Interface Engineering to Enhance the E
fficiency of Conventional
Polymer Solar Cells by Alcohol-/Water-Soluble C
60
Materials Doped
with Alkali Carbonates
Yu-Ying Lai, Ping-I Shih, Yi-Peng Li, Che-En Tsai, Jhong-Sian Wu, Yen-Ju Cheng,* and Chain-Shu Hsu*
Department of Applied Chemistry, National Chiao Tung University, 1001 Ta Hsueh Road Hsin-Chu, 30010, Taiwan
*
S Supporting InformationABSTRACT:
Two new C
60-based n-type materials, EGMC
−
OH
and EGMC
−COOH, functionalized with hydrophilic
triethylene glycol groups (TEGs), have been synthesized and
employed in conventional polymer solar cells. With the
assistance of the TEG-based surfactant, EGMC
−OH and
EGMC
−COOH can be dissolved in highly polar solvents to
implement the polar/nonpolar orthogonal solvent strategy,
forming an electron modi
fication layer (EML) without eroding the underlying active layer. Multilayer conventional solar cells on
the basis of ITO/PEDOT:PSS/P3HT:PC
61BM/EML/Ca/Al con
figuration with the insertion of the EGMC−OH and EGMC−
COOH
EML between the active layer and the electrode have thus been successfully realized by cost-e
ffective solution processing
techniques. Moreover, the electron conductivity of the EML can be improved by incorporating alkali carbonates into the
EGMC
−COOH EML. Compared to the pristine device with a PCE of 3.61%, the devices modified by the Li
2CO
3-doped
EGMC
−COOH EML achieved a highest PCE of 4.29%. Furthermore, we demonstrated that the formation of the EGMC−
COOH
EML can be utilized as a general approach in the fabrication of highly e
fficient multilayer conventional devices. With the
incorporation of the EGMC
−COOH doped with 40 wt % Li
2CO
3, the PCDCTBT
−C8:PC
71BM-based device exhibited a
superior PCE of 4.51%, which outperformed the corresponding nonmodi
fied device with a PCE of 3.63%.
KEYWORDS:
polymer solar cells, interface engineering, hydrophilic fullerene materials, electron-selective layer, dopant,
alkali carbonates
■
INTRODUCTION
Bulk heterojunction (BHJ) solar cells based on polymer/
fullerene blends have attracted enormous attention in the past
decades due to their potential for fabrication onto large areas of
lightweight and
flexible substrates by low-cost solution
processing.
1A conventional BHJ polymer solar cell (PSC)
with an active layer sandwiched by a low-work-function
aluminum cathode and a hole-conducting
poly(3,4-ethylenedioxylenethiophene):poly(styrenesulfonic acid)
(PE-DOT:PSS) layer on top of an indium tin oxide (ITO)
substrate is the most widely used and researched device
con
figuration.
2By reversing the polarity of charge collection in
the conventional cell, stable Ag can substitute for
air-sensitive Al as the anodic electrode to construct a PSC with an
inverted con
figuration.
3In addition to development of p-type/
n-type photoactive materials and control of nanomorphology,
4interface engineering of multilayer conventional and inverted
solar cells plays a critical role in achieving high e
fficiency and
stability.
5One of the directions of interfacial engineering is
focused on the incorporation of an electron-selective layer, such
as ZnO,
6TiO
x,
7LiF,
8Cs
2CO
3,
9poly(ethylene oxide),
10and
conjugated polyelectrolytes
11between the active layer and the
metal cathode for modulating the energy level alignment at the
electrode/active layer interface, inducing electron-extracting/
hole-blocking abilities, and facilitating electron transportation.
Fullerene-based materials, in particular, emerge as the most
superior n-type components to construct an electron-selective
modi
fication layer (EML) because their physical and electrical
properties can be tailored and manipulated by attaching various
addends on the core of fullerene.
12For example, the
fluorinated
fullerene with the lower surface energy tends to self-assemble
on the surface of the active layer during spin-coating to enhance
the device performance.
12a,bMore importantly, the functional
groups incorporated into C
60molecules can be speci
fically
designed for ful
filling the processing requirements. For PSCs,
multilayer structures are fabricated by layer-by-layer
cost-effective spin-coating. One of the encountered challenges for
solution processing techniques is the erosion of the bottom
layer caused by organic solvents used in the subsequent
step.
12g,h,13For inverted PSCs, a solution of active layer needs
to be deposited on top of a fullerene-based EML. Therefore,
the development of chemically self-assembled
12c,dand/or
cross-linked fullerene materials
12e−hto generate a robust and
solvent-resistant EML has overcome the processing difficulties and thus
successfully improved the device characteristics. With opposite
sequence in a conventional device, however, a fullerene-based
Received: March 18, 2013 Accepted: May 20, 2013 Published: May 20, 2013
EML should be formed by spin-coating on top of the active
layer. Considering that hydrophobic active layers such as
P3HT/PC
61BM are soluble in nonpolar organic solvents but
almost insoluble in highly polar solvents, utilization of
orthogonal solvents (i.e., nonpolar/polar) to construct multiple
organic layers is a straightforward and practical strategy.
14In
this research, we have designed and synthesized two fullerene
materials, EGMC
−OH and EGMC−COOH (Scheme 1),
containing hydrophilic triethylene glycol side chains. The two
hydroxyl groups in EGMC−OH are further end-capped by two
phthalic acids to yield EGMC
−COOH. With the aid of
triethylene glycol group (TEG)-based neutral surfactants, these
C
60materials with enhanced hydrophilic nature can be
dissolved in the polar 2-ethoxyethanol/H
2O solvent which
was spin-cast to form an EML without destroying the
underneath active layer. Therefore, we can successfully
intercalate an EML between the active layer and the Ca/Al
electrode in a conventional device. It has been demonstrated
that the electron-transporting properties of organic
semi-conductors can be dramatically enhanced by adding alkali
carbonates as n-dopants.
15Alkali carbonates are ionic and
highly polar compounds that can only be soluble in water. An
organic active layer, such as P3HT/PC
61BM, is not compatible
with the ionic or highly polar compounds. Therefore, the
attempt of spin-coating pure alkali carbonates in aqueous
solution on top of the organic active layer is not successful.
Thermal vacuum coevaporation is the typical way to generate a
thin
film containing an alkali carbonate guest in a host
material.
