7126 Macromolecules 1992,25, 7126-7134
Synthesis and Characterization
of
Ferroelectric Liquid Crystalline
Polysiloxanes and Polymethacrylates Containing
[
(S)-2-Methyl- 1-butoxylphenyl
4-(Alkyloxy) biphenyL4’-carboxylate Side Groups
Chain-Shu Hsu,’ Jhy-Horung Lin, a n d Ling-Rong Chou Department of Applied Chemistry, National Chiao Tung University, Hsinchu, Taiwan 30050, ROCGing-Ho Hsiue
Department of Chemical Engineering, National Tsing Hua University, Hsinchu, Taiwan 30049, ROC
Received May 18, 1992; Revised Manuscript Received September 8, 1992
ABSTRACT The synthesis of side-chain liquid crystalline polysiloxanes and polymethacrylates containing 4- [ (S)-2-methyl-l-butoxy]phenyl4-hydroxybiphenyl-4’-carboxylate moieties as mesogenic units and aliphatic spacers containing respectively three to eleven methylene units is presented. Differential scanning calorimetry, optical polarizing microscopy, and X-ray diffraction measurements reveal chiral smectic mesomorphism for all polymers. Both polysiloxanes which contain respectively six and eleven methylene units in the spacers exhibit smectic A, chiral smectic C, and smectic B phases. Among the polymethacrylates prepared in this study, the polymethacrylate containing eleven methylene units in ita spacers is the only one revealing smectic A and chiral smectic C phases. The results seem to demonatrate that the tendency toward chiral smectic C mesomorphism increases with increasing spacer length, and the thermal stability of the chiral smectic C mesophase is determined by the flexibility of the polymer backbone.
Introduction
In 1975 Mayer’ presented theoretically and then proved experimentally that the chiral smectic C (Sc*) mesophase
was
ferroelectric.A
bistable,fast
switching, electrooptical device which uses the ferroelectric liquid crystals (FLCs)was demonstrated a few years later by Clark and Lager-
well? An increasing interest in the synthesis of low molar mass SC* liquid crystals has since then developed. Nu- merous FLC compounds and room-temperature mixtures have so far been prepared forfast electrooptical appli-
cations.Besides low molar mass FLCs, several side-chain liquid crystalline polymers (LCPs) exhibiting a Sc* mesophase have recently been reported.s-26 Ferroelectric properties, e.g. spontaneous polarization in these polymers, have also been provided in some cases.1J2716-25 This field has been reviewed by LeBarny and D ~ b o i s . ~ ’ The detailed struc- ture-property relationship of
Sc*
LCPs, however, has not been very clear up until now. This has been due to the limited experimental data reported in the literature.Presenting the synthesis of some chiral smectic liquid crystalline polysiloxanes and polymethacrylates containing 4- [ (S)-Bmethyl- 1-butoxy] phenyl 4- (alkyloxy) biphenyl-4’- carboxylate side groups is the goal of this study. Their characterizations by differential scanning calorimetry, optical polarizing microscopy, and X-ray diffraction have been presented.
Experimental Section
A. Materials. Poly(methylhydrosi1oxane)
(Un
= 2270) and divinyltetramethyldiiloxane platinum catalyst were obtained from Patrarch System Inc. and used as received. (S)-(-)-2- Methyl-1-butanol, [ ( r I a 6 ~ = - 6 . 5 O (from Merck), 4-hydroxybi- phenyl-4t-carboxylate (from Tokyo Kaisei Co.), and all other reagents (from Aldrich) were used as received. Toluene used in the hydrosilation reaction and 1,3-dioxane were first refluxed*
To whom all correspondence should be addressed.Scheme I
Synthesis of Monomers IM-VM H , C - C H ~ C H , ~ B ~ + HO-WH
KOH / EtOH
H,C
-
CH(CH,OW
COOHn = 3 , 4 , 6 , 6 , 1 1 I M - V M
over sodium and then distilled under nitrogen. 2,2’-Azobisieo- butyronitrile (AIBN) (from Fluka) was freshly recrystallized from methanol.
B. Techniques. lH NMR spectra (300 MHz) were recorded on a Varian VXR-300 spectrometer. FT-IR spectra were measured on a Nicolet 52OFT-IR spectrometer. Polymer aamplea were cast as films on a KBr tablet for the measurement. Thermal transitions and thermodynamic parameters were determined by using a Seiio SSC/52OO differential scanning calorimeter equipped with a liquid nitrogen cooling accessory. Heating and cooling rates were 10 OC/min. Thermal transitions reported were collected during the second heating and cooling scans. A Nikon
Macromolecules,
Vol.
25,No.
