國立臺中教育大學科學教育與應用學系
科學教育碩士學位暑期在職進修專班碩士論文
指導教授:張嘉麟 博士
2-氟化萘光電子光譜與磷光光譜
的理論研究
A theoretical study of the photoelectron
and phosphorescence spectroscopy of
2-fluoronaphthalene
研究生:黃瑞陽 撰
謝 誌
謹將這篇論文,獻給我摯愛的父親─黃天勝先生(1946.08.30-2015.08.10)。 能夠順利完成碩士班學業,首先要感謝指導教授張嘉麟老師以及本研究室的 黃瓊慧博士,在他們親切且深入淺出的指導下,讓我對量子化學這個陌生的領域, 有了更深一層的認識!從送出論文初稿到論文口試的這段期間,感謝中山醫學大 學的賴金宏教授與本系的陳錦章教授,除了認真仔細的批改論文初稿,更在口試 的時候,熱心提供許多寶貴意見,讓這篇論文的內容能夠去蕪存菁。 感謝中教大的師長─游淑媚老師、黃鴻博老師、許良榮老師、靳知勤老師、 李松濤老師,讓我在就讀碩士班的時候,重溫大學時期嚴謹但溫馨的上課氛圍! 更要感謝葉聰文老師,犧牲許多可以陪伴家人的週末假期,為我們這群暑碩班學 生上課!謝謝當年就讀中師自科所碩士班的指導教授─蘇育任老師,還有劍道與 人生的導師─初教系黃隆民老師,感謝您們對學生的眷顧與提攜! 大學同寢室四年的同窗兼好友─郭文信老師:感謝你 21 年來不曾間斷的鼓勵 與協助,我才能再次回到母校,完成中斷多年的研究所學業!謝謝俐文、偉苓、 明智、素雲、怡茹、星慧、婷莉、美仁!這四年來大家同心協力,終於達成一起 畢業的目標!也由衷祝福本班的老大哥、永遠的班長─馮暐老師身體健康! 感謝陳福順校長、能溪主任、文碩主任、振嘉主任、錦鈴組長、安瑀組長以 及彰化縣興華國小所有同仁的鼓勵和支援,讓我在職進修期間無後顧之憂!謝謝 明璇總是在我疲憊時,貼心的為我準備提神的濃醇咖啡! 感謝我的父親黃天勝先生、母親葉雪犁女士、屘叔黃天祥先生還有家族長輩 的栽培和付出!感謝大哥瑞玄、小妹瑞雯在成長過程中的關懷與扶持!謝謝內人 怡貞以及乖巧體貼的孩子─美芹和安華,讓我可以心無旁鶩順利完成學業! 父親生前一直掛念督促我把論文完成,光榮拿到碩士學位。雖然趕在他過世 前完成論文初稿,但很遺憾未能讓父親看到我拿到畢業證書。最後謹向安息主懷 的父親說:「親愛的爸爸,孩兒已完成您的遺願拿到學位!我愛您!」感謝主!摘要
本研究以量子化學計算的方法,運用密度泛函理論的 B3LYP 及 B3PW91 泛函 數,分別搭配 6-311++G(d,p)、6-311++G(2d,p)及 aug-cc-pVTZ 等基組,計算出
2-氟化萘(C10H7F)分子基態、離子基態及三重態最低能階的平衡結構與諧和振動頻率,
並藉由法蘭克─康登因子的理論計算,模擬 2-氟化萘的光電子光譜與磷光光譜,我 們最後使用完備基組(Complete basis set, CBS)極限的計算方法,計算 2-氟化萘分子 基態、離子基態及三重態的零點能(zero point energy, ZPE),再計算其離子態的絕熱 游離能(adiabatic ionization energy, AIE)和三重態的激發能(excitation energy)。結果 發現計算的分子平衡結構與振動頻率和實驗值相符,模擬光譜與實驗光譜對照後 發現,兩者在相對能量的位置及相對強度的比例上,非常吻合。在能量計算上, 2-氟化萘的絕熱游離能和激發能,不論用 CBS-4M 或 CBS-QB3 這兩種方法,均略 高於實驗值,其中 CBS-QB3 的方法所計算得到的能量,都比 CBS-4M 的方法更接 近實驗值,計算的絕熱游離能與實驗值的最小差距為 0.8%;計算的激發能與實驗 值的最小差距為 2.3%。 關鍵詞:法蘭克-康登因子、2-氟化萘、光電子光譜、磷光光譜、游離能、激發能
II
Abstract
We have performed quantum-chemistry computations to obtain the equilibrium geometries and harmonic vibrational frequencies of 2-fluoronaphthalene (including the neutral molecule, cation and the lowest triplet state) by using the density-functional theory (B3LYP and B3PW91 functionals) associated with the basis sets of 6-311++G(d,p), 6-311++G(2d,p) and aug-cc-pVTZ. The photoelectron and phosphorescence spectra of 2-fluoronaphthalene were simulated by theoretical computations of Franck-Condon factors. The adiabatic ionization energy and the excitation energy of the triplet state were also calculated by the complete basis set method. The computed equilibrium geometries and vibrational frequencies are in agreement with experiment. The simulated photoelectron and phosphorescence spectra are also in harmony with experiment. The calculated ionization and excitation energies by the CBS-QB3 method are closer to the experimental values than those of the CBS-4M method, although both are slightly higher than the experiment. Compared with reported experiment values, the minimum deviation of computed ionization energy is 0.8%, whereas it is 2.3% for the excitation energy.
Keywords: Franck-Condon factor; 2-fluoronaphthalene; photoelectron spectrum;
目次
摘要 ... I Abstract ... II 目次 ... III 表次 ... IV 圖次 ... V 第一章 緒論 ... 1 第一節 研究動機與重要性 ... 1 第二節 文獻探討 ... 3 一、 2-氟化萘的特性及相關文獻 ... 3 二、 螢光 (Fluorescence)與磷光 (Phosphorescence)發光機制 ... 4三、 密度泛函理論 (Density Functional Theory, DFT) ... 7
四、 法蘭克─康登積分 (Franck-Condon integrals, FCIs) ... 9
第二章 研究方法 ... 17 第一節 基組及計算方法 ... 17 一、 基底函數組(basis set) ... 17 二、 計算方法 ... 18 第二節 2-氟化萘分子、離子及三重態的量子計算 ... 20 第三節 模擬光譜 ... 22 第三章 研究結果與討論 ... 25 第一節 量子化學計算結果 ... 25 一、 平衡結構 ... 25 二、 不同基組量子計算結果之比較 ... 30 第二節 振動頻率與振動模式 ... 33 第三節 2-氟化萘的光電子光譜與磷光光譜 ... 35 一、2-氟化萘的光電子光譜 ... 35 二、2-氟化萘的磷光光譜 ... 39 第四節 游離能與激發能 ... 45 第四章 結論 ... 49 參考文獻 ... 51 附錄 ... 59
IV
表次
表 1- 1:2-氟化萘的平衡結構(B3LYP/6-311++G(d,p)) ... 59 表 1- 2:2-氟化萘的平衡結構(B3LYP/6-311++G(2d,p)) ... 61 表 1- 3:2-氟化萘的平衡結構(B3LYP/aug-cc-pVTZ) ... 63 表 1- 4:2-氟化萘的平衡結構(B3PW91/6-311++G(d,p)) ... 65 表 1- 5:2-氟化萘的平衡結構(B3PW91/6-311++G(2d,p)) ... 67 表 1- 6:2-氟化萘的平衡結構(B3PW91/aug-cc-pVTZ) ... 69 表 2- 1:2-氟化萘離子態的振動頻率(B3LYP/6-311++G(d,p)) ... 71 表 2- 2:2-氟化萘離子態的振動頻率(B3LYP/6-311++G(2d,p)) ... 73 表 2- 3:2-氟化萘離子態的振動頻率(B3LYP/aug-cc-pVTZ) ... 75 表 2- 4:2-氟化萘離子態的振動頻率(B3PW91/6-311++G(d,p)) ... 77 表 2- 5:2-氟化萘離子態的振動頻率(B3PW91/6-311++G(2d,p)) ... 79 表 2- 6:2-氟化萘離子態的振動頻率(B3PW91/aug-cc-pVTZ) ... 81 表 2- 7:2-氟化萘三重態的振動頻率(B3LYP/6-311++G(d,p)) ... 83 表 2- 8:2-氟化萘三重態的振動頻率(B3LYP/6-311++G(2d,p)) ... 85 表 2- 9:2-氟化萘三重態的振動頻率(B3LYP/aug-cc-pVTZ) ... 87 表 2- 10:2-氟化萘三重態的振動頻率(B3PW91/6-311++G(d,p)) ... 89 表 2- 11:2-氟化萘三重態的振動頻率(B3PW91/6-311++G(2d,p)) ... 91 表 2- 12:2-氟化萘三重態的振動頻率(B3PW91/aug-cc-pVTZ) ... 93 表 3- 1:2-氟化萘以不同基組與方法所計算之絕熱游離能與實驗值[7,10]對照表 45 表 3- 2:2-氟化萘以不同基組與方法所計算之激發能與實驗值[4,5]對照表 ... 46圖次
