國立交通大學
電子物理學系
碩士論文
氮極性獨立式氮化鎵基板磷酸蝕刻機制與形貌探討
The study of morphology and etching mechanism on N-face
Phosphoric acid treatment free-standing GaN
學生:杜佳豪
指導教授:李威儀 博士
氮極性獨立式氮化鎵基板磷酸蝕刻機制與形貌探討
The study of morphology and etching mechanism on N-face
Phosphoric acid treatment free-standing GaN
學生:杜佳豪 Student:Jia-Hao Du
指導教授:李威儀 advisor:Wei-I Lee
國立交通大學
電子物理學系
碩士論文
A ThesisSubmitted to department of Electro-physics College of Science
National Chiao Tung University In partial Fulfillment of the Requirement
For the Degree of Master
In
Electro-physics
Oct 2014
Hsinchu, Taiwan, Republic of China
中華民國一零三年十月
i
氮極性獨立式氮化鎵基板磷酸蝕刻機制與形貌探討
研究生:杜佳豪 指導教授:李威儀 教授 國立交通大學電子物理研究所 摘要 本論文在探討氮極性獨立式氮化鎵磷酸蝕刻的形貌與機制。磷酸被被認是個很方 便的酸性溶液在於檢驗氮化鎵基板的品質,但是很少團隊對於磷酸去除氧化物的能力 與蝕刻機制的探討。從鎵極性獨立式氮化鎵基板做完 XPS 的結果來看,氧含量的比例 相較於沒做磷酸處理的樣品為 30-50%,且從 XPS 的束縛能往高能階偏移了 0.83 eV, 可以知道電子濃度在做完磷酸蝕刻後增加了,合理地的就是有氮空缺的形,由此推論 氫原子為磷酸蝕刻的主要反應。由拉曼量測結果與其對氫氧化鉀蝕刻的比較後,確立 了磷酸的蝕刻機制。藉此去看氮極性氮化鎵基板的磷酸蝕刻形貌。 從結果來看,磷酸不論是在鎵極性面還是氮極性面,都會從表面的缺陷開始攻 擊。只要是磷酸蝕刻後的形貌,必會出現鎵極性的穩定面。所以在鎵極性面上會出現 V-pits 和氮極性面上出現十二面角錐的情況,亦可以使用這個模型來解釋之。ii
The study of morphology and etching mechanism on N-face
Phosphoric acid treatment free-standing GaN
Student:Jia-Hao Du Advisor:Wei-I Lee Department of electro-physics
National Chiao Tung University
abstract
In this work, we study the morphology and etching mechanism on N-face phosphoric acid treatment free-standing GaN. It's well known that
phosphoric acid is good solution to recognize the surface defect on Ga-polar GaN, but it seldom used for removing native oxide on GaN, and discussed the etching mechanism. On XPS measurement, we found that the oxygen content on acid treatment sample was only 30-50% comparing to un-treatment sample, and the binding energy had been shifted 0.83 eV after H3PO4 treatment, it meant that the electron concentration was raised. We
infer it be caused by the hydrogen atom. And that was the main reactant on H3PO4 etching. This result verified by Raman measurement and KOH etching
mechanism.
Finally, we demonstrated that the pyramids appeared on N-face H3PO4
treatment samples, and v-pits formed on Ga-face by etching pits density experiment. All the facets after H3PO4 etching were Ga-polarity.
iii
誌謝
算一算日子,我在交大也默默地邁向了第四年了。還記得當初李老師在我找他當 指導教授時說:「做我的學生,我不負責幫你找任何儀器。想做什麼研究,需要什麼資 源你都必須自己搞定。」,這剛好是我當初念碩士班的目的之一,所以我就請老師做為 我的指導教授了。這些年走來,發生過大大小小的事,可說是大風大浪都見過了。從 一開始的火箭計畫,中間無數的計畫趕工,這張畢業證書也算是得來不易。套用陳之 藩所說的名言,要謝的人太多,不如就謝天吧。真的很感謝這一路來,幫助我碩士論 文的所有人,也很感謝指導我的博士班學長-彥顯學長、尹豪學長和崇銘學長。以我來 說,顯哥和豪哥真的對實驗室的事務付出很多,所以一直以來我也不遺餘力的達到學 長的要求;崇銘學長,真的很抱歉我沒有幫到你很多,一直想幫你做些事,但是實在 是分身乏術,希望老師新的決定能幫到你。 這個碩士學位,在我心中真的是有兩個遺憾。一是我為人子女的不孝,沒讓我的 母親看到這份畢業證書,一再的讓她失望,僅為了我對於學位和實驗室的風氣的堅 持。從出生以來讓您受的苦,就算是我下輩子再當您兒子也還不清吧。第二個缺憾是 關於順茗你們,不知道為什麼你們和豪哥的關係是我在實驗室這麼久以來,看過學長 與學弟關係最差的一屆。當然我也幫不了你們什麼了,我只能和你們說,若全部的研 究生來交大都只想混個學位,想著偷懶或是只想著自己,那這個社會肯定是進步不了 的。 接著是寫給晟淵你們兩個的。以實驗室目前的現況,我想你們兩個準時畢業絕對 不是問題。晟淵,你看事物的眼光其實不差,對於自身的要求也不太低,我相信你以 後必有所成;巧涵,我大概感覺得到妳對於取得碩士學位與自己對於未來的打算並沒 有很完整的藍圖。加油吧!雖然妳對於機台的學習並沒有很得心應手,老師也突然幫妳 改題目,但是我相信你以後仍然能在台積或是聯電這樣一流的大公司的。在我看來, 你們兩個,也和順茗他們有一樣的問題存在。真的不知道是世代的變遷嗎?還是我和豪 哥對於你們的要求過於嚴苛了。老師也曾說過:「我利用博士班學生,而博士班學長會 要利用碩士班學生」。我們的要求,無非就是在於完成老師的指示與實驗室的運作維持 罷了,作為交換,學長也盡量地幫助你們,讓你們可以準時畢業。 在這三年多一點的日子內,說實在我迷失好多次。也想過要繼續攻讀博士班,不 知道過了十年後的我在看這段日子時,會有什麼想法呢?後悔與否也是無法再回到這時 了。在這裡我真的學到不少東西,這絕對是我當初報考電物所時沒想到的。真的十分 感謝幫助過我的人,指導教授。最後也祝我認識的人,假如有在攻讀碩士或是博士學 位,能順利的取得學歷。 學生 佳豪 寫於民國 103 年 10 月 8 號iv
目錄
第一章 緒論 ... 1 1-1 研究背景、發展 ... 1 1-2 氮化鎵材料特性 ... 1 1-2-1 自發極化(spontaneous polarization) ... 3 1-2-2 壓電極化(piezoelectric polarization) ... 5 1-3 鎵極性與氮極性氮化鎵基本特性比較 ... 7 1-3-1 鎵極性與氮極性氮化鎵的電性比較 ... 7 1-3-2 極化對於氮化鎵的光性影響 ... 10 1-3-3 鎵極性與氮極性氮化鎵物理與化學性質比較 ... 12 1-4 磷酸濕式處理於氮化鎵基板上 ... 14 1-5 實驗動機與目的 ... 15 第二章 實驗流程與機台介紹 ... 16 2-1 實驗流程與參數設計 ... 16 2-2 機台介紹 ... 17 2-2-1 氫化物氣相磊晶 ... 17 2-2-2 雷射剝離 ... 19 2-2-3 化學機械研磨 ... 20 2-3 量測機台介紹 ... 21 2-3-1 原子力顯微鏡 ... 21 2-3-2 x 光繞射分析儀 ... 23 2-3-3 x 射線光電子能譜儀 ... 24 2-3-4 霍爾量測 ... 24 2-3-5 拉曼量測 ... 25 2-3-6 掃瞄式電子顯微鏡 ... 26 第三章 實驗結果與討論 ... 27 3-1 獨立式氮化鎵基板於磷酸處理後的表面分析 ... 27 3-1-1 磷酸處理對於獨立式氮化鎵基板的影響 ... 31 3-1-2 磷酸蝕刻機制於鎵極性氮化鎵基板上 ... 32 3-1-3 KOH 蝕刻機制於氮極性面氮化鎵基板上 ... 33 3-1-4 磷酸蝕刻機制的驗證 ... 35 3-2 氮極性獨立式氮化鎵基板蝕刻機制與形貌探討 ... 37 3-2-1 獨立式氮化鎵基板的磊晶品質討論 ... 38 3-2-2 氮極性氮化鎵蝕刻形貌 ... 39 3-3-3 氮極性氮化鎵的磷酸蝕刻機制探討 ... 42 第四章 結論 ... 44 參考文獻 ... 45v
