國
立
交
通
大
學
電子物理系
博
士
論
文
基板穩定之六方與正交結構
多鐵性稀土元素錳氧化物薄膜的磁與電子特性研究
Magnetic and electronic characteristics of substrate-stabilized
hexagonal and orthorhombic structured
multiferroic rare earth manganese oxide thin films
研 究 生:謝志昌
基板穩定之六方與正交結構
多鐵性稀土元素錳氧化物薄膜的磁與電子特性研究
Magnetic and electronic characteristics of substrate-stabilized
hexagonal and orthorhombic structured
multiferroic rare earth manganese oxide thin films
研 究 生:謝志昌 Student:Chih-Chang Hsieh
指導教授:莊振益教授 Advisor:Prof. Jenh-Yih Juang
國 立 交 通 大 學
電 子 物 理 系
博 士 論 文
A Dissertation
Submitted to Department of Electrophysics College of Science
National Chiao Tung University in Partial Fulfillment of the Requirements
for the Degree of Doctor of Philosophy
in Electrophysics
July 2008
六方與正交結構多鐵性稀土元素錳氧化物薄膜
之磁性與電子結構特性研究
研究生: 謝志昌 指導教授:莊振益教授
國立交通大學
電子物理系
中文摘要
本論文將介紹如何分別製備具有六方與正交結構之多鐵性稀土元素錳氧化物的 粉末、靶材、與薄膜樣品。同時針對不同結構的樣品進行電子結構與磁性特徵的量測。 首先我們利用固態燒結法可以製備出具有六方結構的稀土元素錳氧化物(離子 半徑較小之稀土元素),接著利用摻雜鈣離子或鍶離子取代稀土元素的方式,試圖直 接從材料內部施加應力,以穩定其正交結構。我們發現藉由摻雜鈣超過 30%,就可以 完成結構轉換。藉由摻雜造成了錳氧的結構從 MnO5轉換到 MnO6,同時摻雜之後錳 離子的價數從+3 變成了+3/+4 的混合價數。因為混合價數的雙交換機制,原本的反鐵 磁特性被鐵磁性行為所取代。但是我們發現摻雜鍶離子並沒有造成類似的結構的轉 變,而且造成了磁性特徵的消失。 我們同時利用外部施加應力的方式製備六方與正交結構的薄膜。六方結構的薄 膜可以藉由 YSZ(111)的基板製備或是其他具有表面三角形結構同時晶格常數又不會 差異太大的基板。我們利用 X-ray 繞射儀探討脈衝雷射蒸鍍法製備的單一軸向六方結 構釔錳氧薄膜,我們可以看到薄膜具備六重對稱特性同時基板與薄膜的對稱性亦與我 們的估計相符。 在另一方面正交結構的薄膜亦可利用選擇適當的匹配基板製備。經過預先的比 較計算在這裡我們分別選擇了三種基板鈦酸鍶(110)、鑭鋁氧(110)、與鈦酸鍶(100), 我們可以在這三種基板上分別製備具有單一軸向的 a 軸、b 軸、與 c 軸薄膜。藉由 X-ray繞射儀的判斷我們可以發現薄膜分別具備二重、二重、與四重對稱性。藉由這些判定 我們可以了解薄膜的軸向性。同時我們發現 LAO(110)是我們在探討這類薄膜時最佳的 利器,因為可以利用此類基板製備出軸向分離的薄膜,對於我們探討這些軸向異性的 材料具有相當大的幫助。我們可以在成長於鑭鋁氧基板上的釔錳氧與鈥錳氧薄膜看到 反鐵磁相轉變溫度與第二次磁性相轉變。兩種材料在三個軸向上都可以看到反鐵磁的 相轉變,但是第二次相轉變只會發生在 a 軸(只在釔錳氧看到)與 c 軸(釔錳氧與鈥錳 氧都可看到)。雖然他們的結構特徵非常的相近,但是在這一系列的化合物一點點的 差異性就會改變錳氧間的鍵角與距離,也因為這些微的差異性造成材料間不同的性 質。 最後我們利用線性偏振的同步輻射光譜探討不同結構間的軸向異性行為。在六 方晶系與正交結構的釔錳氧材料,錳光譜與氧光譜分別展現了MnO5與 MnO6正三價 電子結構的差異性。在氧光譜中可以清楚看出能階因晶場與姜-泰勒效應所造成的分 裂:在六方結構中能階分裂為三個,在正交結構中分裂成四個。我們利用軸向異性的 光譜特徵與磁電耦合的理論預測低溫可能造成的光譜變化行為。利用這些方法我們可 以製備出一系列稀土元素錳氧化物薄膜,同時利用這些薄膜可以進行相關的量測確認 磁電耦合的多鐵機制是否存在,並嘗試找出較明顯的磁電極化行為。
Magnetic and electronic characteristics of substrate-stabilized
hexagonal and orthorhombic structured
multiferroic rare earth manganese oxide thin films
Student: Chih-Chang Hsieh Advisor: Prof. Jenh-Yih Juang
Department of Electrophysics
National Chiao Tung University
Abstract
In this dissertation, we present how to prepare hexagonal and orthorhombic structured multiferroic rare earth manganese oxide powders, bulks, and thin films. We also probed the anisotropies in the magnetic behavior and electronic structure of these materials.
Firstly, we prepare hexagonal RMnO3 (R: rare earth and Y) compounds with smaller rare
earth ions. We tried to stabilize the orthorhombic structure by replacing the R-site ions with Ca2+ or Sr2+. This method could provide inner strain force and the material from the thermodynamically stable hexagonal structure to orthorhombic perovskite structure. We find that the entire structure transforms into orthorhombic by doping Ca2+ up to 30%. In the doping process, the MnO5 structure is no longer stable and starts to convert into MnO6
structure, which in turn substantially modifies the magnetoelectric properties and electronic structures of the material. The trivalent manganese converts into +3/+4 mixed valence and induces the double exchange mechanism. Consequently, the antiferromagnetism is replaced by ferromagnetic interaction and revealed traditional spin glass behaviors. The Sr2+ doped sample remains as hexagonal structure and magnetic characteristic was significantly suppressed.
desired crystal structures. We calculated the in-plane mismatch and used the YSZ(111) substrate in preparing hexagonal thin films. The sample revealed six-fold symmetry and in-plane arrangement has corresponded nicely to our estimation.
On the other hand, we deposited thin film on SrTiO3(110), LaAlO3(110), and SrTiO3(100)
substrates and obtained a-, b-, and c-axis thin film of orthorhombic structure. With the X-ray diffraction characterizations, we see the thin films are having 2-fold, 2-fold, and 4-fold symmetry, depending on the substrate chosen. In particular, we find that the a-axis and b-axis films are having distinguishable crystalline axis, while there exists a twin growth behavior in the c-axis film. Owing to the relatively smaller in-plane mismatch, the LaAlO3 (110) is proved
to be the best substrate in stabilizing orthorhombic RMnO3 for smaller ionic size of R. The
films with specific growth directions allow us to directly probe the anisotropies existent in magnetism, polarization, and bonding relations. We observed the antiferromagnetic ordering and spin reordering transition in both YMnO3 and HoMnO3 thin films grown on LaAlO3 (110)
substrates. The antiferromagnetic ordering can be probed with applied field parallel to each crystallographic axis. However, the spin reordering was only observable for field applied parallel to a-axis (only observed in YMnO3) and c-axis (observed in YMnO3 and HoMnO3).
Although the crystal structure of these two films is nearly the same, RMnO3 compound is
sensitive to the Mn-O bonding distance and Mn-O-Mn bond angle, thus might explain the different behaviors exhibited by the two neighboring compounds.
Finally, we used the linear polarized x-ray to probe the anisotropic bonding relation in both of these two crystal structure. In the hexagonal and orthorhombic YMnO3 thin films, Mn
L edge and O K edge x-ray absorption spectra exhibited the MnO5 and MnO6 structure with
trivalent manganese ions in respective crystal structure. We attribute the energy splitting in the O K edge spectra to the effect of crystal field and Jahn-Teller effect. There are three splitting energy levels in hexagonal structure and 4 splitting energy levels in orthorhombic structure. We also utilize the anisotropic spectra and the magnetoelectric theory in E-type magnetic orders to estimate the possible spectra in the lock-in state. By extending these methods, we should be able to prepare a series of rare earth manganese thin films that are potentially able to realize the magnetoelectric state expected in the E-type magnetic structure for more dramatic magnetism-induced polarizations.