圖 1-2- 1:螢光與磷光發光機制(去活化過程) ... 6 圖 3-1- 1:2-氟化萘分子基態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ] ... 26 圖 3-1- 2:2-氟化萘離子基態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ] ... 26 圖 3-1- 3:2-氟化萘三重態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ] ... 27 圖 3-1- 4:不同基組下 2-氟化萘分子基態鍵長比較圖 ... 30 圖 3-1- 5:不同基組下 2-氟化萘離子基態鍵長比較圖 ... 31 圖 3-1- 6:不同基組下 2-氟化萘三重態鍵長比較圖 ... 31 圖 3-1- 7:不同基組下 2-氟化萘分子基態鍵角比較圖 ... 32 圖 3-1- 8:不同基組下 2-氟化萘離子基態鍵角比較圖 ... 32 圖 3-1- 9:不同基組下 2-氟化萘三重態鍵角比較圖 ... 32 圖 3-2- 1:不同基組下 2-氟化萘分子基態振動頻率比較圖 ... 34 圖 3-2- 2:不同基組下 2-氟化萘離子基態振動頻率比較圖 ... 34 圖 3-2- 3:不同基組下 2-氟化萘三重態振動頻率比較圖 ... 34 圖 3-3- 1:模擬的 2-氟化萘光電子光譜比較圖(B3LYP) ... 36 圖 3-3- 2:模擬的 2-氟化萘光電子光譜比較圖(B3PW91) ... 36 圖 3-3- 3:調整不同半高寬後,模擬的光電子光譜圖之比較圖 ... 37 圖 3-3- 4:模擬的 2-氟化萘光電子光譜圖(基組:B3LYP/aug-cc-pVTZ) ... 38 圖 3-3- 5:模擬的光電子光譜與實驗光譜[7]比較圖 ... 38 圖 3-3- 6:模擬的 2-氟化萘磷光光譜比較圖(B3LYP) ... 40 圖 3-3- 7:模擬的 2-氟化萘磷光光譜比較圖(B3PW91) ... 40 圖 3-3- 8:調整不同半高寬後,模擬的磷光光譜圖之比較圖 ... 41 圖 3-3- 9:模擬的 2-氟化萘磷光光譜圖(基組:B3LYP/aug-cc-pVTZ) ... 42 圖 3-3- 10:模擬的磷光光譜與實驗光譜[4]比較圖 ... 43 圖 3-3- 11:模擬的磷光光譜與實驗光譜[5]比較圖 ... 44 圖 3-3- 12:模擬的 2-氟化萘光電子光譜圖(基組:B3LYP/6-311++G(d,p)) ... 95 圖 3-3- 13:模擬的 2-氟化萘光電子光譜圖(基組:B3LYP/6-311++G(2d,p)) ... 95 圖 3-3- 14:模擬的 2-氟化萘光電子光譜圖(基組:B3LYP/aug-cc-pVTZ) ... 96 圖 3-3- 15:不同基組模擬的 2-氟化萘光電子光譜比較圖(B3LYP) ... 96 圖 3-3- 16:調整半高寬後,不同基組模擬的 2-氟化萘光電子光譜比較圖(B3LYP) ... 97 圖 3-3- 17:模擬的 2-氟化萘光電子光譜圖(基組:B3PW91/6-311++G(d,p)) ... 97VI 圖 3-3- 18:模擬的 2-氟化萘光電子光譜圖(基組:B3PW91/6-311++G(2d,p)) ... 98 圖 3-3- 19:模擬的 2-氟化萘光電子光譜圖(基組:B3PW91/aug-cc-pVTZ) ... 98 圖 3-3- 20:不同基組模擬的 2-氟化萘光電子光譜比較圖(B3PW91) ... 99 圖 3-3- 21:調整半高寬後,不同基組模擬的 2-氟化萘光電子光譜比較圖(B3PW91) ... 99 圖 3-3- 22:模擬的 2-氟化萘磷光光譜圖(基組:B3LYP/6-311++G(d,p)) ... 100 圖 3-3- 23:模擬的 2-氟化萘磷光光譜圖(基組:B3LYP/6-311++G(2d,p)) ... 100 圖 3-3- 24:模擬的 2-氟化萘磷光光譜圖(基組:B3LYP/aug-cc-pVTZ) ... 101 圖 3-3- 25:不同基組模擬的 2-氟化萘磷光光譜比較圖(B3LYP) ... 101 圖 3-3- 26:調整半高寬後,不同基組模擬的 2-氟化萘磷光光譜比較圖(B3LYP) 102 圖 3-3- 27:模擬的 2-氟化萘磷光光譜圖(基組:B3PW91/6-311++G(d,p)) ... 102 圖 3-3- 28:模擬的 2-氟化萘磷光光譜圖(基組:B3PW91/6-311++G(2d,p)) ... 103 圖 3-3- 29:模擬的 2-氟化萘磷光光譜圖(基組:B3PW91/aug-cc-pVTZ) ... 103 圖 3-3- 30:不同基組模擬的 2-氟化萘磷光光譜比較圖(B3PW91) ... 104 圖 3-3- 31:調整半高寬後,不同基組模擬的 2-氟化萘磷光光譜比較圖(B3PW91) ... 104
第一章 緒論
本論文共分成四章,第一章為緒論;第二章為研究方法,說明基底函數組、 計算方法、2-氟化萘分子、離子態與三重態的量子計算及光譜模擬的方法;第三章 為研究結果與討論,說明 2-氟化萘分子、離子態與三重態的平衡結構、振動頻率、 法蘭克-康登因子、模擬光電子光譜、磷光光譜與實驗光譜的比對結果,以及絕熱 游離能、激發能的計算並與實驗值的比較結果;最後一章為結論。 本章緒論第一節說明研究動機與重要性,第二節進行文獻探討。第一節 研究動機與重要性
多環芳香烴(Polycyclic aromatic hydrocarbons, PAHs)通常存在於石化產品、橡 膠、塑膠、潤滑油、防銹油、不完全燃燒的有機化合物中;另外則是使用在製造 過程而殘留在產品中,如塑膠、染料及殺蟲劑製造等;而在電子電機產品中之塑 膠及橡膠材質、黑色或深色的硬性聚合物材料、表面塗料與油漆、用於刷毛、皮 革、纖維和木材的防腐劑,均可能含有 PAHs。不但嚴重衝擊環境及生態,對人體 健康更有致癌性與致突變性,是近幾十年來引起學術界及社會大眾高度關注的有 機化合物。另一方面,PAHs 被發現存在於某些湖泊的沉積岩[1]和星際介質[2]中, 與地球上及宇宙中的生命起源有關。萘是最簡單的多環芳香烴,因為其被證實為 致癌物質,列於美國環保署(EPA)及德國 GS 認證之管制 PAHs 名單中,2-氟化萘則 是其衍生物。
2 2-氟化萘是一種近年來發展的藥物中間體,將氟原子取代芳香烴上的氫原子。 研究發現,氟原子取代的芳香烴化合物,具有良好的生物活性,經常應用在藥物 和農業上。 自從顯示器發明之後,為人類生活帶來極大的便利。人們接受訊息的媒介, 從原來的文字、聲音進化到影像。近幾年來,由於科技的蓬勃發展,傳統映像管 顯示器也漸漸被液晶顯示器所取代。由於 2-氟化萘能夠發出磷光的特性,可做為 液晶顯示器的顯示材料,相較於早期的液晶材料,有著更好的穩定性。 在實驗上雖然已經有其光電子光譜和磷光光譜的研究,就理論的觀點而言, 即使是簡單分子的光譜,可能包含基頻、合頻、泛頻等躍遷,使其光譜產生複雜 的結構;受限於實驗儀器的解析度,對於其光譜結構解析的訊息仍然稍嫌不足, 而且常常難以明確的標定其光譜。因此,藉由準確的量子化學理論計算,來模擬 2-氟化萘的光電子與磷光光譜,有助於分析其離子基態與三重態的細微結構。
本研究的目的在於運用密度泛函理論(density functional theory, DFT),對 2-氟 化萘(2-fluoronaphthalene)分子基態、離子基態以及三重態進行量子計算的理論研究; 接著計算分子基態電離成離子基態、三重態的法蘭克-康登因子(Franck-Condon factor, FCF),進而模擬出離子基態的光電子光譜和三重態的磷光光譜;再運用完備 基組(complete basis set, CBS)極限的理論,計算其離子態的游離能與三重態的激發 能。最後將模擬光譜和運算所得的能量,與實驗值進行比對。
第二節 文獻探討
一、2-氟化萘的特性及相關文獻
氟化萘可經由萘的鹵化親電子取代反應可以生成,依據氟在萘環上所取代氫 的位置,可以分為氟連接於α-碳(參見 24 頁圖 3-1-1 的 C2、C5、C10、C11)上面 的 1-氟化萘(1-fluoronaphthalene, α-fluoronaphthalene),以及 β-碳(圖 3-1-1 的 C1、 C6、C14、C15)上的 2-氟化萘(1-fluoronaphthalene, β-fluoronaphthalene)。在萘環上, 由於電子的非定域(delocalized)並不像苯環那樣完全平均化,而是在α-碳原子上的 電子雲密度較高,β-碳原子上的次之,中間共用的兩個碳原子上則更小。因此親 電子取代反應一般發生在α-碳,製備時 1-氟化萘的產量會略高於 2-氟化萘。2-氟 化萘早期在 Thomas H. Kinstle 及 Jeffrey P. Bechner [3]的研究中,作為有機合成反 應的起始物;近年發展成作為液晶顯示器的顯示材料,較先前的液晶材料,有更 好的穩定性。其分子式為 C10H7F,分子量為 146.16。熔點 61℃、沸點 225.9℃ (760 ㎜ Hg)、閃點 65.4℃;密度 1.14 g/cm3 (20℃),在水中的溶解度為 0.01 g/L (25℃), 汽化熱則是 44.36 kJ/mol。在常溫下為固體,燃燒時會釋放有毒煙霧,食入有害人 體健康,對於黏膜、上呼吸道及眼睛具有刺激性,也會造成皮膚的過敏。 歷年來,對於 2-氟化萘的相關研究,條列如下:Ferguson 等人[4]在 1954 年研 究萘及其簡單衍生物的磷光光譜實驗,繪製出 2-氟化萘的磷光光譜同時測得其激 發能;Siman [5]在 1956 年研究β-氟化萘的磷光光譜,繪製出 2-氟化萘的磷光光譜4
並測得其激發能;Bak 等人[6]於 1962 年分析 2-氟化萘的微波光譜探討決定其結構
的可能性;Kinstle等人[3]於 1970 年研究 2-氟化萘的鋰化反應;Kiasinc 等人[7]於 1983 年進行并苯之光電子光譜及電子結構與取代基效應之實驗時,繪製出 2-氟化
萘的光電子光譜,並測得其游離能;Rava 等人[8]於 1985 年研究 1-氟化萘和 2-氟
化萘的二光子螢光蒸汽激發光譜(Two-photon vapor fluorescence excitation spectra); Cassidei 等人[9]在 2008 年對 2-氟化萘上的氫和氟之環間 耦合常數(inter-ring
coupling constants)進行 NMR 實驗與理論研究;Tzeng 等人[10]在 2012 年以共振二 光 子 (Resonant two-photon)游 離 與 質 量 解 析 臨 界 游 離(mass-analyzed threshold
ionization)光譜術研究 1-氟化萘及 2-氟化萘之特性,分析其光譜特性並計算其游離
能。
二、螢光 (Fluorescence)與磷光 (Phosphorescence)發光機制
當分子吸收能量時(通常是光子或電子束),分子內的電子就會受到激發而躍遷 至較高能量的激發態。此一躍遷如果沒有涉及電子自旋狀態的改變,意即由基態 (singlet ground state)躍遷至單態激發態(singlet excited state),其躍遷機率比較高,