表目錄
表 1 針對不同極性面的獨立式氮化鎵基板霍爾量測結果 ... 4 表 2 不同極性面的 SBH 與理想因子(ideality factor) ... 8 表 3 氮化鎵與氮化鋁在不同溶液下的蝕刻率(沒標註溫度則為室溫 25℃) ... 13 表 4 氮極性獨立式氮化鎵基板蝕刻實驗設計 ... 16 表 5 各式氮化鎵基板磊晶優劣比較 ... 17 表 6 獨立式氮化鎵基板規格 ... 27 表 7 有無做過磷酸處理的拉曼量測結果 ... 36vi
圖目錄
圖 1 各式化合物半導體其晶格常數(lattice constant)與能隙(band-gap)分 佈圖 ... 2 圖 2 烏采結構示意圖 ... 2 圖 3 氮化鎵磊晶模式示意圖 ... 3圖 4 (a)HEMT 示意圖 (b)HEMT 於酸蝕刻後的結果 (c)氧(Oxygen, O)濃度縱 深圖 (d)碳(Carbon, C)和矽(Silicon, Si)濃度縱深圖 ... 4
圖 5 氮化鎵單位晶胞與自發極化場方向 ... 5
圖 6 極化方向在鎵極性與氮極性異質結構之示意圖 ... 6
圖 7 n-type 氮化鎵的能帶(band structure)模擬圖 ... 7
圖 8 Ga-face 與 N-face Schottky diode 電壓-電流曲線圖(左)和電壓-電容 曲線圖(右) ... 8
圖 9 XPS Ga 3d 能譜 ... 8
圖 10 能帶示意圖。(a)Ga-face,(b)N-face ... 9
圖 11 XPS Ti 2p 能譜與 Ga 3d 能譜 ... 10
圖 12 (a)極性氮化鎵, (b)非極性氮化鎵的量子井(quantum well)能帶圖 .. 11
圖 13 氮化鎵的極性面、非極性面與半極性面示意圖 ... 11
圖 14 鎵極性與氮極性氮化鎵研磨後照片 ... 12
圖 15 鹽酸(HCl)蝕刻鎵極性氮化鎵,(左)原子力顯微鏡(atomic force microscope, AFM)圖及掃瞄式子顯微鏡(Scanning Electron Microscopy, SEM)影像, (右)穿透式電子顯微鏡(Transmission electron microscopy, TEM)影像 ... 13
圖 16 氫氧化鉀(KOH)蝕刻後形貌。(a)N-face, (b)Ga-face ... 14 圖 17 EPD 結果 ... 14 圖 18 實驗基本流程 ... 16 圖 19 水平式氫化物氣相磊晶系統腔體剖面圖 ... 17 圖 20 水平式氫化物氣相磊晶系統 ... 18 圖 21 LLO 原理 ... 19 圖 22 雷射剝離系統 ... 20 圖 23 化學機械研磨示意圖 ... 20 圖 24 化學機械研磨系統 ... 21 圖 25 原子力顯微鏡示意圖 ... 22 圖 26 原子距離與斥力、引力關係圖 ... 22 圖 27 布拉格繞射示意圖 ... 23 圖 28 x-ray 激發光電子示意圖 ... 24 圖 29 拉曼散射示意圖 ... 25 圖 30 電子與試片的交互作用 ... 26
vii 圖 31 浸泡 HCl 稀釋液後的 I-V curve ... 27 圖 32 獨立式氮化鎵基板在磷酸處理後的 AFM 圖 ... 28 圖 33 獨立式氮化鎵基板在磷酸處理後的 XRC 圖 ... 28 圖 34 XPS Ga 2p 能譜(左), N 1s 能譜(右) ... 29 圖 35 XPS O 1s 能譜 ... 29 圖 36 溫度、壓力、五三比和載氣對長晶模式的影響趨勢 ... 30 圖 37 沒研磨過的氮化鎵基板的 XPS O 1s 能譜 ... 30 圖 38 XPS Ga 2p 量測能帶示意圖 ... 31 圖 39 鎵極性面氮化鎵磷酸蝕刻機制 ... 32 圖 40 有 point defect 的鎵極性氮化鎵基板 ... 33 圖 41 氮極性氮化鎵基板做完 KOH 蝕刻後的形貌 ... 33 圖 42 KOH 蝕刻機制流程 ... 34 圖 43 隨 KOH 蝕刻時間變化的 PL 光譜 ... 34
圖 44 有無做過 KOH 處理的 I-V curve ... 35
圖 45 做完磷酸處理後的樣品電子濃度比較 ... 35 圖 46 拉曼量測中的光學振動模式 ... 36 圖 47 HVPE 厚膜磊晶流程 ... 37 圖 48 有 point defect 的氮極性氮化鎵基板 ... 37 圖 49 不同濕式處理後的表面形貌與其 PL 光譜 ... 38 圖 50 僅做 LT+RT 成長的獨立式氮化鎵基板厚度(unit:㎛) ... 38 圖 51 有無去除 LT+RT layer 的 XRC ... 39 圖 52 不同品質的氮極性氮化鎵基板做 1 分鐘磷酸蝕刻的形貌 ... 39 圖 53 LT+RT N-face polish 樣品隨蝕刻時間變化之形貌 ... 40 圖 54 HT N-face polish 樣品隨蝕刻時間變化之形貌 ... 41 圖 55 HT N-face polish 樣品蝕刻 60 分鐘結果 ... 41 圖 56 N-face 形貌隨著時間變化的示意圖 ... 41 圖 57 氮極性面氮化鎵磷酸蝕刻機制 ... 42 圖 58 氮極性氮化鎵於磷酸蝕刻後的 TEM 圖 ... 42 圖 59 十二面角錐示意圖 ... 43 圖 60 不同濃度的 EPD 蝕刻結果 ... 43
1
第一章 緒論
1-1 研究背景、發展
現今的科技文明發展快速,生活中不乏電子產品,可以歸功於近百年的近代物理 發展。在 1948 年,貝爾實驗室的 W.Schokley、J.Bardeen 和 W.H. Brattain 發明了雙 極性電晶體 (Bipolar transistor),至此半導體技術開始快速發展。以目前的發展 上,矽化物仍是電子業最常使用的材料;而光電產業上,常使用化合物半導體1 ,其共 通特色為高載子遷移率(carrier mobility)。其中氮化物為近年最具潛力的光電材料 1 ,主要應用於高功率高頻元件、藍綠光到紫外光波段的發光元件以及太陽能電池。 氮化鎵成為近年熱門的研究項目可說和發光元件的發展脫不了關係。1960 年,T. Maiman 發明第一支紅寶石雷射後,人們對於這種具高發光強度或是高指向度的光源的 需求就越來越大。根據通訊調變理論,不考慮外界的影響,短波長的發信源是比較好 的,但直到 1990 年代,短波長的發光元件才迅速的發展起來。原因在於 1960 年代, 紅光發光材料如砷化鎵(GaAs)、磷化鎵(GaP)…等的磊晶問題早就被克服。藍綠光的磊 晶材料多為二六族化合物(II-VI compound),像是硒化鋅(ZnSe)、硫化鎂(MgS)、碳化 矽(SiC)及氮化物的半導體系統。二六族半導體系統的材料特性不是很穩定,所以一直 以來並非市場上的主流。而碳化矽則是沒有搭配的材料可以形成異質結構,並且為一 個非直接能隙(in-direct band-gap)的材料,其轉換效率更差。在三五族氮化物半導 體,因為氮的的揮發性很高,因此無法跟砷化鎵、磷化銦和碳化矽一樣,可以直接用 液相磊晶(liquid phase epitaxy, LPE)的方式成長大尺寸的塊材及量產,必須在幾萬 到幾十萬大氣壓之下才能形成基板,且所做出的基板尺寸很小,無法得到發展。1983 年,日本 Yoshida 公司在藍寶石基板(sapphire, Al2O3)上成長高溫氮化鋁(AlN)緩衝層(buffer layer),發現在其上成長的氮化鎵品質較好。之後,名古屋大學 I. Akasaki 教授採用低溫氮化鋁緩衝層隨後再採用高溫(~1000℃)成長氮化鎵(GaN),得到像鏡面 一樣的成品。而 1991 年日亞公司(Nichia)研究員 S. Nakamura 採用低溫氮化鎵緩衝 層,再升到高溫成長,同樣得到了高品質的氮化鎵之後,藍綠光的元件才有了藍寶石 基板可以使用,也因此蓬勃發展。至今這種藍寶石配合緩衝層的成長方式仍然是業界 最常使用的。
1-2 氮化鎵材料特性
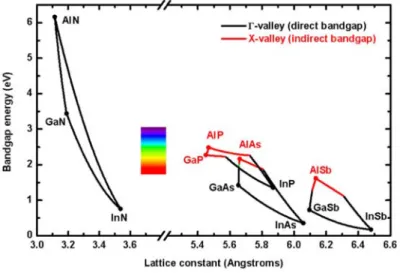
在二六族與三五族化合物中,屬氮化物的特性最好。由圖 1 來看,氮化鋁(AlN)的 能隙為 6.2 eV,氮化銦(InN)為 0.6 eV。若搭配氮化鎵就可以產出四元化合物 (quaternary compound)2 ,其發光範圍可以概括整個可見光範圍。氮化鎵本身的能隙為 3.4 eV,做為吸光材料上已可將太陽發光頻譜中能量最強的部分給概括進去3 ,且氮化2 物系統全為直接能隙(direct band-gap)材料,理論上的光電轉換效率遠比其它材料還 好。此外,因為使用氮為材料,製程中所產生的副產物對環境的影響也少很多。 圖 1 各式化合物半導體其晶格常數(lattice constant)與能隙(band-gap)分佈圖4 氮化物在晶體結構上為六方最密堆積(hexagonal closed-packed, hcp),鍵結方 式為…ABAB…,所以在空間平面可以看到原子以三角形的網狀排列,因此晶胞為一個 六角柱的形狀,我們稱烏采(wurtzite)結構,如圖 2 所示。在烏采結構中,我們定義 卡式座標中的 z 軸為 c 軸(c-axis);x 軸為 a 軸(a-axis)和 y 軸為 m 軸(m-axis)。
圖 2 烏采結構示意圖1
以 c 面(c-plane)的藍寶石基板成長 c-plane 氮化鎵來說明。考量這樣的晶體結構, 氮化鎵可以有兩種初始磊晶模式。一是在基板(substrate)先鍵結氮原子,再進行後續 磊晶,最後終止晶面多會為鎵原子層(Ga-terminated layer),這種結構我們稱鎵極性 面(Ga-face or Ga-polarity)。反之,先鍵結鎵原子,最後終止面多為的氮原子層(N-terminated layer)的就稱為氮極性面(N-face or N-polarity),如圖 3 所示。更細部 地去看的話,可以發現在鎵極性結構上,鎵原子都會被三個氮原子所包圍;而氮極性 結構上會是由三個鎵原子和一個氮原子去形成一個四面體的結構。一般而言,這種互