致 謝
在漫長的研究生涯中完成了這本論文,在這裡要感謝固態實驗室
的指導老師吳光雄教授、林俊源教授、溫增明教授、郭義雄教授的悉
心指導使我在實驗研究及學術的探討上有更深一層的了解。更感謝我
的指導老師莊振益教授,在我研究中遇到困難時認真的指導我正確的
觀念,並導引並修正我的研究核心不至於走過多的冤妄路。
在實驗時與閒暇之餘,更要感謝實驗室夥伴們的幫助。實驗室學
長:世溥、中裕、慧愷、旭禎、世烽、博瑛、小志在實驗上的指導,
讓我對實驗的流程與儀器的架設更為了解﹔同學星哥、璨耀、維仁陪
我度過煩悶的生活;同時也感謝許多學弟妹們宗漢、訓全、政義、家
弘、家恬、珈芸、凱婷……等等在實驗事務上的幫忙。要感謝的人實
在是太多了,讓我不免俗的說一聲感謝大家吧!有你們的陪伴讓我等
生活更加的充實,在科學研究的生活中有充足的生活糧食與豐富的資
源。
最後感謝家人所給予的支持與鼓勵,讓我感受到精神上的支持並
得以完成學業。
Contents
Abstract (in Chinese) i
Abstract (in English) iii
Acknowledgement v
Contents vi
List of Tables viii
List of Figures ix
Chapter 1 Introduction 1
1.1 Introduction to multiferroics 1
1.2 Origin of the multiferroelectricity 2
1.3 Classification of multiferroics 4 1.4 Multiferroic RMnO3 6 1.4.1 Hexagonal phase 8 1.4.2 Orthorhombic phase 10 1.4.2.1 Spiral magnetism 10 1.4.2.2 Collinear magnetism 13 1.5 Motivation 15 1.6 Outline 16 References 19
Chapter 2 Basic physical properties of RMnO3 manganites 23
2.1 Superexchange 23
2.2 Double exchange 24
2.3 Crystal field and Jahn-Teller effect 26
2.4 Crystal and magnetic structure 29
2.4.1 Crystal structure 29
2.4.2 Magnetic structure 31
References 33
Chapter 3 Structural transformation with the doped ionic size effect 35
3.1 Introduction 35
3.2 Experiment 36
3.3 Results and discussions 37
3.4 Summary 48
References 50
Chapter 4 Structure stabilization and magnetic behaviors of hexagonal YMnO3
thin films
53
4.1 Introduction 54
4.2 Experiment 55
4.3 Results and discussions 57
4.4 Summary 63
Contents
Chapter 5 Anisotropic magnetic behavior in substrate-stabilized orthorhombic YMnO3 thin films
65
5.1 Introduction 65
5.2 Experiment 69
5.3 Results and discussions 70
5.4 Summary 85
References 86
Chapter 6 Anisotropic electronic structure in both hexagonal and orthorhombic structured YMnO3 thin films
88
6.1 Introduction 89
6.2 Experiment 93
6.3 Results and discussions 94
6.3.1 Electronic structure of the MnO5 bipyramids in hexagonal
YMnO3 thin films
94 6.3.2 MnO6 octahedral electronic structure in orthorhombic
YMnO3 thin films
96
6.4 Summary 102
References 104
Chapter 7 Summary and future work 106
7.1 Summary 106
7.2 Future work 108
List of Tables
Table 1.1 Classification of ferroelectrics 6
Table 1.2 Crystallographic, polarization, and magnetic properties of RMnO3
compounds for R = Y and Eu to Lu
18
Table 3.1 The fitted lattice constant of Y1-xAxMnO3 (A = Ca, Sr; x= 0, 0.1, 0.3 ,
0.5 )serial powder
38
Table 4.1 The in-plane mismatch of hexagonal YMnO3 and several substrates 55
Table 5.1 The fitting parameters and in-plane mismatch calculations between o-YMO thin films and substrates used in this study.
List of Figures
Fig. 1.1 Illustration of the ABO3 perovskite with ideal cubic and distorted
tetragonal structures
3 Fig. 1.2 The relation between multiferroic and magnetoelectric materials 4 Fig. 1.3 Evolution of the lattice structure in ReMnO3 as a function of the size of
the rare earth
7 Fig. 1.4 The crystal structure of hexagonal YMnO3 in the paraelectric and
ferroelectric phases 8
Fig. 1.5 KNB model in helical spin structure induced magnetoelectric polarization
10 Fig. 1.6 The relation between spiral spin orders and ferroelectric polarization in
an applied field
12 Fig. 1.7 The variation of in-plane Mn-O-Mn bond angle below the lock-in
transition temperature
14
Fig. 2.1 The illustrators of superexchange interaction 23 Fig. 2.2 The illustrators of double exchange interaction 25 Fig. 2.3 Illustration of the energy level split by crystal field and Jahn Teller
distortion
27 Fig. 2.4 The equivalent-density surface of electronic distribution of d orbital 28 Fig. 2.5 The crystal structure of ideal cubic perovskite and real orthorhombic
perovskite in LaMnO3
30 Fig. 2.6 The usual magnetic structure in rare earth manganite compounds 31 Fig. 3.1 The XRD patterns for a series YMnO3 doped with Ca and Sr. 37
Fig. 3.2 The Mn L edge XANES spectra for MnO (standard powder), MnO2
(standard powder), Y1-xAxMnO3 (A = Ca, Sr ; x = 0, 0.1, 0.3, 0.5)
polycrystalline samples, and Mn2O3 (standard powder)
41
Fig. 3.3 The O K edge XANES spectra of Y1-xSrxMnO3 series, intrinsic YMnO3,
and Y1-xCaxMnO3 series (for x = 0, 0.1, 0.3, 0.5)
42 Fig. 3.4 Temperature dependent susceptibility for serial powders measured by
ZFC and FC method with an apllied field 100 Oe, (a) intrinsic YMnO3
polycrystalline powders, (b) Y1-xCaxMnO3 (x = 0, 0.1, 0.3, 0.5)
polycrystalline powders (c) Y1-xSrxMnO3 (x = 0, 0.1, 0.3, 0.5)
polycrystalline powders.
45
Fig. 3.5 Field dependent magnetization (M-H) curves taken at three different temperatures. The data were collected at 30K, 50K, and 80K respectively.
List of Figures
Fig. 4.1 The illustration of pulsed laser vacuum deposition system 56 Fig. 4.2 The XRD θ-2θ patterns of hexagonal YMnO3 thin films grown on
YSZ(111) substrates
58 Fig. 4.3 In-plane arrangements of YSZ(111) substrates and hexagonal YMnO3
(00l) thin film
59 Fig. 4.4 The XRD Φ-scan patterns show both off-normal YSZ(220) and
h-YMO(112) peak with h-YMO(00l)/YSZ(111) thin films deposited at 880 °C
60
Fig. 4.5 Temperature dependent susceptibility of the h-YMO(00l)/YSZ(111) thin films.
61 Fig. 4.6 The field dependent magnetization behavior of the
h-YMO(00l)/YSZ(111) thin films
62
Fig. 5.1 The relation between magnetic and ferroelectric order for E-type commensurate structure (HoMnO3) and the sinusoidal incommensurate
order (YMnO3)
68
Fig. 5.2 The XRD θ-2θ patterns of o-YMO thin films grown on STO(001), STO(110), and LAO(110) substrates, respectively.
71 Fig. 5.3 The schematics of the in-plane arrangements between the o-YMO thin
films and substrates.
73 Fig. 5.4 The XRD Φ-scan patterns of o-YMO thin films deposited at 880 °C on
different substrates.
74 Fig. 5.5 Temperature dependent magnetization along respective crystallographic
orientations of o-YMO(00l)/STO(001), o-YMO(020)/LAO(110), and o-YMO/STO(110) thin films.
77
Fig. 5.6 Temperature dependent magnetization along respective crystallographic orientations of o-HMO(020)/LAO(110) thin films.
83 Fig. 5.7 Magnetic phase diagram for RMnO3 as a function of the ionic radius of
rare earth
List of Figures
Fig. 6.1 Schematic orbital splitting for the majority d states of Mn3+ ion within
orthorhombic and hexagonal crystal field 89
Fig. 6.2 Transition temperatures versus R3+-ion radius (IR) in the perovskite
RMnO3 family.
91 Fig. 6.3 The Mn L edge XANES spectra for LaMnO3(001)/STO(001) and
h-YMO(00l)/YSZ(111) thin films
95 Fig. 6.4 The O K edge XANES spectra for h-YMO(00l)/YSZ(111) thin film 96 Fig. 6.5 Schematic drawing of the crystal structure and the 3d orbitals in the
MnO6 octahedrons for orthorhombic RMnO3
97 Fig. 6.6 The Mn L edge XANES spectra for standard powders (MnO, Mn2O3,
and MnO2), LaMnO3(001)/STO(001), and o-YMO(020)/LAO(110) thin
films
98
Fig. 6.7 Schematic orbital splitting for the majority d states of Mn3+ ion within RMnO3
99 Fig. 6.8 The O K edge XANES spectra for o-YMO(00l)/LAO(110) thin film 100 Fig. 6.9 The illustrations of crystal structure, spin orders, and orbital orders of
LaMnO3 and HoMnO3.
102
Fig. 7.1 Summary of the in-plane mismatch between the orthorhombic RMnO3
with Pbnm space group setting and the two commonly used perovskite substrates.
Chapter 1
Introduction
1.1
Introduction to multiferroics
Multiferroics are materials which exhibit simultaneously long range magnetic and electric orders like ferroelectric, antiferromagnetic (AFM), and ferroelastic behaviors. More interestingly, in this kind of materials, the phase transition of one kind of order could result in another kind of order and hence manifesting itself with strong coupling between the order parameters [12-17]. Namely, the magnetic orders could be changed by an applied electric field and the electric properties could also be affected in the magnetic field when the material is in the multiferroic state. The rich physical characteristics exhibited by these materials not only present great challenges to scientists interested in understanding the prevailing underlying physics but also promise potential in various kinds of novel applications.
