i
國立臺灣大學醫學院臨床醫學研究所 博士論文
Graduate Institute of Clinical Medicine College of Medicine
National Taiwan University Doctoral dissertation
利用新一代 EGFR 酪氨酸激酶抑制劑為放射增敏劑,
用於膀胱癌的治療
Utilizing New-Generation EGFR Tyrosine Kinase Inhibitor as Radiosensitizer in the Treatment of Urinary Bladder Cancer
蔡育傑 Yu-Chieh Tsai
指導教授﹕鄭安理 (Ann-Lii Cheng, M.D., Ph.D.) 郭明良 (Min-Liang Kuo, Ph.D.)
中華民國一百零四年八月 August, 2015
ii
誌謝
這趟旅程,比我原本想像的要漫長。幸好一路上都有貴人相助相伴,才得以踏實 地走下去。最要感謝的是指導老師鄭安理教授。這些年來我很榮幸能在鄭教授領導的 台大醫院腫瘤團隊工作,同時博士班的學業又接受鄭教授指導。鄭教授對於腫瘤學的 基礎和臨床研究有非常深入的見解,總是在重要的時刻提供好的意見,也願意不厭其 煩地提醒我的缺點。同時我也要感謝另一位指導老師郭明良教授,以基礎醫學研究者 的角度指導我如何開始這個計畫,如何把想法和假說實際執行。我的實驗有很多是基 於RTK array的分析結果進行,而這部分就是郭老師的建議。
在博士班的前兩年我對於實驗室的工作一竅不通,也很徬徨要如何找到好的研究 題目。直到在美國羅徹斯特大學的進修期間,接受張傳祥教授的指導才對膀胱癌的研 究有比較好的基礎。本論文的完成要謝謝成佳憲教授的大力協助,剛開始放射生物學 的相關研究對我來說非常陌生,成教授帶我走進這個領域,讓膀胱癌放射增敏劑的研 究可以成為一個完整的計劃,得到重要的結論。我也要謝謝楊志新教授,他是國際知 名的afatinib專家,對這個藥的基礎研究和臨床應用提供精闢的建議。
目前我的臨床工作是以泌尿癌的病人照顧為主,很感謝台大泌尿腫瘤團隊的蒲永 孝教授與林家齊副教授的指導,讓我對膀胱癌有更多的認識。對於實驗室工作我要感 謝前後任助理宗帆、俊宇、媛媛以及何蓓茵博士與劉為麟博士的全力配合,在一次又 一次的嘗試中找到問題的答案。
最後我要感謝家人的犧牲與付出,爸爸、媽媽、姊姊、妻子雅雯和寶貝女兒宜 瑾,你們是最棒的。完成這本論文,只是站上夢想的起點,誠摯地希望未來大家可以 繼續給我鼓勵,繼續結伴前行。
蔡育傑 謹誌於
中華民國104年8月19日
iii
TABLE OF CONTENTS
Page
口試委員會審定書 ... i
Acknowledgement ... ii
Table of Contents ... iii
List of Figures.. ... v
List of Tables... ... viii
Abbreviation… ... ix
Abstract (Chinese) ... xii
Abstract (English) ... xiv
CHAPTER ONE: INTRODUCTION 1.1 Clinical Overview of Bladder Cancer ... 1
1.2 Molecular Biology of Bladder Cancer ... 10
1.3 Radiation Effect and Signal Transduction Pathways ... 14
1.4 New-generation ErbB Family Inhibitors as Radiosensitizers ... 17
1.5 Hypothesis and Experimental Design ... 19
CHAPTER TWO: PILOT STUDY 2.1 Rationale and Approach ... 20
2.2 Materials and Methods ... 20
2.3 Results ... 21
CHAPTER THREE: RADIOSENSITIZING EFFECT OF AFATINIB IN A MURINE BLADDER CARCINOMA MODEL 3.1 Rationale and Approach ... 25
3.2 Materials and Methods ... 26
iv
3.3 Results ... 32
CHAPTER FOUR: RADIOSENSITIZING EFFECT OF AFATINIB IN HUMAN BLADDER CANCER MODELS 4.1 Rationale and Approach ... 41
4.2 Materials and Methods ... 42
4.3 Results ... 49
CHAPTER FIVE: DISCUSSION 5.1 Pilot study ... 67
5.2 Murine Bladder Cancer Model ... 69
5.3 Human Bladder Cancer Model ... 74
5.4 Unfinished Study: Mutant EGFR - Related Research ... 78
CHAPTER SIX: PROSPECT 6.1 Radiosensitizing Activity of Afatinib and Microenvironment ... 83
6.2 Clinical Application ... 87
REFERENCE ... 90
APPENDIX ... 110
v
LIST OF FIGURES
Page
Figure 1-1. The definition of T stage for bladder cancer ... 4
Figure 1-2. The two-pathway model. ... 10
Figure 1-3. Aberrantly activated signal transduction pathways may influence radiosensitivity.... ... 15
Figure 1-4. A model of EGFR-mediated radioprotection ... 16
Figure 2-1. Clonogenic assay of erlotinib +/- RT in T24 cells ... 22
Figure 2-2. Clonogenic assay of erlotinib +/- RT in NTUB1 cells ... 22
Figure 2-3. Clonogenic assay of trastuzumab +/- RT in T24 cells ... 23
Figure 2-4. Clonogenic assay of trastuzumab +/- RT in NTUB1 cells ... 23
Figure 2-5. Clonogenic assay of lapatinib +/- RT in T24 cells ... 24
Figure 2-6. Clonogenic assay of lapatinib +/- RT in NTUB1 cells ... 24
Figure 3-1. Radiosensitization of MBT-2 cells by afatinib. ... 32
Figure 3-2. Radiation activates EGFR and PI3K/Akt pathways at both low and high doses…… ... 33
Figure 3-3. Afatinib inhibits the radiation-activated EGFR phosphorylation and PI3K/Akt pathways……… ... 34
Figure 3-4. Afatinib enhances radiation-induced apoptosis in MBT-2 cells. ... 36
Figure 3-5. Afatinib enhances radiation-induced DNA damage of MBT-2 cells……. ... 34
Figure 3-6. Combined afatinib and irradiation enhances tumor suppressive activity in ectopic murine bladder tumor model. ... 39
Figure 4-1. The effect of combining radiation with EGFR TKIs on radiation-sensitive signals………... ... 50
vi
Figure 4-2. Clonogenic survival analysis shows a difference in the radiosensitizing effect of TKIs that inhibit EGFR/HER2 or EGFR alone. ... 51 Figure 4-3. Tyrosine kinase blockade of both EGFR and HER2 by afatinib, not blockade of EGFR alone, promotes radiation-induced apoptosis. ... 53 Figure 4-4. Western blots show that irradiation increases the expression of the apoptosis markers which are further enhanced by afatinib pretreatment. ... 54 Figure 4-5. The EGFR/HER2 dual inhibitor afatinib, not the EGFR inhibitor erlotinib, promotes radiation-induced DNA damage. ... 56 Figure 4-6A. Tumor volume of mouse xenograft model. ... 57 Figure 4-6B. Representative images of animal PET/CT before treatment and 2 weeks after initial treatment ... 58 Figure 4-6C. Western blot analysis shows that the RT-afatinib combination effectively suppresses radiation-activated EGFR, HER2, and Akt signals and enhances cleaved PARP expression. ... 59 Figure 4-7. The radiosensitizing effect of erlotinib in HER2 knocked-down T24 cells emphasizes the synergism between EGFR and HER2 in determining radiosensitivity. .. 61 Figure 4-8. The radiosensitizing effect of erlotinib in HER2 knocked-down NTUB1 cells emphasizes the synergism between EGFR and HER2 in determining radiosensitivity. .. 63 Figure 4-9A. Western blotting of T24 cell lysates treated with chemical cross-linking. 65 Figure 4-9B. The ratios of EGFR or HER2 dimer formation after different treatment ... 65 Figure 4-9C. Western blotting of T24 cell lysate precipitated with EGFR antibody. ... 66 Figure 5-1. Effects of afatinib on protein phosphorylation after irradiation or afatinib pretreatment+irradiation in PC-9, PC-9-GR and H1975 cells ... 80 Figure 5-2. The effects of afatinib on protein phosphorylations in lung cancer cells... 81 Figure 5-3. Effect of afatinib on clonogenic survival in irradiated lung cancer cells. ... 82
vii
Figure 6-1. Invasion assay of T24 and 5637 bladder cancer cells treated with irradiation +/- afatinib or erlotinib ... 84 Figure 6-2. Western blot of T24 and 5637 bladder cancer cells treated with irradiation +/- afatinib or erlotinib ... 85 Figure 6-3. Gelatin zymography of culture media from T24 and 5637 bladder cancer cells treated with irradiation +/- afatinib or erlotinib ... 85
viii
LIST OF TABLES
Page
Table 1-1. TNM staging of bladder cancer ... 5 Table 5-1. IC50 of anchorage independent cell ... 67 Table 5-2. In vitro inhibitory activity of afatinib ... 68
ix
ARREVIATION
AJCC: American Joint Committee on Cancer
ANOVA: analysis of variance
