國
立 交 通 大 學
生物資訊及系統生物研究所
博士論文
分析微小核醣核酸與目標基因間的作用關係及尋找
標靶基因的應用
In Silico Analysis of microRNA-Target Interaction
and Its Application for miRNA Target
Identification
研 究 生:許勝達
指導教授:黃憲達 博士
分析微小核醣核酸與目標基因間的作用關係及尋找標靶基因的應用
In Silico Analysis of microRNA-Target Interaction and Its Application
for miRNA Target Identification
研 究 生:許勝達
Student:Sheng-Da Hsu
指導教授:黃憲達 博士
Advisor:Hsien-Da Huang
國 立 交 通 大 學
生 物 資 訊 及 系 統 生 物 研 究 所
博 士 論 文
A Thesis
Submitted to Institute of Bioinformatics and Systems Biology
College of the Biological Science & Technology
National Chiao Tung University
in partial Fulfillment of the Requirements
for the Degree of
Ph.D.
in
Bioinformatics and Systems Biology
August 2011
分析微小核醣核酸與目標基因間的作用關係及尋找
標靶基因的應用
學生:許勝達 指導教授:黃憲達 博士
國立交通大學 生物資訊及系統生物研究所
摘要
微小核醣核酸(microRNA/miRNA)是一段長度約為 22 核苷酸的非編碼的核醣核 酸分子,它們可藉由與基因的結合來調控基因的表現,研究發現微小核醣核酸調控許 多的基因與細胞功能有關,例如,細胞凋零、細胞分化與細胞發育。之前的文獻認為 30%或更多的人類基因受到微小核醣核酸的調控。由於微小核醣核酸在生物的重要性, 所以許多相關的資料庫及工具被開發出來。本文主要是分析微小核醣核酸與其目標基 因間的作用關係,並探討此關係的序列及結構特徵,根據此分析結果開發新的方法或 整合系統研究微小核醣核酸的功能。我們總共分析了1,524 個實驗驗證過的微小核醣 核酸與目標基因的作用關係,並以此作為評估預測正確性的資料。通過分析已知的微 小核醣酸與其目標基因的作用關係,我們可以獲得其序列與結構上的特徵。這些分析 與實驗已知的資料對於尋找標靶基因工具的開發是很重要,且生物學家可以透過此結 果選擇適當的預測工具。此外,還有其他因素會影響微小核醣核酸是否會與目標基因 結合,例如,核醣核酸結合的蛋白質(RNA 誘導沉默複合體)、微小核醣核酸及信息核 醣核酸的濃度。未來我們會將這些因素加入分析微小核醣核酸與目標基因的作用關 係。In Silico Analysis of microRNA-Target
Interaction and Its Application for miRNA
Target Identification
Student: Sheng-Da Hsu Advisor : Dr. Hsien-Da Huang
Institute of Bioinformatics and Systems Biology, National
Chiao Tung University
Abstract
MicroRNAs (miRNAs) are small non-coding RNA molecules (~ 22-nt) that can bind to one or more target sites on gene transcript to negatively regulate protein expression and thus control numerous cellular mechanisms. Recent work supports miRNAs downregulate gene expression during various crucial cell processes such as apoptosis, differentiation and development. Previous research has suggested that miRNAs regulate 30% or more of the human protein-coding genes. As the important roles of miRNAs, there were multiple databases storing the miRNA-target interactions (MTIs) identified using different tools. The aim of this work is to systematically analyze the miRNA-target interactions and assess the function of miRNA by developing new methods and resources. In this study, we analyzed 1,524 experimentally verified miRNA-target interactions with strong evidence support in human and elucidate which the more accurate microRNA target prediction database is. Through analyzing the verified MTIs, we could get the overview of relative contribution of sequence and structure features in miRNA targeting. Those analyses are important for identifying putative miRNA targets and are very useful for biologist
factors in future works in order to develop the more reliable miRNA target prediction resources.
誌謝
首先,我要感謝指導教授黃憲達博士在我研究所生涯中對於我的細心指導,使得我 可以在生物資訊這個領域內從無到有的學習到許多知識,也在學術研究上有顯著的進步 及成長。話說當初因緣際會之下進入microRNA 這個領域,唯哲帶著我跟立人著手建構 相關的資料庫,讓我學到如何規劃設計資料庫及相關生物知識分享,透過與陽明大學鄒 安平教授的合作,使我知道原來實驗與計算可以如此緊密結合。最近的兩年內由於致宏、 昭昉、Sirjana、Anas、梁超、維芸、煒志、雯玲、文婷、冠州、明家很認真努力的唸 paper,並從文獻中萃取出實驗驗證過的 microRNA 目標基因、實驗方法、相關疾病, 因此才可以建構如此完整的實驗已知基因資料庫。除此之外,還有一路陪著我成長的熙 淵、博凱、宗夷、威霽、佳宏、緯允、豐茂、詠薷、佳融、致閔,不論是研究或生活都 給予我極大的幫助。此外已經畢業的燕茹、瑞鴻、冠樺、恆嘉、定遠、家慧、伯瑲、在 營、美雪、恆毅、至昶,和大家一起奮鬥的日子,是我成長的動力,實驗室內的點點滴 滴更是美好的回憶。最後,我要特別感謝我的家人及正景給予我的支持,謝謝你們給予 我的支持與鼓勵,才能讓我無後顧之憂的求學。能夠順利完成博士論文並取得博士學位, 是大家的指導、支持、與鼓勵,誠心的謝謝大家,將這份喜悅及成果與關心我的所有人 一同分享。This thesis is based on the following publications:
Hsu, S.D., Lin, F.M., Wu, W.Y., Liang, C., Huang, W.C., Chan, W.L., Tsai, W.T., Chen, G.Z., Lee, C.J., Chiu, C.M. et al. (2011) miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic acids research, 39, D163-169.
Hsu, J.B.K.*, Chiu, C.M.*, Hsu, S.D.*, Huang, W.Y., Chien, C.H., Lee, T.Y. and Huang, H.D. (2011) miRTar: an integrated system for identifying miRNA-target interactions in Human. BMC Bioinformatics, 12.
Tsai, W.C.*, Hsu, S.D.*, Hsu, C.S.*, Hsiao, M.S., Huang, H.D., Chen, S.J., Huang, R.S.Y., Huang, Y., Lai, T.C., Tsai, T.F., Chen, H.C., Hsu, M.T., Wu, J.C., Hsiao, M., Tsou, A.P. (2011) MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. (In preparation)
Lin, F.M., Hsu, S.D., Chou, C.H. and Huang, H.D. (2011) HomoloMTI: Homologous Cluster of MicroRNA Target Interactions. (In preparation)
Hsu, S.D., Chu, C.H., Tsou, A.P., Chen, S.J., Chen, H.C., Hsu, P.W., Wong, Y.H., Chen, Y.H., Chen, G.H. and Huang, H.D. (2008) miRNAMap 2.0: genomic maps of microRNAs in metazoan genomes. Nucleic Acids Res, 36, D165-169.
Hsu, P.W., Lin, L.Z., Hsu, S.D., Hsu, J.B. and Huang, H.D. (2007) ViTa: prediction of host microRNAs targets on viruses. Nucleic Acids Res, 35, D381-385.
Hsu, P.W., Huang, H.D., Hsu, S.D., Lin, L.Z., Tsou, A.P., Tseng, C.P., Stadler, P.F., Washietl, S. and Hofacker, I.L. (2006) miRNAMap: genomic maps of microRNA genes and their target genes in mammalian genomes. Nucleic Acids Res, 34, D135-139.
