國立交通大學
照明與能源光電研究所
碩
士
論
文
離子性聚芴衍生物之合成及其在有機發光元件之應
用
Synthesis of Ionic Polyfluorene Derivatives and Their Applications in
Organic Light-Emitting Devices
研究生:蔡佳昇 Chia-Sheng Tsai
指導教授:楊勝雄 博士 Dr. Sheng-Hsiung Yang
離子性聚芴衍生物之合成及其在有機發光元件之應用
Synthesis of Ionic Polyfluorene Derivatives and Their Applications in
Organic Light-Emitting Devices
研 究 生:蔡佳昇 Student:Chia-Sheng Tsai
指導教授:楊勝雄 博士 Advisor:Dr. Sheng-Hsiung Yang
國 立 交 通 大 學 照明與能源光電研究所
碩 士 論 文
A Thesis
Submitted to Institute of Lighting and Energy Photonics College of Photonics
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master
In
Lighting and Energy Photonics July 2012
Tainan, Taiwan, Republic of China
i 離子性聚芴衍生物之合成及其在有機發光元件之應用 研究生:蔡佳昇 指導教授:楊勝雄 博士 國立交通大學照明與能源光電研究所碩士班 摘要 本研究之目的在於合成出含溴烷側鏈之同聚芴高分子 P1 及共聚芴高分 子 P2,再進行離子交換反應以得到離子性聚芴材料 P1-Br、P1-BF4、P1-PF6、 P2-Br、P2-BF4 及 P2-PF6,探討不同陰離子團基在熱分析、光學及電化學 性質之影響,並作為電子傳輸材料以製作多層高分子發光元件。此外亦摻 入橘光單體苯并硒二唑合成白光高分子 P3,同樣進行離子交換反應,得到 離子性聚芴材料 P3-Br、P3-BF4、P3-PF6以製備白光電化學元件。 含 Br 離子性聚芴 P1-Br、P2-Br 由於產生霍夫曼脫去反應,致使熱穩 定性降低,置換成 BF4及 PF6離子後,可提升高分子之熱穩定性。吸收及放 射光譜顯示含有 Br 之離子性高分子在甲醇會產生藍位移,而含有 BF4及 PF6 離子性高分子在乙腈中會產生藍位移,乃由於高分子在該溶劑之溶解度較 高造成高分子鏈之間距離較遠,分子間作用力降低所致;白光高分子 P3 系 列於薄膜態可得到位於 430 及 560 nm 之雙放射峰。電化學分析結果顯示含
ii BF4及 PF6之離子性高分子其氧化電位明顯下降,因而造成材料 HOMO 以 及 LUMO 提升。 本研究利用離子性聚芴作為電子傳輸層製作多層高分子發光元件,以 MEH-PPV、HMM、HDM、HPM 為發光層,證明離子性聚芴的引入,尤其 是 P1-BF4,確實大幅提升發光元件之亮度及效率。以離子性聚芴製作之藍 光及白光 LEC 元件,亦得到低操作電壓、高亮度及高效率的特點,且不需 額外加入鹽類或聚氧乙烯。以上實驗結果說明這些離子性聚芴材料具有高 度應用於發光元件之潛力。
iii
Synthesis of Ionic Polyfluorene Derivatives and Their Applications in
Organic Light-Emitting Devices
Student: Chia-Sheng Tsai Advisor: Dr. Sheng-Hsiung Yang
Institute of Lighting and Energy Photonics National Chiao Tung University
Abstract
The goal of this research is to synthesize homopolyfluorene P1 and copolyfluorene P2 containing bromoalkyl side chains, followed by ionic exchange reaction to obtain ionic polyfluorene materials including P1-Br, P1-BF4, P1-PF6, P2-Br, P2-BF4, and P2-PF6, and to study the influence of different anionic groups on thermal, optical, and electrochemical properties. Those materials were used as electron transporting layer to fabricate multilayer polymer light emitting devices. Besides, orange emitting monomer
2,1,3-benzoselenadiazole was introduced to synthesis white light polymer P3,
and ionic exchange reaction was carried out to obtain ionic polyfluorene materials P3-Br, P3-BF4, and P3-PF6 for the fabrication of white light electrochemical cells.
The thermal stability of Br-containing ionic polyfluorenes was decreased because of Hofmann elimination. By replacing Br with BF4 and PF6 ions, the
iv
thermal stability of polymers was raised. UV-visible absorption and photoluminescence spectra show that Br-containing ionic polymers generate blue shift in methanol, and BF4 and PF6 ionic polymers generate blue shift in
acetonitrile. This is because higher solubility of polymer was found in the solvent, resulting in longer distance to decrease molecular interaction between polymer chains. Two emission peaks were found for the white light polymer P3 series located at 430 and 560 nm. Electrochemical analysis shows that oxidation potential of BF4 and PF6-containing ionic polymers was significantly
decreased, resulting in increase in HOMO and LUMO of materials. Multilayer polymer light emitting devices were fabricated by using ionic polyfluorenes as electron transporting layer in this research. MEH-PPV, HMM, HDM, and HPM were used as emission layer. Experimental results demonstrated that introduction of ionic polyfluorenes indeed enhanced brightness and efficiency of light emitting devices significantly, especially for P1-BF4. Characters of low operation voltage, high brightness and high efficiency were obtained for blue and white light electrochemical cells by using ionic polyfluorenes, without additional add of salts or poly(ethylene oxide). The above experimental results indicate that these ionic polyflourene materials have highly potential application in light-emitting devices.
v 誌謝 首先要感謝指導教授楊勝雄老師,在兩年的碩士求學生涯裡給了我很 多論文上指導與方向,讓我可以順利完成本論文。於此很感謝口詴期間蘇 海清老師及趙宇強老師對於此碩士論文的指教,讓這本論文更加完善。 在實驗室裡先要感謝學長憲哥提供高分子材料以及實驗上的指導,及 同學國志這兩年來對於實驗上的研究討論,還有生活上的幫助,一路走來 路途艱辛,幸虧有以上兩位的幫助,讓我的碩士生涯更加充實,回新竹本 部的小強,希望你未來在業界闖出一片天地,另外先行脫隊的柯柯,希望 你可以達到你的目標,再來很感謝學弟們的幫忙,謝謝于聖幫忙製作了 LiF 的 PLED 元件,維勝、子軒、國兼、彥廷、徐雍與在職專班的胖威及建璋 讓整間實驗室充滿歡樂的氣息。感謝蘇老師實驗室廖學長在元件量測上的 指導,劉柏村學弟幫忙製作 LEC 元件,有以上學長、學弟幫助下,本人才 得以完成此論文,在此致上萬分感謝。新進來的學弟們也期許你們在未來 研究路上,可以做出亮眼的研究成果,在此共勉之。 謝謝我的爸媽、外婆對於我這二十多年來的培養及照顧,沒有你們就 沒有今天的我,以及謝謝一路上支持我的親友、長輩們對我的鼓勵,感謝 我的女友珈旻多年對我的支持以及打氣,讓我得以度過每個關卡。 最後將本篇論文獻給天上最和藹的外公。
vi 目錄 頁次 摘要(中文) ... i 摘要(英文) ... iii 誌謝 ... v 目錄 ... vi 流程目錄 ... ix 表目錄 ... x 圖目錄 ... xi 第一章 緒論... 1 1-1 前言 ... 1 1-2 有機發光元件簡介 ... 1 1-3 有機共軛高分子發光理論 ... 4 1-3-1 共軛高分子(Conjugated Polymer) ... 4 1-3-2 發光機制 ... 4 1-3-2-1 非輻射形式釋放 ... 6 1-3-2-2 輻射形式 ... 7 1-3-3 光激發光及電激發光 ... 7
vii
1-4 發光電化學電池(Light-emitting Electrochemical Cells, LEC) ... 8
