處理之後,才能 比較各光譜中譜峰的相對變化。 2.8 同步輻射光. 本研究利用國家同步輻射研究中心的同步輻射光做為 XPS 以及 NEXAFS 實驗的光源。同步輻射光為一連續波段的電磁波,涵蓋紅外 線、可見光、紫外線及 X 光等,1947 年首次在美國通用電器公司同 步加速器上意外地被發現,因此命名為「同步輻射」或「同步加速器 光源」。根據電磁學理論,當帶電粒子運動速度或方向改變時,會放 射出電磁波。因此,當電子以接近光速飛行時,受到磁場作用而發生 偏轉,會沿著切線方向放射出電磁波,此電磁波即為同步輻射。同步 輻射光具有以下特性:強度極強、波長連續、準直性佳、光束截面積 29.
(45) sample MCP signal (a.u.). a. substrate MCP signal (a.u.). 260. 280. 300. 320. 340. 280. 300. 320. 340. b. MCPsam / MCPsubs signal (a.u.). 260. c. 260. 270. 280. 290. 300. 310. 320. 330. 340. Photon Energy (eV). 圖 2-6:部分電子產率光譜的處理步驟說明圖。其中圖(a)為樣品的 sam subs ,圖(b)為基材的 MCP 訊號 S MCP ,而圖(c)代表以樣品 MCP 訊號 S MCP sam subs 的 MCP 訊號 S MCP 除以基材的 MCP 訊號 S MCP 後所得到的訊號。. 30.
(46) 小、具有時間脈波性與偏振性。若以 X 光為例,同步輻射光在這個 波段的亮度比傳統 X 光管燈源所產生的光還要強百萬倍以上,所以 可以把原本需要幾個月的實驗時間縮短至幾分鐘。以往因實驗光源亮 度不夠而無法探測的結構,藉由同步輻射光也都可分析得一清二楚。 由於同步輻射光具有高強度與連續可調的特性,所以使用同步輻 射光源可以克服傳統 XPS 的兩大限制:(1)表面靈敏度及(2)能量解析 度不佳的限制。表面靈敏度與電子非彈性碰撞平均自由徑(Inelastic Mean Free Path,IMFP)有很大的關係。IMFP 定義為電子於固態物質 間移動時,發生能量減損的兩連續碰撞間所行的距離。在相同的材料 中,IMFP 會隨著電子動能的變化而改變。若能把激發出的光電子能 量調整於某一範圍之內,使其 IMFP 為最小,則電子動能分析儀所偵 測到的訊號就幾乎全部來自於表面數層的原子,表面靈敏度也隨之提 高。傳統 XPS 所用的光源其能量均偏高(Mg Kα:1253.6eV,Al Kα: 1486.6 eV),而且無法自由調整。這種強度的 X 光會激發出動能約 200 至 1400 eV 的光電子,相對應的 IMFP 為 10 至 20 Å 左右,相當於近 十餘層原子;若使用同步輻射光源則可在一定的範圍之內自由調整光 子能量,使激發出的光電子動能於 50 至 100 eV 之內,其 IMFP 很小 (約 5 Å)。同時,我們亦可以調整激發能量使欲偵測原子的光游離截 面(photo ionization cross section)達到最大,讓光譜訊號(counts)值達到 最高且訊雜比(S/N)值最好。 現今國家同步輻射研究中心的同步輻射光屬於第三代同步加速 器光源,其最大的特色在於儲存環中裝入特別的插件磁鐵,例如增頻 磁鐵或聚頻磁鐵,藉此使電子由偏轉一次變成多次偏轉,同步輻射光 的亮度則可以提高一千倍以上。實驗所使用的光源是由儲存環引出同. 31.
(47) 步輻射光,經由光束線上多種精密光學元件聚焦、選取波長後,再引 進實驗站進行各項實驗。此研究之實驗是在 24A 寬頻(Wide Range)光 束線進行,這條光束線的特點為可調變能量範圍廣,經由六種不同的 球形光柵可使光能量在低能量(15 至 150 eV) 與高能量(130 至 1500 eV) 兩部分做調變。. 32.