For instance, the multiferroic bilayer structures had been demonstrated to be very competitive in application of magnetic sensors. With the effect exerted by the probing magnetic field, the magnetic phase composite was strained and induced a proportional charge in the piezoelectric phase. This sensor shows much more sensitivity than the superconducting quantum Interference Device (SQUID), which has been often used to probing the ultra small field in the laboratory. Unlike the low temperature environment required by operating SQUID, the magnetic sensor can even be more advantageous with the possibility of operating at in ambient environment. The sensitivity of the magnetic
sensor can also be of potential use in magnetic data reading like giant magnetoresistance (GMR) or tunneling magnetoresistance (TMR). For the ultra low field sensitivity in the order of 10-12~10-15 T, the application of the multiferroic devices in hard disk data reader
head could be much more superior as compared to the GMR and CMR sensors. Although the areal density in hard disk with GMR device can reached up to 8Gb/in2, [25] the
magnetic recording might be further increased up to 1Tb/in2 by using the bilayer
multiferroic device schemes. [26-28] Besides, the multiferroic device could be also used in magnetoelectric (ME) gyrators for its giant manetoelastic behavior, [26, 27] and this has been widely used in entertainment business like optical image stabilizer in digital camera, television game platforms in joystick (Wii), GPS assisted system in car driving, and etc. Moreover, the mutiferroic devices can also be prepared as tunable device, resonators, and filters in microwave applications. [26, 27]
1.2
Origin of the multiferroelectricity
Although the multiferroics can be used in many applications, it is a pity that there are only few materials naturally own the multiferroicity. In 1894, P. Curie predicted the existence of coupling between the electric and magnetic fields in materials when performing the theoretical derivation of Electromagnetism [29, 30]. Dzyaloshinskii predicted the magnetoelectric coupling in Cr2O3 in 1959 [31], and the experimental
evidence was observed by Astrov in the Cr2O3 a year later in 1960 [32]. The difficulty of
finding multiferroicity manifested in real materials was owing to the mutually exclusive requirements for the required orders to take place. For instance, the so-called d0-ness
required for ferroelectricity (FE) and the unpaired d-electrons necessary for providing magnetic moments for ferromagnetism (FM) or antiferromagnetism (AFM) apparently
are counteractive to each other. By considering the perovskite oxides with the ABO3
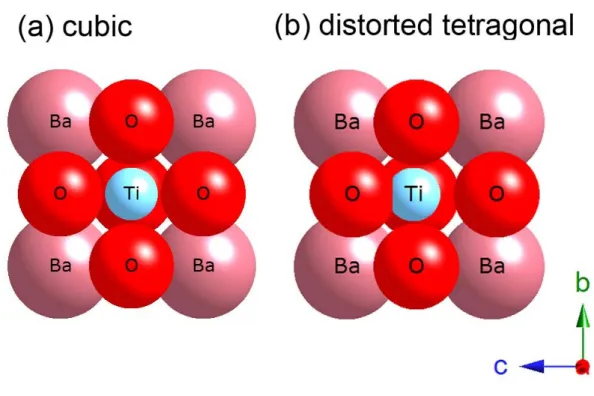
structure with ideal cubic symmetry (Ba and Ti was set to A-site and B-site ions), illustrated in Fig. 1.1 (a) which is typical for the traditional FE materials like tetragonal BaTiO3, the hybridization between empty d orbital in TiO6 octahedrons for the B-site and
fully occupied p orbital in oxygen ions, which in turn would induce the off-center displacements of B-site ions in ABO3 structures and hence the ferroelectricity [33]. On
the other hand, it is well known that half occupied d-orbital electron spins dominate the magnetic behaviors of most of the transition metal oxides. Consequently, the existence of both ferroelectric and magnetic orders in a material imposed a difficult task on B site transition metal ions. Both ferromagnetic and ferroelectric materials require strict limits in exhibiting their special characteristics which happen to play the pivotal roles in digital age with data recording today. The status of polarization or magnetization could produce two states and be assigned to 0 and 1. Each state could be switched with an applied field
Fig. 1.1 Illustration of the ABO
3perovskite structures with
(a) ideal cubic symmetry for paraelectric BaTiO
3and
(b) distorted tetragonal for ferroelectric BaTiO
3and used as non-volatile memory in hard disk or flash disk. Thus despite of the rarity of multiferroic materials as described illustrated in Fig. 1.2 schematically by W. Erenstein [20], the efforts of investigating the existent systems and to explore the new possibilities accordingly are quickly amounted worldwide. Only a few materials own both electric and magnetic orders which is so-called multiferroic, and an even smaller number of them exhibit magnetoelectric characteristics (i.e. displaying strong coupling between the two order parameters). Although the transformation between electricity and magnetism is well known by the Maxwell’s descriptions and used in daily works, it is still exciting and need to be explored in controlling the magnetoelectric phase with both electric and magnetic force in the local structures.
1.3
Classification of multiferroics
Although it was not easy in finding multiferroic materials, many have started to investigate these issues recently. According to the classification proposed in the early report of W. Priller et al. (2005) [18], there are several kinds of multiferroic materials as
listed below.
1. Bi-based compounds (lone pair s electrons)
2. ReMnO3 compounds ( Re = Y, Ho, Er, Tm, Yb, and Lu)
3. ReMn2O5 compounds ( Re = Y, Tb, Dy, and Ho)
4. Artificial multiferroic materials ( mutilayers and nanocomposites films)
More recently, there have been many more multiferroic materials discovered such as LuFe2O4, CoCr2O4 [24] and some systems with reduced dimensions.
Another way of describing these materials is to base on the behavior of coexistent magnetism and ferroelectricity. The traditional ferroelectrics (like BaTiO3) exhibit
tremendous enhancement of polarization with an applied electric field [34], due to the alignment of the permanent dipoles originated from the off-center distortion of B-site ions which is caused by the covalent bonding between 3d0 transition metal
and oxygen. This is termed as the so-called “proper” ferroelectrics. Some kinds of multiferroics have higher Curie temperature like BiFeO3 ( Tc = 1103K), the
ferroelectricity was owing to 6s2 lone pair and the hybridization between Bi 6p and
O 2p orbital along the [111] direction in the rhombohedral distorted perovskite structure [35]. S. W. Cheong has further summarized the classification of “proper” and “improper” [36] ferroelectrics in Table 1.1 by the mechanism of ferroelectricity. [24]. In the proper ferroelectrics, the main driving force toward to the polar state was associated with the electronic pairing. In contrast, the polarization of improper ferroelectricity involves a more complex lattice distortion or other accidental by-product of some other ordering, especially for those induced by magnetic ordering.
1.4
Multiferroic RMnO
3Perhaps, the most intriguing multiferroic system is the special series of compounds made of rare-earth manganites RMnO3 (R = rare earth with smaller ionic size and Y). As
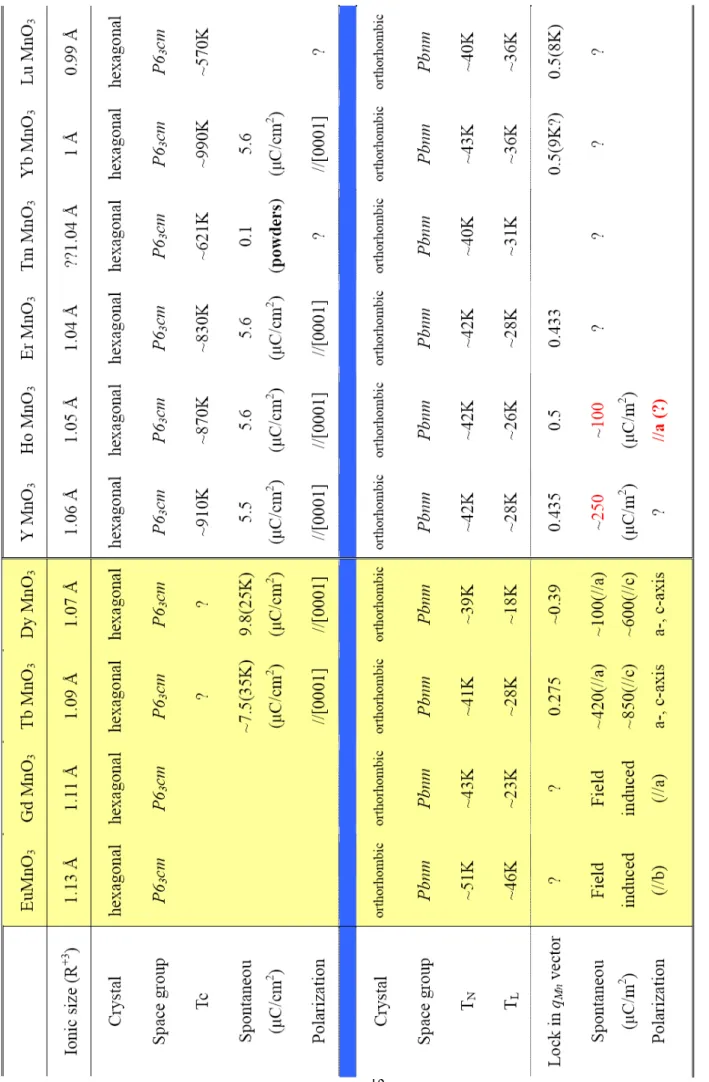
indicated in Table 1.1 RMnO3 has two kinds of ferroelectric behavior. The R3+ ions of the
rare earth element have closely similar chemical characteristics for the outermost electrons with the same 5s25p6 electronic configurations. For the rare earth elements
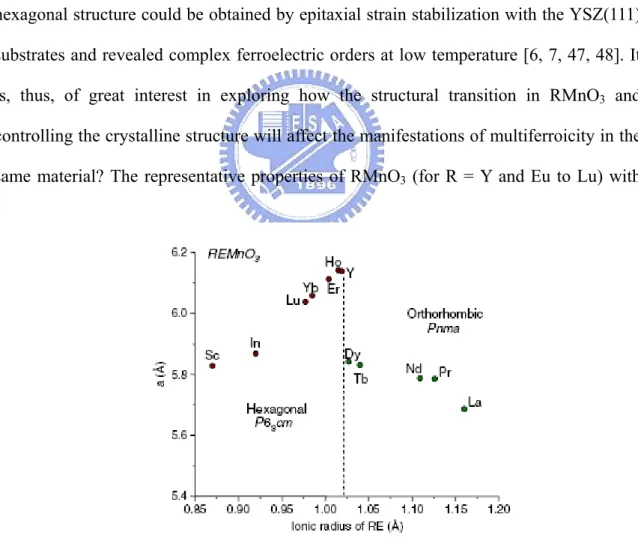
(from La to Lu), on the other hand, the ionic size decreases from 1.22 Å to 0.94 Å with the increasing number of electrons residing in the 4f shell, the so-called lanthanum contraction [34]. With the variation of the ionic size, there is a structural transition in the series of the RMnO3 compounds. As shown in Fig. 1.3 [18], the RMnO3 perovskite
oxides with R= La to Dy have stable orthorhombic structure in Pnma space group and these kinds of compounds fall into “Group I” in RMnO3 [37]. These materials reveal rich
physical properties such as the colossal magnetoresistance (CMR) displayed in Classification of ferroelectrics
Mechanism of inversion symmetry breaking
Materials Covalent bonding between 3d0
transition metal (Ti) and oxygen
BaTiO3
Proper
Polarization of 6s2 lone pair of Bi or Pb BiMnO3, BiFeO3, Pb(Fe2/3W1/3)O3 Structural transition ‘Geometric ferroelectrics’ K2SeO4, Cs2CdI4 hexagonal RMnO3 Charge ordering ‘Electronic ferroelectrics’ LuFe2O4 Improper Magnetic ordering ‘Magnetic ferroelectrics’ Orthorhombic RMnO3, RMn2O5, CoCr2O4
La1-xCaxMnO3 and a large number of related doped manganites. On the other side, the
“Group II” compounds with the decreasing ionic size (for R = Ho, Lu, Y, Sc, In) have stable hexagonal structure with P63cm space group thermodynamically. The critical point
of the structure transition appears to locate near YMnO3 (Y3+ =1.06Å) and HoMnO3
(Ho3+ = 1.05Å). As a result, the crystal structure of Group II compounds has been
managed to transform from hexagonal to orthorhombic via high temperature, high pressure process [38, 39], soft chemistry procedures [2, 40], and the perovskite substrate stabilized thin films [41-46]. Conversely, recent reports have also described that thin films of the Group I manganites such as DyMnO3, TbMnO3, GdMnO3, and EuMnO3 with
hexagonal structure could be obtained by epitaxial strain stabilization with the YSZ(111) substrates and revealed complex ferroelectric orders at low temperature [6, 7, 47, 48]. It is, thus, of great interest in exploring how the structural transition in RMnO3 and
controlling the crystalline structure will affect the manifestations of multiferroicity in the same material? The representative properties of RMnO3 (for R = Y and Eu to Lu) with
Fig. 1.3 Evolution of the lattice structure in REMnO3 as a function of
the size of the rare earth (RE). [18]
both hexagonal and orthorhombic structure reported to date are collected in Table 1.2 [1-11].