Table of Contents
摘要 ... iii
Abstract ... iv
誌謝 ... vi
Table of Contents ... viii
List of Figures ... xi
List of Tables ... xiii
Chapter 1 Introduction ... 1
1.1 Biological background ... 2
1.1.1 miRNA Biogenesis ... 2
1.1.2 Intronic and exonic miRNAs ... 4
1.1.3 Functions of miRNAs ... 4
1.1.4 miRNA-target interactions (MTIs) ... 5
1.2 Principles of miRNA target prediction ... 5
1.2.1 Seed sequence complementary ... 5
1.2.2 Conservation of miRNA targets ... 7
1.2.3 Thermodynamics of miRNA:mRNA duplex ... 7
1.2.4 Site accessibility ... 7
1.3 Motivation ... 8
1.4 Research goals ... 8
1.4.1 Construction of experimentally verified MTIs ... 9
1.4.2 Development of predicted MTIs system ... 9
1.4.3 Identification of homologous MTIs ... 10
1.4.4 Integrated information of MTIs database ... 10
Chapter 2 miRTarBase: a database curates experimentally validated microRNA-target interactions ... 12
2.4.2 Strong experimental evidence of miRNA-target interactions ... 19
2.4.3 Less strong experimental evidence of miRNA-target interactions ... 20
2.5 Results... 20
2.5.1 Statistics ... 20
2.5.2 Gene enrichment and pathway analysis ... 22
2.5.3 Comparisons to other miRNA-target interaction databases ... 26
2.5.4 Web interface... 28
2.6 Conclusion ... 30
Chapter 3 miRTar - an integrated system for identifying miRNA-target interactions in Human ... 32
3.1 Introduction ... 32
3.2 Related works ... 32
3.3 Specific aims ... 35
3.4 Materials and methods ... 36
3.4.1 Data collection ... 37
3.4.2 Identifying miRNA target sites in human ... 38
3.4.3 Exon/Intron boundary recognition ... 40
3.4.4 Identifying different types of alternatively spliced exons ... 40
3.4.5 Alternative splicing effects to miRNA regulation ... 41
3.4.6 GSEA for miRNA-regulated genes ... 42
3.4.7 The approximate runtime of miRTar ... 44
3.5 Utility and discussion ... 44
3.5.1 Case study of alternatively spliced target-containing exon ... 44
3.5.2 Case study of cancer-associated gene group ... 46
3.5.3 Case study of miR-122 target analysis in mouse ... 46
3.5.4 Comparison with other miRNA target prediction web servers ... 47
3.6 Discussion ... 48
3.7 Conclusion ... 50
Chapter 4 Homologous cluster of miRNA target interactions ... 52
4.1 Introduction ... 52
4.2 Specific aims ... 53
4.3 Materials and method ... 53
4.4.1 Statistics ... 54
4.4.2 Case Study of homologous miR-122::ALDOA ... 55
Chapter 5 miRNAMap – integrated database of microRNA-target interactions ... 57
5.1 Introduction ... 57
5.2 Related works ... 58
5.3 The specific aims ... 60
5.4 Improvement ... 61
5.5 Materials and methods ... 63
5.5.1 Annotation resources of Protein-coding gene ... 63
5.5.2 Integration of external human miRNA target prediction databases ... 64
5.5.3 Identification of novel miRNA targets in metazoan ... 66
5.5.4 Expression profiling of microRNAs and target genes ... 68
5.6 Results... 69
5.6.1 miRNA target prediction database similarities ... 69
5.6.2 Comparing miRNA target prediction databses to miRTarBase ... 71
Chapter 6 Conclusion ... 73
References ... 75
Appendix I Supporting material ... 85
Appendix II Experimentally verified miRNA target interactions in human ... 87
Appendix III List of abbreviations ... 116
Appendix IV Supplementary figures ... 117
List of Figures
Figure 1. Growth of miRNA genes in the miRBase database and growth of the keywords with
‘miRNA’ and keyword with ‘miRNA target’ in PubMed. ... 2
Figure 2. The biogenesis of miRNAs. ... 3
Figure 3. miRNA functions. ... 4
Figure 4. miRNA-target interactions (MTIs). ... 5
Figure 5. Seed region of miRNA:mRNA duplex... 6
Figure 6. Type of miRNA target sites (seed type). (Defined by Grimson, A. 2007 and Bartel, D. P., 2009) ... 6
Figure 7. Three types of conserved miRNA targets. ... 7
Figure 8. The accessibility of target site. ... 8
Figure 9. This thesis is consist of four parts: miRTarBase, miRTar, miRNAMap and HomoloMTI. . 9
Figure 10. The concept of miRNA-target interaction validated by GFP reporter assay. ... 13
Figure 11. The result page of TarBase... 16
Figure 12. The web interface of miRecords. ... 17
Figure 13. System flow of miRTarBase. ... 19
Figure 14. The distribution in number of human miRNA target genes ... 22
Figure 15. GO analysis of human miRNA-target interactions, as verified by reporter assay or western blot. (DAVID, grouped according to biological process of level 1) ... 24
Figure 16. Histogram of various features of experimental proven miRNA-target sites. ... 26
Figure 17. The overlap among articles collected in each two manually curated miRNA-target interactions databases. ... 28
Figure 18. The miRTarBase web interface ... 30
Figure 19. Concept that underlies miRTar. ... 37
Figure 20. System flow of miRTar. ... 38
Figure 21. Analysis to identify miRNA target genes in KEGG pathway maps. ... 43
Figure 22. miRNA target genes in KEGG pathway map. ... 44
Figure 23. miR-148 targets protein coding region of DNMT3b in human. ... 45
Figure 24. hsa-let-7a can target on FOXA1 (148) ... 50
Figure 25. The construction diagram of HomoloMTIs. The database integrated three databases and predicted miRNA target sites on the homologous genes genes based on the experimental proved MTIs from miRTarBase. ... 54
Figure 26. The homologous MTI group of miR-122 and target gene ALDOA. ... 56
Figure 27 Graphical web interface of miRNAMap ... 60
Figure 29. Criteria supported in the miRNA target search function for miRNAMap 2.0. (a)
Criterion 1 is to select the potential miRNA target sites, which are predicted by at least two tools; criterion 2 is to select the target gene that contains multiple target sites; (b) RNA accessibility is incorporated into the detection of miRNA target sites. ... 67
Figure 30. The overlap of different datasets provided by the same database. ... 69 Figure S1. The four classes of miRNAs which are categorized by their genomic locations relative
to the known genes (adapt from Kim, V. N. 2009)(61). ...117
Figure S2. Different mechanism of miRNA and siRNA biogenesis. (adapt from Lin, S.L et al 2006)
(178) ...118
Figure S3. The miR-196a target site is conserved in Hoxb8. (adapt from Mansfield, J.H. et al,
2004) (179) ...118
Figure S4. The principle of SILAC. ...119 Figure S5. Matthias Selbach et al developed pSILAC to detect widespread changes in protein
List of Tables
Table 1. The related databases stored experimentally verified MTIs. ... 15
Table 2. Statistics of miRNA-target interactions collected in miRTarBase. ... 21
Table 3. KEGG Pathway annotation of human miRNA-target interactions ... 24
Table 4. Comparison of miRTarBase with other miRNA-target interaction databases. The final column is the number of miRNA-target interactions (MTIs) more than the largest number of records in TarBase, miRecords and miR2Disease. The miRNA-target interactions in TarBase, miRecords and miR2Disease are downloaded from their web sites. ... 27
Table 5. The comparisons of miRNA target prediction tools. ... 34
Table 6. Data statistics and data obtained from databases. ... 38
Table 7. Statistics the various types of alternative splicing exons between two different data sources. ... 41
Table 8. Statistics of miRNA target site locations ... 42
Table 9. Statistics of miRNA target sites within different types of alternatively spliced exons. .... 49
Table 10. Statistics of homologous miRNA-target interactions collected in HomoloMTI ... 55
Table 11. The related resources of MTI. ... 58
Table 12. Numbers of mature miRNAs categorized by type of species in miRNAMap 2.0 and 3.0 ... 61
Table 13. Enhancements and new features of miRNAMap 3.0 ... 62
Table 14. The list of the integrated external data sources in miRNAMap. ... 63
Table 15. The external link of integrated miRNA target prediction databses. ... 65
Table 16. Statistics of predicted human MTIs from different prediction databases. ... 65
Table 17. The statistics of external miRNA expression profiles. ... 69
Table 18. Biological features of miRNA target prediction resources. (176) ... 70
Table 19. Predictive performance across miRNA-target interaction databases by using experimentally verified MTIs with strong evidence support in miRTarBase. ... 72
Chapter 1 Introduction
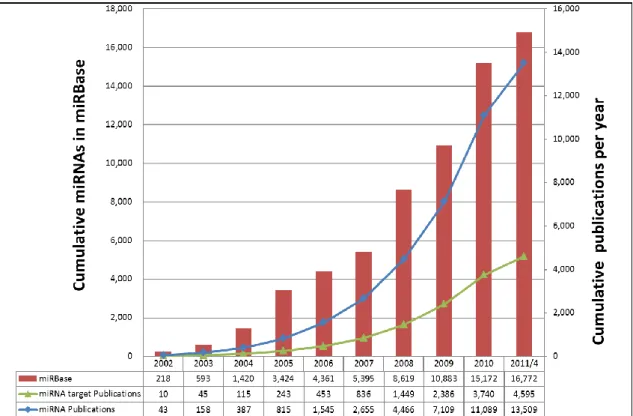
As small non-coding RNAs of approximately 22 nts, microRNAs (miRNAs) regulate gene expression post-transcriptionally through suppressing mRNA translation or inducing mRNA degradation by hybridizing to the 3’-untranslated regions (3’-UTR) of the mRNAs. Discovery of the first miRNA in Caenorhabditis elegans in 1993 (1) ushered in numerous studies on the cellular processes of these tiny regulatory RNAs for a large variety of metazoa. Thousands of miRNAs have been identified in mammalian cells over the past two decades. miRNAs play critical roles in many biological processes, including cell cycle control, cell growth and differentiation, apoptosis, and embryo development. Since their discovery, miRNAs have been found in many organisms. Until now, the miRBase (version 17) (2) contains 16,772 miRNAs which were discovered in 153 different species, the latest amount of miRNAs in miRBase is about ten times larger than the amount in 2004 (Figure 1). In spite of a large number of miRNAs have been identified, most of them are unknown the functions. This thesis is a compilation of the following 5 journal articles (3-6) and of unpublished data. It is consisted of the following chapters: in Chapter 2, the description of miRTarBase, a database which is the most updated collection of miRNA-target interactions (MTI), has accumulated 3,969 experimentally verified MTIs between 625 miRNAs and 2,433 target genes among 17 species by manually surveying pertinent literature. In Chapter 3, we demonstrate the miRNA target prediction system, miRTar, to enable biologists to easily identify the biological functions and regulatory relationships between a group of known/putative miRNAs and protein coding genes. Chapter 4, an application of using these resources or method to extend the experimentally verified miRNA-target interactions from one species to other species is presented. Finally in Chapter 5, presents miRNAMap a database updated to version 3, which was specifically designed to integrate 12 well-known miRNA-target interaction prediction databases and MTIs calculated by miRTar. We further compare
Figure 1. Growth of miRNA genes in the miRBase database and growth of the keywords with
‘miRNA’ and keyword with ‘miRNA target’ in PubMed.