3
為正反關係的的晶體結構,對於材料特性的影響並不大。在氮化鎵中,氮原子的電負 度(electronegativity)和鎵原子的電負度差異很大5,導致氮化鎵鍵(Ga-N bond)就有個偶
極矩(dipole moment)。這種內建極化場(Build-in polarization)的現象在三五族化合物半導 體十分常見,但是影響不大的原因主要可以有以下兩點:一是III 族和 V 族的電負度差 值太小,整體表現上可以忽略它。二是由烏采結構來看,III 族原子必會和 V 族原子形 成一個四面體結構,當 c 軸長度和 a 軸長度比(c0/a0)為 1.63 時6,這個四面體會是一個 正四面體,根據向量和的概念。它的極化場(polarization)總和就會為零。這引申出一個 問題,當晶體並非完美時,內部的極化場總和也會受到影響。一般而言,我們大概把 三五族化合物的極化場分成兩類。 圖 3 氮化鎵磊晶模式示意圖(圖中黃球為鎵原子;藍球為氮原子)
1-2-1 自發極化(spontaneous polarization)
自發極化(PSP)主要是討論在單晶結構上,晶體因結構不對稱且電負度不同所 造成的極化場。氮化鎵的晶軸長度比為 1.625,在沒有外在因素就會有自發極化7 。回 到極化場的基本定義,當一個介電質塊材(dielectric bulk material)有一個淨極化 場時,就表示它內部的電荷分佈是不均的,其電荷的大小可以用(1)式來求出。這就表 示對氮化鎵基板而言,在同一塊基板可能帶來不同的載子濃度。Psp (1)
2010 年,J. Xie, et al.在直徑兩英吋的藍寶石基板上,成長約 20 ㎚的低 溫(~620 ℃)氮化鋁緩衝層後,利用反應式離子式蝕刻(reactive ion etching, RIE) 定義出高電子遷移率電晶體(high electron mobility transistor, HEMT)的源極 (source)與汲極(drain)後,再利用垂直式的有機金屬化學氣相磊晶(metalorganic chemical vapor deposition, MOCVD)成長約 1.3 ㎛的氮化鎵,示意圖如圖 4(a)所示
鎵極性面 氮極性面
4
8
。其中有氮化鋁緩衝層的部分會成長出鎵極性的氮化鎵;而直接成長於藍寶石基板的 氮化鎵會是氮極性,這點可由圖 4(b)酸蝕刻結果來證明。
圖 4 (a)HEMT 示意圖 (b)HEMT 於酸蝕刻後的結果 (c)氧(Oxygen, O)濃度縱深圖 (d)碳 (Carbon, C)和矽(Silicon, Si)濃度縱深圖
J. Xie 指出在他的實驗中,鎵極性和氮極性的氮化鎵為同時成長,應該要有 同樣的載子濃度(carrier concentration),但由圖 4(c)和(d)的二次離子質譜儀 (secondary ion mass spectroscopy)的量測結果來看,卻發現做為施體(donor)的氧 和矽卻都是氮極性區域高出鎵極性氮化鎵,做為主要施體的氧在氮極性氮化鎵上更是 高出鎵極性兩個數量級(order)。
這原因主要在於鎵極性和氮極性氮化鎵的極化方向不同。像是如圖 5一樣的 氮化鎵單位晶胞(unit cell),考慮整體的極化場向量和後會發現無論哪種極性面的氮 化鎵,自發極化場的方向都是由 Ga-terminated layer 指向 N-terminated layer。意 即氮化鎵內部若有自由電子時,會傾向往 N-terminated layer 跑,這導致了氮極性面 的等效電子濃度(effective electron concentration)天生就比鎵極性面高。考慮底 層的緩衝層可能導致電子濃度不一樣。因此我們採用實驗室自產的獨立式氮化鎵基板 針對不同的極性面做了霍爾量測(Hall measurement)。結果如表 1所示:
Polarity Sheet resistance (Ω/
□
) Mobility (cm2/V-s) Concentration (cm-3) Ga-face 2.396 397 -2.621×1017 N-face 1.354 213 -8.665×1017 表 1 針對不同極性面的獨立式氮化鎵基板霍爾量測結果 對於獨立式氮化鎵基板而言,不同極性面只是將同一片樣品反過來做量測。5 若不是內建極化場的影響,就沒有合理的理由能解釋氮極性面的載子濃度高於鎵極性 面。從表 1來看,鎵極性面的載子濃度為2.621×1017cm-3;氮極性面的載子濃度為 2.621×1017cm-3。載子濃度雖有高低差異,但並不會像 J. Xie 的結果一樣,為高出兩個 數量級。 圖 5 氮化鎵單位晶胞與自發極化場方向9
1-2-2 壓電極化(piezoelectric polarization)
壓電效應(piezoelectricity)為外界機械能與電能互相轉換的現象。對晶體 而言,只要壓縮或是擴張晶格相對應的就是改變晶格的內建偶極矩,進而影響電場強 度,反之亦然。在日常生活中最常見到的例子就是裝在喇叭和麥克風上的壓電陶瓷。 由此可知,壓電極化(PPE)在氮化物光電元件上會出現在異質結構(heterostructure)和 晶格受內應力(internal stress)擠壓的情況。 異質結構是常用於提升光電元件效率的方法之一,對 p-n 接面來說,載子和 光子的轉換或是自由載子復合與產生都會在 p 區(p-region)和 n 區(n-region)接面 處,但是對載子來說,要通過空乏區(space charge region)就會造成不少的損失。因 此常利用晶格常數相近的不同材料相接所造成的能隙差來侷限載子能停留在主動區, 使載子生命期延長。對異質結構來看,壓電極化主要產生於不同材料相接,為了讓不 同的晶體能接鍵結在一起,有一方的晶格有所壓縮或擴張而改變了偶極矩。內應力是做磊晶研究的人一個大課題,應力越大,表示成長高厚度塊材的難 度越高。應力的來源有兩個,一是在磊晶過程中,所產生的點缺陷(point defect)、 線差排(line dislocation)和堆疊錯誤(stacking fault)10
,這些缺陷出現除了是磊晶 技術不佳之外,有時是為了平衡整體的晶體成長,這時就會去壓縮或擴張晶格來達到 平衡。二是氮化鎵磊晶多是採用高溫 CVD 系統,晶格常數也隨著溫度變化而擴張,當 溫度降到室溫時,原本在高溫整齊排列的的晶格卻因溫度變化而造成晶格變形,這稱 之為熱壓力(thermal stress)。 Psp Psp
6 整體而言,壓電極化的方向與大小是多種現象競爭的結果。在實質考量上, 內應力隨製程差異的變化較小。線差排密度(dislocation density)隨著不同的磊晶方 法成長,大多落在 107 ㎝-2 的數量級11 ,熱應力的量值也約落在 1.6±0.7 GPa6,12 左右, 幾乎可視為內應力所造成的壓電場為定值,所以一般在討論壓電極化時,以不同材料 相接時所造成的極化場來表示之。 要說明壓電極化場的方向的話,就要從(1)式重新出發,我們看到的電荷不均 不單是自發極化所造成的,故 Psp要修正成 Peff=PSP+PPE,所以(1)式修正如下: Peff ρ PSP PPE (2) 再次回到完美的烏采結構來做討論,晶軸長度比為 1.63。若比值從 1.625 往 1.63 靠近時,表示內部的極化場相互抵消,PSP和 PPE的方向會是相反的。換句話說 c 軸 受到形變拉長或是 a 軸長度受到擠壓都會造成這現象。反之,c 軸長度變短或是 a 軸長 度拉伸就會讓 PSP和 PPE同向。在鎵極性和氮極性的異質結構上,只有 a 軸的長度會變 化,故可以由 a 軸的所受的張或壓應力來評估 PPE的方向。 圖 6 極化方向在鎵極性與氮極性異質結構之示意圖9 有了上述的概念後,我們就可以來解釋圖 6的全部的極化方向。首先 PSP皆為
Ga-terminated layer 指向 N-terminated layer。氮化鋁鎵(AlGaN)的 a 軸長度會介於 aGaN的 3.189 和 aAlN的 3.112 9。圖 6的 a)和 d)考量兩層皆為等大的張應力,所以沒有
PPE的產生。電子分佈由 PSP主導,所以對鎵極性而言,氮化鎵層會感應出正的束縛片電
荷(bound sheet charge),氮極性結構會感應出負的 sheet charge;b)和 e)中 AlGaN layer 受到張應力,所以 AlGaN layer 的 PSP和 PPE同向,整體上會在氮化鎵層有一層正
7 sheet charge,氮極性結構結果會隨之相反;c)和 f)考慮相反情況,為氮化鎵成長在 AlGaN 上,若氮化鎵層受到壓應力,會有相反的極化場。此時感應電荷就由極化場的總 和來判定。一般而言,氮化鎵的壓電極化場會比自發大得多,故結果就會如圖所示。 就以上所述,氮化鎵有許多優點,也有不少的缺點,其中極化對於氮化鎵元件有 一定程度的影響。就目前的研究來看,極性不僅僅影響內部的電子分佈,對於氮化鎵 的電性、物化性、光性都有所影響。
1-3 鎵極性與氮極性氮化鎵基本特性比較
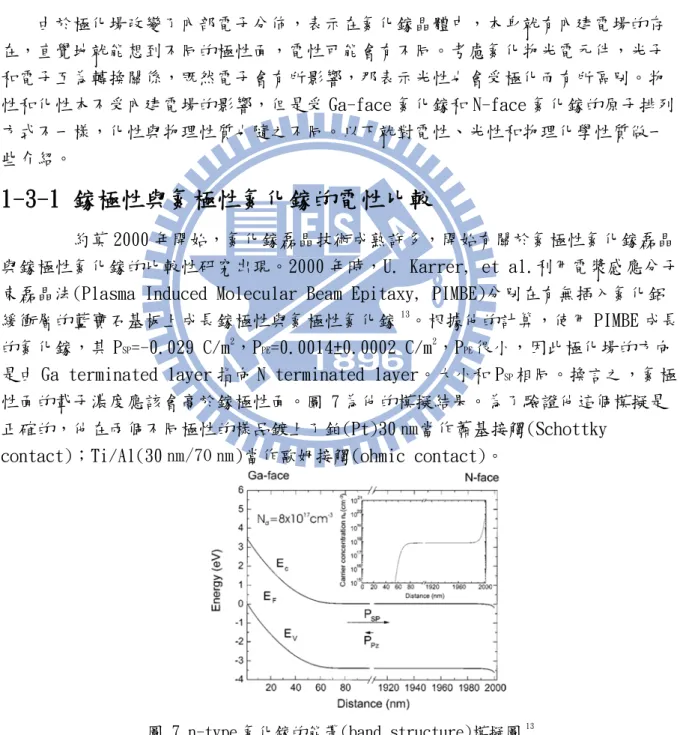