1.4.1 Hexagonal phase
The hexagonal YMnO3 manganites exhibit ferroelectric transition at high
temperature ~910K [1] while become A-type antiferromagnetic below the Neél temperature (TN ~70 K in single crystalline samples) [49, 50]. The paraelectric (PE)
phase above 910K has the hexagonal structure with P63/mmc space group symmetry, and
the ferroelectric (FE) phase has the hexagonal structure in P63cm symmetry. By
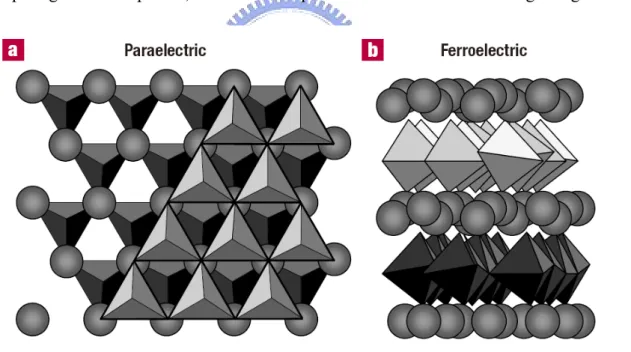
comparing these two phases, one finds a displacement of Y3+ ions occurring along c-axis
Fig. 1.4 These figures illustrate the crystal structure of YMnO3 in the paraelectric and
ferroelectric phases. The trigonal bipyramids depict MnO5 polyhedra and the
spheres represent Y ions. (a) The stacking of two consecutive MnO5 layers
and the sandwiched Y layer looking down the c axis in the paraelectric phase.(b) A view of the ferroelectric phase from perpendicular to the c axis, showing the layered nature of YMnO3. [23]
when undergoing PE-FE transition (Fig. 1.4) [23]. The characteristics for another compound HoMnO3 are very similar to YMnO3. Wherein the ferroelectricity is caused by
the displacements of Ho3+, and the antiferromagnetic (AFM) phase transition occurs at
about 75 K for Mn3+ with an additional AFM ordering occurs at temperature below 5K
for Ho3+ [51, 52].
Owing to the huge difference between the FE Curie temperature (600 ~ 1000K) [1] and AFM Neél temperature (70 ~ 100K) [52], the magnetoelectric coupling between the two order parameters in the hexagonal manganites has been originally regarded as diminishingly small. On the other hand, for the orthorhombic manganites only magnetic orders were observed, albeit it may also be strongly affected by the variation of the rare earth's ionic size and may even induce cooperative Jahn-Teller distortion in the MnO6
octahedrons. Until 2002, M. Fiebig et al. [53] reported the observation of coupled magnetic and electric domains in YMnO3 by imaging the respective domains with optical
second harmonic generation. Later on, they also discovered the magnetielectric effect in HoMnO3 by using magnetooptical techniques [51]. Soon after Fiebig’s observations, it
has been pointed out that, as the Mn-O bond angle deviates more from 180° with decreasing ionic size of the rare-earth ions, the modified AFM magnetic structure can induce FE order and hence provide strongly coupled ferroicities. For HoMnO3, the antiferromagnetic ordering of the Ho3+ has displayed further coupling with an applied
electric field below the Neel temperature of Mn3+ spin orders. This declares the electric
field controlled magnetic phase. Owing to the absence of the spin electrons in rare earth’s 4f orbital, the ability of manipulating the magnetoelectric control at low temperatures is lacking for YMnO3 [51, 53].
1.4.2 Orthorhombic phase
1.4.2.1 Spiral Magnetism
On the orthorhombic side of the perovskite manganites, TbMnO3 was the first to
bring about unprecedented attention for its spontaneous electrical polarization reported by Kimura et al. [14, 22]. The phenomenon was immediately coined with the so-called improper magnetic ferroelectrics (IMF). It was revealed that, in TbMnO3, an AFM
ordering of Mn3+ occurs with a propagated wave vector (0, qMn, 0) below 41K (TN) along
b-axis. In this state, the magnetic ordering is incommensurate with lattice order and the propagation wave vector of the magnetic order is temperature dependent. With the
Fig. 1.5 The cluster model with two transition metal ions M1, M2 with the oxygen atom O between them. The arising spin current rjs was induced by the noncollinear spin
directions between M1 and M2. ( js e1 e2
r r r
×
α ) The direction of the electric polarization
Pr is given by Prα er12 × rjs here er12 is the unit vector connecting M1 and M2
decreasing temperature, the value of qMn increases and is locked into a constant of 0.28
below 28 K (TL, lock-in transition temperature) [4]. This particular spin order transition
was later revised by Kajimoto and was described as an incommensurate to spin spiral AFM transition. It is this spin spiral mechanism that gives rise to spontaneous polarization along the a- and c-axis and the magnetocapacitance behaviors observed in TbMnO3 and DyMnO3 by Kimura et al. [4]. Theoretical studies had further indicated that
the noncollinear spin orders are playing important roles in controlling ferroelectric phase, and vice versa.
The theory of magnetoelectric effect was developed by Landau and Dzyaloshinskii in describing the electric polarization induced by the magnetic field [21, 54, 55]. Katsura, Nagosa, and Balatsky (KNB) [21] developed a theory in considering the spin-orbital interaction, and showed the spin spiral structure was directly originated from the spin current and magnetoelectric effect. Fig. 1.5 illustrates the relations between the interactions among M1-O-M2 (M1, and M2 are the magnetic ions, O is the oxygen ion)
and the induced spin current js
r
. By considering the double exchange and superexchange interaction, the expression was derived below:
s j e V e Pr ~ r12×r ………. (1)
This could be explained the origin in spiral magnets like TbMnO3 and could estimate the
value of polarization. In the later, other equivalent model based on Dzyaloshinskii-Moriya interaction predicts the similar results and shows spin spiral structure may induce the electric polarization. S. W. Cheong and M. Mostovoy further simplified the expression like the form of equation (2)
12 2 1 ) //(e e e
to observe the polarization directly. [24]. In their description, the magnetoelectric effect is mainly originated from the symmetries of polarization and magnetization. Thus, for spontaneous polarization to occur, these two orders can only be non-collinear. Within the context of these scenarios, the polarization will not occur in the incommensutate phase because the collinear neighboring spins will result in zero cross product. Nevertheless, in helical spin array non-zero polarization is expected. Maxim Mostovoy explained the appearance of magnetocapacitance by using the Ginzburg-Landau thermodynamic potential and set up the magnetic easy axis along the x-axis ( ax < ay < az ) [19]. As is
depicted in Fig. 1.6, spins spiraled on the x-y plane result in a spontaneous polarization
Py, consistent with what were observed in TbMnO3 in that the magnetic moments spiral
on the a-b plane with a wave vector propagated along the b-axis and a resultant polarization in the a- or c-axis. Moreover, the polarization was found to be markedly suppressed by an applied field. The reason for this polarization suppression is that the x-y
Fig. 1.6 Magnetic field behavior of electric polarization for the model of Ginzburg-Landau thermodynamic potential. In zero field spins rotate in the x-y plane and P// y. Magnetic field in the x direction suppresses the polarization, while Hy orients P in the z direction. [19]
helix will be broken and replaced by y-z conical helix when a field is applied along the x-axis (Fig. 1.6(a)). The new rotation axis transformed to parallel x-axis and the cross product of spin vector and propagation vector becomes zero. On the contrary, with the field applying parallel to the y-axis, polarization will be induced and parallel to z-axis. However, it is noted that usually a strong magnetic field is needed to change the spin symmetry of AFM state. Thus, the microscopic magnetoelectric behavior could be controlled only with relatively high fields. Similar feature was observed in other noncollinear multiferroics and some of them revealed reversible and memorial effect [56]. The applied field induced polarization could also be five times (DyMnO3: ~2700μC/m2
with an applied field 2T) larger than the spontaneous magnetoelectric effect [4, 57].