26, 1992 Polysiloxanesand
Polymethacrylates 7127 Table ICharacterization of Monomers IM-VM
monomer yield, % [(uIz5D 400-MHz lH NMR (CDCls6, ppm)
IM 77 +4.087 0.88 (t, 3 H, -CHz-CH3), 0.96 [d, 3 H, -CH(CH3)-], 1.21-1.52 (m, 2 H, -CHz--CH3), 1.86 [m, 1 H, -CH(CH+], 3.72 [m, 2 H, -0CHz-CH(CH3)-1,4.54 (d, 2 H, CHz-OPh), 5.03 (m, 2 H, CH&, 5.80 (m, 1 H, =CH-), 6.86-8.17 (m, 12 aromatic protons) 0.88 (t, 3 H, CHZ-cH3), 0.96 [t, 3 H, -CH(CH3)-], 1.19-1.53 (m, 2 H, -C&+H3), 1.80 [m, 1 H, -CH(CH3)-], 2.52 (m, 2 H, -CHz-CH=), 3.73 [m, 2 H, -OCHz-CH(CH3)-1, 4.01 (t, 2 H, -CHZ-OPh), 5.10 (m, 2 H, CH&, 5.88 (m, 1 H,
=CH-),
6.86-8.17 (m, 12 aromatic protons) -CH(CH3)-CH2-], 2.20 (m, 2 H, -CHz-CH=), 3.72 [m, 2 H, -OCHz-CH(CH3)-], 3.97 (t, 2 H, -CHZ-OPh), 4.99 (m, 2 H, CH&, 5.81 (m, 1 H,=CH-),
6.86-8.17 (m, 12 aromatic protons) 0.88 (t, 3 H, CHZ-CH~), 0.95 [d, 3 H, -CH(CH3)-], 1.14-1.81 [m, 7 H, (-CHz-)z and -CH(CH3)-CH2-], 2.07 (m, 2 H, -CHz-CH=), 3.72 [m, 2 H, -OCH~-CH(CHS)-], 3.81 (t, 2 H, -CHZ-OPh), 5.10 (m, 2 H, C H p ) , 5.86 (m, 1 H,=CH-),
6.86-8.16 (m, 12 aromatic protons) 0.86 (t, 3 H, -CHz-CH3), 0.94 [d, 3 H, -CH(CH3)-], 1.18-1.80 [m, 17 H, (-CH2-), and -CH(CH3)-CH2-], 2.03 (m, 2 H, -CHz--CH=), 3.91 [m, 2 H, -OCH2--CH(CH3)-], 4.11 (m, 2 H, CHZ-OPh), 4.90 (m, 2 H, C H p ) , 5.76 (m, 1 H, =CH-), 6.90-8.14 (m, 12 aromatic protons) IIM 74 +4.553IIIM 75 +4.470 0.88 (t, 3 H, CH2-C&), 0.96 [t, 3 H, -CH(CH3)-], 1.17-1.89 [m, 5 H, 4 H . r and
IVM 71 +4.967
VM 43 +4.220
Table I1
Characterization of Compounds VIA-VIIIA
compd yield, % mp, OC 400-MHz 1H NMR (CDC13,6, ppm) VIA VIIA 40 51 136.8 122.3 1.01 (t, 3 H, -CH&H3), 1.07 [d, 3 H, -CH(CH3)-1,1.29-1.62 (m, 2 H, CHz-CHd,
0.87 (t, 3 H, -CH2-CH3), 0.94 [d, 3 H, -CH(CHa)-I, 1.27-1.56 [m, 10 H, -CH&H3 and (-CHr)41,
0.88 (t, 3 H, -CHzCH3), 0.95 [d, 3 H, -CH(CH3)], 1.23-1.52 [m, 20 H, -CH&H3 and (<&-)e],
1.91 [m, 1 H, -CH(CH3)-], 2.31 (m, 2 H, -CHz-), 3.83 [m, 2 H, -O-CHz-CH(CH3)-1, 3.95 (t, 2 H, -CH&Ph), 4.24 (t, 2 H, HO-CHz-), 6.97-8.28 (m, 12 aromatic protons) 1.74 [m, 1 H, -CH(CH3)-], 3.58 (t, 2 H, -CHZ-OPh), 3.71 (m, 2 H, -OCHZ-CH(CH~)-I, 3.95 (t, 2 H, HO-CH2-), 6.84-8.16 (m, 12 aromatic protons)
1.74 [m, 1 H, -CH(CHa)-I, 3.56 (t, 2 H, C H A P h ) , 3.71 [m, 2 H, -OCHzCH(CH3)-1, 3.95 (t, 2 H, HO-CH2-), 6.85-8.15 (m, 12 aromatic protons)
VIIIA 77 113.7
Table 111
Characterization of Monomers VIM-VIIIM
monomer vield. 5% laI2% 400-MHZ 'H NMR (CDCb, 6, DDm)
VIM 55 +5.10 0.86 (t, 3 H, -CH2-cH3), 1.01 [d, 3 H, -CH(C&)-l, 1.21-1.90 [m, 3 H, -CH(CH~)+HZ-I,
1.94 (s,3 H, =(C-)-CH3), 2.20 (m, 2 H, -CH~-CHZ-CHZO-), 3.75 [m, 2 H, -OCHz--CH(CH3)-1, 6.92-8.23 (m, 12 aromatic protons)
0.88 (t, 3 H, -CHz-CH3), 0.98 [d, 3 H, -CH(CH3)],1.22-1.83 [m, 11 H, (<&-)4 and -CH(CH3)-CH2-],
1.87 (8, 3 H, =(C-)-CH3), 3.85 [m, 2 H, -OCH2-CH(CH3)-],3.93 (t, 2 H, -CHZ-OPh-),
4.10 (t, 2 H, -COO-CHz-), 5.47 and 6.02 (d, 2 H, CH&, 6.85-8.20 (m, 12 aromatic protons) 1.91 (s,3 H, =(C-)-CH3), 3.76 [m, 2 H, -0CH~-cH(CH3)-],3.98 (t, 2 H, -CHZ-OPh-),
4.11 (t, 2 H, -COO-CHT), 5.51 and 6.07 (2 d, 2 H, CH&, 6.90-8.21 (m, 12 aromatic protons) 4.12 (t, 2 H, CHZ-OPh), 4.35 (t, 2 H, -COO-CHr), 5.57 and 6.11 (2 d, 2 H, CH&,
VIIM 41 +3.12
VIIIM 60 +4.55 0.93 (t, 3 H, -CH2-CH3), 0.99 [d, 3 H, -CH(CH3)], 1.21-1.83 [m, 21 H, (-CHz-)s and -CH(CH~)--CHZ-],
Microphot-FX optical polarized microscope equipped with a Mettler FP 82 hot stage and a F P 80 central processor was used to observe the thermal transitions and to analyze the anisotropic textures. Preparative gel permeation chromatography (GPC) was run on a Waters 510 LC instrument equipped with a 410 differential refractometer and a preparative GPC column (22.5 mm X 60 cm) supplied by American Polymer Standard Co. X-ray diffraction measurements were performed with nickel-filtered
Cu
Krr
radiation with a Rigaku powder diffractometer. Optical rotations were measured a t 25 OC on a Jasco DIP-140 polarimeter with chloroform as solvent for all compounds.C. Synthesis of Monomers. The synthesis of olefin mono- mers IM-VM for the hydrosilation reaction and methacrylate monomers VIM-VIIIM is outlined in Schemes I and 111. (S)- 2-Methyl-1-butyl tosylate, 10-undecen-1-yl tosylate, 4-bromo- 1-butene, 5-bromo-l-pentene, and 6-bromo-1-hexene were syn- thesized according to literature procedures.avm The optical rotation, [ a ] % ~ of (S)-2-methyl-l-butyl tosylate is +4.6O.
