行政院國家科學委員會專題研究計畫 成果報告
金屬膜表面特性對電極材料附著力的影響研究
計畫類別: 個別型計畫 計畫編號: NSC93-2623-7-009-006- 執行期間: 93 年 01 月 01 日至 93 年 12 月 31 日 執行單位: 國立交通大學電子工程學系暨電子研究所 計畫主持人: 曾俊元 報告類型: 完整報告 處理方式: 本計畫可公開查詢 中 華 民 國 94 年 2 月 17 日國防科技學術合作協調小組研究計畫成果報告
計畫編號 : NSC 93-2623-7-009-006
執行期間 : 93 年 1 月 1 日 至 93 年 12 月 31 日
計畫主持人 : 曾 俊 元
共同主持人 :
執行單位 : 國立交通大學電子工程系所
中華民國 93 年 12 月 31 日
金屬膜表面特性對電極材料附著力的影響研究
摘要
隨著科技進步,能源的發展就更顯重要。二氧化錳為電化學電容 器中極具潛力的電極材料,具有價格低廉、無毒性、安全性高等優點, 但是二氧化錳薄膜與鎳集電板間的附著性不佳,使其充放電循環壽命 衰減,造成應用上的限制。為增加二氧化錳薄膜與鎳金屬板間的附著 性,前半年度研究著重於電鍍條件的最適化,將二氧化錳薄膜電鍍於 鎳金屬板,二氧化錳的合成中,利用三極電解槽結構、醋酸錳為電鍍 液,電鍍於鎳金屬基板上,由循環伏安電性量測、SEM 電子顯微鏡 觀察及 XRD 等材料特性量測,推論其鍍膜最適化條件。本研究於後 半年則將利用前半年所得的最適化條件,將二氧化錳薄膜電鍍於表面 粗操度不同的鎳金屬板,深入研究表面粗操度對電容特性的影響,經 由鎳金屬集電板表面處理過程中,研究鎳金屬表面經處理過後,表面 型態對二氧化錳與鎳金屬板間的附著性及電容特性的影響。此外,本 年度亦引進高比表面積及導電度佳的奈米碳管增加其電極表面粗 度,合成二氧化錳-奈米碳管複合式電極,該電極於掃描速率 5mV/s 的比電容為415F/g,經過 1000 次循環伏安測試後仍保有 79%的電容 值,該複合式電極具有好的電化學循環特性及高比電容,將應用於未 來電化學電容器。第一章 緒論
電 化 學 構 裝 電 容 式 儲 能 元 件 (Electrochemical capacitors 、
ultracapacitors or supercapacitors)擁有優良的化學可逆性、高能量密 度(Power density)、與長使用壽命(Long cycle life)等優點,故可 提供高於介電電容(Dielectric capacitors)的能量密度(Energy density) 與優於電瓶高的功率密度(Power density)的使用壽命,故於未來的 儲能元件中扮演極為重要的角色。超高電容器常被應用各式各樣的攜 帶 或 是 移 動 式 電 器 , 常 見 的 包 括 加 速 時 對 電 瓶 的 均 載 作 用 (Load–leveling function for battery)、啟動電動車及用於行動電話等 電子設備的脈衝能量(Burst-power)產生器等。 過渡金屬氧化物為常見的電化學電容器電極材料,近年以高比電 容電極材料–RuO2廣受研究單位的注目,但其價格高昂且具毒性,故 應用範圍相當有限,在 2000 年時,Goodenough 等人發表以溶膠凝膠 法方式製備完成的MnO2薄膜其比電容高達698F/g,已接近目前電容 值最高的電極材料 RuO2,但 MnO2無論於價格上或化學特性的考量 上,都為一具化學安定性之材料,所以MnO2將可開發成為取代 RuO2 的電化學電容器電極材料。 此外,MnO2 粉體與電極表面的附著力亦影響比電容的高低、循 環特性及其使用壽命,故於本年度計劃中延續前一年度的表面處理對
電容特性的影響,將使用不同的方式製造鎳電極表面的粗糙度,利用
電鍍的方式合成 MnO2 粉體,觀察電鍍 MnO2 粉體與電極間的附著
力,探討附著力的影響,進而製備出具有高比電容值及良好的循環特 性的電化學電容器。
第二章 文獻回顧
2-1 電化學基本原理 電化學(Electrochemistry)反應是一種在電解槽(Electrolytic cell) 中進行的一連串的化學反應,是一種利用外加的電位勢調整電極表面 電 子 能 態 高 低 , 使 電 極 表 面 材 料 亦 即 電 活 性 物 種 (Electroactive species)與電極之間能夠發生電子轉移的化學反應。其主要的研究重 點在於電能與化學反應之間的相互關係。同時探討(1)各種不同電極 上所發生的反應、(2)電極與電位的關係及(3)電池化學反應及電池電 動勢1-3。 電化學反應系統的簡單結構是由外環電路系統、陽極、陰極與電 解質溶液所構成,而電子交換反應是以二種方式來發生,一為電解時 由外加的電位勢提供能量強迫發生,其二則是自發性的發生反應;常 見的電化學反應系統為三極電解槽,其三極分別為工作電極(Working Electrode, WE)、參考電極(Reference Electrode, RE)及對應電極(Counter Electrode, CE),各電極的功能及相互關係如下表 2-1 所列。
當電子從外界獲得能量後,由一電極表面轉移至另一電極表面 時,就會與該電極表面上的材料發生反應,該反應為氧化反應,會使 得該材料由還原態變成氧化態,該電極則稱為陽極。反之,若由氧化
態變為還原態,則為還原反應,該電極則稱為陰極。 表2-1 三極電解槽功能列表 電極種類 功 能 參考電極 (reference electrode) 主要是準確的獲得工作電極上的電位,因此 電極於特定電流範圍下操作時,本身電位需 為一固定值,理想的參考電極的電化學特性 需可逆且穩定。 對應電極 (counter electrode) 相對於工作電極的電極特性;於工作電極上 發生的化學反應通常以不影響工作電極為原 則,白金電極為常見的對應電極。 工作電極 (working electrode) 通常為待測電極,此電極不一定為陽極或陰 極(視反應而定),若電極上發生氧化反應, 此工作電極為陽極;若發生還原反應,則此 工作電極為陰極。 2-2 電極材料 電化學系統的反應速率(電流)及驅動力(電位差)大小決定了 整個電化學反應系統的能量消耗程度,反應電位差需要克服陰極及陽
極的平衡電位,還需考慮電解槽中的電解質、隔膜及外電路間的串並 聯電阻;故電解槽的端電位可以下式表示: ) (t V IR E
Ecell =∆ +ηA+ηC + cell+circuit +∆
其中∆E為熱力學理論計算的電位差值;IRcell+circuit為電解質、膈膜及外 電路電阻造成的電位降大小;ηA及ηc分別為陽極及陰極的反應電位, 但反應電位的大小受電極材料的影響最大,一個良好的電極材料需要 同時具有催化目標反應及降低反應過電位的兩種能力。 於可逆的電子轉移反應中,電極為電子的提供者(Source)或接 受者(Sink),電極本身不會受到反應的影響;但於電催化反應中, 電極亦扮演著觸媒的角色,通常藉由吸附的方式提供一個較低的能 障,及提供新的反應路徑以協助反應的進行。一般來說,電極材料需 具備下列幾個條件4 (1) 高表面積,電催化反應中,電極與介質的接觸面是反應發生的重 要介面,電荷轉移就是在此介面發生,因此電極的表面積大小是 決定反應速率快慢的主要因素,故必須選擇高表面積的多孔性活 性電極(Activated Electrode)來提高電流密度。 (2) 高導電性,導電性會影響端電壓的大小;高導電性電極可以降低 極化阻力來降低過電位;若電阻過大則電場分布情況則會改變, 使得反應的一致性降低。
(3) 物理穩定性,電極材料需具有適當的機械強度、耐浸蝕、不易破 裂及易加工等特性。 (4) 化學穩定性與再現性,電極材料需具有抗腐蝕及防止不必要的氧 化物、氫化物及有機薄膜沉積產生的特性。 (5) 良好的反應速率與選擇性,良好的反應速率及選擇性有助於縮短 反應時間、促進反應進行,並抑制副反應的發生。 (6) 安全性,不論在製作或加工以及應用的過程中,電極材料均要能 夠保有一定的安定性,以提高應用時的安全性。 2-3 金屬氧化物電極種類及製備方法 現今常見的金屬氧化物電極材料多會選擇過渡金屬氧化物,此氧 化物中的過渡金屬多半都擁有半滿或是空的d 軌域,可作為吸附電子 的反應中心,提供較多的軌域給反應中的自由電子。其中最為常見的 為VIIB 族(Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt),像 RuO2、IrO2、Co3O4、