1.4.2.2 Collinear Magnetism
For the Group II compounds of RMnO3, the magnetic orders have been identified to
turn into the E-type AFM magnetic structure which exhibits collinear spins [2, 3, 5, 8, 39, 58]. Within the context of the abovementioned magnetism-induced FE, the collinear AFM shouldn’t be able to result in the complex magnetoelectric polarization described above. In 2004, Lorenz et al. reported the anomaly dielectric behavior in YMnO3 and
HoMnO3 materials [16]. The results have led to the scientists to reconsider the role of
collinear AFM in magnetoelectric effect. Sergienko et al. [22] by considering mechanisms other than spin-orbit interaction have estimated the polarization of the E-type RMnO3 can have up to “2” orders of magnitude enhancement over that of the
spiral phase RMnO3. The polarization in this case is induced along the a-axis in Pbnm
space group setting. In their calculation, the polarization was estimated about 0.5~12 μC/cm2 which is compatible with the hexagonal RMnO3 (see in Table 1.2). As depicted in
Fig. 1.7, in this scenario the ferroelectricity is originated from the oxygen displacement perpendicular to the Mn-Mn bond resulted from the competition between electron hoping and elastic energy within the framework of the double exchange model [22]. For HoMnO3 with E-type AFM, the Mn-O-Mn bond angle is close to 144o(ψo). Since
deviation of the bond angleψo from 180˚ will cost hoping energy while it may reduce the elastic energy by having a smallerψo. This calculation, though was put forth for the E-type AFM system, is also useable for noncollinear TbMnO3 compounds. Picozzi et al.
[59, 60] redo the calculation with first principle calculation and find that the magnetoelectric polarization is induced along mainly c-axis (consistent with the previous report [22]) and partially a-axis. The crystalline axis was set in Pnma space group which
Fig. 1.7 (a) The starting configuration of a Mn-O-Mn bond. Numbers 1–4 enumerate the O atoms surrounding one Mn. (b) An MC snapshot of the IMF E-phase at T = 0.01. The ferromagnetic zigzag chain links are shown as solid lines. The displacements of the oxygen atoms are exaggerated. (c) Left: The local arrangement of the Mn-O bonds with disordered Mn spins (full circles). Right: Oxygen displacements (arrows) within the chains of opposite Mn spins (open and crossed circles) in the E-phase. [22]
could be also transferred to Pbnm space group setting. The polarization was induced by the quantum mechanism in spin-orbital interaction, and the crystal and electronic structure both needed to be considered in magnetoelectric behaviors. The first principle calculation also gave more exact estimation on the strength of the polarization caused by collinear AFM and other mechanisms.
1.5 Motivation
As has been mentioned in length, multiferroic materials have rich physical properties arising from the complicated and yet subtle interactions among the charge, spin, and orbital degree of freedoms of the carriers and the lattice. Especially, the ultimate routes of obtaining effective magnetoelectric coupling between spin and polarization are still needed to be clarified. The rare-earth manganites (RMnO3) in particular are the interesting playgrounds featuring simultaneously the magnetism and ferroelectricity with the Jahn-Teller distortion playing an important role in mingling all these ingredients to result in various emergent physical properties like the magnetoelectric behaviors. As shown in Fig. 1.3, YMnO3 and HoMnO3 are of particular interest because they locate
right at the verge of the crystal transition point. In addition, they were also identified to have the collinear magnetic AFM structure for Mn3+ at low temperature [2, 3], that makes
them ideal for testing the mechanism beyond the DM spin-orbital interaction. It is also interesting to note that YMnO3 does not have the 4f spin electrons, making it unique in
clarifying the prominent role played by the Mn3+ spin orders. Unfortunately, up to now
suitable samples of orthorhombic RMnO3 from the Group II manganites are still lacking for measuring the directly the actual orientations of the spin ordering and the polarization associated with its transition, which couldn’t be decided exactly with polycrystalline ones.
Due to the inherent difficulties of obtaining the single crystalline orthorhombic Group II manganites, thin films seem to be the only choice for resolving these hindrances. Therefore, in this work, we will first concentrate on fabricating suitable thin film samples of the orthorhombic Group II manganites. Then we will show how these films help us in characterizing the magnetism, ferroelecticity, and electronic structure that would eventually demonstrate the effects of anisotropic bonding predicted by Picozzi et al. [59].
1.6 Outline
This dissertation consists of seven chapters. We simply guide the multiferroics and its magnetoelectric coupling and mechanism in chapter 1. In Chapter 2, we will describe the basic physical properties of RMnO3 manganites and origin of the magnetic behaviors.
The relative magnetic and crystal structure in RMnO3 compound will be also described
here. In chapter 3, we will discuss the feasible routes of obtaining the orthorhombic crystal structure from the thermodynamically stable hexagonal RMnO3 phases by substrate stabilization. We will also describe the relation between ionic size, crystal, and electronic structure in structural stabilization. Ca and Sr were used to dope YMnO3 to
investigate the effect of ionic size on the electronic structures and associated magnetic properties. In chapter 4 and 5, we clarify the role of substrate in stabilizing thin film with both hexagonal and orthorhombic structures. The strain dominates the stabilization of metastable orthorhombic structure in Group II RMnO3 compounds. In the temperature
dependence of magnetization behavior, the YMnO3 thin films revealed significant
anisotropic behavior both in hexagonal and orthorhombic structures. The spin reordering behavior was only seen along a- and c-axis which was different from the E-type HoMnO3.
along the primary crystalline axes. Because of the MnO6 octahedron in orthorhombic
structure and MnO5 bipyramid in hexagonal structure, the spectra show entirely different
Ta
ble 1.2 Crystallographic, polarizati
on, and magnetic properties of RMnO
3
compounds for R = Y and Eu to Lu. [1-1
References
[1] N. Fujimura, T. Ishida, T. Yoshimura, T. Ito, Applied Physics Letters. 69 (1996) 1011-1013.
[2] A. Munoz, J.A. Alonso, M.T. Casais, M.J. Martinez-Lope, J.L. Martinez, M.T. Fernandez-Diaz, Journal of Physics-Condensed Matter. 14 (2002)
3285-3294.
[3] T. Kimura, S. Ishihara, H. Shintani, T. Arima, K.T. Takahashi, K. Ishizaka, Y. Tokura, Physical Review B. 68 (2003) 060403.
[4] T. Kimura, G. Lawes, T. Goto, Y. Tokura, A.P. Ramirez, Physical Review B. 71 (2005) 224425.
[5] Y.H. Huang, H. Fjellvag, M. Karppinen, B.C. Hauback, H. Yamauchi, J.B. Goodenough, Chemistry of Materials. 18 (2006) 2130-2134.
[6] J.H. Lee, P. Murugavel, H. Ryu, D. Lee, J.Y. Jo, J.W. Kim, H.J. Kim, K.H. Kim, Y. Jo, M.H. Jung, Y.H. Oh, Y.W. Kim, J.G. Yoon, J.S. Chung, T.W. Noh,
Advanced Materials. 18 (2006) 3125-3129.
[7] J.H. Lee, P. Murugavel, D. Lee, T.W. Noh, Y. Jo, M.H. Jung, K.H. Jang, J.G. Park, Applied Physics Letters. 90 (2007) 012903.
[8] M. Tachibana, T. Shimoyama, H. Kawaji, T. Atake, E. Takayama-Muromachi, Physical Review B. 75 (2007) 144425.
[9] L.J. Wang, S.M. Feng, J.L. Zhu, R.C. Yu, C.Q. Jin, Applied Physics Letters. 91 (2007) 172502.
[10] F. Ye, B. Lorenz, Q. Huang, Y.Q. Wang, Y.Y. Sun, C.W. Chu, J.A. Fernandez-Baca, P.C. Dai, H.A. Mook, Physical Review B. 76 (2007) 060402.
[11] H. Okamoto, N. Imamura, B.C. Hauback, A. Karppinen, H. Yamauchi, H. Fjevag, Solid State Communications. 146 (2008) 152-156.
[12] M. Fiebig, T. Lottermoser, D. Frohlich, A.V. Goitsev, R.V. Pisarev, Nature. 419 (2002) 818.
[13] N. Hur, S. Park, P.A. Sharma, J.S. Ahn, S. Guha, S.W. Cheong, Nature. 429 (2004) 392.
[14] T. Kimura, T. Goto, H. Shintani, K. Ishizaka, T. Arima, Y. Tokura, Nature. 426 (2003) 55-58.