因此所發出的光強度較大,其生命期比較短(約 10−5 至 10−8 秒之間),即是螢光
(Fluorescence)。若牽涉到電子自旋狀態的改變,如躍遷到三重態(triplet excited state),
磷光(Phosphorescence)。 通常激發態電子除了經由發光過程回到基態,還有下列幾種非發光鬆弛過程, 這些過程又稱為非輻射去活化過程,簡述如下: (一)震動鬆弛(vibrational relaxation):主要是起因於激發物種與附近分子或 溶劑碰撞的結果,能量經由碰撞傳遞至其他分子,造成溫度的上升。 (二)內轉換(internal conversion):此過程是因為分子內的電子能階太過密集, 並且有不同電子能階的振動態(vibrational state)相重疊,因此激發態的 電子能在非放光的情況之下,經由重疊的振動態一路下滑,釋出能量 而降至其他較低的電子能階,甚至降到基態。此一去活化過程之可能 性通常大於經由螢光發光去活化的可能性,且速度極快,所放出的能 量通常也可能會造成斷鍵,即所謂的預解離(predissociation)。 (三)外轉換(external conversion):當處在激發態的分子和附近分子(如溶劑等) 作用,而將能量傳遞至其他分子上時,將其他分子激發至激發態,本 身降至基態,並未經由發光方式去活化,稱為外轉換。 (四)系統間跨越(intersystem crossing):所指的是激發態電子的自旋狀態有所 改變,例如由單態激發態跨越至三重態。一般而言三重態的能量會略 低於單態激發態,而這樣的系統間跨越也會造成螢光強度減弱,磷光 強度增強。
6 這些非發光去活化或者經由釋放螢光或磷光去活化的過程,都是激發態分子 重回基態的途徑。但若就觀察發光光譜者的角度而言,非發光方式的去活化過程 是較無利用價值的,因此站在光譜分析的角度,通常會在意經由發光方式去活化 的過程到底佔了多少比例,也就是所量子產率的概念。量子產率: φ= kf kf+ki+kec+kic+kv+kpd+… 其中:kf代表螢光去活化之速率常數;ki乃系統間跨越去活化之速率常數;kec 為外轉換去活化之速率常數;kic 是內轉換去活化之速率常數;kv 為振動鬆弛去活 化之速率常數;kpd 則是預解離之速率常數。簡單的說,就是在計算經由發光方式 去活化的比例。上述的各種去活化過程如圖 1-2-1 所示: 圖 1-2- 1:螢光與磷光發光機制(去活化過程)
S
2S
0S
1T
1三、密度泛函理論 (Density Functional Theory, DFT)
量子力學之波動方程式從二十世紀初開始發展至今,可以用來描述微觀世界 極小粒子(如電子)的行為,到現在已經有非常豐碩的成果。量子化學(quantum chemistry)為其中的一個分支,目的在運用量子力學的理論和方法,來研究並解決 化學問題[11]。科學家認為,量子化學在瞭解物質的基本性質後,藉由薛丁格方程 式(Schrödinger equation)以數學計算的方式求得波函數(wave function),再從波函數 去了解電子的分佈(electronic distribution)情形,就能獲得所有的物理量。 密度泛函理論為本研究採用的計算方法,乃根據1927年波恩-奧本海默近似法 (Born-Oppenheimer approximation)[12]假設。因為原子核的質量比電子大上數千倍, 所以在電子運動的瞬間,原子核的位移相當小,故原子核的座標與動能皆可以視 為常數,加上電子都是電荷、質量、自旋等特徵完全相同的粒子。因此分子結構 的研究可化為N個全同粒子體系的研究,將電子與原子核間的運動,視為分開的獨 立事件,因此密度泛函理論的方法解決了原來多粒子體系的複雜度。 該理論最早可追溯到1927年Thomas和Fermi發展的Thomas-Fermi [12,13]模型, 其原理是先以局部密度近似法(local-density approximation, LDA) [14]求得以電子密 度 分 佈 函 數𝜌(𝑟) 為 變 量 的 能 量 泛 函 E[𝜌(𝑟)] , 進 而 計 算 原 子 的 能 量 。 不 過 Thomas-Fermi理論忽略了電子彼此間交互作用的交換能,因此限制了計算的準確 性而未受重視。
8 Hartree-Fock [15]理論於1958年提出,理論假設多電子體系符合波恩-奧本海默 近似法(Born-Oppenheimer approximation),也就是一個多電子體系,其電子的運動 和能量與原子核的運動和能量,是可以相互分離的。而密度泛函理論就是建立在 Hartree-Fock的理論基礎上。 直到1960年代,Hohenberg和Kohn [16]提出密度泛函理論的基礎雛形而廣受重 視, Hohenberg-Kohn理論主張:多電子系統基態的所有性質,都可視為其電子密 度𝜌(𝑟)的函數。其內容為:一個多粒子(電子)體系的性質(如能量),是由粒子 (電子)密度的空間分布來決定,因此只要解出含有三個空間變數的粒子(電子) 密度函數,而不需解出每個粒子的體系波函數。 Hohenberg和Kohn同時提出了密度泛函理論中兩個重要定理:首先,在一個穩 定 的 量 子 力 學 系 統 中 , 任 一 可 觀 測 的 物 理 量 ( 如 能 量 ) , 都 可 以 藉 由 基 態 (ground-state)的電子密度來計算,換言之,每個可觀測的物理量皆可以基態的 電子密度𝜌(𝑟)泛函來表示;其次,運用變分法,求出以電子密度函數𝜌(𝑟)為變數的 能量泛函𝐸𝐺.𝑆.[𝜌(𝑟)]的最小值,亦即求其最低能量狀態,便能得到基態能量𝐸𝐺.𝑆.。 Kohn與Sham [17]在1965年發表另一種新的計算方法,對密度泛函理論做出巨 大的貢獻,此方法是利用具備等效位能(effective potential)的獨立電子系統,來解決 電子間多體交互效應問題,以交換相關(exchange-correlation)泛函[18,19]來描述電 子間的多體效應,成功說明如何由電子密度函數𝜌(𝑟)求得電子的能量 𝐸𝐺.𝑆.。其計
算過程是利用迭代法,不斷重複運算電荷密度迭代至差異小於設定條件之自洽場 (self-consistent field, SCF) [20]為止,於是形成了密度泛函數理論。
四、法蘭克─康登積分 (
Franck-Condon integrals,
FCIs)
近年來,張嘉麟教授所主持的研究團隊已經開發出一些方法,用來計算諧和 振子(harmonic oscillators, HOs)的法蘭克-康登積分(Franck-Condon integrals, FCIs),
用來解釋或預測電子振動光譜和分子的非輻射過程極具價值性[21-25]。 一維(one-dimensional, 1D) 取代的 FCI 以及扭曲 HO 發表於 2005 年[21];高達 4D 的多維度(multidimensional) FCI 與 HO 則發表於 2008 年[23];在稍後的 2009 年, 得到用來任意轉換兩個電子振動狀態的二維 FCI 和 HO 之通式[24]。而這一連串研 究的終極目標,就是找出任意維度的 FCI 和 HO 通式。然而,因為發生複雜混合 模式的杜辛斯基效應(Duschinsky effect)的繁瑣運算,使得這樣的企圖變得難以實現 [26]。當兩個電子態的幾何形狀完全不同,其中一個電子態在正規座標(normal coordinates)上,相對於另一個電子態有位移、變形或旋轉的情形,便會發生杜辛 斯 基 效 應 。 結 果 , 兩 個 電 子 態 的 正 規 座 標 被 偶 合 於 振 動 波 函 數 (vibrational wavefunctions)的重疊積分。
計算包含杜辛斯基效應的 FCI 和 HO,存在不同的方法[27-52]。Barone 等人將
10
FCIs 用精確的算式計算。 Sharp 和 Rosenstock 首先於 1964 年運用生成函數
(generating function) 導 出 多 維 度 FCIs 的 算 式 [27] 。 Kikuchi 等 人 根 據
Sharp-Rosenstock 的方法,於 2003 年以更簡化的算式,開發多維度 FCIs 的計算方 法[47]。自 1996 年起, Lin 及其夥伴運用各種技術導出 FCIs 和 HO 的分析式[43-45]。 但是實際應用到大量原子組成的分子,目前的分析法無法被證明是可行的。就我 們所知,以分析法直接用來計算諧和振子的法蘭克-康登積分,超過 10 種模式的甚 少。 另一方面,在遞迴法上,特定振動躍遷的 FCI 可由低振動躍遷的遞迴公式獲 得。因此,只有絕熱躍遷的 FCI 需要被精確的計算。 Doktorov 等人首先利用同調 態(coherent-state)的方法獲得多維度度 FCIs 遞迴公式[28]。數十年來已經開發不同 的演算方法,以改善繁瑣運算的效率[29-38]。遞迴法因其簡單而被廣泛採用,在 某種意義上,已經成為計算多原子分子 FCIs 的標準方法。 然而,遞迴法在處理振動狀態被高度激發的龐大的分子,可能會因系統記憶 體飽和[51]而停擺。即使用在每一個繁瑣過程重新計算 FCIs 來避免記憶體不足的 問題,冗長的運算時間讓此方法變得不利。FCIs 的誤差甚至無可避免地從低振動 躍遷,延伸到高振動躍遷,有時更會導致嚴重的偏差[35]。 張嘉麟教授及其研究團隊,在 2013 年提出計算諧和振子的法蘭克─康登積分 的通用分析法[53]。本法可作為最近被 Barone 等人[51]所開發的方法之外的另一項
選擇。Barone 等人結合了分析法與遞迴法來計算分子的振動光譜,其中單激發和
雙重激發的一般模式 FCIs 以 Sharp 及 Rosenstock 的分析法計算[27],較高的激發
模式則使用遞迴法[28,35]。他們所使用的方法已經納入 Gaussian 運算軟體的化學 套件中[54]。 相較於遞迴法,張嘉麟教授的方法主要採分析法,而且在以下幾個面向效果 顯著:首先,這種方法沒有記憶體儲存空間不足的問題;其次,每一個躍遷的 FCI 可被獨立運算,顯示出可有效結合平行演算法(parallel algorithms);最後,此一解 決方案提供評估諸如遞迴法錯誤的數值方法。 研究團隊以張嘉麟教授在 2008 及 2009 年發表,包含杜辛斯基效應並能計算 達到四維度諧和振子 FCI 的方法為基礎[23,24],進一步在 2013 年推導出新的諧和 振子 FCI 公式[53]。開發出包含杜辛斯基效應且適用於任何維度的諧和振子 FCI 之計算程式,介於兩個 n 維度諧和振子間的 FCI 可以寫成:
i i
i i i i i n i i v i i vi n n vv v N H Q H Q Q Q dQ v v v
2 2 1 2 1 2 1 2 1 2 1 exp
(1) 其中 2 1 1 2 ! !