BCG: Bacillus Calmette–Guérin
CI: combination index
CT: computed tomography
DAPI: 4',6-diamidino-2-phenylindole
DMSO: dimethyl sulfoxide
DNA: deoxyribonucleic acid
DNA-PK: DNA-dependent protein kinase
DSB: double-strand break
EGFR: epidermal growth factor receptor
EMT: epithelial-mesenchymal transition
FBS: fetal bovine serum
FDG: fluorodeoxyglucose
FITC: fluorescein isothiocyanate
GC: gemcitabine - cisplatin
HER2: human epidermal growth factor receptor 2
x IC50: half maximal inhibitory concentration
IGFR: insulin-like growth factor receptor
ISUP: International Society of Urological Pathology
MAPK: mitogen-activated protein kinases
MIBC: muscle-invasive bladder cancer
mTOR: mammalian target of rapamycin
M-VAC: methotrexate - vinblastine - adriamycin - cisplatin
NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells
NHEJ: non-homologous end joining
NHUC: normal human urothelial cell
NMIBC: non-muscle-invasive bladder cancer
NSCLC: non-small-cell lung carcinoma
PARP: poly (ADP-ribose) polymerase
PBS: phosphate-buffered saline
PCR: polymerase chain reaction
PET: positron emission tomography
PI3K: phosphoinositide 3-kinase
PVDF: polyvinylidene difluoride
RT: radiotherapy
xi RTK: receptor tyrosine kinase
SD: standard deviation
SNP: single nucleotide polymorphism
STR: short tandem repeat
SUV: standardized uptake value
TCGA: The Cancer Genome Atlas
TGFβ: transforming growth factor beta
TKI: tyrosine-kinase inhibitor
TNM: tumor - node - metastasis
TUR-BT: transurethral resection of bladder tumor
WHO: World Health Organization
xii
摘要
膀胱癌是全世界與台灣男性第九常見的惡性腫瘤。對許患有局部肌肉侵襲性膀胱癌
的病人,標準的膀胱根除手術並不可行。許多研究的焦點放在如何在”膀胱保存”的治
療基準下善用放射線治療。然而相較於膀胱根除手術,放射線治療肌肉侵襲性膀胱癌病
人的長期存活率低了約 10%。傳統上化學治療藥物被用來當成放射增敏劑,但這樣的治
療有許多廣為人知的毒性。因此我們有很強大的需求尋找能夠增進膀胱癌放射治療效果,
又不增加毒性的藥物。
一個可以增進膀胱癌放射治療效果的合理方法,是同時使用抑制放射治療相關信息
傳導路徑的標靶藥物。表皮生長因子受體(EGFR)是其中最重要的。Cetuximab 是一個抑
制 EGFR 的單株抗體,它已被證明在頭頸癌病人身上併用放射治療,可以增加療效。至
於膀胱癌,gefitinib 是一個 EGFR 的酪胺酸激酶抑制劑,它被顯示在膀胱癌的細胞模
式有中等的放射增敏效果,而在膀胱癌的活體模式則只有些微的放射增敏效果。因此這
個主題值得後續的研究。
在前驅性研究中我測試了包括 erlotinib (EGFR 抑制劑)、trastuzumab (HER2 抑
制劑)和 lapatinib (EGFR/HER2 抑制劑) 在膀胱癌細胞的放射增敏效果,可惜沒有一
個有好的發展潛力。反而是 afatinib,一個新一代可以同時抑制 EGFR 和 HER2 的酪胺
酸激酶抑制劑,比較有前途。
在老鼠膀胱癌模式中,我第一次展示了在膀胱癌的細胞及活體模式中,afatinib 都
是一個有效的放射增敏劑。這個部分的動物實驗是在免疫正常的老鼠體內進行,比較類
xiii
似人類的生理狀態。Afatinib 似乎抑制了放射活化的 EGFR 與 HER2 信息,並增加了細
胞 DNA 傷害與凋亡。
基於以上的發現,我假設在膀胱癌細胞,同時抑制 EGFR 和 HER2 酪胺酸激酶活性
的 afatinib,相較於只抑制 EGFR 酪胺酸激酶活性的 erlotinib,會有較佳的放射增敏
性。
為了確認這個假說,第一代的 EGFR 酪胺酸激酶抑制劑 erlotinib 和第二代的
afatinib,第一次在人類膀胱癌細胞中被拿來比較它們的放射增敏性。我展示了在人類
膀胱癌的細胞及活體模式中 erlotinib 的放射增敏性的不足以及 afatinib 優越的放射
增敏效果。在 HER2 被抑制的人類膀胱癌細胞中,可以看到 erlotinib 顯示了放射增敏
效果,所以可能是因為 EGFR 和 HER2 對放射敏感性有協同作用,讓 afatinib 雙重抑制
的特性才會變得有效。我也展示了對 EGFR-HER2 異源雙體的抑制可能是 afatinib 放射
增敏性機轉的證據。
在”展望”中我提到如何繼續這個研究主題,以及怎樣應用到臨床上。我希望這
個研究的結果可以協助達成”增進膀胱癌放射治療效果,又不增加毒性”的目標。
xiv
Abstract
Bladder cancer is the ninth most common cancer in the world and in Taiwanese male
population. For many patients with localized muscle-invasive bladder cancer, radical
cystectomy is not a feasible treatment, and considerable interest was focused on the optimal use of radiotherapy in ”bladder preservation” protocol. However, the long-term survival of
patients receiving radiation-based therapy in muscle invasive bladder cancer is about 10%
inferior to patients receiving standard radical cystectomy. Traditionally chemotherapeutic
agents are used as radiosensitizer but they have many well-known toxicities. Therefore,
there is a strong need to find agents enhancing the radiation effect in urinary bladder
cancer treatment while not increasing the toxicities.
A reasonable way to enhance the outcome of radiotherapy is by concomitantly using
agents that inhibit radiation-activated signaling pathways. Epidermal growth factor receptor
(EGFR) is the most important target. Cetuximab, an anti-EGFR antibody, has shown clinical
benefit in head and neck cancer when combined with radiotherapy. In bladder cancer,
gefitinib, an EGFR tyrosine kinase inhibitor (TKI), has moderate in vitro and marginal in vivo
radiosensitizing activities. Therefore the topic deserves further investigation.
In pilot study, I tested the radiosensitizing activities of erlotinib (EGFR inhibitor),
trastuzumab (HER2 inhibitor) and lapatinib (EGFR/HER2 inhibitor) in bladder cancer cells.
xv
None of them showed good potential. Instead, afatinib, a new-generation EGFR inhibitor
with activity against both EGFR and HER2, is more promising.
In murine bladder cancer model, I demonstrated for the first time the in vitro and in vivo
radiosensitizing activity of afatinib, an EGFR/HER2 dual inhibitor. The animal study was
performed in immunocompetent mice and mimic human physiologic status. Afatinib likely
mediates its effect on bladder cancer cells by suppressing radiation-activated EGFR and
HER2 signals and thereby causing enhanced DNA damage and cell apoptosis.
Based on the findings I hypothesized that in bladder cancer cells, the concomitant
inhibition of EGFR and HER2 tyrosine kinase activity by afatinib has greater
radiosensitizing activity than the inhibition of EGFR tyrosine kinase activity alone by
erlotinib
To confirm the hypothesis, in human bladder cancer model the radiosensitizing effects of
different generations of clinically useful EGFR TKIs were compared for the first time. I
showed the inadequacy of EGFR inhibition alone and the advantage of concomitant blockade
of radiation-activated EGFR and HER2 signaling to inhibit the in vitro and in vivo growth of
bladder cancer cells. The radiosensitizing effect of an EGFR inhibitor was much higher in
HER2 knocked-down than wild-type cells, therefore HER2 may play a synergistic role with
EGFR in determining radiosensitivity. I also showed evidence to support that receptor
heterodimerization plays an important role in the radiosensitizing effect of afatinib.
xvi
In Prospect I mentioned how to continue current project and apply the data to clinical use.
I hope that the results of this study can help to meet the need of enhancing the radiation effect
in urinary bladder cancer treatment while not increasing the toxicities.
CHAPTER ONE: INTRODUCTION
1.1 Clinical Overview of Bladder Cancer
1.1.1 Epidemiology
Bladder cancer is the most common cancer of the urinary tract. According to the
estimation by GLOBOCAN database of World Health Organization (WHO), there will be
468,351 new cases and 179,753 deaths of bladder cancer in 2015 in the world.