NiCo2O4等金屬氧化物電極。為了提高反應的選擇性、電極穩定性及
促進電化學活性,金屬氧化物電極又會添加或混合其他不同種類的金
屬氧化物做其特性改質的媒介,常見的添加組合有 RuO2+TiO2/Ti、
RuO2+SnO2/Ti、RuO2+IrO2/Ti 等二種成分所組成的金屬氧化物電極,
物電極(Rutile Type)、尖晶石型(Spinel Type)及鈣鈦礦結構(Perovskite
Type),表一為常見的金屬氧化物種類(表 2-2):7,8
表2-2 常見的金屬氧化物種類
金紅石結構 RuO2、IrO2、MnO2、PbO2、PtO2、OsO2、ReO2
尖晶石結構
Co2O3、Fe3O4、MCo2O4(M=Ni,Mn,Cr…)
、MFe2O4(M=Ni,Co,Mn…)
鈣鈦鑛結構
NiM2O4(M=La,Pr,Nd)
、M’1-xSrxMO3(M=Mn,Co,Fe,Ni;M’=La,Nd,x=0,1)
其他結構 NiOx, PdO, TiO
金屬氧化物材料製備的方法非常的多樣化與功能化,多半是以其 製作出來產物的功能性做為考量製成的最大因素,一般可以區分為下 列幾種:(1)熱分解法(Thermal deposition)、(2)電化學法(陽極氧化 法、循環伏安沉積法(Cyclic voltammetric deposition))、(3)溶膠凝膠 法(Sol-Gel process)、(4)化學沉積法(Chemical vapor deposition,
CVD)、(5)濺鍍法(Sputtering)及(6)蒸鍍法(Vaporization deposition)
及其他等方法,以下將各種常見的鍍膜方式及其特性與優缺點分別表 列及簡述如下(表 2-3):
表2-3 氧化物電極的製備方式與特點 製備方式 製 備 特 點 熱分解法9 先將欲鍍膜的基板做去脂、酸洗、蝕刻等前處 理,增加基材表面粗糙度,提高金屬氧化物與基 材間的附著力,再將電極表面塗佈調配好的溶 液,蔭乾後於高溫爐中熱分解,即完成電極的製 備。於熱分解法中熱分解溫度、氧化物的種類及 組成、及分解時的氧分壓都會影電極的特性;熱 分解法的缺點為製備之氧化物通常為無水、結晶 的氧化物。 循環伏安法 將基材前處理後至於欲鍍金屬鍍液中,以循環伏 安方式將金屬氧化物成長於基材上,利用循環伏 安法製備的氧化物通常為含水、無結晶的結構。 陽極氧化法 將金屬電極至於電解液中以陽極處理方式,使金 屬表面形成氧化物;或於電極上先沉積金屬層 後,再陽極氧化處理形成氧化物,使用陽極氧化 處理方法製備的氧化物亦為含水的氧化物。 溶膠凝膠法10,11 溶膠凝膠法可以控制材料的結構、組成及均勻
沉澱物形成後需清洗過濾後於高溫下煆燒,使沉 澱物轉變成含水形式的氧化物粉末,於溶膠凝膠 法中,煆燒溫度為決定最終結晶性與比電容值大 小的重要因素。 無電鍍法12 無電鍍法(Electroless deposition)又稱化學鍍法, 是一種沒有電流流過(無外在驅動力),藉由溶 液中的還原劑於電極表面發生自發性氧化還原 反應,使金屬沉積於電極表面的一種批覆方法。 所以無電鍍法是自催化還原沉積金屬的方法,其 反應通式為:
(
)
0 M 由還原劑提供 ne Mn+ + → 無電鍍法具有下列幾項特點: 1.可於各種材料如金屬、半導體和非導體等電極 上批覆金屬。 2.無論電極幾何形狀複雜與否,凡是接觸到鍍液 的地方都能獲得厚度均勻的鍍層。 3.使用無電鍍法可獲的較厚的鍍層。 4.無須外加電源。 5.鍍層常具有特殊的化學及機械性質。2-4 電化學電容器 電化學電容器(Electrochemical capacitor)又被稱為超電容器 (Supercapacitor),其擁有比傳統介電電容器高出 105 倍以上的高比 電容值,異於現今以電極材料儲存能量的傳統介電電容器(Dielectric capacitor)。 電化學電容器也異於一般的充電電池(鎳氫電池或鋰離子二次電 池)除了具有更高的比電容外,並具有較高的循環壽命及較佳的穩定 性。目前除了用於軍事國防科技、行動通訊、航太事業外,也用於電 腦斷電時的電源供應及交通工具燃料電池啟動及加速時的電源供應 之用。鑑於電化學電容器的重要性,各國都給予了高度重視,成為諸 多先進國家重點的科技發展項目之一。如 1996 年歐盟制定了電動汽
車超級電容器的發展計畫(Development of Super-capacitor for Electric vehicle);美國能源部也制定了發展超級電容器的研究計畫,其近期 (1998~2000)的目標達到 500W/Kg,2003 年以後的目標為達到 1500W/Kg 的比功率,使用壽命希望可以突破一萬次以上。 2-4-1 電化學電容器的分類 根據電化學電容器的運作方式區分,電化學電容器可分為兩種: 13一為電雙層電容器(Double-layer capacitor),電雙層電容器是利用
電及與電解質溶液間的庫倫靜電力造成電荷分離的現象,進而形成電 雙層來達到儲能的目的,當電位改變電及表面的電子能量改變,電雙 層的排列相對改變形成電容器電荷分離的現象,圖 2-1 為電雙層電容 器之示意圖,由於電雙層電容器的電極及電解質溶液之間沒有電荷轉 移,只靠靜電力吸引造成電荷分離來儲存電能,故不會有法拉第電流 產生。目前常見的電雙層電容器電即為多孔碳材料,但其微孔洞大小 分布較寬、結晶度低與導電度較差,導致電容值小。當今奈米碳管 (CNTs)的出現可能可以為電雙層式電容器的出現新的開發契機。 電雙層電容器的電化學程序可以下式表示: 正極 − + − + − ⎯ ⎯ ⎯ ⎯ ← ⎯ ⎯ → ⎯ + A Es A e Es e Disch e Ch // arg arg (1) 負極 + − − + ⎯ ⎯ ⎯ ⎯ ← ⎯ ⎯ → ⎯ + +C e Es C Es e Disch e Ch // arg arg (2) 總反應 + − + − + − + ⎯ ⎯ ⎯ ⎯ ← ⎯ ⎯ → ⎯ + +Es C A Es A Es C Es e Disch e Ch // // arg arg (3) 其中 Es:電極表面積 //:電荷堆積所在的電雙層 A-、C+:電解質中的負、正離子。 如(1)、(2)式所示,充電時電子由外部電源從正極向負極移動,此 時正離子與負離子分開且移動至電極表面;放電時,電子經由負載由 負極向正極移動,而離子由電極表面釋出回到電解質中。於總反應式
(3)中,電解質中的鹽類(C+A-)於充電時會被消耗,故電解質被視為 是一種活性材料,故於充放電過程中,電極表面的鹽類濃度改變、電 解質的導電度及電極表面的電荷密度也跟著改變。 圖 2.1 電雙層電容器示意圖 二為準電容電容器(Pseudocapacitor)14,利用電極與電解質間的 可逆且快速法拉第電荷轉移兩個主要因素來儲存電能,即利用電極表 面批覆的電化學活性物質來進行氧化還原(Redox)或電吸附/脫附 (Electrosorption/deposition)的可逆反應。由於牽涉到電荷轉移反應, 因此電荷的儲存能力遠大於傳統的介電電容器及先前的電雙層電容 器。現以RuO2氧化物電極的氧化還原機構來說明準電容的反應機構: 正極 + − − + + ⎯ ⎯ ⎯ ⎯ ← ⎯ ⎯ → ⎯ e H RuO H HRuO e Ch δ δ δ 2 1 arg 2
負極 1 2 arg arg 2 H e H RuO HRuO e Disch e Ch δ δ δ + − + ⎯ ⎯ ⎯ ⎯ ← ⎯ ⎯ → ⎯ + + 總反應 1 2 1 2 arg arg 2
2 HRuO H RuO H RuO
HRuO e Disch e Ch δ δ + − + ⎯ ⎯ ⎯ ⎯ ← ⎯ ⎯ → ⎯ + 其中 0<δ<1 表 2-4 比較了上述的兩種電化學電容器的各項優缺點。 表2-4 電化學電容器的特性 電雙層電容器 準電容電容器 High-voltage operation, High-power operation, 90o phase angle
Low-voltage operation (Limited by electrochemistry & decomposition voltage of solvent.)
Low or zero ESR for vacuum or
gaseous dielectric Phase angle function of frequency
Finite ESR and frequency dependence of phase angle for electrolytic capacitor
Kinetic limitation for high charge/ discharge rate: hence power limited by kinetic factors
Indefinitely reversible High reversible
Capacitance constant with voltage Capacitance not constant with voltage RuO2 極 H2RuO2 分別表示在完全充放電狀況下的正負電極, H2RuO2 代表完全放電狀態下的電極,在充放電的過程中,質子 (Photon)由一側電極穿過隔離膜(Separator)至另一電極,而電子 則是經由電源或外部負載來傳輸。 法拉第電容與電雙層電容在操作上的不同,在於電極與電解質間
沒有淨離子交換行為(Net ion exchange),也就是在整個充放電過程 中,法拉第電容的電解質濃度始終保持一定,電解質主要是扮演質子 導體(Proton conductor)的角色。故要製造高比電容的電化學電容器, 其關鍵在於發展高比電容的電極材料、具化學穩定性及高鹽濃度的電 解質。 2-4-2 電化學電容器的量測 電容(C)定義為單位電壓(V)所含電荷: V Q C = (1) 於 SI 單位下,電容的單位為法拉(F)、電荷的單位為庫倫 (Coulomb)、電流單位為安培(A)、電壓單位為伏特(V)。電容器
的 量 測 可 藉 由(1)Voltage Step、(2)Current step 及(3)Voltage Ramp
(Potential Sweep)等三種方式來量測,其計算方式如下: (1) Voltage step: 量測電路如圖 2-2 所示,E 為施加電壓、ER為電阻 RS所消耗的電 壓、EC為電容 Cd所消耗的電壓。於任何時間內施加的電壓等於電阻 及電容上電壓的總合,它的表示式如下: C R E E E= + (2) 由歐姆定律知E=iR,所以ER可以表示為
ER =iRS (3) 將(1)和(3)代入(2)得 d s C R C q iR E E E= + = + (4) 而電流 qt dq i= 則 s d s R E C R q dt dq + − = (5) 經整理後 dt R E C R q dq s d s = + − 積分後得 t R E C R q C R s d s d s ⎥= ⎦ ⎤ ⎢ ⎣ ⎡ + − − ln 當t=0、q=0 所以得 ⎥ ⎦ ⎤ ⎢ ⎣ ⎡ ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − − = d s d C R t EC q 1 exp 則 ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − = d s s R C t R E i exp (6) 故當施加一電壓於電容器上時,我們可知到某時間下的對應電流 值,在以i、t、E、RS代入(6)是可得 Cd值。 (2) Current step: 量測電路如圖 2-3 所示。當 RsCd電路以一固定電流充電,因
∫
= idt q ,i 為一常數。代入(4)得 = +∫
dt C i iR E t d S 0 或 ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ + = d S C t R i E (7) 圖2-4 為(7)式中的 E 對 i 作圖而得,利用其斜率 d C i 就可得電容。(3) Voltage ramp(Potential Sweep):
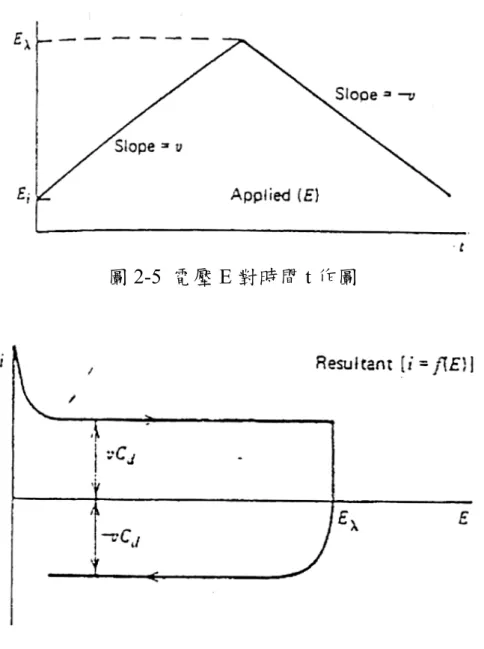
量測電路也與2-1 相同,不同的是給的電壓不是定值而是隨其電位 起始點和時間及掃描速度的乘積合所示: E=Ei+vt (8) 則 d s i C q dt dq R vt E + = + (9)
將(9)式取 Laplace Transform 整理後在 Inverse 回來則得:
⎥ ⎦ ⎤ ⎢ ⎣ ⎡ ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − + = d s d S i d C R t vC R E vC i exp (10) 當t = 0 可得 s i R E ,而隨時間的增加其電流值會成曲線遞減到一個 固定的電流值vCd,由此電流即可知電容值大小。假如將電壓從起始 電壓Ei以固定的電壓掃描速率v 增加至 Eλ後,再將電壓由Eλ以-v 的 掃描速率減少至Ei。其電壓 E 對時間 t 的作圖會是一個等邊三角形如 圖2-5,而電流 i 對電壓 E 作圖會得一長方形如圖 2-6。故我們可由充 放電圖或是CV 圖來判斷該材料是否適合作為電容器應用。
圖2.2 Voltage step 量測電路 圖 2.3 Current step 量測電路 圖2-4 於 current step 量測中 E 對 i 作圖而得,利用其斜率 d C i 就可得 電容。
圖2-5 電壓 E 對時間 t 作圖
圖 2-6 電流 i 對電壓 E 作圖
2-5 錳系氧化物在超電容(電化學電容)上的應用
準電容的電容量主要來自於兩種主要反應:(1)在低電位能沉積 (UPD, Underpotential deposition)反應過程中所產生的吸附/脫附 (Electrosorption/deposition)準電容。(2)不同活性物種的不同氧化態 所能進行的三維(3D)、擬二維(quasi-2D)及二維(2D)氧化還原
反應所產生的氧化還原準電容反應。以吸附/脫附準電容而言,常見 的例子為:氫原子在白金電極上或鉛原子在金電極上的二維 UPD 反 應。然而由於鉑電極的價格太高,故該系統多半只能用於實驗室的規 模而不適用於商業上的電化學電容器,工業與商業的考量下,也試圖 使用其他較低廉的金屬來取代昂貴的鉑電極,但是其他種類的金屬又 不易產生 UPD 的吸附/脫附反應。故開發可產生 UPD 反應且價格便 宜的新一代材料也就成為該電容器的重要課題。因此準電容器的發展 也就偏重於尋找較大氧化還原電位的準電容的電極材料,近年來文獻 指出無水及含水氧化物(如:氧化釕、氧化鎳)都具有相當高的氧化 還原準電容量,其中又以 RuO2最受矚目而備廣泛研究。因 RuO2於 電雙層電容外於其表面上進行可逆的快速氧化還原反應所造成的準 電容,可使其電容量大為增加。除了RuO2外,研究顯示IrO2也具有 準電容特性,能提供高比電容量及可逆性等等。15,16但由於RuO2、IrO2 皆為貴重金屬氧化物,起始原料較為昂貴。因此,其他氧化物如 NiO17-19、CoOx20,21、V2O522及 MnO223的電化學特性開始被研究及探
討,希望能研發出成本低廉的電極材料。
二氧化錳(MnO2)最早是由 Goodenough 等人在 1999 年提出做為準
電容製作材料 24,文中指出 a-MnO2˙nH2O 型材料具有極高的準電容
貴無法商用化的缺點,因此是一個相當有潛力的電化學電容器電極材 料。為達到較高的比電容量及較佳的電容特性,許多研究都著重於如 何製作出較高純度的 a-MnO2˙nH2O 材料,而在許多不同的製備方式 上尋求突破。 目前常用的幾個方式有溶膠凝膠法(Sol-Gel method),利用溶膠凝 膠法所製備的MnO2可提供高達698F/g 的比電容量25,其製備方法為 使用水溶性之氯化錳作為前趨物溶於水中,再加入含OH 基的的化合 物改變溶液的 pH 值,使形成膠體或沉澱的奈米級 MnO2金屬氧化物 粉體顆粒。形成 MnO2 後再以浸泡披覆(Dip-coated)於鎳金屬板上 沉積一層MnO2金屬氧化物薄膜後,完成電極的製作,利用此方法形 成的電極雖然可以提供高達 698F/g 的比電容,但使用計時電位法 (Chronopotentiometry)定電流進行 0.1mA 充放電測試時,其所儲存 的電容於1.1 秒即放電完畢,顯示其電荷儲存能力不佳;這個原因在 於使用溶膠凝膠法製備而得金屬氧化物薄膜電極的導電度較差與電 阻太高導致無法擁有電容器的行為,同時經由實驗也發現,溶膠凝膠 法的製膜方式其於鎳金屬板上的附著性也不佳,對於比電容值也因為 附著性太差的原因而不能有更顯著的提升,無法符合電化學電容器的 需求。
2-6 複合式電極材料
奈米碳管由於具有高化學穩定性、低質量密度、高比表面積及高
導電度等優點,被廣泛的應用於各種儲能元件如二次電池 26、燃料電
池 27 及 準 電 容 28-30。 為 改 善 電 容 特 性 , 複 合 式 電 極 如 activate
carbon-indium tin oxide31, polyaniline-gold nanocluster32及RuO2-VO233
被廣泛的作為準電容電及應用。Park34等人亦提出使用二氧化釕-奈米 碳管複合式電極可提升二氧化釕比電容至900F/g,故過度金屬氧化物 -奈米碳管複合式店及為一具有潛力的準電容電極材料。 利用熱裂解法合成出純二氧化錳再加以塗佈,此一方法雖然改善 了溶膠凝膠法的凝團現象,但是由於結晶型態的二氧化錳導電度並不 高,而使用熱裂解方式雖然可以得到純度極高的二氧化錳粉體,但是 其結晶型態過多,而連帶的使電容特性難以提升。 MnO2 是一個具有商業潛力的亟待開發電化學電容器電極材料, 在製程上的研發改進,可使製程方法趨於簡單化,且能夠改進電化學 特性上的缺點,現今的改進重點在於(1)增加二氧化錳的導電性,這 個重點也就在於如何有效的控制其結晶型態,最好使多晶甚且是完全 非結晶的型態,如此可以增強導電度並提升其準電容特性。(2)改善 層間的附著性,因為較差的附著性將會使電容的表層與底層產生完全
不同的化學反應機制,這樣會使得所能夠達到預期的比電容值大為下 降。(3)增加氧化錳層的表面及底面微孔洞,設法將製程裡可能增加 薄膜表面積,增加店化學反應的表面積。 (4)利用複合式電極材料, 引進奈米碳管的優點,這樣可以在進一步的提升其電容表現,期望可 以在未來得到較高、較佳的電容特性及比電容量。
第三章 實驗步驟與研究方法
鑒於第一、二年度之實驗結果及分析得知:(1)利用溶膠凝膠法所 合成之二氧化錳其存在著凝團現象嚴重、成份組成不易及以及比表面 積不足的缺點。(2)利用熱裂解法合成出的純二氧化錳雖改善凝團現 象,但導電度不高,使電容特性難以提升。(3)二氧化錳粉體與鎳金 屬之表面附著性極差。 (4)溶膠凝膠法或旋轉塗佈的方式均無法有效 的作大面積均勻性的控制。(5)二氧化錳粉體之導電特性不佳導致電 極內外層的電容效應不佳等。針對這些缺失促使我們使用幾個方式, 逐一克服並加以改進二氧化錳的超電容特性。 3-1 二氧化錳薄膜製備及特性研究 3-1-1 陽極電鍍法 針對先前所提之二氧化錳粉體的製備方式,此次我們採用陽極電鍍方 式,嘗試製作非晶相之二氧化錳奈米薄膜。二氧化艋奈米薄膜的製備 如下:使用三極電鍍法,其中鎳基板為工作電極(working electrode)、 白金板為對相電極(counter electrode)及銀/氯化銀(Ag/AgCl)為標準電極(reference electrode) ,於酸鹼值 5.6 的 0.16M 硫酸錳(MnSO4)水溶
液,外加一直流電,二氧化錳薄膜即可電鍍於鎳金屬板上。不同電鍍 條件電鍍的二氧化錳薄膜,利用各種不同的分析工具,觀察其特性間 的相關性,尋求良好的電容特性,在分析所有的製備流程時我們使用
(1) X 光繞射儀(XRD)分析電鍍薄膜的晶相特性。(2)粒徑分析儀 (PDA)分析二氧化錳的粉體幾何大小與分佈。(3)掃描式電子顯微 鏡(SEM)觀察金屬前處理與二氧化錳的附著情形及充放電過程中對 電極附著力的影響。(4)穿透式電子顯微鏡(TEM)觀察所製備之二
氧化錳粉體的結晶型態。(5)循環伏安法及循環伏安計(EG & AG 270A)
分別依序量測不同製成條件的二氧化錳薄膜之循環伏安曲線充放電 性與疲勞度測試。 3-1-2 電極表面前處理 為加強鎳金屬與二氧化錳材料間的表面附著性,我們引進不同的 蝕刻方式,以加強金屬表面的粗糙度,來使得其二者間的潤濕角降 低,並且進一步降低鎳金屬與二氧化錳材料的表面能。這個不同的蝕 刻方式為分別使用不同的有機及無機溶液(DI water、ACE、HCl、 H2SO4、HNO3)作鎳金屬表面的前處理,以改進其表面粗糙度。利用 光學顯微鏡(OM)觀察金屬前處理與二氧化錳的附著情形及充放電 過程中對電極附著力的影響,並使用循環伏安法及循環伏安計(EG & AG 270A)分別依序量測不同表面處理條件的二氧化錳薄膜之循環伏 安曲線充放電性與疲勞度測試。
3-2 使用複合式電極材料增加附著力及電容特性
為加強二氧化錳材料間的電容特性,更進一步將奈米碳管電泳法
電 鍍 於 鎳 基 板 上 , 電 泳 法 所 使 用 的 溶 液 為 0.5 mg CNTs/ 1 ml
dimethylformamide,於直流電壓 20V 下電鍍 5 分鐘,增加金屬表面 的粗糙度及比表面積,再將二氧化錳薄膜使用前半年所得最佳化條件
電鍍於基板表面。分別使用XRD、SEM 、TEM 及 EDX 等分析法觀
察複合電極的材料特性,再用循環伏安法及循環伏安計(EG & AG 270A)量測複合材料電極之循環伏安曲線充放電性(掃描速率 5~100 mV/s)、疲勞度測試與阻抗頻譜分析(振幅 10mV、頻率 10mHz~ 100kHz),觀察奈米碳管電鍍前後對二氧化錳的附著情形及充放電過 程中對電極附著力的影響。本實驗所有的電化學測試均於三極電池結
構下測試,reference electrode 為甘汞電極,counter electrode 為白金電
第四章 實驗步驟與研究方法
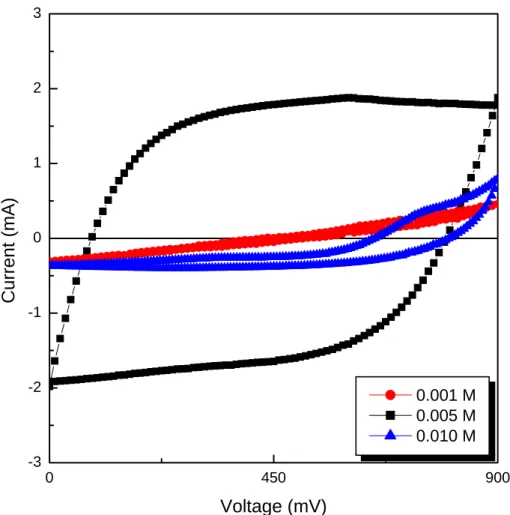
4-1 以電鍍方式製作 MnO2薄膜 二氧化錳粉體除了第一年的溶膠凝膠法及第二年的熱分解法合 成後,塗佈於鎳金屬表面外,使用電鍍法也可有效增加二氧化錳薄膜 與鎳金屬集電板間的附著力,進而增加電化學電容器之電容特性及電 容循環特性。故於本年度前半年的實驗中,嘗試改變電鍍方式,觀察 其循環特性及循環前後電極表面微結構狀態,進而探討電鍍時的參數 與表面附著力之關係。並於下半年度使用不同的表面處理方式,對鎳 金屬表面做表面處理,期望以表面處理的方式,更進一步的增加二氧 化錳薄膜與鎳金屬集電板間的附著力。 4-1-1 電鍍液對 MnO2薄膜的影響 製備二氧化錳薄膜時,電鍍液濃度影響二氧化錳薄膜的成長速 度,進而影響旋轉塗佈二氧化錳薄膜的附著性、厚度及集電板至二氧 化錳薄膜導電性。故本次實驗中,選擇 0.001M、0.005M 及 0.01M 三 種不同濃度的醋酸錳溶液,藉由不同濃度的醋酸錳溶膠凝膠,於鎳金 屬箔上製備二氧化錳薄膜。為了解二氧化錳薄膜與金屬表面的附著性 關係,本次實驗中選擇使用循環伏安法觀察不同方式所鍍之二氧化錳薄膜的電容特性,配合使用掃描式顯微鏡觀察二氧化錳電極於充放電 前後的表面情況,了解醋酸錳電鍍液濃度對二氧化錳薄膜製備時的影 響,及其相對應之電容特性及附著力。 圖 4.1 是循環伏安法觀察使用 0.001M、0.005M 及 0.01M 三種不 同濃度的醋酸錳溶液所鍍的二氧化錳薄膜,由圖 4.1 可看出以濃度 0.005M 製備而得的二氧化錳薄膜之循環伏安圖形較接近方形也就是 形 狀 上 較 接 近 理 想 的 電 容 曲 線 , 且 其 電 容 量 比 醋 酸 錳 溶 液 濃 度 0.001M 及 0.01M 製備而得的較大。形成該電極試片的電容特性差異 在於使用較低濃度的醋酸錳製備的二氧化錳薄膜厚度較薄、均勻度不 佳也具有較多孔洞,使得其電容特性較差;當使用濃度較高的醋酸錳 溶液電鍍製備二氧化錳薄膜,也發現其電容特性不佳,推測其可能原 因應為當鍍液濃度較高,二氧化錳薄膜成長速率較快,導致二氧化錳 薄膜厚度雖然較厚,但薄膜組織鬆散、導電度低,故循環伏安特性差。 再分別使用光學顯微鏡觀察二氧化錳薄膜充放電前表面型態,進 而了解醋酸錳鍍液濃度對附著性的影響。圖4.2 為以不同鍍液濃度製 備的二氧化錳薄膜之光學顯微鏡表面型態,圖 4.2(a)為以醋酸錳鍍液 濃度 0.001M 製備而得的二氧化錳薄膜表面型態,由照片可知此二氧 化錳薄膜表面孔隙較多且薄膜厚度較薄,圖4.2(b)為醋酸錳鍍液濃度 0.005M 製備而得的二氧化錳薄膜,觀察其二氧化錳薄膜表面較為緻
密且其薄膜厚度增加,圖 4.2(c)為醋酸錳鍍液濃度 0.010M 製備而得 的二氧化錳薄膜,觀察其二氧化錳薄膜表面緻密度增加且其薄膜厚度 增加。 圖 4.3 為以二氧化錳薄膜經充放電後之光學顯微鏡表面型態,圖 圖 4.3(a)為以醋酸錳鍍液濃度 0.001M 製備而得的二氧化錳薄膜經充 放電過表面型態,二氧化錳表面有輕微脫落現象,表面孔隙較充放電 前多,圖 4.3(b)為醋酸錳鍍液濃度 0.005M 製備而得的二氧化錳薄膜, 觀察二氧化錳薄膜也相同出現剝落現象,因其薄膜厚度較厚,故剝落 的整體效應不如 0.001M 製備而得的二氧化錳薄膜的薄膜嚴重,圖 4.3(c)為醋酸錳鍍液濃度 0.010M 製備而得的二氧化錳薄膜,觀察二氧 化錳薄膜剝落現象為上述三種條件下最嚴重的。整體而言,以醋酸錳 鍍液濃度 0.001M 製備而得的二氧化錳薄膜因厚度較薄、表面孔隙較 多,二氧化錳薄膜厚度較薄、電容特性不佳,而醋酸錳鍍液濃度0.01M 製備而得的二氧化錳薄膜因沉積速率較高,厚度較厚但組成鬆散、經 充放電循環後表面孔隙較多,故電容特性不佳。故之後的實驗選擇使 醋酸錳鍍液濃度0.005M 作為之後電鍍鍍液濃度。
圖4.1 使用鍍液醋酸錳濃度 0.001M、0.005M 及 0.010M 電鍍製備二 氧化錳薄膜的循環伏安曲線 0 450 900 -3 -2 -1 0 1 2 3 0.001 M 0.005 M 0.010 M Current (mA) Voltage (mV)
圖4.2 使用鍍液醋酸錳濃度(a) 0.001M (b) 0.005M 及(c) 0.010M 電鍍 製備二氧化錳薄膜的光學顯微鏡照片,觀察黏製係數對表面附著力影 (a) (b) (c) 200μm 200μm 200μm
圖4.3 使用鍍液醋酸錳濃度(a) 0.001M (b) 0.005M 及(c) 0.010M 電鍍 製備二氧化錳薄膜於充放電循環後的 SEM 照片,觀察黏製係數對表 面附著力影響 (a) (c) (b) 200μm 200μm 200μm
除了醋酸錳溶液濃度會影響二氧化錳薄膜物理及化學特性外,使 用不同的電鍍溫度,亦會電鍍而得的二氧化錳薄膜的附著性、厚度及 電極的電化學特性。故於電鍍溫度對MnO2薄膜的影響研究中,使用 溫度 3oC、23oC 及 50oC 的醋酸錳溶液,分別於鎳金屬箔上電鍍二氧 化錳薄膜後,以循環伏安法及掃描式電子顯微鏡觀察其電容特性及充 放電前後的表面型態,了解電鍍液溫度對電容特性及附著力的影響。 圖4.4 是循環伏安法觀察使用鍍液溫度 3oC、23oC 及 50oC 的醋酸 錳溶液所鍍之二氧化錳薄膜,由圖 4.4(a)發現以鍍液溫度 3oC 的醋酸 錳溶液所鍍之二氧化錳薄膜,此二氧化錳薄膜幾乎無電容特性,圖 4.4(b)發現以鍍液溫度 23oC 的醋酸錳溶液所鍍之二氧化錳薄膜,其電 容特性呈長方形曲線接近理想電容特性,圖 4.4(c)發現以鍍液溫度 50oC 的醋酸錳溶液所鍍之二氧化錳薄膜,其電容值增加,但整體電 容特性不佳,故以鍍液溫度 23oC 的醋酸錳溶液所鍍之二氧化錳薄膜 具有較佳的電容特性,鍍液溫度過高或過低,都不適合用來沉積二氧 化艋薄膜。 推測鍍液溫度對電容特性的影響,其可能原因為使用鍍液溫度 3oC 的醋酸錳溶液所鍍之二氧化錳薄膜,因鍍液溫度過低的原因,電 鍍時的沉積速率低,故留於鎳金屬箔表面的二氧化錳薄膜則較薄、孔 洞較多,造成電容特性差。而當甲基纖維素重量百分比逐漸增加,二
氧化錳薄膜厚度也逐漸提升,所製備的二氧化錳薄膜逐漸變厚,使電 容特性提升;當鍍液溫度逐漸提升,二氧化錳薄膜沉積速率較快,雖 然二氧化錳薄膜厚度增加,薄膜組成鬆散,但電鍍法合成二氧化錳導 電度不佳,使薄膜可視為內外兩部分,較靠近集電板二氧化錳由於無 法與水溶液接觸,造成電容特性又逐漸下降。 圖 4.5 為以不同渡液溫度製備的二氧化錳薄膜之電子顯微鏡表面 型態,圖 4.5(a)為鍍液溫度 3oC 所鍍的二氧化錳薄膜,此二氧化錳薄 膜表面孔隙多薄膜、厚度較薄且均勻度差,圖4.5(b)及(c)為鍍液溫度 23oC 及 50oC 所鍍之的二氧化錳薄膜,觀察二氧化錳薄膜表面較為緻 密且其薄膜厚度逐漸增加。 圖 4.6 為以二氧化錳薄膜經充放電後之光學顯微鏡表面型態,圖 4.6(a)為使用鍍液溫度 3oC 所鍍的二氧化錳薄膜表面,此二氧化錳薄 膜經充放電過後,二氧化錳表面雖有脫落現象,大致上來說並不嚴 重,圖4.6(b)及(c)為鍍液溫度 23oC 及 50oC 所鍍之的二氧化錳薄膜, 觀察二氧化錳薄膜表面也出現剝落現象,尤以鍍液溫度50 oC 所鍍之 的二氧化錳薄膜特性明顯,因其薄膜厚度較厚,故薄膜剝落較為嚴 重。綜觀各項結果,雖然以鍍液溫度 3oC 所鍍的二氧化錳薄膜,具有 較佳的附著特性,但由於其二氧化錳薄膜厚度較薄,故其電容特性較 差,以鍍液溫度 50oC 所鍍的二氧化錳薄膜,具有較厚的薄膜厚度,
但由於其二氧化錳薄膜厚度增厚,二氧化錳與集電板間的附著能力降
低,故其電容特性較差。故以鍍液溫度 23oC 所鍍的二氧化錳薄膜因
厚度較適當,所形成的二氧化錳薄膜厚度雖不厚,但其電容特性為最
佳,故之後的實驗選擇使用鍍液溫度 23oC 作為電鍍二氧化錳薄膜的
圖4.4 使用鍍液溫度 3oC、23oC 及 50oC 的醋酸錳溶液所鍍之二氧化 錳薄膜循環伏安曲線 0 450 900 -3 -2 -1 0 1 2 3 3 oC 23oC 50oC Current (mA) Voltage (mV)
圖4.5 使用鍍液溫度 (a)3oC (b) 23oC 及(c) 50oC 的醋酸錳溶液所鍍 100μm (a) (b) (c) 200μm 200μm 200μm
圖4.6 使用鍍液溫度 (a)3oC (b) 23oC 及(c) 50oC 的醋酸錳溶液所鍍 之二氧化錳薄膜的光學顯微鏡照片,觀察黏製係數對表面附著力影響 100μm (a) (b) (c) 200μm 200μm 200μm
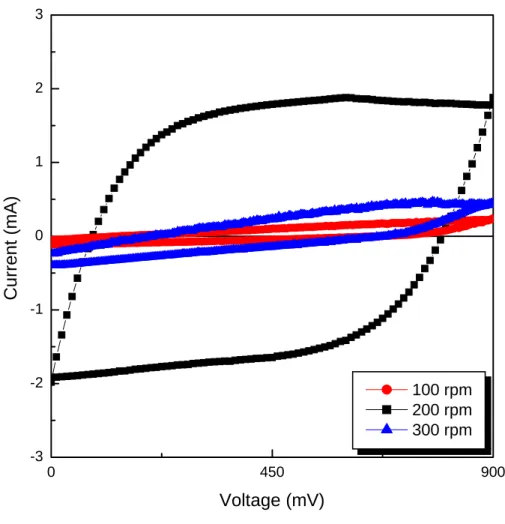
4-1-2 鍍液參數對 MnO2薄膜的影響 將二氧化錳電鍍於鎳金屬箔表面時,除了前述的鍍液溶液特性會 影響影響二氧化錳薄膜的附著力及電容特性外,電鍍時的幾項重要參 數如:電鍍液轉速及電鍍時間亦影響二氧化錳薄膜的附著性、厚度、 集電板至二氧化錳薄膜導電性及薄膜電容特性。故本次實驗中,選擇 電鍍時的鍍液旋轉轉速及電鍍時間,將二氧化錳薄膜利用電鍍的方式 鍍於鎳金屬箔後,觀察二氧化錳薄膜電極的電容特性及表面型態。 圖 4.7 是循環伏安法量測電鍍鍍液旋轉轉速為 100rpm、200rpm 及300rpm 的所鍍二氧化錳薄膜,由圖 4.7 知當電鍍鍍液轉速為 100rpm 所製備出的二氧化錳薄膜電極雖具有電容器雛型,但其電容值小於 200rpm 所製備之薄膜,電容特性又以 200rpm 為極大值,當轉速增至 300rpm 所測得之電容值又隨之縮小。對應觀察電鍍時電極表面的情 形,當鍍液轉速為 100rpm 時,電極表面有許多氣泡附著,由於轉速 過慢電極表面氣泡逐漸累積,二氧化錳薄膜於金屬表面沉積速率不 一,故導致於二氧化錳薄膜均勻度差外,更由於電鍍時表面氣泡過 多,使該二氧化錳薄膜結構鬆散,故於乾燥時以可明顯看出剝落現 象。當轉速過快時,雖可以將附著於電極表面的氣泡去除,但二氧化 錳薄膜於電鍍時受到溶液攪拌時的應力,使該薄膜於金屬表面的附著 力不足,導致製備出來的二氧化錳薄膜厚度較薄、孔洞較多,為電容
特性差的主要因素。 再分別使用光學顯微鏡觀察二氧化錳薄膜充放電前表面型態,進 而了解電鍍時攪拌轉速對附著性的影響。圖4.8 為以不同電鍍渡液攪 拌轉速電鍍製備的二氧化錳薄膜之光學顯微鏡表面型態,圖 4.8(a)為 以轉速為 100rpm 所製備的二氧化錳薄膜,此二氧化錳薄膜表面平整 度較差且厚度較薄,圖4.8(b)及(c)分別為轉速 200rpm 及 300rpm 所製 備的二氧化錳薄膜,二氧化錳薄膜表面隨轉速增加其均勻度增加。 圖 4.9 為以二氧化錳薄膜經充放電後之光學顯微鏡表面型態,由 圖 4.9(a)可知此二氧化錳薄膜經充放電過後,除了轉速為 200rpm 之 二氧化錳表面脫落現象較不明顯外,其他電鍍鍍液轉速所鍍之薄膜皆 有有脫落現象,表面孔隙較充放電前多。故當電鍍鍍液轉速太慢,所 製備的二氧化錳薄膜厚度較薄且其附著力不佳,故此二氧化錳薄膜電 容特性不佳,當電鍍時鍍液轉速過快時,所製備的二氧化錳薄膜表面 孔隙變多及二氧化錳薄膜的附著力較差、也會造成電容特性不佳,故 之後的實驗選擇200rpm 作為此電鍍鍍液攪拌時的最佳旋轉速度。 利用電鍍方法製備二氧化錳薄膜時,除了鍍液攪拌轉速會影響薄 膜特性外,電鍍沉積二氧化錳薄膜時間也會影響薄膜的物理化學特 性。圖4.10 是循環伏安法量測旋轉塗佈時轉速 200rpm 費時 1 分鐘、 3 分鐘及 5 分鍾所製備的二氧化錳薄膜,由圖 4.10 發現以不同旋轉時
間的電極電容器特性隨旋轉時間增加而變好,尤以 200rpm 3 分鍾所 製備之薄膜電容值為最大,當旋轉時間增至5 分鍾所測得之電容值又 隨之縮小。其可能原因為當電鍍時間短時,於集電板上所鍍的二氧化 錳薄膜較薄,造成電容特性較差,當電鍍時間逐漸增加,二氧化錳溶 膠於金屬表面形成一最適化厚度時期電容值達到最大,當旋轉時先再 度增加,該二氧化錳薄膜厚度雖然增加但其附著力不足,導致製備出 來的二氧化錳薄膜電容特性差的主要因素。 為驗證循環伏安結果,使用光學顯微鏡觀察二氧化錳薄膜充放電 前表面型態。圖 4.11 為以不同電鍍時間所製備的二氧化錳薄膜之光 學顯微鏡表面型態,圖4.11(a)、(b) 及(c)為電鍍時間分別為 1 分鐘、 3 分鐘及 5 分鍾所製備的二氧化錳薄膜,觀察其表面型態,當電鍍時 間較短,其薄膜厚度較薄、二氧化錳薄膜表面均勻度差,將電鍍時間 延長,則薄膜厚度逐漸增加,二氧化錳薄膜的內聚力增加,故電極表 面形成裂縫,電極表面的孔洞也逐漸變多。 圖4.12 為以二氧化錳薄膜經充放電後之光學顯微鏡表面型態,由 圖 4.12 可知此二氧化錳薄膜經充放電過後,當所鍍之二氧化錳薄膜 厚度較厚者,表面脫落現象較不明顯外,薄膜厚度減低時則出現表面 剝落現象,表面孔隙較充放電前多。於電容特性優良且電及表面附著 力優良的前提下,之後的實驗選擇電鍍液攪拌轉速200rpm 電鍍時間
圖4.7 電鍍液攪拌轉速 100rpm、200rpm、300rpm 製備二氧化錳薄 0 450 900 -3 -2 -1 0 1 2 3 100 rpm 200 rpm 300 rpm C u rrent (mA) Voltage (mV)
圖4.8 電鍍液攪拌轉速 (a) 100rpm (b) 200rpm (c) 300rpm 製備二氧 化錳薄膜的電極表面之光學顯微鏡照片 (a) (b) (c) 200μm 200μm 200μm
圖4.9 電鍍液攪拌轉速 (a) 100rpm (b) 200rpm (c) 300rpm 製備二氧 (a) 200μm (c) 200μm (b) 200μm
圖4.10 使用電鍍沉積二氧化錳薄膜 1 分鐘、3 分鐘及 5 分鐘製備的 循環伏安曲線 0 450 900 -3 -2 -1 0 1 2 3 1 min 3 min 5 min Current (mA ) Voltage (mV)
圖4.11 使用電鍍沉積二氧化錳薄膜(a)1 分鐘 (b)3 分鐘及(c) 5 分鐘製 (a) (b) (c) 200μm 200μm 200μm
圖4.12 使用電鍍沉積二氧化錳薄膜 (a) 1 分鐘 (b) 3 分鐘及(c) 5 分鐘 製備二氧化錳薄膜循環伏安測試後的電極表面之光學顯微鏡照片 (a) (b) (c) 200μm 200μm 200μm
4-1-3 電鍍最適化薄膜之物性分析 為了解電鍍所得之二氧化錳基本物理特性,可將該方法製備而得 之二氧化錳薄膜置於酒精溶液中,再將該溶液經超音波震盪 30 分鐘 後,將混濁的溶液乾燥以獲得二氧化錳粉末。圖 4.13 使用電鍍方法 製備而得之二氧化錳粉末的 X 光繞射圖譜,由圖上沒有觀察到如熱 分解法所製備之二氧化錳峰,故知經由電鍍方法製備的二氧化錳為非 晶相之二氧化錳粉末。圖 4.14 為該粉末之掃描式電子顯微鏡照片及 其對應之選區繞射結果,由照片上明顯發現使用電解法製備的二氧化 錳為球狀結構,不同於熱分解法製備的長方形二氧化錳粉末形狀,由 照片知球狀之二氧化錳粉體其晶體結構為非晶相之結構,其結果與 X 光繞射圖譜結果相符。
圖4.13 使用電鍍法合成之二氧化錳粉末之 X 光繞射分析圖譜
20
30
40
50
60
70
Ni
Int
e
ns
it
y (
a
.u.)
2
Θ(Degrees)
Ni
4-2 集電板表面處理對電鍍二氧化錳薄膜的影響 由第二年的實驗知集電板的表面處理,的確有助於二氧化錳薄膜 與鎳金屬表面集電板間的附著特性。故於本年度的前半年先使用電鍍 方法,獲得最適化電鍍參數後,利用此最適化之鍍膜參數,探討表面 處理對電鍍二氧化錳薄膜於鎳金屬箔上的附著力及電容特性的影響。 圖4.16 為鎳極電板先經過一般去脂處理後,放入去離子水溶液後 經超音波震盪 20 分鐘後,再使用電鍍方法製備電極層後,分別經過 不退火處理、100oC、200oC 及 300oC 退火處理,以形成的二氧化錳 薄膜。由圖 4.16 發現未經退火處理及 300oC 退火處理之二氧化錳薄 膜其電容特性最差,經過100oC 及 200oC 退火處理的二氧化錳薄膜, 其循環伏安曲線為紡錘形,雖有電容特性但電容值不高。 將二氧化錳薄膜鍍於經去離子水表面處理的鎳箔上,經由不同溫 度退火處理後,再使用光學顯微鏡觀察其表面型態。圖 4.17 分別為 未經退火、100oC、200oC 及 300oC 退火處理的二氧化錳表面附著性 不佳,推測其可能原因為鎳箔表面曾經過去脂處理,但未能將表面油 脂及部分鎳金屬氧化物去除;且薄膜經由 300oC 退火處理後,表面孔 洞分布變多,且出現剝落的現象,推測其可能原因是當加熱溫度較高 時,當二氧化錳薄膜中的水分快速蒸散,使薄膜表面形成裂縫及孔 隙,則薄膜整體附著性降低。
圖 4.18 分別為未經退火、100oC、200oC 及 300oC 退火處理的二 氧化錳經充放電處理後的表面型態,由圖可知無論薄膜經循環伏安充 放電處理後,表面孔洞分布變大,表示二氧化錳薄膜與鎳金屬板間的 附著性差,電極表面的孔洞現象也尤以經300oC 退火處理過後最為嚴 重,推測退火溫度對二氧化錳薄膜的影響,當未退火或退火溫度較低 時,二氧化錳薄膜內部含水量較高,二氧化錳結構過於鬆散導致經由 充放電循環後,二氧化錳薄膜脫落;當退火溫度升至 300oC,二氧化蜢 薄膜的含水量銳減,整體結構破壞導致孔洞生成,附著力下降。 總而言之,於鎳金屬板經由去離子水處理鎳金屬,鎳金屬表面的 仍可保持平整。於電鍍備備二氧化錳薄膜的過程中,基板平坦度越 高,電鍍所得之二氧化錳薄膜上的孔隙越多,且二氧化錳薄膜與鎳極 電板間的附著力也就越差。故使用去離子水處理的鎳電極板,既缺乏 電極與極電板間的附著力,也不具有好的電容特性。
圖 4.16 將去離子水處理的鎳箔表面鍍上二氧化錳薄膜分別經 未經 退火、100oC、200oC 及 300oC 退火處理 的循環伏安曲線 0 450 900 -3 -2 -1 0 1 2 3 RT 100oC 200oC 300oC Current (mA ) Voltage (mV)
圖 4.17 將去離子水處理的鎳箔表面鍍上二氧化錳薄膜分別經 (a)未 (a) (b) (c) (d) 200μm 200μm 200μm 200μm
圖 4.18 將去離子水處理的鎳箔表面鍍上二氧化錳薄膜分別經 (a)未 經退火 (b) 100oC (c) 200oC (d) 300oC 退火處理 及循環伏安測試後的 表面型態 (a) (b) (c) (d) 200μm 200μm 200μm 200μm
將鎳極電板先經過一般去脂處理後,放入去丙酮溶液中經超音波 震盪20 分鐘後,旋轉塗佈製備電極層,分別經過不退火處理、100oC、 200oC 及 300oC 退火處理,以形成的二氧化錳薄膜後,分別進行表面 型態觀察極循環伏安測試。由圖 4.19 發現未經退火處理的二氧化錳 薄膜幾乎沒有電容特性,100oC 及 300oC 退火處理之二氧化錳薄膜則 具有非理想電容圖形的紡錘狀且電容值不高,經過200oC 退火處理的 二氧化錳薄膜循環伏安圖形也成紡錘形電容值仍不高。 將二氧化錳薄膜鍍於經去丙酮表面處理的鎳箔上,經由不同溫度 退火處理後,再使用掃描式電子顯微鏡觀察其表面型態。圖 4.20 分 別為未經退火、100oC、200oC 及 300oC 退火處理的二氧化錳表面, 由照片知經丙酮處理過後的鎳金屬表面與二氧化錳薄膜的附著性增 加,但由圖4.1 知經丙酮處理的表面與去離子水處理過的平坦度佳, 推測其附著性提升的原因在於去脂處理過程中,只使用乙醇清洗過後 表面仍殘留一部分的油脂及其他物質,當去之處理過後的極電板,在 製入丙酮中,則可進一步的清潔,故表面清潔度亦為影響鎳極電板與 二氧化錳薄膜間的附著性。且如前所述,當退火溫度低於200oC,二 氧化錳薄膜表面的孔隙較少,薄膜經由 300oC 退火處理後,表面孔洞 分布變多,則薄膜整體附著性降低,其可能原因為部分存在於 a-MnO2 中的水因退火而逸出,使電鍍的二氧化錳薄膜結構改變,導致孔洞生
成、附著性降低。 圖 4.20 分別為未經退火、100oC、200oC 及 300oC 退火處理的二 氧化錳經充放電處理後的表面型態,由圖可知無論薄膜經循環伏安充 放電處理後,表面孔洞分布大且多,但剝落現象不如表面使用去離子 水處理的電極嚴重,表示使用丙酮處理鎳金屬表面的確有助於增加二 氧化蜢薄膜與鎳金屬集電間的附著性。 總而言之,於鎳金屬板經由丙銅處理鎳金屬,鎳金屬表面平坦度 高。但丙酮有助於更進一步清洗鎳金屬表面,故可提升二氧化錳薄膜 與鎳金屬板的附著性,但由循環伏安曲線可知,其電容特性仍待改進。
圖 4.19 丙酮處理的鎳箔表面鍍上二氧化錳薄膜分別經 未經退火、 0 450 900 -3 -2 -1 0 1 2 3 RT 100oC 200oC 300oC Current (mA ) Voltage (mV)
圖 4.20 經丙酮處理的鎳箔表面鍍上二氧化錳薄膜分別經 (a)未經退 火 (b) 100oC (c) 200oC (d) 300oC 退火處理 的表面型態 (b) 200μm (c) 200μm (d) 200μm 200μm (a)
圖 4.21 經丙酮處理的鎳箔表面鍍上二氧化錳薄膜分別經 (a)未經退 火 (b) 100oC (c) 200oC (d) 300oC 退火處理 及循環伏安測試後的表面 型態 (a) (b) (c) (d) (a) (b) (c) (d) 200μm 200μm 200μm 200μm
將鎳極電板先經過一般去脂處理後,放入20 % 氯化氫溶液中經 超音波震盪20 分鐘後,旋轉塗佈製備電極層,分別經過不退火處理、 100oC、200oC 及 300oC 退火處理,以形成的二氧化錳薄膜後,分別 進行表面型態觀察極循環伏安測試。由圖4.22 發現無論是未經退火、 100oC、200oC 及 300oC 退火處理的二氧化錳的循環伏安特性或電容 值,都比前述使用去離子水及丙酮清洗者佳,推測其可能原因為鎳金 屬板於酸性溶液中,除了表面被輕微腐蝕,也有效的去除表面鎳金屬 氧化物,使二氧化錳與鎳金屬表面的接觸更好,故有較佳的循環伏安 表現。 將二氧化錳薄膜鍍於經20 %氯化氫溶液表面處理的鎳箔上,經由 不同溫度退火處理後,再使用光學顯微鏡觀察其表面型態。圖 4.23 分別為未經退火、100oC、200oC 及 300oC 退火處理的二氧化錳表面, 由照片知經 20%氯化氫溶液處理過後的鎳金屬表面與二氧化錳薄膜 的附著性增加,故表面粗糙度影響鎳極電板與二氧化錳薄膜間的附著 性。且如前所述,薄膜經由300oC 退火處理後,表面孔洞分布變多, 也可於薄膜表面發現許多的裂縫,則薄膜整體附著性降低。 圖 4.24 分別為未經退火、100oC、200oC 及 300oC 退火處理的二 氧化錳經充放電處理後的表面型態,由圖可知無論薄膜經循環伏安充 放電處理後,表面孔洞分布雖變多,但剝落現象已稍有效改善,表示
使用 20%氯化氫溶液處理鎳金屬表面的確有助於增加二氧化錳薄膜 與鎳金屬集電間的附著性。
總而言之,於鎳金屬板經由20 %氯化氫溶液處理鎳金屬,鎳金屬
表面略有侵蝕孔洞,且可有效清除鎳金屬表面氧化物,增加二氧化錳 薄膜與集電板間的接觸及附著性。
圖4.22 經 20 %氯化氫溶液處理的鎳箔表面鍍上二氧化錳薄膜分別經 未經退火、100oC、200oC 及 300oC 退火處理 的循環伏安曲線 0 450 900 -3 -2 -1 0 1 2 3 RT 100oC 200oC 300oC Current (mA ) Voltage (mV)
圖4.23 經 20 %氯化氫溶液處理的鎳箔表面鍍上二氧化錳薄膜分別經 (a) (d) (c) (b) 200μm 200μm 200μm 200μm
圖4.24 經 20 %氯化氫溶液處理的鎳箔表面鍍上二氧化錳薄膜分別經 (a)未經退火 (b) 100oC (c) 200oC (d) 300oC 退火處理 及循環伏安測試 後的表面型態 (d) (a) (c) (b) (d) 200μm 200μm 200μm 200μm
將鎳極電板先經過一般去脂處理後,放入20 % 硫酸水溶液中經 超音波震盪 20 分鐘後,經電鍍製備二氧化錳電極層,分別經過不退 火處理、100oC、200oC 及 300oC 退火處理,以形成的二氧化錳薄膜 後,分別進行表面型態觀察極循環伏安測試。由圖 4.25 發現無論是 未經退火、100oC、200oC 及 300oC 退火處理的二氧化錳的循環伏安 特性,都較使用去離子水及丙酮清洗者佳,其原因為鎳金屬板於酸性 溶液中,表面被輕微腐蝕,也去除表面鎳金屬氧化物,使二氧化錳與 鎳金屬表面的接觸及附著性更好,故有較佳的電容特性表現。 將二氧化錳薄膜鍍於經20 %硫酸水溶液表面處理的鎳箔上,經由 不同溫度退火處理後,再使用光學顯微鏡觀察其表面型態。圖 4.26 分別為未經退火、100oC、200oC 及 300oC 退火處理的二氧化錳表面, 由照片知經 20%硫酸水溶液處理過後的鎳金屬表面與二氧化錳薄膜 的附著性增加,配合圖 4.1 知經 20 %硫酸水溶液處理的表面有輕微侵 蝕的孔洞存在,且經由電鍍方法製備的二氧化錳薄膜表面亦有孔洞存 在,故表面粗糙度影響鎳極電板與二氧化錳薄膜間的附著性。 圖 4.27 分別為未經退火、100oC、200oC 及 300oC 退火處理的二 氧化錳經充放電處理後的表面型態,由圖可知無論薄膜經循環伏安充 放電處理後,表面孔洞分布雖變多,但剝落現象已有效改善,表示使 用 20%硫酸水溶液處理鎳金屬表面的確有助於增加二氧化錳薄膜與
鎳金屬集電間的附著性。故鎳金屬板經由 20%硫酸水溶液處理鎳金 屬,鎳金屬表面略有侵蝕孔洞,且可有效清除鎳金屬表面氧化物,增 加二氧化錳薄膜與集電板間的接觸及附著性,進而改善循環伏安特 性。
圖4.25 經 20 %硫酸水溶液處理的鎳箔表面鍍上二氧化錳薄膜分別經 0 450 900 -3 -2 -1 0 1 2 3 RT 100oC 200oC 300oC Current (m A) Voltage (mV)
圖4.26 經 20 %硫酸水溶液處理的鎳箔表面鍍上二氧化錳薄膜分別經 (a)未經退火 (b) 100oC (c) 200oC (d) 300oC 退火處理 的表面型態 (a) (b) (d) (c) 200μm 200μm 200μm 200μm
圖4.27 經 20 %硫酸水溶液處理的鎳箔表面鍍上二氧化錳薄膜分別經 (a)未經退火 (b) 100oC (c) 200oC (d) 300oC 退火處理 及循環伏安測試 (a) (d) (c) (b) 200μm 200μm 200μm 200μm
將鎳極電板先經過一般去脂處理後,放入20 %硝酸水溶液中經超 音波震盪 20 分鐘後,旋轉塗佈製備電極層,分別經過不退火處理、 100oC、200oC 及 300oC 退火處理,以形成的二氧化錳薄膜後,分別 進行表面型態觀察極循環伏安測試。由圖4.28 發現無論是未經退火、 100oC、200oC 及 300oC 退火處理的二氧化錳的循環伏安特性為最佳, 推測其可能原因為鎳金屬板於硝酸溶液中,除了表面腐蝕,也有效的 去除表面鎳金屬氧化物,使二氧化錳與鎳金屬表面的接觸更好,故有 較佳的循環伏安表現。 將二氧化錳薄膜鍍於經20 %硝酸溶液表面處理的鎳箔上,經由不 同溫度退火處理後,再使用掃描式電子顯微鏡觀察其表面型態。圖 4.29 分別為未經退火、100oC、200oC 及 300oC 退火處理的二氧化錳 表面,由照片知經 20%硝酸水溶液處理過後的鎳金屬表面與二氧化錳 薄膜的附著性優於使用其他酸性水溶液,由圖4.1 知經 20 %硝酸水溶 液處理的表面粗糙度提升,故鎳極電板與二氧化錳薄膜間的附著性變 好。 圖 4.30 分別為未經退火、100oC、200oC 及 300oC 退火處理的二 氧化錳經充放電處理後的表面型態,由圖可知無論薄膜經循環伏安充 放電處理後,但剝落現象已有效改善,表示使用20%硝酸水溶液處理 鎳金屬表面,大幅增加金屬電極板表面粗操度,的確有助於增加二氧
化錳薄膜與鎳金屬集電間的附著性。 總而言之,於鎳金屬板經由20 %硝酸溶液處理鎳金屬,鎳金屬表 面有明顯侵蝕孔洞,可有效增加二氧化錳薄膜與集電板間的接觸、附 著性及循環伏安特性。故使用硝酸對鎳金屬表面進行表面處理的確有 助於二氧化錳薄膜與鎳金屬板間的附著力,除外電容特性也應而有效 提升。
圖4.28 經 20 %硝酸水溶液處理的鎳箔表面鍍上二氧化錳薄膜分別經 未經退火、100oC、200oC 及 300oC 退火處理 的循環伏安曲線 0 450 900 -3 -2 -1 0 1 2 3 RT 100oC 200oC 300oC Current (m A) Voltage (mV)
圖4.29 經 20 %硝酸水溶液處理的鎳箔表面鍍上二氧化錳薄膜分別經 (a) (b) (d) (c) 200μm 200μm 200μm 200μm
圖4.30 經 20 %硝酸水溶液處理的鎳箔表面鍍上二氧化錳薄膜分別經 (a)未經退火 (b) 100oC (c) 200oC (d) 300oC 退火處理 及循環伏安測試 後的表面型態 (a) (b) (c) (d) 200μm 200μm 200μm 200μm
4-3 奈米碳管-二氧化錳複合式電極材料 圖 4.31 為將奈米碳管電鍍於鎳基板表面的 SEM 照片,由圖上知 碳管的線徑約為20 至 30 奈米,電鍍於基板表面的奈米碳管呈現隨機 分佈,相互交錯於鎳基板表面增加表面粗糙度,基板表面積可使用 BET 比表面積儀計算而得,鎳基板的表面積為 0.7 m2/g、奈米碳管/ 鎳基板的表面積則為14.9 m2/g,表面積增加的原因為高比表面積的奈 米碳管(298.7 m2/g)電鍍增加基板表面積。 圖 4.32 為二氧化錳電鍍於鎳基板及奈米碳管/鎳基板表面的 SEM 照片,由圖可知將二氧化錳薄膜電鍍於鎳基板上得一相對平整的薄膜 表面,其晶粒大小約為25 奈米,其 BET 比表面積為 6.0 m2/g(二氧化 錳比表面積根據二氧化錳質量為58.9 m2/g);將二氧化錳薄膜電鍍於 奈米碳管/鎳基板上得分散良好的奈米二氧化錳粒子,其晶粒大小約 為10~25 奈米,其 BET 比表面積為 20.2 m2/g(二氧化錳比表面積根據 二氧化錳質量為82.0 m2/g)。故將二氧化錳電鍍於奈米碳管/鎳基板可 得一高比表面積的奈米二氧化錳粒子,這樣的二氧化錳薄膜將有助於 電極操作於快速的充放電速率。 圖 4.33 為二氧化錳/鎳基板及二氧化錳/奈米碳管/鎳基板電極經 100 循環伏安測試後的 SEM 照片,由圖可知將二氧化錳薄膜電鍍於 鎳基板上經循環伏安測試後,部分晶粒增加至 45 奈米,而部分的晶
粒及結成更大的二次粒子(如區域 A),其 BET 比表面積降至 3.4 m2/g; 而二氧化錳薄膜/奈米碳管/鎳基板上晶粒成長至 30 奈米,其 BET 比 表面積為19.8 m2/g;由比表面積的改變得之大量的二氧化錳粒子經由 循環伏安測試後由鎳基板表面脫落,二氧化錳薄膜/奈米碳管/鎳基板 仍可保有大部分二氧化錳粒子。故將二氧化錳電鍍於奈米碳管/鎳基 板可得一由於附著力較佳,故可於多次電容測試後仍可保有其電容 特。 圖4.34 為奈米碳管、二氧化錳/奈米碳管的 TEM 照片選區繞射圖 譜,由圖知碳管的平均線徑約為20 奈米,電鍍於奈米碳管/鎳基板的 二氧化錳粒子約為10~25 奈米,經由選區繞射知電鍍的二氧化錳粒子 為非晶相結構;為進一步了解二氧化錳粒子的化學組成,將此二氧化 錳粒子做 EDX 分析(圖 4.34(c)),由圖譜知錳及氧為該粒子的主要組 成,銅及碳的訊號是由 TEM 鍍碳銅網造成,經由該圖譜的分析計算 得Mn:O 為 1:1.7。
圖4.32 (a),(b) 將二氧化錳電鍍於鎳基板表面的 SEM 照片; (c), (d) 將 二氧化錳電鍍於奈米碳管/鎳基板表面的 SEM 照片
200nm
(a)
200nm
(b)
200nm
(c)
200nm
(d)
圖 4.33 經 100 次循環伏安測試後的表面型態(a),(b)二氧化錳/鎳基板;
200nm
(a)
200nm
(c)
200nm
(d)
200nm
(b)
A
圖4.34 (a) 奈米碳管的 TEM 照片 (b)二氧化錳/奈米碳管的 TEM 照片 集選區繞射圖譜 (c) 二氧化錳的 EDX 圖譜
(a)
(b)
CNT
MnO
x 0 5 10 Cu Cu Cu Mn Mn Mn O Inte n s ity (a .u .) Energy (keV) C(c)
圖4.35 為二氧化錳/鎳基板及二氧化錳/奈米碳管/鎳基板的電化學 特性測試。圖 4.35 (a)為循環伏安測試,循環伏安測試操作於室溫、 電解液為 0.1M 硫酸鈉水溶液、電壓範圍 0~1V 及掃描速率 5mV/s, 兩種電極的CV 曲線都接近正方形,也就是說此兩種電極都呈現電容 特性,經計算其平均比電容量分別為233(二氧化錳/鎳基板)及 415(二 氧化錳/奈米碳管/鎳基板)F/g。圖 4.35 (b)為此二電極的定電流放電測 試曲線,平均電容量可經由定電流放電測試的平均斜率計算,經計算 其平均比電容量分別為 241(二氧化錳/鎳基板)及 418(二氧化錳/奈米 碳管/鎳基板)F/g。此二種電化學測試都顯現出具有較大比表面積的二 氧化錳/奈米碳管/鎳基板的電容量都優於二氧化錳/鎳基板。 圖 4.36(a)為二氧化錳/奈米碳管/鎳基板的不同 CV 掃描速率的電 化學特性測試,由圖所知所有的 CV 曲線都呈現電容特性,於 10、20、 40、100 mV/s 掃描速率下,平均電容值為 410、407、403 及 386 F/g, 由此知二氧化錳/奈米碳管/鎳基板複合式電極具有分散良好的奈米極 粉體大小的二氧化錳,提供良好的氧化還原循環特性。圖 4.36(b)為 二氧化錳/鎳基板及二氧化錳/奈米碳管/鎳基板的不同 CV 掃描速率對 應的電容量,二氧化錳/鎳基板的電容量隨掃描速度的增加快速減 少,當掃描速度增加至 100 mV/s,其電容值降至 126 F/g;二氧化錳/ 奈米碳管/鎳基板複合式電極具有良好的電容特性是由於高導電度的
奈米碳管降低電極接觸電阻,使二氧化錳發揮良好的電容特性。 圖4.37 二氧化錳/鎳基板及二氧化錳/奈米碳管/鎳基板的阻抗頻譜 分析曲線,阻抗頻譜分析量測於開路電壓約 300 mV 0.1M 的硫酸鈉水 溶液中,由圖知此二頻譜皆由兩個部份組成,在高頻區頻譜為一弧 狀、而低頻區則呈直線,由於電雙層反應速度比法拉第反應快,故高 頻區顯示了電雙層反應特性,兩電極於高頻時(100k Hz)交實數軸於 0.863(二氧化錳/鎳基板)及 0.347(二氧化錳/奈米碳管/鎳基板)Ωcm2, 皆表現出電阻的特性,高頻時的電阻特性可視為系統的串聯電阻造 成,系統的串聯電阻可由電極材料電阻、電解液電阻及導線電阻造 成,電解液電阻及導線電阻於此二系統中為一定值,故較低的電極電 阻材料為影響串聯電阻大小的主因,猜測其可能原因為將奈米碳管電 鍍於鎳基板上時提供大面積的集電極面積,降低其地接觸電阻,故二 氧化錳/奈米碳管/鎳基板具有較低的串聯電阻。於高頻區域時,電雙 層電容特性由於較大面積及分散良好的二氧化錳奈米粒子造成,故二 氧化錳/奈米碳管/鎳基板的虛數值均高於二氧化錳/鎳基板;於低頻區 域時,法拉第電容特性由活性大的二氧化錳奈米粒子造成,故二氧化 錳/奈米碳管/鎳基板的斜率均高於二氧化錳/鎳基板。 圖4.38 二氧化錳/鎳基板及二氧化錳/奈米碳管/鎳基板的電化學穩 定測試曲線,二氧化錳/鎳基板在 100 及 600 次循環伏安測試後,電
容值降至原先的83 %及 70 %,經由 1000 次循環後更降至原先的 11 %;而二氧化錳/奈米碳管/鎳基板在 100 及 600 次循環伏安測試後,
電容值降至原先的97 %及 91 %,經由 1000 次循環後則保有原先的
79 %,由此知二氧化錳/奈米碳管/鎳基板電極除了具有良好可逆的電 容特性外,也具有足夠的電化學穩定特性。







![HPSH [ 氧化數平衡反應式係數 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)