1-4-1 LEC 的起源 ... 8 1-4-2 LEC 的發展 ... 10 1-5 離子性共軛高分子材料簡介 ... 14 1-6 研究動機 ... 17 第二章 實驗方法與步驟 ... 19 2-1 詴藥 ... 19 2-2 鑑定儀器... 19 2-3 元件製作... 22 2-3-1 ITO 基板清洗步驟 ... 22 2-3-2 發光二極體製作流程 ... 22 2-4 材料合成 ... 23 2-4-1 單體合成 ... 23 2-4-2 高分子聚合 ... 29 2-4-3 高分子離子化 ... 30 2-4-4 離子置換 ... 31 第三章 實驗結果與討論 ... 37 3-1 單體及高分子結構鑑定 ... 37 3-1-1 NMR 光譜分析 ... 37
viii 3-1-2 ESCA 光譜分析 ... 39 3-1-3 FTIR 分析 ... 41 3-2 高分子分子量測定 ... 43 3-3 高分子熱性質分析 ... 44 3-4 高分子光學性質分析 ... 47 3-4-1 吸收光譜分析 ... 47 3-4-2 螢光放射光譜分析 ... 49
3-4-3 螢光量子效率(PL Quantum Yield, PLQY) ... 53
3-5 高分子電化學性質分析 ... 54 3-6 高分子之溶解度測詴 ... 57 3-7 PLED 元件表現 ... 58 3-7-1 比較離子材料增加元件特性之探討 ... 59 3-7-2 以離子材料 P1-BF4增進 HMM、HDM、HPM 元件特性 ... 64 3-8 LEC 元件表現 ... 68 第四章 結論... 78 第五章 參考文獻 ... 80 附錄 ... 85
ix 流程目錄 頁次 Scheme 1 單體 M1~M5 之合成途徑 ... 33 Scheme 2 高分子 P1~P4 之合成途徑 ... 34 Scheme 3 離子性高分子 P1-Br、P2-Br、P3-Br 之合成途徑 ... 35 Scheme 4 離子性高分子 P1-BF4、P1-PF6、P2-BF4、P2-PF6、P3-BF4、P3-PF6 之合成途徑 ... 36
x 表目錄 頁次 Table 3-1 高分子 P1~P3 之 FTIR 訊號 ... 42 Table 3-2 高分子 P1~P3 之分子量及分子量分佈 ... 44 Table 3-3 高分子 P1~P3 系列 Td值 ... 45 Table 3-4 高分子 P1~P3 之 Tg及 Tc值 ... 46 Table 3-5 高分子 P1~P3 之吸收與螢光放射最大峰值 ... 52 Table 3-6 離子高分子 P1-Br~P3-Br 之吸收與螢光放射最大峰值 ... 52 Table 3-7 離子高分子 P1-BF4~P3-BF4吸收與螢光放射最大峰值 ... 53 Table 3-8 離子高分子 P1-PF6~P3-PF6吸收與螢光放射最大峰值 ... 53 Table 3-9 高分子於薄膜下的螢光量子效率 ... 54
Table 3-10 各高分子之氧化及還原電位、HOMO、LUMO 及 Bandgap ... 56
Table 3-11 高分子之溶解度比較表 ... 57
Table 3-12 MEH-PPV、HMM、HDM、HPM 之分子量及其分佈 ... 59
Table 3-13 MEH-PPV 元件特性一覽表 ... 64
Table 3-14 HMM、HDM 及 HPM 元件特性一覽表 ... 68
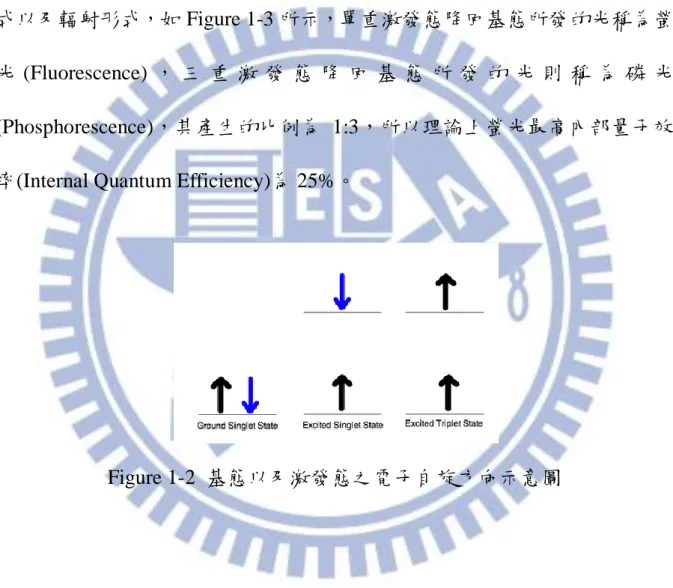
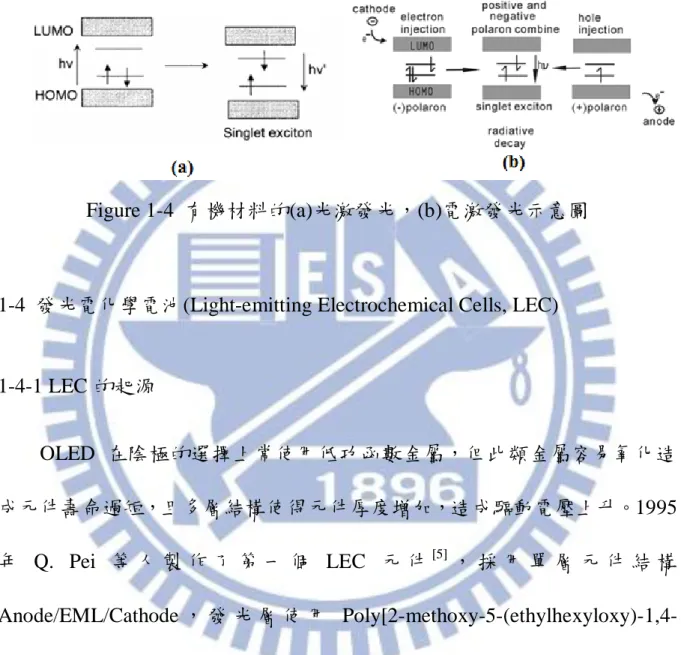
xi 圖目錄 頁次 Figure 1-1 多層結構之有機發光元件 ... 4 Figure 1-2 基態以及激發態之電子自旋方向示意圖... 5 Figure 1-3 電子躍遷移示意圖 ... 6 Figure 1-4 有機材料的(a)光激發光,(b)電激發光示意圖 ... 8
Figure 1-5 (a) MEH-PPV,(b) Lithium trifluoromethanesulfonate,(c) PEO 之化學結構 ... 9
Figure 1-6 LEC 的操作機制 ... 9
Figure 1-7 能帶示意圖:(a) p-i-n 接面形成前,(b) p-i-n 接面形成後能帶彎 曲 ... 10
Figure 1-8 離子性液體(a) ATOAAS,(b) MATS ... 11
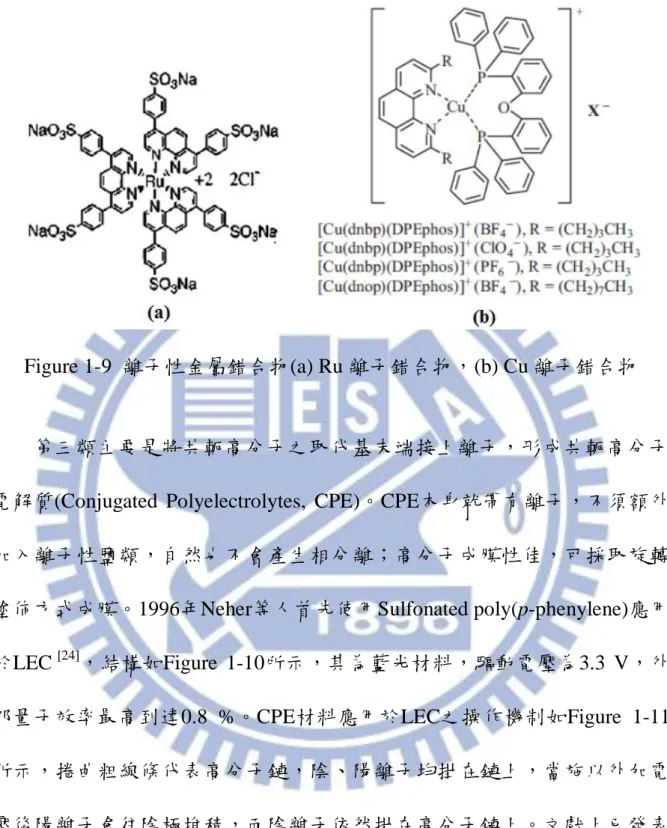
Figure 1-9 離子性金屬錯合物(a) Ru 離子錯合物,(b) Cu 離子錯合物 ... 12
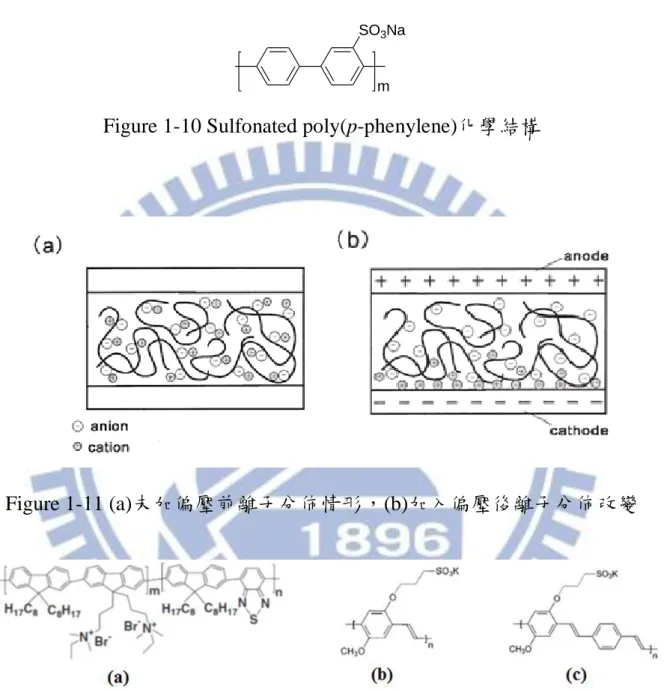
Figure 1-10 Sulfonated poly(p-phenylene)化學結構 ... 13
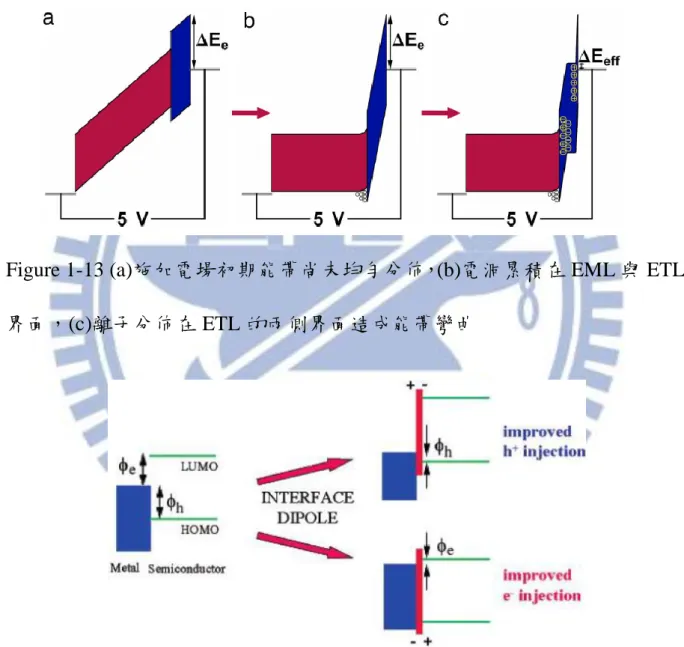
Figure 1-11 (a)未加偏壓前離子分佈情形,(b)加入偏壓後離子分佈改變 ... 13
Figure 1-12 離子性高分子(a) PFNBr-BTDZ,(b) MPS-PPV,(c) CO-MPS-PPV ... 13
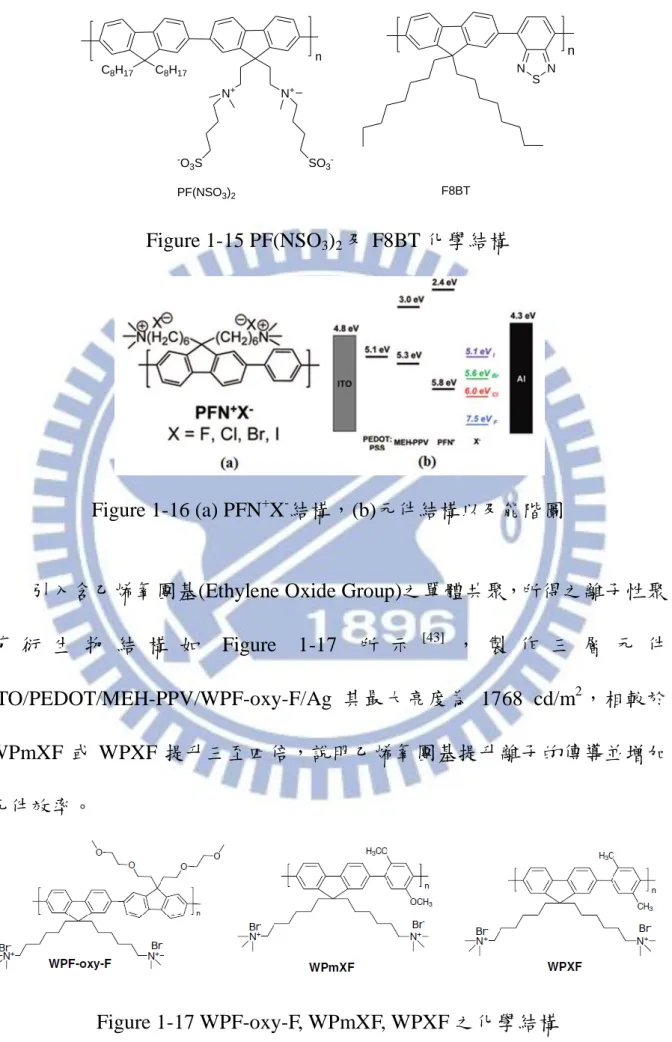
Figure 1-13 (a)施加電場初期能帶尚未均勻分佈,(b)電洞累積在 EML 與 ETL 界面,(c)離子分佈在 ETL 的兩側界面造成能帶彎曲 ... 15
xii Figure 1-14 界面偶極產生內建電場誘使載子注入有機層示意圖 ... 15 Figure 1-15 PF(NSO3)2及 F8BT 化學結構 ... 16 Figure 1-16 (a) PFN+X-結構,(b)元件結構以及能階圖 ... 16 Figure 1-17 WPF-oxy-F, WPmXF, WPXF 之化學結構 ... 16 Figure 1-18 離子性聚芴或聚噻吩衍生物 ... 17 Figure 1-19 本研究擬合成之離子性聚芴衍生物 ... 18 Figure 3-1 單體 M3 及高分子 P2、P2-Br 在苯環區上的 NMR 光譜圖 ... 38 Figure 3-2 單體 M3 及高分子 P2、P2-Br 在長碳鏈區上的 NMR 光譜圖 .. 38 Figure 3-3 高分子 P3-Br 之 ESCA 光譜 ... 40 Figure 3-4 高分子 P3-BF4之 ESCA 光譜 ... 40 Figure 3-5 高分子 P3-PF6之 ESCA 光譜 ... 41
Figure 3-6 高分子(a) P1 及 (b) P3 之 FTIR 光譜 ... 42
Figure 3-7 高分子 P1 系列之 TGA 曲線圖... 45
Figure 3-8 Hofmann elimination 反應機制示意圖 ... 45
Figure 3-9 高分子 P1 之 DSC 曲線圖 ... 46
Figure 3-10 高分子 P2 系列溶液及薄膜態之吸收光譜(a) P2,(b) P2-Br, (c) P2-BF4,(d) P2-PF6 ... 48
xiii Figure 3-12 高分子 P2 系列溶液及薄膜態之螢光放射光譜(a) P2,(b) P2-Br,(c) P2-BF4,(d) P2-PF6 ... 50 Figure 3-13 高分子 P3 系列溶液及薄膜態之螢光放射光譜(a) P3,(b) P3-Br,(c) P3-BF4,(d) P3-PF6 ... 51 Figure 3-14 高分子 P3 系列於薄膜態之螢光放射光譜 ... 52 Figure 3-15 高分子 P1 薄膜在積分球中光譜 ... 54 Figure 3-16 高分子 P1 系列之氧化曲線圖 ... 56 Figure 3-17 MEH-PPV、HMM、HDM 及 HPM 之化學結構 ... 58 Figure 3-18 雙層元件 ITO/PEDOT/MEH-PPV/Al 之 EL 光譜 ... 59
Figure 3-19 雙層元件 ITO/PEDOT/MEH-PPV/Al 之(a) J-V-B (b) E-J 曲線圖 ... 60
Figure 3-20 三層元件 ITO/PEDOT/MEH-PPV/P1-Br/Al 之 EL 光譜 ... 60
Figure 3-21 三層元件 ITO/PEDOT/MEH-PPV/P1-Br/Al 之(a) J-V-B (b) E-J 曲線圖 ... 61
Figure 3-22 三層元件 ITO/PEDOT/MEH-PPV/P1-BF4/Al 之(a) J-V-B (b) E-J 曲線圖 ... 61
Figure 3-23 三層元件 ITO/PEDOT/MEH-PPV/P1-PF6/Al 之(a) J-V-B (b) E-J 曲線圖 ... 62
Figure 3-24 三層元件 ITO/PEDOT/MEH-PPV/P1-BF4/LiF/Al 之(a) J-V-B (b) E-J 曲線圖 ... 63
xiv
Figure 3-25 三層元件 ITO/PEDOT/HMM/P1-BF4/Al 之(a) EL 光譜 (b)
J-V-B (c) E-J 曲線圖 ... 65 Figure 3-26 三層元件 ITO/PEDOT/HDM/P1-BF4/Al 之(a) EL 光譜 (b)
J-V-B (c) E-J 曲線圖 ... 66 Figure 3-27 三層元件 ITO/PEDOT/HPM/P1-BF4/Al 之(a) EL 光譜 (b)
J-V-B (c) E-J 曲線圖 ... 67 Figure 3-28 以 P1-BF4為發光層之 LEC 元件於不同電壓下之(a) EL 光譜(b)
J- T-B (c) E-T 曲線圖 ... 69 Figure 3-29 以 P1-PF6為發光層之 LEC 元件於不同電壓下之(a) EL 光譜(b)
J- T-B (c) E-T 曲線圖 ... 70 Figure 3-30 以 P2-BF4為發光層之 LEC 元件於不同電壓下之(a) EL 光譜(b)
J- T-B (c) E-T 曲線圖 ... 71 Figure 3-31 以 P2-PF6為發光層之 LEC 元件於不同電壓下之(a) EL 光譜(b)
J- T-B (c) E-T 曲線圖 ... 72 Figure 3-32 以 P3-BF4 (0.5 %)為發光層之 LEC 元件於不同電壓下之(a) EL
光譜(b) J- T-B (c) E-T 曲線圖 ... 73 Figure 3-33 以 P3-PF6 (0.5 %)為發光層之 LEC 元件於不同電壓下之(a) EL
xv
Figure 3-34 以 P3-BF4 (0.25 %)為發光層之 LEC 元件於不同電壓下之(a) EL 光譜(b) J- T-B (c) E-T 曲線圖 ... 75 Figure 3-35 以 P3-PF6 (0.25 %)為發光層之 LEC 元件於不同電壓下之(a)
1
第一章 緒論
1-1 前言
有機半導體材料與元件近年來的研究如雨後春筍般發展,主要優勢為 低成本、可撓性、可採大面積製程,主要應用於有機太陽能電池(Organic Solar Cells, OSCs)、有機場效電晶體(Organic Field-effect Transistors, OFETs)、 有機發光二極體(Organic Light Emitting Diodes, OLEDs)等,其中 OLED 的發 展最引人注目,因目前市面上平面顯示器以液晶顯示器(Liquid Crystal Display, LCD)為主,其存在許多缺點,例如:反應時間慢(液晶分子受電場 轉動導致應答時間落在毫秒)、耗電量大(額外採用背光模組與彩色濾光片 來產生色彩效果),在此情況下有機發光顯示器就因應而生,其具備自發光、 廣視角、反應時間快、高發光效率、較輕薄、高對比度、可撓性、製程較 簡單等優點,也被期待可以取代液晶顯示器成為下一代顯示器的主流。 1-2 有機發光元件簡介 有機發光元 件 運 作的 基本 原理 是 利用 有機 物通 電後 產生 電激 發 光 (Electroluminescence, EL)現象,其研究肇始於 1963 年,Pope、Kallmann 等 人製作膜厚 10~20 微米的單晶蔥 (anthracence)通以 400 伏特電壓產生發光