(http://globocan.iarc.fr/Default.aspx). By World Cancer Report 2014, it is the 9th most
common cancer on the world. In Taiwan, according to the 2012 Cancer Registry Annual
Report, there were 2,003 new cases and 807 deaths of bladder cancer this year. The median
age of new case and death were 72 and 79, respectively. (http://www.hpa.gov.tw/
Bhpnet/Web/Stat/StatisticsShow.aspx?No=201504290001) Bladder cancer is also the 9th
most common cancer in male population of Taiwan.
1.1.2 Risk factors
Risk factors for the development of bladder cancer can be classified into: (a) genetic and
molecular abnormalities, (b) chemical or environmental exposures, and (c) chronic irritation
(Kaufman, Shipley, & Feldman, 2009). Genetic and molecular factors include oncogenes,
such as TP63, the epidermal growth factor receptors (EGFR), and Ras p21 proteins. Tumor
suppressor genes, including TP53 and RB1 are also important in the pathogenesis of bladder
cancer. Chemical and environmental exposures include aromatic amines, aniline dyes, nitrites
and nitrates, acrolein, coal, and arsenic, but the most important environmental factor is
cigarette smoking. Other causal factors include chronic irritation, indwelling catheters,
Schistosoma haematobium infection, and pelvic irradiation.
1.1.3 Pathologic classifications and grading
According to the WHO/International Society of Urological Pathology (ISUP) the
histologic types of bladder cancer are (Miyamoto et al., 2010):
- Urothelial carcinoma (also known as transitional cell carcinoma).
- Squamous cell carcinoma
- Adenocarcinoma
- Undifferentiated carcinoma and other rare types
By far the most common histology type of bladder cancer is urothelial carcinoma, and they
are graded as low-grade or high-grade according to their cellular characteristics (Lokeshwar,
Ruiz-Cordero, Hupe, Jorda, & Soloway, 2015).
1.1.4 Staging
Like most solid tumors, bladder cancer is staged using the Tumor - Node - Metastasis
system (TNM system). In anatomy, urinary bladder consists of three layers: the epithelium
and the subepithelial connective tissue, the muscularis, and the perivesical fat (peritoneum
covering the superior surface and upper part) (Fig. 1-1). According to the seventh edition
of staging system by American Joint Committee on Cancer (AJCC), the TNM stage of
bladder cancer is defined as:
Primary Tumor (T)
TX: Primary tumor cannot be assessed
T0: No evidence of primary tumor
Ta: Noninvasive papillary carcinoma
Tis: Carcinoma in situ: “flat tumor”
T1: Tumor invades subepithelial connective tissue
T2: Tumor invades muscle
pT2a - Tumor invades superficial muscle (inner half)
pT2b - Tumor invades deep muscle (outer half)
T3: Tumor invades perivesical tissue
pT3a - Microscopically
pT3b - Macroscopically (extravesical mass)
T4: Tumor invades any of the following: prostate, uterus, vagina, pelvic wall, abdominal wall
T4a - Tumor invades prostate, uterus, vagina
T4b - Tumor invades pelvic wall, abdominal wall
Figure 1-1. The definition of T stage for bladder cancer
(Adopted from AJCC Cancer Staging Manual, Seventh Edition, 2010)
Regional Lymph Nodes (N)
NX: Regional lymph nodes cannot be assessed
N0: No regional lymph node metastasis
Nl: Metastasis in a single lymph node, 2cm or less in greatest dimension
N2: Metastasis in a single lymph node, more than 2cm but not more than 5cm in greatest
dimension; or multiple lymph nodes, none more than 5cm in greatest dimension
N3: Metastasis in a lymph node, more than 5 cm in greatest dimension
Distant Metastasis (M)
MX: Distant metastasis cannot be assessed
M0: No distant metastasis
M1: Distant metastasis
The stage grouping is defined as Table 1:
0a Ta N0 M0
0is Tis N0 M0
I T1 N0 M0
II T2a - T2b N0 M0
III T3a - T4a N0 M0
IV T4b
Any T
Any T
N0
N1 - N3
Any N
M0
M0
M1 Table 1-1. TNM staging of bladder cancer
(Adopted from AJCC Cancer Staging Manual, Seventh Edition, 2010)
1.1.5 Muscle-invasive versus non-muscle-invasive tumors
At diagnosis the majority of tumors are non-muscle-invasive bladder cancer (NMIBC,
including Ta, Tis, T1), and the patients with stage 0 and stage I diseases account for two-
thirds of bladder cancer patients in Taiwan (2012 data). Most of them have a favorable
prognosis but a high rate of relapse can be found in high-grade disease. In contrary, patients
with muscle-invasive bladder cancers (MIBC, including T2, T3 and T4) only comprise one-
third of all patients in Taiwan and has a relatively poor prognosis. In particular, 25% of pT2
tumors (defined by invasion of muscle layer of the bladder), 50% of pT3 tumors (defined by
invasion of perivesical fat) and 80% of pT4 tumors (defined by invasion into nearby organs
and structures) will eventually develop into metastatic disease, while 5-year survival is 67%
for pT2 tumors, 35% for pT3 tumors and 27% for pT4 tumors. (Prasad, Decastro, &
Steinberg, 2011).
1.1.6a Treatment of non-muscle-invasive bladder cancer
For NMIBC, transurethral resection of the bladder tumor (TUR-BT) is the standard
treatment. Due to high rate of recurrence, patients should have cystoscopy and voided urine
cytology every 3 months for 2 years, then 6 monthly for 2 years, and then once yearly
indefinitely (Kaufman et al., 2009). Upper tract imaging is suggested because the lifetime risk
of upper tract tumor after a diagnosis of bladder cancer is about 5% (Smith, Weaver,
Barjenbruch, Weinstein, & Ross, 1989). Intravesical therapy including the immunomodulator
Bacillus Calmette–Guérin (BCG) and chemotherapeutic agents such as mitomycin,
doxorubicin, thiotepa and gemcitabine was developed to reduce recurrence (Patel, Cohen,
Weiner, & Steinberg, 2015).
1.1.6b Challenge in the treatment of localized muscle-invasive bladder cancer
Currently radical cystectomy (radical prostatocystectomy for male patients) is the
standard treatment for patients with MIBC and a chance to cure (Herr, Dotan, Donat, &
Bajorin, 2007). Patients who undergo radical cystectomy or prostatocystectomy have an
inevitable requirement of urinary tract diversion which results in impaired quality of life
(Singer et al., 2013). In addition, the median age of bladder cancer at diagnosis is over 70
years old and patients of this age often have substantial comorbidities. Altogether, curative
surgery is not usually a viable option for patients with MIBC, especially in old population.
As for the alternative treatment for patients who are not suitable for radical cystectomy,
considerable interest focuses on the optimal use of radiotherapy. The “bladder preservation”
protocols usually consist of aggressive TUR-BT, concurrent chemoradiation and several
cystoscopic examinations (Kaufman et al., 2009; Prasad et al., 2011). However, the long-term
prognosis of this multimodality therapy is still inferior to standard radical cystectomy, with 5-
year cancer-specific survival rate 36% versus 40% and 5-year overall survival rate 26%
versus 35% (Booth et al., 2014). Moreover, salvage cystectomy may still be warranted for
local failure after radiotherapy. The advance of bladder-preservation treatments could provide
patients with a chance to improve treatment outcome and quality of life (James et al., 2012).
1.1.6c Systemic chemotherapy of bladder cancer
For metastatic bladder cancer, cisplatin-based combination chemotherapy is the only
modality that demonstrated a survival benefit in randomized phase III trials (Loehrer et al.,
1992). However the previous standard of methotrexate, vinblastine, doxorubicin and cisplatin
(M-VAC) is too toxic for many patients. The doublet of gemcitabine and cisplatin (GC) have
similar effects on survival with median overall survival 14.0 months for GC and 15.3 months
for M-VAC (von der Maase et al., 2005) Since GC regimen is less toxic, currently it is widely
used as first-line treatment of metastatic bladder cancer.
As the rate of eventual metastatic disease is very high in patients with high-risk bladder
cancer, neoadjuvant chemotherapy before radical cystectomy had been studied to reduce
occult micrometastases. Meta-analyses have demonstrated a small (5%), but sustainable
benefit with neoadjuvant platinum-based combination chemotherapy on overall survival at 5
years (Advanced bladder cancer (ABC) meta-analysis collaboration, 2005; Winquist,
Kirchner, Segal, Chin, & Lukka, 2004).
1.2 Molecular Biology of Bladder Cancer
1.2.1 The two-pathway model
In addition to the difference of anatomic location and prognosis between muscle-
invasive versus non-muscle-invasive bladder cancers, several lines of evidence support the
general concept that the distinct clinical outcomes of NMIBC versus MIBC reflect their
distinct molecular causes and potentially their distinct cells of origin (X. R. Wu, 2005).