[15] B. Lorenz, Y.Q. Wang, C.W. Chu, Physical Review B. 76 (2007) 104405. [16] B. Lorenz, Y.Q. Wang, Y.Y. Sun, C.W. Chu, Physical Review B. 70 (2004)
212412.
[17] T. Lottermoser, T. Lonkai, U. Amann, D. Hohlwein, J. Ihringer, M. Fiebig, Nature. 430 (2004) 541.
[18] W. Prellier, M.P. Singh, P. Murugavel, Journal of Physics-Condensed Matter. 17 (2005) R803.
[19] M. Mostovoy, Physical Review Letters. 96 (2006) 067601.
[20] W. Eerenstein, N.D. Mathur, J.F. Scott, Nature. 442 (2006) 759-765.
[21] H. Katsura, N. Nagaosa, A.V. Balatsky, Physical Review Letters. 95 (2005) 057205.
[22] I.A. Sergienko, C. Sen, E. Dagotto, Physical Review Letters. 97 (2006) 227204.
[23] B.B. Van Aken, T.T.M. Palstra, A. Filippetti, N.A. Spaldin, Nature Materials. 3 (2004) 164-170.
[24] S.W. Cheong, M. Mostovoy, Nature Materials. 6 (2007) 13-20.
[25] R.L. Comstock., Introduction to magnetism and magnetic recording, 1999. [26] M.I. Bichurin, D. Viehland, G. Srinivasan, Journal of Electroceramics. 19
(2007) 243-250.
[27] C.W. Nan, M.I. Bichurin, S.X. Dong, D. Viehland, G. Srinivasan, Journal of Applied Physics. 103 (2008) 031101.
[28] Y. Zhang, Z. Li, C.Y. Deng, J. Ma, Y.H. Lin, C.W. Nan, Applied Physics Letters. 92 (2008) 152510.
[29] P. Curie, J. Phys. 3(Ser. III). 393 (1894) 415. [30] Editorial, Nature Materials. 6 (2007) 1.
[31] I.E. Dzyaloshinskii, Sov. Phys. JETP. 10 (1959) 628-629. [32] D.N. Astrov, Sov. Phys. JETP. 11 (1960) 708–709
[33] N.A. Hill, A. Filippetti, Journal of Magnetism and Magnetic Materials. 242 (2002) 976-979.
[34] C. Kitel, Introduction to Solid State Physics - 7th ed., 1996.
[35] J. Wang, J.B. Neaton, H. Zheng, V. Nagarajan, S.B. Ogale, B. Liu, D.
Viehland, V. Vaithyanathan, D.G. Schlom, U.V. Waghmare, N.A. Spaldin, K.M. Rabe, M. Wuttig, R. Ramesh, Science. 299 (2003) 1719-1722.
[36] A.P. Levanyuk, D.G. Sannikov, Sov. Phys. Usp. 17 (1974) 199-214. [37] J.S. Zhou, J.B. Goodenough, J.M. Gallardo-Amores, E. Moran, M.A.
Alario-Franco, R. Caudillo, Physical Review B. 74 (2006) 014422.
[38] M.N. Iliev, M.V. Abrashev, H.G. Lee, V.N. Popov, Y.Y. Sun, C. Thomsen, R.L. Meng, C.W. Chu, Physical Review B. 57 (1998) 2872-2877.
[39] J.S. Zhou, J.B. Goodenough, Physical Review Letters. 96 (2006) 247202. [40] J.A. Alonso, M.J. Martinez-Lope, M.T. Casais, M.T. Fernandez-Diaz,
Inorganic Chemistry. 39 (2000) 917-923.
[41] T.H. Lin, C.C. Hsieh, H.C. Shih, C.W. Luo, T.M. Uen, K.H. Wu, J.Y. Juang, J.Y. Lin, C.H. Hsu, S.J. Liu, Applied Physics Letters. 92 (2008) 132503.
[42] P.A. Salvador, T.D. Doan, B. Mercey, B. Raveau, Chemistry of Materials. 10 (1998) 2592.
[43] X. Marti, F. Sanchez, J. Fontcuberta, M.V. Garcia-Cuenca, C. Ferrater, M. Varela, Journal of Applied Physics. 99 (2006) 08p302.
[44] X. Marti, V. Skumryev, V. Laukhin, F. Sanchez, M.V. Garcia-Cuenca, C. Ferrater, M. Varela, J. Fontcuberta, Journal of Materials Research. 22 (2007) 2096.
[45] C.C. Hsieh, T.H.S. Lin, H. C. , C.-H. Hsu, C.W. Luo, J.-Y.W. Lin, K. H., T.M. Uen, J.Y. Juang, submit to Journal of Applied Physics (2008).
[46] H.C. Shih, T.H. Lin, C.C. Hsieh, C.W. Luo, J.-Y. Lin, J.L. Her, H.D. Yang, C.-H. Hsu, K.H. Wu, T.M. Uen, J.Y. Juang, submit to New Jounal of Physics
[47] D. Lee, J.H. Lee, S.Y. Jang, P. Murugavel, Y.D. Ko, J.S. Chung, Journal of Crystal Growth. 310 (2008) 829-835.
[48] A.A. Bosak, C. Dubourdieu, J.P. Senateur, O.Y. Gorbenko, A.R. Kaul, Journal of Materials Chemistry. 12 (2002) 800-801.
[49] M.N. Iliev, H.G. Lee, V.N. Popov, M.V. Abrashev, A. Hamed, R.L. Meng, C.W. Chu, Physical Review B. 56 (1997) 2488-2494.
[50] T. Lonkai, D. Hohlwein, J. Ihringer, W. Prandl, Applied Physics a-Materials Science & Processing. 74 (2002) S843-S845.
[51] T. Lottermoser, T. Lonkai, U. Amann, D. Hohlwein, J. Ihringer, M. Fiebig, Nature. 430 (2004) 541-544.
[52] H. Sugie, N. Iwata, K. Kohn, Journal of the Physical Society of Japan. 71 (2002) 1558-1564.
[53] M. Fiebig, T. Lottermoser, D. Frohlich, A.V. Goitsev, R.V. Pisarev, Nature. 419 (2002) 818-820.
[54] L.D. Landau, E. Lifshitz, Electrodynamics of Continuous Media (Butterworth-Heinemann, Oxford), 2002.
[55] I.E. Dzyaloshinskii, Sov. Phys. JETP. 10 (1960) 628.
[56] N. Hur, S. Park, P.A. Sharma, J.S. Ahn, S. Guha, S.W. Cheong, Nature. 429 (2004) 392-395.
[57] N. Aliouane, D.N. Argyriou, J. Strempfer, I. Zegkinoglou, S. Landsgesell, M.V. Zimmermann, Physical Review B. 73 (2006) 020102.
[58] B.B. van Aken, A. Meetsma, T.T.M. Palstra, Acta Crystallographica Section E-Structure Reports Online. 57 (2001) I101-I103.
[59] S. Picozzi, K. Yamauchi, G. Bihlmayer, S. Blugel, Physical Review B. 74 (2006) 094402.
[60] S. Picozzi, K. Yamauchi, B. Sanyal, I.A. Sergienko, E. Dagotto, Physical Review Letters. 99 (2007) 227201.
Chapter 2
Basic physical properties of RMnO
3manganites
2.1 Superexchange
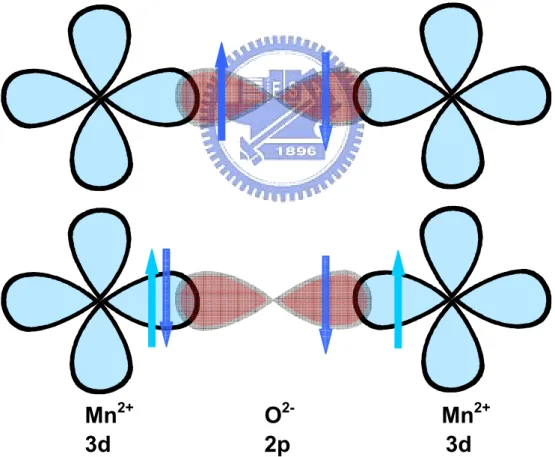
The indirect interaction describes the virtual hoping of electrons between the oxygen ions and neighboring metal ions. Fig. 2.1 illustrates the spin superexchange interaction
Mn
2+O
2-Mn
2+3d
2p
3d
Fig. 2.1 Top: The wave function of O-2 2p and Mn+3 3d overlapping in the superexchange interactions. Bottom: The excited state in the superexchange interaction. [reproduced with Reference [1]]
taking place along the Mn-O-Mn chain of the MnO6 octahedron crystal structure [1]. The
3d orbital of Mn2+ couples with the p-orbital of O2-. The overlap of the wave functions of
the p- and d-orbitals leads to the so-called p-d rehybridization. The electrons locate at the left-hand-side of the oxygen p-orbital could mediate to the neighboring manganese. According to the Pauli’s exclusion principle, the electrons in manganese must be antiparallel to the mediating one. The O1- becomes the excited state with an unpaired
electron, which can be, in turn, paired with the other neighboring manganese. These two neighboring manganese ions are thus effectively coupled by virtue of the bridging of the oxygen ions and result in an antiferromagnetic ordering.
2.2 Double exchange
Intrinsic perovskite manganese LaMnO3 is a well known parent manganite compound exhibiting A-type AFM originated from the orbital ordering (3x2-r2, 3y2-r2)
staggered behavior with TN = 140K [2]. La1-xAxMnO3 (A = Ca, Sr, Ba, Ce, Sn, and etc.)
are having the mixed valence state in manganese ions by replacing La3+ with the “A” ions.