15a,b,d,g,hEncouragingly, by the combination of water-/
alcohol-soluble EGMC
−OH and EGMC−COOH as the host
materials with water-soluble alkali carbonates as the dopants, an
n-doped C
60-based EML can be easily generated by
cost-e
ffective spin-coating processing. The P3HT:PC
61BM-based
and
poly(carbazole-dicyclopentathiophene-alt-benzothiadia-zole) (PCDCTBT
−C8, Scheme 1):PC
71BM-based devices
integrating alkali carbonate-doped EGMC
−COOH as the
EMLs exhibited 19% and 24% improvement in e
fficiency,
respectively, compared to their corresponding nonmodi
fied
cells.
■
EXPERIMENTAL SECTION
All chemicals were obtained from commercial sources and used as received unless otherwise specified. Anhydrous tetrahydrofuran, toluene, and dichloromethane were obtained from an SD-300 solvent purification system by AsiaWong Enterprise. NMR measurements are reported for Varian Unity-300 and UI-500 spectrometers (1H, 300
MHz;13C, 75 MHz). Chemical shifts (δ values) are reported in parts
per million with respect to Me4Si (δ = 0 ppm) for13C and1H NMR.
Coupling constants (J) are given in Hertz. 13C NMR was proton
broad-band-decoupled. Multiplicities of peaks are denoted by the following abbreviations: s, singlet; d, doublet; t, triplet; m, multiplet; br, broad. Thermogravimetric analysis (TGA) was recorded on a PerkinElmer Pyris analyzer under nitrogen atmosphere at a heating rate of 10°C min−1. Absorption spectra were collected on a HP8453 UV−vis spectrophotometer. Electrochemical cyclic voltammetry (CV) was conducted on a CH Instrument model 611D analyzer. Pt coated with a thin fullerenefilm was used as the working electrode, Ag/Ag+
electrode as the reference electrode, and Pt as the counter electrode, while 0.1 M tetrabutylammonium hexafluorophosphate in o-dichlor-obenzene was the electrolyte. CV curves were calibrated using ferrocence as the standard, whose oxidation potential is set at−4.8 eV with respect to zero vacuum level. The HOMO energy levels were obtained from the equation HOMO = −(Eoxonset − E(ferrocene)onset +
4.8) eV. The LUMO levels were obtained from the equation LUMO = −(Eredonset− E(ferrocene)onset+ 4.8) eV
Synthesis of Tri(ethylene glycol) tert-Butyldimethylsilyl Ether (1). To an anhydrous CH2Cl2(100 mL) solution of triethylene
glycol (49.56 g, 0.33 mol), 4-dimethylaminopyridine (1.613 g, 13 mmol), and triethylamine (6.68 g, 66 mmol) was added dropwise an anhydrous CH2Cl2(25 mL) solution of tert-butyldimethylsilyl chloride
(10 g, 66 mmol) via an addition funnel under nitrogen atmosphere. The mixture was stirred at room temperature for 3 h and extracted with CH2Cl2/NH4Cl(aq)three times. The collected organic layer was
dried over MgSO4,filtered, and evaporated in vacuo. The resultant
residue was then submitted to column chromatography on silica gel (ethyl acetate:hexane = 1:3, v/v) to give a transparent liquid (13.45 g, 77%):1H NMR (CDCl
3, 300 MHz)δ 0.04 (s, 6H, Si(CH3)2), 0.86 (s,
9H, C(CH3)3), 2.60 (br, 1H, OH), 3.53−3.77 (m, 12H, OCH2);13C
NMR (CDCl3, 75 MHz) δ −5.3 (2C, SiCH3), 18.4 (1C, (Si−
C(Me)3), 25.9 (3C, (Si−C(CH3)3), 61.8 (1C, OCH2), 62.7 (1C,
OCH2), 70.4 (1C, OCH2), 70.7 (1C, OCH2), 72.5 (1C, OCH2), 72.7
(2C, OCH2).
Synthesis of Bis(TBDMS-triethylene glycol)-malonate (2). To an anhydrous CH2Cl2 (50 mL) solution of malonic acid (1 g, 9.60
mmol), 4-dimethylaminopyridine (0.234 g, 1.914 mmol), and 1 (5.69 g, 21.5 mmol) was added 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (4.128 g, 26.60 mmol). The mixture was stirred at room temperature under nitrogen atmosphere for 15 h, evaporated to remove CH2Cl2, and extracted with ethyl acetate/H2O three times.
The collected organic layer was dried over MgSO4, filtered, and
evaporated. The resultant residue was then purified by column chromatography on silica gel (ethyl acetae:hexane = 1:6, v/v) to furnish a transparent liquid (3.5 g, 62%): 1H NMR (CDCl
3, 300
MHz)δ 0.03 (s, 12H, Si(CH3)2), 0.86 (s, 18H, Si−C(CH3)3), 3.42 (s,
2H, (OC)CH2(CO)), 3.53 (t, J = 5.19 Hz, 4H, OCH2), 3.62−
3.63 (m, 8H, OCH2), 3.69 (t, J = 4.8 Hz, 4H, OCH2), 3.74 (t, J = 5.5
Hz, 4H, OCH2), 4.27 (t, J = 4.2 Hz, 4H, ((OC)OCH2);13C NMR
(CDCl3, 75 MHz)δ −5.3 (4C, Si−(CH3)2), 18.3 (2C, (Si−C(Me)3),
25.9 (6C, (Si−C(CH3)3), 41.2 (1C, (OC)CH2(CO)), 62.7 (2C,
OCH2), 64.6 (2C, OCH2), 68.8 (2C, OCH2), 70.6 (2C, OCH2), 70.7
(2C, OCH2), 72.7 (2C, OCH2), 166.5 (2C, CO).