n i i i v v i i v v N i i (2) i i (3)12
i
代表第 i 種模式的角頻率, 是普朗克常數(Planck’s constant)除以 2π,Hv( x)為 厄 米 多 項 式 (Hermite polynomial) 。 假 設Q 可 根 據Q 藉 由 和 杜 辛 斯 基 矩 陣
(Duschinsky matrix) J 和位移向量(shift vector) D 表示[23,24,26],
D JQ Q (4) 其中 T L L J (5) 𝐿′代表正常模式起始態的位移矩陣(displace matrix)
𝐿𝑇代表最終態位移矩陣的轉置矩陣(transpose of the displace matrix)
0 0
2 1 X X M L D (6) M 為由原子量構成的對角矩陣(diagonal matrix) 𝑋0及𝑋0′分別為最終態及起始態平衡結構的直角座標(Cartesian coordinates) 然後,將上述方程式進行配方
n i j i j ij i i Q B Q C x 1 (7)
n i j i j ij i i x B x C Q 1 (8) 可求得
i i
i i n i j j ij n i v i n i j j ij vi n n vv v NE H a x d H a x d Ax dx v v v i 2 1 2 1 2 1 exp
(9)其中
1 1 2 1 2 2 1 i k ki k n k ki k i i J AB A (10)
n k i k kj ki k kj ki k i ij J J AB B A B 1 1 1 2 1 1 (11)
n k i k k ki k ki k k i i D J A B C A C 1 1 1 2 1 1 (12) 1 , 1 , i ii i B B (13)
1 1 j i k kj ik ij ij B B B B , ji1 (14)
n i k k ik i i C B C C 1 (15) i ii a (16) ij i ij B a , ji (17) 1 1 i i i J (18)
1 1 j k kj ik ij i ij J J B , j1 (19) i i i C d (20)
n k i k ik i i J C D d 1 (21) 以及
n i n i i i i iD AC E 1 1 2 2 2 1 exp (22) 方程式(10)-(15)中,係數 A、B、C 必須從低到高的𝑖值計算,而係數𝐵′和𝐶′必須以 相反的順序求得,程式(9)的厄米多項式可以反覆使用下面的方程式進行擴展[55]:
n k k k n n H d x k n d x H 0 2 (23)14 其中 k n 是二項式係數,展開之後的積分式剛好是多重高斯積分的乘積,故可由下 列高斯積分關係式進一步簡化,成為可供計算求解的方程式[55]:
2 1 2 2 2 ! ! 1 2 exp a a s dx ax x s s (24) 其中 (2s-1)!! = 1×3×5×(2s-1)是雙階乘,代數運算後可得 V HI v v v v v v n n 0 2 1 2 1 (25) 其中 2 1 1 0 00 0 00 0
n i i i i A E I
(26)是絕熱躍遷(adiabatic transition)的 FCI
12 1 ! ! 2
n i i i v v v v V i i (27) 以及
ij ij ij ij u k u k F F F H 0 0 3 2 1 (28) 其中
n i n j n j k ij ij ij k ij ij ij ij ij a k u a k u F 1 1 1 1 (29)
n i i m i m d H d H F i i 1 2 (30)
n j j K j K A F j 1 2 3 1!! 2 (31)在方程式(31)
j i n i ij ij j k k 1 1 K (32) 其值必須為偶數。在程式(28)中,𝑢𝑖𝑗和𝑢𝑖𝑗′ 分別是𝑘 𝑖𝑗和𝑘𝑖𝑗′ 的上限,可由下式確認
j1 i p ip i ij v k u (33)
1 1 j p ip i ij v k u (34) 在方程式(30) in in iu
k
m
(35) in in iu
k
m
(36) 這種方法的優點在於,上述方程式的每一個表達式各有一個封閉且通用的形 式,而且可以被輕易編碼。結合平衡結構、振動頻率、正常模式、激發能和躍遷 偶極矩(可選擇)的量子化學計算。而且可以用來預測或解釋各種分子的吸收光 譜、螢光光譜、磷光光譜和光電子光譜等電子振動光譜(vibronic spectrum)。第二章 研究方法
本研究採用 Gaussian 09 套裝軟體,選用密度泛函理論之 B3LYP 以及 B3PW91 兩種方法,分別計算出 2-氟化萘離子基態及三重態的平衡結構及振動頻率,再以 本研究室所發展的方法計算 FCF(法蘭克-康登因子),然後將量子化學計算所得之 結果及模擬的光電子光譜與磷光光譜,與實驗光譜進行比對與討論;最後計算出 游離能和激發能,與實驗值進行比對與討論。本章共分成三節,第一節介紹基組 的理論基礎及量子化學計算方法;第二節介紹 2-氟化萘分子、離子及三重態的量 子化學計算;第三節介紹光譜模擬的方法。第一節 基組及計算方法
一、 基底函數組(basis set)
我們在進行量子計算時須先選擇一基底函數組(basis set,簡稱基組),用來表 示分子軌域之波函數。基組是由ㄧ群基底函數組合而成,而基底函數則是由許多 高斯函數(primitive Gaussian) 組合而成,主要藉由高斯函數的線性組合(linear combination) 而成。而不同大小的基組將對運算的精準度以及所需的時間產生影響, 所以須針對研究所需的準確性以及計算時間上取得折衷點,來進行基組的選擇。本 研 究 則 採 用 Dunning 等 人 [56-59] 所 提 出 基 組 中 , aug-cc-pVTZ (augment correlation-consistent valence triple-zeta)的基組進行2-氟化萘分子、離子及三重態的
18 平衡結構及振動頻率的計算,另外選用Pople等人[60]所提的6-311++G(d,p) 以及 6-311++G(2d,p)共3組基組。為了求得更精確的計算結果,可加入擴散函數(diffuse function)來涵蓋距離原子核較遠的電子,讓軌域擁有較大空間可供電子佔據。且 為了使分子結構中每個原子可調的位置更具彈性,可在基組中加入極化函數 (polarization function),使整個基組能更接近真正的分子軌域性質。而在基底函 數之前,加上aug-前置字串,表示增加擴散函數;在基底函數之前,加上cc-p前置 字串,表示增加極化函數。以Dunning所提的基組aug-cc-pVTZ為例,基底函數VTZ 表示價殼層為三重分裂函數,除了極化函數cc-p之外,還加上擴散函數aug-。而以 Pople系列選用的6-311++G(2d,p)基組為例,6表示此內殼層基底函數用6個初始函數 線性組合展開;311表示價殼層的用三組函數展開,每組各有3、1、1個函數;二 個「+」代表重原子(本研究為C和F)、氫原子皆加入擴散函數,2d為重原子加 入兩組d軌域型態的極化函數,p則代表將氫原子加入極化函數。
二、 計算方法
本研究的分子特性計算採用 Gaussian 09 [54]軟體,它是量子化學計算的專業 軟體, 它是利用量子力學的原理以數值方法來預測化學分子的性質。Gaussian 軟 體的創始人是 John Pople (1998 年諾貝爾化學獎得主),是目前無論在學術界或工 業界使用最廣泛的量子化學計算軟體,只需提供想要計算的分子之三度空間結構, 電荷等資料並選擇一種理論計算方法,Gaussian 程式即進行大量的數值運算求出在此理論方法下之電子波函數(或電子密度)及能量,並藉此計算出分子的其他各 種化學性質:如最低能量結構、振動頻率、光電性質、以及光譜特性等。 本研究採用B3LYP以及B3PW91密度泛函數的計算方式。B3LYP以及B3PW91 都是屬於混合型泛函數(hybrid functional),都是廣泛被使用的計算方法,對於分子 結構和振動頻率的計算上具有足夠的準確性,且由於採用電子密度的估算方法, 在計算速度上具有其優勢。 本研究所採用的B3LYP是指使用Becke三參數交換泛函數[61]與LYP相關泛函 數[61,62]做計算,其中LYP是描述由 Lee、Yang、Parr 三人所提出的相關泛函數, 包含侷域(local)和非侷域(non-local)項,其計算公式如下: 𝐸𝑋𝐶𝐵3𝐿𝑌𝑃 = 0.72 × 𝐵88 + 0.08 × 𝑆 + 0.81 × 𝐿𝑌𝑃88 + 0.19 × 𝑉𝑊𝑁 + 0.2 × 𝑒𝑥𝑎𝑐𝑡 𝑒𝑥𝑐ℎ𝑎𝑛𝑒𝑡 𝑒𝑛𝑒𝑟𝑔𝑦