Certain molecular alterations, such as gain of function mutations in fibroblast growth factor
receptor 3 (FGFR3), are prevalent in low-grade NMIBC, whereas other alterations, such as
p53 loss or mutation, are prevalent in high-grade MIBC (Esrig et al., 1994; Goebell &
Knowles, 2010) (Fig. 1-2). Analyses of gene expression profiling (Dyrskjot et al., 2003)
and/or genomic alterations (Hurst, Platt, Taylor, & Knowles, 2012; Lindgren et al., 2010)
have supported this concept, although it is difficult to establish a mutual-exclusivity model,
since some superficial bladder tumors can progress to invasive disease.
Figure 1-2. The two-pathway model. The blue and purple pathways indicate the two major
pathways with distinct histopathological and molecular features that have been recognized
(Knowles & Hurst, 2015).
Up to 60% of stage Ta tumors have activating point mutations in FGFR3 (Cappellen et
al., 1999; Hernandez et al., 2006), which is associated with favourable outcome. In contrary,
FGFR3 mutation is less common in MIBC (10-20% in tumors of stage T2 or above) (Billerey
et al., 2001). In cultured normal human urothelial cells (NHUCs), mutant FGFR3 activates
the RAS-Mitogen activated protein kinase (MAPK) pathway and phospholipase Cγ,
leading to increased survival and proliferation to high cell density (di Martino, L'Hote,
Kennedy, Tomlinson, & Knowles, 2009). This in vitro phenotype suggests that FGFR3
mutation could contribute to early clonal expansion within the urothelium in vivo.
FGFR3 is also implicated in contributing to the risk of bladder cancer development. A
single-nucleotide polymorphism (SNP) in an intron of TACC3, which is 70 kb from FGFR3,
is associated with bladder cancer risk and with higher risk of recurrence in stage Ta disease,
particularly for FGFR3-mutant NMIBC (Kiemeney et al., 2010). One possible mechanism is
that altered chromatin structure associated with increased expression of FGFR3 could
increase the probability of mutation and/or increase the expression and impact of mutated
proteins (Knowles & Hurst, 2015).
In MIBC, the phosphoinositide 3-kinase (PI3K) pathway deserves more attention.
Upstream activators include ErbB family receptors. For example, EGFR induces PI3K
activation via RAS activation. ErbB3 interacts with p110α, the catalytic subunit of PI3K, and
conveys signals from human epidermal growth factor receptor 2 (HER2)-EebB3
heterodimers. Overexpression of EGFR, HER2 and/or ErbB3 in subsets of bladder cancer is
associated with higher grade, stage and worse outcome (Jimenez et al., 2001; Kassouf et al.,
2008; Kruger et al., 2002). HER2 amplification or overexpression is more common in
metastases than in the corresponding primary tumor, implying a role in the metastatic process
(Fleischmann, Rotzer, Seiler, Studer, & Thalmann, 2011).
1.2.2 Molecular subtypes of bladder cancer
With the advance of new technology, more sophisticated classifications of bladder
cancers are proposed recently. Sjödahl et al. studied 308 bladder tumors and defined five
urothelial carcinoma subtypes: urobasal A, genomically unstable, urobasal B, squamous cell
carcinoma like, and an infiltrated class of tumors (Sjödahl et al., 2012). A further analysis
identified that urobasal A subtype shares features with normal urothelium such as keratin 5,
P-cadherin, and EGFR expression confined to basal cells, and cell cycle activity restricted to
the tumor-stroma interface. In contrast, the squamous cell cancer-like subtype uniformly
expresses keratin 5, P-cadherin, EGFR, and cell cycle genes throughout the tumor
parenchyma. The genomically unstable subtype shows proliferation throughout the tumor
parenchyma and high HER2 and E-cadherin expression but absence of keratin 5, P-cadherin,
and EGFR expression. Urobasal B tumors demonstrate features shared by both urobasal A
and squamous cell cancer-like subtypes (Sjödahl et al., 2013). A major transition in tumor
progression seems to be loss of dependency of stromal interaction for proliferation.
The Cancer Genome Atlas (TCGA) reported an integrated analysis of 131 urothelial
carcinomas to provide a comprehensive landscape of molecular alterations. There were
statistically significant recurrent mutations in 32 genes, including multiple genes involved in
cell-cycle regulation, chromatin regulation, and kinase signalling pathways. RNA sequencing
revealed four expression subtypes, two of which (papillary-like and basal/squamous-like)
were also evident in microRNA sequencing and protein data.
TCGA study also identified potential therapeutic targets in 69% of the tumors, including
42% with targets in the PI3K/AKT/mammalian target of rapamycin (mTOR) pathway and
45% with targets (including HER2) in the receptor tyrosine kinases (RTK) /MAPK pathway.
Chromatin regulatory genes were more frequently mutated in urothelial carcinoma than in
any other common cancer, indicating the future possibility of targeted therapy for chromatin
abnormalities (Cancer Genome Atlas Research Network, 2014).
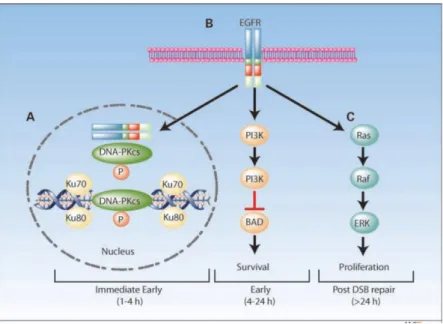
1.3 Radiation Effect and Signal Transduction Pathways
Ionizing radiation induces a variety of deoxyribonucleic acid (DNA) lesions, including
oxidized base damage, abasic sites, single-strand breaks and double-strand breaks. These
lesions, if unrepaired, will result in cell death through mitotic catastrophe and apoptosis.
Radiation also induces complicated biologic responses to cancer cells including repairing
DNA damage and counteracting the propagation to apoptosis. Traditionally chemotherapeutic
agents are used to enhance radiation effect because these agents can augment DNA damage
to cancer cells (Boeckman, Trego, & Turchi, 2005), but the toxicities usually hinder their use
in patients with old age or underlying medical diseases.
There are evidences showing that aberrantly activated signal transduction pathways can
influence radiosensitivity of cancer cells by modulating apoptotic response and DNA repair
mechanism (Begg, Stewart, & Vens, 2011). For example, AKT (Schlessinger, 2000), MAPK
(Dent, Yacoub, Fisher, Hagan, & Grant, 2003) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-B) (Beg & Baltimore, 1996) signaling can inhibit the apoptotic
response after DNA damage by activation of the anti-apoptotic protein MCL1, or inactivation
of the pro-apoptotic protein BAD and BIM. On the other hand, links between the AKT,
MAPK and transforming growth factor beta (TGFβ) pathways and DNA repair have been
found. Activation of the AKT and MAPK pathways leads to the activation of the catalytic
subunit of DNA-dependent protein kinase (DNA-PK) (Golding et al., 2007) which is a
central protein in double-strand break repair by non-homologous end-joining (NHEJ) (Kriegs
et al., 2010). This protein can also be activated by EGFR after it is translocated to the nucleus
(Dittmann, Mayer, Fehrenbacher, et al., 2005). TGFβ is necessary for the full activation of
ATM to DNA damage (Kirshner et al., 2006). (Fig.1-3)
Figure 1-3. Aberrantly activated signal transduction pathways may influence radiosensitivity
(Begg et al., 2011)
Receptor tyrosine kinases such as EGFR and insulin-like growth factor receptor (IGFR)
share common downstream signaling pathways which may influence radiosensitivity.
Multiple lines of evidence indicate that EGFR is an important determinant of radioresponse
and has a radioprotective function. Based on current evidence, EGFR-mediated
radioprotection can be conceptually divided into three phases (D. J. Chen & Nirodi, 2007):
(a) an immediate early phase that involves DNA repair; (b) suppression of DNA damage-
induced apoptosis before and after cell cycle arrest, and (c) a tumor repopulation step that
offers a proliferative advantage to tumors emerging from radiation-induced cell cycle arrest
(Fig. 1-4).
Figure 1-4. A model of EGFR-mediated radioprotection (D. J. Chen & Nirodi, 2007)
The role of EGFR in radioprotection is best demonstrated in a large phase III clinical trial.
In this study patients with locoregionally advanced head and neck cancer were randomly
assigned to treatment with high-dose radiotherapy alone (213 patients) or high-dose
radiotherapy plus weekly cetuximab, an EGFR-specific antibody (211 patients). The median
duration of locoregional control was 24.4 months among patients treated with cetuximab plus
radiotherapy and 14.9 months among those given radiotherapy alone. Overall survival was
49.0 months among patients treated with combined therapy and 29.3 months among those
treated with radiotherapy alone (Bonner et al., 2006). More molecular agents targeting
EGFR/ErbB family receptors, angiogenesis and histone deacetylase are under investigation in
Phase III studies in combination with radiation therapy (Morris & Harari, 2014).