Synthesis of Bis(TBDMS-triethylene glycol)-malonate Full-erene (3). To an anhydrous toluene (270 mL) solution of C60(0.3 g,
0.416 mmol) was added sequentially 2 (0.25 g, 0.419 mmol), I2(0.102
g, 0.402 mmol), and 1,8-diazabicyclo[5.4.0]undec-7-ene (0.159 g, 1.04 mmol). The reaction mixture was stirred for 20 h and then quenched with sodium thiosulfate aqueous solution. The organic layer was collected, evaporated, and purified by column chromatography on silica gel (toluene:methanol)ato give a crude product, which was then
reprecipitated from toluene/methanol to afford a brown solid (0.24 g, 44%):1H NMR (CDCl
3, 300 MHz,)δ 0.06 (s, 12H, Si(CH3)2), 0.89
(s, 18H, Si−C(CH3)3), 3.55 (t, J = 5.4 Hz, 4H, OCH2), 3.63−3.69 (m,
8H), 3.76 (t, J = 5.4 Hz, 4H, OCH2), 3.88 (t, J = 4.5 Hz, 4H, OCH2),
4.64 (t, J = 5.1 Hz, 4H, (OC)OCH2);13C NMR (CDCl3, 75 MHz)
δ −5.2 (4C, Si(CH3)2), 18.4 (2C, Si−C(Me)3), 25.8 (6C, Si−
C(CH3)3), 62.67, 66.2, 68.8, 70.7, 71.4, 72.7, 139.1, 140.9, 141.8,
142.2, 142.9, 143.0, 143.1, 143.9, 144.6, 144.7, 144.9, 145.2, 145.3, 163.5 (2C, CO). aInitially, only toluene was used as the eluent to remove pristine C60. Afterward, the eluent was changed to
toluene:methanol = 98:2, v/v for collecting crude product.
Synthesis of Bis(triethylene glycol) Malonate C60 (4). To a
THF (10 mL) solution of 3 (0.1 g, 0.076 mmol) was slowly added HCl (2 N, 1 mL). The reaction mixture was stirred vigorously at room temperature under nitrogen atmosphere for 30 min. An amount of 50 mL of CH2Cl2 was added, and the mixture was then washed by
saturated NaHCO3(aq)and H2O several times. The organic layer was
collected, dried over MgSO4, evaporated, and purified by column
chromatography on silica gel (CH2Cl2:methanol = 30:1, v/v). The
collected residue was then reprecipitated from CH2Cl2/hexane to
furnish a brown solid (60 mg, 72%):1H NMR (CDCl
3, 300 MHz)δ
3.02 (s, 2H, OH), 3.60−3.63 (m, 4H, OCH2), 3.67−3.70 (m, 4H,
OCH2), 3.72−3.75 (m, 8H, OCH2), 3.91 (t, J = 4.8 Hz, 4H, OCH2),
4.67 (t, J = 4.8 Hz, 4H, OC−OCH2);13C NMR(CDCl3, 75 MHz)δ
61.7, 66.1, 68.7, 70.4, 70.7, 72.7, 139.1, 140.9, 141.8, 142.2, 142.9, 143.0, 143.1, 143.9, 144.6, 144.7, 144.9, 145.1, 145.2, 145.3, 163.5 (2C, CO); MS (FAB, C75H26O10+•) calcd, 1086.1526; found, 1086.1520
Synthesis of Bis(triethylene glycol phthalic acid)-malonate C60(5). A mixture of 4 (0.05 g, 0.046 mmol), phthalic anhydride
(0.136 g, 0.92 mmol), and benzene (30 mL) was heated to the refluxing temperature, and 2.5 mL of pyridine was then added. The refluxing was continued for 20 h. The reaction mixture was evaporated and purified by column chromatography on silica gel (CH2Cl2:methanol = 30:1, v/v). The collected residue was then
reprecipitated from CH2Cl2/methanol to furnish a brown solid (43
mg, 67%):1H NMR (CDCl 3, 300 MHz)δ 3.68−3.70 (m, 4H, OCH2), 3.76 (t, J = 4.2 Hz, 4H, OCH2), 3.82−3.83 (m, 4H, OCH2), 4.02 (t, J = 3.9 Hz, 4H, OCH2), 4.44−4.46 (m, 4H, (OC)OCH2), 4.72 (t, J = 4.5 Hz, 4H, (OC)OCH2), 7.52−7.55 (m, 4H), 7.74−7.76 (m, 4H); 13C NMR (CDCl 3, 75 MHz)δ 64.7 (2C, OCH2), 65.7 (2C, OCH2),
68.8 (2C, OCH2), 68.9 (2C, OCH2), 69.7 (2C, OCH2), 70.9 (2C,
OCH2), 71.3, 128.9, 129.3, 130.9, 131.3, 131.5, 132.8, 138.9, 140.9,
141.8, 142.2, 142.9, 143.0, 143.8, 144.5, 144.6, 144.9, 145.1, 145.2, 145.2, 163.4 (2C, CO), 167.6 (2C, CO), 169.8 (2C, CO). MS (HR-FAB, C91H34O16+•) calcd, 1382.1847; found, 1382.1859.
Device Fabrication. An indium tin oxide (ITO)-coated glass substrate was ultrasonically washed by detergent, deionic water, acetone, and isopropanol sequentially for 15 min each and then cleaned by UV-ozone for another 15 min. PEDOT:PSS (Baytron PVP AI-4083) wasfiltered and spin-coated on a cleaned ITO-coated glass to produce a ca. 35 nm thick interlayer, which was then baked at 150 °C for 1 h. For active-layer fabrication, P3HT was mixed with PC61BM
in 1:1 weight ratio, and PCDCTBT-C8 was mixed with PC71BM in
1:3 weight ratio, respectively, in o-dichlorobenzene. The weight percent of P3HT:PC61BM in o-dichlorobenzene was 2 wt %, and that
of PCDTCBT:PC71BM was 1 wt %. The individual solutions were
heated and spin-coated on top of the PEDOT:PSS interlayer at 600 rpm. The resultantfilm was covered in a Petri dish to allow the solvent to slowly evaporate (solvent annealing) until thefilm was dried and then baked at 140°C for 10 min (thermal annealing). EGMC−OH or EGMC−COOH (2 mg) was then dissolved in mixed solvents (200 mg) and doped with an inorganic salt in some of the tested devices. The fullerene mixture was sonicated for 20 min,filtrated via a PVDF filter (45 μm), and spin-coated on top of the active layer at 3000 rpm for 60 s to give an 8 nm thick film. The top electrode was then prepared by sequential thermal evaporation of Ca (35 nm) and Al (100 nm) at reduced pressure below 10−6 Torr to furnish the BHJ solar cell devices. All devices contain an active area of 0.04 cm2, and
the photovoltaic parameters were measured at room temperature under air atmosphere with a Xenon lamp coupled to an AM 1.5G solar filter (SAN-EIXES-301S solar simulator). J−V characteristics were recorded in a Keitheley 2400 Source Measurement Unit.