These doped LaMnO3 have been shown to exhibit ferromagnetic behavior accompanied
by the metal-insulator transition around the Curie temperature. This characteristic was explained by the double exchange mechanism proposed by Clarence Zener [3]. With the crystal field the degeneracy of the d-orbital of the transition metal manganese is lifted and splits into a doublet eg (dx2−y2andd3z2−r2) and a triplet t2g (d ,xy d , and yz d ) state. As a zx
result, the Mn3+ and Mn4+ ions will have the ground state configuration of t2g3eg1 and
t2g3eg0, respectively, as illustrated schematically in Fig. 2.2(b). For instance, in Ca-doped
LaMnO3, the manganese becomes Mn3+ and Mn4+ mixed valence and results in finite
ions. The hoping process is also illustrated in Fig. 2.2. On the top row of Fig. 2.2(a), the initial state can be expressed as (Mn3+-O2--Mn4+). On the bottom of Fig. 2.2(a), the
left-hand-side electron in manganese hops to the right-hand-side next neighboring manganese and the state configuration is now expressed as (Mn4+-O2--Mn3+). There may
be also metastable states, such as (Mn3+-O1--Mn3+) between these two processes. All the
processes must also obey the Hund’s rule, thus, the lowest energy state is achieved with all the spins in parallel between the neighboring transition metal ions, leading to the ferromagnetic state in Zener’s report. This indirect electronic mediated magnetic ordering
eg t2g eg t2g θ teff Mn3+(S1) Mn4+(S2) eg t2g Mn3+ O2- Mn4+ Mn4+ O2- Mn3+ Mn3+ O1- Mn3+ (a) (b) (c)
Fig. 2.2 (a) and (b) depict the hoping processes of the double exchange. (c) Effective hoping between the two noncollinear spins. [reproduced with rederence [3-5]]
process has been termed as the double exchange mechanism. Later, P. W. Anderson and H. Hasegawa further considered the real hopping integral might be obtained in double exchange [4, 5]. In Fig. 2.2(c), the effective hoping between two neighboring transition metal ions is defined as )
2 cos(θ ⋅ = t
teff where θ was the angle between the two spin orientations. The Pauli’s exclusion principle requires that the eg and t2g electrons possess
the same spin state and the angle could also be simply described as t2g electronic spins.
For the antiparallel spin situation, θ = 180˚ the system is antiferromagnetic, the effective hoping will be zero and electrons cannot hop in this situation. In contrast, hoping will be the largest whenθ= 0˚.
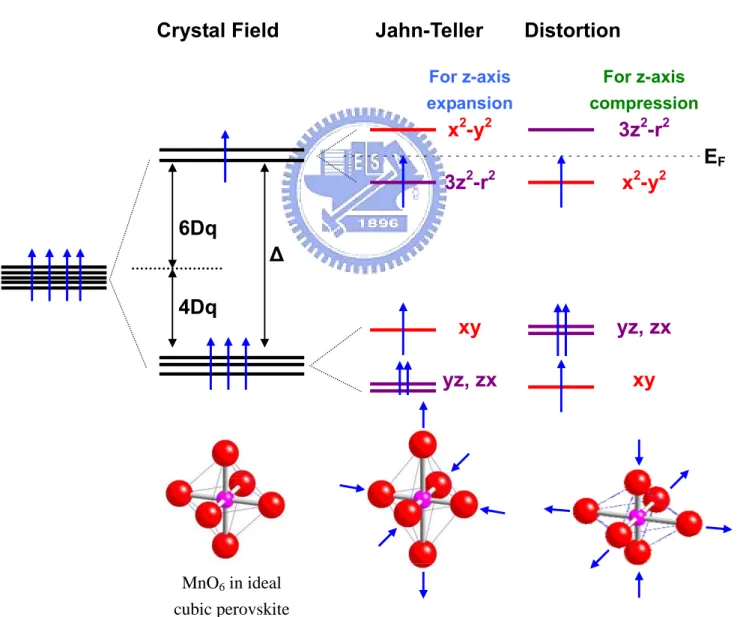
2.3 Crystal field and Jahn-Teller effect
As mentioned above, in transition metal compounds, there exists five-fold degeneracy in the 3d orbitals. Fig. 2.3 shows the electronic structure and the most prominent atomic arrangement of a representative ideal cubic perovskite (e.g. LaMnO3). The Mn ion locates
at the center of the MnO6 octahedron and is surrounded by six oxygen anions. Ideally, the
bond length of the six Mn-O bonds is equivalent. Nevertheless, the 3d shell experiences strong inhomogeneous field originated from the neighboring ions. As a result, the five fold degeneracy is lifted and splits into eg and t2g orbitals. The respective electronic
distribution of the d-orbitals are schematically illustrated in Fig 2.4 [6]. The t2g orbitals
(d , xy d , and yz d ) will be hybridized by the oxygen p orbital and form the π-bond zx
while the eg orbitals (dx2−y2and d3z2−r2) will hybridize with the oxygen p-orbital to form
the σ bond. The energy splitting between the t2g and eg states has been usually set to be
by 6Dq and that of the t2g orbital will be lowered by 4Dq when the crystal field
interaction is considered [5, 7].
For Mn3+, there are four 3d electrons. Three electrons occupy each triplet t2g orbital
and the final electron occupies the eg orbital. In real case, the spatial symmetry of the
ideal cubic perovskite is broken by doping. Replacing the La-ions by ions with different ionic size and electronic valences induces a noncubic potential and distorts the crystal lattice. Further energy splitting is induced by the distortion of MnO6 and such a
spontaneous displacement is the so-called Jahn-Teller effect. Fig. 2.3 shows two
For z-axis expansion For z-axis compression
x
2-y
23z
2-r
23z
2-r
2x
2-y
2xy
yz, zx
yz, zx
xy
6Dq
Δ
4Dq
Crystal Field
Jahn-Teller Distortion
E
FMnO6 in ideal
cubic perovskite
Fig. 2.3 Illustration of the energy level split by crystal field and Jahn Teller distortion. Pink and red ball denoted the Mn and O ions respectively. [reproduced with rederence [5-7]]
distortion modes of the MnO6 octahedrons. By taking into account the Jahn-distortion, eg
and t2g energy levels are split when the bond length of the Mn-O bonds is no longer
equivalent. If the octahedron is expanded along the z-axis, the eg orbitals are split into the
higher dx2−y2 and the lower d3z2−r2 levels and the t2g orbitals are split into the higher
xy
d and the lower energy level for d and yz d . The energy splitting distribution is zx also shown in Fig. 2.3 for the situation when the z-axis is under compression. The basic forms of Jahn-Teller distortion in influencing electron phonon coupling has been
formulated by Kanamori [5, 8, 9]. There are different kinds of distortion and are usually classified as 3 different modes Q1, Q2, and Q3. Q1 mode means that the x-, y-, z-axis are
Fig. 2.4 The equivalent-density surface of electronic distribution of d orbital. With the crystal field splitting, the degenerate was separated to doublet eg (3z2-r2 and
stretching or contracting as a whole and is usually called as the breathing mode. Q2 mode
describes the x-axis expansion and y-axis compression or vice versa. Q3 mode describes
the z-axis compression accompanied with expansion in x-y plane or z-axis expansion accompanied with compression in x-y plane. The Q3 mode usually involves the Q2 mode.
2.4 Crystal and magnetic structure
2.4.1 Crystal structure
The serial compounds of RMnO3 (for La to Eu) with orthorhombic structure have
the same space group setting Pbnm(62). Figure 2.5(a) shows the ideal cubic perovskite structure of LaMnO3 with a MnO6 octahedron specifically identified [9]. Figure 2.5(b)
shows the similar crystal structure of the perovskite LaMnO3 which has been slightly
distorted by the Jahn Teller distortions. The lattice constants have also been correspondently modified by the compressive and expansive strain leading to the variations in the Mn-O bond length and Mn-O-Mn bond angles. There is a trend showing the decrease in bond angle with the decreasing ionic size. To quantify the distortion with the competition between the A-site atom and Mn element, the tolerance factor t has been defined as: ) ( 2 ) ( O Mn O A d d t − −
= , where dA-O is the bond length of the A-site element and
oxygen and dMn-O is the bond length between Mn and oxygen [10]. For t = 1, it means the
cubic perovskite with the Mn-O-Mn bond angle towards 180° and Mn-O bond length being isotropically equivalent.
There also has been another space group setting Pnma frequently used to describe the crystal symmetry of these compounds. These two space groups can be easily converted to each other with the relation [11].
Pbnm Pnma a a’ b b’ c c’
In this work, we use Pbnm space group setting in that the c-axis lattice constant is the longest lattice constant of the crystal structure.
(a) (b)
Fig. 2.5 The crystal structure of (a) ideal cubic perovskite LaMnO3 and (b) real
orthorhombic perovskite LaMnO3. Green, pink, and red ball denoted the La, Mn, O
atoms in the crystal structure respectively. The MnO6 octahedron was distorted by Jahn-Teller effect with combination complicated in-plane and c-axis distortion. The apical Mn-O-Mn chain in MnO6 wouldn’t parallel to c-axis.