由於極化場改變了內部電子分佈,表示在氮化鎵晶體中,本身就有內建電場的存 在,直覺地就能想到不同的極性面,電性可能會有不同。考慮氮化物光電元件,光子 和電子互為轉換關係,既然電子會有所影響,那表示光性也會受極化而有所區別。物 性和化性本不受內建電場的影響,但是受 Ga-face 氮化鎵和 N-face 氮化鎵的原子排列 方式不一樣,化性與物理性質也隨之不同。以下就對電性、光性和物理化學性質做一 些介紹。1-3-1 鎵極性與氮極性氮化鎵的電性比較
約莫 2000 年開始,氮化鎵磊晶技術成熟許多,開始有關於氮極性氮化鎵磊晶 與鎵極性氮化鎵的比較性研究出現。2000 年時,U. Karrer, et al.利用電漿感應分子 束磊晶法(Plasma Induced Molecular Beam Epitaxy, PIMBE)分別在有無插入氮化鋁 緩衝層的藍寶石基板上成長鎵極性與氮極性氮化鎵13。根據他的計算,使用 PIMBE 成長 的氮化鎵,其 PSP=-0.029 C/m2,PPE=0.0014±0.0002 C/m2,PPE很小,因此極化場的方向
是由 Ga terminated layer 指向 N terminated layer。大小和 PSP相同。換言之,氮極
性面的載子濃度應該會高於鎵極性面。圖 7為他的模擬結果。為了驗證他這個模擬是 正確的,他在兩個不同極性的樣品鍍上了鉑(Pt)30 ㎚當作蕭基接觸(Schottky
contact);Ti/Al(30 ㎚/70 ㎚)當作歐姆接觸(ohmic contact)。
圖 7 n-type 氮化鎵的能帶(band structure)模擬圖13
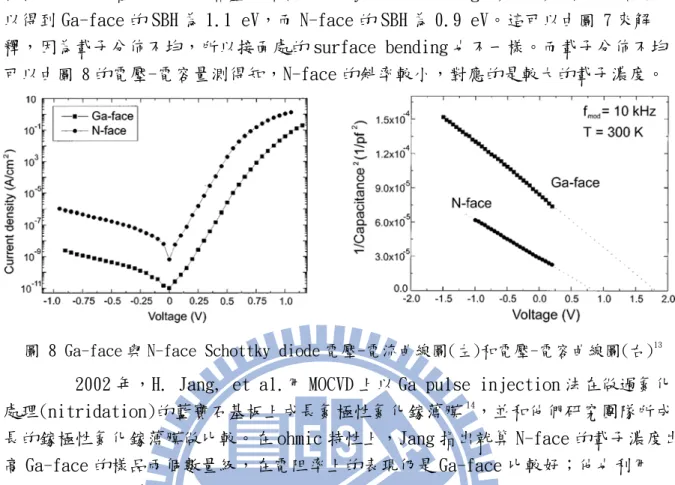
量測結果如圖 8,Karrer 發現 N-face 的樣品,在逆偏 1 伏(voltage)時,漏 電流約大出 Ga-face 的樣品三個數量級。而在順偏情況下,N-face 的樣品的電流爬升
8
較快,可以知道 N-face 的蕭基能障(Schottky barrier height, SBH)比較低,計算可 以得到 Ga-face 的 SBH 為 1.1 eV,而 N-face 的 SBH 為 0.9 eV。這可以由圖 7來解 釋,因為載子分佈不均,所以接面處的 surface bending 也不一樣。而載子分佈不均 可以由圖 8的電壓-電容量測得知,N-face 的斜率較小,對應的是較大的載子濃度。
圖 8 Ga-face 與 N-face Schottky diode 電壓-電流曲線圖(左)和電壓-電容曲線圖(右)13
2002 年,H. Jang, et al.用 MOCVD 上以 Ga pulse injection 法在做過氮化 處理(nitridation)的藍寶石基板上成長氮極性氮化鎵薄膜14
,並和他們研究團隊所成 長的鎵極性氮化鎵薄膜做比較。在 ohmic 特性上,Jang 指出就算 N-face 的載子濃度出 高 Ga-face 的樣品兩個數量級,在電阻率上的表現仍是 Ga-face 比較好;他也利用 Schottky diode 來評估兩個極性的不同,結果如表 2。
表 2 不同極性面的 SBH 與理想因子(ideality factor)14
Jang 的實驗結果和 Karrer 的結果趨勢相同。為了驗證不同極性面真的會有不 同的 surface bending,Jang 利用表面元素分析常用的 X 射線光電子能譜(X-ray photoelectron spectroscopy, XPS)來分析不同極性面的能階情況,結果如圖 9。
圖 9 XPS Ga 3d 能譜14
9 eV。考慮非簡併(non-degenerated)半導體的情況,載子濃度(Nd)與費米能階(EF)可表 示成下式: (3) Jang 將他的實驗結果代入計算後,得到 ≅ 0.1 ,再考慮上 Ga-face 的 SBH 是 1.43 eV,可以畫出如圖 10的示意圖。 圖 10 能帶示意圖。(a)Ga-face,(b)N-face14 圖 10(b)的情況就是 binding energy 的移動,所以造成 SBH 下降了,一般認 為有可能有下列的原因: 1) Fermi level 往高能階移動,表示載子濃度變高,這與前面的討論相同。 2) Fermi level 不動,但受到 polarization 的影響,有一個極化產生的內建 電場。它和原本的內建電場方向相反,所以等效電場變小,空乏區隨之變 小,surface bending 變小。
這也能解釋 Karrer 的結果,和 ohmic 特性的結果來對照,為什麼鋁金屬能在 GaN 上形成好的 ohmic contact。原因就在於 Al 會和 GaN 的氮原子做反應形成 AlN,又 AlN 的 Band 遠大於 GaN。使得接面處有大量的二維電子氣(two dimension electron gass, 2DEG)。在 Ga-face 上,因為極化場,所以 2DEG 會一直往介面跑,但在 N-face 上因為極化場,電子會一直跑離介面,所以反倒在 Ga-face 會有比較好的 ohmic 特 性。 以上的討論,可以對於鎵極性與氮極性的氮化鎵的電性有較深入的見解,後 來的研究也都以此為理論基礎來做討論15 。Karrer 和 Jang 的研究對於薄膜的氮化鎵電 性有所幫助,但是以確定極化的真正影響程度來看,仍有所不足。一為 MBE 和 MOCVD 所成長的薄膜最多在 5 ㎛下,與獨立式氮化鎵基板的厚度(~300 ㎛)有很大的差距,整 體上的影響會較小,還是表現上就有根本上的差異。二是他們的研究都是在兩種不同 緩衝層的藍寶石基板上成長,並不像直接拿氮化鎵塊材來做討論這麼有力,而能做這 樣的研究的團隊也必須要有能力能生產高品質的獨立式氮化鎵基板。
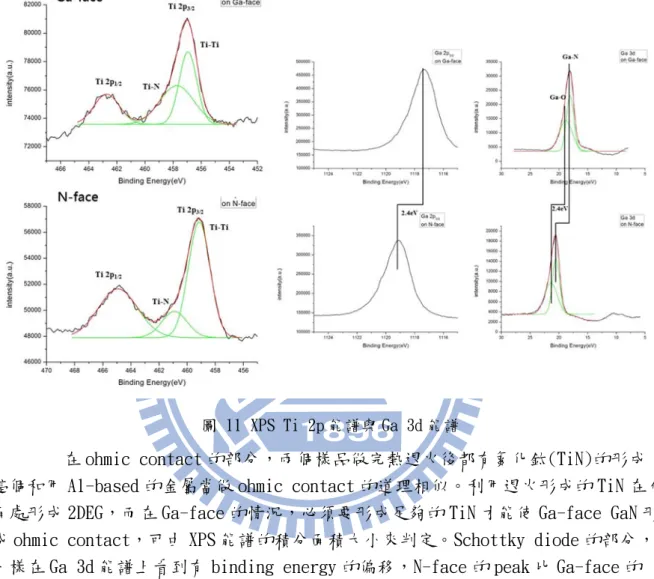
2010 年,交通大學氮化物光電元件實驗室(NCTU NOD lab)的陳則銘等在實驗 室自產的獨立式氮化鎵基板使用鎳(Ni)/金(Au)(50 ㎚/100 ㎚)當作 Schottky contact 和 Ti/Al/Ni/Au(30 ㎚/100 ㎚/50 ㎚/100 ㎚)當作 ohmic contact 對極化對比較性研究。他發
10
現在獨立式氮化鎵基板上,氮極性面的樣品ohmic contact 的形成較容易,接觸電阻 (contact resistance)可以達到 1.8×10-2 Ωcm2,而鎵極性的樣品則必須在氮氣(nitrogen,
N2)環境以750℃以上溫度做熱退火才能有較好的電性表現,但是仍差氮極性樣品的接
觸電阻一個數量級。反之,在鎵極性面上去形成Schottky diode 是比較容易的。這點和 以前的文獻相同,也可以利用XPS 量測來證明。
圖 11 XPS Ti 2p 能譜與 Ga 3d 能譜
在 ohmic contact 的部分,兩個樣品做完熱退火後都有氮化鈦(TiN)的形成, 這個和用 Al-based 的金屬當做 ohmic contact 的道理相似。利用退火形成的 TiN 在介 面處形成 2DEG,而在 Ga-face 的情況,必須要形成足夠的 TiN 才能使 Ga-face GaN 形 成 ohmic contact,可由 XPS 能譜的積分面積大小來判定。Schottky diode 的部分, 一樣在 Ga 3d 能譜上看到有 binding energy 的偏移,N-face 的 peak 比 Ga-face 的 peak 高出了 2.4 eV,雖然這個值比 H. Jang 的結果高出不少,但是卻是比較接近 U. Karrer 的理論 3.5 eV 的13 。 就結果來說,在不同磊晶技術成長的 Ga-face 與 N-face 氮化鎵在極化的討論 可能有些不同,但是趨勢和現象的解釋是大家所認同的。
1-3-2 極化對於氮化鎵的光性影響
根據半導體能帶理論,發光性質應只決定於晶體能隙,不會受極性的影響16 。 如 1-3-1 節所討論的,極化會形成內建電場進而對電子的分佈造成 contact 的品質不 一。對發光二極體來說,有內建電場就表示沒外加偏壓時,能帶就已經歪斜。如此本 來是直接能隙的氮化鎵反而變成非直接能隙,這種將導帶(conduction band)的電子和 價帶(valence band)的電洞各自侷限在能帶不同地方的現象,我們稱之量子侷限史塔11
克效應(quantum confinement stark effect, QCSE),下面將簡單說明之。
`
圖 12 (a)極性氮化鎵, (b)非極性氮化鎵的量子井(quantum well)能帶圖1
以 GaN/InGaN/GaN 的量子井結構來說明,正常的能帶圖應該像圖 12(b)一樣, 電子波函數(wave function)會和電洞波函數上下對稱,不需要聲子(phonon)補助就可 以復合產生光子,但圖 12(a)的能帶因氮化物極化場造成的內建電場而歪斜。量子井內 部載子也受極化場影響傾斜,所以波函數就沒有對稱,導致載子復合效率變差。對於 氮化鎵光電元件來說,要降低 QCSE 的影響最好的方式就是採用半極性(semi-polar)或 是非極性(non-polar)的氮化鎵。 圖 13 氮化鎵的極性面、非極性面與半極性面示意圖 從圖 12(b)來看 QCSE,只要內建電場不是平行磊晶面,就不會有嚴重的波函
12 數分離現象。對於非極性氮化鎵來說,如圖 13(b)和(c),極化場剛好垂直磊晶面,從 一個單位晶胞來看非極性氮化鎵,可以發現磊晶成長面的原子排列永遠為鎵原子和氮 原子各半,這可以便於極性面的判讀,由磊晶面的終止面原子分佈就可以判定晶體內 部的極化大致往哪個方向。 考慮如圖 12一樣的量子井結構,若 PPE很小或是與 PSP同向的話。可以人為地 減低 QSCE 的影響。只要電流方向是和 PSP反向的話,這樣在操作時能帶歪斜反倒會降 低,由此來看,會發現量子井結構必須成長於 N-face 氮化鎵上,這也是氮極性氮化鎵 的磊晶研究一直有團隊在致力研究的一個原因。這種加入電場來減輕 QCSE 的概念,也 已經有團隊實現出來17 。