1.1 Biological background
1.1.1 miRNA Biogenesis
microRNAs (miRNAs) are small non-coding RNAs of ~22 nt sequences that could regulate gene expression via hybridizing to 3’ untranslated regions (3’-UTR), resulting in mRNA degradation and inhibiting mRNA translation (1,7-9). The major function of miRNAs is to repress the gene expression at post-transcriptional level (10). Recent work supports miRNAs downregulate gene expression during various crucial cell processes such as apoptosis (11-23), differentiation, development (24-47) and tumor growth (37-48).
The general biogenesis of the miRNA is shown in Figure 2. These microRNA (miRNA) genes are typically transcribed as primary miRNA (pri-miRNA) by RNA polymerase II (Pol II) (49,50) or RNA polymerase III (Pol III) (50,51) in the nucleus. If the miRNAs are transcribed by Pol II, the kind of primary miRNA transcripts (pri-miRNAs) contain cap structures as well as the poly(A) tails, which are the unique properties of class II gene transcripts. Then, the pri-miRNAs are processed into the
precursor of miRNAs (pre-miRNAs) by a protein complex - RNase III enzyme Drosha and DGCR8 (Pasha) (52). It’s an essential process for the most miRNAs to release pre-miRNA, but a small group of miRNAs located within introns can bypass this step (53,54). Figure
S2 shows the possible biogenesis of intronic miRNAs. The pre-miRNA (~70 nts) is folded
as a stem-loop (hairpin) structure which contains a short nucleotide (~17-24 nts) sequences embedded in the stem part of hairpin. The pre-miRNA is exported from the nucleus to the cytoplasm by Exportin 5. The pre-miRNA is then processed by the enzyme DICER (55-57) into a dsRNA (double strand RNA) that includes the ~22 nts mature miRNA and miRNA*. This dsRNA is further processed to the mature sequence, which becomes part of the RNA-induced silencing complex (RISC) (58-60).
1.1.2 Intronic and exonic miRNAs
According to the miRNA locations of mRNA transcripts, four full-length pri-miRNAs have been characterized in Figure S1 (61). Figure S1 (a) miR-15a~16-1 cluster are intronic miRNAs which locate on non-protein-coding transcript (DLEU2 is a well-defined non-coding RNA gene) (62). Figure S1 (b) miR-155 is located in the exon on non-protein-coding transcript (BIC). Figure S1 (c) miR-25~93~106b cluster is embedded in the intron of MCM7 transcript. Figure S1 (d) miR-985 was found in the last exon of CACNG8. The possible intronic miRNA biogenesis is shown in Figure S2.
1.1.3 Functions of miRNAs
The mature miRNA then binds to complementary sites in the mRNA target to negatively regulate gene expression through two major mechanism. Figure 3 shows the major function of miRNAs in two mechanisms: mRNA degradation and translation repression. In plant, many miRNA target sites have perfect hybridization between miRNAs and their sites, and they cause mRNA degradation (7,63,64). However, not all the miRNAs in plants induce mRNA degradation; some of them may inhibit the mRNA translation through hybridizing to their target genes imperfectly. In animals, miRNAs are imperfectly complementary to the target mRNA which usually locates in 3’-untranslated region (3’-UTR). As shown in Figure 3, when miRNA-target interactions with perfect complementarity tend to result in mRNA cleavage and degradation; and miRNA-target interactions with imperfect complementarity tend to result in blocking ribosome processing and inhibiting mRNA translation.
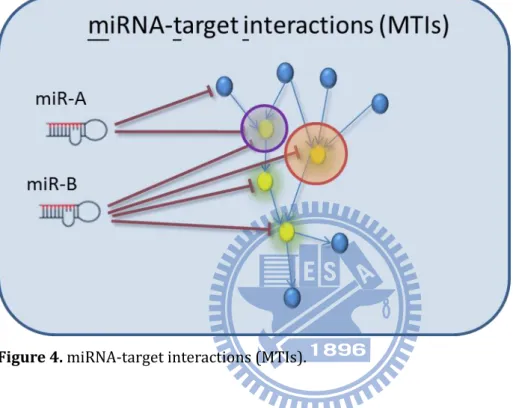
1.1.4 miRNA-target interactions (MTIs)
Figure 4 shows the definition of miRNA-target interaction (MTI). Each red line denotes
the repressed relationship between miRNA and its target gene. As shown in this figure, miR-A repress two target genes; miR-B repress four target genes. In other words, there are six miRNA-target interactions shown in this figure.
Figure 4. miRNA-target interactions (MTIs).
1.2 Principles of miRNA target prediction
1.2.1 Seed sequence complementary
There are a lot of tools or resources developed to identify the miRNA target genes. Almost all of them majorly consider this rule – perfect seed complementary to its target site. The seed region is located at nucleotides 2-7 or 2-8 of the 5’ end of miRNA. The site of mRNA hybridizes to seed region called “seed match”. Figure 5 clearly shows the cartoon picture of “seed region” and “seed match”. Previous works report that the perfect
Figure 5. Seed region of miRNA:mRNA duplex.
Lewis et al discovered that conserved seed matches in vertebrates often have A anchors which maybe pair to the first position of miRNA (66). Grimson et al clearly defined the seed types of miRNA target sites – 6mer, 7mer-A1, 7mer-m8 and 8mer (67).
Figure 6 shows the seed types of miRNA target sites. 6mer site is the 6 continues perfect
Watson-Crick pairs hybridize to miRNA seed region (nucleotides 2-7 of miRNA). If nucleotides 2-8 of miRNA perfectly hybridize to its target site, we call this site as 7mer-m8 (seed match + match at position 8). 7mer-A1 site means that seed match flanked A anchor which may pair to first nucleotide of miRNA or not. The 8mer site comprises the seed match flanked by both the match at position 8 and the A at position 1. It is quite reasonable that considering 8mer site could increase specificity, whereas searching for 6nt seed pairing (6mer) yields greater sensitivity.
