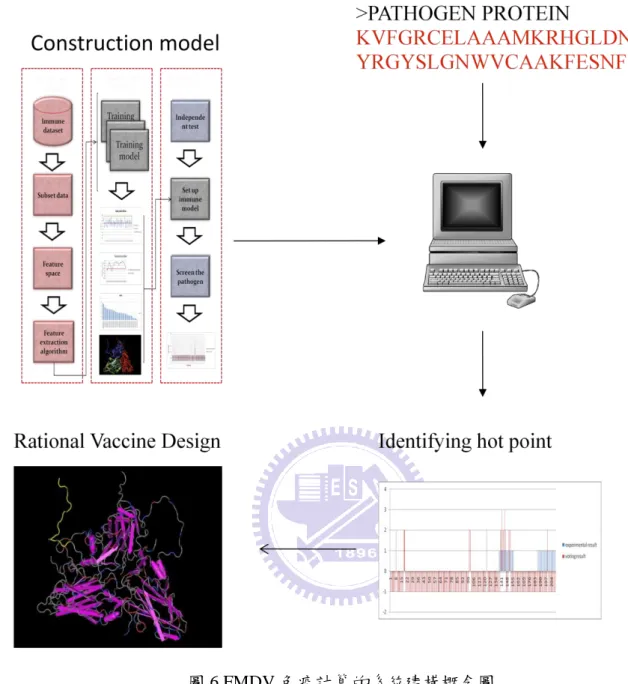
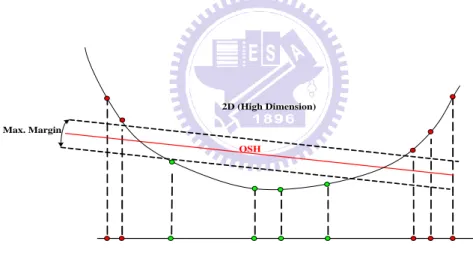
1
國 立 交 通 大 學
生物資訊及系統生物研究所
碩 士 論 文
辨識口蹄疫病毒抗原決定位之計算系統
Computational system for identifying antigenic determinant
site of foot-and-mouth diseases virus
研 究 生:黃泰欽
指導教授:何信瑩 教授
2
辨識口蹄疫病毒抗原決定位之計算系統
Computational system for identifying antigenic
determinant site of foot-and-mouth diseases virus
研 究 生:黃泰欽 Student:Tai-Chin Huang
指導教授:何信瑩 Advisor:Shinn-Ying Ho
國 立 交 通 大 學
生物資訊及系統生物研究所
碩 士 論 文
A Thesis Submitted to Institute of Bioinformatics and Systems Biology Department of Biological Science
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master in Bioinformatics
July 2011
Hsinchu, Taiwan, Republic of China
i
辨識口蹄疫病毒抗原決定位之計算系統
學生:黃泰欽
指導教授:何信瑩
國立交通大學生物資訊研究及系統生物研究所所碩士班
摘 要
口蹄疫疾病主要影響偶蹄類動物是一個高度傳染性的疾病,被認為是世界上經濟方 面最重要的家畜動物疾病。此疾病由口蹄疫病毒所造成,相關的研究認為疫情爆發時使 用接種疫苗方式被視為一種更合理的手段。為了達到上述的目標,定義出具意義的抗原 決定位及瞭解其重要的物化性質扮演一個重要的角色在保護性疫苗的設計、免疫的診斷 及抗體的生產上。由於決定抗原上之 B 細胞表頂有賴於實驗的方式定義出來,但是此工 作是耗時且昂貴的,因此發展計算的方式來幫助病原體自抗原的序列中定義出可靠性的 表頂是必須地。 在此研究欲建立一個免疫的模型基於 B 細胞特異性的次分類群。我們擷取來自 IEDB 註解有關口蹄疫病毒病原體 B 細胞表頂之實驗數據做為我們的訓練資料。將訓練序列轉 換成物化特性的特徵空間指標,並結合特徵選取的方式來改善預測準確度。使用繼承式 雙目標基因演算法,其可最大化我們研究問題分類的準確度,同時最小化選取特徵數, 來幫助擷取重要的資訊自我們的目標資料集之中。然後擴展所選出的物化性質組合來定 義出抗原決定位的熱點藉由掃描病原體蛋白質。使用滑動窗口給予每一個詢問片段中心 位點一個抗原性質倾向依據所選出的物化性質組合,結合投票的方式及利用智慧型基因 演算法調整參數來達到一致性的預測結果。此外分析所選出物化性質來找尋重要的生物 意義幫助改善疫苗的設計。 在這個研究中,我們發展一個計算的系統對於預測抗原決定位基於使用病原體特異 性的次分類群及特徵選取的策略。結果顯示出,此建立的預測模型不僅能達到較高的預 測準確度(訓練 89.33%及測詴 72%),也能自病原體蛋白質序列中定義出抗原決定位的熱 點。此外基於特徵選取也能提供有用的生物訊息供分析之用。此系統不僅可以被使用當 做研究口蹄疫病毒新興病原體的工具而且可以提供一個概念對於改善 B 細胞表頂的預 測上。 關鍵字:口蹄疫病毒、FMDV、B 細胞表頂、抗原決定位ii
Abstract
Foot-and-mouth disease (FMD) is a highly contagious disease affecting cloven-hoofed animals and it is deemed as economically important diseases of livestock worldwide. The causative agent is the foot-and-mouth disease virus (FMDV). The first priority of suggestion in the outbreak was to develop effective FMD vaccines. Accordingly, identifying significant antigenic determinant sites and understanding its important physicochemical properties play an important role in protective vaccine designs, immunodiagnostic tests and antibody production. The experimental methods for determining B-cell epitopes are time-consuming and expensive. Therefore, it is desirable to develop computational methods for reliable identification of putative B-cell epitopes from antigenic sequences.
This study aims to establish a computational system for identifying antigenic determinant sites of foot-and-mouth diseases virus based on specific subclasses of B-cell epitopes of FMDV. We retrieved training data from the IEDB database and used the annotation of B-cell epitopes experimental data about FMDV. We transferred the training sequences to feature vectors based on the physicochemical feature index and then combined a feature selection method to improve prediction accuracy. An inheritable bi-objective genetic algorithm is used to maximize classification accuracy of the investigated problem and minimize the number of selected features to draw out significant information from our objective dataset. Then the selected feature set was spanned to identify hot points of the antigenic determinant sites by screening pathogen proteins. This method assigns a scale tendency value using the selected feature set and sliding windows of the query fragment. Moreover, we analyze the
physicochemical feature set to mine significant biological findings to aid improve vaccine designs.
The results showed that the prediction system could obtain high performance (training accuracy 89.33%, and test accuracy 72%) and identify promising putative antigenic hot points. Moreover, the feature selection method could provide much useful information for biological analysis. The prediction system is capable of identifying antigenic determinant sites from pathogen proteins. The system not only could be used as a tool for investigation of emerging pathogen strain of FMDV but also provides a conception to improve B-cell epitopes
prediction effectiveness.