1.4 New-generation ErbB Family Inhibitors as Radiosensitizers
Preclinical studies showed that cetuximab, an anti-EGFR monoclonal antibody, induces
cell cycle arrest in the more radiosensitive G1 and G2-M phases, suppressing growth,
radiation-induced DNA damage repair, and tumor angiogenesis (Huang & Harari, 2000).
Meanwhile, although EGFR tyrosine kinase inhibitors (TKIs) such as erlotinib enhance the
radiation response in preclinical models (Chinnaiyan et al., 2005), their clinical role in
radiosensitization is not settled. It has been speculated that the radiosensitizing effect may
differ between different classes of EGFR inhibitors (Baumann et al., 2007). EGFR tyrosine
kinase inhibition alone may be inadequate to overcome radioresistance.
For bladder cancer, gefitinib, an EGFR specific tyrosine kinase inhibitor, was shown to
enhance the growth inhibition of bladder cancer by ionizing radiation. In in vitro model,
gefitinib has moderate in vitro radiosensitizing activity in bladder cancer cells, and there is a
differential response in different bladder cancer cell lines, probably related to EGFR
expression (Dominguez-Escrig, Kelly, Neal, King, & Davies, 2004). In in vivo model,
gefitinib has at best marginal in vivo radiosensitizing activity in bladder cancer cells, based
on single literature figure (Colquhoun, McHugh, Tulchinsky, Kriajevska, & Mellon, 2007),
and the effect of combination therapy lasts only for the duration of gefitinib administration.
One strategy to increase the radiosensitization in urinary bladder cancer treatment is to
utilize new-generation ErbB family inhibitors which have more potent activity against EGFR
as well as other ErbB family members such as HER2. Both EGFR and HER2 are key
signaling molecules activated by ionizing radiation in literature (Bowers et al., 2001). It is
also noteworthy that both EGFR and HER2 are important in bladder cancer progression for
their frequent overexpression and association with poor prognosis in bladder cancer
(Lipponen, Eskelinen, Syrjanen, Tervahauta, & Syrjanen, 1991; Neal et al., 1990).
Afatinib (BIBW2992) is an anilino-quinazoline TKI designed to irreversibly bind EGFR
and HER2, and potently suppresses the kinase activity in wild-type and activated mutant cells
(Bean et al., 2008; Li et al., 2008). It is approved as the first-line treatment of patients with
metastatic non-small cell lung cancer (NSCLC) whose tumors have EGFR exon 19 deletions
or exon 21 (L858R) substitution mutations.
However it remains unclear whether new generation EGFR inhibitors with broader
blockade of Erb-B family tyrosine kinase activities like afatinib is superior to first generation
inhibitors (which block EGFR kinase activity alone) in enhancing radiosensitivity of bladder
cancer cells.
1.5 Hypothesis and Experimental Design
In my PhD work, I first used molecular agents targeting EGFR and/or HER2 to
evaluate whether further radiosensitizer experiment is feasible. Meanwhile RTK array was
done to detect the change of signaling pathways after irradiation with or without drugs. In
murine bladder cancer model, I tested the in vitro and in vivo radiosensitizing activity of
afatinib.
In human bladder cancer cells, I used in vitro and in vivo models of to test the
hypothesis that in bladder cancer cells, the concomitant inhibition of EGFR and HER2
tyrosine kinase activity by afatinib has greater radiosensitizing activity than the
inhibition of EGFR tyrosine kinase activity alone by erlotinib. To genetically verify the
role of HER2, we also tested whether the radiosensitizing activity mediated by EGFR
inhibition can be improved by downregulating HER2 expression. Finally I showed evidence
to support that receptor heterodimerization plays an important role in the radiosensitizing
effect of afatinib.
CHAPTER TWO: PILOT STUDY
2.1 Rationale and Approach
Based on literature and clinically available targeted agents, I chose EGFR and HER2
inhibitors to test the potential of their radiosensitizing activity. First I examined the EGFR
mutation status of bladder cancer cells and then performed clonogenic assay to evaluate the
radiosensitizing activity of different targeted agents.
2.2 Materials and Methods
Cell Cultures
The NTUB1 cells were cultured with RPMI1640 medium; T24 cells were cultured with
DMEM medium with 10% fetal bovine serum (FBS). All of the cells were incubated at 37℃
and 5% CO2. The cells were trypsinized by trypsin-EDTA and collected for further studies.
EGFR mutation sequencing
The cDNA of each cell lines was sequenced their EGFR mutation at exon 18-21, the
tyrosine kinase domain of EGFR, by AutoSequencing system. The result sequences were
aligned with wild-type EGFR sequence and checked if there have any mutation on this
region.
Clonogenic assay
The cells for radiotherapy with or without drug treatment were seeded 1000 cells/well in
6-well plates (in triplicates). Drugs with different dosages as 0, 1nM, 10nM, 100nM, 1μM and 10μM, along with different radiation dosages 0, 2.5Gy, 5Gy, 7.5Gy and 10Gy were
tested. Each drug was given three times, namely 24, 48 and 72 hours after the starting time.
Radiation was given 48 hours after starting time. After 7 days in culture, colonies were fixed
with 10% buffered formalin and stained with 2% crystal violet. The number of colonies were
determined and normalized to the number of colonies in controls.
2.3 Results
2.2.1 EGFR mutation analysis
The EGFR mutation status was tested by polymerase chain reaction (PCR) and
sequencing. No mutation was found in exon 18, 19, 20 and 21 of NTUB1 or T24 cells.
2.2.2. Clonogenic assay
For erlotinib, the radiosensitizing activity is not significant. (Fig. 2-1 and Fig. 2-2)
Figure 2-1. Clonogenic assay of erlotinib +/- RT in T24 cells
Figure 2-2. Clonogenic assay of erlotinib +/- RT in NTUB1 cells
For trastuzumab, the radiosensitizing activity is not significant, either. (Fig. 2-3 and Fig. 2-4)
Figure 2-3. Clonogenic assay of trastuzumab +/- RT in T24 cells
Figure 2-4. Clonogenic assay of trastuzumab +/- RT in NTUB1 cells
For lapatinib, there was no effect of lapatinib in both cell lines. (Fig. 2-5 and Fig. 2-6)
Figure 2-5. Clonogenic assay of lapatinib +/- RT in T24 cells
Figure 2-6. Clonogenic assay of lapatinib +/- RT in NTUB1 cells
CHAPTER THREE: RADIOSENSITIZING EFFECT OF AFATINIB IN A MURINE BLADDER CARCINOMA MODEL
3.1 Rationale and Approach
As the mechanism of EGFR-mediated radioprotection may differ in various time
sequence, we will test the radiosensitivity after afatinib pretreatment of bladder cancer cells
in (a) immediate early phase (1-4 h) by immunofluorescence detection of γH2AX foci, (b)
early phase (4-24 h) by flow cytometry analysis, and (c) post double-strand break (DSB)
repair phase (>24 h) by clonogenic assay according to a model of EGFR-mediated
radioprotection (D. J. Chen & Nirodi, 2007). The efficacy of afatinib will also be validated in
mouse xenografts. The advantage of murine bladder cancer model is that the in vivo
experiments can be performed in immunocompetent animals. The tumor graft in mice will be
validated by animal imaging and immunohistochemistry.
3.2 Materials and Methods
Murine bladder tumor cell line
The murine (C3H/HeN) bladder tumor cell line, MBT-2, was obtained from the Japanese
Collection of Research Bioresources (Okayama, Japan). Cells were cultured in RPMI-1640
supplemented with 10% fetal bovine serum and 50 U/ml penicillin/streptomycin. Cells were
cultured at 37 °C in a humidified atmosphere of 5% CO2 and 95% air.
Reagents
Afatinib was purchased from Selleck Chemicals (Houston, TX, USA). For in vitro studies,
stock solutions of afatinib were prepared in dimethyl sulfoxide (DMSO) and diluted in
culture medium containing 10% fetal bovine serum. For in vivo studies, afatinib was
suspended in a vehicle (0.5% methylcellulose [wt/vol] and 0.4% Tween 80 [vol/vol] in sterile
water) for oral administration to C3H/HeN mice bearing xenograft tumors.
Irradiation of cells
MBT-2 cells in culture flasks were irradiated with different doses of radiation, using a 6-MV
photon linear accelerator. The distance from the radiation source to the bottom of the flask
was set at 100 cm.
Colony formation assay
Cells (500/well) were seeded in six-well plates and treated with different doses of radiation
following 30-min pretreatment with various doses of afatinib (200–1000 nM) or DMSO
vehicle. Cells were then cultured for an additional 7 days, after which the number of colonies
(clusters of more than 50 cells) was counted in each well using an inverted phase-contrast
microscope at 100X magnification and photographed. The effect on colony number was
analyzed using CompuSyn software (ComboSyn, Inc., Paramus, NJ, USA).