2.4.2
Magnetic structure
In an early report, Wollan and Koehler determined the magnetic structure of the La1-xCaxMnO3 system with neutron powder diffraction [12, 13]. In Fig. 2.6(a), the circles
are the Mn ions and the sign of the notation indicates the orientation of the z-axis spin projection. For example, the A-type magnetic structure has the spins ordering ferromagnetically in plane and the interplanar cpupling reveales an antiparallel antiferromagnetic ordering. The macroscopic bulk characteristic will exhibit the A-type as an antiferromanetic material in magnetization behavior. On the other hand, the B-type reveals the ferromagnetic behavior for their parallel spin orders no mater in the x-y plane
(b)
(a)
(c)
Fig. 2.6 (a) The classification of magnetic structure labeled by E. O. Wollan and W. C. Koehler.[13] (b) The illustration of comparing incommensurate spin orders and atomic orders in crystalline structure. [17] (c) The illustration of spin spiral AFM phase for TbMnO3.[18]
or along z-axis. An E-type magnetic order has a special spin arrangement and has been theoretically predicted to be capable of inducing strong magnetoelectric polarization [14, 15]. It is also vividly described as a “up-up-down-down” type magnetic spin ordering and usually observed in RNiO3 (R= trivalent lanthanoids but without La) systems [16]. The
E-type ordering has two up spins followed by two down spins along the principal axes of the cubic unit cell. This is different from another spin spiral phase that has induced most of the multiferroicities observed to date.
Fig. 2.6(b) shows the incommensurate spin spiral AFM magnetic orders in multiferroic TbMnO3 [17, 18]. The term of incommensuration (ICM) means that the spin
ordering periodicity is not compatible in a rational manner to the periodicity of the underlying crystal structure, as sketched schematically in Fig. 2.6(b). The ICM AFM ordering has been observed ubiquitously in the entire family of RMnO3 (R = Y and rare
earth Eu to Lu) compounds in their magnetic phase diagram. The spin density wave propagates along the b-axis with a propagation vector (0, ks, 0). The parameter ks
describes the periodic relation between the magnetic and crystal structure. For HoMnO3, the ks parameter increases ( 0.4≦ks<0.5 for the incommensurate state) with Mn3+ spin
orders below TN and finally locked into ks = 0.5 when transforming into the
commensurate (CM) state and becomes temperature independent. The temperature at which the transition occurs is called the locked-in transition temperature (TL).
Recently, with the improvement of the instrumentation and sample preparation techniques, the magnetic structure of TbMnO3 was revised by Kajimoto [19]. In his
report, TbMnO3 was found to involve magnetic structures of ICM A-, G-, C-, and F-type
below TN, and the G-, C-, F-type were further enhanced in the CM state below TL. The G-,
AFM mechanism has since then played a key role in magnetoelectric materials.
References
[1] R.L. Comstock., Introduction to magnetism and magnetic recording, 1999. [2] M. Tachibana, T. Shimoyama, H. Kawaji, T. Atake, E. Takayama-Muromachi,
Physical Review B. 75 (2007) 144425. [3] C. Zener, Physical Review. 82 (1951) 403.
[4] P.W. Anderson, H. Hasegawa, Physical Review. 100 (1955) 675. [5] T. Chatterji, Colosslal Magnetoresistive Manganites, 2004. [6] Y. Tokura, N. Nagaosa, Science. 288 (2000) 462-468. [7] C. Kitel, Introduction to Solid State Physics - 7th ed., 1996. [8] J. Kanamori, Journal of Applied Physics. 31 (1960) S14-S23.
[9] S. Satpathy, Z.S. Popovic, F.R. Vukajlovic, Physical Review Letters. 76 (1996) 960-963.
[10] E. Dagotto, T. Hotta, A. Moreo, Physics Reports-Review Section of Physics Letters. 344 (2001) 1-153.
[11] International tables for crystallography, Dordrecht, Holland ;D. Reidel Pub. Co. ;c2004.Boston, U.S.A. :Sold and distributed in the U.S.A. and Canada by Kluwer Academic Publishers Group,Hingham, MA :, 2004.
[12] E. Dagotto, Nanoscale Phase Sepration and Colossal Magnetoresistance, 2002.
[13] E.O. Wollan, W.C. Koehler, Physical Review. 100 (1955) 545.
[14] I.A. Sergienko, C. Sen, E. Dagotto, Physical Review Letters. 97 (2006) 227204.
[15] S. Picozzi, K. Yamauchi, B. Sanyal, I.A. Sergienko, E. Dagotto, Physical Review Letters. 99 (2007) 227201.
[16] T. Kimura, S. Ishihara, H. Shintani, T. Arima, K.T. Takahashi, K. Ishizaka, Y. Tokura, Physical Review B. 68 (2003) 060403.
[17] T. Kimura, T. Goto, H. Shintani, K. Ishizaka, T. Arima, Y. Tokura, Nature. 426 (2003) 55-58.
[18] Y. Yamasaki, H. Sagayama, T. Goto, M. Matsuura, K. Hirota, T. Arima, Y. Tokura, Physical Review Letters. 98 (2007) 147204.
[19] R. Kajimoto, H. Yoshizawa, H. Shintani, T. Kimura, Y. Tokura, Physical Review B. 70 (2004) 012401.
[20] T. Kimura, G. Lawes, T. Goto, Y. Tokura, A.P. Ramirez, Physical Review B. 71 (2005) 224425.
Chapter 3
Structural transformation with the doped ionic size
effect
3.1 Introduction
As discussed in chapter 1, the materials exhibiting multiferroic characteristics are usually
classified into three types: (i) the Bi-based compound like BiFeO3 or BiMnO3, in that the lone
pair of Bi 6s electrons are responsible for the ferroelectricity (FE) [1]; (ii) the ReMnO3
perovskites (Re = Tb, Dy, Y, Ho, Er, Tm, Yb, Lu) [1-7]; and (iii) the ReMn2O5 double-layer
perovskites (Re = Y, Tb, Dy, Ho) [1, 8]. Interestingly, many of the ReMnO3 perovskites are also
representative materials exhibiting the colossal magnetoresistance (CMR) [9, 10].
It has been established that, in this family, decreasing the ion size of the rare earth elements
will gradually drive the structure of ReMnO3 from orthorhombic into hexagonal. However, the
ferroelectric Curie temperature of the hexagonal manganites usually is much higher than the magnetic ordering temperature and the linear magnetoelectric effect is usually not allowed by symmetry at zero-field [3]. Nevertheless, evidences of coupling between the c-axis polarization and in-plane staggered antiferromagnetic (AFM) magnetization have been demonstrated in
HoMnO3. [12, 13] In this respect, with the ionic size sitting right at the orthorhombic and
hexagonal phase boundary of perovskite manganites, YMnO3 (h-YMO) (with Y3+ (1.06Å)) is of
particular interest. Indeed, extraordinary spin-phonon interaction was observed through the thermal conductivity measurements on this so-called geometrically frustrated FE system. [16] Since the ferroelectric polarization along the c-axis in h-YMO comes from the structure
distortion such as tilted MnO5 and displacement of Y3+ ions [5], it is, thus, interesting to conduct
a systematic study to see how the lattice distortion induced by ion size difference affects the relative ionic displacement and the associated modifications in the staggered frustrated magnetic moments and electronic structure. In this paper, we report the magnetic properties, O K-edge X-ray absorption near edge spectroscopy (XANES) and Mn L edge XANES on a series of
doped multiferroic materials Y1-xAxMnO3 (A = Ca, Sr; x = 0, 0.1, 0.3, 0.5) (YAMO) obtained by
the solid-state reactions and discuss how the ionic size and doping level of A-element affect the magnetic properties and electronic structure of h-YMO.
3.2 Experiment
All the samples of Y1-xAxMnO3 (A = Ca, Sr ; x = 0, 0.1, 0.3, 0.5) were prepared by solid
state reaction. Briefly, the nominal stoichiometry composition of Y2O3(99.999%),
MnCO3(99.95%), and CaCO3(99.95%) (or SrCO3(99.99%)) powders were ground and heated at
1300 °C. First, Y2O3 were preheated to 500℃ for 5h to remove the adsorptive mist and the
desired stoichiometric mixture of Y2O3, MnCO3, CaCO3, and SrCO3 were heated for 1300℃ for
18h. The mixture was pressed and sintered at 1300 °C for 24 hours, then ground and pressed again for the subsequent sintering. After repeating the process for 3-4 times, it was pressed into disk for the final sintering carried out at 1300 °C for 24 hours. We compared the density of these bulks with that calculate from the ideal bulk that all form with unit cell without grain boundary of defeat. We broke these bulks, pressed and reheat until the density rate of the bulk had trends of slow down. This process usually cost two to three times. For future measurements referred below, parts of the bulk were ground to minute powders again. The crystalline structure, magnetization behaviors, and electronic structure of these samples were examined by x-ray
diffraction (XRD), Superconducting Quantum Interference Device (SQUID), and (XANES), respectively. The XANES spectra were collected by the fluorescence yield (FY) and total electron yield (TEY) mode with resolution of 0.15 eV.
3.3 Results AND Discussions
Figure 3.1 shows the XRD θ-2θ scans of all the YAMO samples presented in this study. It
is clear that the undoped YMnO3 is of pure hexagonal structure. The Sr2+-doped YMnO3 also
shows hexagonal structure for various doping levels, as well. Nevertheless, the structure of the
Y1-xCaxMnO3 (YCMO) samples appears to change from the hexagonal structure for the undoped
FIG. 3.1: The XRD patterns for a series YMnO3 doped with Ca and Sr. Both pristine YMnO3
and Y1-xSrxMnO3 samples show the same hexagonal crystal structure. The structure
of Y1-xCaxMnO3 changes from hexagonal to orthorhombic with the increasing
![Fig. 1.2 The relation between multiferroic and magnetoelectric materials. [20]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8394547.178859/17.892.159.761.532.830/fig-relation-multiferroic-magnetoelectric-materials.webp)
![Table 1.1 Classification of ferroelectrics. [24]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8394547.178859/19.892.120.783.114.510/table-classification-of-ferroelectrics.webp)