iii
誌謝
由衷的感謝我的指導教授何信瑩老師平時對我們的關心指導,當我們產生放棄題目 的念頭時告訴我們不要殘壘,鼓勵並給予我們研究方向的指引及提供我們許多寶貴的實 驗室資源讓我們無後顧之憂地學習研究,此外平時除了研究上,何老師也教導我們做人 的道理要常懷感恩的心,我也常常放在心上做為我的座右銘,在我們眼中老師彷彿是無 所不能地。還有要感謝黃慧玲老師對於我們的關心,黃老師會用較為詼諧的方式指導我 們,在課堂上耐心的引導我們程式的要訣。感謝黃憲達主任對我有關於論文架構上的建 議讓我的論文內容能更臻完善。還有感謝論文研討時楊進木所長、尤禎祥老師及林勇欣 老師給予我的指導及鼓勵讓我有勇氣在台上報告,也感謝楊所長對我的推薦雖然覺得受 之有愧但是還是由衷地感謝楊所長。最後在這個所上的所有老師,無論是直接或間接的 給予我們許多寶貴的資源及平時的言行都是我所敬佩的也十分地感謝他們。 對於實驗室的所有成員感謝你們在我的研究過程中於公於私的關心及鼓勵我會永遠 放在心上的。最後我要感謝我的家人給予我的支持讓我能完成學業,也願所有我關心及 關心我的人在將來的日子裡都能平安幸福!iv
目錄
頁次 摘要 ... i Abstract ... ii 誌謝 ... iii 目錄 ... iv 表目錄 ... vi 圖目錄 ... vii 一、緒論 ... 1 1.1 研究動機 ... 1 1.2 研究背景 ... 3 1.3 研究題目定義 ... 7 1.4 研究目標 ... 7 二、計算上之相關研究 ... 10 2.1 資料庫 ... 10 2.2 研究方法 ... 10 三、最佳化演算法 ... 14 3.1 基因演算法 ... 14 3.2 直交實驗設計與因素分析 ... 15 3.2.1 直交表 ... 15 3.2.2 直交實驗設計 ... 16 3.3 智慧型基因演算法 ... 16 3.3.1 智慧型交配 ... 18 3.3.2 突變運算及演化終止條件 ... 18 3.4 繼承式雙目標基因演算法 ... 18 3.5 向量學習機 ... 20 四、資料的建構及物化性質指標 ... 21 4.1 使用資料的建立 ... 21 4.1.1 口蹄疫病毒資料 ... 21 4.1.2 獨立測詴資料 ... 26 4.1.3 病原體中表頂位置的資料 ... 27 4.1.4 評估演算法的資料 ... 27 4.2 物化性質指標 ... 28 五、免疫模型建構 ... 29 5.1 免疫模型建構方法及評估的方法 ... 29 5.2 自病原體蛋白質序列中定義出表頂位置及評估的方法 ... 30 5.3 物化特性的分析準則 ... 34 5.4 評估最佳化演算法效益 ... 35 六、結果與分析 ... 36v 6.1 評估最佳化演算法與相關研究之效益 ... 36 6.2 針對 FMDV 預測模型的訓練及測詴 ... 36 6.3 決定滑動窗口使用 IGA-投票的方式及比較其它的方法 ... 39 6.4 物化性質分析 ... 43 七、結論 ... 50 7.1 討論 ... 50 7.2 未來研究 ... 50 參考文獻 ... 51
vi
表目錄
頁次 表 1 為相關研究中定義的表頂 ... 6 表 2 表示使用表頂預測結果一致性的概念 ... 13 表 3 為兩水準三因素完全實驗 ... 15 表 4 為兩水準三因素直交實驗表 ... 15 表 5 此研究所用的資料集 ... 21 表 6 蹄疫病毒訓練資料分佈 ... 25 表 7 為獨立測詴資料集 ... 26 表 8 為 aaindex 註解格式... 34 表 9 表示 IBCGA 基於 EL-Manzalawy 資料的分類比較結果 ... 36 表 10 獨立測詴資料庫針對口蹄疫病毒抗原辨識上 ... 38 表 11 為獨立測詴資料庫針對口蹄疫病毒設計胜肽辨識上 ... 38 表 12 為比較獨立測詴集中不同病原體之序列在使用不同工具之下 ... 42 表 13 大於 MED 大於 30 的分析結果 ... 45 表 14 呈現出此統計在 100 批次下,大於 30 的物化性質 ... 46 表 15 為物化性質分群 ... 49vii
圖目錄
頁次 圖 1 為 2011 年 1~6 月間爆發口蹄疫疫情之區域 ... 1 圖 2 抗體結合至表頂與 B 細胞表頂類型示意圖 ... 3 圖 3 (a)口蹄疫病毒在單一細胞之生活史 (b) 口蹄疫病毒蛋白質體組成 ... 4 圖 4 口蹄疫病毒的 protomer 及 β-Barrels 結構單元 ... 5 圖 5 為 B 細胞表頂映射概念 ... 7 圖 6 為 FMDV 免疫計算的系統建構概念圖 ... 9 圖 7 表示使用性質指標預測的概念 ... 11 圖 8 為 BcePred server 提供圖型化的輸出結果 ... 11 圖 9 基因演算法示意圖 ... 14 圖 10 直交實驗設計 ... 16 圖 11 表示智慧型基因演算法架構 ... 17 圖 12 為 IBCGA 染色體編碼的方式 ... 19 圖 13 二維 SVM 概念示意圖 ... 20 圖 14 為 IEDB 網站... 22 圖 15 分類學上的層級 ... 23 圖 16 主要使用的資料項目 ... 23 圖 17 進入 epitope ID 的項目 ... 24 圖 18 為 epitope ID 內含的項目 ... 24 圖 19 表示 B cell assay 項目 ... 25 圖 20 表示本研究最佳化演算法使用的部分 ... 29 圖 21 利用 IBCGA 結合物化性質建立免疫模型示意圖 ... 30 圖 22 物化性質判斷抗原位點的概念 ... 31 圖 23 處理完成的資料格式 ... 32 圖 24 評估定義出表頂位置效能的概念 ... 33 圖 25 表示物化性質分析的流程 ... 35 圖 26 在 100 批次實驗下所建立的免疫預測模型 ... 37 圖 27 最高訓練準確度其物化特性組別 ... 37 圖 28 為 IGA 在 100 代內的準確度 ... 39 圖 29 使用 IGA 選定滑動窗口定義出的熱點 ... 39 圖 30 呈現病原體編號為 no 6318188 之結構 ... 40 圖 31 呈現病原體編號為 no 6318188 之結構 ... 40 圖 32 獨立測詴集中為定義出來的熱點位置 ... 41 圖 33 為獨立測詴集中定義出的位置顯示在同源結構下 ... 42 圖 34 為所選出的物化性質單一指標所得到的值 ... 44 圖 35 為 MED 排名最高準確度訓練結果的那組物化性質 ... 45 圖 36 在 100runs 物化性質出現的頻率 ... 46viii
圖 37 為 G-H loop 結構上的特徵 ... 47
圖 38 為表示出極性物化性質的指標與實驗結果 ... 48
圖 39 訓練資料中表頂及非表頂組成 ... 48
1
一、緒論
1.1、研究動機
口蹄疫疾病(Foot-and-mouth diseases, FMD)是一個高度傳染性的疾病,其主要影響偶 蹄類動物(cloven-hoofed animals),如:牛,綿羊,山羊,豬和鹿。此疾病由口蹄疫病毒 (Foot-and-mouth diseases virus, FMDV)所造成,此病毒具有七種血清型,分別為 O、A、 C、SAT-1、SAT-2、SAT-3 及 Asia-1,各血清型之間無交互保護力或是其保護力有限, 因此防治上難度極大。此疾病造成許多嚴重的影響,如:直接增加年幼動物的致死率、 間接影響感染動物所有方面的產率以及造成重要的貿易限制及減少觀光上的價值等。基 於以上的理由,口蹄疫疾病被認為是世界上經濟方面最重要的家畜動物疾病。由圖 1 可 以看出口蹄疫疾病至今仍然在世界各地爆發尤其是資源較為不足的國家,所以這是一個 及需解決且重要的問題[1]。 圖 1 為 2011 年 1~6 月間爆發口蹄疫疫情之區域 From World Animal Health Information Database (WAHID)
目前兩種主要的策略被用來控制疾病的擴散,分別為使用大規模的撲殺感染之動物 以及在口蹄疫病爆發的期間使用疫苗提供動物保護力。在 2001 年期間英國爆發大規模 的口蹄疫病疫情及其後爆發的疫情之後,相關的研究認為使用接種疫苗方式被視為一種 更合理的手段來替代大規模的撲殺感染之動物,其後並建議認為應優先考慮發展改進口 蹄疫病疫苗效益。然而,如果疫苗的手段被用來作為首選的控制措施,我們必須擴展我 們對疫苗的理解範圍來改善疫苗的效益。尤其是傳統的不活化疫苗具有數種缺點,包括: 需要具有大量培養有毒性的病毒、短的保質期(short shelf life)、需要冷凍保存以及不易 於某些血清型及亞型細胞培養疫苗之生產[2]。基於這些原因,發展其不需要活病毒培養
2
的疫苗是必然地方向,例如:亞單位疫苗(subunit vaccines)、合成胜肽疫苗(synthetic peptides)[3-4]、DNA 疫苗(DNA vaccines)[5]以及重組病毒疫苗(recombinant virus vaccine)[6]等,均已被廣泛探討其效用在對抗口蹄疫病毒上。