44 (S)-2-Methyl-l-butoxy]phenyl Benzyl Ether. p-(Ben- zyloxy)phenol(20.8 g, 0.104 mol) was added to a stirred solution of KOH (5.9 g, 0.104 mol) and KI (1.0 g) in 250 mL of 95% ethanol. (S)-2-Methyl-l-butyl toeylate (25.2 g, 0.104 mol) was added when dissolution was complete. The solution was refluxed for 3 h and cooled to room temperature. The solvent was then removed in a rotovap. The residue was washed with water and extracted with diethyl ether. The ether layer was dried over
anhydrous MgS04, fiitered, and evaporated to dryness. The crude product was recrystallized from methanol to yield 23.9 g (85% )
of white crystals; mp = 33.6 "C. lH
NMR
(CDC13,6, ppm): 0.90- 1.01 (m, 6 H, CHs-), 1.18-1.63 (m, 2 H, -CHz-), 1.82 (m, 1H,
>CH-), 3.70 (m, 2 H, -CHZOPh), 5.00 (8, 2 H, Ph-CH2-OPh), 6.81-7.42 (m, 9 aromatic protons).
4-[(S)-2-Methyl-l-butosy]phenol. Sodium (18.4g,0.80 mol) was added rapidly but in small pieces to a hot solution of 4-[(S)- 2-methyl-1-butoxylphenyl benzyl ether (21.6 g, 0.80 mol) in anhydrous t-BuOH (150 mL). The solution was heated to reflux for 20 h. A small amount of cold water was added after the sodium had all reacted. This was followed by the addition of a cold, dilute hydrochloric acid solution. The t-BuOH was removed in a rotovap, and the residue was extracted with diethyl ether. The collected diethyl ether solution was washed with water, dried over anhydrous MgSO4, and then evaporated to dryness. The obtained product was purified by column chromatography (silica gel, ethyl acetateln-hexane = 1:lO as eluent) to yield 13.35 g (92.7%) of white crystals; mp = 45.6 OC; [a]%D = -8.44'. NMR (CDCl3,6, ppm): 0.89-0.98 (m, 6 H, CH3-),1.18-1.56 (m, 2 H, -CHz-), 1.80 (m, 1 H, >CH-), 3.69 (m, 2 H, -CH20Ph), 6.01
(s, 1 H, -OH), 6.71-6.77 (2d, 4 aromatic protons).
4-(Allyloxy)biphenyl-4'-carboxylic Acid, 4-(3-Buten-l- ylosy)biphenyl-4'-carbosylic Acid, 4-(4-Penten-l-ylosy)- biphenyl-4'-carbolylic Acid, 4-(6-Hesen-l-yloxy)biphenyl- 4'-carboxylic Acid, a n d 4 4 10-Undecen- 1-y1oxy)biphenyl-
7128 H s u e t a l . Macromolecules, Vol. 25, No. 26,1992 I \I 1 8 1 1 3 0 5 1 - 1 ” 50 1 o n I 5 0 200 2 5 0 0 Temp ( “ C l
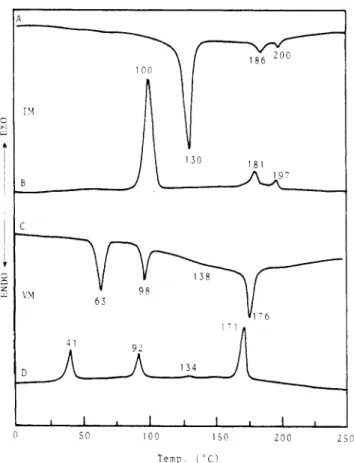
Figure 1. DSC thermogram (10 OC/min): (A) IM,second heating scan;
(B)
IM, cooling scan; (C) VM, second heating scan;(D)
VM, cooling scan.
Table I V
Phase Transitions and Phase-Transition Enthalpies for Monomers IM-VM
phase transitions, “C (corresponding enthalpy changes, kcal/mol)
monomer no
-+%-
K 130 (3.89) SA 186 (0.37) N* 200 (0.14) I I 197 (0.11) N* 181 (0.38) SA 100 (4.08) K IM K 131 (4.56) SA 187 N* 194 (0.84) I I193 (0.14) N* 191 (0.54) SA 92 (3.79) K IIM IIIM 5 K 139 (5.98) S A 183 (0.22) N* 200 (1.64) I I199 (0.67) N* 195 (0.67) SA 101 (2.89) K K 116 (5.25) S A 193 (1.26) I IVM 6 I 187 (1.48) SA 109 (0.71) SB 86.6 (0.66) K K 63 (0.93) SB 98 (0.61) Sc’ 138 ( - ) b SA 176 (1.56) I I171 (1.34) S A 134 (-)* Sc* 92 (0.61) S, 41 (1.05) K VM 11a n according to Scheme I. Enthalpy is very small.
4’-carboxylic Acid.