Figure 6. Type of miRNA target sites (seed type). (Defined by Grimson, A. 2007 and
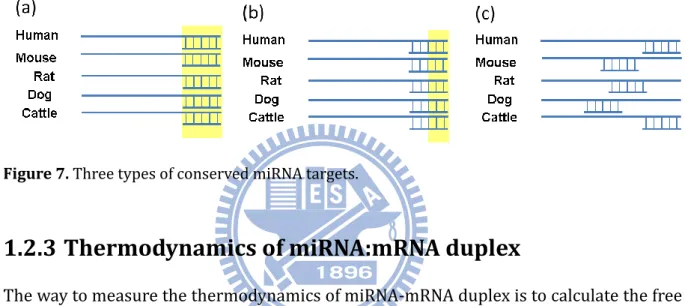
1.2.2 Conservation of miRNA targets
Conserved miRNA target is the other important rule for identifying miRNA target genes. miRNA target sites that are conserved across species are likely to be biologically significant miRNA target sites. However, the different reports sometimes took into account slightly different conservation of miRNA targets. We could conclude that three types of conserved miRNA targets as shown in Figure 7. Figure S3 shows the highly conserved miR-196a target site in Hoxb8.
Figure 7. Three types of conserved miRNA targets.
1.2.3 Thermodynamics of miRNA:mRNA duplex
The way to measure the thermodynamics of miRNA-mRNA duplex is to calculate the free energy of miRNA:mRNA targets site. The Free Energy of the microRNA:mRNA duplex (∆G), is often calculated with the Vienna RNA package (69) or RNAhybrid (70).
1.2.4 Site accessibility
The conventional target prediction tools consider the complementarity between the miRNA and its target sequence, the conservation of the target sites, and the kinetics and thermodynamics of miRNA::target duplex. Although these properties are important factors to determine the miRNA target sites, the sequence context surrounding miRNA
modified lin-41 3’-UTR sequences. miRNAs hybridize to the target sites, which is within more accessible regions, are with more possibility to be real, as shown in Figure 8.
Figure 8. The accessibility of target site.
1.3 Motivation
Although there were more than one thousands human miRNA genes have been discovered, the functions of most of them are unknown. Figure 1 shows that the exponential growth of miRNA related publications in Pubmed, it is quite urgent to extract the useful information from those articles. An up-to-date curated collection of miRNA-target interactions (MTIs) with experimental support is crucial to provide effective information for investigating miRNA functions at different conditions and in different species.
1.4 Research goals
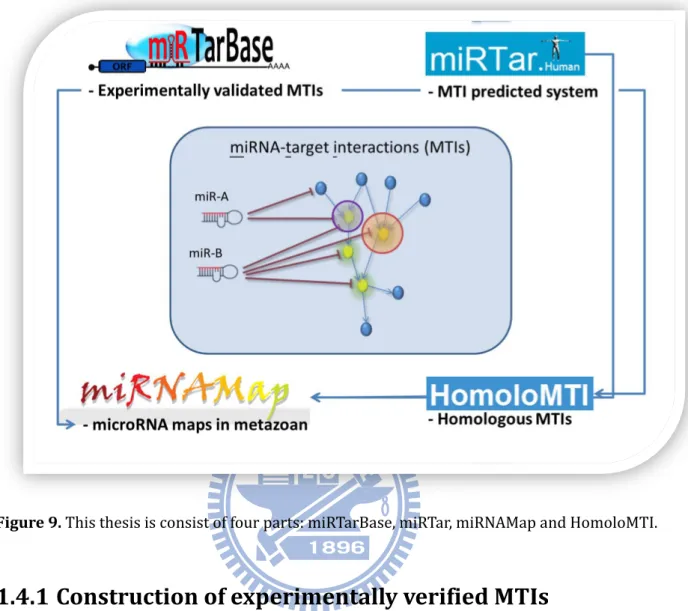
The goal of this work is to systematically analyze the miRNA-target interactions and assess the function of miRNA by developing new methods and resources. In this dissertation, we focus on the manually curated miRNA-target interactions with experimental support, a system for miRNA-target interations prediction, identification of homologous miRNA-target interactions and incorporated miRNA-target interactions database. Figure 9 summarizes the major aims of this dissertation and is described in the following sections.
Figure 9. This thesis is consist of four parts: miRTarBase, miRTar, miRNAMap and HomoloMTI.
1.4.1 Construction of experimentally verified MTIs
First to all, we aim to develop a frequently updated database by continuously surveying research articles with the pre-screening by text-mining programs and intend to make the database become a major repository for experimentally confirmed miRNA-target interactions. Through analyzing more than seven hundreds verified MTIs with the target sequences, we could get the overview of relative contribution of sequence and structure features in miRNA targeting. The MTIs collection in the proposed database can also become a bigger amount of positive samples for the developments of computational methods to identify miRNA-target interactions.
the regulatory relationship between one miRNA and one gene, one miRNA and multiple genes, multiple miRNAs and one gene, and multiple miRNAs and multiple genes. Besides, miRTar identifies miRNA target sites against 3'UTR, as well as the coding regions and 5'UTR. This resource provides the information concerning that miRNA-target interactions are regulated by alternative splicing. Additionally, miRTar performs a gene set enrichment analysis for miRNA-regulated gene set to decipher possible roles in biological process and pathways.
1.4.3 Identification of homologous MTIs
MicroRNA plays important roles in post-translational gene regulation among various species. Nowadays many miRNA target interaction are revealed by experiments and miRNA target site prediction tools. In order to provide more evidences for predicted miRNA target interactions and discover real miRNA target interactions, a database, HomoloMTIs, was contructed based on three databases, miRTarBase, miRBase and HomoloGene, to reveals the miRNA target interactions might be shared in homologous genes. The homologous MTIs profiles could reveal the homologous genes regulated by miRNA with at least one experimental validation in one of the homologous genes and predicted miRNA target interactions. The novel miRNA target interactions and miRNAs could be pioneer studied by HomoloMTIs. HomoloMTI aims to provide a comprehensive comparative perspective on the metazoan repertoire of miRNA-target interactions as complementary to miRTarBase, the database of experimentally verified miRNA-target interactions.
1.4.4 Integrated information of MTIs database
The main contribution of this work is the extended development to miRNAMap version 3.0. We make the focus on the investigation of miRNA-target interaction. To make miRNAMap more comprehensive, we integrate the experimentally verified MTIs, the MTIs predicted by 11 predicted miRNA target databases and more expression profiles of miRNA and protein-coding gene. A useful feature specially designed to human genome is the comparison between miRNA expression profiles and expression profiles of target genes. We also analyzed 1,524 experimentally verified miRNA-target interactions with
strong evidence support in human and elucidate which the more accurate microRNA target prediction database is. Those analyses are important for identifying putative miRNA targets and are very useful for biologist to choose the proper tool for miRNA research.
Chapter 2 miRTarBase: a database curates
experimentally
validated
microRNA-target
interactions
2.1 Introduction
As small non-coding RNAs of approximately 22 nts, microRNAs (miRNAs) regulate gene expression post-transcriptionally through suppressing mRNA translation or inducing mRNA degradation by hybridizing to the 3’-untranslated regions (3’-UTR) of the mRNAs. Discovery of the first miRNA in Caenorhabditis elegans in 1993 (1) ushered in numerous studies on the cellular processes of these tiny regulatory RNAs for a large variety of metazoa. Thousands of miRNAs have been identified in mammalian cells over the past two decades. miRNAs play critical roles in many biological processes, including cell cycle control, cell growth and differentiation, apoptosis, and embryo development.
Literature on miRNA research has recently grown exponentially (Figure 1). The accelerate rate of miRNA gene discovery has led to the need to elucidate the functions of these miRNAs. Additionally, more than 20 databases and computational methods have been developed for identifying candidates of miRNA-target interactions. A curated collection of up-to-date miRNA-target interactions (MTIs) with experimental support is crucial to provide effective information for investigating miRNA functions at different conditions and in different species. In this work, we propose a database, miRTarBase, which has accumulated more than three thousand miRNA-target interactions collected by manually surveying literature after a systematic text-mining process to select research articles related to functional studies of miRNAs. Generally, the collected MTIs were experimentally validated by reporter assay, western blot, or microarray experiments with overexpression or knockdown of miRNAs.