其中,B88是Becke 1988交換泛函數[61],S是局域自旋密度交換(local spin density exchange)泛函數[16,17,62],LYP88是Lee、Yang、Parr 1988相關泛函數[63,64], VWN是局域自旋密度(LSD)相關泛函數[65]。 而本研究另一計算方法B3PW91 [66]是指Becke三參數交換泛函數和PW91相 關泛函數[67]做計算,其中 PW91 是 1991 年由 Perdew-Wang提出的非侷域 (non-local)相關泛函數。 𝐸𝑋𝐶𝐵3𝑃𝑊91 = 𝐸𝑋𝐶𝐿𝑆𝐷𝐴+ 𝑎0(𝐸𝑋𝑒𝑥𝑎𝑐𝑡− 𝐸𝑋𝐿𝑆𝐷𝐴) + 𝑎𝑋∆𝐸𝑋𝐵88+ 𝑎𝐶∆𝐸𝑋𝑃𝑊91
20
其中,𝑎0、𝑎𝑋、𝑎𝐶是半經驗係數(semiempirical cofficients),𝐸𝑋𝑒𝑥𝑎𝑐𝑡是確切交換
能量,LSDA是侷域自旋密度交換(Local Spin Density exchange)泛函數,∆𝐸𝑋𝐵88是
Becke’s 1988相關泛函數,∆𝐸𝑋𝑃𝑊91是1991年Perdew和Wang提出做梯度修正的相關
泛函數。
Becke 三參數交換泛函數是指:Becke 利用 Hartree-Fock 的交換泛函數與 DFT 的交換相關能量(exchange-correlation-energy)泛函數混合所得的新函數方法,所以 B3LYP 與 B3PW91 計算所得修正能量結果比 Hartree-Fock 方法更準確,且由於採 用電子密度的估算方法,故其計算速度更快。
第二節 2-氟化萘分子、離子及三重態的量子計算
本研究採用用量子化學計算套裝軟體Gaussian 09,此軟體屬於電子結構計算程 式, 使用者在選擇了量子計算方法與基組後,輸入分子或離子的電荷、多重性 (multiplicity)、原子種類與初始結構(鍵長、鍵角)等參數,便可透過程式的計算 獲得分子或離子的平衡結構、振動模式、振動頻率以及能量等數據。再利用分子 與離子間在平衡結構以及振動頻率這兩種數據的差異性,即可計算出法蘭克-康登 因子的光譜數據,最後再利用OriginPro7 繪圖程式,將理論計算後所得數據點描 繪成光譜圖。 我們以B3LYP及B3PW91兩種計算方法,採用6-311++G(d,p)、6-311++G(2d,p)和 aug-cc-pVTZ 為 基 組 , 來 計 算 2- 氟 化 萘 分 子 、 離 子 以 及 三 重 態 的 平 衡 結 構 (equilibrium structure) 與 正 規 振 動 模 式 (vibrational normal mode) 的 振 動 頻 率
(vibrational frequency)。 本研究採用部分幾何優選(geometry optimization)之策略,將分子設定為Cs的結 構,尋找2-氟化萘分子、離子及三重態的穩定平衡結構,在進行幾何優選的過程中, 分子中每一個原子位置會做些許的微調,使其幾何結構持續不斷地變化,直到位 能面達到穩定點(stationary point)為止。2-氟化萘分子、離子及三重態的性質均屬於 Cs點群,為簡化計算,在結構計算的鍵長、鍵角等初始值,如果給予的數值接近 該分子的平衡結構,其計算結果將容易收斂且縮短計算時間。 最後,計算 2-氟化萘分子、離子及三重態的絕熱游離能(adiabatic ionization energy, AIE),我們首先採用 CBS-QB3 及 CBS-4M 兩種方法來運算 2-氟化萘分子態、 離子態及三重態的零點能(zero point energy, ZPE),再計算其離子態的游離能和三重 態的激發能。Petersson 等人於 1998 年開發 CBS-QB3 的化學計算模型[68],使用密 度泛函數 B3LYP 的幾何形狀和頻率來進行能量計算,最大誤差從 3.9 降到 2.8 kcal/mol,其絕對誤差從 0.98 降到 0.87 kcal/mol,大幅提高能量計算結果的可靠性 與精確性,此模型適用於化學反應的過渡態。CBS-4M 是 Petersson 等人於 1999 年 開發的最新化學計算模型[69],使用最低群體定位方法,雖然這種方法的精確度比 其他模型低,但可適用於更大的系統,在修改了一些問題後,進而提升 CBS-4M
22
和 CBS-QB3 的計算可靠性。
第三節 模擬光譜
本研究在不同計算基組的條件下,計算出兩個電子態之間振動躍遷機率的法 蘭克─康登因子,在模擬光電子光譜時,將每根光譜線的高度設定為法蘭克-康登 因子,把每個躍遷的貢獻疊加起來,並且以高斯線型函數(Gaussian line shape function)來進行光譜模擬,即可得到整個分子躍遷的模擬光電子光譜。其計算公式 如下: 𝐼(𝜔) = ∑|⟨𝑣|𝑣′⟩|2𝑒𝑥𝑝 [−4𝑙𝑛2 ( 𝜔 − 𝜔(𝑣,𝑣′) 𝜔1 2 ⁄ ) 2 ] 𝑣 其中,𝐼(𝜔)是相對強度,𝜔是相對於絕熱躍遷的激發能量,𝜔(𝑣,𝑣′)是指𝑣 → 𝑣′能 階的振動能量,𝜔1 2
⁄ 是半高寬(full-width at half maximum, FWHM)。由於本研究模
擬光譜所預設之半高寬為30 cm-1,在與實驗光譜比較時,模擬光譜的半高寬需視 實驗的解析度做適當的調整,為了能與實驗光譜進行更精確的比對,本研究模擬 之光電子光譜調整半高寬為500 cm-1,模擬之磷光光譜則將半高寬調整為200 cm-1。 將本研究的模擬光電子光譜和實驗光譜進行比較前,首先要先將實驗光譜運 用繪圖軟體(如 Photoshop 軟體)進行數位化處理,在不改變實驗光譜原圖形的原 則下,利用 Origin Pro7 軟體將其光譜圖形轉換成座標的數值,並繪製成可供精確
讀取的電腦曲線圖,再來與理論模擬光譜圖做圖形的疊合。其中需注意的是,模 擬光譜圖的橫軸單位是以波數(cm-1 )表示,而文獻中光電子光譜的實驗光譜圖橫坐 標(x 軸)對應的電子動能單位為電子伏特(eV),磷光光譜的實驗光譜圖之橫坐標所 對應的能量單位則是波數(cm-1 ),因此對於實驗的光電子光譜,必須將電子伏特轉 換成波數的單位(1 eV = 8065.64465 cm-1 ),再進行兩者比較。
第三章 研究結果與討論
本章將分成四節,第一節呈現 2-氟化萘的分子基態、離子基態及三重態的最 佳平衡結構的量子計算結果,同時標定原子標號及鍵長、鍵角;第二節比較不同 基組下,三種狀態的振動頻率與振動模式;第三節探討 2-氟化萘的光電子光譜與 磷光光譜;第四節討論其游離能與激發能。第一節 量子化學計算結果
本研究利用 Gaussian 09 套裝軟體計算出 2-氟化萘的分子基態、離子基態與三 重態的最佳化平衡結構、諧和振動頻率和正規振動模式後,再以 FCF 公式進行光 電子光譜與磷光光譜的模擬,最後用 CBS-4M 及 CBS-QB3 兩種方法計算出 2-氟化 萘的游離能與激發能,並和實驗值進行比對。一、 平衡結構
本 研 究 分 別 以 密 度 泛 函 數 B3LYP 及 B3PW91 兩 種 計 算 法 , 同 時 使 用 6-311++G(d,p)、6-311++G(2d,p)與 aug-cc-pVTZ 等三種不同基組進行 2-氟化萘的幾 何優選(geometry optimization)計算。由於資料繁多,各基組間的計算結果差異不大, 在此主要以 B3LYP/aug-cc-pVTZ 的計算基組進行結構分析,各基組所計算的平衡 結構列於附錄表 1-1 至表 1-6,振動頻率則列於附錄表 2-1 至表 2-12。26
圖 3-1- 1:2-氟化萘分子基態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ]
圖 3-1- 3:2-氟化萘三重態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ] 2-氟化萘分子基態的結構如圖 3-1-1 所示,是由 10 個碳、7 個氫和 1 個氟,三 種非金屬原子所組成,彼此間的鍵結都是共價鍵,所有原子都位於同一個平面, 其點群對稱性為 CS,共有 19 組鍵長、30 組鍵角。有 4 個 C=C 雙鍵;離子基態的 平衡結構如圖 3-1-2 所示,除了鍵長、鍵角產生變化外,C=C 雙鍵剩下 1 個;三 重態的平衡結構如圖 3-1-3 所示,鍵長與鍵角對照分子基態仍有些微差異,有 2 個 C=C 雙鍵,除此之外,三者的結構大致相似。 分析與比較平衡結構的鍵長,原子之間的鍵結主要是由 C 原子分別與 H 原子、 F 原子形成的單鍵,以及 C 原子間所形成的雙鍵和共振雙鍵。整體而言,2-氟化萘 分子基態、離子基態與三重態的鍵長由小至大排列依序為:C─H 單鍵(107.95 至