All
five compounds were prepared by the etherification of alkenyl bromides or undecenyl tosylate with 4-hydroxybiphenyl-4’-carboxylic acid. The synthesis of 4-(allyl- oxy)biphenyl-4‘-carboxylic acid is described below.4-Hydroxybiphenyl-4’-carboxylic acid (6 g, 0.028 mol) was added to a solution of
KOH
(3.76 g, 0.067 mol) and KI (0.2 g) in90% ethanol (150 mL). The solution was refluxed for 1 h, and allyl bromide (4.56 g, 0.034 mol) was added dropwise. The resulting solution was refluxed for 20 h and cooled to room temperature, and 100 mL of water was added. The solution was acidified with dilute hydrochloric acid. The precipitate was filteredandrecrystallizedfromaceticacidtoyield 4.76g (66.9%) of white crystals; mp = 232.9 O C .
lH
NMR (CDCls, 6, ppm): 4.60Scheme I1
Synthesis of Polysiloxanes IP-VP
7%
+ Me,%- Of ,?i-
Oj-SiMe,
Hn = 3 , 4 , 5 , 6 , 1 1 IP-VP
Table
V
Thermal Transitions and Phase-Transition Enthalpies for Polvmers IP-Vp
phase transitions, O C (corresponding enthalpy change, kcallmrub)
heating polymer na cooling 3 g 25 SA 117 (1.82) I IP I99 (1.80) SA IIP 4 g 18 SA 84 (0.78) I I80 (0.42) S A 5 g 22 SB 99 (0.89) S A 212 (0.86) I I204 (0.68) SA 89 (0.82) SB IIIP IVP 6 g 16 SB 120 (1.73) Sc* 166 (-)‘SA 244 (1.24) I I236 (0.98) S A 164 (-)‘Sc* 109 (1.47) SB g 20 S, 109 (0.78) Sc* 143 (-)’ SA 218 (0.86) I I210 (0.89) S A 142 (-)‘Sc* 103 (0.74) S,
VP
11a n a c m r d i i to Scheme 11.
*
mru = mole repeating unit. e Enthalpyis very small.
(d, 2
H,
-CHz-),5.40 (m, 2H,
CHF), 6.08 (m, 1H,
=CH-), 6.98-8.16 (m, 8 aromatic protons).4-[ (5)-2-Methyl-l-butoxy]phenyl 4-(Allylo.y)biphenyl- 4’-carboxylate (IM), 4-[ (S)-2-Methyl-l-butoxy]phenyl4-(3- Buten-l-yloxy)biphenyl-4’-carboxylate (IIM), 4-[(5)-2- Met hy 1- 1-butoxy ] pheny 1 4- (4-Penten- 1 -ylo.y)biphenyl-4’-
carboxylate (IIIM), 4-1 (s)-2-Methyl-l-butoxy]~he~14-(5- Hexen-l-yloxy)biphenyl-4’-corboxyla~ (IVM), and
&[(e-
2-Methyl- l-butoxylphenyl4-( 10-Undecen-l-ylo.y)biphenyl- 4’-carboxylate(VM).
All
five olefin monomers(IM-VM)
were prepared by the same method. The synthesis of monomerIM
is described below.
4-(Allyloxy)biphenyl-4’-carboxylic acid (1.524 g, 0.006 mol) was reacted a t room temperature with excess thionyl chloride (4 mL) containing a few drops of dimethylformamide in methylene chloride (7 mL) for 2 h. The solvent and exceaa thionyl chloride were removed under reduced p r w u r e to give the corresponding acid chloride. The product was dissolved in 20 mL of methylene chloride and slowly added to a cold solution of I-[(S)-a-methyl- l-butoxy]phenol(l.O8 g, 0.006 mol) and triethylamine (1.67 mL) in 100 mL of methylene chloride. The solution was stirred at
Macromolecules, VoI. 25,
No.
26, 1992VP
Polysiloxanes and Polymethacrylaten 7129
3 . 7 8 A " ( A ) s m e c t i c A 170°C 1 9 . 4 1 A " 4 . 6 5 A " 6 . 0 3 A " ( B l s m e c t i c C' 120°C 4 . 6 0 A ' I 8 . 9 8 A ' (Cl s m e c t i c B 80°C 4 . 4 7 A " 1 9 . 4 I A " ( D ) s m e c t i c B 25'C 4 . 4 4 A ' I I I I I I 0 5
i n
1 5 2 0 2 5 30 Figure 2. Temperature-dependent X-ray measurements for polymerIVP
a t (A) 170 "C,(B)
120 "C. (C) EO "C, and(D)
25"C.
roomtemperature. Thesolvent wasthendistilled. The obtained crude product was dissolved in methylene chloride and passed through silica gel. The solvent was removed in a rotovap. The productwaa recrytallizedfromamixtureofmethanoland benzene to yield 1.93 g (77.2%) of white crystals. The yields, melting points. optical rotations, and
'H
NMR chemical shifts of all synthesized monomers have been summarized in Table I.4-[(S)-2-Methyl-l-butoxy]phenyl 4-[(3-Hydroxyprop-l- yl)oxy]biphenyl-4'-carboxylate (VIA), 4-[(S)-2-Methyl-l- bntoxy]phenyl4-[(6-Aydnyhex-l-yl)oxy]biphenyl-4'-car- boxylate (VIIA), a n d 4-[(S)-2-Methyl-l-butoxy]phenyl 4-[(ll-Hydroxyundec-l-yl)oxy]biphenyl-4'-carboxylate (VIIIA). All three compounds were prepared by the same method. The synthesis of compound VI11 A is described below. Into a dry three-neck flask equipped with a thermometer, a pressure-equilibrated dropping funnel, and a reflux condenser fitted with a nitrogen adaptor and connected to a silicon oil bubbler was placed a solution of 4-[(S)-2-methyl-l-butoxylphenyl 4-(l0-undecen-l-yloxy)hiphenyl-4'-carhaylate (1.32g, 2.5 mmol) in dry THF (6 mL). 9-BBN (6 mL, 3.0 mmol), 0.5 M solution in THF, was charged via a syringe into the dropping funnel and then added dropwise into the stirring olefin solution. Themixture
was oxidized after the additon was completed. A solution of 3 N NaOH (1 mL) was injected into the flask, followed by 3 mL of 30% H202 solution, which was added dropwise over 15 min.