以往的研究表明,合成肽或重組蛋白,其包含一個或兩個免疫原性(immunogenic)抗 原決定位(antigenic determinant site),可以誘導中和抗體(neutralizing antibodies)顯著的對 抗口蹄疫病毒,並賦予充分的保護對於以口蹄疫病毒攻毒的小動物[7]。然而,這些類型 的免疫疫苗在以口蹄疫病毒進行攻毒針對自然宿主(natural host)的實驗上,其效益大大 低於傳統的滅活疫苗且只能給予自然宿主有限的免疫保護。有研究指出增加抗原表位的 數量並找到更多具免疫抗原性的抗原表位可以增加疫苗免疫的效力[6, 8]。此外由於口蹄 疫病毒具有抗原多變性的特性,可經由突變(mutation)及基因重組(gene reconstruction)兩 種方式產生新型病毒,點突變(point mutation)只會造成抗原小部份改變稱為抗原漂移 (antigenic drift),此包括一連串漸進式自發的點突變,引起區域性的小流行,基因重組 則為不同來源的病毒株同時感染同一宿主,複製過程中產生基因段的交換(crossover)和 重新排列組合(reassortment)稱之為抗原轉移(antigenic shift),則會引起全球性的大流行。 目前觀察病毒的變異與演化主要分為兩個方向,一為透過病毒之親緣關係,觀察不 同品系(strain)之病毒與流行病學上(Epidemiology)的關聯性[9];而另一方向為透過觀察 病毒隨著時間推進,在序列各個位置上異同之處[10]。在瞭解產生變異的原因後,則需 建立模型,模擬病毒演化的過程。例如:目前有一派的理論是病毒是以準種(quasi-species) 的形式存在,而不是單一的病毒株,因此病毒株的擴增,是與病毒和宿主免疫系統的交 互作用有關。一旦能預測到可能的病毒株,則要預測具有抗原性的區域(antigenic region), 做為以發展檢驗詴劑與疫苗的參考。在病毒演化時,有些胺基酸的改變可能會與鄰近胺 基酸一起變化,以維持其結構。這些區段的單獨變異可能會造成結構上大的變化,因而 成為新的抗原。為了達到上述的目的,定義出重要的抗原決定位以及找出影響抗原決定 位重要的物化特性(physicochemical properties),就扮演相當重要的角色,在針對保護性 疫苗的設計、免疫的診斷和測詴以及抗體的生產上。 病毒感染的免疫防禦涉及既有先天(native)及適應(adaptive)免疫階段。從歷史上看,
特異性的體液免疫反應(humoral immune response)被認為是最重要的因素在賦予保護對
抗口蹄疫疾病上[11]。因此找出重要的抗原決定位就相當的重要在口蹄疫病毒的防治以 及診斷上。然而,決定抗原上之 B 細胞表頂(b cell epitope )位置有賴於實驗的方式定義 出可能的抗原決定位,但是此工作是耗時且昂貴的,尤其是面對此口蹄疫病毒多變的特 性,在針對不同的病毒株上定義其抗原決定位經由實驗更是不易,因此發展計算的方式 來幫助疫苗從病原體抗原的序列中定義出具可靠的推論表頂(deduced epitope)是必須地。 最近,已經有研究結合實驗以及生物資訊(bioinformatics)的方式,在病原體疫苗研究上 來幫助定義出口蹄疫病毒之病原體上可能的表頂位置,達到減少實驗成本的耗損的效益 [12]。此外研究高變異性之病原體的研究中指出,使用生物資訊的方式來幫助疫苗的研
3 究及擴大我們對病原體的理解似乎是一個必然地方向[13]。因此,在此我們想要利用病 原體相關之免疫資訊,研究口蹄疫病毒 B 細胞抗原決定位的物化特性,建立此病原體的 免疫模型及預測系統,更進一步地獲取更多表頂的資訊達到幫助改善相關疫苗的研究 上。
1.2 研究背景
B 細胞表頂或稱為抗原決定位(b-cell epitopes or antigenic determinants ),其定義為一 抗原分子上的某個區域可以被免疫球蛋白(immunoglobulin)上的結合位所辨識稱之。此 蛋白上的表頂分通常劃分為連續(continuous)或不連續的(discontinuous),主要取決於是否 列入表頂的胺基酸序列是連續的鏈或非連續的(圖 2)。研究指出大部份的 B 細胞抗原決 定位為非線性的[14],但是對於疫苗的研究以及免疫診斷的應用方面,針對連續性的 B 細胞抗原決定位研究是值得關注的方向。近年來,由於生物資訊免疫學的蓬勃發展已經 發展出許多可應用的預測工具,在線性 B 細胞抗原決定位預測方面[15-16]。 圖 2 為抗體結合至表頂與 B 細胞表頂類型示意圖[17] 註:a.為不連續型表頂、b.為連續型表頂 接下來針對我們欲研究的對象—口蹄疫病病毒,其生化背景做一個簡單探討。口蹄 疫病毒是屬於小病毒科(Picronaviridiae)中的鵝口瘡病毒(Aphthovirus),其為一種無 封套(non-envelop)的病毒,其結構為二十面體對稱(icosahedral symmetry)結構直徑 約25nm;外面由結構上的衣殼(capsid)包覆著其基因體,其基因體由單股正股的RNA 所組成,約有8500個鹼基,可直接作為mRNA轉譯成蛋白質。病毒的開放式閱讀框架(open reading framework,ORF)編碼一個大的單鏈聚合蛋白(single-chain polyprotein),該聚合蛋 白在轉譯過程中被不斷裂解產生病毒的結構性蛋白(structural protein, SP)包括1A、1B、 1C和1D和非結構性蛋白(non-structural protein,NSP)包括2A、2B、2C、3A、3B、3C、
4 3D等,所以實際上並不存在一個完整的聚合蛋白,所以ORF可分為L、P1、P2、P3 四 個區(如圖3)。 圖3 (a)口蹄疫病毒在單一細胞之生活史 (b) 口蹄疫病毒蛋白質體組成[18] L區編碼LB和LaB兩種重疊的非結構性蛋白,這兩種蛋白是從2個不同的功能性起始 密碼子轉譯而成的;LB和LaB催化自身從單鏈聚合蛋白上裂解下來,並裂解宿主細胞 eIF24F和eIF24G,而阻止宿主細胞蛋白質合成,其中eIF24F是cap結合蛋白複合體中的成 分,是宿主細胞蛋白質轉譯過程中所必需的起始因子。L區為病毒複製的非必需區,缺 失L區的突變株仍能在宿主細胞內複製。 P1區依次編碼VP4(1A)、VP2(1B)、VP3(1C)和VP1(1D)四種結構性蛋白,最後組裝 成病毒的衣殼蛋白。口蹄疫病毒的結構性蛋白(SP)組成的衣殼由60個複本組成,每個 複本由四個蛋白質所組裝而成,分別為VP1(1D)、VP2(1B)、VP3(1C)、VP4(1A)組成一 蛋白質次單元(protomer),其為構成蛋白質衣殼的最小單位,而蛋白質次單元再組成衣 殼蛋白亞單位(capsomere)進而組裝成蛋白質衣殼(capsid)。而VP1(1D)、VP2(1B)、 VP3(1C)在病毒顆粒表面組成八股的β-Barrels結構,而VP4(1A)則在內部(如圖4)。
5 圖4 口蹄疫病毒的protomer及β-Barrels結構單元[19] P2區依次編碼2A、2B、2C三種非結構性蛋白,2A為一16個胺基酸所組成的多肽, 能夠自我催化P1-2A與2C解離,且從胺基酸序列分析推估2A可能是一種Tyr-Gly特異性的 蛋白酶(protease)。2B和2C的功能尚不清楚,2C可能與RNA的複製有關,2B可能與病毒 的感染有關。(雖然2A屬於P2區,但是有證據顯示其先形成P1-2A前驅物後再由3C所轉 譯的蛋白酶將之裂解開)。 P3區編碼4種非結構性蛋白分別為3A、3B、3C和3D。3A被認為是小RNA病毒複製 複合體與膜架構結合的錨定蛋白,與病毒誘導的細胞病理效應和阻斷細胞內蛋白的分泌 有關。3A似乎與FMDV的致病性有密切的關係,不同血清型FMDV之3A編碼區的改變或 缺失均會減弱其致病性。3B蛋白是由編碼區3個串聯重複的非等同的基因編碼,因而可 以產生3種不同的3B蛋白(VPg)。VPg基因的複本數與RNA病毒的感染力有關。攜帶有 pUpU架構的VPg蛋白可以與3′端的poly(A)結合作為病毒RNA合成時的引導蛋白,這種特 殊的RNA複製模式與宿主mRNA轉錄不同,因而在宿主RNA合成受到抑制時,並不影響 病毒RNA的合成。3C蛋白為Gln-Gly特異的蛋白酶,催化大部分聚蛋白的裂解;3C也參 與組蛋白H3的裂解,可能對宿主細胞的基因轉錄有抑制的作用。3D為RNA倚賴的RNA 聚合酶(RNA polymerase),又稱病毒感染相關抗原(VIAA)催化病毒RNA的合成。病毒 RNA就以VPg為引導物,以3D為RNA聚合酶,以3A、2B、2C及一些宿主蛋白形成複製 複合體,附著於胞質內的膜架構上合成負鏈RNA,再以負鏈RNA為模板合成正鏈RNA, 新合成的正鏈RNA包被完整的衣殼即成為一個完整的病毒顆粒[20]。 基於上述分子生物學方面的研究,蛋白質衣殼由四個部分VP4(1A)、VP2(1B)、 VP3(1C)及VP1(1D)組成,由文獻可以得知對於定義出口蹄疫抗原決定位主要的研究在其 結構蛋白方面,利用抗體定義出具保護能力的B細胞抗原表頂,現今已經有許多相關的 研究定義出許多此病原體上保護性抗原決定位,例如:在結構上GH loop、BC loop、EF loop、C β-sheet、H β-sheet 等VP1上二級結構上[21-22]。其次,有必要提高診斷能力, 對於感染動物及免疫動物的辨別上,因此近來也針對非結構蛋白部位單株抗體辨識抗原