2.1.1 Experimental
approaches
for
identifying
the
miRNA-target interactions
involves using computational methods to identify target sites of miRNAs. These putative miRNA-target interactions are then validated by molecular experiments, including reporter assay and western blot. Reporter assay and western blot are the conventional means of confirming the interaction between miRNA and its target mRNA. Besides, Northern blot analysis, quantitative real-time PCR (qPCR), or in situ hybridization is often performed to examine the co-expression of predicted miRNA and mRNA target gene. In contrast with traditional validation, genome wide screenings approaches, including microarray experiments with overexpression or knockdown of miRNAs, stable isotope labeling with amino acids in culture (SILAC) or pulsed SILAC (pSILAC; Figure S5), have been developed. For instance, Selbach et al. determined the complement of all genes targeted by five miRNAs induced independently in HeLa cells using microarrays and pSILAC (73), and more than 400 miRNA-target interactions were identified.
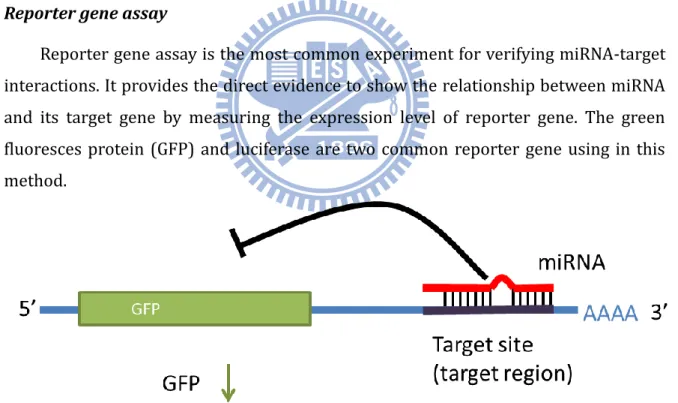
Reporter gene assay
Reporter gene assay is the most common experiment for verifying miRNA-target interactions. It provides the direct evidence to show the relationship between miRNA and its target gene by measuring the expression level of reporter gene. The green fluoresces protein (GFP) and luciferase are two common reporter gene using in this method.
Stable isotope labeling with amino acids in culture (SILAC) or pulsed SILAC (pSILAC) SILAC (stable isotope labeling with amino acids in cell culture, Figure S4 and
Figure S5) is a technique which is a popular method for quantitative proteomics, based
on mass spectrometry that detects differences in protein abundance among samples using non-radioactive isotopic labeling.
2.2 Related works
Many miRNA-related database systems have been developed in recent years to provide information on miRNAs and their target genes. miRBase (74) is the most complete repository for miRNA annotation and nomenclature. Until now, the miRBase (version 17.0) contains 16,772 miRNA entries and many more new sequences are added regularly. miRGen (75), miRGator (76), miRDB (77), microRNA.org (78) and miRNAMap (3,4) provide miRNA targets based on combinations of extensively adopted target prediction programs. Furthermore, TarBase (79), miRecords (80), and miR2Disease (81) contain experimentally validated miRNA-target interactions. TarBase is the first resource that provides experimentally verified miRNA-target interactions by surveying literature (79). miRecords collects both experimentally validated miRNA targets and computationally predicted miRNA targets (80). miR2Disease contains relationships among miRNAs, target genes and diseases in human (81). miRSel (82) utilizes a text-mining method to systematically extract miRNA-target relationships from the PubMed abstracts. Additionally, several computational methods and web-based programs were developed for computationally identifying target genes of miRNAs, e.g., miRanda (78), TargetScan (66), RNAhybrid (83), Pictar (84) and PITA (85). These tools are widely used by researcher, and these candidates of miRNA-target interactions are then confirmed experimentally.
There are 5 databases have been developed for storing experimentally verified miRNA-target interactoins (MTIs) showed in Table 1, including TarBase, miRecords, miR2Disease, miRSel and miRWalk.
Table 1. The related databases stored experimentally verified MTIs.
Name Organism Remark Ref
TarBase Plants, Flies, Nematodes,
Virus, Vertebrates, 1. First database stored experimental verified miRNA targets. (79) miRecords Nematodes, Vertebrates, Flies 1. Incorporated 11 known miRNA target
prediction tools.
2. Experimentally verified miRNA targets.
(80) miR2Disease Human 1. miRNA deregulation in various human
diseases. (81)
miRSel Human, mouse and rat 1. Text-mining method to extract the
experimental MTIs. (82)
miRWalk Human, mouse and rat 1. Incorporated 8 known miRNA target prediction tools.
2. Experimental miRNA targets
(86)
TarBase
TarBase is a first database that provides information of experimentally support miRNA targets in several animal species, plants and viruses. In version 5.0 (release in October 2008), there are about 1,300 experimentally verified microRNA-target interactions stored in their database. The information of MTIs was curated from about 160 articles. Additionally, the result page of TarBase is functionally linked to other databases such as Ensembl, Hugo, UCSC Genome Browser and SwissProt (87). The TarBase 5.0 database can be queried or downloaded from http://microrna.gr/tarbase.
Figure 11. The result page of TarBase.
miRecords
miRecords, a resource for animal miRNA-target interactions, consists of two components including Validated Targets and Predicted Targets. The Validated
Targets component houses about 1,600 experimentally validated miRNA targets curated
from meticulous literature. The Predicted Targets component of miRecords integrated the pre-compiled data identified by 11 established miRNA target prediction tools. The miRecords is available at http://miRecords.umn.edu/miRecords.
Figure 12. The web interface of miRecords.
miR2Disease
miR2Disease, the other manually curated miRNA information database, provides the relationship between deregulated micorRNAs and various human diseases. It
miRSel
miRsel, text-mining method for automatic extracting miRNA-target interactions from PubMed abstracts, is different from the manually curated miRNA-target interactions datbases mentioned above. It is the only one database that stores the miRNA-target interactions by extracting them from miRSel is freely available online at
http://services.bio.ifi.lmu.de/mirsel.
2.3 Specific aims
During last two years, a continuously growing number of identified miRNAs and their targets, combined with their major roles in biological systems, explains why it is crucial to have an accurate, up-to-date, easily accessible and centralized information repository. In this work, we aim to develop a frequently updated database by continuously surveying research articles with the pre-screening by text-mining programs and intend to make the database become a major repository for experimentally confirmed miRNA-target interactions. The miRTarBase contains the largest amount of validated MTIs and provides the most up-to-date collection by comparing to other similar databases previously developed, such as TarBase, miRecords, and miR2Disease. Moreover, we investigated the biological features of miRNA/target duplex based on more than seven hundreds validated miRNA-target interactions in human, where the miRNA target sites of MTIs were reported in the source articles. The MTIs collection in the proposed database can also become a bigger amount of positive samples for the developments of computational methods to identify miRNA-target interactions.
2.4 Materials and methods
2.4.1 Database content
All entries in the database are collected manually that describe how a miRNA and its target genes are related with experimental support (Figure 13). Initially, all fields in the
PubMed database are searched based on the keywords ‘microRNA targets’ or ‘miRNA targets’, followed by downloading all full-text of these articles. Next, a text-mining system is developed to allow for screening of full-text literature that potentially describes miRNA-target interactions, as verified by various experimental methods. Each research article was carefully reviewed by at least two of our developers to extract the miRNA-target interactions, which experimentally confirmed by reporter assay, western blot, microarray experiments, pSILAC or qRT-PCR, as well as to extract other effective information, including the species of miRNAs, the species of target genes, and experimental conditions.
Figure 13. System flow of miRTarBase.
levels and protein expression levels at conditions of miRNA overexpression or miRNA knock-down cells. Although these methods are capable of accurately identifying miRNA target genes, other experimental methods are required to determine the location of regions targeted by miRNAs. Luciferase reporter assay is adopted conventionally. Here, we view the miRNA-target interactions with strongly support when they are validated by western blot, qPCR, or reporter assay.
2.4.3 Less strong experimental evidence of miRNA-target
interactions
The high-throughput miRNA target identification methods, including pSILAC and microarray experiments, can be used to determine the genome wide changes in the mRNA expression levels or protein expression levels when the miRNA is present or not (79). Given our inability to understand whether the over-expressed miRNAs cause the changed expression patterns directly or not, these technologies only provide less strong experimental evidence for the collected miRNA-target interactions.