1-3-3 鎵極性與氮極性氮化鎵物理與化學性質比較
氮化鎵晶體其實十分穩定,摩氏硬度(Mohs hardness)約 6,與石英(quartz) 的硬度相似,可以承受約 19 GPa 外力11
。在化學機械研磨(chemical mechanism polishing, CMP)中的製程中,可以發現鎵極性氮化鎵研磨不容易碎裂,而氮極性氮化 鎵卻很容易碎裂,導致大面積的 epi-ready 氮極性獨立式氮化鎵基板較難取得。研磨 完後,也可以發現相同的製程下,表面的形貌也會有所不同。圖 14為鎵極性和氮極性 獨立式氮化鎵基板研磨後的照片與光學顯微鏡(optic microscope, OM)影像。可以看 到鎵極性樣品會是很整齊的直角,而氮極性樣品卻有很嚴重的邊緣效應,推測是研磨 過程中有蝕刻的現象。從 OM 影像來看可以發現 Ga-face 會有向下凹的刮痕,這在 CMP 製程中十分常見,N-face 卻有類似月球表面的平台。AFM 的表面粗糙度(surface roughness)結果為鎵極性的方均根(root-mean-square, RMS)值為 0.6 ㎚,氮極性的 RMS 為 0.55 ㎚。皆為十分平整表面。 圖 14 鎵極性與氮極性氮化鎵研磨後照片 從化性來說,氮化鎵也是幾乎不會被蝕刻。在 HCl 環境下的爐管中加熱到 800 50 ㎛ 50 ㎛
13
℃,才會有輕微的蝕刻現象 18
。2005 年,D. Zhuang 整理單晶 AlN 與 GaN 在不同酸鹼 溶液下的蝕刻率,如表 3所示: 表 3 氮化鎵與氮化鋁在不同溶液下的蝕刻率(沒標註溫度則為室溫 25℃)19 在實質層面上,考慮有表面缺陷的氮化鎵上,酸鹼溶液會從這些缺陷上攻擊 進而開始蝕刻,原因在於這類缺陷的晶格穩定度低,容易和溶液起化學反應。一般而 言,在濕式處理後,多半會發現鎵極性面的氮化鎵會有凹向下的洞,而氮極性面會有 凸起的角錐出現,如圖 15和圖 16。詳細的現象將在下一節做簡單的介紹。
14
及掃瞄式子顯微鏡(Scanning Electron Microscopy, SEM)影像, (右)穿透式電子顯微鏡 (Transmission electron microscopy, TEM)影像20
圖 16 氫氧化鉀(KOH)蝕刻後形貌。(a)N-face, (b)Ga-face21
1-4 磷酸濕式處理於氮化鎵基板上
如 1-3-3 節所說,氮化鎵化學性質穩定,多數的酸和鹼都無法與之反應。因此晶 圓(wafer)的清洗也相對不容易22 ,無法使用矽晶圓常用的 RCA cleaning 來做清洗。通 常使用有機溶劑和 HCl:H2O(1:1)做清洗即可23。磷酸在氮化物上一般不用來 wafer 清 洗,主要是利用高於 100℃時,磷酸會優先攻擊 dislocation 的特性來評估磊晶品質。 如圖 15和圖 17所示,只要能在有效地攻擊表面缺陷,使之出現 pits,再計算單位面 積有幾個 pits,這類檢定法都稱之為 etching pit density(EPD)。15 EPD 實驗主要目標是在本來看起來沒任何缺陷的表面找出 dislocation,所以對於 溫度和時間皆無特別要求。從表 3可以知道磷酸在低溫時和氮化鎵的反應不佳,一般 而言會希望 EPD 實驗能操作在 220℃左右的溫度24 。因為磷酸的沸點約在 160℃,所以 常使用硫酸:磷酸為 3:1 來當作實驗條件。 磷酸蝕刻的結果,基本上和 1-3-3 節說的相似。在鎵極性面會有六面的凹洞24 ,在 氮極性面上,會出現十二面的角錐。對於判定氮化鎵的極性面,這也是一個方便快速 的方法。
1-5 實驗動機與目的
對於元件磊晶或是打線封裝,為了確保不會有任何氧化物或是其它問題干擾後續 製程。多半會在進爐管前做濕式清潔或是電漿處理8 。以 ohmic contact 的例子來說 明,多半的研究都知道不同的溶液對於後續的 contact 有影響25 ,但是對於原因大多只 是不明確的推測,為了讓 contact 的品質固定,常在不同的退火條件下在做實驗。本 論文的目標就是想從磷酸處理是否可以去除表面氧化物,且評估為何不同的溶液可以 讓電性出現這些差異。 氮化鎵在目前在研究上有個有趣的主題,就是在表面上形成角錐,然後在角錐上 去成長量子井或是量子點(quantum dot)的結構26 。由於角錐的斜面為 semi-polar 面, 所以就不需要用 non-polar 或是 semi-polar 的氮化鎵當作基板。對獨立式氮化鎵基板 而言,氮極性面只要經過磷酸處理過後,就會在表面出現十二面的角錐,不需要使用 (epitaxial lateral overgrowth, ELOG)或是感應耦合電漿離子蝕刻(inductively coupled plasma reactive ion etching, ICP-RIE)的技術。相對而言是個相當方便的 製程,所以了解磷酸在氮極性面如何去形成這個形貌為本論文的重點。16
第二章 實驗流程與機台介紹
2-1 實驗流程與參數設計
圖 18 實驗基本流程 圖 18 為氮化物光電元件實驗室的樣品產出流程。所有的樣品皆是在用 MOCVD 成長 2 ㎛氮化鎵薄膜的 c-plane sapphire 磊晶,因為這只是取得獨立式氮化鎵基板的一個 暫時基板,所以我們稱這個為 template。磊晶的主要機台為本實驗室自組式的氫化物 氣相磊晶(hydride vapor phase epitaxy, HVPE)機台。完成 HVPE 磊晶後,我們稱這 種 GaN on sapphire 的架構為厚膜(thick film),接著會做雷射剝離(Laser-lift off, LLO)將 bulk GaN 與 sapphire 分離。這樣的 bulk GaN 其實並不平整,無法在上 面成長元件架構或是晶體特性研究,所以會利用 CMP 將表面研磨至平整。若 bulk GaN 的翹曲(bowing)太大的話,則在研磨製程前會先利用 ICP 或是濕式處理將 bowing 降低 再開始研磨。完成後,我們這稱這個 epi-ready 的氮化鎵基板為獨立式氮化鎵基板 (free-standing GaN)。 實驗就是在這樣的流程下取得的獨立式氮化鎵基板做磷酸處理,全部的條件皆為 150℃的磷酸環境,根據不同的實驗需來更改浸泡時間。結果與討論的第一部分是在鎵 極性面在做表面分析的討論,接著第二部分是在不同品質下的氮極性獨立式氮化鎵基 板做蝕刻形貌與機制的討論,條件如表 4。 表 4 氮極性獨立式氮化鎵基板蝕刻實驗設計17
2-2 機台介紹
由 2-1 節可知,所有的實驗結果皆是在獨立式氮化鎵基板上。如何去產出這樣的 基板就十分重要,以下將對圖 18中的 HVPE 系統、LLO 系統和 CMP 系統做一些簡單的介 紹。2-2-1 氫化物氣相磊晶
獲得 bulk GaN 的方法普遍來說有以下幾種方法:HVPE、high pressure nitrogen solution growth、波蘭的 Ammono 公司所提出的 ammonothermal 法和鈉流 法,在此不詳細 HVPE 以外的磊晶方法,以表 5簡單地說明各法的優劣。 表 5 各式氮化鎵基板磊晶優劣比較 HVPE 早先應用在砷化鎵材料的成長。而在氮化鎵單晶厚膜的應用上,由於 Ga 與 NH3無法直接形成 GaN,所以主要是利用氯化氫氣體(HCl)與液態鎵金屬(Ga) 反應生 成氯化鎵(GaCl),GaCl 再與氨氣(NH3)反應生成氮化鎵沈積到基板上。其主要反應式為 (4)和(5)式,示意圖如圖 19所示。
2HCl(g) + 2Ga(melt) ↔ 2GaCl(g) + H2(g) (>850 ) (4)
GaCl(g) + NH3(g) ↔ GaN(s) + HCl(g) + H2(g) (1000 ~1100 ) (5)
18
反應的主要產物是 GaN,磊晶源(Source)是五族的 NH3,跟三族的 HCl 與 Ga 形
成的 GaCl。HVPE 跟 MOCVD 的成長方式類似,都是有相同的五族來源-NH3,而不同的是 三族的磊晶源。在 HVPE 是使用液態鎵與 HCl 合成 GaCl,而在 MOCVD 使用的則是三甲基
鎵(Trimethylgallium, TMGa)或者是三乙基鎵(triethylgallium, TEGa)這類有機金屬化
合物來做成長。因為五族磊晶源容易揮發,所以 HVPE 或 MOCVD 在成長的時候,五族與 三族流量比(V/III ratio)都遠大於 1。 HVPE 磊晶過程中,除氮化鎵之外,尚有其他副產物生成,反應式如下: HCl(g) + NH3(g) → NH4Cl(s) (6) GaCl(g) + HCl(g) → GaCl3(g) + H2(g) (7) GaCl3(g) + NH3(g) → GaN(s) + 3HCl(g) (8) 由(6)(7)(8)式可知副產物包括 NH4Cl(s)、GaCl3(g)以及 HCl(g)等,其中氯化銨為白 色粉末,容易大量沉積在反應爐管的末端管壁和管口上而造成腔體真空度下降或是微 塵吸附器堵塞等副作用;而 GaCl3(g)亦可和氨氣反應生成氮化鎵,但由於 GaCl3(g)在高溫 下為極度不穩定的狀態,故非生成氮化鎵的主要反應。
以 HVPE 成長 GaN 的最主要優點就是 HVPE 擁有極高的成長速率(~100μ