Western blot analysis
Aliquots of cell lysates containing 90 μg of protein were separated by SDS–PAGE (6–15%
polyacrylamide) and then transferred onto polyvinylidene difluoride membranes and
immunoblotted with various antibodies. Bound antibodies were detected using appropriate
peroxidase-coupled secondary antibodies followed by enhanced chemiluminescence (ECL,
Boehringer Mannheim, Mannheim, Germany). Antibodies to the phospho-HER2 and
phospho-EGFR were obtained from Epitomics, Inc. (Burlingame, CA, USA), EGFR and
HER2 from GeneTex, Inc. (Irvine, CA, USA), poly(ADP-ribose) polymerase (PARP) and
cleaved PARP from Cell Signaling Technology (Danvers, MA, USA), beta-actin from Santa
Cruz Biotechnology (Santa Cruz, CA) and histone variant H2AX, phospho-H2AX (Ser139)
and clone JBW301 from Millipore Corporation (Billerica, MA, USA).
Cell cycle phase analysis
The distribution of cells among the phases of the cell cycle was determined by quantifying
the cellular content of propidium iodide-stained DNA. Cells (106/ml) were treated as
indicated, harvested by centrifugation, stained with propidium iodide (PBS containing 0.5%
Tween 20, 15 μg/ml propidium iodide and 5 μg/ml DNase-free RNase), and analysed using a
Becton Dickinson FACScan flow cytometer equipped with Cell Quest software (Becton
Dickinson Immunocytometry Systems, San Jose, CA, USA).
γH2AX immunofluorescence microscopy
Cells were plated on polylysine-coated coverslips, allowed to attach overnight and exposed to
ionizing irradiation of 2.5 Gy either alone or combined with 100 nM afatinib. After treatment,
cells were incubated for 30 min, washed three times with ice-cold phosphate-buffered saline
(PBS), fixed in 4% formaldehyde/PBS for 30 min, permeabilized in 0.5% Triton X-100 in
PBS for 1 h, blocked in 5% bovine serum albumin for 1 h at room temperature, incubated
with the antibody (fluorescein isothiocyanate [FITC] conjugated anti-phospho-Histone γH2AX [Ser139; 1:1500; Millipore, Billerica, MA, USA]) for 2 h at room temperature in the
dark, washed with PBS and mounted in Vectashield mounting medium containing diamidino-
2-phenylindole (Vector Laboratories, Burlingame, CA, USA). γ-H2AX foci were examined
using a Zeiss Axio Imager A1 fluorescence microscope. In each sample, the number of γ-
H2AX foci per nucleus was counted by focicounter under high power field and the average of
150 nuclei was calculated. The average number of γ-H2AX foci per nucleus represents the
amount of double strand breaks.
In vivo studies
Female C3H/HeN mice (6 weeks of age) were obtained from the National Laboratory Animal
Center and used for ectopic (subcutaneous) xenograft implantation. Body weights were
measured weekly. Mice from each group were sacrificed on day 8. The tumor was fixed in
10% neutral buffered formalin and processed for histopathological and immunohistochemical
evaluations. Tumor volumes were measured with a set of calipers and calculated using a
standard formula: width2 × length/2. All experimental procedures using these mice were
performed in accordance with protocols approved by the National Taiwan University
Institutional Animal Care and Use Committee.
Ectopic tumor model
Ectopic tumors were established by subcutaneous injection of MBT-2 cells (2 × 106) into the
right hind leg of mice. As the tumors became established (mean starting tumor volume = 162
mm3), the mice were randomized into 4 groups to receive the following treatments: (1)
methylcellulose/Tween 80 vehicle; (2) afatinib (10 mg/kg/day of body weight) on day 1–7;
(3) methylcellulose/Tween 80 vehicle plus 15 Gy of radiotherapy on day 4; (4) afatinib plus
radiotherapy. Small animal positron emission tomography/computed tomography (PET/CT)
scans with [18F]-2-fluoro-2-deoxy-D-glucose (FDG) were performed on the 8th day of
treatment. The mice were intravenously injected with 14 MBq (378 Ci) of FDG in saline via
the tail vein.
Irradiation of mice
Mice were immobilized using a customized harness. With the body shielded, the thigh tumor
was irradiated with a half-beam rectangular field. A 6-MV photon beam from a linear
accelerator was used to irradiate the thigh tumor with 15 Gy on day 4.
Histological evaluation
After fixation, tumor tissues were embedded in paraffin blocks and sectioned (5 μm). Tumor
cells were detected in representative stained sections. The expressions of phospho-EGFR
(Cell Signaling Technology, Inc., Danvers, MA, USA) and phospho-HER2 (Abcam PLC,
Cambridge, UK) were evaluated after immunohistochemical staining using specific
antibodies.
Statistical analysis
The tumor volume data satisfied the assumptions of normality and homogeneity of variance
for parametric analysis; thus, group means on day 21 for the ectopic tumor models were
compared with a one-way analysis of variance (ANOVA) followed by Fisher’s least
significant difference method for multiple comparisons. Differences were considered
significant at p < 0.05.
3.3 Results (Tsai et al., 2013)
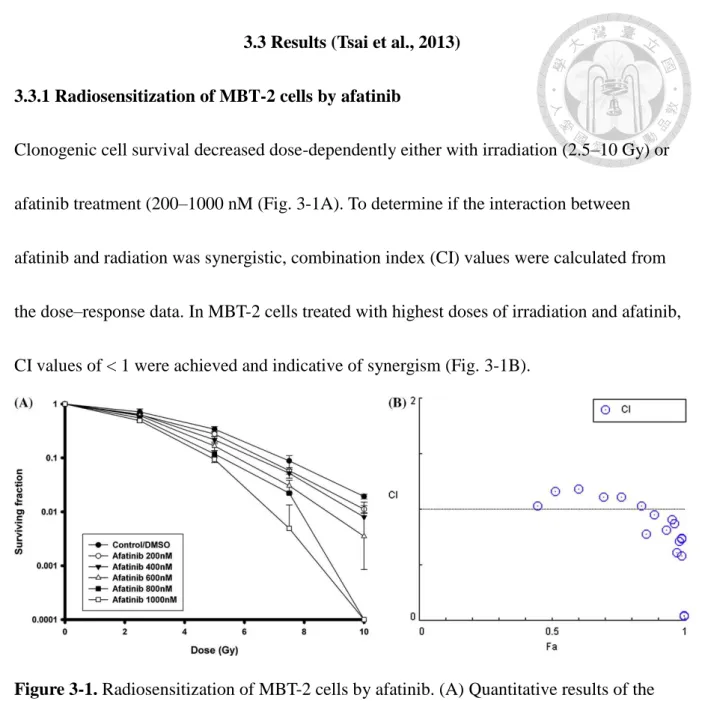
3.3.1 Radiosensitization of MBT-2 cells by afatinib
Clonogenic cell survival decreased dose-dependently either with irradiation (2.5–10 Gy) or
afatinib treatment (200–1000 nM (Fig. 3-1A). To determine if the interaction between
afatinib and radiation was synergistic, combination index (CI) values were calculated from
the dose–response data. In MBT-2 cells treated with highest doses of irradiation and afatinib,
CI values of < 1 were achieved and indicative of synergism (Fig. 3-1B).
Figure 3-1. Radiosensitization of MBT-2 cells by afatinib. (A) Quantitative results of the
clonogenic assays after combination treatment with afatinib and irradiation. Cells were
cultured at a density of 500 cells per well in six-well plates and pretreated with different
doses of afatinib (200–1000 nM for 30 min and then irradiated with different doses (2.5–10
Gy). After 7 days, the cells were fixed, stained and photographed (100X). The images were
used to count the number of colonies containing more than 50 cells in each well. The number
of MBT-2 colonies at each dose level is expressed as a percentage of those in the
corresponding control group. Lines, mean (n = 3); Bars, S.D. (B) CI for each dose level of
irradiation and afatinib were calculated and plotted as a function of the MBT-2 cell fraction
affected (Fa). CI values < 1 indicate synergism.
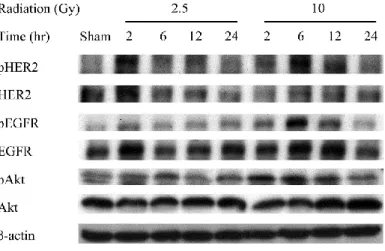
3.3.2 Radiation activates EGFR/HER2 and Akt protein expressions in a time-dependent
manner
It has been reported that receptor tyrosine kinases, such as Erb-B family proteins, are
activated by irradiation. Besides, activation of the PI3K/Akt pathway is associated with
radioresistance. We found in Western blotting assays that levels of both HER2 and EGFR
proteins increased time-dependently, starting at 2 h and 6 h after irradiation in MBT-2 cells
with 2.5 Gy and 10 Gy, respectively (Fig. 3-2). Similarly, the increased expression of
phospho-Akt was induced in a time-dependent manner.