20 u A 2um B C
Figure3. Opticalpolarizingmicrographa displayed by Ivp: (A) SA texture obtained after cooling to 198.2 "C;
(B)
Sc* texture obtainedcoolingto 161.8'C; (C) Sstertureohtainedaftercooli~ to 98.0 "C.The reaction mixture was stirred for 1 additional h and poured into water. The precipitated product was filtered, washed with water, dried, and finally recrystallized from toluene to yield 1.05 g (77%) of white crystals. The yields, melting points, and
'H
NMRchemical shifts of compounds VIA-VIIIA are summarized in Table 11.
4-[(S)-2-Methyl-l-hutoxy]~henyl 4-[(3-Methacryloyl- prop-l-yl)oxy]biphenyl-4'-carboxylate (VIM), 4-[(@-2- Methyl-l-butoxy]phenyl4-[(6-Methacryloylhex-l-yl)oxy]- biphenyl-4'-carboxylate (VIIM), a n d 4-[(S-Z-Methyl-l- hutoxy]phenyl4-[ (1 1-Methacryloylundee-l-yl)olylbipbenyl- 4'-carboxylate (VIIM). All methacrylate monomers VIM- VIIIM weresynthesized hytheesterification ofthe corresponding alcohols VIA-VIIIA, with methacryloyl chloride. An example of their synthesis is as follows. Compound VIA (1.26 g, 2.90 mmol) wasdissolvedinamixtureofdriedTHF (2mL)and triethylamine
7130
Hsu
e t a l . Macromolecules, Vol. 25,No.
26, 1992t
1 1 I Iremp i = c 1
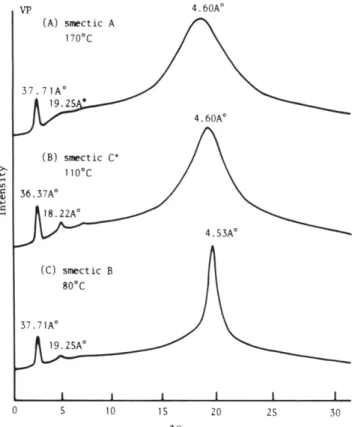
Figure 4. DSC thermogram of VF' (10 W m i n ) : (A) second heating scan; (B) cooling scan.
W 4.60.4'
B
S IO IS 2Q 25 30
1 0
Figure 5. Temperature-dependent X-ray measurements for polymer VF' at (A) 170 "C, (B) 110 "C. and ( C ) 80 " C .
(0.5 mL, 3.3 mmol). The resulting solution was cooled in an ice-water bath to 0 "C, and methacryloyl chloride (0.25 mL, 3.19 mmol) was added dropwise. The reaction mixture was allowed to warm slowly to room temperature and then stirred overnight. After the solution was poured into water, the precipitated product was filtered, dried under vacuum, and purified by column chromatography (silica gel, chloroform aa eluent)toyield0.8g(55%)ofwhitecrystals. Theyields,optid rotationsand'H NMRchemicalshiftsofmonomers VIM-VIIIM are summarized in Table 111.
D. Synthesis of Polysiloranes IP-W. The synthesis of liquid crystalline polysiloxanes is outlined in Scheme 11. Ageneral synthetic procedure is described below.
The olefin derivative, 0.8 g (10 mol % excess versus the Si-H groups present in polysiloxane), was dimolved in 80 mL of dry,
freshlydistilled toluenetogetherwith the proper amountof poly-
C
Figure 6. Optical polarizing micrographs displayed by
VP:
(A) SA texture obtained after cooling to 180.9 OC; (B) Sc* texture obtained after cooling to 134.3 OC; (C) Ss texture obtained after cooling to 106.5 "C.(methylhydrosiloxane). The reaction mixture was heated to 110 OC under nitrogen, and 100 pg of divinyltetramethyldisiloxane platinum catalyst was tehn injected with a syringe as a solution in toluene (1 mg/mL). The reaction mixture was refluxed (110 OC) under nitrogen for 24 h. After this reaction time the FT-IR analysis showed that the bydrosilation reaction was complete. The polymers were separated and purified by several reprecig itations from tetrahydrofuran solution into methanol, further purified by preparative GPC, and then dried under vacuum.
E.
Synthesis of Polymethacrylates VIP-VIIIP. The radical polymerizations of the monomers were carried out in Schlenk tube containing the dioxane solution of the monomer (lo%, wtivol) and the initiator (AIBN, 1 w t % VB monomer) was f i t degassed by several freezepump-thaw cyclesundervacuum and then filled with argon. All polymerizations were carried out at 60 O C for 15 h. After the polymerization time, the polymersMacromolecules, Vol. 25, No. 2S,
1992n - 3 , 6 , 1 1
Polysiloxanes and Polymethacrylates 7131
VIM-VIIW
Scheme I11
Synthesis of Polymethacrylates VIP-VIIIP
1) 9
-
BBN /THF2) KO, I O H
n = 3 , 6 , 1 1
Table VI
Phase Transitions and Phase-Transition Enthalpies for
Monomers VIM-VIIIM
~~
phase transitions,
"C
(correspondingenthalpy changes, kcal/moi)
heating monomer no cooling
VIM
3K
81 (6.65) SB 86 (0.58) SA 147 (1.07) I I 145 (1.01) SA 85 (0.55) SB 34 (4.08)K
VIIM
6K
101 (1.97) SB 123 (0.58) SA 149 (0.82) 1 I142 (0.97) S A 119 (0.53) SB 96 (2.00) KK
88 (1.45) Sc* 125 (-Ib SA 169 (0.66) II
165 (0.61) S A 120 (-)b Sc* 81 (0.63) SB 65 (0.07) KVIIIM
110 n according to Scheme 111. Enthalpy is very small.
were precipitated into methanol, fiitered, and purified by reprecipitation from THF solutions into methanol. The po- lymerization results have been summarized in Table VII.