m/hr),對於成長厚膜來說相當有利;而其長晶的品質亦可接受,故對於成長獨立式氮 化鎵基板是最適合的一種長晶方式。而外觀上的缺點主要就是表面平整度不佳,容易 長出六角形結構的小丘(Grain),但是此一情況可藉由後續的研磨拋光來獲得平整度較 高的表面。 在本論文中,獨立式氮化鎵基板皆是利用本實驗室所自行組裝的水平式氫化 物氣相磊晶機台所成長的。本實驗室的 HVPE,大致可區分為:高溫爐管、氣體供應系 統、溫控系統、真空系統和冷卻水循環系統等,如圖 20。 圖 20 水平式氫化物氣相磊晶系統 高溫爐管前端是連接氣體供應系統,而末端則連接氣壓計和真空系統。其中 爐管的材質為石英,由於安全考量所以最高操作溫度我們設定為 1100℃。石英管外圍 是以碳化矽棒來加熱升溫爐管,外圍並覆蓋絕熱性佳的氧化鋁(aluminum oxide,
19 Al2O3)保溫磚,因此具有極高的升溫效率。石英管兩端與不鏽鋼架連接,而接縫處則使 用抗酸且耐高溫的氟化橡膠(viton)材質的 O 型環(O-ring)封住以保持爐管的真空度 氣體供應系統經由外管、上管、下管和前管四條管路將氣體送入高溫爐管, 其中前管固定通入低流量的氮氣,用以保護位於爐管末端的氣壓計(pressure gauge) 免於被實驗產生的微粒阻塞。而外管、上管和下管三條管路則將氣體輸送入高溫爐管 前端。外管通入較大量的氮氣以穩定整個爐管的氣體流場,並且避免基板因上下氣壓 差而吹飛。上下管分別通入實驗所需之反應氣體,上管可選擇性的通入氮氣、氫氣 (hrydrogen, H2)和 HCl 氣體,而下管則可通入 N2、H2、NH3和 HCl,主要是看實驗內容
來調整氣體的成份。所有氣體流量皆以電子式質量流量控制器(electronic mass flow controller, MFC)控制和調整,共計有 9 個 MFC。
溫控系統控制高溫爐管六個區域的溫度,分別以六支白金探頭之 k 型熱電耦 (k-type thermal couple)偵測。區域 I 設定 HCl 和金屬鎵(gallium, Ga)反應的溫度 —850℃。區域 II 至 V 為實驗反應進行區,設定 GaN 磊晶溫度。以四個區域控制溫 度,能使反應區可大範圍地維持溫度均勻及穩定;區域 VI 並非反應區且靠近抽氣區, 因此溫度通常固定設為 950℃。
真空系統由一台機械幫浦(mechanical pump)進行抽氣。爐管的氣壓值由位於 爐管末端的氣壓計(pressure gauge)偵測,配合幫浦節流閥(throttle valve)開合的 程度來達到控制及穩定氣壓。由於實驗操作在高溫並會產生微粒,因此在爐管與幫浦 中間須連接一冷卻過濾器(chiller)和微粒吸附器(particle trap),前者用於降低來 自高溫氣體的溫度,後者則過濾實驗微粒。 冷卻水循環系統分佈於爐管前末端、chiller 和 trap,目的使高溫氣體降 溫,避免機台的零組件因高溫而損壞。
2-2-2 雷射剝離
雷射剝離技術 1998 年由 W.S.Wong, et al.應用在光電元件上,他們利用雷射 由藍寶石基板面入射,而大部分的雷射能量被吸收於氮化鎵緩衝層與藍寶石基板界面 處,最後此界面於雷射能量的照射下,溫度可高達 1000℃,在此熱能之作用下,氮化 鎵將被分解為氮氣及金屬鎵,因此達到氮化鎵與藍寶石基板剝離的目的。 圖 21 LLO 原理由於氮化鎵能隙 3.4 eV,可以吸收 360㎚附近的光。為了將 thick film 可以 完全的剝離,故採用 Nd:YAG laser 以三倍頻的方式得到波長 355 ㎚的雷射光。剝離
20 時,會將樣品升溫至 1070℃,並以螺旋的方式從外側向樣品中心打,以防止樣品破 裂,機台架構如圖 22。 圖 22 雷射剝離系統
2-2-3 化學機械研磨
化學機械研磨,最早是在 1980 年代中期由 IBM 公司提出的概念,是使用與待 磨物會產生化學反應的研磨粒子和液體進行研磨拋光,圖 23為化學機械研磨示意圖。 過程中這些反應,將研磨粒子以摻和加工液,進行拋光加工,當試片與研磨粒子的接 觸點,局部因摩擦能量發生高溫、高壓,在接觸時間內,隨著兩者之間的相互影響產 生反應。然後,這個反應部位由於摩擦力,而漸漸以微小單位量被去除,而進行作 用。 圖 23 化學機械研磨示意圖 基本上,使用較高硬度研磨粒子的場合,可以發現是較有效率的拋光加工。21 在對力學作用依存的拋光加工情況下,硬度小的研磨粒子加工時,其有效率地加工較 低。故可以整理移除厚度(removed thickness, ∆t)為(9)式, 是移除系數, 為樣品 所受壓力,∆ν為轉速變化。 ∆t ∆ν (9) 然而考慮晶圓和酸鹼溶液之間化學反應,研磨粒子的選擇,對研磨粒子的硬 度就沒有依存性存在,可以得到特別高的效率。因此,在反應拋光作用中,是可以使 用比受拋物材料軟的研磨粒子。由於在接觸點研磨粒子側會變形,因此由於粒子的壓 入、拉伸作用,所以不會造成刮痕或機械損傷層。另外,化學機械拋光法的主要特 徵,是研磨粒子介於受拋物表面與拋光盤之間,施與表面過大的能量使研磨粒子變形 破壞,而吸收能量,所以,不一定需要使用軟質的拋光盤。其中影響 CMP 製程的變數 包括有:研磨置具所施的壓力與晶圓的平坦度、晶圓與研磨墊的旋轉速度、研磨漿與 研磨顆粒的化學成份、溫度、以及研磨墊的材質與磨損性等等。 實驗室使用的研磨機台為正越公司之 M-15 機型,配有鑽石液噴灑系統、拋光 液控制系統,搭配四種磨盤及真空治具和空壓機,如圖 24所示。 圖 24 化學機械研磨系統
2-3 量測機台介紹
在本論文中,會使用到原子力顯微鏡、x 光繞射分析儀、,下面逐一介紹各機台的 原理與量測分析2-3-1 原子力顯微鏡
在 1982 年早期,由 IBM 公司蘇黎士(Zurich)研究實驗室的兩位科學家 Gerd Binning 和 Heinrich Rohrer 利用原子之間的穿隧效應(tunneling effect)發明掃描穿
22
隧顯微鏡(Scanning Tunneling Microscope,STM)。雖然 STM 的發明解決了物理學中 的很多問題,不過 STM 能分析的材料只局限於導體或半導體。在 1986 年由 IBM 公司的 Binnig 與史丹佛大學的 Quate 所開發原子力顯微鏡發明後,利用探針針尖和欲測試樣 本間凡得瓦爾力的強弱,得知樣本表面的起伏高低和幾何形狀,且樣本可為導體或非 導體,AFM 成功解決 STM 在材料上的限制。圖 25為原子力顯微鏡的簡單示意圖。 圖 25 原子力顯微鏡示意圖 (資料來源:科榮股份有限公司訓練資料) AFM 除了擁有其他表面分析儀器無法達到的原子級解析度能力外,其量測的環 境可不必在真空的環境中直接可在大氣中或者在液相中,因為這兩項的特點其應用性 就更加的廣泛了。AFM 之探針一般由成份為 Si 或 Si3N4懸臂樑及針尖所組成,針尖尖端 直徑介於 20 至 100 nm 之間。主要原理是藉由針尖與試片間的原子作用力,使懸臂樑 產生微細位移,可以測得表面結構形狀,如圖 26。AFM 操作模式主要可區分為接觸式 (contact)、非接觸式(non-contact)及間歇接觸式或稱為輕敲式(intermittent contact or tapping)三大類操作。特性分述如下: 圖 26 原子距離與斥力、引力關係圖 (資料來源:http://web1.knvs.tp.edu.tw/AFM/ch2.htm) 1.接觸模式(Contact Mode) :
探針與樣品交互作用力為排斥力,利用定力模式(Constant Force Mode)維持 探針與樣品間的交互作用力為定值。系統回饋電路讓偵測訊號維持在原先的設定 值。當偵測訊號大於或小於設定值時,回饋電路會以誤差值訊號轉成驅動掃描器 的電壓訊號,掃描器以延展或收縮的動作,使探針與樣品間的作用力維持在設定 值。在掃描成像過程中,探針容易刮傷樣品。
23 2.非接觸模式(Non-Contact Mode) : 探針與樣品交互作用力為吸引力,其操作模式是將探針的懸臂以一振幅振 動,當探針與樣品間產生交互作用力時懸臂的振幅會衰減,此振幅衰減的大小與 交互作的梯度有關,即可得到樣品表面形貌。頻率與相位也可做為回饋訊號。在 掃描成像過程中,探針不接觸樣品減少對樣品的傷害,在大氣中解析度易受水膜 影響。 3.輕敲模式(tapping mode): 探針和樣品問的距離介於接觸式和非接觸式,即 0.5 至數十奈米之間,探針 微懸臂樑振動的振幅較大,在振幅最低點的位置,探針恰敲擊試件表面。敲擊模 式和非接觸模式的操作方式類似,屬於動態量測。其成像模式是採定振幅方式, 當探針與樣品問產生交互作用力時,微懸臂樑的振幅會增加或衰減,利用壓電掃 瞄平台的上下調整可得到定振幅,因此得知樣品表面形像,解析度較非接觸模式 的解析度高。
2-3-2 x 光繞射分析儀
1913 年 W.L.Bragg 父子在晶體結構分析實驗中,從散射 X 光的分佈情 形,他們認為可以將繞射現象視為入射光被晶面反射。此種反射如同鏡面反射,入射 角等於反射角。在某些散射角下,從相鄰晶面散射之波彼此相位相同,光程差為波長 的整數倍,因而產生建設性干涉。滿足此條件便可產生繞射,稱為布拉格定律 (Bragg's law)如圖 27。 圖 27 布拉格繞射示意圖27 不同的晶體結構晶面間距(d)會有所差異,其中 hkl 是各晶面之米勒指數 (Miller index),因此會有不同組合之繞射角(2θhkl)。藉由 θ-2θ 掃描,可以得到 晶體的晶格常數,並可進一步做應力的分析。本實驗所使用的量測方式為 ω-real scanning,亦稱為 x 射線搖擺曲線(Rocking Curve, XRC)量測,其量測方式為固定 2θ 角,藉由微動 ω 軸來看磊晶品質。當晶體為一個完美單晶時,只會在一個 θ 值有繞 射,其它地方應皆無訊號,為一個 delta function,所以若 XRC 為一個高斯分佈,則 可以利用半高寬來評估晶體品質。繞射的發生除了必須滿足布拉格條件外,也會受晶 體對稱性影響。當晶胞內所含原子數目不只一個時,由於這些原子彼此的對稱關係, 而限制了某些繞射的發生,稱為消光條件。所以當 X 光照射晶體時,只有在某些特定24