(48) 第三章、實驗藥品、儀器設備清單與實驗步驟. 3.1 藥品與儀器設備. 藥品. 芳杯(5,17-dipropylthiolcalix[4]arene):鍾文聖教授實驗室何怡婷製備. H S. * * O. O H. 2 H. 賽吩(Thiophene):C4H4S,Fluka,純度 99.5% 二賽吩(2,2'-Bithiophene):C8H6S2,Fluka,純度 97% 四氫呋喃(Tetrahydrofuran):C4H4O,TEDIA,純度 99.9% 乙醇(Ethanol):C2H5OH,Fluka,純度 99.8% 過氯酸(Perchloric acid):HClO4,Fluka,純度 70% 硝酸(Nitric acid):HNO3,Fluka,純度 65% 超純水(ultrapure water):H2O,Millipore,電阻值達 18.2 MΩ‧cm. 氣體. 氮氣(nitrogen gas):N2,健仁氣體,純度 99.999% 33.
(49) 金屬. 金線(gold wire):Au,純度 99.99%. 儀器設備. 機械幫浦(mechanical pump):Alcatel. 離子幫浦(ion pump)與鈦昇華幫浦(titanium sublimation pump):Perkin Elmer 各式渦輪分子幫浦(turbo-molecular pumps):Seiko Seiki、Balzer 與 Varian 掃描穿隧顯微鏡(scanning tunneling microscope)與電化學掃描穿隧顯 微鏡(electrochemical scanning tunneling microscope):Digital Instruments,Nanoscope E. 差比抽壓離子槍(differentially pumped sputtering ion gun):Vacuum Generator,EX05. 差比抽壓四極質譜儀(differentially quadrupole mass spectrometer): UTI,100C. 電子動能分析儀(Triple-channeltron electron energy analyzer,. 34.
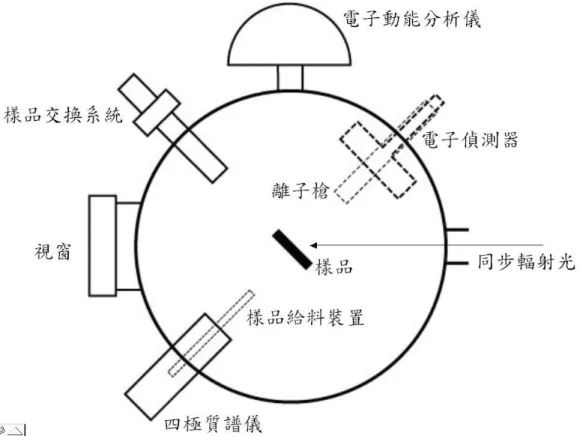
(50) CLAM2):Vacuum Generator. 電子產率偵測器(electron yield detector):自行組裝. 同步輻射光源(synchrotron radiation source):同步輻射研究中心. 3.2 超高真空系統. XPS、NEXAFS 與 TPD 實驗均在可以遮蔽地磁的µ-metal 超高真 空(ultra-high vacuum, UHV)球型腔體內進行,而超高真空腔體中各儀 器的配置如圖 3-1 所示。於腔體上方開口接有一樣品操作平 台 (manipulator)作為移動樣品位置與角度之用,下方接上 T 型通管,T 型通管兩開口端分別接離子幫浦及分子渦輪幫浦。腔體其餘開口端分 別接上電子動能分析儀、差比抽壓離子鎗、樣品交換系統 (load-locksystem)、四極質譜儀、樣品給料裝置(doser)等裝置。腔體前 方開口則與光束線相連接,引進同步輻射光源來進行實驗。 隨著實驗中樣品製備方式的不同,樣品平台(sample holder)的設 計也必須隨之變更。當進行溶液浸泡製備所得的樣品量測時,樣品是 裝載於一可加熱的樣品平台(圖 3-2)上,可利用樣品交換手臂(sample transporter)將樣品平台由一小真空腔傳送至超高真空腔,並與樣品操 作平台上的套件(圖 3-3)組合,使樣品平台能固定於樣品操作平台上。 當樣品是利用真空吸附製備時,樣品平台則選用一前端焊有兩根銅棒 的中空不銹鋼管(圖 3-4(a)、(b)),樣品可以兩橫跨於銅棒之間的鎢線 固定,而此鎢線亦可作為加熱樣品的電熱線,除此之外,中空不銹鋼 管中可加入並儲存液態氮,以冷卻樣品。利用以上方法對樣品進行加 熱與冷卻,可使樣品溫度被控制於 100 至 1100 K 之間。 35.