Figure 3-2. Radiation activates EGFR and PI3K/Akt pathways at both low and high doses.
MBT-2 cells were treated with irradiation (2.5 Gy and 10 Gy). Cell lysates were prepared for
Western blotting of the phosphorylation forms of EGFR, HER2 and Akt, and the effects of
irradiation can be seen to unfold in a time-dependent manner.
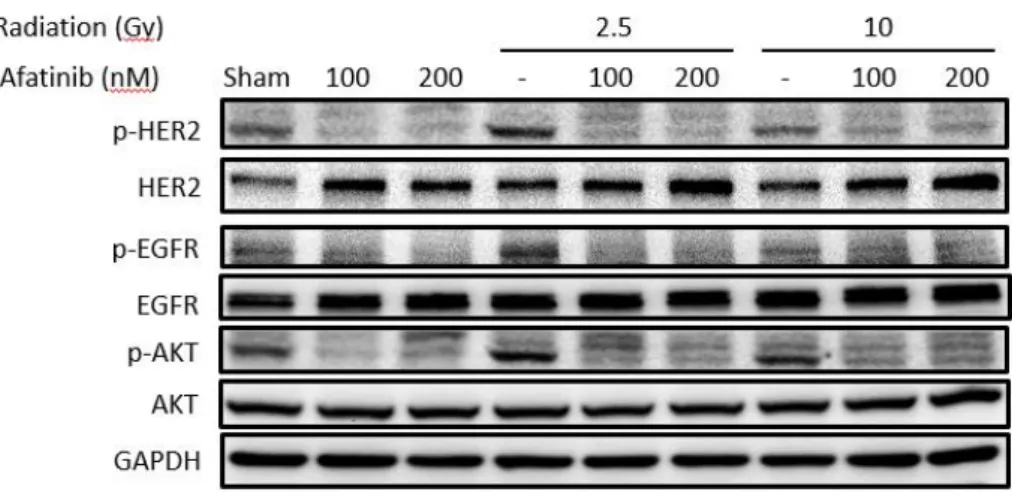
3.3.3 Afatinib inhibits radiation-induced EGFR/HER2 and Akt protein expressions in
MBT-2 cells
Since radiation induces Erb-B family protein expression, we investigated whether the dual
EGFR/HER2 inhibitor, afatinib, can suppress induced expression of these proteins. In MBT-2
cells that received irradiation (either alone or in combination with afatinib), the increased
expression of HER2 and EGFR proteins as well as expression of activated phospho-Akt were
inhibited by afatinib at 6 h (Fig. 3-3).
Figure 3-3. Afatinib inhibits the radiation-activated EGFR phosphorylation and PI3K/Akt
pathways. MBT-2 cells were pretreated with afatinib (100 nM and 200 nM for 24 h and then
irradiated (2.5 Gy and 10 Gy). After 6 h, cell lysates were prepared for Western blotting of phosphorylated forms of EGFR, HER2 and Akt, with β-actin as a loading control.
3.3.4 Afatinib combined with irradiation increases the apoptosis of MBT-2 cells
Our analysis of the cell cycle distribution of MBT-2 cells at 6 h after irradiation (10 Gy) with
or without pre-treatment of afatinib (100 nM, 30 min) revealed that the combination
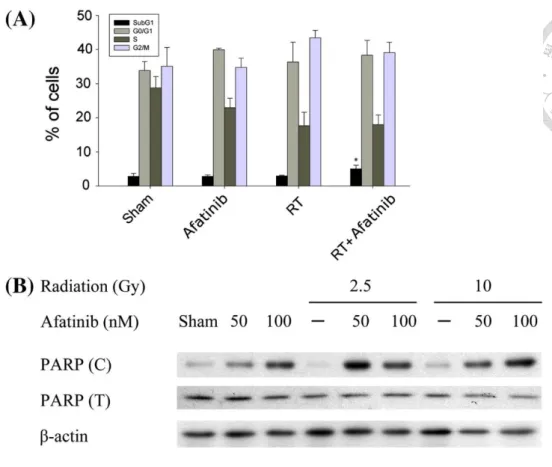
significantly increased the sub-G1 population (p < 0.05), indicating apoptotic cell death (Fig.
3-4A). Radiation alone failed to cause a statistically significant increase in the sub-G1
population, but it did insignificantly increase the proportion of cells in G2/M phase arrest and
insignificantly decrease the proportion of cells in S phase. Afatinib alone, however, did not
cause any significant change in the cell cycle phases. Moreover, Western blot analysis of
cleaved PARP revealed that pretreatment with afatinib strongly increases the expression of
this apoptotic marker in response to irradiation (Fig. 3-4B).
Figure 3-4. Afatinib enhances radiation-induced apoptosis in MBT-2 cells. (A) MBT-2 cells
were pretreated with afatinib (200 nM for 30 min and then with radiation (RT; 10 Gy). The
cell cycle distributions were assessed 6 h after afatinib alone, RT alone and in combination.
Columns, mean (n = 3); Bars, S.D. ∗, p < 0.05. (B) MBT-2 cells were pretreated with afatinib
(50 nM and 100 nM for 30 min and then with RT (2.5 Gy and 10 Gy). After 6 h, cell lysates
were prepared for Western blotting to detect the apoptotic marker PARP (cleavage form).
3.3.5 Afatinib combined with irradiation increased DNA damage of MBT-2 cells
Fig. 3-5A and 3-5B show the result of immunofluorescence staining of γ-H2AX, a marker of
DNA double-strand breaks. While sham-irradiated cells exhibited a minimal number of γ- H2AX foci (0 ± 0.05/cell), radiation alone induced immediate increases in γ-H2AX foci (13.0
± 0.28/cell) that were evident at 30 min as a result of cellular DNA damage. In contrast, treatment with afatinib had no effect on γ-H2AX foci (0 ± 0.03/cell). However, in cells
pretreated with afatinib prior to irradiation, the number of γ-H2AX foci was significantly
increased over that observed after irradiation alone (20.0 ± 0.46/cell versus 13.0 ± 0.28/cell, p
< 0.001). Western blot assay revealed dose-dependent changes in γ-H2AX levels in MBT-2
cells pre-treated with afatinib (50 or 100 nM for 30 min followed by irradiation (2.5 or 10
Gy; Fig. 3-5C).
Figure 3-5. Afatinib enhances radiation-induced DNA damage of MBT-2 cells. (A)
Micrographs (1000X) of γ-H2AX foci, a marker of DNA double-strand breaks, of MBT-2
cells at 30 min after pretreatment with afatinib (100 nM for 30 min and then with radiation (RT; 2.5 Gy) show the 4’,6-diamidino-2-phenylindole (DAPI) staining for cells, FITC for γ-
H2AX (green foci) and the merged images. (B) The number of γ-H2AX foci counted in 150
cells per group. Data presented are the mean number of foci per cell in each group. Columns,
mean; Bars, S.D. ∗, p < 0.05. (C) MBT-2 cells were pretreated with afatinib (50 and 100 nM
for 30 min and then RT (2.5 Gy and 10 Gy). After 30 min, cell lysates were prepared for
Western blotting to detect phospho-γ-H2AX (p-H2AX) and H2AX (loading control).
3.3.6 The combination of afatinib and radiotherapy exhibits an enhanced ability to
control the growth of ectopic MBT-2 xenograft tumors
Daily oral treatment of mice with afatinib (10 mg/kg for 7 days) in combination with
radiotherapy on day 4 suppressed the growth of xenograft tumors to a greater extent than
radiotherapy alone (Fig. 3-6A). Afatinib itself did not satisfactorily control growth. Treatment
with afatinib enhanced radiation-induced suppression of MBT-2 tumor growth by 64%.
One day after the treatment (day 8), thigh tumors were imaged by micro-PET/CT with
18F-FDG. Tumor viability was decreased after combined afatinib and radiotherapy, when
compared to either modality alone or sham treatment (Fig. 3-6B). The treatment with afatinib
alone failed to reduce metabolic tumor volume, but radiotherapy to thigh tumors (15 Gy) by
itself partially reduced tumor size. Importantly, co-treatment with afatinib at 10 mg/kg
significantly improved this radiotherapeutic effect.
The expressions of HER2 and EGFR were assessed immunohistochemically in MBT-2
tumors harvested at 8 days after initiation of treatments. Radiotherapy itself increased the
expressions of both HER2 and EGFR (Fig. 3-6C). Moreover, combined treatment with
radiotherapy and afatinib suppressed radiation-activated expression of HER2 and EGFR in
tumor tissues.