2
並不受到重視;直到 1987 年 Kodak 的 C. W. Tang 與 S. A. VanSlyke 等人 採用了 Tris(8-hydroxyquinolinato)aluminum (Alq3)做為發光層與電子傳輸層 [2]
,Diamine 做為電洞傳輸層,採用鎂銀合金當作電極,並使用真空蒸鍍法 來製作元件,此結構增加電子電洞在有機層中再結合的比率,在操作電壓 10 V 下,外部量子效率(External Quantum Efficiency)達到 1 %,亮度為 1000
cd/m2,自此有機發光元件獲得極大的進展,也引起了研究的熱潮。
在 發 光 高 分 子 的 研 究 方 面 , 最 早 於 1982 年 Patridge 使 用 Poly(N-vinylcarbazole) (PVK)利用溶液旋轉塗佈 (spin coating)的方式製作第 一個高分子電激發光元件[3];之後 1990 年英國劍橋大學 J. H. Burroughes 等 人利用 Poly(1,4-phenylenevinylene) (PPV)做為發光層[4],製作出 ITO/PPV/Al 的元件結構,量子效率為 0.05 %。此後有機發光元件也就分成兩類,其一 為有機染料小分子作為主動層,稱為 OLED,製作方式採用真空蒸鍍;其 二為有機共軛高分子,稱為高分子發光二極體(Polymer Light Emitting Diode, PLED),製作方式採用溶液製程。比較兩種材料的優缺點,小分子材料合成 較為簡單且純度較高,但真空蒸鍍設備的成本頗高;高分子材料合成較繁 雜且分子量大小分佈不均,但成膜性佳且可大面積製作,成本較為便宜。 有機發光元件經過演進後形成多層結構,如 Figure 1-1 所示: 1. 陽極(Anode): 通常選用透明導電度高的材料、具有良好的熱穩定性、功函數(Work
3
Function)頇與電洞傳輸層(Hole Transporting Layer, HTL)或是電洞注入層 (Hole Injection Layer, HIL)的最高填滿分子軌域(Highest Occupied Molecular Orbital, HOMO)匹配,一般元件製作選用氧化銦錫(Indium Tin Oxide, ITO) 為陽極。
2. 電洞注入層(HIL):
主要目的為幫助電洞注入至 HTL 或是發光層(Emission Layer, EML), 所以材料選擇主要為 HOMO 與陽極功函數相匹配。
3. 電洞傳輸層(HTL): 用途為幫助電洞傳輸並且阻擋電子,選取高電洞遷移率以及最低未填 滿分子軌域(Lowest Unoccupied Molecular Orbital, LUMO)高於發光層之 LUMO。
4. 發光層(EML):
優良的發光材料需具備良好的熱穩定性、高螢光強度、良好的成膜性。 5. 電子傳輸層(Electron Transporting Layer, ETL):
ETL 幫助元件中電子傳輸,必頇有良好的電子遷移率,也需兼具阻擋 電洞能力,所以其 HOMO 必頇低於發光層之 HOMO。
6. 電子注入層(Electron Injection Layer, EIL):
其 LUMO 需與陰極功函數匹配,幫助電子從陰極注入。 7. 陰極(Cathode):
4 一般來說要有良好的熱穩定性以及附著性,為了減低電子注入能障, 其功函數頇與 EIL 或是 ETL 相匹配,可選擇低功函數的金屬如鈣、鎂,但 低功函數金屬的活性較大,容易與氧及水氣反應,故頇具備良好的封裝技 術克服此一缺點。 Figure 1-1 多層結構之有機發光元件 1-3 有機共軛高分子發光理論 1-3-1 共軛高分子(Conjugated Polymer) 共軛高分子利用化學結構上的單鍵—雙鍵交替,p 軌域上的電子可沿著 分子軌域形成非定域化(Delocalized) π 電子雲,相鄰原子之電子雲互相接觸, 使得電子可以沿著主鏈移動。 1-3-2 發光機制 有機分子受到外在能量激發,當能量大於能隙(Energy Gap)時使得電
5
子從基態(Ground State, S0)躍遷至激發態(Excited State, S1)。在激發態時電子
自旋方向相反於基態,此稱為單重激發態(Singlet Excited State, Sn),而電子
自旋方面與基態相同時,稱為三重激發態(Triplet Excited State, Tn),如 Figure
1-2 所示。電子從激發態落回基態會釋放出能量,釋放的方式分為非輻射形 式以及輻射形式,如 Figure 1-3 所示,單重激發態降回基態所發的光稱為螢 光 (Fluorescence) , 三 重 激 發 態 降 回 基 態 所 發 的 光 則 稱 為 磷 光 (Phosphorescence),其產生的比例為 1:3,所以理論上螢光最高內部量子效 率(Internal Quantum Efficiency)為 25%。
6 Figure 1-3 電子躍遷移示意圖 1-3-2-1 非輻射形式釋放 1. 振動鬆弛(Vibrational Relaxation) 受激發的電子由於分子間的碰撞,從較高的振動能階降至較低的振動 能階,並以熱的方式釋放能量;生命週期(Life Time)在 10-12秒內。 2. 內轉換(Internal Conversion) 屬於分子內非輻射轉換過程,當激發態的電子能階非常接近,致使振 動能階相互重疊,此時電子可以在 S2與 S1之間躍遷。 3. 外轉換(External Conversion) 激發態分子與溶劑分子或其他物質相互作用,產生能量轉移並降低發 光強度;低溫或是高黏度可以降低外轉換導致的能量散失。
7 4. 系統間跨越(Intersystem Crossing) 從單重激發態轉換到三重激發態(S1→T1),材料本身常含有重原子如碘 或溴原子。 1-3-2-2 輻射形式 1. 螢光:激發電子從單重激發態回到基態的放光形式,生命週期在 10-7~10-9 秒之間。 2. 磷光:電子從三重激發態降至基態的放光過程,生命週期在 10-4至數秒, 甚至數小時。由於衰退時間太長,常導致非輻射方式釋放能量。 1-3-3 光激發光及電激發光 有 機 共 軛 高 分 子 會 受 到 兩 種 激 發 形 式 而 放 光 , 其 一 為 光 激 發 光 (Photoluminescence, PL),係使用光為激發源,有機分子吸收光使得電子由 基態躍遷至激發態,而形成激子(Exciton);由於此高能狀態不穩定,其落回 基態過程以光的形式釋放,如 Figure 1-4 (a)所示。其二為電激發光(EL),使 用電為激發源,於有機薄膜兩端加上電極,當施以順偏壓時電子從陰極注 入發光層的 LUMO,而電洞從陽極注入發光層的 HOMO,此時電子與電洞 分別跨越能障(Energy Barrier),在發光層中相遇並再結合(Recombination)形 成激子,再由高能狀態降至基態並以光的形式釋放,如 Figure 1-4 (b)所示。
8
元件發出的光色由發光層的能隙決定,針對不同材料可發出紫外光、可見 光或是紅外光。
Figure 1-4 有機材料的(a)光激發光,(b)電激發光示意圖
1-4 發光電化學電池(Light-emitting Electrochemical Cells, LEC)
1-4-1 LEC 的起源
OLED 在陰極的選擇上常使用低功函數金屬,但此類金屬容易氧化造 成元件壽命過短,且多層結構使得元件厚度增加,造成驅動電壓上升。1995 年 Q. Pei 等 人 製 作 了 第 一 個 LEC 元 件[5], 採 用 單 層 元 件 結 構 Anode/EML/Cathode , 發 光 層 使 用 Poly[2-methoxy-5-(ethylhexyloxy)-1,4- phenylene vinylene] (MEH-PPV),摻混離子性鹽類 Lithium trifluoromethane sulfonate 以提供離子,並加入聚氧乙烯(Polyethylene Oxide, PEO)幫助離子傳 導,化學結構如 Figure 1-5 所示。施以偏壓後在電極附近產生氧化還原反應, 並使得陽離子往陰極移動,而陰離子往陽極移動,在陽極形成 p 型摻雜, 而陰極形成 n 型摻雜,最終形成 p-i-n 接面,如 Figure 1-6 所示,造成能帶
9
的彎曲並降低能障,如 Figure 1-7 所示。LEC 的製作可選擇銀、金等活性較 低的金屬當作電極,其驅動電壓甚低,約等於材料的能隙;最後發光區域 則被侷限於本質 區 (Intrinsic Region), 此種機制也被認 為是一種自 組裝 (self-assembled) p-i-n 高分子接面[6]。
Figure 1-5 (a) MEH-PPV,(b) Lithium trifluoromethanesulfonate,(c) PEO 之 化學結構
10
Figure 1-7 能帶示意圖:(a) p-i-n 接面形成前,(b) p-i-n 接面形成後能帶彎曲
1-4-2 LEC 的發展
目前 LEC 的發展主要分為三類,第一類是以共軛高分子加入離子性鹽 類與 PEO [7-13]。高分子與離子性鹽類有時會產生相分離(Phase Separation)而 減少元件壽命,後續研究針對離子性鹽類加以改良,採用離子性的液體材 料 如 allyltrioctylammonium allylsulfonate (ATOAAS) [14]、 methyltrioctyl- ammonium trifluoromethanesulfonate (MATS)等[15-17],結構如 Figure 1-8 所示, 減少了相分離且不頇再加入 PEO,惟仍存在離子移動造成反應時間過慢的 問題。為改善上述缺點,選用可聚合離子團基通電排列後,再進行自由基 聚合將 p-i-n 接面固定住,可以增快反應時間[18-19]。
11
Figure 1-8 離子性液體(a) ATOAAS,(b) MATS
第二類是採用離子性過渡金屬錯合物(Ionic Transition Metal Complex, ITMC)。1996年Handy、Lee、Rubner使用Ruthenium的錯合物[20],結構如Figure 1-9 (a)所示,其本身具有離子團基,不需要加入離子性鹽類,同時引入磷光, 整 體 的 發 光 效 率 有 大 幅 提 升[21-22]。 2006 年 Zhang 等 人 使 用 [Cu(2,9- di-n-octyl-1,10-phenanthroline)(bis[2-(diphenylphosphino)phenyl]ether)]+X- (X = BF4, ClO4, PF6)製作單層LEC [23],結構如Figure 1-9 (b)所示,以鋁做為電極 並施以4 V電壓,LEC之量子效率達16 %,電流效率為56 cd/A;以鈣做為電 極時,最高亮度達4100 cd/m2 (以25 V電壓驅動)。
12
Figure 1-9 離子性金屬錯合物(a) Ru 離子錯合物,(b) Cu 離子錯合物