28 108.26 pm)<C─F 單鍵(131.81 至 135.26 pm)<C=C 雙鍵(134.94 至 138.40 pm)<C 與 C 的共振雙鍵(138.71 至 144.55 pm)。從原子的大小來看,H 原子<C 原子<F 原子,H 原子的體積最小,因此與 C 原子的鍵結長度最短;F 原子雖然是 2-氟化 萘組成原子中體積最大的,但在週期表中具有最高的電負度,高度的電子親和力 凌駕立體空間效應,形成 C─F 鍵長會比 C=C 雙鍵及共振雙鍵來得短的現象;由 於 C=C 雙鍵的鍵能較大,原子間作用力較強,另外共振雙鍵的 π 電子會受到氫的 吸引而游離,因此共振雙鍵的鍵長會大於 C=C 雙鍵。 觀察三種狀態 C─H 鍵的鍵長,相較於分子基態,離子基態的 C─H 鍵呈現鍵 長微幅縮短了 0.01 至 0.13 pm 的現象,唯獨 C11與 H16的鍵長由分子基態的 108.11 pm 增長為 108.18 pm。推測是由於 C11與 C14的雙鍵打開後拉大兩個碳原子間的距 離,加上與 C14連接的氟原子的影響,導致離子基態中,C11與 H16的鍵長不減反 增。而三重態 C─H 鍵的鍵長均呈現比分子基態縮短了 0.01 至 0.11 pm 的趨勢。綜 觀之,因為 C15在三種狀態中都有 C=C 雙鍵連接,加上與 C15相鄰的 C14和具有 最高電負度的氟相連,在這雙重效應的影響之下,C15 與 H17 的 C─H 鍵長最短 (107.95 至 108.04 pm);而三種狀態 C2與 H9都有最長的 C─H 鍵長(108.17 至 108.26 pm)。 而 C─F 鍵中,離子基態的 2-氟化萘因為帶正電,與最高電負度的 F 原子相互 吸引,形成了 131.81 pm 的最短鍵長,其次為三重態的 134.93 pm,最長的則是分
子基態的 135.26 pm。 至於 C=C 雙鍵的部分,分子基態有 4 組,鍵長介於 136.32 至 137.07 pm;離 子基態有 1 組,鍵長為 138.40 pm,較分子基態同位置的鍵長增加了 1.42 pm;三 重態有 2 組,鍵長分別是 134.94 及 135.83 pm,是 2-氟化萘三種狀態中,C=C 雙 鍵鍵長最短的。就 C=C 雙鍵的鍵長來看,三重態的鍵長最短、分子基態次之、離 子基態最長;在相同狀態的 2-氟化萘中,越靠近 F 原子的 C=C 雙鍵,其雙鍵的 鍵長越短。 觀察 C 與 C 的共振雙鍵中,分子基態的鍵長分布在 140.44 到 142.85 pm 之間; 離子基態則是 138.71 至 142.88 pm;而三重態是介於 140.28 與 144.55 pm 間。鍵長 的變化主要受到 C=C 雙鍵之打開或形成的影響,當雙鍵打開時,會拉開雙鍵兩側 C 彼此的距離,相對地推向與雙鍵兩側 C 相鄰的其他原子,形成雙鍵時則相反。 三種狀態中,C3與 C4間共振雙鍵的鍵長都是最長的。 就鍵角而言,大多在 120∘附近。三種狀態最小與最大的鍵角都在距離 F 原子 最近的 C14旁,像分子基態的∠C15C14F18 = 117.6∘、∠C11C14C15 = 122.9∘;離子 基態∠C11C14F18 = 117.9∘、∠C11C14C15 = 122.7∘;三重態的∠C11C14F18 = 116.3∘、 ∠C11C14C15 = 122.9∘。推測是因為受到 F 原子體積較大的立體空間效應、高電負 度造成強拉電子,以及鄰近 C=C 雙鍵的打開或形成的影響。 除此之外,當 C=C 雙鍵形成時,除了拉近雙鍵兩側 C 原子的距離,也會使 C
30 =C 雙鍵外側的鍵角變小。舉例來說,相較於分子基態,C1與 C6在三重態形成雙 鍵,C1上的∠C2C1H8從分子基態的 120.2∘縮小為 119.1∘、C6上的∠C5C6H13則 是從分子基態的 119.9∘縮小為 119.4∘。 觀察 2-氟化萘三種狀態的平衡結構,不論鍵長或者是鍵角的變化,我們可以 歸納受到下列主要因素的影響: (一)F 原子的高電負度造成的強拉電子現象。 (二)C=C 雙鍵的形成或打開。 (三)氫、碳、氟三種原子大小的立體空間效應。
二、 不同基組量子計算結果之比較
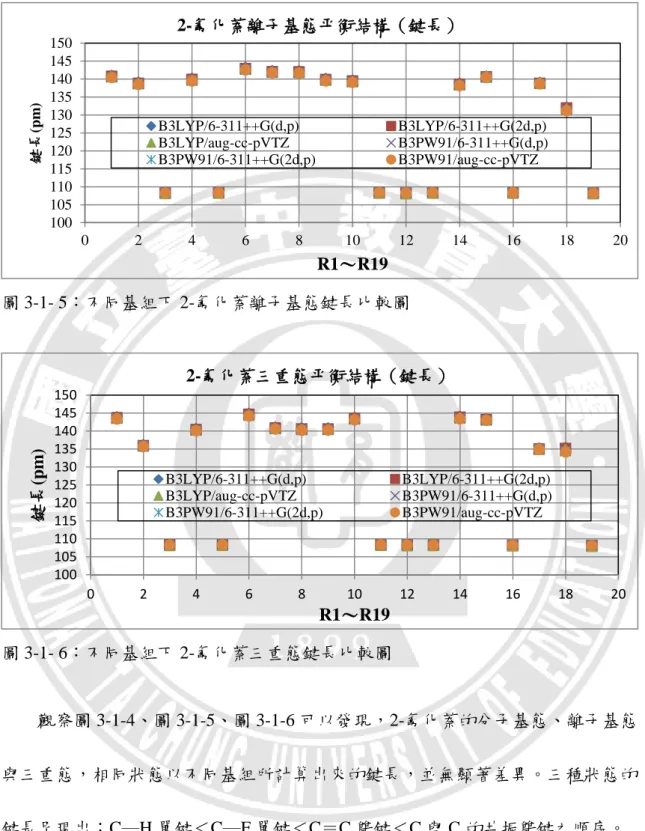
我們將附錄的表 1-1 至表 1-3,2-氟化萘分子基態、離子基態與三重態的鍵長, 分別以六種基組計算後,分別整理繪製成圖 3-1-4 至圖 3-1-6,橫坐標代表 19 組鍵 長,即 R1~R19;縱坐標則代表鍵長長度。 圖 3-1- 4:不同基組下 2-氟化萘分子基態鍵長比較圖 100 105 110 115 120 125 130 135 140 145 150 0 2 4 6 8 10 12 14 16 18 20 鍵長 (pm ) R1~R19 2-氟化萘分子基態平衡結構(鍵長) B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ圖 3-1- 5:不同基組下 2-氟化萘離子基態鍵長比較圖 圖 3-1- 6:不同基組下 2-氟化萘三重態鍵長比較圖 觀察圖 3-1-4、圖 3-1-5、圖 3-1-6 可以發現,2-氟化萘的分子基態、離子基態 與三重態,相同狀態以不同基組所計算出來的鍵長,並無顯著差異。三種狀態的 鍵長呈現出:C─H 單鍵<C─F 單鍵<C=C 雙鍵<C 與 C 的共振雙鍵之順序。 100 105 110 115 120 125 130 135 140 145 150 0 2 4 6 8 10 12 14 16 18 20 鍵長 (pm ) R1~R19 2-氟化萘離子基態平衡結構(鍵長) B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ 100 105 110 115 120 125 130 135 140 145 150 0 2 4 6 8 10 12 14 16 18 20 鍵長 (pm ) R1~R19 2-氟化萘三重態平衡結構(鍵長) B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ
32 圖 3-1- 7:不同基組下 2-氟化萘分子基態鍵角比較圖 圖 3-1- 8:不同基組下 2-氟化萘離子基態鍵角比較圖 圖 3-1- 9:不同基組下 2-氟化萘三重態鍵角比較圖 117.0 118.0 119.0 120.0 121.0 122.0 123.0 124.0 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 鍵角 ( º) A1~A30 2-氟化萘分子基態平衡結構(鍵角) B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ 117.0 118.0 119.0 120.0 121.0 122.0 123.0 124.0 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 鍵角 ( º) A1~A30 2-氟化萘離子基態平衡結構(鍵角) B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ 115.0 116.0 117.0 118.0 119.0 120.0 121.0 122.0 123.0 124.0 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 鍵角 ( º) A1~A30 2-氟化萘三重態平衡結構(鍵角) B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ
同樣將附錄表 1-1 至表 1-3,不同基組運算得到 2-氟化萘三種狀態平衡結構的 鍵角,整理繪製成圖 3-1-7 至圖 3-1-9,橫坐標代表的是 30 組鍵角、縱坐標則代表 鍵角的角度。六個基組所計算出來的鍵角沒有顯著差異,三種狀態中,最大鍵角 都在 A25(∠C11C14C15),最小鍵角除了分子基態位於 A27(∠C15C14F18)外,離子基 態與三重態則都位於 A26(∠C11C14F18)。
第二節 振動頻率與振動模式