6 表頂的辨識上用來幫助檢測感染與否[23]。 表 1 為相關研究中1不同血清型所定義出來的抗原決定位 O A C Asia 1 G-H loop (RGD) VP1 133-160 G-H loop (RGD) VP1 133-160 G-H loop (RGD) VP1 133-160 G-H loop (RGD) VP1 133-160 VP1 C terminus VP1 198-213 VP1 C terminus VP1 198-213 VP1 C terminus VP1 198-213 5 fold axis B-C loop VP1 43-48 H-I loop around VP1 170 G-H loop (RGD) VP1 135~167 B-C loop VP2 70-79 B-C loop VP2 70-79 B-C loop VP2 70-79 B-C loop VP2 70-79 E-F loop VP2 131-134 3 fold axis B-B "knob" VP3 56-61 B-B "knob" VP3 56-61 B-B "knob" VP3 56-61 B-B "knob" VP3 56-61 C terminus VP3 56-61 註:表表示不同血清型(O、A、C 及 Asia1)其實實驗中被定義出的 B 細胞表頂,如:G-H loop 表示其結構上的名稱,VP1 表示其在蛋白質體上的部位,133-160 則是序列的位置 在 VP1 上。 迄今為止,大多數商業化的抗病毒疫苗已被來自滅活或減毒活病毒或重組蛋白。雖 然他們至今已成功對一某範圍的病毒病原體,但是此類型疫苗的有效性對高變病毒 (hypervariable viruses)是有限的。這樣的高變RNA病毒逃避免疫機制的宿主物種由於高 突變率的基因組上,它導致的轉錄錯誤。免疫系統無法趕上變化率與病毒逃避識別和控 制,從而導致疾病或死亡。因此,迫切需要改進的疫苗能夠解決這些僵局是顯而易見的。 事實上,正在採取各種辦法在發展這種改進的疫苗,其中的策略是採用反向疫苗的方法。 逆向疫苗學涉及使用的計算方法來識別所有潛在候選人的免疫原基因組序列內的病原 體[13]。 在有關定義出重要的口蹄疫抗原決定位來對抗新爆發的疫情,使用生物資訊的方式 來對幫助增加實驗的效益及減少成本的耗損已經被使用,此研究篩選並確定B細胞抗原 表頂的結構蛋白的口蹄疫病毒血清型Asia1,採用生物信息學方法結合分子生物學方法, 以取得進一步的了解抗原的蛋白質結構中的應用設計的疫苗。 在此研究中,描述了分析 完整結構的胺基酸序列的使用DNAStar,使用多項物化性質找出潛在的B細胞抗原表頂, 利用表現載體表達出此序列,然後用西方點墨法(Western Blot)檢測和酶聯免疫吸附詴驗 (ELISA)法分別評價其抗原性。此預測出潛在的17個 B細胞抗原表頂,找出了6個可能具 有誘導免疫反應的序列,待進一步實驗進行確認其免疫原性[12]。
7
1.3 研究目標定義
當動物的免疫系統遭遇到一個新的抗原時,免疫系統會針對抗原上的抗原決定位 (epitope)的數目,產生許多相對應的抗體。動物抗體被誘發後,可藉由採血後經由被誘 導免疫動物之血清(serum)部分純化後獲得,亦即所謂的抗血清(antiserum),此抗血清為 多株抗體(polyclonal antibodies),在許多免疫方面的研究,利用此抗血清來當作決定抗原 決定位的位置以及抗原上是否有抗體能辨識的位置。一般來說可以被此靠血清所辨識的 片段,我們可以合理地認為其具有抗原性(antigenicity),但是是否具有能真正誘導欲免 疫動物產生免疫原性(Immunogenicity)則是需要更進一步的動物實驗來認定。 圖 5 為 B 細胞表頂映射概念[24] 圖 5 為利用圖表式描繪抗血清定義出病原上抗原決定位實驗,宿主暴露在某受質下 產生抗體由 B 細胞,此受質即為免疫原(immunogen),此受質被抗體體辨識可被當做一 抗原,當抗原或免疫原被抗體特異性地辨識出來此位置為表頂。我們的題目為使用此經 由表頂映射實驗所定義出來的實驗數據,找出此類型資料的物化特性來達到預測的效 果。1.4 研究目標
我們的目標是利用相關研究所建立的知識下,使用病原體(FMDV)相關B細胞抗原表 頂免疫資訊,結合機械學習的方式,建立口蹄疫病毒的免疫模型來改善預測的準確度在 此B細胞抗原表頂的子群當中。為了達到上述的目標必須從病原體資訊中萃取重要的特 徵及降低特徵的維度達到減小有害的訊息及冗贅及不恰當的特徵,此需要針對不同次分 類資料庫中,擷取出免疫相關重要資訊並使用此資訊來幫助免疫問題的研究。 我們必須瞭解我們所面對及欲解決的問題-即預測 FMDV 線性的抗原決定位自免疫8 序列的資訊。由於此病毒具有易突變及多種血清型的特性,此趨同或趨異性演化缺乏顯 著的序列相似性,但是其可能分享相同的結構及生物性質。在這種情況下,使用一般序 列相似度的方式例如:排序(alignment)基礎的方式可能會導致預測的偏差。此外,抗原 性是一種性質,也許被一種微妙的且深奧的方式編碼成序列,因此無法應用排序的方式 來處理這類的問題。另一方面,由流行病學方面的研究顯示出,雖然大部分突變雖然傾 向於負選擇(隨機突變),但是口蹄疫病毒其抗原的變異受到達爾文正選擇的影響(定向突 變),其胺基酸變異上傾向於突變或取代成具有相似物化性質的胺基酸來逃避宿主的免 疫系統[25]。 由此,在此我們使用物化特性(physicochemical properties)當做我們的特徵向量,我們 轉換訓練序列成特徵空間基於物化特性的指標,並結合特徵選取(feature selection)的方式 來改善預測準確度。由於此類問題需要由大量特徵向量中選擇出一組最佳的解集合,此 為一 NP-hard 問題。為了解決此大量參數最佳化問題,我們使用 IBCGA 可最大化我們 研究問題分類的準確度,同時最小化選取特徵數,來幫助擷取重要的資訊自我們的目標 資料集之中。我們的目標是找出一組重要的物化特性特徵結合 SVM 分類器,達到改善 分類準確度在口蹄疫病毒之 B 細胞抗原表頂的預測上。更進一步地把我們在此選出的物 化特性特徵進行分析比較與先前相關的研究及找出其重要的生物意義。 另一方面為了擴展我們所選出物化性質的應用,我們使用此物化性質來定義出表頂 的位置自病原體蛋白質體的序列中,我們使用不同長度的滑動窗口(sliding windows)結合 免疫實驗數據去找出病原體上具此組物化性質所涵蓋的位置,然後再利用智慧型基因演 算法找出一組最佳的滑動窗口組合達到結果的一致性,來決定可能的表頂位點。這部分 的重點是我們使用一組由相關抗原物化性質來幫助找到重要的抗原決定位之熱點,此不 同於各別物化性質的統計結果所定義出的位點而是使用綜合物化性質決定的結果。 在我們的研究中,我們發展一個計算的系統對於預測抗原決定位基於使用病原體特 異性的次分類群及特徵選取的策略,找出重要的一組物化特性。此系統可以給予一段胜 肽片段對其是否為具 B 細胞表頂性質做判別,也可以給予一蛋白質體序列利用此投票方 式找出抗原的熱點。我們的結果顯示出,我們所建立的預測模型不僅能達到較高的預測 準確度在口蹄疫病毒 B 細胞表頂的預測上,也能提供有用的資訊對於病原體蛋白質體抗 原決定位的定位上。使用測詴資料針對我們所預測的結果結合其它預測抗原決定位的工 具所得到的結果,證實可以定義出可能的抗原決定位來減少實驗的成本及幫助疫苗的設 計上。此外藉由分析此組物化性質可以更進一步瞭解及確認病原體抗原決定位的特性。
9
10
二、計算上相關研究
2.1 資料庫
最初在 B 細胞表頂的的研究及預測基於研究 B 型肝炎表面抗原(hepatitis B surface antigen)、流感血凝素(influenza hemagglutinins)、雞痘病毒的血凝素(fowl plague virus hemagglutinin)、人類組織相容複合體抗原(human histocompatiBility antigen HLA-B7)、人 類干擾素(human interferons)、大腸桿菌(Escherichia coli)、霍亂腸毒素(cholera
enterotoxins)、豚草過敏原(ragweed allergens Ra3 and Ra5)及鏈球菌(streptococcal M protein),針對抗原胺基酸組成物化性質[26]。其後,發展了許多資料庫用來研究線性 B 細胞表頂的研究,在此我們介紹兩個主要用來發展線性 B 細胞表頂預測的資料庫。
Bcipep:
Bcipep 被建立用於發展線性 B 細胞表頂相關研究,Bcipep 是一個資料庫使用實驗的 方式來測定線性 B 細胞表頂,包含免疫原性資料收集自不同文獻和其他公開資料庫中 而來,此資料庫提供的有關單抗與多抗的抗體所產生的表位資料。其包含的 2479 個項 目,每個項目包含胺基酸序列來源蛋白病原體分類免疫原性、中和性、實驗方法、模式 生物、資料參考來源、抗體抗原結構等,並且涵蓋了範圍廣泛的病原微生物如病毒,細 菌,原生動物和真菌[27]。此資料庫建立於 2005 年,其後許多B 細胞表頂預測研究接 使用此資料並用不同的機械學習型是建立模型。IEDB:
IEDB (Immune Epitope DataBase, http://www.immuneepitope.org)此資料庫是由美國 LIAI 研究所(La Jolla Institute for Allergy and Immunology)協同其他學術研究機構共同合作,動 員數十名研究人員,花費數年時間閱讀文獻蒐集、整理建製而成,為目前世界上資料量 最豐富的抗原決定位資料庫,此外其會定期更新資料內容[28]。
2.2 研究方法
最初企圖預測B細胞抗原決定位研究建立在由相關實驗結果所得到物理化學特性 (physicochemical properties)的分析上,其使用性質尺度的方法(propensity scale method), 將序列轉換為性質尺度值來衡量每個胺基酸的傾向,為了減少結果的波動,每個目標胺 基酸序列在在滑動窗口(sliding window)計算平均性質傾向的胺基酸值,然後給予滑動窗 口的中心目標的胺基酸殘基此性質尺度分數的值,以此為基礎預測是否給予胺基酸殘基 序列可能是線性B細胞表頂(如圖7)。