第三類主要是將共軛高分子之取代基末端接上離子,形成共軛高分子 電解質(Conjugated Polyelectrolytes, CPE)。CPE本身就帶有離子,不頇額外 加入離子性鹽類,自然也不會產生相分離;高分子成膜性佳,可採取旋轉 塗佈方式成膜。1996年Neher等人首先使用Sulfonated poly(p-phenylene)應用 於LEC [24],結構如Figure 1-10所示,其為藍光材料,驅動電壓為3.3 V,外 部量子效率最高到達0.8 %。CPE材料應用於LEC之操作機制如Figure 1-11 所示,捲曲粗線條代表高分子鏈,陰、陽離子均掛在鏈上,當施以外加電 壓後陽離子會往陰極堆積,而陰離子依然掛在高分子鏈上。文獻上已發表 Polyfluorene (PF) [25-32],以及Poly(p-phenylenevinylene) (PPV) 離子性衍生物 [33-34],如Figure 1-12 (a)~(c)所示。雖然目前CPE應用於LEC其元件效率無法 與ITMC相比,然而在調整光色方面較具優勢,只需要在聚合時調整單體比
13 例,或是更換不同光色之單體即可達成。
SO3Na
m
Figure 1-10 Sulfonated poly(p-phenylene)化學結構
Figure 1-11 (a)未加偏壓前離子分佈情形,(b)加入偏壓後離子分佈改變
Figure 1-12 離子性高分子(a) PFNBr-BTDZ,(b) MPS-PPV,(c) CO-MPS-PPV
近年來LEC的發展除了以上三類外,還可在LEC中摻入量子點(Quantum
Dot, QD)材料[6],利用p-i-n結構造成發光區侷限於本質區的特性,減少元件
14 作多層元件結構,但此處依靠LEC的單層結構,即可得到接近純的QD光色。 LEC的結構也可以運用在主客體的發光機制[35-36],也都得到不錯的量子效 率。 1-5 離子性共軛高分子材料簡介 CPE除了用於製作LEC元件外,還有多種光電及生醫方面應用,例如用 於探測RNA-Protein [37-39]、Biosensors [40-41],此外還可利用CPE溶於高極性溶 劑之特性以製作多層元件。一般有機發光層通常溶於極性較低的苯環類溶 劑 如 : 甲 苯 (Toluene) 、 氯 苯 (Chlorobenzene, CB) 等 , 而 CPE 可 溶 於 甲 醇 (Methanol)、乙腈(Acetonitrile, CH3CN)等極性較高之溶劑,故可直接塗佈於
主動層上,並應用於各式有機光電元件。
在PLED的應用方面[42-49],CPE可以作為ETL或是EIL,降低能障並提升 電子的注入及傳輸,以及利用能階的匹配阻擋電洞,對於元件效果有明顯 的提升。究其原因主要有以下兩點:其一著重於離子移動,造成能帶的彎 曲[50-51],如Figure 1-13所示;其二乃形成界面偶極(Interface Dipole),內建電
場誘使載子容易注入[52-53],如Figure 1-14所示。這兩種機制的劃分點在於
ETL的膜厚,Nguyen等人指出膜厚大於8 nm應採用離子的移動論[54],小於8
nm則採用界面偶極來解釋。以聚芴的離子性衍生物PF(NSO3)2為例[42],其化
15
於6 V電壓驅動時亮度可達80,000 cd/m2,電流效率達到10 cd/A。Nguyen等 人曾探討鹵素陰離子對於ETL的影響[48],其化學結構及能階圖如Figure 1-16 所示,歸納出元件亮度與效率順序為氟離子 氯離子 溴離子 碘離子。
Figure 1-13 (a)施加電場初期能帶尚未均勻分佈,(b)電洞累積在 EML 與 ETL 界面,(c)離子分佈在 ETL 的兩側界面造成能帶彎曲
16 N S N n C8H17 C8H17 N+ N+ -O 3S SO3 -n PF(NSO3)2 F8BT Figure 1-15 PF(NSO3)2及 F8BT 化學結構 Figure 1-16 (a) PFN+X-結構,(b)元件結構以及能階圖
引入含乙烯氧團基(Ethylene Oxide Group)之單體共聚,所得之離子性聚 芴 衍 生 物 結 構 如 Figure 1-17 所 示 [43] , 製 作 三 層 元 件 ITO/PEDOT/MEH-PPV/WPF-oxy-F/Ag 其最大亮度為 1768 cd/m2,相較於 WPmXF 或 WPXF 提升三至四倍,說明乙烯氧團基提升離子的傳導並增加 元件效率。
17
在 OSC 應 用 方 面[55-57], CPE 導 入 有 助 於 提 升 開 路 電 壓 (Open-circuit Voltage)以及元件之光電轉換效率(Power Conversion Efficiency);在OTFT應 用方面[58-59],加入CPE可提升遷移率(mobility)並降低驅動電壓(Threshold Voltage, Vth),還可以提升元件之開關電流比(On/Off Ratio)。相關離子性聚
芴或是聚噻吩衍生物如Figure 1-18所示。 Figure 1-18 離子性聚芴或聚噻吩衍生物 1-6 研究動機 聚芴衍生物之合成及其作為發光層的研究相當廣泛,而離子性聚芴之 開發相對上較為稀少,多侷限於鹵素類離子;其置換為它種離子以應用於 有機光電元件(例如 LEC),文獻上則尚未被報導過。本研究擬合成三種不同 聚芴高分子,並置換三種離子包括 Br –、BF4 –及 PF6 –, 探討其做為 ETL 以 評估元件性質增強效益;本研究合成之離子性聚芴衍生物亦將應用於 LEC 製作,不需額外加入離子性鹽類,預期能提升 LEC 元件特性。 除做為發光元件應用外,本研究亦將探討離子種類對於聚芴本身之光
18 學、電化學及熱性質所造成之變化。本研究擬使用紫外-可見光吸收光譜儀 及螢光光譜儀探測不同離子材料之吸收及螢光放射光譜,以循環電位儀分 析其能階,並以熱重量分析儀及示差掃描卡計檢驗離子種類是否會造成熱 性質或相行為的改變。 有鑑於白光在照明以及顯示器應用的重要性,本研究同時將合成出可 發白光之離子性聚芴衍生物,並將其應用於雙層LEC元件,設計策略乃在聚 合過程中引入少量2,1,3-Benzoselenadiazole橘光材料,以達到調整光色[60]之 目的。其屬於單一發白光材料,不會有傳統摻混引起相分離之問題,亦由 於主鏈上已掛著離子團基,製作LEC過程中不需再加入鹽類,故能簡化製程 並獲得低電壓驅動之白光LEC元件。 本研究所合成之離子性聚芴衍生物結構如Figure 1-19所示。 C8H17 C8H17 R R X=Br,BF4, PF6 R R N N N R R N Se N R R R=(CH2)6NMe3X 1-y y y=0.5% or 0.25% n n n n Figure 1-19 本研究擬合成之離子性聚芴衍生物
19
第二章 實驗方法與步驟
2-1 詴藥
本實驗詴藥及溶劑皆自聯工、Merck、Aldrich、Matrix Scientific、SHOWA 與 Alfa Aesar 購入,不經純化直接使用。發光層高分子 MEH-PPV、HMM、 HPM 、 HDM 由 實 驗 室 楊 譔 憲 學 長 提 供 。 反 應 用 之 無 水 四 氫 呋 喃 (Tetrahydrofuran, THF)是在氮氣環境下加入鈉除水,並加入 Benzophenone 作為指示劑,經除水 2 天後蒸出使用。反應使用之無水甲苯(Toluene)及二氧 陸圜(Dioxane)於氮氣環境中加入氫化鈣,經除水 2 天後蒸出使用。
2-2 鑑定儀器
1. 核磁共振光譜儀(Nuclear Magnetic Resonance Spectrometer, NMR)
係 使 用 BRUKER AVANCE 600 MHz NMR , 並 使 用 d-DMSO 與
d-Chloroform 當作溶劑,Tetramethylsilane (TMS)作為標定基準點。在光譜
中 s 代表單峰(singlet),d 代表雙重峰(doublet),t 代表三重峰(triplet),m 代 表多重峰(multiplet)。
2. 化學分析電子光譜儀(Electron Spectroscopy for Chemical Analysis, ESCA) 係使用 PHI 5000 VersaProbe 型。將待測物溶於 Methanol 或 CH3CN,再
20
3. 示差掃描卡計(Differential Scanning Calorimeter, DSC)
係使用 SII DSC 6200 及液態氮冷卻系統,測量樣品之相轉移溫度及其 焓熱值,熔點(Melting Point, Tm)取其極值,玻璃轉移溫度(Glass Transition
Temperature, Tg)則取其反曲點。溫度以 Indium 及 Tin 作校正,取樣品 5-10 mg,
於氮氣流量 100 mL/min 升溫量測,加熱速率為 10 oC/min。 4. 熱重分析儀(Thermogravimetric Analyzer, TGA)
係使用 SII TG/DTA 7200 型。實驗時取樣品 5-10 mg,在氮氣流量 100 mL/min 下測量其熱裂解溫度(Decomposition Temperature, Td),加熱速率為
10 oC/min。
5. 凝膠滲透層析儀(Gel Permeation Chromatography, GPC)
係使用 Viscotek VE3580 GPC 型。使用 Polystyrene (PS)標準品製作分子 量檢量線,THF 為沖提液,流速為 1 mL/min,管柱保持在 32 oC 之恆溫槽 內。樣品溶液配置濃度為 4 mg/2 mL。
6. 紫外-可見吸收光譜儀(Ultraviolet-visible Absorption Spectrophotometer)與 螢光光譜儀(Fluorescence Spectrophotometer)
此兩種光譜儀皆採用 Princeton Instruments Acton 2150 機型。薄膜樣品 的製備步驟:將高分子以濃度 1.0 wt%分別溶於 Toluene、Methanol、 Acetonitrile,再以旋轉塗佈法成膜於已清洗之玻璃基板上。溶液樣品的製備: 將高分子以濃度 0.1 mg/20 ml 分別溶於 Toluene、CB、THF、Methanol、
21
N,N-Dimethylformamide (DMF) 、 N,N-Dimethylsulfoxide (DMSO) 及
Acetonitrile 中,並將溶液滴入石英槽中量測。 7. 循環伏安計量法(Cyclic Voltammetry, CV)
係使用 AUTOLAB PGSTAT30 機型。配製 0.1 M Tetrabutylammonium tetrafluoroborate 溶於 Acetonitrile 或 Methanol 之電解液,參考電極為 Ag/AgCl 之玻璃電極,另準備兩片面積相同之 ITO 基板作為工作電極與對應電極。 將待測高分子溶液以滴乾成膜法(Drop Casting)塗佈於 ITO 工作電極上,測 量範圍從 3 V 到-3 V。
8. 橢圓偏光儀(Ellipseometer)
係使用 J. A. Woollam 公司製造的 ɑ-SETM機型。薄膜樣品之製備步驟: 將高分子溶液以旋轉塗佈法成膜於已清洗之 ITO 基板上,置入真空烘箱以 50 oC 烘烤三十分鐘。
9. 氣相層析質譜儀(Gas Chromatography-Mass Spectrophotometer, GC-MS) 係使用 Micromass TRIO-2000 GC-MS。氣相層析質譜儀經由氣相層析 之高分離效果與質譜之高辨別性的機器,氣相層析分離之成分蒸氣進入質 譜儀分析獲得分子結構之資訊。
10. 傅立葉紅外光譜儀(Fourier Transform Infrared Spectrometer, FTIR)