■
RESULTS AND DISCUSSION
The synthetic procedure for the hydrophilic C
60derivatives is
described in Scheme 1. Triethylene glycol was treated with
tert-butyldimethylsilyl chloride (TBDMS-Cl) in the presence of
4-dimethylaminopyridine (DMAP) and triethylamine in
dichloro-methane to give tri(ethylene glycol) tert-butyldimethylsilyl
ether 1. Ester
fication of malonic acid with 1 by using
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and a catalytic amount
of DMAP yielded 2. Bingel reaction of C
60with 2 in the
presence of 1,8-diazabicyclo[5.4.0]undec-7-ene and I
2led to the
formation of bis(TBDMS-triethylene glycol)-malonate
full-erene 3. Desilylation of 3 with hydrochloric acid (2 M)
furnished bis(triethylene glycol) malonate C
60(EGMC
−OH,
4) which was reacted with phthalic anhydride to a
fford
bis(triethylene glycol phthalic acid)-malonate C
60(EGMC
−
COOH, 5). Their identities are fully characterized by NMR
spectroscopy and mass spectrometry.
The cyclic voltammograms of PC
61BM, EGMC
−OH, and
EGMC
−COOH are shown in Figure 1 and summarized in
Table 1. All voltammograms exhibit three well-de
fined,
single-electron reversible waves in the scanning range. The LUMO
energy levels of EGMC
−OH (−3.88 eV) and EGMC−COOH
(
−3.91 eV) are both lower than that of PC
61BM (
−3.85 eV).
The presence of the n-type EMLs can provide an extra donor
−
acceptor interface with the p-type material in the active layer for
exciton dissociation. On the other hand, the intrinsically
deep-lying HOMO energy level of EGMC
−OH and EGMC−
COOH
e
ffectively imparts the hole-blocking ability toward
p-type material to reduce the charge recombination at the
interfaces. The absorption spectra of EGMC
−OH and
EGMC
−COOH are depicted in Figure 2. Both spectra are
similar to each other with three distinct bands of ca. 258, 326,
and 426 nm in the CHCl
3solution.
The strategy of using hydrophilic EGMC
−OH and EGMC−
COOH
to construct the EML was then examined in
conventional solar cells. The devices are fabricated on the
basis of ITO/PEDOT:PSS/P3HT:PC
61BM/EML/Ca/Al
con-figuration. The current density−voltage characteristics of the
EGMC
−OH-based devices are illustrated in Figure 3, and their
device parameters are summarized in Table 2. For comparison,
the device without the EML was fabricated under otherwise
identical conditions and yielded an open-circuit voltage (V
oc) of
0.6 V, a short-circuit current (J
sc) of 9.02 mA cm
−2, a
fill factor
(FF) of 67%, and a PCE of 3.61%. The choice of polar solvents
for processing of the hydrophilic C
60plays an important role in
determining the
film-forming properties and the device
characteristics, as shown in Table 2. Employing a less polar
2-ethoxyethanolfurnished a low PCE of 0.02%, while the device
using isopropanol (IPA) as the solvent to deposit the EGMC
−
OH
layer gave a PCE of 2.41%. Both 2-ethoxyethanoland
isopropanol have a certain ability to dissolve PC
61BM in the
underlying active layer during the spin-coating of EGMC
−OH,
thus resulting in erosion and mixing of the active layer. The
PCE was optimized to 3.71% when 2-ethoxylethanol/H
2O
(10:1 v/v) was used as the processing cosolvent. The increased
polarity as a result of H
2O strengthens the orthogonal solvent
Figure 1. Cyclic voltammograms of PC61BM (black), EGMC−OH
(red), and EGMC−COOH (blue).
Table 1. Redox Potential and LUMO Energy Levels for
PC
61BM, EGMC−OH, and EGMC−COOH
compound E1pca(V) E1pab(V) E1redc(V) LUMOd(eV) EGMC−COOH −0.886 −0.736 −0.811 −3.91 EGMC−OH −0.910 −0.780 −0.845 −3.88 PC61BM −0.987 −0.752 −0.870 −3.85 aFrom the maximum of reduction potential.bFrom the maximum of oxidation potential.cFrom the equation Ered= 0.5(Epc+ Epa).dFrom
the equation LUMO =−(4.72 + Ered,onset).
Figure 2. Absorption spectra of EGMC−OH (red) and EGMC− COOH(blue).
Figure 3. Current density−voltage characteristics of ITO/PE-DOT:PSS/P3HT:PC61BM/EGMC−OH/Ca/Al devices under
effect, thus forming the EML layer without altering the
underlying active layer. Therefore, the formation of an
EGMC
−OH-based EML between the electrodes and the active
layers enhanced the electron-transporting characteristics at the
interfaces. A similar observation was found for the case of
EGMC
−COOH. The best efficiency of 3.80% was achieved
when 2-ethoxylethanol/H
2O (10:1 v/v) was used as the
cosolvent, indicating again the importance of utilizing highly
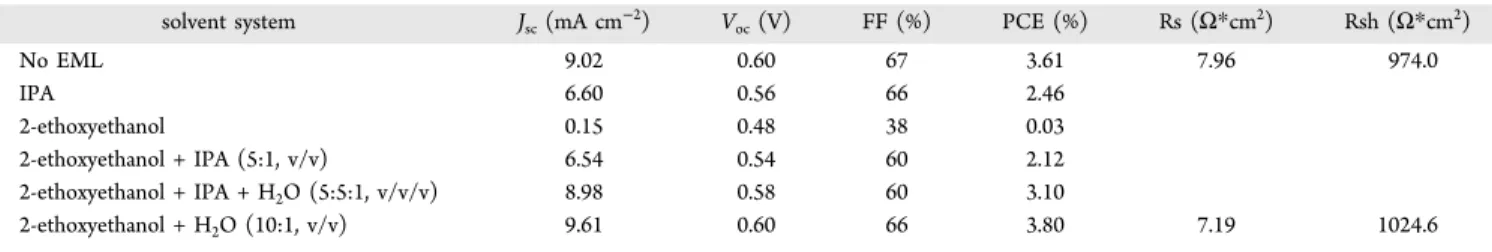
polar solvents (Figure 4, Table 3). The devices with EGMC
−
COOH
showed higher PCEs than those with EGMC
−OH.