Results and Discussion
The synthetic route used for the preparation of
4-[(S)-
2-methyl-l-butoxy]phenyl4-(alkenyloxy)biphenyl-4'-car-
boxylates (IM-VM) is outlined in Scheme
I.
The chiral group was inserted into these mesogenic compounds starting with the commercially available (-)-2-(S)-methyl- 1-butanol.This
was done by asequence of reactions which avoided ita recemization. The monomers IM-VM were characterized by differential scanning calorimetry and optical polarizing microscopy. Representative DSC traces of monomers IM and VM are presented in Figure 1. Monomer IM exhibits a melting transition at 130 "C, a smectic A to cholesteric phase transition a t 186 "C, and a cholestericto
isotropic phase transition a t 200"C
on the heating scan (curve A). Crystallization temperature is more supercooled on the cooling scan (curve B) than the other two phase-transition temperatures. Curves C and'IIIP 8 . 8 5 A " ( A ) smectic A 1 6 0 ° C
r
f
r
r
4 . 7 6 A " 1 5 . 8 3 A " ! 7 . 2 4 A u c(B)
smectic C* 1 1 0 ° C 4 . 60Ae 1 4 . 0 2 A " 18.03A" (C) crystalline 7 0 ° C 4 . 6 7 A " 5 . 8 3 A "A.
1 1 .41A" 47A" 8 . 0 3 A "(D)
crystalline 25°C 4 . 6 7 A " 1 5 . 8 3 A " I I I 1 IL
5 10 15 20 25 30 2 0Figure 7. Temperature-dependent X-ray measurements for polymer VIIP at (A) 160
"C,
(B)
110 O C ,(C)
70 O C , and (D) 25O C .
D from Figure 1 are typical
DSC
traces for monomerVM.
It displays smectic
B,
chiral smectic C, and smecticA
mesophases
on
both heating and cooling scans. The mesophase identifications have been achieved by optical polarizing microscopic observation and X-ray diffraction measurements. The thermal transitions and thermody- namic parameters of monomers IM-VM aresummarized
in Table
IV.
As
can be seen from Table IV the tendency toward chiral smectic C mesomorphism increases by increasing the length of alkenylosy spacer.The synthesis of polymers
IP-VP
is describedin
Scheme11.
An excess amount of olefin monomers was usually used to cany the hydrosilation reaction to completion. The unreacted monomers were removed by several reprecip- itations from tetrahydrofuran solution into methanoland
by preparative GPC. Therefore the polymers were isolated with high purity. Table V summarizes the thermal transitions and thermodynamic parameters of the obtained polymers
IP-VP.
All
five polymers present smectic mesomorphism. Both polymers IP and IIP show respec- tively an enantiotropic smectic A phase, while polymer7132 Hsuetal. Macromolecules, Vol. 25, No. 26, 1992
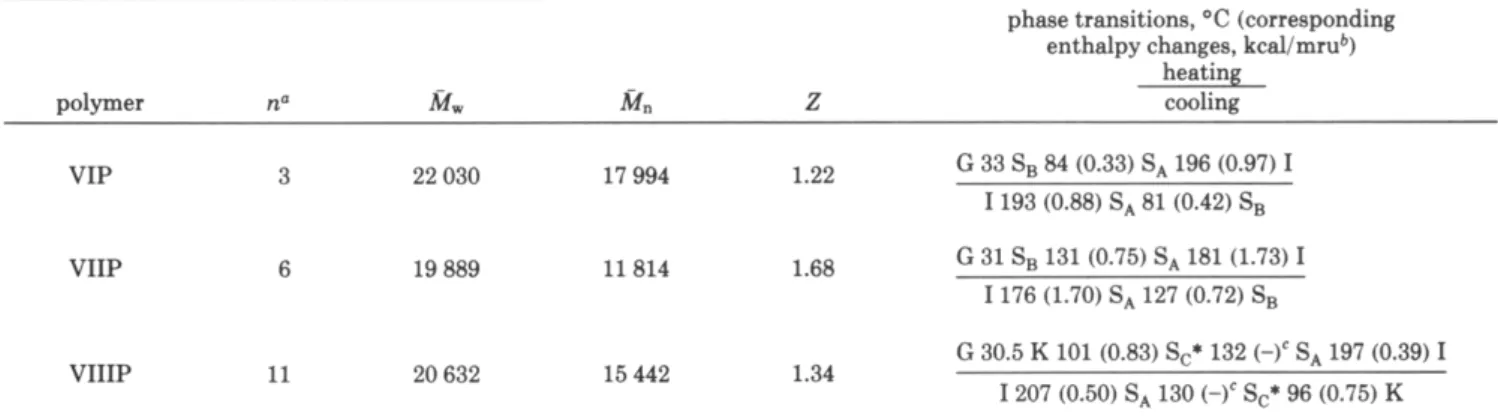
Table VI1
Molecular Weiehts. P h a E Transitions. and PhasaTransition Enthaloies for Polrmers VIP-VIIIP
phase transitions, 'C (corresponding enthalpy chanaes. .. kcalImm4
heating polymer n'
M,
M n Z cooling G 33 S, 84 (0.33) SA 196 (0.97) I I 193 (0.88) SA 81 (0.42) SB VIP 3 22 030 17 994 1.22 VIIP G 31 SB 131 (0.75) SA 181 (1.73) I I176 (1.70) SA 127 (0.72) SB 6 19 889 11 814 1.68 G 30.5 K 101 (0.83) Sc* 132 (3' SA 197 (0.39) I I207 (0.50) SA 130 (-)' Sc* 96 (0.75) K VIIIP 11 20 632 15 442 1.34n according to Scheme 111. mru = mole repeating unit. Enthalpy is very small.