由於 2-氟化萘有 18 個原子,其振動自由度依據 3N-6(N 為原子數)的計算公 式可算出,2-氟化萘有 48 個振動自由度,亦即有 48 種振動模式。在經過 Gaussian 09 套裝軟體計算後,三種狀態由不同基組計算出來的振動頻率整理成附錄表 2-1 至表 2-12。各表中 48 組振動頻率都是實數,可以證明理論計算的平衡結構並非過 渡態。 依據附錄表 2-1 至表 2-12 的資料,我們將六種基組計算出 2-氟化萘三種不同 狀態的振動頻率,整理成為圖 3-2-1 至圖 3-2-3 的比較圖。從這三張圖可以看到, 2-氟化萘的分子基態、離子基態與三重態,分別使用 B3LYP/6-311++G(d,p)、 6-311++G(2d,p) 、 aug-cc-pVTZ 及 B3PW91/6-311++G(d,p) 、 6-311++G(2d,p) 、 aug-cc-pVTZ 六種基組所計算出來的振動頻率,彼此間的差異並不顯著。34 圖 3-2- 1:不同基組下 2-氟化萘分子基態振動頻率比較圖 圖 3-2- 2:不同基組下 2-氟化萘離子基態振動頻率比較圖 圖 3-2- 3:不同基組下 2-氟化萘三重態振動頻率比較圖 0 500 1000 1500 2000 2500 3000 3500 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 振 動 頻 率 (cm -1) 振動模式1~48 2-氟化萘分子基態 B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ 0 500 1000 1500 2000 2500 3000 3500 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 振 動 頻 率 (cm -1) 振動模式1~48 2-氟化萘離子基態 B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ 0 500 1000 1500 2000 2500 3000 3500 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 振 動 頻 率 (cm -1) 振動模式1~48 2-氟化萘三重態 B3LYP/6-311++G(d,p) B3LYP/6-311++G(2d,p) B3LYP/aug-cc-pVTZ B3PW91/6-311++G(d,p) B3PW91/6-311++G(2d,p) B3PW91/aug-cc-pVTZ
第三節
2-氟化萘的光電子光譜與磷光光譜
本節將呈現 2-氟化萘的光電子光譜與磷光光譜的研究結果。在本研究使用的 程式中,光譜是以每個躍遷的 FCF 為高度、高斯函數為線形,並將全部的訊號強 度疊加而得,最後將光譜數據利用 Origin Pro7 進行繪圖。由於本研究採用程式所 設定的半高寬(full-width at half maximum, FWHM)為 30 cm-1,然而實驗的光譜圖解
析度通常不是很高,於是我們參考了本研究室之前所作的相關研究,將半高寬從 100 cm-1開始,以 100 cm-1的幅度做調整,逐步遞增到 600 cm-1,再與實驗光譜進 行比對,分別選出最合適的半高寬。接著對齊二者相對能量的數值,調整光譜訊 號的相對強度,使二者位於000的訊號強度一致,讓模擬光譜的圖形與實驗光譜圖 盡量吻合,以便和實驗光譜圖進行比較。
一、2-氟化萘的光電子光譜
我們依據六種不同基組所計算的結果,以 Origin Pro7 繪製成模擬的光電子光 譜,相互對照比較如圖 3-3-1 及圖 3-3-2,橫坐標代表相對能量、縱坐標則是相對 強度。從兩張比較圖可以發現,不同基組所模擬出的光電子光譜,彼此之間的差 異不大。由於資料眾多,在此僅呈現 B3LYP/aug-cc-pVTZ 的基組所模擬的光譜圖, 並與實驗光譜圖[7]進行比較,其餘模擬光譜圖則置於附錄圖 3-3-12 至圖 3-3-21。36
圖 3-3- 1:模擬的 2-氟化萘光電子光譜比較圖(B3LYP)
圖 3-3- 3:調整不同半高寬後,模擬的光電子光譜圖之比較圖 為了與低解析度的實驗光譜圖[7]做比較,我們將半高寬從 100 cm-1的位置, 以 100 cm-1的幅度調整,逐步遞增到 600 cm-1。如圖 3-3-3 之比較圖所示,為避免 模擬之光譜訊號過於密集造成不易辨識,僅列出預設的 30 cm-1、以及調整後的 200、 400、500 及 600 cm-1之光譜訊號。經過與實驗光譜圖比對後,我們採用與實驗光 譜圖最為接近,半高寬為 500 cm-1的紅色訊號。
38
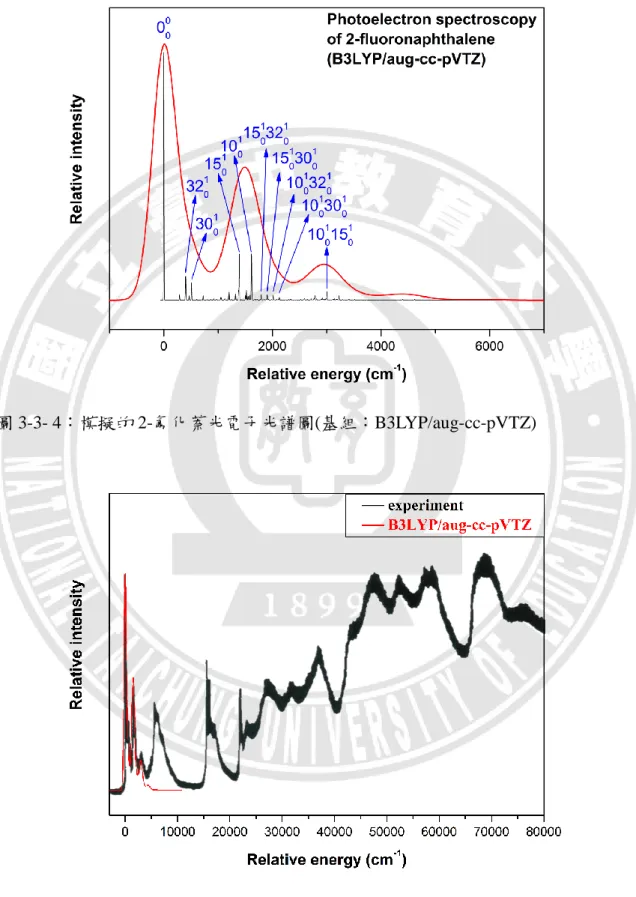
圖 3-3- 4:模擬的 2-氟化萘光電子光譜圖(基組:B3LYP/aug-cc-pVTZ)
圖 3-3-4 是以 B3LYP/aug-cc-pVTZ 的基組模擬 2-氟化萘光電子光譜圖,黑色訊 號為本研究之預設半高寬(30 cm-1 ),紅色的訊號則是將半高寬調整成 500 cm-1的低 解析度,以配合實驗光譜圖。從不同的半高寬,我們可以明顯看出躍遷譜帶所呈 現的訊號強弱關係。 如圖 3-3-5 所示,我們將模擬的光電子光譜圖與實驗光譜圖進行疊合比較,其 中相對激發能高於 5000 cm-1的訊號是離子激發態的訊號,我們模擬的離子基態相 對激發能約小於 5000 cm-1。從比較圖可以發現,本研究模擬出來的 2-氟化萘光電 子光譜,與實驗光譜都有三個較明顯的躍遷譜帶,同時相對能量位置一致,兩者 非常相似;另外在強度的分布上也幾乎雷同,僅有第三個譜帶高度略低於實驗值。 可見本研究室所研發的計算方法,用來模擬 2-氟化萘的光電子光譜,具有極高的 可靠性。
二、2-氟化萘的磷光光譜
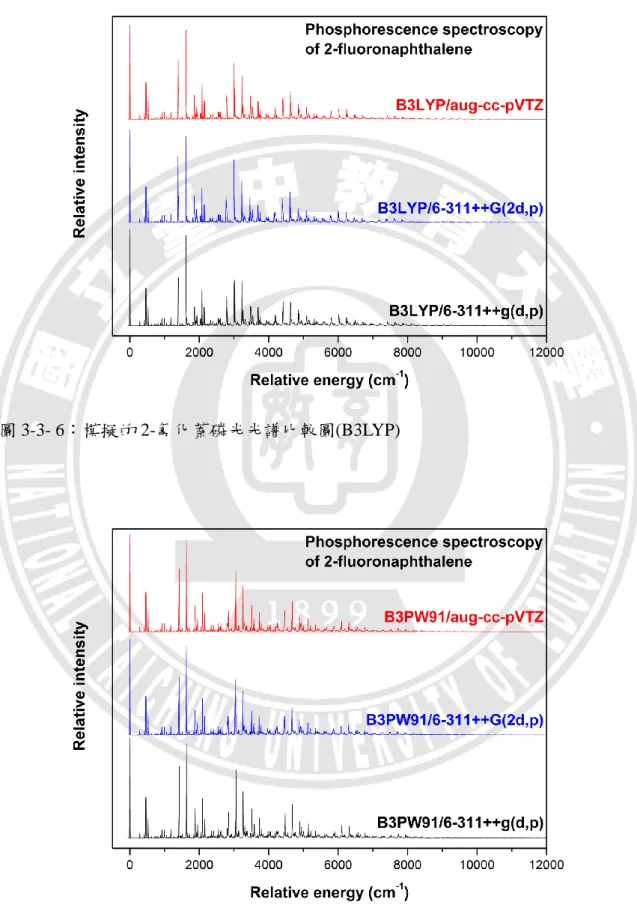
接下來我們將六種不同基組所計算的結果,分別模擬 2-氟化萘的磷光光譜, 調整適當的半高寬後,再和實驗的磷光光譜圖[4,5]進行比較。不同基組模擬出來的 磷光光譜,繪製比較圖於圖 3-3-6 及圖 3-3-7,我們可以發現,彼此之間並沒有太 大的差異。因此僅以 B3LYP/aug-cc-pVTZ 的基組所模擬的光譜圖來呈現,其餘模 擬光譜圖則置於附錄圖 3-3-22 至圖 3-3-31。40
圖 3-3- 6:模擬的 2-氟化萘磷光光譜比較圖(B3LYP)
圖 3-3- 8:調整不同半高寬後,模擬的磷光光譜圖之比較圖 為了與實驗的磷光光譜圖做比較[4,5],我們同樣將半高寬從 100 cm-1 起,以 100 cm-1的幅度調整,逐步遞增到 600 cm-1。如圖 3-3-8 所示,為避免模擬的磷光 光譜訊號過於密集造成不易判讀,僅列出預設的 30 cm-1、以及調整後的 100、200、 300 及 500 cm-1之光譜訊號。在與實驗的磷光光譜圖比對之後,我們採用與實驗光 譜圖最為接近,半高寬為 200 cm-1的紅色訊號。
42 圖 3-3- 9:模擬的 2-氟化萘磷光光譜圖(基組:B3LYP/aug-cc-pVTZ) 圖 3-3-9 是以 B3LYP/aug-cc-pVTZ 的基組計算結果繪製的模擬 2-氟化萘磷光光 譜圖,黑色訊號為本研究之預設半高寬(30 cm-1 ),而紅色訊號則是將半高寬調整為 200 cm-1的低解析度,以配合實驗光譜圖。我們仍可從不同的半高寬,看到躍遷譜 帶所呈現的訊號強弱關係。 我們從相關的研究文獻中,找到兩篇有關 2-氟化萘磷光光譜圖的實驗研究, 1954 年由 J. Ferguson 等人研究萘及其簡單衍生物的磷光光譜[4],實驗光譜圖與模 擬的磷光光譜之比較圖繪製於圖 3-3-10;1956 年,Jerome W. Siman 研究β-氟化萘 的磷光光譜[5],實驗光譜圖與模擬的磷光光譜之比較圖則呈現於圖 3-3-11。
圖 3-3- 10:模擬的磷光光譜與實驗光譜[4]比較圖 在圖 3-3-10 中,J. Ferguson 等人於 1954 年實驗所得的磷光光譜[4],訊號以黑 色線條呈現,其光譜所測得的能量從 21268 記錄到 18080 cm-1 ,其中 21268 cm-1為000, 也就是相對能量 0 的位置。轉換成相對能量後,分別在 464、948、1390、1586、 1843、2053、2768 及 2983 cm-1等位置,均有明顯的躍遷譜帶。對照紅色線條,是 本研究以 B3LYP/aug-cc-pVTZ 基組所模擬出來的磷光光譜訊號,可以觀察到在相 關的相對能量位置,也有類似的躍遷譜帶。
44 圖 3-3- 11:模擬的磷光光譜與實驗光譜[5]比較圖 在圖 3-3-11 中,黑色線條是 1956 年,由 Jerome W. Siman 所實驗得到的磷光 光譜[5],在該實驗光譜中,能量值 18554 cm-1以下的訊號為溶質萘的磷光光譜, 因此 2-氟化萘的磷光光譜訊號是在 20791 至 18554 cm-1之間,轉換為相對能量後, 則為 0 到 2237 cm-1。因此在圖 3-3-11 中所呈現之黑色的實驗光譜,我們可以忽略 在相對能量 2237 cm-1之後的光譜訊號。該實驗分別在 513、1023、1160、1382 還 有 1572 cm-1觀察到振動的躍遷譜帶。對照我們以 B3LYP/aug-cc-pVTZ 的基組所計 算繪製出來之紅色模擬光譜圖,在相對能量 0 到 2237 cm-1之間,其躍遷譜帶與實 驗的磷光光譜也極為相似。可見本研究使用的計算方法,不僅適用於光電子光譜, 在磷光光譜的模擬上,同樣具有相當的可靠性。
第四節
游離能與激發能