These results may be presumably associated with the carboxylic
acid functionality on EGMC−COOH. The two carboxylic
groups might interact with the metal electrode and decrease the
ohmic contact between the EML and the electrode, thus
lowering the energy barrier for electron transporting from the
EML to the electrode.
16Furthermore, as can be concluded
from Table 2 and Table 3, after the installation of the EML, the
improvement of PCE mainly comes from the increase of J
sc,
while the V
ocand FF values remained generally unchanged. The
enhancement of J
scis in resonance with the diminution of the
device series resistance (Rs), indicating that the presence of the
EML can improve the device conductivity.
In an attempt to further optimize the electron-transporting
properties of the EML, n-dopant alkali carbonates were added
into the EGMC−OH and EGMC−COOH hosts.
15a−d,g,hAgain, a 2-ethoxylethanol/H
2O (10:1, v/v) cosolvent system
was used as the processing solvent for the following tests. As
demonstrated in Table 4 and Figure 3, the device performance
increased slightly upon the addition of Cs
2CO
3in EGMC
−
OH-based EMLs. A PCE of 3.80% was achieved when 40 wt %
Cs
2CO
3was doped. The presence of Cs
2CO
3can strengthen
the electron-transporting ability of the EML by increasing the
electron conductivity, thus resulting in the enhancement of J
scand PCE. The doping e
ffect is much more pronounced in the
case of EGMC
−COOH. An improved PCE of 4.21% was
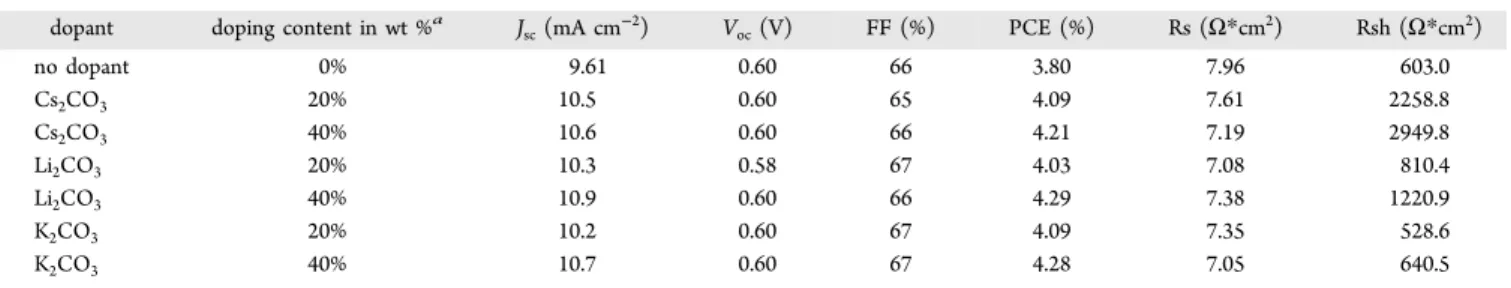
obtained as the doping content was increased to 40 wt %
(Figure 5, Table 5). Encouraged by these results, we further
evaluated di
fferent carbonates, i.e., Li
2CO
3and K
2CO
3, which
were incorporated in the EGMC
−COOH-based EMLs as the
n-dopants. As summarized in Figure 5 and Table 5, 40 wt %
Li
2CO
3is capable of increasing the J
scfrom 9.61 mA cm
−2(without dopant) to 10.9 mA cm
−2and the PCE from 3.80%
(without dopant) to 4.29%. Similar results can also be found for
K
2CO
3(Figure 5, Table 5). The signi
ficant improvement of
PCE mainly comes from the increase of J
sc, while the V
ocand
FF values remained generally unchanged. These results suggest
again that alkali carbonates may enhance the conductivity of the
EML, which is supported by comparing the conductivity of
EGMC−COOH with and without doping of Cs
2CO
3(see
Figure S3 in the Supporting Information). Notably, this e
ffect is
more prominent in EGMC
−COOH than EGMC−OH. The
carboxylic acid groups of EGMC
−COOH may interact with
the carbonate bases from the ionic EGMC
−carboxylate salt
which is expected to have strong a
ffinity with the metal
Table 2. Photovoltaic Performances of the Devices Using EGMC
−OH as the EML
solvent system Jsc(mA cm−2) Voc(V) FF (%) PCE (%) Rs (Ω*cm2) Rsh (Ω*cm2)
no EML 9.02 0.60 67 3.61 10.00 603.0 IPA 6.52 0.56 66 2.41 2-ethoxyethanol 0.14 0.36 38 0.02 2-ethoxyethanol + IPA (5:1, v/v) 6.48 0.54 59 2.01 2-ethoxyethanol + IPA + H2O (5:5:1, v/v/v) 9.22 0.56 59 3.06 2-ethoxyethanol + H2O (10:1, v/v) 9.43 0.60 66 3.71 8.23 862.9
Figure 4. Current density−voltage characteristics of ITO/PE-DOT:PSS/P3HT:PC61BM/EGMC−COOH/Ca/Al devices under
illumination of AM1.5G at 100 mW cm−2.
Table 3. Photovoltaic Performances of the Devices Incorporating EGMC
−COOH as the EML
solvent system Jsc(mA cm−2) Voc(V) FF (%) PCE (%) Rs (Ω*cm2) Rsh (Ω*cm2)
No EML 9.02 0.60 67 3.61 7.96 974.0 IPA 6.60 0.56 66 2.46 2-ethoxyethanol 0.15 0.48 38 0.03 2-ethoxyethanol + IPA (5:1, v/v) 6.54 0.54 60 2.12 2-ethoxyethanol + IPA + H2O (5:5:1, v/v/v) 8.98 0.58 60 3.10 2-ethoxyethanol + H2O (10:1, v/v) 9.61 0.60 66 3.80 7.19 1024.6
Table 4. Photovoltaic Performances of the Devices
Incorporating EGMC
−OH Doped with Cs
2CO
3dopant doping content in wt %a Jsc (mA cm−2) Voc (V) FF (%) PCE (%) no dopant 0% 9.43 0.60 66 3.71 Cs2CO3 20% 9.64 0.60 65 3.74 Cs2CO3 40% 9.71 0.60 65 3.80 aWt % is the weight percentage of Cs
electrode. The ohmic contact between the EML and the
electrode is therefore reduced, leading to the enhancement of
J
scand PCE.