2.5 Results
2.5.1 Statistics
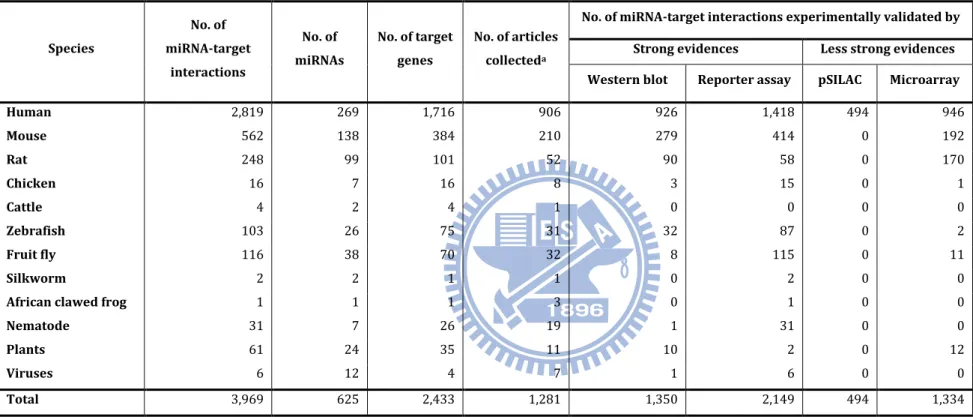
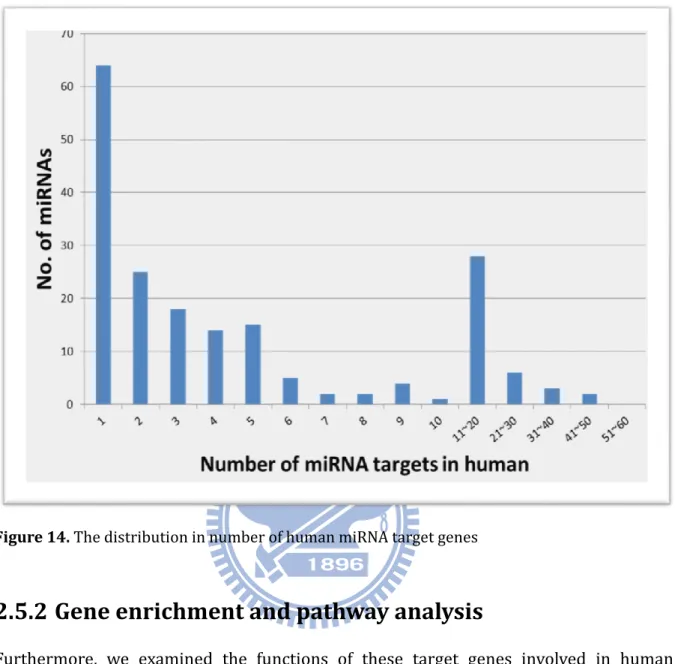
In the release 2.4 (Apr. 15, 2011) of miRTarBase, 3,969 curated miRNA-target interactions between 625 miRNAs and 2,433 target genes were collected from 1,211 articles. Table 2 lists the number of collected miRNA-target interactions in each species. For instance, 2,819 human MTIs were collected between 269 miRNAs and 1,716 target genes with the experimental support from 906 articles, and 926 and 1,418 interactions were experimentally confirmed by western blot and reporter assay, respectively. Each human miRNA can target to five target genes in average, and Figure 14 gives the distribution of miRNAs categorized by the number of target genes for each miRNA which are supported by reporter assay or western blot. In miRTarBase, hsa-miR-122 was recorded to have 45 target genes, which were experimentally validated by luciferase reporter assay or western blot. hsa-miR-122 is a liver-specific miRNA in human and is significantly down-regulated in liver cancers (88).
Table 2. Statistics of miRNA-target interactions collected in miRTarBase. Species No. of miRNA-target interactions No. of miRNAs No. of target genes No. of articles collecteda
No. of miRNA-target interactions experimentally validated by
Strong evidences Less strong evidences
Western blot Reporter assay pSILAC Microarray
Human 2,819 269 1,716 906 926 1,418 494 946 Mouse 562 138 384 210 279 414 0 192 Rat 248 99 101 52 90 58 0 170 Chicken 16 7 16 8 3 15 0 1 Cattle 4 2 4 1 0 0 0 0 Zebrafish 103 26 75 31 32 87 0 2 Fruit fly 116 38 70 32 8 115 0 11 Silkworm 2 2 1 1 0 2 0 0
African clawed frog 1 1 1 3 0 1 0 0
Nematode 31 7 26 19 1 31 0 0
Plants 61 24 35 11 10 2 0 12
Viruses 6 12 4 7 1 6 0 0
Total 3,969 625 2,433 1,281 1,350 2,149 494 1,334
Figure 14. The distribution in number of human miRNA target genes
2.5.2 Gene enrichment and pathway analysis
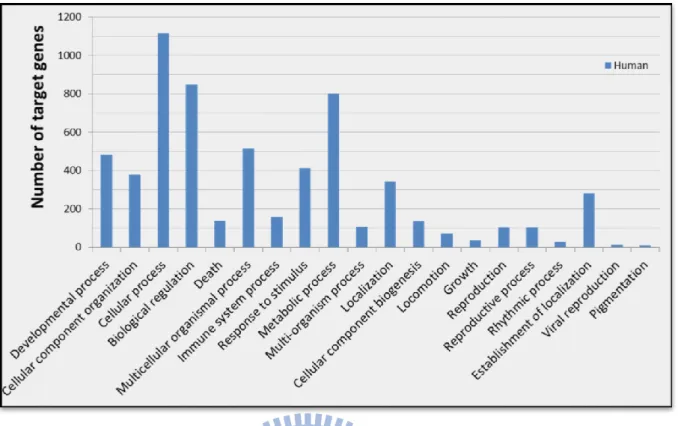
Furthermore, we examined the functions of these target genes involved in human miRNA-target interactions collected in the database by performing Gene Ontology (GO) and KEGG (89) pathway enrichment annotation using the DAVID gene annotation tool (90). GO enrichment analysis indicates that the cellular process, biological regulation and metabolic process are the most significantly enriched GO terms for this selection of
Figure 15. GO analysis of human miRNA-target interactions, as verified by reporter
assay or western blot. (DAVID, grouped according to biological process of level 1)
Table 3 lists the top 20 pathways significantly enriched in these human target
genes, and most of which are involved in cancer, including pancreatic cancer, colorectal cancer, prostate cancer, small cell lung cancer, bladder cancer, non-small cell lung cancer and endometrial cancer. Interestingly, above analysis provides an overview of the possible functions of human miRNAs based on this curation of miRNA-target interactions although the data should be biased due to miRNAs have attracted more attentions in cancer research recently.