11 圖7 表示使用性質指標預測的概念 第一個傾向規模預測方法線性B細胞表頂是由Hopp及Woods利用Levitt所提出的親水性 性質尺度(hydrophilicity propensity)分配的傾向值給予每個胺基酸[26]。這種方法是基於 假設抗原決定位對應的蛋白質序列通常含有大量的帶電荷性和極性以及親水性殘基。其 後,其他幾個傾向量表陸續被提出對於預測線性B細胞表頂,包括:親水性、易曲性、 轉位、以及溶液的可接近性質[29-32]。之後,PREDITOP、PEOPLEE、PITOPE及BcePred 預測線性B細胞表頂基於組合多項物理化學性質指標,而不是依賴個別性質指標的方式 [33-35]。(如圖8) 圖8 為BcePred server提供圖型化的輸出結果 註:圖縱座標表示其性質指標給的性質數值,橫坐標表示在抗原上的胺基酸位置 對於上述的研究根據Blythe and Flower研究中指出性質尺度的方法預測的結果只比 隨機猜測略佳,其評估484個胺基酸的傾向量表之間的相關性研究傾向規模為基礎的輪 廓和位置的線性B細胞表頂的數據集50蛋白質,其證實使用單一種胺基酸的性質傾向不 能用來預測表頂的位置,並建議使用人工智能的技術幫助改善預測的方式[36]。根據相
12 關研究的瞭解,可以知道使用單一性質無法準確地預測出可能的B細胞表頂但是如果使 用多項指標來找出可能的B細胞表頂,又不易確認出確切的邊界,因而產生過多的預測 結果。 為了改善以性質指標的方式預測的準確度提出了一些改善的方式,例如:提出根據 序列傾向的指標使用AAP指標[37]。BepiPred則是建立一預測模型使用隱藏馬可夫模型 (HMM)結合親水性質指標的方式改善預測的準確度[38]。ABCPred使用倒傳遞類神經網 路(RNN)對於預測線性表頂使用700個B細胞表頂及700個非B細胞表頂使用5倍交叉驗證 訓練,輸入序列長度從10至20個胺基酸殘基,進行了測詴和評估效益,得到66%的訓練 準確率,在使用一個長度大小的16個胺基酸下為最佳的預測結果[39]。BCPred及FBCPred 則是預測線性及彈性長度的表頂各自地,使用向量學習機使用string Kernels達到預測的 準確度為67.90%及65.49%各自地[40-41]。COBEpro使用兩階段的過程預測線性B細胞表 頂,在第一階段一個SVM分類器被使用來給予分數對於詢問的抗原片段,此SVM的輸 入一個向量相似度對於在輸入的片段以及所有訓練的片段。在第二階段,一個預測的分 數被產生關聯性與每一殘基在詢問抗原性質基於SVM分數對於每一片段。然而, COBEpro無法被使用針對分辨抗原從非抗原[42]。對於B細胞表頂預測的工具缺少一個 一致性的比較方式,難以比較預測工具預測結果的優劣,如何定義出真正非表頂的特徵 是此難以比較的原因之一,在無實驗驗證預測結果的研究之下[15]。 所以由以上相關研究的瞭解知道主要分為兩種類型的預測方式,一種為根據抗原的 性質指標的方式給予每一個滑動窗口的中心點一個分數然後再利用訓練的結果決定表 頂的位置及範圍,另一種則是給予一片段決定可能為表頂或非表頂。 以上相關研究的努力下已經有很大的改善在B細胞表頂的預測上,但是對於改善保護 性的B細胞表頂在減少實驗成本的效益上仍值得努力。Söllner et al.最近研究了實用的預 測的抗原性、序列變異和保護轉譯後修飾模體在預測具保護性線性B細胞表頂,他們的 分析表明在查詢蛋白質序列使用重點領域的一個子集有可能提高預測性能線性B細胞表 頂預測結果。這指出針對B細胞表頂的研究結合演化方面的資訊亦即設計分類器結合使 用特異性次分類當做訓練資料,具有改善預測效率的潛力[15, 43]。 另一方面,有研究指出為了增加敏感度及特異性,一致性的方法被利用結合各種工 具。下面的表格示範此一致性如何產生效益,理論上來說這是簡單及容易瞭解的概念。 但是實際上,很多問題存在在這方面的研究,像是電腦計算的限制,計算預測的速度, 不同的輸出格式以及困難的整合預測的結果。例如:一個序列片段在預測中被給予高的 分數,但此預測結果與實驗結果所認同的表頂不同,困難地對於決定是否有高的分數為 表頂或是預測中多次出現是否應當被包括在此一致性的輸出中。以上所述均增加困難在 建構一致性的方法上,此外很少一致性預測軟體具有增加預測準確度可以被使用[16]。
13 表 2 為表示使用表頂預測結果一致性的概念 Prediction method M1 M2 M3 M4 Consensus method Prediction result
Non-epitope Non-epitope Non-epitope Non-epitope 0% epitope epitope Non-epitope Non-epitope Non-epitope 25% epitope epitope epitope Non-epitope Non-epitope 50% epitope epitope epitope epitope Non-epitope 75% epitope epitope epitope epitope epitope 100% epitope
14
三、最佳化演算法
3.1 基因演算法
基因演算法(Genetic Algorithm )是 John Holland 及其學生於 1970 年代左右共同研 究出來的。經過近年來的不斷發展,現今已被廣泛的應用到求解最佳化問題、資料搜尋、 人工智慧以及機器學習的領域上[44]。其以達爾文演化論為基礎,模擬生物界的 ―適者 生存,不適者淘汰‖ 的生存演化法則,每一物種在生存環境中會彼此相互競爭,只有適 應性強的物種才得以存活與繁衍,這種自然淘汰機制會逐漸發展出來最優秀的品種。 Holland 認為自然界的演化是發生在生物染色體的基因中,每一種生物的特徵係來自於 該種生物上一代的基因排列,而演化是指每一代間的基因所發生的變化情形。所謂適者 生存是指這一代的基因排列優於上一代的基因排列而產生比上一代更能適應環境生存 的世代。 圖 9 基因演算法示意圖 因此基因演算法是強調基因型的轉變,將問題的可能解答經過編碼成為基因型式, 利用遺傳運算子演化來找到最佳解,其中基因型的改變主要經由交配、突變達到演化的 效果。在此部分我們使用實數型(real-code)基因演算法來設計染色體,此具有減低如二 進位(binary)及符號型(symbol)基因演算法實際操作時需要經過編碼及解碼的步驟,只要 針對問題的解空間進行染色體設計便可達到演化式計算的效果。
15
3.2 直交實驗設計與因素分析
3.2.1 直交表
直交表 (orthogonal array) 是由 R. A. Fisher 最先提出的,直交所代表的意思是平衡 (balance) 而不混合 (mix),亦即統計上的獨立 (statistically independence),因此直交表 中每一欄的各水準值 (level) 出現次數是相同的,使用直交表事實上僅是進行部份因素 實驗 (fractional-factorial experiment) ,因此能較完全因素實驗 (full-factorial experiment) 節省大量執行的時間,且直交實驗具有系統推理的特性,因此只需進行部份因素實驗就 可以求得最佳解的近似解 (near optimum)[45]。 以兩水準,三因素直交表說明,若要進行完全因素實驗,需要八次實驗(238),如 果需要在八次實驗中選擇只作四次實驗,如何選擇能達到均勻取樣的目的,建構一個 3 4(2 ) L 的直交表,如下表所示,可以縮減成四次實驗,這樣的取法使用均勻且對稱的取 樣來推測全部實驗的最佳解。 表 3 為兩水準三因素完全實驗 實驗批次 因素 F1 F2 F3 1 0 0 0 2 0 0 1 3 0 1 0 4 0 1 1 5 1 0 0 6 1 0 1 7 1 1 0 8 1 1 1 表 4 為兩水準三因素直交實驗表 實驗批次 因素 F1 F2 F3 1 0 0 0 2 0 1 1 3 1 0 1 4 1 1 0
16
3.2.2 直交實驗設計
直交實驗設計(Orthogonal Experimental Design, OED),實驗的過程中通常利用假設來 減少實驗結果的因素個數,以節省成本的損耗但是又要接近真實的結果,直交實驗設計 是為了解決此問題而被提出的方法之一。其主要包含兩個重要的部份:直交表
(Orthogonal Array, OA)與因素分析(Factor Analysis)來達到此效果[45-47]。首先透過直交 表,產生出獨立且均衡的每一個因素,再藉由每一個因素分析出的主效果(Main Effect), 由主效果推論每一個因素對於該實驗結果的優劣。因此以直交實驗來解最佳化問題時, 問題的一個參數可視為直交實驗中的一個因素,而參數視為因素的水準(Level)值。完全 因素實驗(Complete Factorial Experiment)會以全部水準值的排列組合進行實驗,而 OED 僅取全部排列組合中的一部份來進行分析實驗,也就是部份因素實驗(Fractional Factorial Experiment),因此直交陣列系統推理化的特性只需進行部份因素實驗就可以推測出所有 搜尋空間中最佳的近似解(Near Optimum),可節省大量執行的時間。 圖 10 為直交實驗設計 例如:f1 的| Sj1 – Sj2 |計算 Sj1的值為 Y1+ Y2+ Y3+ Y4 ,Sj2的值為 Y5+ Y6+ Y7+ Y8的 計算出相對應的| Sj1 – Sj2 |值,然後以此數值大小決定此因素的影響力。 3.3 智慧型基因演算法 為解決得到最佳的 window size 組合投票結果符合真實的實驗結果,此為參數最佳 化問題,必須使用最佳化演算法幫助解決此問題,在此本文使用智慧型基因演算法 (Intelligent Genetic Algorithm, IGA)[48]做為最佳化的工具,此最佳化演算法具有收斂速 度快,精確度高的優點。