Figure 3-6. Combined afatinib and irradiation (RT) enhances tumor suppressive activity in
ectopic murine bladder tumor model. (A) C3H/HeN mice bearing subcutaneous MBT-2
tumors were randomized into 4 groups (n = 5 in each group) to receive RT alone (15 Gy on
day 4), oral afatinib (10 mg/kg/day from day 1–7) alone, combined afatinib and RT or control
treatment (sham). Data presented are the mean tumor volume for each group measured on the
indicated days. Points, mean; bars, S.D. (B) Mice bearing ectopic MBT-2 tumors were
randomized as in (A) to receive afatinib (10 mg/kg/day) alone, RT alone, combined RT and
afatinib or sham treatment. Mice were scanned by positron emission tomography/computed
tomography to determine tumor metabolism on day 8. Images of representative mice from
each group are shown. Arrows indicate viable right thigh tumors. The standardized uptake
value (SUV) and volume of each tumor were shown on top of the panel. (C) Mice bearing
ectopic tumors were sacrificed on day 8. Microscopic images (200X) of tumor tissue
sectioned and immunohistochemically stained with antibodies against HER2 (left panel) and
EGFR (right panel) from a representative mouse in each group are shown.
CHAPTER FOUR: RADIOSENSITIZING EFFECT OF AFATINIB IN HUMAN BLADDER CANCER MODELS
4.1 Rationale and Approach
Erlotinib (Maemondo et al., 2010) is a first-generation EGFR tyrosine kinase inhibitors
and have well-established efficacy in non-small cell lung cancer patients with EGFR
activating mutations. Afatinib, on the other hand, is a second-generation EGFR tyrosine
kinase inhibitor and found to be of benefit to patients with advanced lung adenocarcinoma
who failed previous gefetinib or erlotinib (Miller et al., 2012). First RTK array was done to
detect the change of signaling pathways after irradiation with or without drugs. Then the
experimental design is similar to murine bladder cancer model and includes: (a) clonogenic
assay, (b) flow cytometry analysis, (c) immunofluorescence detection of γH2AX foci and (d)
animal study. The advantage of human bladder cancer model is that the result may benefit
patients clinically. Finally the mechanism was explored.
4.2 Materials and Methods
Cell lines
The human bladder urothelial carcinoma cell line, T24, was purchased from the American
Type Culture Collection / Bioresource Collection and Research Center (Hsinchu, Taiwan) in
2011. The cells were authenticated in BCRC by short-tandem repeat (STR)-PCR profiling.
They were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal
bovine serum. Another human bladder carcinoma cell line, NTUB1, was established from
human bladder cancer tissue and kindly provided by Dr. Hong-Jeng Yu (Yu, Tsai, Hsieh, &
Chiu, 1992). It was not authenticated. NTUB1 cells were cultured in RPMI-1640 medium
with 10% fetal bovine serum. Both cell lines were incubated at 37°C in a humidified
atmosphere of 5% CO2 and 95% air. We sequenced the cDNA of both cell lines, and none of
the common EGFR mutations was found.
Reagents
Afatinib and erlotinib were purchased from Selleck Chemicals (Houston, TX). For in vitro
experiments, afatinib and erlotinib stock solutions were prepared in DMSO and 50%
acetonitrile, respectively. Both compounds were diluted in culture medium before dosing. For
in vivo experiments, afatinib and erlotinib were suspended in a vehicle (0.5% methylcellulose
[wt/vol] and 0.4% Tween 80 [vol/vol] in sterile water) for oral administration to ICR nude
mice (BioLASCO, Ilan, Taiwan) bearing tumor xenografts.
Irradiation of cells and animals
T24 and NTUB1 cells cultured in flasks were irradiated with various doses of ionizing
radiation, using a 6-MV photon beam from a Siemens Primus linear accelerator (Siemens
Oncology Medical Systems, Inc., Concord, CA). Mice were immobilized using a customized
harness. With the body shielded, the thigh tumor was irradiated with a half-beam rectangular
field. The distance from the radiation source to the bottom of the flask or the thigh tumor of
nude mice was set at 100 cm.
RTK signaling antibody array
The PathScan® RTK signaling antibody array kit from Cell Signaling Technology (Danvers,
MA) contained 39 antibodies against phosphorylated forms of receptor tyrosine kinases or
key signaling proteins. T24 cells were first treated with 100 nM afatinib or erlotinib for 30
min, and then with 10 Gy of radiation. After 24 h of incubation, the cells were processed for
RTK array analysis according to the manufacturer's instructions. The membrane was
developed with LumiGLO® and Peroxide reagent (Cell Signaling Technologies), and RTK
spots were visualized using a UVP imaging system and densitometrically quantified with
ImageProPlus software. Each kinase array dot was manually selected, and an average
intensity for each kinase was calculated. For comparison of different stimulation conditions,
sets were normalized to allow equal intensities of positive controls.
Clonogenic assays
T24 or NTUB1 human bladder cancer cells (1×103 per well) were cultured in 6-well plates,
treated with different doses of radiation following 1-h pretreatment with afatinib or erlotinib
on day 1, re-treated with the drugs on day 2 and day 3 using the same concentrations,
incubated for 7 days, and stained with 0.5% crystal violet (Sigma-Aldrich; St. Louis, MO) in
10% methanol for 30 min at room temperature. Colonies with more than 50 cells were
counted. At each drug concentration, the surviving fraction was determined by dividing the
total number of colonies after irradiation by the number of colonies without irradiation. Each
point on the survival curve represents the mean surviving fraction from 3 independent
experiments.
Cell-cycle analysis
Cell cycle stages were analyzed using a BD FACSCan Flow Cytometer (Becton Dickinson;
Franklin Lakes, NJ). In brief, T24 or NTUB1 bladder cancer cells were pretreated for 30 min
with vehicle, 200 nM afatinib, or 200 nM erlotinib, irradiated (2.5 Gy), incubated 24 h, fixed in 70% ethanol, and stained with a solution containing 50 μg/mL propidium iodide and 0.1
mg/mL RNAase (both from Sigma-Aldrich) in the dark for 30 min. Ten thousand events were
examined for each determination. The relative proportions of cells in different cell cycle
phases were determined using WinMDI software.
Determination of apoptosis with fluorescence microscopy
Apoptotic cells were detected using the annexin V/FITC apoptosis detection kit (AVK050,
Strong Biotech, Taipei, Taiwan) according to the manufacturer’s instructions. The annexin V-
positive cells were examined using a Zeiss Axio Imager A1 fluorescence microscope.
Representative images from different treatment groups were taken into account and at least 50
cells were calculated in every group. The portion of annexin V-positive cells was calculated as
the ratio of positively stained cells divided by the total cell numbers.
Western blotting and immunoprecipitation
Aliquots of T24 and NTUB1 bladder cancer cell lysates containing 50 μg of protein were
separated by SDS-PAGE (8–15% polyacrylamide), and the separated proteins were
transferred to polyvinylidene difluoride (PVDF) membranes and immunoblotted with various
antibodies. For immunoprecipitation experiments, we used the Catch and Release v2.0
Reversible Immunoprecipitation System (Millipore) according to the manufacturer's instructions. The immunoprecipitates (50 μg) of cells were eluted, resolved by 8% SDS-
PAGE, electrotransferred to PVDF membranes and incubated with primary antibodies. For
whole-cell preparations, tumor tissue from individual animals was homogenized with a motor
driven pestle and then lysed in 0.2 ml of RIPA lysis buffer/20 mg tissue. The homogenate was
then centrifuged (13,000 g) for 10 min and the supernatant was used as whole-cell extract.
Bound antibodies were detected using appropriate peroxidase-coupled secondary antibodies
followed by enhanced electrochemiluminescence (Roche Diagnostics; Basel, Switzerland).
The antibodies used were EGFR, HER2, phosphor-Akt (Ser473), caspase-3, PARP, cleaved
PARP (Cell Signaling Technology), phospho-EGFR (pY1086), phospho-HER2 (pY1139)
(Epitomics, Burlingame, CA), and beta-actin (Santa Cruz Biotechnology, Santa Cruz, CA).
Chemical cross-linking
Samples for the cross-linking analysis were obtained at 60 minutes after various treatments.
The cells were washed with PBS three times and incubated for 60 min at room temperature
with 5 mM Suberic acid bis(3-sulfo-N-hydroxysuccinimide ester) sodium salt (Sigma-
Aldrich) in PBS, and the reaction was terminated using 20mM Tris-HCl, pH 8.5 for 15 min at
room temperature. Subsequently, cells were washed with PBS and solubilized in lysis buffer.
The collected proteins were subjected to western blotting using EGFR or HER2 antibody.
HER2 RNA interference and stable transfection





![HPSH [ 氧化數平衡反應式係數 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)