16To assess the applicability of the hydrophilic C
60materials
doped with alkali carbonates as an EML, a low band polymer
PCDCTBT
−C8 (Scheme 1) blended with PC
71BM was then
used as the active layer in a conventional device.
17The
reference device without the EML gave a V
ocof 0.72 V, a J
scof
8.72 mA cm
−2, a FF of 57%, and a PCE of 3.63% (Table 6).
The PCDCTBT
−C8:PC
71BM-based device using EGMC
−
COOH
as the EML doped with 40 wt % of Li
2CO
3delivered a
V
ocof 0.72 V, a J
scof 11.1 mA cm
−2, a FF of 56%, and a PCE of
4.51%, which represents a 24% enhancement. Analogous to the
P3HT:PC
61BM system, the improvement of PCE mainly
results from the increase of J
sc. This example demonstrates
the general applicability of using alcohol-/water-soluble C
60materials for improving the e
fficiency of conventional solar
cells.
■
CONCLUSIONS
We have rationally designed and synthesized two new C
60-based n-type materials, EGMC
−OH and EGMC−COOH,
functionalized with hydrophilic triethylene glycol groups. With
the assistance of the TEG-based surfactant, EGMC
−OH and
EGMC−COOH can be dissolved in highly polar solvents to
implement orthogonal solvent strategy, forming an electron
modi
fication layer without eroding the underlying active layer.
We found that 2-ethoxylethanol/H
2O (10:1, v/v) with higher
polarity is the best cosolvent to deposit the EGMC
−OH and
EGMC
−COOH EMLs. Multilayer conventional solar cells
(ITO/PEDOT:PSS/P3HT:PC
61BM/EML/Ca/Al) with
inser-tion of the C
60-modi
fier between the active layer and electrode
have been successfully realized by a cost-e
ffective solution
processing technique. Compared to the pristine device with a
power conversion e
fficiency (PCE) of 3.61%, the device
modi
fied by the EGMC−OH and EGMC−COOH EMLs
improved the PCEs to 3.71% and 3.8%, respectively. Moreover,
the electron-transporting properties of EGMC
−OH and
EGMC
−COOH can be further enhanced by the incorporation
of alkali carbonates as the n-dopants. Regardless of the
counterions, Cs
2CO
3, Li
2CO
3, as well as K
2CO
3carbonates
are all e
ffective to obtain higher efficiency. The device using
EGMC
−COOH doped with 40 wt % Li
2CO
3achieved a
highest PCE of 4.29%. The superior performance of EGMC
−
COOH
to EGMC
−OH might be associated to the existence of
the carboxylic functionality. Most importantly, the alcohol-/
water-soluble EGMC
−COOH can be applicable and effective
to other conventional devices. With the incorporation of the
EGMC
−COOH EML doped with 40 wt % Li
2CO
3, the
PCDCTBT-C8:PC
71BM-based device exhibited a superior
PCE of 4.51%, which outperformed the corresponding
nonmodi
fied device with a PCE of 3.63%.
■
ASSOCIATED CONTENT
*
S Supporting InformationThermal gravimetric analysis, cyclic voltammograms,
measure-ment of conductivity of EGMC
−COOH,
1H and
13C NMR
spectra, and mass spectrometry of the new materials. This
material is available free of charge via the Internet at http://
pubs.acs.org.
■
AUTHOR INFORMATION
Corresponding Author
*E-mail: [email protected] and [email protected].
tw.
Notes
The authors declare no competing
financial interest.
■
ACKNOWLEDGMENTS
We thank the National Science Council and the
“ATU
Program
” of the Ministry of Education and the Center for
Interdisciplinary Science (CIS) of the National Chiao Tung
University Taiwan for
financial support.
Figure 5. Current density−voltage characteristics of ITO/PE-DOT:PSS/P3HT:PC61BM/EGMC−COOH/Ca/Al devices doped
with alkali carbonates.
Table 5. Photovoltaic Performances of the Devices Incorporating EGMC
−COOH Doped with Alkali Carbonates
dopant doping content in wt %a J
sc(mA cm−2) Voc(V) FF (%) PCE (%) Rs (Ω*cm2) Rsh (Ω*cm2) no dopant 0% 9.61 0.60 66 3.80 7.96 603.0 Cs2CO3 20% 10.5 0.60 65 4.09 7.61 2258.8 Cs2CO3 40% 10.6 0.60 66 4.21 7.19 2949.8 Li2CO3 20% 10.3 0.58 67 4.03 7.08 810.4 Li2CO3 40% 10.9 0.60 66 4.29 7.38 1220.9 K2CO3 20% 10.2 0.60 67 4.09 7.35 528.6 K2CO3 40% 10.7 0.60 67 4.28 7.05 640.5 aWt % is the weight percentage of alkali carbonates relative to EGMC−COOH.
Table 6. Photovoltaic Performances of the PCDCTBT
−
C8:PC
71BM-Based Devices Using EGMC
−COOH Doped
with 40 wt % Li
2CO
3as the EML
device Jsc(mA cm−2) Voc(V) FF (%) PCE (%) without EML 8.72 0.72 57 3.63 with EML 11.10 0.72 56 4.51
■
REFERENCES
(1) (a) Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J. Science 1995, 270, 1789−1791. (b) Günes, S.; Neugebauer, H.; Sariciftci, N. S. Chem. Rev. 2007, 107, 1324−1338. (c) Thompson, B. C.; Fréchet, J. M. J. Angew. Chem., Int. Ed. 2008, 47, 58−77. (d) Chen, J.; Cao, Y. Acc. Chem. Res. 2009, 42, 1709−1718. (e) Cheng, Y.-J.; Yang, S.-H.; Hsu, C.-S. Chem. Rev. 2009, 109, 5868−5923. (f) Zhou, H.; Yang, L.; You, W. Macromolecules 2012, 45, 607−632. (g) Li, Y.; Zou, Y. Adv. Mater. 2008, 20, 2952−2958. (h) Li, Y. Acc. Chem. Res. 2012, 45, 723−733.