(51) 圖 3-1:超高真空腔體配置俯視圖。. 36.
(52) 圖 3-2:可進行樣品交換的樣品平台。. 圖 3-3:可進行樣品交換的樣品操作平台。 37.
(53) 圖 3-4(a):中空不銹鋼管的樣品平台。. 圖 3-4(b):樣品平台近照。橫跨兩銅棒間的鎢線可用來固定樣品,通 電後亦可進行樣品的加熱。. 38.
(54) 3.3 STM 實驗方法與步驟. 在開始 STM 的量測前,必須要先準備一金(111)晶面來作為基 材,而本實驗室則是自行製備金(111)晶面以供實驗所需。除此之外, 實驗中所使用的 STM 探針也都是由實驗室自行製作。完成製備的單 晶面還需要經過清理步驟,除去表面上的有機物等不純物之後,才能 浸泡於樣品溶液中開始進行自我組裝薄膜的製備。. STM 探針的製作. 常見的 STM 探針材料為鎢與鉑銥合金,而常見的製備方法則有機 械處理與電化學蝕刻法兩種。鉑銥合金探針的機械處理法是取一段適 當長度且乾淨的鉑銥合金金屬線,用斜口鉗剪出尖銳的斷面,之後再 用去離子水與丙酮沖洗,最後用氮氣吹乾。此製造方式相當簡便,但 缺點是無法有效的控制探針的形狀與尖銳度。理想的探針應為圓錐 狀 , 尖 銳 度 不 夠 或 是 形 狀 不 好 的 探 針 會 造 成 多 重 影 像 (multiple imaging)的產生。鉑銥合金探針的電化學蝕刻法所使用的電解液毒性 高,所以本實驗室沒有採用。由於鎢的硬度大,所以利用機械處理法 並不易得到理想的探針。本實驗室是利用電化學蝕刻法來製造鎢探 針,所使用的器具如圖 3-5(a)、(b)所示。製作探針時,先滴數滴 2 M 的氫氧化鈉電解質於鑽有孔洞的不銹鋼薄板上,並在板上抹開,使洞 上有一液膜形成。以適當長度且清理過的鎢絲穿過洞中的液膜,分別 以鎢絲為陽極,金屬板為陰極,施加 5 至 10 V 的直流電壓來進行反 應。. 39.
(55) 圖 3-5(a):利用電化學蝕刻法製作探針時所使用的裝置。. 圖 3-5(b):探針製作裝置的近照。圖中所使用的孔洞直徑為 5 mm。 40.
(56) 下列為鎢絲的電化學蝕刻法反應式:. 陰極. 6H2O + 6e– → 3H2 + 6OH–. 陽極. W + 8OH– → WO42– + 4H2O + 6e–. 總反應 W + 2OH– + 2H2O → WO42– + 3H2. 於蝕刻過程中,液膜會因為劇烈反應而不斷產生氣泡,隨著反應 時間的增加,氣泡的產生會漸漸減少,表示液膜中的電解質濃度因為 反應而降低,此時應該暫停電壓的提供,補充數滴電解質之後,再開 始進行反應,以維持其反應速率,並避免液膜的破裂。隨著蝕刻反應 的進行,浸泡於液膜中的鎢絲會因為反應而漸漸向內凹陷變細,最後 因為無法承受液膜下方鎢絲的重量而斷裂,而鎢絲上下尖銳的斷裂處 均可作為探針使用。由於剛製作完成的探針上可能會有氫氧化鈉等化 合物的殘餘,所以需要以去離子水與丙酮沖洗,之後再用氮氣吹乾, 才能拿來使用。. 金(111)晶面的製備. 用丁烷焰小心的將金線末端熔融,熔融的金線會聚合成一小球, 若熔融的量夠多,則小球會呈水滴狀,直徑大小為 2 至 3 mm 左右。 經過反覆的熔融與凝結過程,在金球表面上可以清楚觀察到金(111) 與金(100)小晶面的形成,其中金(111)晶面直徑約 0.6 mm 左右,可直 接作為 STM 實驗的基材來使用。. 金(111)晶面的清理 41.