的入射角才會出現繞射波,這主要是決定於晶胞的形狀、大小及對稱性。
2-3-3 x 射線光電子能譜儀
X 射線光電子能譜一般又稱作電子能譜化學分析儀(electron spectroscopy for chemical analysis, ESCA),利用 X 光光束照射固態表面,若 X 光能量大於內層 Z 軌域電子束縛能(EB),則該 Z 軌域的電子可被游離成為自由電子,此即是光電子,如 圖 28所示。 圖 28 x-ray 激發光電子示意圖28 偵測器可量測光電子(photoelectron)的動能,並從而推算該光電子的束縛 能,一般而言,可以表示成: (10) 因此可以應用於辨識發射光電子的元素種類和化學鍵結情形。由於是以 X 光 激發光電子,所以此光電效應稱之為 X 光光電子能譜,如果入射光是使用紫外光(UV light,4< hν <50 eV),則稱之為深紫外光光電子能譜量測。 X 光光電子能譜術之訊號主要來自於光電子,以光電子能譜圖的能量值進行元 素分析,以強度分析元素濃度,以化學位移分析狀態。
2-3-4 霍爾量測
在 1879 年,霍爾(Edwin H.Hall)利用於導體中導入電流,將導體置於外加磁 場中量測霍爾電壓(Hall Voltage)來判斷傳導載子的極性與濃度,稱為霍爾效應(Hall effect)。此方式廣泛的被利用於半導體中參雜載子性質的量測上。 以 n-type 半導體為例,於材料左右端施加一偏壓,因此材料內的載子會順著 偏壓流動,此時再施加一與載子流動方向垂直之磁場 B,流動中的載子因受磁力(F=qVd ×B)的影響,而產生一與偏壓及磁場方向皆垂直的電壓 VH,此電壓即為霍爾電壓。此時 可藉由量測電壓之正負與大小,並推得半導體的參雜載子性質、濃度與電子遷移率 等。 一般來說,在半導體薄膜上,不是使用霍爾量測來判定載子濃度的,而是利25
用 Van der Pauw 法來量得載子濃度,基本概念與霍爾量測相同,但是是分別在薄膜表 面的四點 contact 上分別加入電流訊號與電壓訊號,藉由改變磁場來測出載子濃度, 片電阻…等。
2-3-5 拉曼量測
單色光進入另一介質傳播,若傳播方向產生變化,稱為散射。若散射光的能 量與入射光相同,則稱為雷利散射(Rayleigh scattering),表示能量無耗損,此為彈 性散射。拉曼散射為 C. V. Raman 於 1928 年發現了與入射光不同的散射光,此乃光 子與分子發生了非彈性碰撞,光子散射後除行進方向改變且有能量的轉換,如。 圖 29 拉曼散射示意圖 散射光子能量減少其頻率變小,且形成一個聲子並使分子振動,假若分子裡 含有數種振動的能階,就會產生數種頻率位移的拉曼光譜線。拉曼散射所測量的光子 能量變化以波數(wave number)表示,拉曼位移與能量差的關係為: E(雷射光能量) – E'(散射光能量) = ΔE/hc = Raman shift (11) 拉曼光譜最主要是用來量測倒晶格中心(zone center)的聲子震動模態,對於 烏采結構的氮化鎵材料,其單位晶胞屬於空間群(space group)的對稱結構。由於動量 守恆定律,一階拉曼散射是指波向量(wavevector)為零,也就是在Γ點。由群論 (group theory)分析預測在Γ位置所不能縮減的表示量:
Γ =2A1+2E1+2B1+2E2 (12)
其中,一個 A1 與一個 E1 是聲子震動模態(acoustic),所以只有六種是光學
震動模態(optical):A1+E1+2B1+2E2。而其中兩個 B1模態無論是在拉曼或是紅外光譜都
是不活耀的(silent mode),A1與 E1對於拉曼與紅外光譜都是活耀的(active mode)。
對於 E2 模態而言,其對拉曼光譜最為活耀。因長距離庫侖電場的作用,會使 A1和 E1模
態分裂成 TO 和 LO 兩種模式,TO 表示橫向光學振動模式(transverse optical vibrational mode);LO 模式代表縱向光學振動模式(longitudinal optical vibrational mode),故全部共有六種拉曼活躍的振動模態:A1(LO)、A1(TO)、 E1(LO)、E1(TO)、E2(low)、E2(high)。
26
2-3-6 掃瞄式電子顯微鏡
掃描式電子顯微鏡原理與光學顯微鏡相似,僅是由電子替代了光子;因為入 射電子的物質波波長較可見光波長短,所以 SEM 可得到較高的解析度。此外,SEM 試片 製備方式容易、檢測速度快,並且空間解像能力很好、景深長,可以顯示清晰的三度 空間影像,因此 SEM 被廣泛地應用於各材料領域。SEM 的操作為利用加熱燈絲(thermionic emission)或強電場發射(field emission)的方式使電子槍產生高能電子束,再藉由電磁透鏡系統在試片上聚焦,並以 電磁偏折線圈控制電子束的行進方向在試片上,作二度空間的掃描。
電子與試片交互作用會產生各種電子和光子訊號,如圖 30:歐傑電子(Auger electrons)、二次電子(secondary electrons)、背向散射電子(backscattered electrons)、吸收電子(absorbed electron)、陰極螢光(cathode luminescence)和特 徵 X 射線(X-rays)等。偵測器測得這些訊號後,可用來分析試片材料的特性。二次電 子和背向散射電子可作為 SEM 的成像訊號,其中以二次電子為最主要;二次電子是試 片表面被高能電子束重擊後釋出的電子,是能量較低的訊號(能量約 50eV),因此只有 距離試片表面深度約 50-500Å 範圍內產生的二次電子才有機會逃離試片表面而被偵測 到,所以二次電子訊號對於試片的表面非常敏感,並且訊號產生的數量會受到試片表 面凹凸起伏的影響,所以二次電子帶有表面形貌的訊息,可用以觀察試片表面的形 貌。另一方面,背向散射電子是高能電子在試片內部受原子核彈性散射作用,形成大 角度散射之後再逸出表面的電子。在散射過程中並無太多的能量損耗,所以對表面平 整的試片來說,背向散射電子影像不會有明顯的對比,除非試片材料成份是屬於不同 的原子序,因為背向散射係數隨原子序增加而增大,故可用以觀測不同元素的組成相 和成份元素分佈情形。由於背向散射電子影像的陰影對比效果良好,故能充分反應試 片表面凹凸形狀,但因其屬於高能量電子(其能量約等於入射電子的能量),所以偵測 到的背向散射電子可產生於距試片表面約 5000Å 的深度範圍內;由於訊號產生位置較 深,所以訊號產生後須在試片內部行進一段距離後才能脫離試片而被偵測到,因此背 向散射電子影像解析度較差。 圖 30 電子與試片的交互作用29
27
第三章 實驗結果與討論
3-1 獨立式氮化鎵基板於磷酸處理後的表面分析
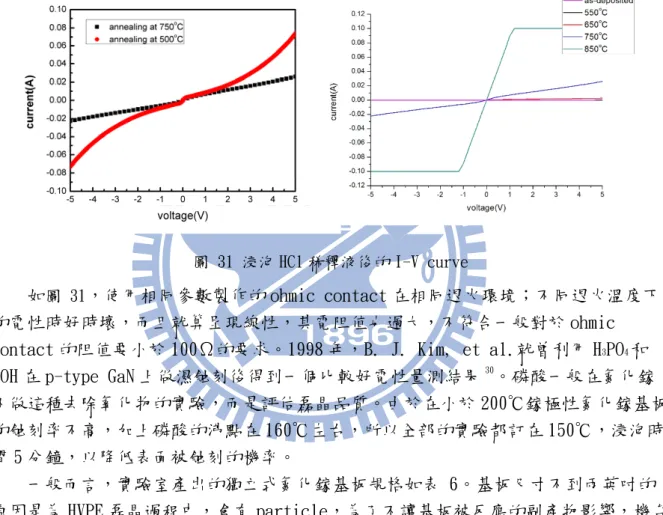
傳統上,移除 native oxide 會用加熱到 50℃的 HCl:H2O=1:1 稀釋液,是因為它
表現出來的電阻率最低,可以小於 10-5 Ω㎝2 。25 但是結果顯示這個配方在我們自產的 獨立式氮化鎵基板並不好,而且品質不夠穩定。 圖 31 浸泡 HCl 稀釋液後的 I-V curve 如圖 31,使用相同參數製作的 ohmic contact 在相同退火環境;不同退火溫度下 的電性時好時壞,而且就算呈現線性,其電阻值也過大,不符合一般對於 ohmic contact 的阻值要小於 100Ω的要求。1998 年,B. J. Kim, et al.就曾利用 H3PO4和
KOH 在 p-type GaN 上做濕蝕刻後得到一個比較好電性量測結果30
。磷酸一般在氮化鎵 不做這種去除氧化物的實驗,而是評估磊晶品質。由於在小於 200℃鎵極性氮化鎵基板 的蝕刻率不高,加上磷酸的沸點在 160℃左右,所以全部的實驗都訂在 150℃,浸泡時 間 5 分鐘,以降低表面被蝕刻的機率。 一般而言,實驗室產出的獨立式氮化鎵基板規格如表 6。基板尺寸不到兩英吋的 原因是為 HVPE 磊晶過程中,會有 particle,為了不讓基板被反應的副產物影響,機台 設計為磊晶面向下。 Thickness ~330μm Size 1.5 inch Roughness ~1.5 nm XRC FWHM(002) <120 arcsec 表 6 獨立式氮化鎵基板規格
28
由圖 32的 AFM 圖來看,做完磷酸處理的樣品的 RMS 變大了,但是從紅線處切出剖 面圖來看,其實扣除刮痕的地方仍是平整,而且蝕刻前後的 RMS 值皆在我們的基板規 格內。
圖 32 獨立式氮化鎵基板在磷酸處理後的 AFM 圖
從 x 射線搖擺曲線(x-ray rocking curve, XRC)來看,磷酸處理前的半高寬(full width half maximum, FWHM)為 112 arcsec;處理後的 FWHM 為 102 arcsec。考慮前後 量測的不會在同一點上量測,可以視為以 150℃浸泡 5 分鐘的條件下,磷酸處理並不會 對獨立式氮化鎵基板的表面有任何的改變。
圖 33 獨立式氮化鎵基板在磷酸處理後的 XRC 圖
接著我們利用 XPS 對沒做過 CMP 研磨的 bulk GaN(圖 34和圖 35中的
29
34的 acid treatment 和圖 35的 H3PO4 treatment)做表面分析。根據文獻,氮化鎵的