A C
D B
Figure 8. Wide-angle X-ray diffraction patterns for polymer VIIIP obtained at (A) 160 OC,
(B)
120"C,
(C) 70"C,
and(D)
25 OC.IIIP
shows enantiotropic smectic A and smecticB
phases. diagrams obtained from powder samples ofIVF'
a t 170, Polymers IVP andVF'
are the only two polymers which 120, 80, and 25 OC. A broad reflection at wide angles revealanenantiotropic chiral smectic C phase besides the (associatedwiththe1ateralpackmgs)andasharpreflection smectic A and smecticB
phases. The phase assignment at low angles (associated with the smectic layers) are was conducted by optical polarizing microscopic ohser- respectively shown by all curves. Curve A presents a vation and X-ray diffraction measurements. Figure 2 diffuse reflection a t about 4.65A,
which corresponds to presents the temperature-dependent X-ray diffraction lateral spacing of two mesogenic side groups, and a sharpMacromolecules. Vol. 25, No. 26, 1992
rW. ~ Zl- 3 - 1 /A%*. . W I * l
*
a
'1B
C
Figure 9. Optical polarizing micrographs displayed by VIIIP (A) SA texture obtained after cooling to 160.9
'C;
(B) Sc* texture obtained after cooling to 127 O C ; (C) crystalline texture obtainedafter cooling to 80 O C .
first-order reflection at 36.78
A
and a second-order reflection at 19.41A,
which correspond to smectic layers. The optical polarizing micrograph (Figure 3A) reveals a focal-conic fan texture for polymer IVP at this temperature range. Both results are consistent with a smectic A structure. When the measuring temperature bas been lowered from 170 to 120 "C, the d spacing of the first- order reflection decreases from 36.78 to 36.03A
( c w e B). This gives strong evidence for the formation of the tilted chiral smectic C phase. This result is also in agreement with the optical microscopic observation which reveals a broken fan texture (Figure 3B). When the measuring temperature hasbeenfurthercooledtoSO'C, thedspacing of first-order reflection changes back to 36.78A
and thePolysiloxanes and Polymethacrylates 7133 wide-angle reflecton becomes verysbarp (curve
0.
These results indicate the formation of a smectic B phase. Curve D which bas been measured a t room temperature, is eventually identical to curve C. This demonstrates that the smectic B phase has been quenched to room temper- ature. Figure 3C displays the smectic B texture exhibited by IVP.Representative DSC traces for polymer
VP
are presented in Figure 4. A glass transition a t 20 OC is displayed here, followed byasmecticB tochiralsmecticCphase transition at 109 "C, a chiral smectic C to smectic A phase transition a t 143 "C, and a smectic A to isotropic phase transition a t 218 "Con the heating scan (curve A). The cooling scan (curveB)
looks almost identical to the heating scan, except that a verysmall supercooling (less than 10 " C ) is observed for three exothermic transitions. The temperature- dependent X-ray diffraction diagrams obtained from powdersamplesofVPat 170, 110,and8O0Carepresented in Figure 5. These results are very similar to those presented in Figure 2. The d spacings of first-order reflections show respectively a value of 37.71A
at 170 "C (curve A), 36.37A
a t 110 "C (curve B), and 37.71A
a t 80O C ( n w e c ) . Thesevaluesareveryclosetothoseexhibited by polymer
IVP.
However, when the chemical structures of both polymers are compared,VP
is five methylene unita longer in its spacer. This result seems due to larger amount of overlapping of side groups in polymerVP.
Figure 6 presents the typical textures exihibited by the smectic A, chiral smectic C, and smectic B phases of VP.The synthesis of methacrylate monomers and polymers isdescribed in Scheme 111. The hydroboration of the vinyl groups of monomers IM, IVM, and VM with 9-BBN gave only the primary alcohol, which by esterification with methacryloyl chloride led to the corresponding monomers VIM-VIIIM. All reaction steps were performed with retention of configuration of the chiral end groups. The thermal transitions and thermodynamic parameters of monomers VIM-VIIM are summarized in Table VI. Both monomers VIM and VIIM show enantiotropic smetic A and smectic B phases, while monomer VIIIM presented enantiotropic smectic A and chiral smectic C phases and a monotropic smectic B phase.
The results of the radical polymerization of the mono- mers are summarized in Table VII. All polymers were purified by several precipitations until GPC measurments could not detect traces of unreaded monomers. The molecular weights of these polymers were determined by GPC using a calibration based on polystyrene standards and therefore have only a relative meaning. The thermal transitions and thermodynamic parameters of polymers VIP-VIIIP have also been reported in Table VII. All three polymers show smectic mesomorphism. Both polymers VIP and VIIP, respectively, show a glass transition temperature a t 33 and 31 "C and enantiotropic smectic A and smectic B phases. Polymer
VIIP,
which contains 11 methylene units in the spacers, presents a glass transition temperature a t 30.5 OC and enantiotropic smectic A and chiral smectic C phases and a crystalline phase. Figure 7 presents the temperature-dependent X-ray diffraction diagram of polymer VIIIP. Curves A and B are consistent respectively withsmectic A and chiral smectic C structures. BothcurvesC andDshowaseriesofsmall-anglereflections at 28.03, 15.83, 11.41, and 8.47A
and two wide-angle reflections at 4.67 and 4.27A.