Figure 15. GO analysis of human miRNA-target interactions, as verified by reporter assay or
western blot. (DAVID, grouped according to biological process of level 1)
Table 3. KEGG Pathway annotation of human miRNA-target interactions
KEGG Pathway No. of target genes Ratio P-Value
Pathways in cancer 120 0.70 2.91E-32
Pancreatic cancer 42 0.25 4.30E-20
Chronic myeloid leukemia 42 0.25 3.37E-19
Colorectal cancer 40 0.23 4.11E-15
Prostate cancer 41 0.24 7.27E-15
Small cell lung cancer 38 0.22 1.69E-13
Bladder cancer 26 0.15 2.73E-13
Melanoma 32 0.19 2.20E-11
Cell cycle 44 0.26 4.30E-11
Neurotrophin signaling pathway 43 0.25 1.29E-10
Focal adhesion 58 0.34 1.91E-10
MAPK signaling pathway 70 0.41 2.07E-10
Non-small cell lung cancer 26 0.15 4.31E-10
Renal cell carcinoma 30 0.18 4.60E-10
Glioma 28 0.16 7.43E-10
Endometrial cancer 25 0.15 1.05E-09
p53 signaling pathway 29 0.17 1.10E-09
Acute myeloid leukemia 25 0.15 1.55E-08
Adherens junction 28 0.16 1.25E-07
Epithelial cell signaling in Helicobacter pylori
Only 709 human miRNA-target interactions in miRTarBase have miRNA target site annotations, which can be extracted from the articles. Of these target site sequences, 9 of them only provide the sequence of seed region (< 10 nucleotides); 667 of them contain the target site sequences (10 ~ 50 nucleotides), while the others (29) provide cloned partial UTR sequences (> 50 nucleotides). Next, an attempt is made to summarize the data distributions of twelve biological features of the miRNA/target duplex in these 709 known human miRNA-target interactions, as shown in Figure 16. The miRNA target sites were mapped to the 3’UTR of the corresponding target gene; in addition, 70 nucleotides around the target site were extracted. Additionally, the miRNA target sites were selected when the alignment score of miRNA/target duplex is greater than 100 and the number of base-pairs within the seed region is more than 5. Notably, 1,610 miRNA target sites are obtained from the 709 miRNA-target interactions. Figure 16 gives the histograms of various features of these miRNA-target duplex. Figure 16 (A) and (B) show the longest consecutive matches (excluding or including wobble pairing (GU pairing) in a seed region), which is a subsequence from nucleotide 1 to 8 in the 5’ end of the miRNA, respectively. More than 55% of all binding sites have more than 7 bases of consecutive pairings. The minimum free energy of the seed regions and the binding sites is also calculated, as shown in Figure 16 (C) and (D), respectively. The mean value of the free energy of the binding site is approximately -14 kcal/mol. The free energy of most of the seeds is smaller than -6 kcal/mol. Next, analysis is performed of the number of nucleotides matches, GU matches, and mismatches in the seed regions and the target sites. Figure 16 (E) and (J) summarize these statistics. More than 80% of all target sites have at least 6 matches in the seed region; in addition, the GU matches rarely occur in the seed region, which is smaller than 40%. The number of matches is significantly larger than the number of mismatches. GU matches in the target sites are substantially smaller than the quantity of matches and mismatches. The target site accessibility is also estimated based on the calculation of Kertesz M. et al (85). According to our results,
Figure 16. Histogram of various features of experimental proven miRNA-target sites.
2.5.3 Comparisons to other miRNA-target interaction
databases
Comparing the other manually curated databases such as TarBase, miRecords, and miR2Disease, miRTarBase accumulates a bigger collection and a more up-to-date curation of miRNA-target interactions than other resources (Table 4), especially around nine hundreds research articles were collected. It also reveals that our miRTarBase has the most abundant miRNA-target interactions, even if only considering the entries supported by reporter assay or western blot experiments. Furthermore, the Venn diagrams show the intersection of articles collected in different databases (Figure 17). miRTarBase almost covers all the research articles collected in TarBase, miRecords and miR2Disease.
Table 4. Comparison of miRTarBase with other miRNA-target interaction databases. The final
column is the number of miRNA-target interactions (MTIs) more than the largest number of records in TarBase, miRecords and miR2Disease. The miRNA-target interactions in TarBase, miRecords and miR2Disease are downloaded from their web sites.
Database names TarBase (79) miRecords (80) miR2Disease (81) miRTarBase
Number of records added
Last update date 2008/06 2010/05/05 2010/06/02 2011/04/15
Support species Metazoa x 6 Viridiplantae
Viruses
Metazoa x 11
Viruses x 2 Human Metazoa x 9 Viridiplantae x 3
Viruses x 5
Total No. of miRNAs 223 381* 179 625 + 244
Total No. of target genes 1028 1057* 394 2433 +1376
Total No. of articles 154 410 421* 1211 + 790
Total No. of miRNA-target
interactions (MTIs) 1264 1513* 635 3969 +2456
Supported by strong experimental evidences No. of MTIs validated by
"Reporter assay" 305 672* 635 2149 + 1477
No. of MTIs validated by
"Western blot" 27 295* 0 1350 + 1055
No. of MTIs validated by
“Reporter assay and
Western blot” 25 123* 0 1092 + 969
No. of MTIs validated by "Reporter assay or Western
blot" 307 747* 635 2407 + 1660
Supported by less strong experimental evidences No. of MTIs validated by
"pSILAC experiments" 455* 0 0 494 + 39
No. of MTIs validated by
"Microarray experiments" 343 380* 0 1334 + 954
Figure 17. The overlap among articles collected in each two manually curated miRNA-target
interactions databases.
Text-mining method is an alternative approach which can retrieve the information of associations between miRNAs and target genes; however, miRNA-target interaction is generally described in natural language and is not easy to be extracted correctly by only computational methods. For example, DICER1 and Drosha are very important genes which are involved in the biogenesis of miRNA, but they are usually not the target genes of a miRNA when they are discussed along with the miRNA in an article. However, text-mining methods may identify the association between a miRNA and DICER1, and incorrectly annotate the association as a miRNA-target interaction. Therefore, manually reviewing the articles potentially containing miRNA-target interactions is inevitable for extracting those experimental evidences to support a miRNA-target interaction. Here, we do not compare the contents in miRTarBase with other databases established by only text-mining methods without manual review.
2.5.4 Web interface
facilitate the access of miRNA-target interaction data (Figure 18). Several search functions for retrieving miRNA-target interactions are designed, including search by miRNA accessions, search by target genes, and search by literature. Alternatively, miRTarBase provide keyword search in all fields of all data entries. We designed a result page to present a miRNA-target interaction, where each MTI was assigned a miRTarBase accession. The result page majorly comprises three main parts: miRNA information, target gene information and evidence support. Generally, web pages of the miRTarBase contain many effective quick links to several other web resources, including NCBI Entrez (91), UCSC Genome Browser (92), miRBase (74), BioGPS (93), iHOP (94) and HGNC (95). Detailed descriptions of web pages are provided below.
The ‘miRNA information’ page contains the characteristics of a miRNA such as accession, synonyms, descriptions, the sequence of miRNA, and links to other putative miRNA-target interaction databases. Especially, in this page all the miRNA-target interactions of the miRNA are presented as a network, which can depict the relationships between a miRNA and multiple target genes. In the ‘Target Gene’ page, the basic information of a target gene is provided, including gene symbol, description, genomic location, transcript sequence, and links to other resources. The information of target sites located in the transcript are carefully examined and provided on the web page. Notably, many articles only report the regulatory relationship between a miRNA and its target genes without providing the exact regions of miRNA target sites. Here, we utilized miRanda to computationally identify the potential target sites belongs to a miRNA-target interaction, which is supported by experimental evidences.
In the “Evidences” page, the experimental information to support a miRNA-target interaction from one or multiple articles is provided by presenting the experimental validation methods, the experimental conditions, the location of target sites, computational tools used in article, partial key descriptions extracted from the article, and article abstract. Additionally, this resource also provides data submission page that allows users or researchers to submit information of miRNA-target interactions, which
Figure 18. The miRTarBase web interface
2.6 Conclusion
This work presents a more comprehensive collection of miRNA-target interactions, which were experimentally validated. The biological features of miRNA/target duplex were observed based on largest collection of human miRNA-target interactions currently available. Various web interfaces are designed to facilitate the presentation of miRNA-target interactions. A pipeline combining text-mining and manual review was established to extract MTI information from research articles.
Application involving the proposed database is to extend the human miRNA-target interaction to mouse, rat and other mammalian genome based on evolutionary conservation of miRNA and its target sites. More probable miRNA-target interactions can be provided as the candidates for experimental confirmation. We will describe the more detail in Chapter 4.
According to the more high-throughput experimental technology such as high-throughput sequencing of RNAs isolated by crosslinking immunoprecipitation (HITS-CLIP) was introduced to validate miRNA-target interactions, we should also curate
Chapter 3 miRTar - an integrated system for
identifying miRNA-target interactions in Human
3.1 Introduction
Before starting this section, we should note that this part was also done by our co-group member, Justin, Bokai, Hsu.
MicroRNAs (miRNAs) are small non-coding RNA molecules that are ~22 nts sequences capable of suppressing protein synthesis. Deriving from ~70–120 nts precursor transcripts that fold into stem-loop structures and thought to be highly conserved in genome evolution, miRNAs regulate 30% or more of the human protein-coding genes (65,66). Moreover, previous investigation suggested that miRNA target sites in mammalians are preferentially conserved in the mRNA sequences, especially in 3' UTR (96). Since these miRNA-regulated genes are involved in various crucial cell processes including apoptosis, differentiation and development, Gene Ontology (GO) or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enrichment analysis of them are helpful in understanding the biological functions of miRNA (97-100). For instance, the target genes of miR-124a such as ephrins B1, B2, and B3, ephrin receptors A2, A3, and B4, semaphorins 5A, 6A, 6C, and 6D, and plexins A3 and B2 are involved in nervous system development in the axon guidance pathway.