係使用 Perkin Elmer instruments Spectrum One 光譜儀。利用干涉波照射 至樣品後,將干涉光譜作富利葉轉換可得材料之紅外線光譜,使用於有機
22 材料中可用來鑑定化學鍵。將待測高分子使用滴乾成膜法均於分佈在 KBr 壓片上,並使用氮氣緩緩吹乾 KBr 樣品形成薄膜量測。 2-3 元件製作 2-3-1 ITO 基板清洗步驟 1.將 ITO 基板切割為 1.7 cm × 1.7 cm。 2.配製 Detergent 與去離子水(體積比 1:10 )的溶液。 3.以牙刷沾上述溶液清潔 ITO 玻璃基板。 4.將 ITO 基板置入 Detergent 溶液中施以超音波震洗 20 分鐘。 5.取出 ITO 基板置入去離子水以超音波震洗 20 分鐘 6.依此類推,將溶液依序換為丙酮與異丙醇震洗之。 7.取出 ITO 基板置入真空烘箱以 120 oC 烘烤半小時。 8.待其冷卻後施以 UV-ozone 照射半小時。 2-3-2 發光二極體製作流程 1.將 PEDOT(Clevios CH8000)溶液配置(重量比為異丙醇:Detergent:去 離子水:PEDOT=1:1:1:3 )以 6500 rpm/30 sec 之參數設定塗佈於 ITO
基板上,再放入真空烘箱中以 120 o
C 烘烤半小時。
23 再放入真空烘箱中以 50 oC 烘烤半小時。 3.將電子傳輸層材料溶液 (濃度為 0.05 wt%)以 2000 rpm/30 sec 的參數 設定塗佈於發光層上,再放入真空烘箱中以 50 oC 烘烤半小時。 4.以熱蒸鍍方式蒸鍍鋁電極 (腔體壓力在 8×10-6 torr 以下)。 2-4 材料合成 2-4-1 單體合成 各中間物及單體 M1~M5 之詳細合成途徑如 Scheme 1 所示。 2,7-Dibromofluorene (1)
取 Fluorene (20.0 g, 120.48 mmole) 及 N-Bromosuccinimide (42.0 g, 235.96 mmole)置入雙頸瓶中,依序加入 Glacial acetic acid (240 mL)及 48 % Hydrobromic acid (4 mL),於室溫下攪拌 16 小時。反應完成後加入大量純水 使固體析出,減壓抽濾收集米白色粗產物,再使用乙醇進行再結晶純化, 得白色固體 26.5 g,產率 68%。
1
H-NMR (600 MHz, CDCl3, ppm): 3.79 (s, 2H, -CH2), 7.47-7.48 (d, J = 8.4 Hz,
2H, aromatic protons), 7.53-7.54 (d, J = 8.4 Hz, 2H, aromatic protons), 7.62 (s, 2H, aromatic protons). 13C-NMR (600 MHz, CDCl3, ppm): 36.49, 120.89,
24 2,7-Dibromo-9,9-dioctylfluorene (2)
取化合物(1) (2.0 g, 6.17 mmole)、n-Bromooctane (9.0 g, 46.6 mmole)與 Toluene (35 ml)置入雙頸瓶中,再加入 50 wt% 氫氧化鈉水溶液(160 mL)及 Tetrabutylammonium bromide (0.5 g, 1.55 mmole),於 60 oC 氮氣環境下反應 12 小時。反應完成後加入乙酸乙酯及純水萃取,有機層以無水硫酸鎂除水, 並利用旋轉濃縮儀除去溶劑,粗產物利用乙醇進行再結晶純化,得白色固 體 1.4 g,產率 41%。 1 H-NMR (600 MHz, CDCl3, ppm): 0.61-0.62 (t, J = 7.2 Hz, 4H, -(CH2)6CH2CH3), 0.83-0.85 (t, J = 7.1 Hz, 3H, -(CH2)7CH3), 1.06-1.18 (m, 16H, -CH2CH2(CH2)4CH2CH3), 1.20-1.28 (m, 4H, -CH2CH2(CH2)5CH3), 1.91-1.94 (m,
4H, Fluorene-CH2), 7.45-7.46 (d, J = 6.7 Hz, 4H, aromatic protons), 7.51-7.53
(d, J = 8.6 Hz, 2H, aromatic protons). 13C-NMR (600 MHz, CDCl3, ppm): 14.46, 22.57, 23.61, 29.70, 29.84, 31.74, 40.13, 55.67, 121.08, 121.47, 126.17, 130.13, 139.05, 152.54. MASS (EI): m/z 548. Tm = 53 oC. 2,7-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dioctylfluorene (M1) 取化合物(2) (3.3 g, 6 mmole)置入乾燥雙頸瓶中,以針筒抽取無水 THF (50 ml)打入雙頸瓶中,待(2)溶解後置於-78 oC 下持續攪拌 10 分鐘,之後以 針筒抽取 1.6M n-Butyllithium (9.5 mL, 15.2 mmole)緩慢滴入雙頸瓶中,於-78 o C 下持續攪拌 1 小時;再以針筒抽取 2-Isopropoxy-4,4,5,5-tetramethyl-1,3,2- dioxaborolane (8.25ml, 40.4 mmole)滴入上述溶液,之後緩慢回升至室溫並攪
25 拌反應 12 小時。反應完成後使用旋轉濃縮儀除去溶劑,再加入乙酸乙酯與 純水萃取,有機層以無水硫酸鎂除水,濃縮後之粗產物使用正己烷進行再 結晶純化,得白色固體 2.3 g,產率 60%。 1 H-NMR (600 MHz, CDCl3, ppm): 0.55-0.56 (d, J = 6.6 Hz, 4H, -(CH2)6CH2CH3), 0.80-0.82 (t, 6H, J = 7.2 Hz, -(CH2)7CH3), 1.00-1.13 (m, 16H, -CH2CH2(CH2)4CH2CH3), 1.16-1.20 (m, 4H, -CH2CH2(CH2)5CH3),1.39 (s, 24H, -CH3), 1.98-2.01 (m, 4H, -CH2(CH2)6CH3), 7.71-7.72 (d, J = 7.5 Hz, 2H,
aromatic protons), 7.75 (s, 2H, aromatic protons), 7.80-7.81 (d, J = 7.7 Hz, 2H, aromatic protons). 13C-NMR (600 MHz, CDCl3, ppm): 14.05, 22.58, 23.59,
24.93, 29.18, 29.92, 31.77, 40.08, 55.17, 83.70, 119.36, 128.91, 133.64, 143.91, 150.47. MASS (EI): m/z 642. Tm = 128 oC.
9,9-Bis(bromohexyl)-2,7-dibromofluorene (M2)
取化合物(1) (4.0 g, 12.3 mmole)、1,6-Dibromohexane (30 g, 123 mmole)、 Tetrabutylammonium bromide (0.8 g, 2.48 mmole)與 50%氫氧化鈉水溶液(100
mL)置入雙頸瓶中,於 75 oC 氮氣環境下攪拌 30 分鐘。反應完成後加入乙酸 乙酯與純水萃取,濃縮後得到黃色黏稠液體,再利用減壓蒸餾去除未反應 之 1,6-Dibromohexane,粗產物以凝膠管柱層析法純化(沖提液為二氯甲烷: 正己烷=1:20),得白色固體 4.9 g,產率 61%。 1 H-NMR (600 MHz, CDCl3, ppm): 0.52-0.62 (m, 4H, -(CH2)3CH2-(CH2)2Br), 1.06-1.11 (m, 4H, -(CH2)2CH2(CH2)3Br), 1.18-1.23 (m, 4H, -CH2CH2(CH2)4Br), 1.65-1.69 (m, 4H, -(CH2)4CH2CH2Br), 1.91-1.94 (m, 4H, Fluorene-CH2-), 3.28-3.30 (t, J = 6.8 Hz, 4H, -CH2-Br), 7.43-7.44 (d, J = 1.3 Hz, 2H, aromatic
26
protons), 7.46-7.47 (dd, J1 = 8.2 HZ, J2 = 1.7 Hz, 2H, aromatic protons),
7.52-7.53 (d, J = 8.0 Hz, 2H, aromatic protons). 13C-NMR (600 MHz, CDCl3, ppm): 23.44, 27.74, 28.93, 32.59, 33.83, 40.02, 55.54, 121.21, 121.55, 126.07, 130.32, 139.06, 152.15. MASS (EI): m/z 650. Tm = 66 oC. 2,7-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-bis(6-bromohexyl) fluorene (M3) 取化合物(M2) (4.0 g, 6.1 mmole )、Bis(pinacolato)diboron (4.0 g, 15.7
mmole)、Potassium acetate (4.6 g, 46.9 mmole)、[1,1'-Bis(diphenylphosphino) ferrocene]palladium(II) chloride, complex with dichloromethane (0.32 g, 0.39 mmole)及無水 Dioxane (60 mL)置入乾燥雙頸瓶中,進行除氣 3 次後,於氮 氣環境下加熱至 85ºC 攪拌 24 小時。反應結束後使用旋轉濃縮儀去除溶劑, 再加入二氯甲烷與純水萃取,有機層以無水硫酸鎂除水,濃縮後再以凝膠 管柱層析法純化(沖提液為二氯甲烷:正己烷=1:1),得白色固體 2.7 g,產 率 59 %。 1 H-NMR (600 MHz, CDCl3, ppm): 0.52-0.58 (m, 4H, -(CH2)3CH2-(CH2)2Br), 1.01-1.06 (m, 4H, -(CH2)2CH2(CH2)3Br), 1.13-1.18 (m, 4H, -CH2CH2(CH2)4Br), 1.39 (s, 24H, -CH3), 1.61-1.64 (m, 4H, -(CH2)4CH2CH2Br), 2.00-2.02 (m, 4H, Fluorene-CH2-), 3.24-3.26 (t, 4H, J = 6.8 Hz, -CH2-Br), 7.716-7.729 (d, J = 7.7
Hz, 2H, aromatic protons), 7.733 (s, 2H, aromatic protons), 7.807-7.819 (d, J = 7.6 Hz, 2H, aromatic protons). 13C-NMR (600 MHz, CDCl3, ppm): 23.37, 27.72,
28.95, 32.65, 33.93, 39.91, 83.77, 119.46, 128.77, 133.80, 143.90, 150.08. MASS (EI): m/z 744. Tm = 123 oC.