17 智慧型基因演算法與傳統基因演算法最大之不同,乃是以智慧型交配取代一般的單 點交配或多點交配,因為傳統基因演算法的交配方式無法評估染色體中參數個別的優劣, 且交配點是由隨機方式產生,得到的後代染色體品質不容易有顯著地提昇。此智慧型基 因演算法將染色體交配過程視為一種因素實驗(factor experiment)。將來自親代的兩個染 色體中已經被切割好將要進行交配的片段,視為直交實驗設計的因素,並以此染色體片 段「互換」或「不換」做為兩種水準值(1/0),以此兩水準直交實驗產生出優良品質染色 體的機率便可大幅提昇,進行步驟如下: 1.令產生染色體中的交配點所切割出的基因片段為實驗因素;假設因素數目為 n, 即有 n 個基因片段,選擇 Lβ(2β-1)直交表的前 n 欄作為實驗之用,其中 β = 2[log(n+1)]。 2.令因素 j 的水準 1 與水準 2 分別表示來自親代染色體 P1與 P2第 j 個基因片段。 3.根據直交表,計算各因素組合實驗的評估值 yt,t =1, 2,….,β。 4.計算主效果 Sjk,其中 j = 1, 2,…,n,k = 1, 2。 5.決定各因素的最佳水準。在評估函數望大時,則選擇主效果值較大之水準;在評 估函數望小時,則各因素的最佳水準為主效果值較小之水準。如評估函數望大且 Sj1>Sj2,則因素 j 的最佳水準為 1;反之則最佳水準為 2。 6.根據各因素的最佳水準,選擇對應親代染色體中的基因片段,組合成第一個子代 染色體。 7.將各因素的主效果差值( | Sj1 – Sj2 |)排名,差值越大者排名越高。 8.以類似第一個子代染色體的方式來組合因素,將差值排名最差的因素,選擇與第 一個子代相反的水準,則可產生第二個子代染色體。 圖 11 表示智慧型基因演算法架構
18
3.3.1 智慧型交配
智慧型交配運算結合了兩水準直交實驗與交配運算,能夠有效率的產生具有優秀評 估值的新染色體。假設我們使用 LN+1(2N) 直交表來做智慧型交配運算,本染色體交配運 算的詳細進行步驟如下: 步驟一:假設即將進行交配運算的兩條染色體為 P1、P2,比對 P1、P2內的基因,並 將重複出現在兩條染色體內的基因移動至染色體末端,其相對應的控制基 因。 步驟二:隨機將基因切割成[ N / 2 ]個基因片段。每個基因片段及代表直交表的一個 因素。P1中第 j 個基因片段,及代表因素 j 的第一個水準值;P2則代表第二 個水準值。 步驟三:計算直交表中每個染色體排列組合方式的適應值 ft ,t = 1,2,3,…,N+1。 步驟四:計算各因素之主效果 Sjk,j = 1,2,3,…N,k = 1,2。 步驟五:決定各因素的最佳水準。在評估函數望大時,則選擇主效果值較大之水準; 在評估函數望小時,則各因素的最佳水準為主效果值較小之水準。如評估 函數望大且 Sj1>Sj2,則因素 j 的最佳水準為 1;反之則最佳水準為 2。 步驟六:根據各參數的最佳水準,選擇對應親代染色體中的參數,組合出第一個子 代染色體。 步驟七:將各參數的主效果差值排名,差值越大者排名越高。 步驟八:以類似第一個子代染色體的方式組合參數,除了盤明最差的參數選擇的水 準與第一個子代染色體相反,可產生第二個子代染色體。3.3.2 突變運算及演化終止條件
突變運算使用任意合理之亂數運算。假設一條染色體編碼之總長為 N 個位元,突變 率為 Pm,則每次的突變運算,隨機由染色體中選出「N *Pm 」個位元,然後以亂數變 化所選中的物化性質參數。 基因演算法所需的演化時間必須視問題的複雜度而定,較一般化的作法是設定演算 法的評估次數與問題中的參數數目成正比。一但終止條件達到設定值,演化即停止,並 輸出所搜尋過的最佳解、用最佳解當模型參數。3.4 繼承式雙目標基因演算法
針對欲選擇一組最小的可提供資訊的特徵向量(informative feature),但是又要最大化 預測的準確度此為一個雙目標最佳化的問題。一個有效率的繼承式雙目標基因演算法 (Inherit Bi-oBjective Genetic Algorithm, IBCGA)被[49]我們用來解決這問題。IBCGA 包含一個智慧型基因演算法具有繼承式的機制。此智慧型基因演算法使用分 割解決問題的策略( divide-and-conquer strategy)及直交表(orthogonal array)針對基因的互 換步驟解決大尺度參數最佳化的問題。在這個研究中智慧型基因演算法可以有效地探索 及利用搜尋空間 C(n, r). IBCGA 可以搜尋 C(n, r1)藉由繼承一個好的解空間 C(n, r)。所
以,IBCGA 可以獲得一組高品質的解在單一批次實驗下,在這裡 r 是指一個我們有興趣 的範圍內。
19 染色體編碼: 此被提出來的染色體被編碼成一個二進位的基因用來特徵選取之用,此外此染色體 還包括一參數基因用來調整 SVM 的參數,在這裡基因( gene)及染色體(chromosome)即 為一般被使用在基因演算法中所描述的。此染色體由 n=531 個二進位的基因所組成,每 一個基因(bi)代表可被選擇含有訊息的性質,此外還包含 4 位元的基因此被用來調整 SVM 的參數 C 及 γ。假如 bi=0 此第 i 項的性質被排除在 SVM 的分類器中,反之則被包 含在內。對於 SVM 的及 C 參數編碼為 16 個值 2-7 , 2-6…, 28 圖 12 為 IBCGA 染色體編碼的方式 此特徵向量對於 SVM 分類器,被編碼成一個含多個基因的染色體使用下面的步驟。 確認一個蛋白質或胜肽序列,然後將此序列轉換成該序列內含胺基酸所對應 aaindex 中 物化性質胺基酸的數值,然後使用平均的方式把此相關的指標轉成此染色體中的基因此 代表此序列所包含物化性質的數值,由於每一個 aaindex 中所對應不同的物化性質,所 以每一條序列可以得到 531 個物化性質的特徵向量。然後最後使用正規化的方式把以上 得到各染色體的特徵向量正規化成數值[-1, 1]之間。 然後此適應函數針對 IBCGA 得到令人滿意的解,此外選擇適當的參數對於 SVM 在 10 倍的交互驗證下(10-CV),因此此最終染色體被獲得,IBCGA 使用適應函數 f(X)可以 同時獲得一組解 Xr 在這裡 r=rstart, rstart+1, …, rend 在一次運算中同。此 IBCGA 演算法被
給予 rstart 及 rend依照下列描述: 步驟一:起始,隨機產生一起始族群 Npop所有染色體中 n 個二位元的基因具有 r 1’s 及 n-r 0’s 在此 r = rstart。 步驟二:評估此適應函數值對於個體使用 f(X) 。 步驟三:選擇傳統的競爭選擇其在隨機選取的兩個中,選擇較優良的自交配池中。 步驟四:選擇 pc·Npop親代自交配池中執行直交表互換在選擇一對親代當中,在此 pc 為互換的機率。 步驟五:隨機選擇 pm·Npop應用在互換的個體進行突變的操作,在此 pm為突變的機 率。為了避免最好的適應函數值被破壞,此步驟不針對最好的適應函數值 個體進行。 步驟六: 終止條件,假設停止條件對於獲得解 Xr被滿足,輸出最好的個體 Xr。否 則回到步驟二。 步驟七:繼承是假如 r < rend隨機改變一個位元在二位元的基因上對於每一個個體由 0 到 1,增加數量對於 r 為 1 然後繼續步驟 2 否則停止演算法。
20
3.5 向量學習機
向量學習機(Support vector machine, SVM),是一個學習的模型處理兩種分類的問題。 SVM是藉由找出一個超平面(hyperplane),使之將兩個不同的集合分開使用最大距離在兩 個向量組成之間[50]。為了使要本更容易地分離開來,SVM使用各種功能的kernel來轉 換樣本至更高維度的空間或是特徵空間(feature space)。在這邊我們使用常用的radial based function (RBF)當作我們投射樣本的kernel。此RBF為一非線性的Kernel,定義如下 面所示: 0 ), exp( ) , (xi xj xi xj K 此 kernel 參數 γ 決定樣本如何轉換到更高維度的空間。而成本參數 C(cost)為給有誤 差的資料一點懲罰,讓它多一點成本,亦即 C 是我們決定要給多少懲罰的權重。此兩個 參數 C 及 γ 必須被調整找到最佳的值在得到最好的預測效益上。一般的做法是把訓練 資料拆成兩個部分,一個部分用來訓練,另一個部分用來驗證準確度,若準確度不夠的 話,換參數再做一次。在這部份我們是利用 IBCGA 在利用此 SVM 分類器情況下,同時 選取 feature 下幫助選取此 feature set 的最佳的參數值。
圖 13 為二維 SVM 概念示意圖
1D (Low Dimension) 2D (High Dimension)
OSH Max. Margin
21
四、資料的建構及物化性質指標
4.1 使用資料的建立
在這個研究中,我們使用了三個資料集,分別為口蹄疫病毒資料集、獨立測詴資料 集以及 EL-Manzalawy 資料集(FMDV-dataset、independent test dataset、EL-Manzalawy dataset)分別做為免疫模型建立、獨立的測詴資料及用來評估演算法的資料各自地。其中 口蹄疫病毒資料集及獨立測詴資料集由網路上的免疫資料庫(IEDB)及相關研究文獻中 收集得到,EL-Manzalawy 資料集則是使用 Bcipep 中的資料而來(如表 5)。 表 5、此研究所用的資料集 資料集名稱 序列來源 生物體 資料數目 (正反應/負反 應) 序列長度 (分別為最短長 度、平均長度 以及最長長度) 資料庫描述 口蹄疫病毒資 料集 口蹄疫病 毒 806(214/592) 4,9.76,50 此病原體(FMDV)免疫實 驗數據來自於 IEDB 獨立測詴資料 集 口蹄疫病 毒 118(83/35) 6,15.87,41 由此病原體研究所得到 的數據及 IEDB 得到(此 不包含於口蹄疫病毒資 料集中之序列) EL-Manzalawy 資料集 數種不一 樣病原體 範圍的表 頂,包含 口蹄疫病 1868(934/934) 4,14.19,56 此為降低同源性的資料 集由 EL-Manzalawy et al 自 Bcipep 得到