These results indicate the formation of a crystalline phase. Figure8
shows the wide- angle diffraction patterns of VIIIP. Figure 9 presents the textures exhibited by VIIIP. Both figures also demon- strate the formation of smectic A, chiral smectic C, and7134 Hsuet al.
Macromolecules, Vol. 25,
No.
26,
1992 (7) Dubois,J.
C.; Decobert, G.; LeBarny, P.; EBeelin, S.; Friedrich,C.; Noel, C. Mol. Cryst. Liq. Cryst. 1986, 137,349. (8) Eeselin,
S.;
Bosio, L.; Noel, C.; Decobert, G.; Duboii, J. C. Lip.Cryst. 1987,2, 505.
(9) Zentel,
R.;
Rekert, G.; Reck B. Liq. Cryst. 1987,2, 83. (10) Hahn, B.; Percec, V. Macromolecules 1987,20, 2961. (11) Bualek,S.;
Kapitza, H.; Meyer, J.; Schmidt, G. F.; Zentel R. (12) Uchida,S.;
Morita, K.; Miyoshi, K.; Haehimoto, K.; Kawasaki, (13) Eeaelin. S.: Noel. C.: Decobert, G.: Dubia, J. C. Mol. Cwst. Lio.Mol. Cryst. Liq. Cryst. 1988, 155,47.
K.
Mol. Cryst. Liq. Cryst. 1988,155,93. crystdine phases. Upon comparison of the thermaltransitions and corresponding enthalpy change data of polymer
VIIIP
to those of polymerVP,
a flexible polymer backbone can be seen hereto
have a tendency toward having a lower glass transition, wider mesomorphic ranges, and larger enthalpy changes. The most important ten- dency is that the flexible polymer backbone also leads to a wider temperature range of the chiral smectic C phase. In conclusion, aseriesof new side-chain liquid crystalline
polysiloxanes and polymethacrylates containing 4-[(S)- 2-methyl- 1-butoxyl phenyl 4- (alkyloxy) biphenyl-4’-car- boxylate side groups has been prepared.All
the obtained polymers have exhibited smectic mesomorphism. The tendency toward chiralsmectic C mesomorphism increases by increasing the lengthof
the alkyloxy spacers. Flexible polymer backbones enhance the decoupling of the motions of the side chain and main chain and therefore tendto
give riseto
a higher thermal stability of the mesophases, including the chiral smectic C phase.Acknowledgment.
The authors are grateful to the National Science Council of the Republic of China for financial support of this work (Grants NSC79-0416-EW9- 02 and 80-0416-Em-02).References and
Notes
(1) Mayer,
R.
B.; Liebert, L.; Strzeleckl, L.; Leller, P. J. Phys. Lett. (2) Clark, N.A.;
Lagerwall, S. T. Appl. Phys. Lett. 1980,36,899. (3) Shibaev,V. P.; Kozlovsky,M. V.;Beresnev, L. A.;Blinov, L. M.; (4) Decobert,G.;
Soyer, F.; Dubois, J. C. Polym. Bull. 1985,14,179.( 5 ) Guglielminetti,
J.
M.; Decobert, G.; Dubois, J. C. Polym. Bull. (6) Decobert, G.; Dubois, J. C.; Esselin, S.; Noel, C. Liq. Cryst.1975 36, L-69.
Plate, N. A. Polym. Bull. 1984,12,299.
1986, 16, 411. 1986, 1, 307.
. .
Cryst. igS8, 155,371.
(14) Kapitza, H.; Zental, R. Makromol. Chem. 1988,189,1793. (15) Zentel,
R.
Liq. Cryst. 1988.3, 531.(16)
Zentel,R.;Rackert,G.;Bualek,S.;Kapitza,H.
Makromol.Chem. 1989,190. 2869.(17) Val1erien;S.
U.;
Zentel, R.; Kremer, F.; Laptiza, H.; Fischer, E. W. Makromol. Chem., Rapid Commun. 1989,10,33. (18) Scherowsky, G.; Schliwa, A.; Springer, J.; Kuhnpast, K.; Trapp,W . Liq. Cryst. 1989, 5, 1281.
(19) Shibaev, V. P.; Kozlovsky,
M.
V.; Plate, N. A. Liq. Cryst. 1990,8, 1281.
(20) Dumon, M.; Nguyen, H. T.; Mauzac, M.; Destrade, C.; Achard, M. F.; Gasparoux, H. Macromolecules 1!W, 23,355. (21) Vallerien, 5.
U.;
Kremer, F.; Fischer, E. W . Makromol. Chem.,Rapid Commun. 1990,11,593.
(22) Vallerien,
S.
U.; Kremker, F.; Kapitza, H.; Zentel, R.; Fischer, E. W . Ferroelectrics 1990,109, 273.(23) Brand,
H.
R.;
Pleiner, H. Makromol. Chem., Rapid. Common. 1990,11,607.(24) Endo, H.; Hachiya, S.; Uchida, S.; Hashimoto K.; Kawasaki, K. Liq. Cryst. 1991, 9, 635.
(25) Kapitza, H.; Zental, R. Makromol. Chem. 1991,192,1859. (26) Bomelburg,
J.;
Heppke G.; Hollidt, J. Makromol. Chem.,RapidCommun. 1991,12,483.
(27) LeBarny,
P.;
Dubois, J. C. In Side Chain Liquid Crystal Polymers; McArdle, C. B., Eds.; Blackie: Glasgow and London, 1989; p 130.(28) Percec, V.; Hahn, B. J . Polym. Sci., Polym. Chem. Ed. 1989,27, 2367.
(29) Percec,V.; Hau, C. S.; Tomazos,D. J . Polym. Sci., Polym. Chem. Ed. 1988, 26, 2047.