27 2,1,3-Benzoselenadiazole (3)
取 1,2-Phenylenediamine (5.0 g, 46.2 mmole)、Selenium dioxide (5.6 g,
50.4 mmole)於常溫下研磨 30 分鐘,之後加入大量純水,過濾收集淡褐色固 體,再使用乙醇進行再結晶純化,得淡褐色針狀晶體 5.6 g,產率 66 %。
1
H-NMR (600 MHz, CDCl3, ppm): 7.44-7.46 (dd, J1= 7.0 Hz, J2= 3.2 Hz, 2H,
aromatic protons), 7.82-7.83 (dd, J1 = 7.0 Hz, J2 = 3.2 Hz, 2H, aromatic protons). 13 C-NMR (600 MHz, CDCl3, ppm): 123.45, 129.42, 160.53. MASS (EI): m/z 184. Tm = 75 o C. 4,7-Dibromo-2,1,3-benzoselenadiazole (M4)
取化合物(3) (0.5 g, 2.7 mmole)、Silver sulfate (0.86 g, 2.7 mmole)、濃硫 酸(6 mL)置入雙頸瓶中並攪拌均勻,再緩慢加入 Bromine (0.97 g, 6 mmole) 並攪拌 1 小時。反應完成後使用 G4 漏斗進行減壓抽濾,收集濾液並滴入冰 水中,收集固體再使用乙酸乙酯進行再結晶純化,得黃色針狀晶體 0.3 g, 產率 32%。 1 H-NMR (600 MHz, CDCl3, ppm): 7.64 (s, 2H, aromatic protons). 13 C-NMR (600 MHz, CDCl3, ppm): 132.14. MASS (EI): m/z 341. Tm = 282 oC 1,2-Bis(4-bromobenzoyl)hydrazine (4)
取4-Bromobenzoyl chloride (2.0 g, 9.1 mmole)、Hydrazine monohydrate
(0.25 g, 5 mmole)與1-Methyl-2-pyrrolidone (15 mL)置入雙頸瓶中,於室溫下 攪拌5小時。反應結束後加入純水(50 mL),收集固體並以乙酸乙酯清洗,得
28 白色固體1.5 g,產率82%。
1
H-NMR (600 MHz, d-DMSO, ppm): 10.611 (s, 2H, -NHCO-Ph-Br), 7.73~7.74 (d, J = 8.3 Hz, 4H, aromatic protons), 7.84-7.85 (d, J = 8.34 Hz, 4H, aromatic protons).13C-NMR (600 MHz, d-DMSO, ppm): 125.93, 129.75, 131.78, 131.83, 165.20. MASS (EI): m/z 397. Tm = 76 o C. 1,2-Bis[chloro(4-bromophenyl)methynyl]hydrazine (5) 取化合物(4) (2.0 g, 5 mmole)、Phosphorus pentachloride (2.4 g, 11.5 mmole)與Toluene (30 mL)置入雙頸瓶中,於氮氣下加熱至120 oC迴流3小時。 反 應 結 束 後 降 溫 , 於 冰 浴 下 緩 慢 加 入 純 水 去 掉 多 餘 的 Phosphorus pentachloride,濃縮去除溶劑後,粗產物以乙醇進行再結晶純化,得銀白色 固體1.66 g,產率76%。 1 H-NMR (600 MHz, d-DMSO, ppm): 7.80-7.81 (d, J = 8.64 Hz, 4H, aromatic protons), 7.98-8.0 (d, J = 8.58 Hz, 4H, aromatic protons).13C-NMR (600 MHz,
d-DMSO, ppm): 126.76, 130.23, 131.87, 132.36, 143.44. MASS (EI): m/z 435.
Tm = 144 o
C.
3,5-Bis(4-bromophenyl)-4-phenyl-1,2,4-triazole (M5)
取 化 合 物 (5) (1.0 g, 2.3 mmole) 、 Aniline (0.22 g, 2.3 mmole) 與
N,N-Dimethylaniline (20 mL)置入雙頸瓶中,於氮氣下加熱至135 oC迴流12
29
洗,再使用乙醇進行再結晶純化,得白色固體0.71 g,產率68 %。
1
H-NMR (600 MHz, CDCl3, ppm): 7.17-7.19 (d, J = 7.32 Hz, 2H, aromatic
protons), 7.29-7.3 (d, J = 8.1 Hz, 4H, aromatic protons), 7.44-7.45 (d, J = 8.0 Hz, 4H, aromatic protons), 7.47-7.50 (t, J = 7.26 Hz, 2H, aromatic protons), 7.52-7.55 (t, J = 7.38 Hz, 1H, aromatic proton).13C-NMR (600 MHz, CDCl3, ppm): 124.87, 124.99, 127.70, 130.25, 130.31, 130.41, 131.88, 153.88.MASS (EI): m/z 454. Tm = 66 oC. 2-4-2 高分子聚合 高分子P1~P4之合成途徑如Scheme 2所示。詳細合成步驟以P1為例, 說明如下: Poly[9,9-dioctylfluorene-alt- 9,9-bis(6-bromohexyl)fluorene](P1) 將 化 合 物 M1 (0.644 g, 1 mmole) 、 M2 (0.650 g, 1 mmole) 及 Tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) (0.04 g, 0.034 mmole)置入 乾燥雙頸瓶中(瓶壁以鋁箔紙包覆避免光線照射),於氮氣環境下依序打入無 水 Toluene (10 mL)與 2M K2CO3 (aq) (10 mL),再升溫至 90 o C 反應 72 小時。 反應完成後把溶液滴入 Methanol 中,收集之固體以 THF 溶解,再滴入 THF 與 Methanol (體積比為 2:1 )的混合溶液進行再沉澱,重複上述步驟二至三次, 得黃綠色固體 0.6 g,產率 60 %。
30 Poly[9,9-bis(6-bromohexyl)fluorene] (P2):取 M2 (0.325 g, 0.5 mmle )、M3 (0.372 g, 0.5 mmole)及 Pd(PPh3)4 (0.02 g, 0.017 mmole),並參照 P1 之合成步 驟,得黃綠色固體 0.34 g,產率 69 %。 Poly[9,9-bis(6-bromohexyl)fluorene-co-2,1,3-benzoselenadiazole] (P3) : 取 M2 (0.3218 g, 0.495 mmole)、M3 (0.372 g, 0.5 mmole)、M4 (0.0017 g, 0.005 mmole)及 Pd(PPh3)4 (0.02 g, 0.017 mmole),並參照 P1 之合成步驟,得黃綠 色固體 0.35 g,產率 72 %。 Poly[9,9-bis(6-bromohexyl)fluorene-alt-3,4,5-triphenyl-1,2,4-triazole] (P4): 取 M3 (0.372 g, 0.5 mmole)、M5 (0.2275 g, 0.5 mmole)及 Pd(PPh3)4 (0.02 g, 0.017 mmole),並參照 P1 之合成步驟。惟反應結束後所得之黃色固體,無 法溶解於 THF、DMF、Chlorobenzene、Dichloromethane,故無法進行後續 離子交換及化學分析。 2-4-3 高分子離子化 離子性高分子 P1-Br、P2-Br 及 P3-Br 之合成途徑如 Scheme 3 所示。 詳細合成步驟以 P1-Br 為例,說明如下:
31 P1-Br 取 100 mg 之 P1 溶於 THF (10 mL)中,於-78 oC 氮氣環境下緩慢加入 30 % Trimethylamine (aq) (5 mL),再回升至室溫反應 24 小時(如有沉澱物析 出可重複上述步驟一次)。反應完成後濃縮去除溶劑,粗產物以純水溶解後 再滴入丙酮進行再沉澱,得綠色固體 90 mg,產率 79 %。 P2-Br:取 100 mg 之 P2 及 30 % Trimethylamine (aq) (10 mL),並參照 P1-Br 之合成步驟,得綠色固體 96 mg,產率 77 %。 P3-Br:取 100 mg 之 P2 及 30 % Trimethylamine (aq) (10 mL),並參照 P1-Br 之合成步驟,得土黃色固體 92 mg,產率 74 %。 2-4-4 離子置換 離子性高分子 P1-BF4、P1-PF6、P2-BF4、P2-PF6、P3-BF4、P3-PF6之 合成途徑如 Scheme 4 所示。詳細合成步驟以 P1-BF4及 P1-PF6為例,說明 如下: P1-BF4
取 100 mg 之 P1-Br 溶於 Methanol (20 mL),另取 Sodium tetrafluoroborate (NaBF4) (0.264 g, 2.4 mmole)溶於純水(20 mL),將上述水溶液緩慢加入
32 P1-Br/Methanol 溶液中,於室溫下反應 48 小時。反應完成後濃縮去除溶劑, 再重複上述步驟四至五次以達到理想的離子交換率。最後加入去離子水並 攪拌 8 小時,過濾得黃色固體 80 mg,產率 79%。 P1-PF6 取 100 mg 之 P1-Br 溶 於 Methanol (20 mL) , 另 取 Ammonium
hexafluorophosphate (NH4PF6) (0.2 g, 2.4 mmole)溶於 Methanol20 mL,將上
述溶液緩慢加入 P1-Br/Methanol 溶液中,於室溫下反應 48 小時。反應完成 後濃縮去除溶劑,再重複上述步驟四至五次以達到理想的離子交換率。最 後加入去離子水並攪拌 8 小時,過濾得到黃色固體 90 mg,產率 87 %。 P2-BF4:取 100 mg 之 P2-Br 及 NaBF4 (0.528 g, 4.8 mmole),並參照 P1-BF4 之合成步驟,得黃色固體 85 mg,產率 83 %。 P2-PF6:取 100 mg 之 P2-Br 及 NH4PF6 (0.4 g, 4.8 mmole),並參照 P1-PF6 之合成步驟,得黃色固體 90 mg,產率 75 %。 P3-BF4:取 100 mg 之 P3-Br 及 NaBF4 (0.528 g, 4.8 mmole),並參照 P1-BF4 之合成步驟,得黃色固體 80 mg,產率 78 %。
33 P3-PF6:取 100 mg 之 P2-Br 及 NH4PF6 (0.4 g, 4.8 mmole),並參照 P1-PF6 之合成步驟,得黃色固體 79 mg,產率 77 %。 HBr/HOAC Br Br (1) NaOH (aq)/TBABr Toluene (2) O O B O O B O n-Butylithium THF (M1) Br (1) O B O B KOAc/Pd(dppf)Cl2 Br Br KOH (aq)/TBABr Br Br Br O O (M2) Br Br O B O (M3) NH2 H2N SeO2 N Se N N Se N Br Br Br2 H2SO4 /Ag2SO4 (M4) (3) NBS Br Br H17C8 C8H17 B O O H17C8 C8H17 Br Dioxane B O O Br C Cl O NH 2NH2.H2O Br C O H N HN C O Br PCl5 Br C Cl N N C Cl Br N N N Br Br Toluene NMP (4) (5) (M5) NH2 N,N'-Dimethylaniline Scheme 1 單體 M1~M5 之合成途徑