4.1.1 口蹄疫病毒資料
此病毒主要分為七種血清型,各血清型之間無交互保護力或是其保護力很有限,在 此我們不欲針對每一個血清型做詳細的分類,而是採取找出可能的共同的物化性質針對 病毒抗血清辨識特異性序列上。找出抗血清抑或單株抗體中,所能辨識出病原體上抗原 決定位的物化性質是我們的目標。於此我們自 IEDB 資料庫提取出相關 B 細胞表頂的實 驗資料,包含所有有關口蹄疫病毒此病原體生物體,但不包含非此病毒病原體上的序列。 此資料集被建立用來建構我們想要的免疫模型。 我們在此研究中使用此病原體抗原上能與此病毒抗血清在免疫實驗上產生反應的序22 列當作我們免疫正反應資料(positive data)為表頂的資料。另一方面,先前在免疫研究中 針對 B 細胞表頂的研究使用分類方式來建立免疫的模型,其使用表頂的部分是基於抗體 抗原交互作用免疫實驗上有反應的序列,而非表頂部分是利用結構的訊息選擇非表面蛋 白的部分當做訓練的資料,有文獻指出此並非真正的負反應資料(negative data)[15]。在 此我們同樣地使用基於實驗所得到的結果,使用此病原體中經由免疫實驗無法與此抗血 清產生特異性結合的當作我們的負反應資料為非表頂資料,在這樣的設定之下,我們自 IEDB 此重要的免疫資料庫中取出我們的訓練集來建立我們此病原體相關的 B 細胞表頂 免疫模型。 FMDV 資料篩選及建立步驟如下:
我們依照 IEDB Curation Manual 2.0 所描述來獲得我們的訓練資料避免錯誤的擷取資 料。來源生物體(Source Organism)表頂的識別是基於病毒抗血清或單株抗體來辨識此病 原體序列。由 Browse 選項由來源生物體選項(by source organism),進入後可以得到一個 生物體分類的樹狀表,此表為生物學上的分類,其由上而下的層次依照分類學的次序, 依照對於口蹄疫病毒分子生物學背景的瞭解,使用此表選擇依序由病毒-單股 RNA 病毒 -單股正股 RNA 病毒-小核醣體病毒-小核醣體病毒科-鵝口瘡病毒屬-口蹄疫病毒。 ( ssRNA viruses - ssRNA positive-strand viruses - Picornavirales- Picornaviridae -
Aphthovirus - Foot-and-mouth disease virus ),選擇 Foot-and-mouth disease virus 此項為與 口蹄疫病毒相關的免疫資訊。
23
圖 15 為分類學上的層級
進入此 FMDV 病毒選項後,此與 FMDV 相關免疫研究資料項目包含 structure(1083)、 reference(140)、source antigen(75)、MHC Binding(21)、B cell arra(2540)及 T cell array(590), 每一項目後面的數字代表其所包含資料數量。標示紅色的部分為我們所主要使用的資訊 部分。分別簡單說明其所包含的內容 structure 代表此病毒免疫實驗所代表的序列此序列 有一個獨一無二的表頂序號(epitope ID),選擇 structure 可以看見此但是此獨一無二的表 頂相關的資訊,此表頂可能來自不同的免疫接受器所定義出來的表頂,包含 T 細胞、B 細胞及 MHC Binding,例如:若為 T 細胞免疫陣列所辨識具有實驗上正反應我們可以稱 之為 T 細胞表頂,每一表頂可能具有多種特性,若選擇進入某表頂 ID 後,可查閱其為 何種免疫實驗所鑑定出來的表頂以及參考文獻。Reference 則是此資料項目相關文獻的 彙集。source antigen 則是這個資料庫中用來實驗用的病原體不同的品系(strain)數目。 MHC Binding(21)、B cell arra(2540)及 T cell array(590)則各是用不同的免疫接受器來定義 出相關抗原的項目。
24
由 structure 的 Epitope ID 選項進入,我們可以看到有幾個重要的訊息,包含 source accession 此代表序號為此 FMDV 在 NCBI 中的序號、B cell assay 在 B 細胞微陣列實驗 中與此序列有關的實驗數目、3D 同源結構、source antigen 指在此病原蛋白質為其蛋白 質體上的那一部分,source organism 則是此病原體名稱及品系,reference 則是此序列所 參考的文獻。
圖 17 為進入 Epitope ID 的項目
由下圖可以看出進入 Epitope ID 的項目選擇 B cell assay 可以看到多個實驗結果每個 B cell 實驗均有一個 B cell ID,在這邊我們可以看到此表頂實驗結果均為正值,但是在 其它表頂可能有正有負,在此我們假設只要多次實驗中有正反應的實驗其為具有表頂可 能性,我們把此序列歸為正反應的資料在我們的訓練的資料中。
25
此外也可以直接進入 B cell assay 中由 B cell ID 去得到我們所要的資訊,但是要移除 重複的序列資訊及確認實驗的結果。 圖 19 為表示 B cell assay 項目 由上所述我們確認我們要收集的 B 細胞表頂資訊,由表頂序號進入,確認每一序列 實驗的類型及實驗的反應,擷取我們要的 B 細胞表頂相關資訊。此部分,由 B 細胞表 頂序號(B cell ID)去下載其此病原體相關所有與 B 細胞免疫反應相關的序列,此 B 細胞 表頂序號是指不同實驗下或不同文獻所鑑定的 B 細胞表頂,因此同一表頂序號序列下可 能有不同的實驗數據。此相關的資料中移除冗贅的資料然後留下 845 個 B 細胞表頂資料。 由於線性表頂的長度是不明確的,我們移除了只一個胺基酸的序列為 39。最後剩下 806 個獨一無二的序列當作我們的訓練集,此包含了 214 個具有抗體反應的及 592 個無抗體 反應的資料,並確認與表頂序號所得相同序列資訊,交叉確認,使用此資料當作我們的 訓練集來進行接下來物化特性特徵萃取的工作。此外為了瞭解實驗資料定義出表頂序列 的分佈在此病原體蛋白質體上,我們也統計了用來訓練的資料庫中的分佈數量(如表 6)。 表 6 口蹄疫病毒訓練資料分佈 蛋白質體 組成次單元 序列數量 病毒的結構蛋白 VP4 5 VP2 17 VP3 14 VP1 662 病毒的非結構蛋白 2B 108 2C 3A 3C 3D
26
4.1.2 獨立測詴資料
為了評估我們所建立的免疫模型獨立的測詴是必須的,我們收集了相關研究由免疫 實驗方式定義出 FMDV 上之 B 細胞表頂之數據當作我們的測詴資料集,此包含兩個部 分。第一個測詴集,此實驗數據包含 FMDV 結構及非結構蛋白中定義出的 B 細胞表頂, 共 50 條相關的序列,這部分得到的資料是以關鍵字:FMDV、B cell epitope 來進行資料 的探索。所以本質上與我們得到的訓練資料一致,但是其建立於我們免疫模型建立之後, 目的是為了測詴是否可以預測出病原體中具有免疫特性的片段。此外,也收集 IEDB 中 研究 FMDV 但是非天然來源序列(no natural source),所得到的資料當做我們第二個測詴 集子集,此包含 71 個序列我們移除包括非 20 個胺基酸之序列,最後得到 68 條。第二 個測詴集則是是否能預測出利用此致免疫部位設計的胜肽疫苗是否具有免疫反應(如表 5)。此兩個子集的最大差異在於是否與病原體上的序列完全一致,但是相同地是其序列 實驗的結果乃基於病毒的抗血清來確認此是否具有抗原性。 表 7 為獨立測詴資料集 抗原來源 數量(正反應資料/負反應資料) 參考資料 合成胜肽 68(57/11) IEDB 病毒結構蛋白 病毒非結構蛋白 參考文獻 病原體 VP0~VP3 17 (6/11) Χ [12] VP1 10 (8/2) Χ [51] Χ 2C 13(2/11) [23] VP1 (4/0) 3B 1/0 [52] VP1 (2/0) Χ [53] VP1 (1/0) Χ [54] VP2 (1/0) Χ [55] VP1 (1/0) Χ [56]27
4.1.3 病原體中表頂位置的資料
欲針對 FMDV 使用我們所選出的物化性質來辨識出可能的表頂位置由病原體蛋白質 體序列中,因此我們必須自所得到的訓練及測詴的資料中延伸相關資訊找出其原始病原 體抗原位置的資訊,目的是確認出實驗所得到表頂序列位置的資訊,用來進行進一步地 測詴並決定我們所使用的投票機制結合智慧型基因演算法。 這個部份可以由我們所擷取出來的表頂來進行資料的收集依照下列的步驟: 1. 首先,藉由表頂序號我們可以得到一組 source accession,此為此表頂針對此病原體 株。2. 由此 source accession 可以連接到 NCBI 原始資料建立的序列,我們依此原則建立一 個檔案其包含 Source Molecule Accession(GI)、GeneBank No,並針對實驗上所使用 的序列在此病原體蛋白質體上結構部份及非結構部份及對象予以註解。 3. 接下來由表頂序號中的 B 細胞表頂序號註解可以用來找出此段表頂序列是針對此病 原體蛋白質體上的那一個位置。 4. 把以上的病原體序列自 NCBI 中下載 fasta 格式,並加上免疫研究位置的註解,最後 得到我們所要的序列及實驗的位置。