(57) 清理晶面所用的酸液是硝酸與過氯酸以體積比 1:1 的比例所配 置而成的。將單晶面置於酸液中數小時,以除去吸附於單金面上的有 機雜質。單晶面從酸槽中拿出之後,以去離子水充分的沖洗,接著以 丁烷焰進行回火(annealing)步驟,加熱單晶至紅熱狀態,並維持 10 到 30 分鐘,使晶面更為平整。結束回火步驟後,立刻以去離子水冷 卻。由圖 3-6(a)、(b)的 STM 掃描影像中可以觀察到進行回火步驟前 後表面狀況的顯著差異。從圖中可以發現,經過回火步驟後的表面平 台較寬廣,而且沒有島嶼(island)的出現。. STM 自我組裝薄膜樣品的製備. 完成單金的清理後,將單晶面浸泡於樣品溶液中。當達預定的浸 泡時間後,將單金從溶液中拿出,以溶劑充分的沖洗,最後再用氮氣 吹乾。. STM 實驗步驟. 盡速將完成自我組裝薄膜製備的單晶面安置於自行設計製作的 樣品平台(sample holder)上後,如圖 3-7 所示,再將樣品平台裝置於 STM 中,如圖 3-8 所示。把製作好的探針置於探針揷管(tip holder)中, 並使之垂直貼近單晶面。為了避免空氣中的水氣吸附於樣品表面上形 成水膜所造成的干擾,而把 STM 封於透明壓克力箱中,並且通以氮 氣,使箱內溼度降至 15%以下,再把壓克力箱置於彈簧懸吊平台上, 以去除環境震動以及噪音的影響。完成上述步驟後,即可開始 STM 的影像掃描。 42.
(58) 圖 3-6(a):未進行回火步驟,直接進行 STM 掃描所得的影像。. 圖 3-6(b):為經過回火步驟後再進行 STM 量測所得的影像。圖中縱 向的細小規則條紋為雜訊,並非表面上的規則結構。. 43.
(59) 圖 3-7:裝置於 STM 樣品平台上的金(111)晶面。. 圖 3-8:裝載於 STM 中的樣品。. 44.
(60) 3.4 EC-STM 實驗方法與步驟. EC-STM 除了所使用的單晶面的製備與 STM 有些微的不同之 外,在單晶面的清理與探針的製作上均使用相同的方法。. EC-STM 專用金(111)晶面的製備. 利用 STM 單晶的製作方法製作出一具有金(111)晶面的水滴狀金 球,再調整金球的方向,使實驗時所用的金(111)晶面水平朝上,最後 再以點焊機固定於小金薄片上,依此方式製備好的 EC-STM 專用單 晶如圖 3-9 所示。. EC-STM 探針的前處理. 目前已知有多種的材料可使用作為探針上的絕緣層,而本實驗室 選擇以一主要成份為硝化纖維(nitrocellulose)的透明指甲油作為絕緣 材料。將適當量的透明指甲油塗附於製作完成的鎢探針上(實驗中所 使用的 EC-STM 探針均為電化學蝕刻法所製造出的鎢探針),並以針 尖朝上的方式放置一段時間,則於針尖端的指甲油會因為重力而向下 流動,最後在針尖裸露出一小區金屬表面,以供穿隧電流的產生。等 到指甲油完全凝固之後,即完成探針絕緣層的塗附步驟。. EC-STM 實驗步驟. 盡速將清理過後的金(111)晶面裝置於自行製作的電化學樣品試 45.