34 (M1) Br Br 2M K2CO3 (aq) Toluene Pd(PPh3)4 C8H17 C8H17 (M2) + (M3) Br Br (M2) + n 2M K2CO3 (aq) Toluene Pd(PPh3)4 n P1 P2 Br Br 1-y y=0.5% or 0.25% y Br Br (M2) + (M3) 2M K2CO3 (aq) Toluene Pd(PPh3)4 (M4) + N SeN n P3 (M3) + (M5) 2M K2CO3 (aq) Toluene Pd(PPh3)4 N N N n P4 Scheme 2 高分子 P1~P4 之合成途徑
35 N+ N+ Br-NMe3 C8H17 C8H17 P1-Br P2 P2-Br THF/H2O y=0.5% or 0.25% P3 P3-Br P1 n NMe3 THF/H2O n N+ Br-N+ Br-NMe3 THF/H2O 1-y y N Se N n N+ Br-N+ Br-Scheme 3 離子性高分子 P1-Br、P2-Br、P3-Br 之合成途徑
36 P1-BF4, X=BF4 P1-PF6, X=PF6 P1-Br P2-Br P3-Br NaBF4 or NH4PF6 NaBF4 or NH4PF6 P2-BF4, X=BF4 P2-PF6, X=PF6 P3-BF4, X=BF4 P3-PF6, X=PF6 NaBF4 or NH4PF6 MeOH (H2O) MeOH (H2O) MeOH (H2O) y=0.5% or 0.25% N+ N+ C8H17 C8H17 n X- X -n N+ N+ X- X -1-y y N SeN n N+ N+ X- X -Scheme 4 離子性高分子 P1-BF4、P1-PF6、P2-BF4、P2-PF6、P3-BF4、P3-PF6 之合成途徑
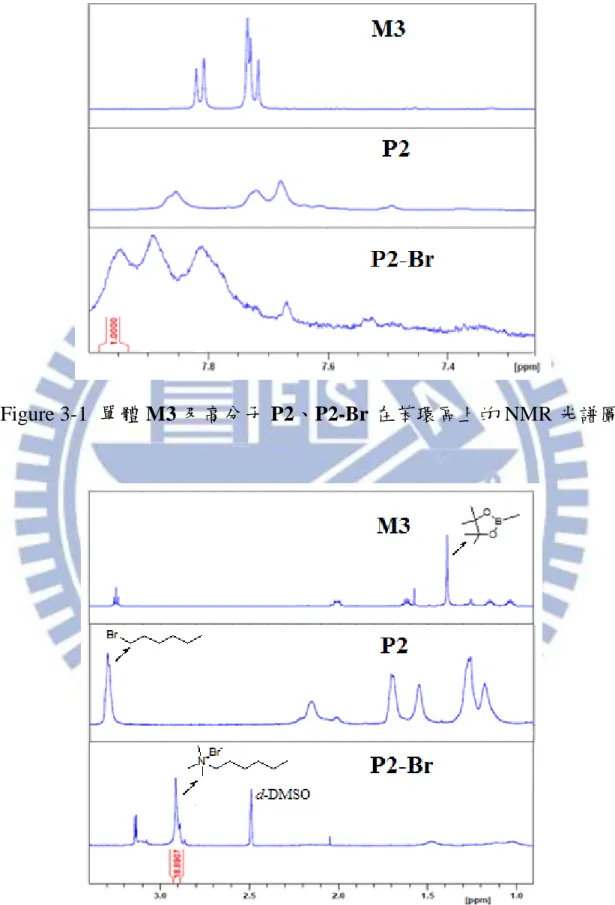
37 第三章 實驗結果與討論 3-1 單體及高分子結構鑑定 3-1-1 NMR 光譜分析 本研究首先利用 NMR 光譜分析材料結構及聚合結果。Figure 3-1 為單 體 M3 及高分子 P2、P2-Br 在苯環區上的 NMR 光譜圖,所有訊號皆為芴環 上之氫訊號,而高分子 P2、P2-Br 之氫訊號相較於單體 M3 顯得更寬,為 高分子氫譜特色。三者在長碳鏈區上的 NMR 光譜圖如 Figure 3-2 所示,單 體 M3 硼酯上之甲基訊號位於 δ 1.39 ppm,而在高分子 P2 中該訊號消失, 其餘氫訊號顯得更寬,可得知硼酯已脫去並聚合成高分子鏈。此外,離子 性高分子 P2-Br 在 δ 2.91 ppm 觀察到明顯的單重峰訊號,此為四級銨鹽團 基上甲基的氫訊號,且其積分值為 18,代表共有 18 個氫原子(芴環 9 號位 上接有兩條長鏈),與預測結構吻合,表示 P2 已成功離子化。其餘 P1-Br 及 P3-Br 之 NMR 光譜亦觀察到相同結果。
38
Figure 3-1 單體 M3 及高分子 P2、P2-Br 在苯環區上的 NMR 光譜圖
39 3-1-2 ESCA 光譜分析 本研究同時藉由 ESCA 光譜分析離子化及離子置換結果。ESCA 之原理 為利用 X-Ray 照射材料,使材料內層電子(可稱為光電子)受激發而脫離,再 收集光電子以得到其束縛能(Binding Energy);由於不同原子之內層電子其 束縛能皆不相同,可藉此得知材料的原子種類,並進一步推測化學結構。 Figure 3-3 為離子化高分子 P3-Br 之 ESCA 光譜,於 400 eV 上有 N 1s 訊號,於 80、180、260 eV 分別為 Br 3d、3p 及 3s 訊號,顯示可得溴與氮 離子已經接在高分子上。Figure 3-4 為離子置換後高分子 P3-BF4 之 ESCA 光譜,N 1s 訊號依然存在,另於 20、700 eV 觀察到 F 2s、1s 訊號,於 190 eV 觀察到 B 1 s 訊號,而 Br 之訊號則消失,表示 BF4團基已成功置換 Br 原子。 Figure 3-5 為離子置換後高分子 P3-PF6之 ESCA 光譜,N 1s 訊號依然存在, 同時於 20、700 eV 觀察到 F 2s、1s 訊號,另於 150、195 eV 觀察到 P 2p、 2s 訊號,而 Br 之訊號則消失,再度顯示離子交換已完成,PF6團基成功接 在高分子上。其餘離子化高分子之 ESCA 光譜亦觀察到相同現象。
40
Figure 3-3 高分子 P3-Br 之 ESCA 光譜
41 Figure 3-5 高分子 P3-PF6之 ESCA 光譜 3-1-3 FTIR 分析 本研究利用 FTIR 光譜分析所合成之高分子結構,以高分子 P1 以及 P3 為例,其 FTIR 光譜如 Figure 3-6 所示,芳香環上 C-H 吸收峰位於 3049 cm-1, 長碳鏈上 C-H 吸收峰則位於 2926、2851 cm-1,芳香環上 C=C 吸收峰位於 1636、1458 cm-1;另外 P3 於 1610 cm-1觀察到 2,1,3-Benzoselenadiazole 之 C=N 吸收峰,於 1130 cm-1觀察到 N-Se 吸收峰,顯示該單體的確引入高分 主鏈中。高分子 P1~P3 之 FTIR 吸收波峰值列表於 Table 3-1 中。
42 4000 3000 2000 1000 0 20 40 60 80 100 T ra n smi tta n ce (% ) Wavenumber (cm-1 ) C-H =C-H C=C (a) 4000 3500 3000 2500 2000 1500 1000 500 0 20 40 60 80 100 T ra n smi tta n ce (% ) Wavenumber (cm-1 ) C=N N-Se C-H C-H =C-H (b)
Figure 3-6 高分子(a) P1 及 (b) P3 之 FTIR 光譜
Table 3-1 高分子 P1~P3 之 FTIR 訊號 Polymer Ar C-H Aliphatic
C-H
Ar C=C C=N -CH3 N-Se
P1 3032 2926, 2851 1636, 1458 N/A 1380 N/A P2 3039 2929, 2852 1638, 1458 N/A N/A N/A P3 3034 2929, 2854 1640,1459 1610 N/A 1133
43 3-2 高分子分子量測定
本研究藉由 GPC 測定高分子 P1~P3 之分子量及分子量分佈。離子化高 分子因僅涉及到離子置換,高分子主鏈不受影響,再加上離子化後高分子 難溶於 THF,無法進行 GPC 量測,在此不做離子性高分子之報導。GPC 結 果顯示,高分子 P1~P3 之數量平均分子量(Number-average Molecular Weight,
Mn)分別為 3.41×104、8.0×103、7.1×103 g/mol,而重量平均分子量(Weight -average Molecular Weight, Mw)分別為 1.06×105、2.99×104、2.8×104 g/mol, 分子量分佈 (Polydispersity Index, PDI)分別為 3.1、3.7、3.9。高分子 P1 之 分子量相較於 P2 為高,乃因 9 號取代長碳鏈尾端帶有溴原子時,溶解度會 變差,造成分子量降低;吾人在合成過程中,亦觀察到含溴原子單體 M2 之溶解度相較於一般碳鏈中間物(2)為低,此特性即影響了聚合反應。高分 子 P3 聚合使用不具可溶性側鏈之 2,1,3-Benzoselenadiazole 單體 M4,其分 子量相較於 P1 為低。高分子 P4 的聚合引入 Triazole 單體 M5,反應結束收 集的產物亦無法溶於 THF 或其它溶劑,無法進行後續探討,文獻上亦報導 過類似結構無法得到可溶性高分子產物[61]。所有高分子之 GPC 數據整理於 Table 3-2 中。
44
Table 3-2 高分子 P1~P3 之分子量及分子量分佈
Polymer Mn×103 (g/mol) Mw×104 (g/mol) PDI
P1 34.1 10.60 3.1
P2 8.0 2.99 3.7
P3 7.1 2.80 3.9
P4 N/A N/A N/A
3-3 高分子熱性質分析 本研究利用 TGA 及 DSC 測定高分子之熱性質。在 TGA 量測方面,定 義 Td為重量損失 5 %之溫度。Figure 3-7 為高分子 P1 及其離子性高分子之 TGA 曲線圖,P1、P1-Br、P1-BF4及 P1-PF6之 Td分別為 334、209、274、 224 oC,離子性高分子其 Td相較於原本高分子下降 60~135 o C,顯示這類材 料在加熱過程中會提早裂解,熱穩定性下降,此現象與文獻記載結果一致 [43,62]。究其原因,2010 年 Nguyen 等人提出解釋[54],離子性高分子加熱超過 180 oC 後會產生 Hofmann elimination,四級銨鹽團基會先行斷裂,產生三甲 胺與氫氟酸等小分子,而長碳鏈末端則形成雙鍵,其反應機制如 Figure 3-8 所示。比較三種離子團基熱穩定性,C-Br 最先斷裂,依序則為 C-PF6與 C-BF4, 推測後兩者較能抑制 Hofmann elimination 的產生,故將陰離子置換為 BF4 與 PF6,對於熱穩定性的提升很有幫助。P2 與 P3 系列高分子亦呈現相同熱 穩定性傾向。至於來源高分子 P1~P3 之熱穩定性比較,以 P1 的 Td最高,
45 因其分子量最大。所有高分子材料之 Td整理於 Table 3-3 中。 100 200 300 400 500 600 0 20 40 60 80 100 W e ig h t R e si d u e (% ) Temperature (oC) P1 P1-Br P1-BF4 P1-PF6 Figure 3-7 高分子 P1 系列之 TGA 曲線圖
Figure 3-8 Hofmann elimination 反應機制示意圖
Table 3-3 高分子 P1~P3 系列 Td值
Polymer Series Td (oC)
P1, P1-Br, P1-BF4, P1-PF6 334, 209, 274, 224 P2, P2-Br, P2-BF4, P2-PF6 297, 239, 303, 292 P3, P3-Br, P3-BF4, P3-PF6 298, 211, 289, 274
46 在 DSC 量測方面,Figure 3-9 為高分子 P1 的 DSC 曲線圖,於 70 oC 觀 察到明顯 Tg,另於 116 oC 發現一個放熱峰,表示 P1 於加熱過程中產生再 結晶之相行為。高分子 P2 及 P3 的 Tg經測定分別為 92 及 82oC,其中 P2 為同聚物(Homopolymer),主鏈結構規則易產生堆疊,而 P3 僅引入極少量 平面性 2,1,3-Benzoselenadiazole 單體,主鏈大致仍呈現規則結構,兩者的 Tg均較 P1 為高。此外, P2 與 P3 並未發現其它吸、放熱峰,是為非晶態 高分子。高分子 P1~P3 的 Tg及 Tc列表在 Table 3-4。 40 60 80 100 120 140 -800 -600 -400 -200 0 200 400 600 116oC H e a t F lo w (u W ) Temperature (o C) 70oC Figure 3-9 高分子 P1 之 DSC 曲線圖 Table 3-4 高分子 P1~P3 之 Tg及 Tc值 Polymer Tg (oC) Tc (oC) P1 70 116 P2 92 N/A P3 82 N/A
47 3-4 高分子光學性質分析
3-4-1 吸收光譜分析