導帶會在鎵原子的 4s 軌域;價帶會在氮原子的 2p 軌域1 ,所以在做 XPS 表面分析時, 為了避免外界的雜訊都會去掃能階較深的 Ga 2p 能譜和 N 1s 能譜,如圖 34(左)和 (右)。為了能評估氧化物的影響,也針對 O 1s 能譜去做量測。 圖 34 XPS Ga 2p 能譜(左), N 1s 能譜(右) 圖 35 XPS O 1s 能譜 一開始,從 O 1s 能譜開始討論。因為在氮化鎵中,氧原子只有兩個作用,一是磊 晶過程中作為 donor 自然混入的氧原子,來源主要是因為我們採用的是石英爐管、載 台(holder)和藍寶石基板作為 template,這些石英製品被認為在高溫中會有氧原子獲 得能量往氮化鎵摻雜的能力31 ;二是 HVPE 機台的特性。一般來說,磊晶機台會要求操 作時在接近真空(~10-6 torr)讓環境不會有其它原子混入讓磊晶品質不佳;或是在高壓 操作,此時就算爐管不是完全的氣密,也因為內部壓力高於外部壓力而不會跑進去雜 質。對 HVPE 系統來說,它僅需要使用機械幫浦來抽氣,真空度大約在 10-3 torr,只要 一分鐘不會上升超過 0.2 mbar 的氣壓,我們就認定機台的真空度正常。再由圖 36可 以知道,HVPE 系統的磊晶趨勢,在圖 36的左半部為垂直成長速度較快的 3D 成長模 式;在圖 36的右半邊屬於水平成長速度較快的 2D 成長模式。一般而言,希望得到足 夠厚,且表面較平整的氮化鎵基板,所以在實驗室的 HVPE 磊晶參數多半會接近常壓的 700 torr,溫度由 950℃升到 1050℃,載氣為氮氣與氫氣的混合氣體來成長。在這樣 的情況下,腔體內的氧氣和水氣含量其實是不夠低的,也就造成了氮化鎵基板天生為 n-type 的基板,這點可以從表 1的霍爾量測結果看到。這也表示,在 XPS O 1s 能譜
30 中,它的訊號是相對單純好分析的,只有受到載子濃度和表面的氧化物影響。 圖 36 溫度、壓力、五三比和載氣對長晶模式的影響趨勢 首先,從沒有做過任何研磨的 Bulk GaN 的 O 1s 能譜來看,如圖 37所示。由於在 XPS 中,強度表示單位時間內收到的計數,所以氧元素的濃度越高,對應的強度越高。 所以在做數值擬合(fitting)時,必須扣除掉背景訊號的值。圖 37的 fitting 結果為 531.82 eV,背景訊號的強度大約都維持在 27 萬 5 左右。 圖 37 沒研磨過的氮化鎵基板的 XPS O 1s 能譜
一般認為,最常出現在氮化鎵表面的 native oxide 為氧化鎵(Ga2O3),它的束縛能
多半認為在 529.8 eV 到 532.0 eV32,33
。由此可以很合理的判定,出現在獨立式氮化鎵 基板上的 native oxide 為 Ga2O3。將三種樣品放在一起比較,如圖 37。可以發現未做
研磨的樣品的 oxide 的訊號最強,做完 CMP 研磨的強度其次,最弱的為做過磷酸處理 的樣品。原因大致為 thick film 在做 LLO 時必須升溫到 1070℃,使表面的氧化物大量 增加。在 CMP 的製程中,我們採用 2M 的次氯酸鈉(NaOCl)和檸檬酸的混合溶液去氧化 氮化鎵基板,由於溶液為弱酸性,就可以把表面被氧化的氮化鎵用絨布墊移除掉,藉 以降低研磨造成的細刮痕,所以做完 CMP 製程的獨立式氮化鎵雖然也會有表面氧化 物,但是含量就比未做 CMP 製程的低。從強度的比值來看,做完磷酸處理的樣品的 peak intensity 只有獨立式氮化鎵基板的 51.37%,考慮 binding energy 可以是個區 間,改由積分面積來算的話,訊號強度更只佔了沒做過磷酸處理的獨立式氮化鎵基板
31 的 13.03%,積分面積這個結果雖然很好,但是在此並不認為真的有減少這麼多的氧含 量,由 Ga 2p 能譜的計算結果來看的話,大概只減少了 35.63%的比例。再來是由圖 35 的背景訊號來看,三個樣品的背景訊號完全不同,利用積分面積來計算只會把比例拉 得很誇張。至於背景訊號的不同,只能猜測是因為我們的機台並沒有旋轉的功能,所 以除了厚度不均勻之外,載子濃度的分佈也應該要不均勻才是。 N 1s 能譜一般上不認為和氧原子會有強烈的反應,在這邊僅做為一個 binding energy 峰值偏移的參考標的。 Ga 2p 能譜上,意外地發現有兩個 peak 分別落於 1120 eV 與 1145 eV 的區間。一 般認為接近 1120eV 的峰為氮化鎵鍵(Ga-N bond)34
和氧化鎵(Ga-O bond)35
的混合訊號, 1145 eV 那組 peak 無文獻指出為何出現這個訊號,考慮不太可能是 Ga-N bond 和 Ga-O bond 混成的訊號在獨立式氮化鎵基板上完全分開了,所以只針對 1120 eV 這個區間做 討論。圖 34的趨勢比較合乎一般對於化學機械研磨的認知,研磨的過程並不會大幅地 增加氧化層的厚度,取而代之的是會有一層損害層36,37
。圖 35和圖 34的 Ga 2p 能譜看 似差異很大,但是如前所述,O 1s 能譜僅代表電子濃度和表面氧化物的訊號,但是 Ga 2p 能譜必須為 Ga-N bond 與 Ga-O bond 的混成,若差異十分大,就表示氮化鎵基板內 部的組成十分絮亂,那基板所量到的 XRC 半高寬就不會達到<120 arcsec 這個值。 結論上,利用 XPS 評估磷酸去除獨立式氮化基板的表面氧化物的能力約可以把氧 化物移除 30-50%的含量。
3-1-1 磷酸處理對於獨立式氮化鎵基板的影響
有趣的是,我們在對作 Ga 2p 能譜做 fitting 時,發現獨立式氮化鎵基板的 峰值在 1118.63 eV;磷酸處理後的樣品峰值在 1119.46 eV,相比之下,binding energy 往高能階偏移 0.83 eV,根據(10)式,我們可以畫出XPS Ga 2p 量測能帶示意圖: 圖 38 XPS Ga 2p 量測能帶示意圖 考慮這三片樣品都是在取自同一片樣品,它們的 2p 軌域應該要在相同位置, 所以可以推論做完磷酸蝕刻後,樣品的 Fermi level 上升了,這也表示樣品中的等效 電子濃度變大。Binding energy 往高能階偏移的情況一樣可以由 N 1s 能譜與 O 1s 能 譜中看到。根據利用 XPS 在氮化鎵電性量測的分析經驗來看,比較有可能的是做完磷32
酸處理後,造成了氮空缺(nitrogen vacancy, VN)。一般認為氮空缺是屬於
donor-like 的缺陷,原因在於 Ga-N 的一組結構就已滿足原子八隅體的規則。氮空缺即表示這 樣一組 Ga-N 的氮原子不見了,相對來說多了三個電子,與其去抓住 5 個電子來維持八 隅體,不如就把三個電子釋出,這時就可以將其視為 donor。這點其實亦有研究提及, 有氮空缺的氮化鎵基板,其 valence band maximum(VBM)會比沒有氮空缺的高出約 0.7 eV10
。VBM 的定義為 EF到 EV的差值,因為為相同來源,2p 軌域與 EV的位置當然相同,
這也就是表示 Fermi level 提高了 0.7 eV,與我們量測結果相似。
3-1-2 磷酸蝕刻機制於鎵極性氮化鎵基板上
由氮空缺的想法出發,可以來推測為什麼磷酸只會在鎵極性面的 dislocation 做蝕刻,而其它地方不會有蝕刻的現象。我們猜想,磷酸在接近沸點時,會解離成氫 離子和磷酸根,如(13)。 H3PO4 → 3H+ + PO43- (at 150℃) (13) 一般來說,酸鹼溶液能蝕刻氮化鎵的話,皆是認為溶液中的陰離子扮演重要 的角色,如 KOH 蝕刻中的氫氧根離子38 和 HCl 蝕刻中的氯離子39 。在此,我們假設氫離 子才是磷酸蝕刻的主要反應,而氫原子對於氮化鎵的氮原子有極高的攻擊性是眾所皆 知的40 ,這樣磷酸蝕刻就可以用圖 39說明之。 圖 39 鎵極性面氮化鎵磷酸蝕刻機制 在圖 39中,我們猜想磷酸解離後,氫原子會去和氮原子反應成 NH3,磷酸根 則不做任何反應。如此,磷酸蝕刻的反應式就可以寫成(14)式 GaN + H3PO4 → Ga3+ + PO43- + NH3↑ (14) 會考慮如圖 39的蝕刻機制主要有兩點,一是磷酸根的分子量 95,是個非常大 的分子團,想和氮原子反應就必須擠進兩個鎵原子中間才有機會和氮原子反應,二是 磷酸根為主要反應,對在鎵極性面氮化錚而言,Ga-terminated layer 習慣會是在最外33 層裸露的,所以有大量的 dangling bond,相對於氮原子是五族元素,磷酸根應該更傾 向和三族的鎵原子反應才是。反過來想若不是磷酸根為主要反應,那一切就比較合理 了。對於氫原子來說,不僅是容易和氮化鎵的氮原子反應而已。因為分子量才 1,要擠 進氮化鎵的晶格就相對容易許多。再者,氫離子和有 dangling bond 的鎵原子都是帶 正電,本來就會互斥。這也能解釋會什麼在表 3的磷酸蝕刻率會是 0。以這樣的想法 回到實驗設計,只要是如圖 40一樣,有 point defect 的話,那氫原子和氮原子反應 完全,就有可能會將反應停止,此時就會造成 VN。 圖 40 有 point defect 的鎵極性氮化鎵基板
3-1-3 KOH 蝕刻機制於氮極性面氮化鎵基板上
在此,心中其實已有一個很簡單的假設了,若是氫原子是磷酸蝕刻中的主要 反應,然後造成氮空缺,應該可以從其它溶液的蝕刻得到相同的驗證才是。 一般而言,要判斷氮化鎵是鎵極性還是氮極性並不容易,比較精確的方法是 用 TEM 的 convergent beam electron diffraction(CBED)繞射圖形來確定極性41,另 一種就是利用 KOH 判斷極性,KOH 對鎵極性並不會蝕刻,在氮極性上會蝕刻出如圖 41 的形貌。
圖 41 氮極性氮化鎵基板做完 KOH 蝕刻後的形貌21
一直以來,KOH 的蝕刻機制是比較明確的。2001 年,Dongsheng Li, et al. 就在他的研究中說明了 KOH 溶液對於氮極性氮化鎵蝕刻的流程38 。 首先,KOH 並不扮演蝕刻的角色,而是一個催化劑。它會使水解離成 H+ 和 OH -。OH -會和鎵原子反應形成 Ga2O3或是相關的氧化物,會溶於酸鹼溶液中,接著露出