(61) 圖 3-9: EC-STM 專用金單晶電極,左邊的單晶為金(111)晶面,而右 邊的單晶為經過研磨處理過後的金(100)晶面。. 46.
(62) 槽中,再將樣品試槽安置於 EC-STM 中,放入氫參考電極與白金相 對電極,如圖 3-10 所示,之後調整探針使其垂直貼近晶面,並且在 試槽中加入電解液(於本實驗中是使用 0.1 M 過氯酸作為電解液),如 圖 3-11 所示,最後將 EC-STM 放置於彈簧懸吊平台上,待平台穩定 之後,即可開始影像的掃描。此時掃描所得到的是加入樣品之前乾淨 基材的表面影像。當要加入樣品溶液於試槽中時,首先要降低探針的 掃描速度,之後再滴入數滴適當濃度的樣品溶液於電解液中,然後恢 復原本的探針掃描速度,繼續影像的掃描。. 3.5 超高真空的準備. XPS、NEXAFS 與 TPD 實驗均在超高真空系統中進行,所以在 實驗之前,需要先將真空腔中的壓力抽低至 10-10 Torr 左右,將金(111) 晶面於真空腔中清理乾淨後,再把單晶面拿出真空腔並浸泡於樣品溶 液中,來進行自我組裝薄膜的製備,或是於真空腔中進行樣品的吸 附。完成薄膜的製備後,再於真空腔中進行 XPS、NEXAFS 與 TPD 實驗的量測。 確定腔體上的各項儀器裝置均銜接無誤之後,開始使用機械幫浦 作初抽的工作,直到腔體壓力從一大氣壓抽至 10-3 Torr 左右,再改以 渦輪分子幫浦輔助抽氣,壓力可迅速抽至 10-6 Torr 左右。為了避免裝 置銜接失誤而造成氣體滲漏,導致無法達到超高真空,因此使用氦氣 來進行測漏。氦氣由於分子量小,擴散速率大,易通過細小孔洞,所 以是極好的測漏工具。使用氦氣進行檢查並且確定腔體無滲漏之後, 則可利用加熱帶對腔體進行加熱烘烤(bake out)。腔體暴露在大氣壓下 時,腔壁表面會吸附水氣及其他氣體分子,而這些分子會在低壓下緩 47.
(63) 圖 3-10:電化學樣品試槽,其中包括有金工作電極、氫參考電極與白 金相對電極。. 圖 3-11:裝置完成後的 EC-STM。. 48.
數據


+7



Outline
相關文件
‧此模型亦能解釋Stokes shlft的緣 由。.. 許多陽離子都具有兩種以之價態,如果價態 錯誤且較穩定存在於主體晶格內,就會由活
在上圖中,最上層的物件是 Root,代表電腦的桌面(Desktop),而 每個桌面可以有多個 MATLAB 圖形視窗(Figures),所以我們通常 定義 Figure 是 Root 的孩子(Child),而 Root
(十二) 裁判長資料袋整理、封條及成績彙收作業(評分結束後收取評分 表、競賽總成績表、優勝前 5 名及佳作成績表及競賽場紀錄表
十七、申訴:申訴應以書面報告由單位領隊或教練簽名,並在該項比 賽結束成績公告後 30 分鐘內附保證金 5,000
在1980年代,非晶矽是唯一商業化的薄膜型太 陽能電池材料。非晶矽的優點在於對於可見光
2.本系學生須於大二學年結束前參加英文檢定 考試,成績並應達 CEF 之 B2 高階級(相當於 TOEIC 成績 750
理解並欣賞幾何的性質可以透過坐標而轉化成數與式的 關係,而數與式的代數操作也可以透過坐標產生對應的
有能生得者購千金。於是有縛 廣武君而致戲下者,信乃解其