(2) (a) Ma, W.; Yang, C.; Gong, X.; Lee, K.; Heeger, A. J. Adv. Funct. Mater. 2005, 15, 1617−1622. (b) Mayer, A. C.; Scully, S. R.; Hardin, B. E.; Rowell, M. W.; McGehee, M. D. Mater. Today 2007, 10, 28−33. (c) Li, G.; Shrotriya, V.; Huang, J.; Yao, Y.; Moriarty, T.; Emery, K.; Yang, Y. Nat. Mater. 2005, 4, 864−868. (d) Reyes-Reyes, M.; Kim, K.; Carroll, D. L. Appl. Phys. Lett. 2005, 87, 083506. (e) Woo, C. H.; Thompson, B. C.; Kim, B. J.; Toney, M. F.; Fréchet, J. M. J. J. Am. Chem. Soc. 2008, 130, 16324−16329.
(3) (a) Li, G.; Chu, C. W.; Shrotriya, V.; Huang, J.; Yang, Y. Appl. Phys. Lett. 2006, 88, 253503. (b) Liao, H.-H.; Chen, L.-M.; Xu, Z.; Li, G.; Yang, Y. Appl. Phys. Lett. 2008, 92, 173303. (c) Mor, G. K.; Shankar, K.; Paulose, M.; Varghese, O. K.; Grimes, C. A. Appl. Phys. Lett. 2007, 91, 152111. (d) Waldauf, C.; Morana, M.; Denk, P.; Schilinsky, P.; Coakley, K.; Choulis, S. A.; Brabec, C. J. Appl. Phys. Lett. 2006, 89, 233517. (e) White, M. S.; Olson, D. C.; Shaheen, S. E.; Kopidakis, N.; Ginley, D. S. Appl. Phys. Lett. 2006, 89, 143517. (f) Steim, R.; Choulis, S. A.; Schilinsky, P.; Brabec, C. J. Appl. Phys. Lett. 2008, 92, 093303. (g) Yang, T.; Cai, W.; Qin, D.; Wang, E.; Lan, L.; Gong, X.; Peng, J.; Cao, Y. J. Phys. Chem. C 2010, 114, 6849−6853. (4) (a) Cheng, Y.-J.; Hsieh, C.-H.; Li, P.-J.; Hsu, C.-S. Adv. Funct. Mater. 2011, 21, 1723−1732. (b) Kim, B. J.; Miyamoto, Y.; Ma, B.; Fréchet, J. M. J. Adv. Funct. Mater. 2009, 19, 2273−2281. (c) Gholamkhass, B.; Holdcroft, S. Chem. Mater. 2010, 22, 5371− 5376. (d) Drees, M.; Hoppe, H.; Winder, C.; Neugebauer, H.; Sariciftci, N. S.; Schwinger, W.; Schaeffler, F.; Topf, C.; Scharber, M. C.; Zhu, Z.; Gaudiana, R. J. Mater. Chem. 2005, 15, 5158−5163.
(5) (a) Li, C.-Z.; Yip, H.-L.; Jen, A. K.-Y. J. Mater. Chem. 2012, 22, 4161−4177. (b) Ma, H.; Yip, H.-L.; Huang, F.; Jen, A. K.-Y. Adv. Funct. Mater. 2010, 20, 1371−1388. (c) Yip, H.-L.; Jen, A. K.-Y. Energy Environ. Sci 2012, 5, 5994−6011. (d) Huang, F.; Jen, A. K.-Y. In Organic Electronics; CRC Press: Boca Raton, FL, 2009; pp 243−261.
(6) Hau, S. K.; Yip, H.-L.; Baek, N. S.; Zou, J.; O’Malley, K.; Jen, A. K. Y. Appl. Phys. Lett. 2008, 92, 253301.
(7) (a) Lee, K.; Kim, J. Y.; Park, S. H.; Kim, S. H.; Cho, S.; Heeger, A. J. Adv. Mater. 2007, 19, 2445−2449. (b) Gilot, J.; Wienk, M. M.; Janssen, R. A. J. Appl. Phys. Lett. 2007, 90, 143512. (c) Hayakawa, A.; Yoshikawa, O.; Fujieda, T.; Uehara, K.; Yoshikawa, S. Appl. Phys. Lett. 2007, 90, 163517.
(8) Brabec, C. J.; Shaheen, S. E.; Winder, C.; Sariciftci, N. S.; Denk, P. Appl. Phys. Lett. 2002, 80, 1288−1290.
(9) (a) Liao, H.-H.; Chen, L.-M.; Xu, Z.; Li, G.; Yang, Y. Appl. Phys. Lett. 2008, 92, 173303. (b) Chen, F.-C.; Wu, J.-L.; Yang, S. S.; Hsieh, K.-H.; Chen, W.-C. J. Appl. Phys. 2008, 103, 103721.
(10) (a) Zhang, F.; Ceder, M.; Inganäs, O. Adv. Mater. 2007, 19, 1835−1838. (b) Chen, F.-C.; Chien, S.-C. J. Mater. Chem. 2009, 19, 6865−6869.
(11) (a) Oh, S.-H.; Na, S.-I.; Jo, J.; Lim, B.; Vak, D.; Kim, D.-Y. Adv. Funct. Mater. 2010, 20, 1977−1983. (b) He, Z.; Zhang, C.; Xu, X.; Zhang, L.; Huang, L.; Chen, J.; Wu, H.; Cao, Y. Adv. Mater. 2011, 23, 3086−3089. (c) Luo, J.; Wu, H.; He, C.; Li, A.; Yang, W.; Cao, Y. Appl. Phys. Lett. 2009, 95, 043301. (d) Duarte, A.; Pu, K.-Y.; Liu, B.; Bazan, G. C. Chem. Mater. 2010, 23, 501−515. (e) He, C.; Zhong, C.; Wu, H.; Yang, R.; Yang, W.; Huang, F.; Bazan, G. C.; Cao, Y. J. Mater. Chem. 2010, 20, 2617−2622. (f) Seo, J. H.; Gutacker, A.; Sun, Y.; Wu, H.; Huang, F.; Cao, Y.; Scherf, U.; Heeger, A. J.; Bazan, G. C. J. Am. Chem. Soc. 2011, 133, 8416−8419. (g) He, Z.; Zhong, C.; Huang, X.; Wong, W.-Y.; Wu, H.; Chen, L.; Su, S.; Cao, Y. Adv. Mater. 2011, 23, 4636−4643. (h) He, Z.; Zhong, C.; Su, S.; Xu, M.; Wu, H.; Cao, Y. Nature Photon. 2012, 6, 591−595. (i) Duan, C.; Zhang, K.; Guan, X.;
Zhong, C.; Xie, H.; Huang, F.; Chen, J.; Peng, J.; Cao, Y. Chem. Sci. 2013, 4, 1298−1307. (j) Guan, X.; Zhang, K.; Huang, F.; Bazan, G. C.; Cao, Y. Adv. Funct. Mater. 2012, 22, 2846−2854.