3.2 Related works
Our previous work, miRTarBase (101), which is the most updated collection of miRNA-target interactions (MTI), has accumulated 3,969 experimentally verified MTIs between 625 miRNAs and 2,433 target genes among 17 species by manually surveying pertinent literature. Moreover, numerous computational programs are available for identifying miRNA target sites. TargetScan (102), miRanda (103) and RNAhybrid (70) are three computational tools for determining the most energetically favored hybridization sites of small to large RNAs. PicTar (84) is capable of identifying common targets of known miRNAs. DIANA-microT (104) system utilizes experimentally derived miRNA/mRNA binding rules. miRNAMap (3,4) and miRecords (80), miRGen (75,105) and GOmir (106) provide the putative miRNA-target interactions by combining
prediction from multiple programs.
The miRU (107), MicroInspector (108), RNA22 (109), EIMMO (99), StarMir (72) and MMIA (110) are web-based tools for identifying miRNA binding sites. MicroInspector can search miRNA binding sites for a user-defined target RNA sequence that is potentially regulated by a miRNA. MicroInspector allows for variations in temperature and energies and allows the selection of various miRNA databases to identify miRNA binding sites of different strengths. The miRU tool was developed to predict plant miRNA target genes in any plant that is likely to be regulated by a user-defined miRNA. The pattern-based approach incorporated in the RNA22 program identifies putative target sites independent of miRNA target conservation and calls these sites as ‘target islands’. The EIMMO considers evolutionary distance and branching when scoring the degree of miRNA target conservation. Otherwise, Dang et al. posited the target structure-accessible model for predicting miRNA targets and could also be accessed on system called StarMir. MMIA combines the inverse expression profiles of miRNA and mRNA data and then predicts the target genes by TargetScan, PicTar and PITA. Not only the aforementioned targeting of the 3' UTR of transcripts, but also the possibility of the targeting by miRNA of the coding sequence (CDS) and 5'UTR regions of the transcripts, are the subject of extensive research (66,67,109,111-120). Indeed, more than twenty miRNA target prediction tools were developed to identify potential candidates for miRNA-target interactions. However, most of them do not provide convenient functions for biologists in exploring the biological functions and regulatory relationships between miRNAs and protein coding genes. The comparisons among miRNA target prediction tools are given in Table 5.
Table 5. The comparisons of miRNA target prediction tools.
Features miRTar microT/miRPath DIANA-
(121,122)
EIMMO
(99) miRU (107) RNAhybrid (70) STarMir (72) RNA22 (109) MMIA (110)
Species Human Human and mouse Vertebrates, nematode,
fly Plants Human, nematodes, flies - Vertebrates, nematode, fly Human Possible relation between miRNA and gene group * 1 to 1 + + + - + + + - * 1 to N + 1 to All genes + - + + - + * N to 1 + - + - + + - - * N to M + - + - + + - +
* All to M + All miRNAs to 1 + - - - - -
* 1 to KEGG + + - - - - - -
miRNA targets on alternatively
splicing exon + - - - -
miRNA targets from mRNA 3'UTR, CDS, and 5'UTR 3’UTR 3’UTR 3'UTR, CDS, and 5'UTR 3’UTR 3'UTR, CDS, and 5'UTR 3'UTR, CDS, and 5'UTR 3’UTR
Known miRNAs miRBase V15 - miRBase V12 - - - - -
Accessibility of target site + - - - - Sfold - -
Conservation of target site + + + + - - - -
Expression profile of
miRNA - - - +
Target - - + - - - - +
* 1 to 1 means the relation of one miRNA and one gene; 1 to N means the relation of one miRNA to multiple interesting genes; N to 1 means the relation of N miRNAs and one gene; N to M means the relation of N miRNAs and M genes; All to M means the relation of all miRNAs and M genes; 1 to KEGG means the relation of one miRNA and the genes of the selected KEGG map.
RNA alternative splicing plays important roles to regulate the gene expression in many biological processes among eukaryotic species. Recent studies have shown that more than 50% of genes undergo alternative splicing in humans (123-125). Additionally, some researchers have observed that appropriate splice variants are involved in several cellular and developmental processes, including gender determination, apoptosis, axon guidance, cell excitation and contraction (126). Relatedly, inappropriate alternative splicing causes the genetic disorders, because the expression of disease-related genes, many of which encode influential proteins in cancer biology, including those that govern cell cycle control, proliferation, differentiation, signal transduction pathways, cell death, angiogenesis, invasion, motility and metastasis, become abnormal (126-129). Moreover, generated spatio-temporal splicing variants can be divided into five classical forms, which are cassette exons, alternative 5' splice sites, alternative 3' splice sites, mutually exclusive exons and retained introns (126,130). Furthermore, the variety of combinations of cis-elements and trans-factors make understanding this mechanism difficult (126,129,130).
3.3 Specific aims
In this work, we aims to provide an integrated resource to allow biologists to elucidate miRNA-target interactions affected by the alternative splicing, thus the location of miRNA target sites may locate in the exons, which are alternatively spliced. Several previous investigations have studied the miRNA-target interactions affected by alternative splicing (111,112,116,117,119). For instance, Duursma et al. reported that human DNA methyltransferase 3b (DNMT3b) gene can be repressed by miR-148 family (116) and the miR-148 target sites are located in the DNMT3b exons, which is alternatively spliced. Furthermore, the gene set enrichment analysis (GSEA) for a group of genes, which are targeted by one or more miRNAs, can provide effective viewpoint to elucidate the miRNA functions in different biological process and pathways (131,132).
including the regulatory relationship between one miRNA and one gene, one miRNA and multiple genes, multiple miRNAs and one gene, and multiple miRNAs and multiple genes. Besides, miRTar identifies miRNA target sites against 3'UTR, as well as the coding regions and 5'UTR. This resource provides the information concerning that miRNA-target interactions are regulated by alternative splicing. Additionally, miRTar performs a gene set enrichment analysis for miRNA-regulated gene set to decipher possible roles in biological process and pathways.
3.4 Materials and methods
The miRTar is a web-based system that runs on an Apache web server with a Linux operating system. Figure 19 presents in brief the intention that underlies miRTar, which is to design an analytical platform that allows researchers to focus on all possible scenarios to discuss the regulatory relationships between miRNAs and genes. After data are submitted to the system, miRTar identifies the miRNA target sites using TargetScan, miRanda, PITA, and RNAHybrid. The miRTar identifies the target sites against 3' UTR, 5' UTR and coding regions. Thus, the potential miRNA-target interactions between miRNAs and genes are constructed. For a gene set that may be regulated by single miRNA, based on gene set enrichment analysis (GSEA), a p-value is calculated to estimate the overrepresentation of genes in which the KEGG pathways, to estimate the biological function of miRNA. Additionally, miRTar can provide the information of miRNA target sites within exons, which are alternatively spliced (AS) or constitutively spliced (CS).
Figure 19. Concept that underlies miRTar.
3.4.1 Data collection
Figure 20 depicts the system flow of miRTar. miRTar utilizes several well-known resources, including the miRNA sequences, obtained from miRBase database Release 15 (74), gene information and relevant annotations, based on ASTD database Release 1.1 (133) and GenBank database Release 167 (134). The splice variants of transcripts are obtained from this ASTD (133), UniGene database Release 217 (135) and GenBank database (134). The biological pathways are extracted from the KEGG/PATHWAY database Release 53.0 (136). Table 6 lists all versions and data types obtained from
Figure 20. System flow of miRTar.
Table 6. Data statistics and data obtained from databases.
Data source Version Data descriptions Data amount
miRBase (74) V.15 MicroRNA information (name, sequences, ...) 1100
KEGG (136) V. 53 The pathway maps 195
ASTD (133) V. 1.1
Gene annotation 16,715
mRNA sequences 93,467
Protein information 34,545
Alternative splicing events 78,165
GenBank (134) V. 167 Gene annotation 32,123 Genomic sequences 32,123 Protein sequences 125,259 UniGene (135) V. 217 mRNA sequences 137,654
protein information (mRNA gi to protein gi) 125,259
3.4.2 Identifying miRNA target sites in human
First, TargetScanS was utilized to detect perfect Watson-Crick base pairing against all mRNA transcripts with lengths of at least six nucleotides. Four seed types, 8mer,