運用量子化學計算的結果,我們除了模擬 2-氟化萘的光電子光譜與磷光光譜, 還進行絕熱游離能(adiabatic ionization energy, AIE)以及激發能(excitation energy)的 理論計算。完備基組極限(CBS)乃多重步驟的運算,可算出非常精細的能量數值, 採用的計算方法為 CBS-4M 及 CBS-QB3。本研究先將不同基組量子計算所得數據, 運用上述兩種計算方法,分別算出 2-氟化萘分子基態、離子基態與三重態在絕對 溫度 0 度(0 K)的零點能(zero point energy, ZPE),再計算出由分子基態游離到離子 基態的絕熱游離能,以及從三重態發出磷光回到分子基態的激發能,最後和實驗 值進行比較。由於實驗值所測得之能量分別有 eV 及 cm-1兩種單位,而本研究所計 算出來的能量單位為 hartree,故利用下列公式進行單位換算: 1 eV = 0.03674932379 hartree = 8065.64465 cm-1 表 3- 1:2-氟化萘以不同基組與方法所計算之絕熱游離能與實驗值[7,10]對照表 能量 方法 狀態 ZPE (hartree) 絕熱游離能(AIE) Experiment (最小差距) hartree eV cm-1 CBS-4M 分子態 -484.406912 0.307343 8.3632 67455 8.23 eV [7] (1.4%) 66771 cm-1 [10] (0.8%) 離子態 -484.099569 CBS-QB3 分子態 -484.319343 0.306682 8.3452 67310 離子態 -484.012661 表 3-1 列出 2-氟化萘將六種基組的量子計算結果,以 CBS-4M 及 CBS-QB3 這 兩種方法所計算出來的絕熱游離能,兩者計算的結果均略大於實驗值。CBS-4M 計 算得到的絕熱游離能為 8.3632 eV (67455 cm-1 ),與實驗值[7]的差距為 0.1332 eV,與
46 實驗值[10]則為 684 cm-1;CBS-QB3 則是 8.3452 eV (67310 cm-1 ),與實驗值的差距 分別為 0.1152 eV [7]及 539 cm-1 [10]。兩相比較之下,CBS-QB3 所計算的結果比 CBS-4M 更接近實驗值。 表 3- 2:2-氟化萘以不同基組與方法所計算之激發能與實驗值[4,5]對照表 能量 方法 狀態 ZPE (hartree) 激發能 Experiment (最小差距) hartree eV cm-1 CBS-4M 三重態 -484.293922 0.112990 3.0746 24799 21268 [4] (2.3%) 20791 [5] (4.6%) 分子態 -484.406912 CBS-QB3 三重態 -484.220251 0.099092 2.6964 21748 分子態 -484.319343 在激發能方面,由於實驗值[4,5]使用的能量單位都是 cm-1,在表 3-2 所呈現的 激發能則從 hartree 換算成 cm-1,與絕熱游離能的計算有類似的結果,理論計算得 到的激發能都稍高於實驗值。CBS-4M 計算所得的激發能為 24799 cm-1,與實驗值 之差距為 3531 [4]及 4008 cm-1 [5];CBS-QB3 則是 21748 cm-1,對照實驗值,其差 距分別是 480 [4]及 957 cm-1 [5]。可見以 CBS-QB3 的方法所計算出來的激發能仍然 與實驗值較為接近。 相較於絕熱游離能,激發能所計算出來的數值與實驗值之差距,看似比較大。 探究其原因,推測其可能性有兩個:首先是實驗時的溫度、其次是溶劑(雜質, impurity)的存在。本研究是分別以 2-氟化萘分子基態、離子基態及三重態在絕對 溫度 0 度(0 K)的零點能,來計算絕熱游離能與激發能;Fergus 等人於 1954 年的實 驗中,溫度為 77 K,以純化的石油醚(light petroleum)當溶劑;而 Sidman 在 1955
年的實驗則是在 20 K 的溫度下,混雜了萘晶體作為溶劑所測得的磷光光譜。 在能量計算上,2-氟化萘的絕熱游離能和激發能,不論用 CBS-4M 或 CBS-QB3 這兩種方法,均略高於實驗值。其中 CBS-QB3 的方法所計算得到的能量,都比 CBS-4M 的方法更為接近實驗值。其中計算的絕熱游離能與實驗值的最小差距僅有 0.8 [10]及 1.4% [7];激發能的最小差距則稍大,分別為 2.3 [4]及 4.6% [5]。 本研究對於 2-氟化萘的量子計算,不論是平衡結構、振動頻率、絕熱游離能 與激發能,與實驗值的差異都不大;而且模擬的光電子光譜圖和磷光光譜圖,與 實驗光譜圖的吻合度也非常高,足見本研究使用的方法,對於多環芳香烴及其衍 生物,具有相當高的實用性與可靠性。
![圖 3-1- 1:2-氟化萘分子基態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ]](https://thumb-ap.123doks.com/thumbv2/9libinfo/7443831.109454/34.892.130.769.147.1088/圖3112氟化萘分子基態的平衡結構及原子標號B3LYPaugccpVTZ.webp)
![圖 3-1- 3:2-氟化萘三重態的平衡結構及原子標號[B3LYP/aug-cc-pVTZ] 2-氟化萘分子基態的結構如圖 3-1-1 所示,是由 10 個碳、7 個氫和 1 個氟,三 種非金屬原子所組成,彼此間的鍵結都是共價鍵,所有原子都位於同一個平面, 其點群對稱性為 C S ,共有 19 組鍵長、30 組鍵角。有 4 個 C=C 雙鍵;離子基態的 平衡結構如圖 3-1-2 所示,除了鍵長、鍵角產生變化外,C=C 雙鍵剩下 1 個;三 重態的平衡結構如圖 3-1-3 所示,鍵長與鍵角對照分子基態仍有些](https://thumb-ap.123doks.com/thumbv2/9libinfo/7443831.109454/35.892.128.770.138.957/三重態及原子萘分子個氫和有原子一個平面其點群對稱性如圖照分子.webp)

![圖 3-3- 3:調整不同半高寬後,模擬的光電子光譜圖之比較圖 為了與低解析度的實驗光譜圖[7]做比較,我們將半高寬從 100 cm -1 的位置, 以 100 cm -1 的幅度調整,逐步遞增到 600 cm -1 。如圖 3-3-3 之比較圖所示,為避免 模擬之光譜訊號過於密集造成不易辨識,僅列出預設的 30 cm -1 、以及調整後的 200、 400、500 及 600 cm -1 之光譜訊號。經過與實驗光譜圖比對後,我們採用與實驗光 譜圖最為接近,半高寬為 500 cm -1 的紅色訊號。](https://thumb-ap.123doks.com/thumbv2/9libinfo/7443831.109454/45.892.129.769.178.991/光電子之光譜訊號過於密集造成不易辨識僅列出預設以及後的訊號.webp)

![圖 3-3- 8:調整不同半高寬後,模擬的磷光光譜圖之比較圖 為了與實驗的磷光光譜圖做比較[4,5],我們同樣將半高寬從 100 cm -1 起,以 100 cm -1 的幅度調整,逐步遞增到 600 cm -1 。如圖 3-3-8 所示,為避免模擬的磷光 光譜訊號過於密集造成不易判讀,僅列出預設的 30 cm -1 、以及調整後的 100、200、 300 及 500 cm -1 之光譜訊號。在與實驗的磷光光譜圖比對之後,我們採用與實驗光 譜圖最為接近,半高寬為 200 cm -1 的紅色訊號。](https://thumb-ap.123doks.com/thumbv2/9libinfo/7443831.109454/49.892.128.769.176.984/磷光光譜訊號過於密集造成不易判讀僅列出預設的以及調整之光訊號.webp)
![圖 3-3- 10:模擬的磷光光譜與實驗光譜[4]比較圖 在圖 3-3-10 中,J. Ferguson 等人於 1954 年實驗所得的磷光光譜[4],訊號以黑 色線條呈現,其光譜所測得的能量從 21268 記錄到 18080 cm -1 ,其中 21268 cm -1 為0 00 , 也就是相對能量 0 的位置。轉換成相對能量後,分別在 464、948、1390、1586、 1843、2053、2768 及 2983 cm -1 等位置,均有明顯的躍遷譜帶。對照紅色線條,是 本研究以 B3LYP/aug](https://thumb-ap.123doks.com/thumbv2/9libinfo/7443831.109454/51.892.129.772.180.973/年實磷光光譜號以色線條呈光譜所測能量記錄其中也就是相量後分別.webp)