(12) (a) Wei, Q.; Nishizawa, T.; Tajima, K.; Hashimoto, K. Adv. Mater. 2008, 20, 2211−2216. (b) Jung, J. W.; Jo, J. W.; Jo, W. H. Adv. Mater. 2011, 23, 1782−1787. (c) Hau, S. K.; Cheng, Y.-J.; Yip, H.-L.; Zhang, Y.; Ma, H.; Jen, A. K. Y. ACS Appl. Mater. Interfaces 2010, 2, 1892−1902. (d) Duan, C.; Zhong, C.; Liu, C.; Huang, F.; Cao, Y. Chem. Mater. 2012, 24, 1682−1689. (e) Chang, C.-Y.; Wu, C.-E.; Chen, S.-Y.; Cui, C.; Cheng, Y.-J.; Hsu, C.-S.; Wang, Y.-L.; Li, Y. Angew. Chem., Int. Ed. 2011, 50, 9386−9390. (f) Cheng, Y.-J.; Hsieh, C.-H.; He, Y.; Hsu, C.-S.; Li, Y. J. Am. Chem. Soc. 2010, 132, 17381− 17383. (g) Cheng, Y.-J.; Cao, F.-Y.; Lin, W.-C.; Chen, C.-H.; Hsieh, C.-H. Chem. Mater. 2011, 23, 1512−1518. (h) Hsieh, C.-H.; Cheng, Y.-J.; Li, P.-J.; Chen, C.-H.; Dubosc, M.; Liang, R.-M.; Hsu, C.-S. J. Am. Chem. Soc. 2010, 132, 4887−4893.
(13) (a) Cheng, Y.-J.; Liao, M.-H.; Shih, H.-M.; Shih, P.-I. Macromolecules 2011, 44, 5968−5976. (b) Huang, F.; Cheng, Y.-J.; Zhang, Y.; Liu, M. S.; Jen, A. K. Y. J. Mater. Chem. 2008, 18, 4495− 4509. (c) Cheng, Y.-J.; Liu, M. S.; Zhang, Y.; Niu, Y.; Huang, F.; Ka, J.-W.; Yip, H.-L.; Tian, Y.; Jen, A. K. Y. Chem. Mater. 2007, 20, 413−422. (d) Müller, C. D.; Falcou, A.; Reckefuss, N.; Rojahn, M.; Wiederhirn, V.; Rudati, P.; Frohne, H.; Nuyken, O.; Becker, H.; Meerholz, K. Nature 2003, 421, 829−833.
(14) (a) Ayzner, A. L.; Tassone, C. J.; Tolbert, S. H.; Schwartz, B. J. J. Phys. Chem. C 2009, 113, 20050−20060. (b) Hagemann, O.; Bjerring, M.; Nielsen, N. C.; Krebs, F. C. Sol. Energy Mater. Sol. Cells 2008, 92, 1327−1335. (c) Zakhidov, A. A.; Lee, J.-K.; Fong, H. H.; DeFranco, J. A.; Chatzichristidi, M.; Taylor, P. G.; Ober, C. K.; Malliaras, G. G. Adv. Mater. 2008, 20, 3481−3484. (d) Sax, S.; Rugen-Penkalla, N.; Neuhold, A.; Schuh, S.; Zojer, E.; List, E. J. W.; Müllen, K. Adv. Mater. 2010, 22, 2087−2091.
(15) (a) Barbot, A.; Di, B. C.; Lucas, B.; Ratier, B.; Aldissi, M. J. Mater. Sci. 2013, 48, 2785−2789. (b) Xu, Z.-Q.; Yang, J.-P.; Sun, F.-Z.; Lee, S.-T.; Li, Y.-Q.; Tang, J.-X. Org. Electron. 2012, 13, 697−704. (c) He, M.; Jung, J.; Qiu, F.; Lin, Z. J. Mater. Chem. 2012, 22, 24254− 24264. (d) Cai, Y.; Wei, H. X.; Li, J.; Bao, Q. Y.; Zhao, X.; Lee, S. T.; Li, Y. Q.; Tang, J. X. Appl. Phys. Lett. 2011, 98, 113304. (e) Huang, F.; Shih, P.-I.; Shu, C.-F.; Chi, Y.; Jen, A. K. Y. Adv. Mater. 2009, 21, 361− 365. (f) Huang, F.; Shih, P.-I.; Liu, M. S.; Shu, C.-F.; Jen, A. K. Y. Appl. Phys. Lett. 2008, 93, 243302. (g) Wu, C.-I.; Lin, C.-T.; Chen, Y.-H.; Chen, M.-H.; Lu, Y.-J.; Wu, C.-C. Appl. Phys. Lett. 2006, 88, 152104. (h) Chen, S.-Y.; Chu, T.-Y.; Chen, J.-F.; Su, C.-Y.; Chen, C. H. Appl. Phys. Lett. 2006, 89, 053518.
(16) (a) O’Malley, K. M.; Li, C.-Z.; Yip, H.-L.; Jen, A. K. Y. Adv. Energy Mater. 2012, 2, 82−86. (b) Li, C.-Z.; Chueh, C.-C.; Yip, H.-L.; O’Malley, K. M.; Chen, W.-C.; Jen, A. K. Y. J. Mater. Chem. 2012, 22, 8574−8578.
(17) Cheng, Y.-J.; Wu, J.-S.; Shih, P.-I.; Chang, C.-Y.; Jwo, P.-C.; Kao, W.-S.; Hsu, C.-S. Chem. Mater. 2011, 23, 2361−2369.