4.1.4 評估演算法的資料
這部份的資料是想瞭解在綜合的病原體資料下此演算法分類的效益是否有顯著地差 異,並比較此資料之下基於SVM分類器的研究比較彼此的分類效率。 由於B 細胞表頂序列長度非固定,我們使用資料集來自 EL-Manzalawy et al.,此包含彈性長度之表頂(flexible length)。此 EL-Manzalawy et al.所包含的 B 細胞表頂之序列 來自於 Bcipep。基於此資料庫提取的資料集來比較分類的準確度與相關研究中,評估所 使用演算法效益,此彈性長度的資料及建立的準則如下: 1. 在Bcipep中所有與B細胞表頂相關的當作正反應資料,但是要取大於4個胺基酸長度 的長度的資料因為大部分表頂長度範圍在此資料庫中為4~20內得到1223條序列。 2. 移除重複的序列及降低其相似性54至80%下,因此最後剩下934條序列。 3. 隨機自SwissProt選取等量於正反應資料當作負反應資料。 4. 最後得到1868(934/934)條序列。 以上所描述的部分來自此Bcipep資料庫引用的文獻中處理資料的方式[40]。此為我們 EL-Manzalawy dataset的資料取得方式。
28
4.2 物化性質指標
針對此一目的分析了 B 細胞表頂胜肽(peptide)片段胺基酸的物理化學性質 (physicochemical properties),根據這些特性建立規則,設計分析 B 細胞表頂發生位置的 預測程式,預測沒有實際實驗資料驗證的病毒蛋白的 B 細胞表頂位置,提供研究人員進 一步的參考資訊,協助加速病毒疫苗研究的進展。我們使用 AAindex 當作我們的物化特性指標,此胺基酸指標資料庫Amino Acid Index Database (AAIndex),包含數以百計的各種物理化學性質及生物的胺基酸性質,每一個 指標表現出一種胺基酸的性質其使用數位矩陣的型式呈現此性質。
在最新版的 AAIndex 9.0 包含 562 個指標。這些物化性質包含分子量( molecular
weight)、疏水性( hydrophobicity)、親水性(hydrophilicity)、水合潛力( hydration potential)、 易接近表面面積( average accessible surface area)、自由能轉移( free energy transfer)、彈性 ( flexibility)、殘基體積( residue volume, mutability)、熔點( melting point)、光學活性(optical activity)、側鏈體積( side chain volume)、極性( polarity)及等電點 isoelectric points。在此 我們在這些物化性質指標中移去缺少 20 個胺基酸的部分,最後剩下 531 個物化性質, 使用這些物化性質當作我們的物化特性指標[57]。
29
五、免疫模型建構
此章針對本研究中所收集之材料及如何應用最佳化演算法建立免疫模型做一詳細的 說明,並描述我們選取之物化性質所建立的模型與相關研究比較的準則,接著說明如何 使用我們所選出的物化性質來定義出表頂位置自病原體中以及其評估的方式。最後討論 我們所分析物化性質的準則。 圖 20 表示本研究最佳化演算法使用的部分5.1 免疫模型建構方法及評估的方法
在這個研究中我們利用由免疫資料庫所得到的由此口蹄疫抗血清所定義出病毒病原 體蛋白質中表頂的部位,當作我們的訓練資料,依照下列步驟建立我們的免疫模型: (1) 將所得到的序列資料依註解 positive 定為 1 類,negative 定為 0 類。 (2) 把所得到的訓練資料轉成 aaindex 中的 531 個物化特性。 (3) 由於每一物化特性值轉換後大小不一,接著將其利用正規化公式轉換每一物化特 性值為-1~1 之間。 (4) 以上前處理完後,使用繼承式雙目標基因演算法幫助選取重要的物化特性,設定 的參數如下: Npop = 30、Pc= 0.8、Pm = 0.05、rstart = 40 及 rend = 10 及 10 倍的交互驗證下(cross-validation)。
(5) 在以上的相同的設定之下,跑 100 批次實驗,最後選取訓練準確度最高的當作我 們的免疫模型。
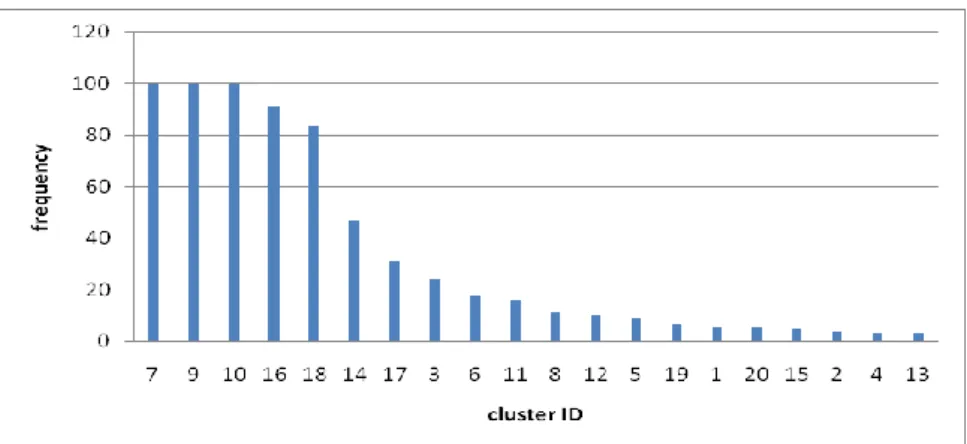
30 我們認為在最好的訓練結果中所代表的含意,是指在使用機械學習方式分類上,此 組特徵集合可以正確地幫我們分辨出此基於實驗所的訓練資料中表頂及非表頂的序列。 另一方面,由於高的準確度(accuracy)其所代表的物化性質,我們合理的相信可以更加接 近 FMDV 病原體中 B 細胞表頂所蘊含的物理化學性質。 圖 21 利用 IBCGA 結合物化性質建立免疫模型示意圖 依照所得到訓練的免疫模型,依照得到的值計算下面的值,可做為評估比較的標準: 敏感度 (sensitivity or true positive rate, TPR):TPR = TP / P = TP / (TP + FN)
準確度 (accuracy, ACC):ACC = (TP + TN) / (P + N)
特異性 (specificity or True Negative Rate, TNR):SPC = TN / N = TN / (FP + TN) = 1 − FPR Matthews correlation coefficient (MCC):MCC=(TP*TN−FP*FN)/(PN*P′N′)1/2
此外為了比較我們基於 B 細胞表頂次分類群所萃取出重要的物化性質所建立的模型 之實際效益,我們使用獨立測詴資料集來測詴建立模型的效益,並與其它兩篇相關研究 基於不同病原體表頂範圍使用分類的方式所建立的預測 B 細胞表頂的模型做比較 (BCPred 及 FBCPred)。
5.2 自病原體蛋白質序列中定義出表頂位置及評估的方法
此研究的另一個重點為,利用我們所挑出病原體中重要的物化性質來幫助定義出病 原體上表頂的位置。由先前研究中可以瞭解欲定義某病原體蛋白質序列中表頂的位置, 一般使用與表頂相關的物化性質來定義出來,但是有研究指出其只略佳於隨機選取,因 為表頂所具有之物化性質無法以單一物化性質定義出來,也有研究使用多種物化性質來31 交互比對定義出表頂的位置,然後利用訓練的結果決定閾值(threshold),以此來決定抗 原決定位的位置。 在此我們使用所選出的物化性質來定義出表頂的位置自病原體序列中,針對每個目 標胺基酸序列在在滑動窗口(sliding window)計算平均性質傾向為表頂或非表頂,然後給 予滑動窗口的中心目標的胺基酸殘基此性質尺度的傾向值,由於多種不同的滑動窗口所 定義出的抗原位置波動的結果,我們欲從變動的結果中找尋一致性的結論並且降低偽陽 性的結果因此在此使用訓練資料中原始實驗位置的資訊結合投票的機制並使用智慧型 基因演算法幫助找出最佳的投票組合。 決定病原體蛋白質序列中抗原決定位依照下列的三個步驟來進行: 步驟一:資料的處理,此部份要得到我們步驟二最佳化演算法所需要的格式。 (1) 利用訓練資料中的序列資訊,找到此抗原決定位在病原體序列中的位置,由先前 建立訓練資料庫中,自 NCBI 中下載病原體蛋白質序列,切成不同的滑動窗口分 別為 3、5、7、9、11、13、15、17、19、21,此滑動窗口範圍大小被決定依照 大部份抗原決定位範圍,為小於等於 20 長度大小。 (2) 將切好的序列轉成所選出的物化性質,然後假設每一序列均具有抗原決定位性 質。 (3) 將處理完成的序列,使用我們所建立 FMDV 免疫模型來決定其具抗原性質或不 具抗原性質。依照假設每一滑動窗口均假設具有抗原性質,其與 FMDV 免疫模 型預測相符合給予該滑動窗口中心位點一個值 1,若不符合給予值 0,此時在相 同位點上由於不同滑動窗口大小下,其胺基酸殘基組成的差異將造成物化性質的 差異,因此在同一位點不同滑動窗口此位點上有不同的性質尺度傾向值(圖 22)。 圖 22 為物化性質判斷抗原位點的概念
32 (4) 然後再依照真實實驗中定義出 FMDV 表頂的位置給每一位置相對應的值,具有 抗原性質給 1,不具抗原性及無實驗結果均給予 0,最後可以得到如下列例子的 資料格式(圖 23)。 圖 23 為處理完成的資料格式 實驗結果左側部分為位置右側為真實實驗的結果 不同滑動窗口的值為其評估物化性質的結果 步驟二:決定適當的抗原位點,我們必須瞭解,在同一位點上所得到抗原性質及非抗原 性質物化性質的差異是由於不同滑動窗口下胺基酸序列組成的不同導致的差異,而此差 異乃基於此物化性質綜合的結果,為了避免過度變動的結果,我們必須決定適當的方式 確認表頂的位置,此外我們欲確認出抗原表頂位置,但是又要減少偽陽性的值,因此我 們利用在訓練集中多個實驗結果所定義的位置資訊,以及我們物化性質的組合相近的位 點來找出最好的組合,並由多組不同滑動窗口找出最一致性的結果來確認表頂的位置 (圖 22)。 (5) 在此我們使用投票的方式來決定哪幾個滑動窗口值之一致性的結果與真實實驗 結果相符合,使用智慧型基因演算法幫助決定最佳的組合。 (6) 此智慧型基因演算法的適應函數(fitness function)在此設定為針對陽性預測的值 PVV=TP/TP+NP,針對所得到預測結果與真實結果的一致性為我們此投票結果所 需求的。 (7) 我們設定得到投票結果相對高票數的位置,其為表頂的可能性最大,此外若投票 得到最高票且其相近位置也都有相對高票我們認為此位置為表頂的機會相對地 也很大當作我們的假設。 (8) 由以上結果可以定義出我們要的抗原熱點(hot point)。此位點意味著自多組滑動窗 口中決定可能的抗原決定位之位置,並減少偽陽性情況下得到的位點。