由於離子團基掛於 9 號取代長鏈末端且不影響主鏈之吸收,預期各類 離子高分子之光學性質差異不大。以 P2 系列高分子為例,其溶液及薄膜態 之吸收光譜整理於 Figure 3-10 (a)~(d)中。P2 在三種溶劑 Toluene、CB、THF 及薄膜態下差異不大,如 Figure 3-10 (a)所示,最大吸收峰位於 383~388 nm, 屬於主鏈之 π-π* transition,僅薄膜態之吸收範圍增加約 12 nm。P2-Br 在三 種溶劑 Methanol、DMF、DMSO 及薄膜態下之最大吸收峰位於 388~402 nm, 如 Figure 3-10 (b)所示,其在 Methanol 中有明顯的藍位移(約 10 nm),推測 原因為 Methanol 分子介入 P2-Br 高分子鏈之間,造成高分子彼此間隔較遠, 不致使堆疊產生,吾人在實驗過程中亦發現 Methanol 對其溶解力最高;另 外值得注意的是,P2-Br 薄膜態吸收曲線出現強度接近的雙峰,分別位於 396 與 411 nm,此現象與文獻記載有相似結果[49]。P2-BF4之最大吸收峰位 於 389~396 nm,P2-PF6之最大吸收峰位於 392~402 nm,如 Figure 3-10 (c)、 (d)所示,兩者在 CH3CN 中有明顯的藍位移(約 7~10 nm),此現象與 P2-Br 溶於 Methanol 的情況相似;而薄膜態吸收曲線並未出現雙峰。至於 P1 及 P3 系列高分子之吸收特性更為接近,在此僅將最大吸收峰值整理於 Tables 3-5 ~ 3-8 中,不詳列所有吸收光譜。Figure 3-11 為高分子 P1~P3 於薄膜態
48 之吸收光譜比較圖,三者的最大吸收峰(384~385 nm)及波形差別並不明顯, 乃因高分子主鏈以聚芴結構為主,即使 P3 引入少量2,1,3-Benzoselenadiazole 結構,也不影響吸收光譜。 300 350 400 450 500 0.0 0.2 0.4 0.6 0.8 1.0 (a) Ab so rb an ce (a .u .) Wavelength (nm) CB THF Toluene Thin Film 300 350 400 450 500 0.0 0.2 0.4 0.6 0.8 1.0 Ab so rb a n ce (a .u .) Wavelength (nm) MeOH DMF DMSO Thin Film (b) 300 350 400 450 500 0.0 0.2 0.4 0.6 0.8 1.0 Ab so rb a n ce (a .u .) Wavelength (nm) CH3CN DMF DMSO Thin Film (c) 300 350 400 450 500 550 0.0 0.2 0.4 0.6 0.8 1.0 Ab so rb a n ce (a .u .) Wavelength (nm) CH3CN DMF DMSO Thin film (d) Figure 3-10 高分子 P2 系列溶液及薄膜態之吸收光譜(a) P2,(b) P2-Br,(c) P2-BF4,(d) P2-PF6
49 300 350 400 450 500 0.0 0.2 0.4 0.6 0.8 1.0 Ab so rb a n ce (a .u .) Wavelength (nm) P1 P2 P3 Figure 3-11 高分子 P1~P3 於薄膜態之吸收光譜 3-4-2 螢光放射光譜分析 高分子 P2 及其離子性衍生物於溶液及薄膜態之螢光放射光譜整理於 Figure 3-12 (a)~(d)中。P2 在溶液態並無太大差別,如 Figure 3-12 (a)所示, 最大螢光放射波長位於 419~422 nm,薄膜態時紅位移至 430 nm,均為主鏈 的 S10→S00的放射峰;另於薄膜態時發現肩峰位於 457 nm,此為主鏈 S10→ S01躍遷[63]。離子高分子 P2-Br 溶液態最大放射峰位於 423~429 nm,如 Figure 3-12 (b)所示,最大螢光放射波長位於 428~437 nm,其在 Methanol 溶液中 最為藍位移,而薄膜態時最為紅位移,與吸收光譜的傾向一致。P2-BF4 之 最大放射峰位於 428~435 nm,P2-PF6之最大吸收峰位於 420~428 nm,如 Figure 3-12 (c)、(d)所示,兩者在 CH3CN 中有明顯的藍位移(約 7~8 nm),此 現象與 P2-Br 溶於 Methanol 的情況相似;所有材料最大放射峰值整理於
50 Tables 3-5 ~ 3-8 中,不詳列所有螢光放射光譜。仔細比較表中數據,可看出 離子化高分子(帶有團基 Br、BF4者)的放光波長,相較於未離子化前有略微 的紅位移。 400 450 500 550 0.0 0.2 0.4 0.6 0.8 1.0 N o rma lize d PL In te n si ty (a .u .) Wavelength (nm) CB THF Toluene Thin Film (a) 400 450 500 550 0.0 0.2 0.4 0.6 0.8 1.0 N orma lize d PL in te nsi ty (a .u .) Wavelength (nm) MeOH DMF DMSO Thin Film (b) 400 450 500 550 0.0 0.2 0.4 0.6 0.8 1.0 N o rma lize d PL In te n si ty (a .u .) Wavelength (nm) CH3CN DMF DMSO Thin Film (c) 400 450 500 550 0.0 0.2 0.4 0.6 0.8 1.0 N orma lize d PL In te nsi ty (a .u .) Wavelength (nm) CH3CN DMF DMSO Thin Film (d) Figure 3-12 高分子 P2 系列溶液及薄膜態之螢光放射光譜(a) P2,(b) P2-Br, (c) P2-BF4,(d) P2-PF6 高分子 P3 及其離子性衍生物於溶液及薄膜態之螢光放射光譜整理於 Figure 3-13 (a)~(d)中。四者在溶液態時僅放出藍光,其最大放射波長介於 419~429 nm;而在薄膜態時可同時發出藍、黃橘光,形成單一發白光高分 子,其最大放射波長分別位於 430、560 nm 左右。仔細觀察圖譜可發現,
51 其在 Methanol 或 CH3CN 中最為藍位移,薄膜態時最為紅位移,且離子化 高分子(帶有團基 Br、BF4者)的放光波長略微的紅位移,均與前段敘述類似。 另外發現到當 P3 形成離子高分子後,其黃橘光的放射會被抑制,如 Figure 3-14 所示,推測原因如下:2,1,3-Benzoselenadiazole 為一傳輸電子材料,其 光色為黃橘光,亦是高分子 P3 能發出白光的主要原因,而陰離子團基 Br、 BF4、PF6亦具有拉電子特性,將之引入後會與 2,1,3-Benzoselenadiazole 競 爭,減少載子在該發光團的結合比率,因此抑制其放光強度。 400 450 500 550 600 650 700 0.0 0.2 0.4 0.6 0.8 1.0 N o rma lize d PL In te n si ty (a .u .) Wavelength (nm) CB THF Toluene Thin Film (a) 400 450 500 550 600 650 700 0.0 0.2 0.4 0.6 0.8 1.0 N o rma lize d PL In te n si ty (a .u .) Wavelength (nm) MeOH DMF DMSO Thin Film (b) 400 450 500 550 600 650 700 0.0 0.2 0.4 0.6 0.8 1.0 N o rma lize d PL In te n si ty (a .u .) Wavelength (nm) CH3CN DMF DMSO Thin Film (c) 400 450 500 550 600 650 0.0 0.2 0.4 0.6 0.8 1.0 N o rma lize d PL In te n si ty (a .u .) Wavelength (nm) CH3CN DMF DMSO Thin Film (d) Figure 3-13 高分子 P3 系列溶液及薄膜態之螢光放射光譜(a) P3,(b) P3-Br, (c) P3-BF4,(d) P3-PF6
52 400 450 500 550 600 650 0.0 0.2 0.4 0.6 0.8 1.0 N orma lize d PL In te nsi ty (a .u .) Wavelength (nm) P3 P3-Br P3-BF4 P3-PF6 Figure 3-14 高分子 P3 系列於薄膜態之螢光放射光譜 Table 3-5 高分子 P1~P3 之吸收與螢光放射最大峰值 Polymer UV-vis λmax (nm) PL λmax (nm)
Film CB Toluene THF Film CB Toluene THF P1 384 390 387 392 429 423 419 422 P2 388 385 383 386 430 422 419 422 P3 385 383 383 383 431 422 419 420
Table 3-6 離子高分子 P1-Br~P3-Br 之吸收與螢光放射最大峰值 Polymer UV-vis λmax (nm) PL λmax (nm)
Film MeOH DMF DMSO Film MeOH DMF DMSO P1-Br 389 390 400 403 434 423 428 429 P2-Br 396 388 398 402 437 423 428 430 P3-Br 400 384 392 395 438 420 426 429
53
Table 3-7 離子高分子 P1-BF4~P3-BF4吸收與螢光放射最大峰值 Polymer UV-vis λmax (nm) PL λmax (nm)
Film CH3CN DMF DMSO Film CH3CN DMF DMSO
P1-BF4 388 392 402 401 432 427 431 436
P2-BF4 393 389 395 396 435 428 433 434
P3-BF4 388 385 392 395 432 420 425 428
Table 3-8 離子高分子 P1-PF6~P3-PF6吸收與螢光放射最大峰值 Polymer UV-vis λmax (nm) PL λmax (nm)
Film CH3CN DMF DMSO Film CH3CN DMF DMSO
P1-PF6 391 395 401 403 430 419 426 429
P2-PF6 393 392 399 402 428 420 426 428
P3-PF6 391 385 392 396 430 419 426 426
3-4-3 螢光量子效率(PL Quantum Yield, PLQY)
所有高分子激發波長設定為 360 nm,不採取最大吸收波長為激發波長, 乃為了避免與放光範圍重疊。利用 PL 光譜儀搭配積分球,分別量測基板與 樣品(以 P1 為例)之光譜,如 Figure 3-15 所示。PLQY 計算方式如下:: PLQY= (A3 / A1-A2) 100 % 其中 A1為激發光於 320~400 nm 的積分面積,A2為激發光於 320~400 nm 被 樣品吸收後的積分面積,A3為高分子 P1、P2 系列於 400~600 nm 的放光面 積,或是高分子 P3 系列於 400~660 nm 的放光面積。所有高分子於薄膜下 之 PLQY 整理於 Table 3-9 中,其數值在 7~16%之間。
54 300 350 400 450 500 550 600 0.0 0.2 0.4 0.6 0.8 1.0 A3 A2 PL In te n si ty (a .u .) Wavelength (nm) Excitation Light P1 Emission A1 Figure 3-15 高分子 P1 薄膜在積分球中光譜 Table 3-9 高分子於薄膜下的螢光量子效率
Polymer PLQY in film (%) PLQY in solvent (%)
P1, P1-Br, P1-BF4, P1-PF6 9, 8, 7, 12 64, 67, 62, 61 P2, P2-Br, P2-BF4, P2-PF6 14, 8, 14, 13 58, 82, 65, 74 P3 ,P3-Br, P3-BF4, P3-PF6 13, 10, 13, 16 58, 80, 72, 85 3-5 高分子電化學性質分析 使用 CV 往正偏壓掃描可得氧化曲線,往負偏壓掃描可得還原曲線,但 還原電位通常並不準確,所以只取氧化起始電位(Eox)所得之 HOMO,搭配 高分子薄膜態吸收光譜推算出之 Bandgap,兩者相加求得 LUMO。其計算 方式如下: