行政院國家科學委員會專題研究計畫 成果報告
新穎多嵌段功能性高分子材料之合成及應用(3/3) 研究成果報告(完整版)
計 畫 類 別 : 個別型
計 畫 編 號 : NSC 95-2221-E-011-067-
執 行 期 間 : 95 年 08 月 01 日至 96 年 10 月 31 日 執 行 單 位 : 國立臺灣科技大學化學工程系
計 畫 主 持 人 : 廖德章
計畫參與人員: 博士班研究生-兼任助理:黃慶成、陳文祥 碩士班研究生-兼任助理:劉志峰、林繼正
報 告 附 件 : 出席國際會議研究心得報告及發表論文
處 理 方 式 : 本計畫涉及專利或其他智慧財產權,2 年後可公開查詢
中 華 民 國 96 年 10 月 22 日
行政院國家科學委員會補助專題研究計畫 □ 成 果 報 告
□期中進度報告
新穎多嵌段功能性高分子材料之合成及應用
計畫類別:□ 個別型計畫 □ 整合型計畫
計畫編號:NSC 95-2221- E - 011 - 067 -
執行期間:93 年 8 月 1 日至 96 年 10 月 31 日
計畫主持人:廖德章 共同主持人:
計畫參與人員:黃慶成、陳文祥、劉志峰、林繼正
成果報告類型(依經費核定清單規定繳交):□精簡報告 □完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:國立台灣科技大學
中 華 民 國 96 年 10 月 22 日
目錄
摘要… … … . p.3 計畫緣由與目的… … … . p.5 儀器與設備… … … . p.8 實驗… … … . p.9 結果與討論… … … . p.16 結論… … … . p.23 計畫成果自評 … … … . p.25 致謝… … … . p.25 參考文獻 … … … . p.26 反應式、表格、圖… … … . p.30
摘要
本計畫主要在合成新型兩親媒性多嵌段共聚合物,利用三種不同分子量之 二醇化合物(PEG 600, PEG 400 和PTMEG 2000)、兩種聚二胺化合物(D2000和 ED2003)及一種聚二醇化合物(PCL 2000)與2-溴異丁醯溴或2-氯丙醯氯反應,合成 得到巨起始劑,此巨起始劑分別與甲基丙烯酸甲酯(MMA)、甲基丙烯酸三氟乙酯 (TFMA)及五氟苯乙烯(PFS)單體在2,2’-二? 啶及溴化亞銅(或氯化亞銅)為觸媒的條 件下進行原子轉移自由基聚合反應(Atom Transfer Radical Polymerization, ATRP),
如此可得到一系列低分子量分佈之新型兩親媒性多嵌段共聚合物。本計劃在聚合 物中導入親水基團及疏水基團,使其成為兩親媒性多嵌段共聚合物,此新型兩親 媒性多嵌段共聚合物具有極佳的溶解度,在常溫下可溶於氯仿、二氯甲烷、苯、
甲苯、二甲苯、二甲基乙醯胺、? 啶及四氫? 喃等有機溶劑。這些新型兩親媒性 多嵌段共聚合物之熔點介於18.2-23.2℃之間及其數目平均分子量和重量平均分子 量分別最高可達61,500及90,700,廣分布指數介於1.16-1.51之間。這些新型兩親媒 性多嵌段共聚合物在氮氣及空氣之環境下,10%熱重損失溫度分別介於255-355℃
以及254-313℃之間。在 GPC分析圖譜中,可以很明顯的看出此新型兩親媒性多嵌 段共聚合物中並沒有巨起始劑及單體存在。
本 計 畫 主 要 在 合 成 新 型 ATRP 之 巨 起 始 劑 [poly(NBMECl) 和 poly(HNBMECl)],利用原冰片烯甲醇(NBCH2OH) 與 2-氯丙醯氯反應所得之雙官 能基單體(NBMECl)經由開環複分解反應而得到此巨起始劑。經由熱重分析實驗可 得知氫化後之巨起始劑 [poly(HNBMECl) ] (Td10=331oC) 相較於氫化前之巨起始劑 [poly( HNBMECl) ] (Td10=286oC) 有較佳的熱穩定性及較高的熱重損失溫度。新型梳 狀共聚合物 [poly(NBMECl-g-MMA)(Mn=1.41× 105) 和 poly(HNBMECl-g-MMA)
(Mn=1.15× 105)] 藉由結合開環複分解聚合及原子轉移自由基聚合技術成功地被合
成出來。新型巨起始劑及其梳狀共聚合物具有極佳的溶解度,在室溫下可溶於四 氫? 喃、? 啶、1,2-二氯苯、二氯甲烷、氯仿、甲苯、二甲苯、苯、二甲基乙醯胺、
N-甲基-2-? 咯 酮及二甲基甲醯胺等有機溶劑。
關鍵詞:開環複分解聚合,原子轉移自由基聚合,兩親媒性多嵌段共聚合物,巨 起始劑,梳狀共聚合物,親水基團,疏水基團,分子量及其分佈。
英文摘要
Amphiphilic triblock copolymers of methyl methacrylate (MMA)、trifluoroethyl methacrylate (TFMA) and pentafluorostyrene (PFS) were synthesized by atom transfer radical polymerization (ATRP) in organic solvents using a Cu(I)X/2,2’-bipyridine catalyst system (X = Cl or Br) with poly(ethylene glycol) (Y-PEG 600-Y and Y-PEG
400-Y), poly(tetramethylene ether glycol) (Y-PTMEG 2000-Y)(Y = Cl or Br),
poly(propylene oxide) (Br-D2000-Br), poly(propylene oxide- ethylene oxide) (Br-ED2003-Br) and polycaprolactone (Br-PCL2000-Br) macroinitiator. Most of the amphiphilic triblock copolymers exhibited excellent solubility in various solvents such as chloroform, methylene chloride, benzene, toluene, xylene, N,N-dimethylacetamide (DMAc), pyridine, and tetrahydrofuran (THF). The crystalline phase-transition temperatures of the amphiphilic triblock copolymers were determined by differential scanning calorimetry (DSC) and they were in the range of 18.2-23.2 ℃. Amphiphilic triblock copolymers with molecular weight up toMn
= 61,500,Mw
= 90,700 and low polydispersities (Mw
/Mn
= 1.16-1.51) were prepared. The temperatures of 10 % weight loss of these triblock copolymers were in the range of 255-355 ℃ and 254- 313 ℃ in nitrogen and air, respectively. GPC analysis of the triblock copolymers indicated no signal of residual macroinitiator and the monomer in the elution traces.Novel macroinitiators for ATRP, poly(NBMECl) and poly(HNBMECl), were derived from a difunctional monomer NBMECl, which was synthesized from the reaction of 5-(hydroxymethyl)bicyclo[2.2.1]hept-2-ene (NBCH2OH) and 2-chloropropionyl chloride. The highly stable macroinitiator, poly(HNBMECl), indicated better thermalstability than poly(NBMECl). The Td10 value of poly(HNBMECl) (Td10=331oC) is higher than poly(NBMECl) (Td10=286oC). Novel comb-shaped copolymers such as poly(NBMECl-g-MMA)(Mn=1.41× 105) and poly(HNBMECl-g-MMA) (Mn=1.15× 105) were successfully obtained by combination of ROMP and ATRP. Novel macroinitiators and their comb-shaped copolymers showed excellent solubility in THF, pyridine, 1,2-dichlorobenzene, methylene chloride, chloroform, toluene, xylene, benzene, DMAc, NMP, and DMF.
Keyword : Ring-Opening Metathesis Polymerization , Atom Transfer Radical Polymerization , Amphiphilic triblock copolymers , Macroiniator , Comb-shaped copolymers, Hydrophilic group ,Hydrophobic group , Molecular weight and Polydispersity.
計畫緣由與目的
自由基聚合(Radical Polymerization)是工業上廣泛使用來製造高分子材料的 重要方法之一。傳統自由基聚合最主要的問題是在高分子的結構上控制不易,所 得到的高分子聚合物會有較高的分子量及較寬的分子量分佈。這些特徵會明顯的
影響聚合物之物性及機械性質。陰離子聚合技術最早由Szwarc 研究群所發展1) ,
利用此技術可控制高分子鏈、末端官能基以及嵌段共聚合物2-5)。此技術之聚合條
件必須在無水與嚴苛的條件下操作。並且,陰離子聚合技術只適用於某些特定的
單體,若是單體中含有其它反應性官能基,有可能會導致不必要的副反應1-5)。這
些 條 件 及 特 徵 大 大 限 制 了 此 技 術 在 工 業 上 應 用 的 潛 力 。控 制 性 自 由 基 聚 合 (Controlled Radical Polymerization)反應是一種較新的方法來合成聚合物或共聚合
物6-9)。目前已有相當多種方法來控制分子量及功能性末端基團10-31),其中最成功的
技 術 為 近 幾 年 所 發 展 出 來 的 原 子 轉 移 自 由 基 聚 合 (Atom Transfer Radical Polymerization,簡稱ATRP)32,33)技術。這個控制性自由基聚合(Controlled Radical Polymerization) 的方法[原子轉移自由基聚合技術(ATRP)]32,33)能夠適用於相當多的 單體來進行聚合反應,諸如苯乙烯(Styrenes)34-36)、丙烯酸酯(Acrylates) 37,38)以及甲 基丙烯酸酯(Methacrylates)39,40)等。因為原子轉移自由基聚合(ATRP)是一種活性 (Living)控制的自由基聚合,所以聚合物之分子量可藉由反應單體(M)以及反應起始 劑(I)之濃度比例([M]/ [I])來控制,且分子量分佈(Polydispersity)約可控制在低於1.3 的水準。因此,原子轉移自由基聚合(ATRP)提供了精準聚合的技術與方法來合成
各種新材料41)。各種型態的聚合物也可順利的藉由此方法合成出,如線性聚合物、
分枝聚合物,超分枝聚合物及星狀聚合物等。藉由這個製程,聚合物中經由起始
劑導入的末端官能基能被完好的保留下來42,43)。因此,各種不同的起始劑搭配上不
同的功能性末端基團,都將可被導入聚合物中42,43)。
原子轉移自由基聚合(ATRP)技術對於嵌段共聚合物之合成提供了精準的聚合 技術。原子轉移自由基聚合(ATRP)反應可應用於相當多種官能基之單體,例如苯 乙烯(Styrenes)、丙烯酸酯(Acrylates)、甲基丙烯酸酯(Methacrylates)、甲基丙烯醯 胺(Methacrylamide)以及丙烯? (Acrylonitrile)35,41,44,45),其中特別是利用(甲基)丙烯 酸酯(Methacrylates)及(甲基)丙烯醯胺(Methacrylamide)等單體所得聚合物更是有許 多方面的應用46,47),如生物感測器(Biosensors)、穿透膜(Membranes),藥物傳輸系 統(Drug Delivery Systems)、細胞培殖基材(Substrates for Cell Culture)、生物分子隔
離(Isolation of Biomolecules)及酵素活性控制(Enzyme Activity Control)等48-50),因為 它 們 顯 示 出 相 當 特 別 的 溶 液 性 質 , 像 是 微 胞 化 (Micellization) 、 熱 感 應 性 (Thermosensitivity)、pH 感應性(pH Sensitivity)等48-50)。已經工業化的三嵌段共聚合 物,如聚乙二醇-聚丙二醇-聚乙二醇 (PEG-PPG-PEG),是一種非離子性兩親媒性 (Amphiphilic)嵌段共聚合物之界面活性劑,而兩親媒性嵌段共聚合物,已被廣泛地
運用於改變塗料的界面性質、清潔劑、印刷以及製藥等51-59)。改變聚乙二醇及聚丙
二醇之比例使所製備之聚合物能依據不同的需要而產生不同功能,例如洗淨力 (Detergency) 、 分 散 穩 定 化 (Dispersion Stabilization) 、 發 泡 (Foaming) 、 乳 化 (Emulsification) 、 潤 滑 (Lubrication) 等 等51,52)。 其 他 特 別 的 應 用 有 如 製 藥 (Pharmaceutics) 53-56)、生化程序(Bioprocessing) 57)、和分離(Separations) 58,59)等。本 計 畫 利 用 各 種 不 同 分 子 量 之 親 水 性 二 醇 及 二 胺 化 合 物 合 成 新 型 巨 起 始 劑 (Macroinitiator),此巨起始劑再和丙烯酸基或含乙烯基型單體,在存在觸媒的條件 下,藉由原子轉移自由基聚合(ATRP)反應合成出一系列不同鏈段長之新型低分子 量分佈的兩親媒性(Amphiphilic)多嵌段共聚合物(Multi-block Copolymer)。另外,本 計劃更利用環戊二烯與丙烯醇進行狄耳士-阿得爾反應 (Diels-Alder Reaction) 合 成原冰片烯甲醇,原冰片烯甲醇再與2-溴異丁醯溴或2-氯丙醯氯反應,來合成新型 含 原 冰 片 烯 起 始 劑 , 接 著 利 用 開 環 複 分 解 聚 合 (Ring Opening Metathesis Polymerization,簡稱ROMP)技術,將含原冰片烯起始劑聚合形成新型聚原冰片烯 巨起始劑,進一步,再藉由原子轉移自由基聚合(ATRP)技術,進行接枝聚合反應 得到各種新型多嵌段接枝共聚合物高分子材料。本計劃並利用元素分析、傅立葉 轉換紅外線光譜、核磁共振光譜鑑定各種巨起始劑及多嵌段共聚合物材料之結 構。並探討各種巨起始劑及多嵌段共聚合物之溶解特性、固有黏度、分子量及分 子量分佈、熱性質(包括玻璃轉移溫度及熱裂解溫度等)。進一步,可利用掃描式電 子顯微鏡觀察所得多嵌段共聚合物的自組裝性結構。本計劃利用原子轉移自由基 聚合(ATRP)反應技術,其具有操作容易且所使用觸媒之價格便宜的優點,大大提 高了應用上的潛能。因此,執行本計劃所得之各種新型巨起始劑及多嵌段共聚合 物新材料具有相當高的學術及應用價值。
儀器與設備
(1) 紅外線光譜儀(IR),型號為 JASCO IR-9700 測試範圍為 4000-400cm-1,測試 法為利用 KBr 打片測試而得。
(2) 13C 及1H NMR 核磁共振光譜儀,型號為 Bruker DRX-500,測試範圍為 125 MHz 到 500 MHz 之間。所使用的溶劑為重氫氯仿(CDCl3)。
(3) 示差熱分析儀(DSC),型號為 Du pont 9000 DSC system,設定升溫速度為每 分鐘 10℃。
(4) 毛細管熔點測定儀,型號為 Model BüCHI 535。
(5) 元素分析儀(Elemental analzer),型號為 Perkin- Elmer 2400。
(6) 熱重分析儀(TGA),型號為 ULVAC (Sinku-Riko) model 7000,升溫速率為每 分鐘 10℃,氮氣流率為每分鐘 60 立方公分。
(7) 凝膠滲透層析儀(GPC),由五根 Waters (Ultrastyragel) 所構成 [300 × 7.7 mm (guard, 500, 103, 104, 105 Å in a series)],四氫? 喃為移動相,使用 RI 偵測器測 定樣品之分子量,聚苯乙烯(Polystyrene)作為標準品。
實驗:
第一年度
Preparation of PEG Macroinitiators
The Macroinitiators (Cl-PEG 600-Cl and Br-PEG 600-Br) were synthesized as outlined in Scheme 1. A mixture of PEG 600 (9.0 g, 15 mmol) in anhydrous methylene chloride (60 mL) and triethylamine (TEA) (4.6 g, 45 mmol) was charged into a round-bottom flask (150 mL) and maintained on an ice bath and nitrogen atmosphere.
2-Chloropropionyl chloride (9.5 g, 75 mmol) or 2-bromoisobutyryl bromide (17.2 g, 75 mmol) was dissolved in anhydrous methylene chloride (25 mL), then added to the above reaction mixture from a dropping funnel over a period of 1 hr under dry nitrogen;
subsequently the temperature was allowed to rise to room temperature. The reaction was continued under stirring for 24 hrs. After completion of reaction, the solution was filtered then was poured into water to wash three times and extracted with methylene chloride. The organic extracts were washed successively with 1 N HCl, 1 N NaOH and 1 % Na2CO3 solution and extracted by using methylene chloride. The collected organic layer was dried over MgSO4 overnight then filtered. The solvent was removed under reduced pressure. After drying, the pure macroinitiators were obtained (Cl-PEG 600-Cl and Br-PEG 600-Br). Yield: 2.87 g (27 %) for Br-PEG 600-Br. The IR spectrum of
Br-PEG 600-Br exhibited absorptions at 1732 cm
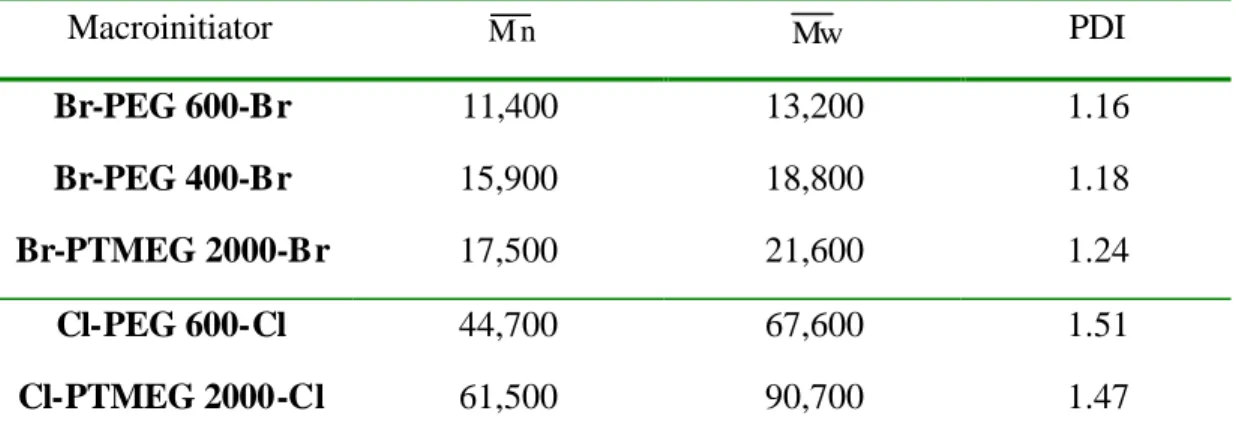
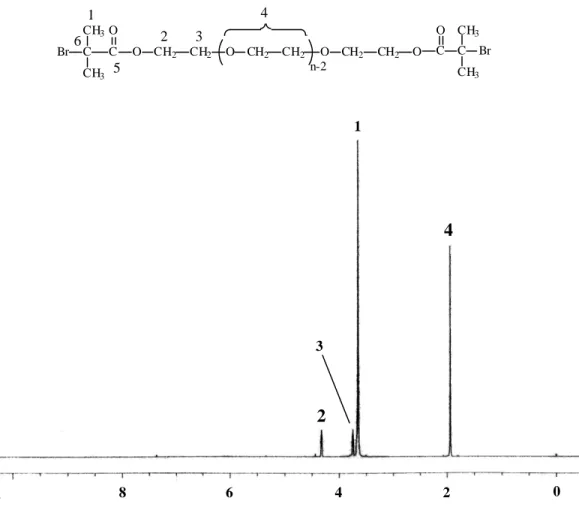
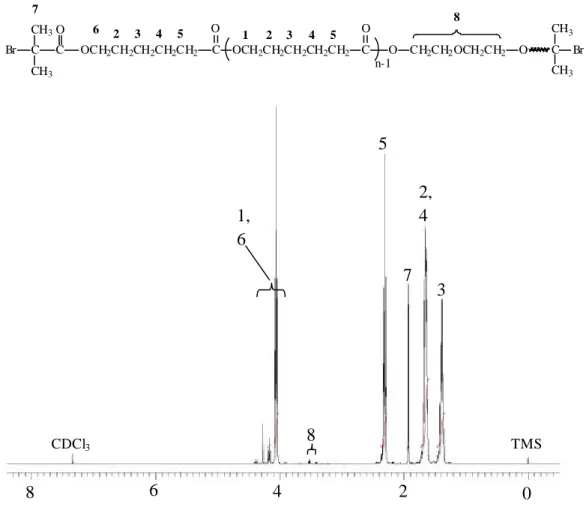
-1 (C=O) and 1103 cm-1 (C-O-C).1H-NMR (500 MHz, CDCl3) of Br-PEG 600-Br: d(ppm)=4.33 (4H, H2), 3.75 (4H, H3), 3.65 (40H, H4), 1.93 (12H, H1); 13C-NMR (125 MHz, CDCl3): d(ppm)=175.25 (C5), 70.34 (C4), 68.44 (C3), 64.83 (C2), 55.47 (C6), 30.48 (C1).
6 5
4 3
2 1
O CH2 CH2 O CH2 CH2 O C O
C CH3
CH3 CH2 Br
CH2 O C O C CH3
CH3 Br
n-2
1H-NMR and 13C-NMR spectrum (CDCl3) of Br-PEG600-Br was shown in
Figure 1 and 2, respectively. Br-PEG 400-Br, Cl-PEG 400-Cl, Br-PTMEG 2000-Br
and Cl-PTMEG 2000-Cl were prepared analogously.Br-PEG 600-Br
Synthesis of Triblock Copolymer with PMMA Segment Derived from Br-PEG 600-Br as a Macroinitiator via ATRP
The synthetic route of
Cl-PMMA-b-PEG 600-b-PMMA-Cl and Br-PMMA-b-PEG 600-b-PMMA-Br is outlined in Scheme 2. To an ampoule,
Cu(I)Br (0.0143 g, 1 × 10-4 mol), 2,2’-bipyridine(bipy)(0.0312 g, 2 × 10-4 mol),Br-PEG 600-Br [molecular weight = 841.79 (Obtained from
1H-NMR, n = 11.94)](0.0842 g, 1 × 10-4 mol) and methyl methacrylate (1 mL, 93.5 × 10-4 mol) were added in 3.5 mL anhydrous xylene. The heterogeneous mixture was frozen and placed under vacuum and the n degassed via a freeze-pump-thaw cycle thrice. After degassing three times, the ampoule was stirred at 90 ℃ for 20 hrs. After completion the reaction and the reaction mixture cool down the ambient temperature, methylene chloride was added to the ampoule and then was passed through a silica column (SiO2) to remove the copper complex. The resulting colorless polymer, Br-PMMA-b-PEG 600-b-
PMMA-Br, was precipitated from methanol, dissolved in CH
2Cl2 and reprecipitated from methanol three times, after which the polymer was collected and dried under vacuum for 1 day at 30 ℃. Yield: 0.5 g (49 %). The IR spectrum ofBr-PMMA-b-PEG 600-b-PMMA-Br exhibited absorptions at 1723 cm
-1 (C=O) and 1141 cm-1 (C-O-C).1H NMR (CDCl3):
δ
(ppm) = 4.18, 3.64, 3.60, 3.45, 1.97-1.81, 1.43, 1.02, 0.84. 13C NMR (CDCl3):δ (ppm) = 178.27-176.86, 70.50, 68.87, 63.54, 53.36-51.16, 45.60,
44.80, 44.46, 18.64, 16.39.Cl-PMMA-b-PEG 600-b-PMMA-Cl, X-PMMA-b-PEG 400-b-PMMA-X and X-PMMA-b-PTMEG 2000-b-PMMA-X (X = Cl and Br) were prepared analogously.
第二年度
Preparation of D2000 Macroinitiators
The Macroinitiators (Br-D2000-Br and Br-ED2003-Br) were synthesized as outlined in Scheme 3. A mixture of D2000 (10.0 g, 5 mmol) in anhydrous methylene chloride (60 mL) and triethylamine (TEA) (2.0 g, 20 mmol) was charged into a round-bottom flask (150 mL) and maintained on an ice bath and nitrogen atmosphere.
2-Bromoisobutyryl bromide (6.9 g, 30 mmol) was dissolved in anhydrous methylene chloride (25 mL), and then added to the above reaction mixture from a dropping funnel over a period of 1 hr under dry nitroge n; subsequently the temperature was allowed to
rise to room temperature. The reaction was continued under stirring for 24 hrs. After completion of reaction, the solution was filtered then poured into water to wash thrice times. The organic extracts were washed successively with 1 N HCl, 1 N NaOH and 1
% Na2CO3 aqueous solution and extracted by using methylene chloride. The collected organic layer was dried over MgSO4 overnight. The solution was filtered, half of the solvent was evaporated, and the PEG macroinitiator was precipitated in cold diethyl ether. The macroinitiator was filtered, washed with cold ether, and dried in vacuum oven at ambient temperature. After drying, the pure macroinitiator was obtained (Br-D2000-Br). Yield: 9.08 g (79 %). The IR spectrum of Br-D2000-Br exhibited absorptions at 1717 cm-1 (C=O) , 3318 cm-1 (N-H) and 1157 cm-1 (C-O-C). The
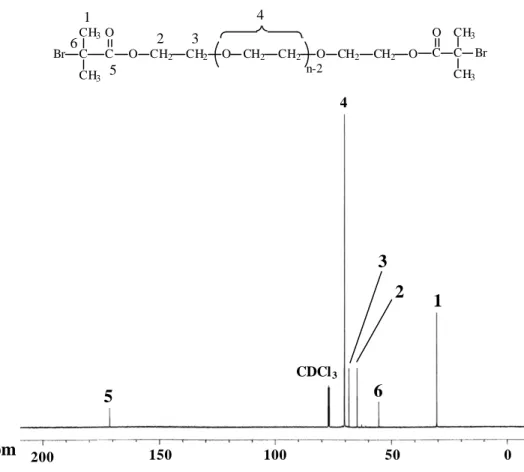
1H-NMR spectrum (500 MHz, CDCl3) of Br-D2000-Br and Br-ED2003-Br was shown in Figure 3. Br-ED2003-Br was prepared analogously.
Preparation of PCL 2000 Macroinitiators
The Macroinitiator (Br-PCL2000-Br) was synthesized as outlined in Scheme 4.
A mixture of PCL2000 (10.0 g, 5 mmol) in anhydrous methylene chloride (50 mL) and triethylamine (TEA) (2.1 mL, 25 mmol) was charged into a round-bottom flask (150 mL) and maintained on an ice bath and nitrogen atmosphere. 2-Bromoisobut yryl bromide (3.1 mL, 25 mmol) was dissolved in anhydrous methylene chloride (20 mL), then added to the above reaction mixture from a dropping funnel over a period of 1 hr under dry nitrogen; subsequently the temperature was allowed to rise to room temperature. The reaction was continued under stirring for 24 hrs. After completion of reaction, the solution was filtered then poured into water to wash thrice times. The organic extracts were washed successively with 1 % Na2CO3 aqueous solution and extracted by using methylene chloride. The collected organic layer was dried over MgSO4 overnight. The solution was filtered, half of the solvent was evaporated, and the PCL macroinitiator was precipitated in methanol. The macroinitiator was filtered and dried in vacuum oven at ambient temperature. After drying, the pure macroinitiator was obtained (Br-PCL 2000-Br). Yield: 5.02 g (43.7 %). The IR spectrum of Br-PCL
2000-Br exhibited absorptions at 1713 cm
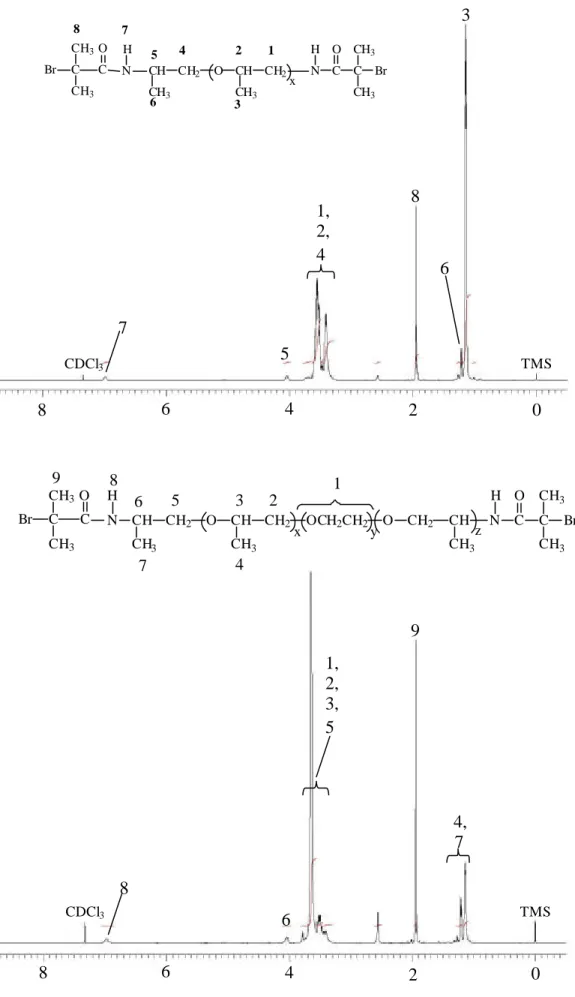
-1 (C=O) and 1101 cm-1 (C-O-C). The1H-NMR spectrum (500 MHz, CDCl3) of Br-PCL2000-Br was shown in Figure 4.
Synthesis of Triblock Copolymer with PMMA Segment Derived from Br-D2000-Br as a Macroinitiator via ATRP
The synthetic route of amphiphilic triblock copolymer (Br-D2000-Br 、
Br-ED2003-Br and Br-PCL2000-Br) is outlined in Scheme 5. To an ampoule,
Cu(I)Br (0.0536 g, 3.74 × 10-4 mol), 2,2’-bipyridine(bipy)(0.1168 g, 7.48 × 10-4 mol),Br-D2000-Br (0.7475 g, 3.74 × 10
-4 mol) and methyl methacrylate (2 mL, 18.69 × 10-3 mol) were added in 4 mL anhydrous toluene. The heterogeneous mixture was frozen and placed under vacuum and then degassed via a freeze-pump-thaw cycle thrice.After degassing three times, the ampoule was stirred at 100 ℃ for 10 hrs. After completion the reaction and the reaction mixture cool down the ambient temperature, tetrahydrofuran (THF) was added to the ampoule and then was passed through a silica column (SiO2) to remove the copper complex. The resulting colorless polymer,
PMMA-b-D2000-b-PMMA, was precipitated from n-hexane, dissolved in THF and
reprecipitated from n-hexane three times, after which the polymer was collected and dried under vacuum for 1 day at 30 ℃. Yield: 0.3623 g (25.8 %). The IR spectrum ofPMMA-b-D2000-b-PMMA exhibited absorptions at 1726 cm
-1 (C=O) and 1165 cm-1 (C-O-C). The 1H NMR (500 MHz, CDCl3) of PMMA-b-D2000-b-PMMA was shown in Figure 5.第三年度
Synthesis of 5-(2-chloropropionate methylene)bicycle[2,2,1]hept- 2-ene (NBMECl)
The monomer NBMECl was synthesized as outlined in Scheme 6. A mixture of NBCH2OH (12.4 g, 0.1 mol) in dry tetrahydrofuran (60 mL), and triethylamine (TEA) (13.1 g, 0.13 mol) was charged into a flask (150 mL) and maintained on an ice bath.The 2-chloropropionyl chloride (13.9 g, 0.11 mol) was dissolved in tetrahydrofuran (5 mL), then added to the above reaction mixture from a dropping funnel over a period of 30 mins, and stirred for another 1 hour. After completion of reaction, tetrahydrofuran was removed under reduced pressure. Obtained dark crude product was purified by column chromatography using methylene chloride as an eluant. Subsequently, the yellowish liquid was distilled under reduced pressure (95–97 ℃ / 3 mmHg) and the final product (NBMECl) was obtained. The ratio of exo/endo-isomer was determined from the 1H NMR spectrum and found to be 20 / 80. ANAL. Calculated for
C11H15O2Cl: C, 61.52 %; H, 7.04 %; found: C, 60.87 %; H, 6.93 %. The spectra agree satisfactorily with the proposed structure. 1H NMR (CDC l3): d (ppm) = 0.53 (Hn6n), 1.13–1.35 (Hx6x, Hx6n, Hn7a, Hx7a, Hx7s) , 1.42 (Hn7s), 1.66 (H10), 1.72 (Hx5), 1.82 (Hn6x), 2.40 (Hn5), 2.68 (Hx1), 2.79 (Hn1), 2.81 (Hx4), 2.86(Hn4), 3.70–3.95 (Hn8), 4.01–4.30 (Hx8),4.36 (H9), 5.90 (Hn3), 6.05 (Hx2,Hx3), 6.13 (Hn2)[Figure 6(A)]. 13C NMR (CDCl3): d (ppm)=21.4 (C10), 28.7 (Cn6), 29.3 (Cx6), 37.5 (Cn5), 37.7 (Cx5), 41.5 (Cx4), 42.1 (Cn1), 43.4 (Cx1), 43.7 (Cn4), 44.8 (Cx7), 49.2 (Cn7), 52.5 (C9), 69.2 (Cn8), 69.8 (Cx8), 131.9 (Cn3), 136.0–136.8 (Cx2, Cx3), 137.5 (Cn2), 169.5 (C11) [Figure 6(B)].
Ring-opening metathesis polymerization of NBMECl
The synthetic route of poly(NBMECl) is outlined in Scheme 7. A solution of catalyst was prepared by dissolving RuCl2(CHPh)[P(C6H11)3]2 (I) (0.0197 g, 2.4 × 10-5 mol) in 0.5 mL of anhydrous methylene chloride under argon- filled dry box. The NBMECl (1.0264 g, 4.8×10-3 mol) was dissolved in 96 mL of methylene chloride and then degassed via a freeze-pump-thaw cycle. After complete degassing, the catalyst solution was injected into the monomer solution by a syringe. The pink solution was vigorously stirred at ambient temperature for 24 hours, and the color changed from pink to yellow. The polymerization reaction was terminated by the addition of a small amount of ethyl vinyl ether (0.8 mL) and stirred for a further 15 mins. After termination, the solution was concentrated to 10 mL and poly(NBMECl) was precipitated in vigorously stirred methanol under nitrogen atmosphere and dried for several hours under vacuum at ambient temperature and gave a flaky white solid.
The spectra agree satisfactorily with the proposed structure. 1H NMR (CDCl3):
δ
(ppm) = 1.15–2.99 (H4, H5, H6, H1, H7, H10), 3.95–4.10 (H8), 4.33 (H9), 5.19–5.33 (H2, H3). 13C NMR (CDCl3):δ (ppm) = 21.49 (C
10), 35.53–44.18 (C4, C5, C6, C1, C7), 52.55 (C9), 67.62 (C8), 129.23–134.90 (C2, C3), 170.00 (C11).Hydrogenation of poly(NBMECl)
The synthetic route of poly(HNBMECl) is outlined in Scheme 7.
Poly(NBMECl) (0.49 g) was dissolved in 20 mL of xylene in an ampoule. To the above solution, 2.98 g (7 equiv. relative to the repeating unit) of p-toluenesulfonhydrazide as a hydrogenation agent and a trace of 2,6-di-tert-butyl-4- methylphenol were added. The ampoule containing the polymer, solvent and hydrogenation age nt was then degassed thrice via a freeze-pump-thaw cycle
and sealed. Subsequently, it was gradually heated to 110 ℃ and stirred for another 12 hours. The solution was cooled to ambient temperature and precipitated in excess methanol. The polymer was purified by dissolving in CH2Cl2 and reprecipitating from methanol. The hydrogenated polymer [poly(HNBMECl)] was dried under vacuum overnight at ambient temperature. The spectra agree satisfactorily with the proposed structure. 1H NMR (CDCl3): δ (ppm) = 0.71–2.25 (H1, H2, H3, H4, H5, H6, H7, H10), 3.94–4.34 (H8), 4.34 (H9). 13C NMR (CDCl3): δ (ppm) = 21.44 (C10), 29.78–43.28 (C1, C2, C3, C4, C5, C6, C7), 52.56 (C9), 67.72–69.48 (C8), 170.11 (C11).
Synthesis of comb-shaped copolynorbornene with PMMA segments derived from poly(NBMECl) as a macroinitiator via ATRP
The synthetic route of poly(NBMECl- g-MMA) is outlined in Scheme 8. To an ampoule, Cu(I)Cl (0.0495 g, 5 × 10-4 mol), 2,2’-bipyridine (0.0781 g, 5×10-4 mol), poly(NBMECl) [poly(NBMECl),
Mn
= 6.93 × 104, PDI = 1.63, Figure 7(A)] (0.1074 g, 5 × 10-4 mol) and methyl methacrylate (1.0000 g, 1 × 10-2 mol) were added in 2 mL anhydrous toluene. The heterogeneous mixture was frozen and placed under vacuum and then degassed via a freeze-pump-thaw cycle thrice. After degassing three times, the ampoule was stirred at 90 ℃ for 30 mins. The polymer, poly(NBMECl-g-MMA), was precipitated from methanol, dissolved in CH2Cl2 and reprecipitated from methanol three times under nitrogen atmosphere. The spectra agree satisfactorily with the proposed structure. 1H NMR (CDCl3): δ (ppm) = 0.82–1.00 (H13), 1.19–2.00 (H6, H7, H10, H12), 2.11–3.10 (H4, H5, H1), 3.40 –3.81 (H14), 3.97–4.11 (H8), 4.35 (H9), 5.21–5.40 (H2, H3). 13C NMR (CDCl3):
δ
(ppm) = 16.45–18.69 (C13), 21.51 (C10), 36.35–44.85 (C4, C5, C6, C1, C7), 51.75 (C14), 52.57 (C9), 54.41 (C12), 67.65 (C8), 128.85–134.95 (C2, C3), 169.97 (C11), 176.92–178.04 (C15).Synthesis of comb-shaped copolynorbornene with PMMA segments derived from poly(HNBMECl) as a macroinitiator via ATRP
The synthetic route of poly(HNBMECl-g-MMA) is outlined in Scheme 9. To an ampoule, Cu(I)Cl (0.0495g, 5×10-4 mol), 2,2’-bipyridine (0.0781g, 5×10-4 mol), poly(HNBMECl) [poly(HNBMECl),
Mn
= 7.95×104, Figure 7(B)] (0.1084g, 5 ×10-4 mol) and methyl methacrylate (1.0000g, 1×10-2 mol) were added in 2 mL anhydrous toluene. The heterogeneous mixture was frozen and was placed under vacuum andthen degassed via a freeze-pump-thaw cycle thrice. After complete of degassing, the ampoule was stirred at 90 ℃ for 30 mins. The polymer, poly(HNBMECl-g-MMA), was precipitated from methanol, dissolved in CH2Cl2 and reprecipitated from methanol three times. The spectra agree satisfactorily with the proposed structure. 1H NMR (CDCl3): δ (ppm) = 0.72–2.50 (H1, H2, H3, H4, H5, H6, H7, H10, H12, H13), 3.40 –3.81 (H14), 3.97–4.11 (H8), 4.35 (H9). 13C NMR (CDCl3):
δ
(ppm) = 16.45–18.69 (C13), 21.45 (C10), 29.80–44.83 (C1, C2, C3, C4, C5, C6, C7), 51.72 (C14), 52.57 (C9), 54.20 (C12), 67.73 (C8), 170.13 (C11), 176.89–178.02 (C15).結果與討論:
第一年度
Synthesis and Characterization of Macroinitiator Br-PEG 600-Br and Cl-PEG 600-Cl
In Scheme 1, the synthesis of Br-PEG 600-Br and Cl-PEG 600-Cl is outlined.These macroinitiators were synthesized by coupling poly(ethylene glycol) to 2-chloropropionyl chloride or 2-bromoisobutyryl bromide. The detail of experiments was presented in Experimental Section. Also, the structure of the macroinitiators was confirmed by IR and NMR spectroscopies. The IR spectrum of Br-PEG 600-Br showed absorption bands at 1723 cm-1 (ester C=O stretching) and 1103 cm-1 (C-O-C stretching vibration), confirmed the presence of poly(ethylene glycol) and ester groups in the structure. The 1H and 13C NMR spectra and assignment of the signals for the macroinitiator Br-PEG 600-Br is depicted in Figure 1 and 2. In 1H NMR the hydroxyl group (-OH) disappears, and the new signals at 4.33 and 1.93 ppm appear for the
Br-PEG 600-Br macroinitiators in CDCl
3 after the esterification. To enable the hydroxyl groups to be completely substituted, the feed molar ratio of 2-chloropropionyl chloride or 2-bromoisobutyryl bromide to PEG was five times. Also, completely quantitative ester substitution was identified by integral values of signals at 4.33, 3.65, and 1.93 ppm of the macroinitiator Br-PEG 600-Br in 1H NMR. From the 1H NMR spectra of PEG 600 and Br-PEG 600-Br, the integral values at 3.65 ppm (peak 4) were around 9.90 and it is nearly the same. The integral value was around 3 at 1.93 ppm (peak 1) and 1 at 4.33 ppm (peak 2) for the macroinitiator Br-PEG 600-Br. So, these evidences can decide that the hydroxyl group is completely substituted and the macroinitiator is a difunctional initiator. The spectra agree satisfactorily with the proposed structure.Synthesis and Characterization of triblock Copolymers
ATRP allowed the preparation of well-defined amphiphilic triblock copolymers via PEG-base and PTMEG-base initiator. The CuBr or CuCl/bipy catalyst system was used in the polymerization of MMA. The choice of the appropriate initiator/CuX (X =Br or Cl) system is a key parameter, when polymerizing monomer such as MMA with high observed propagation rate constants (kpobs = kqKeq according to the terminology defined by Matyjaszewski 60)) to avoid slow initiation (low observed initiation rate kiobs
= kiKeq
compared tokpobs
) and/or possible side reactions.
Solubility
Solubility of various amphiphilic triblock copolymers (X-PMMA-b-PEG
600-b-PMMA-X, X-PMMA-b-PEG 400-b-PMMA-X and X-PMMA-b-PTMEG 2000-b-PMMA-X, X = Cl and Br) was checked in various organic solvents and
presented in Table 1. All the amphiphilic triblock copolymers exhibited excellent solubility in various solvents such as methylene chloride, chloroform, pyridine, benzene, toluene, xylene, N,N-dimethylacetamide (DMAc) and tetrahydrofuran (THF).NMR Analysis
The 1H NMR spectrum of amphiphilic triblock copolymers was obtained after ATRP of macroinitiator (Br-PEG 600-Br) and MMA exhibited signals at 3.60 ppm due to proton of OCH3 group, 1.02 ppm and 0.84 ppm due to proton of PMMA segment, and the 13C NMR spectra also exhibited signals at 51.70 ppm due to carbon of OCH3
groups and 18.64 ppm and 16.39 ppm due to carbon of PMMA segments. This observation confirmed the blocking of macroinitiator with MMA. Because the peak of poly (ethylene glycol) moiety was overlapped the peak of OCH3 of MMA unit in the 1H NMR spectrum, the quantity of MMA incorporated can not calculate.
GPC Analysis
GPC analysis of the purified triblock copolymers indicated no signal of residual
Br-PEG 600-Br macroinitiator and the monomer MMA in the elution traces.
Number-average molecular weight ( M n ), weight-average molecular weight (
Mw
) and polydispersity index (PDI) of the amphiphilic triblock copolymers (X-PMMA-b-PEG 600-b-PMMA-X, X-PMMA-b-PEG 400-b-PMMA-X andX-PMMA-b-PTMEG 2000-b-PMMA-X, X = Cl and Br) were found to be in the range
of 1.14 × 104-6.15 × 104, 1.32 × 104-9.07 × 104 and 1.16-1.51, respectively (Table2).
The system in Br- macroinitiator-Br was more easily controlled polymerization than that in Cl- macroinitiator-Cl. This was indicated that initiation is faster in Br-macroinitiator-Br /CuBr than that in Cl- macroinitiator-Cl /CuBr due to a weaker C-X bond in the former. Although halide exchange occurs relatively rapidly, during the initiation step there is not enough Cu(II) species formed to ensure substantial exchange, and so initiator is consumed before exchange is significantly completed (i.e., ki[M] >
kd[CuII]). 61) Therefore,a fast rate of initiation relative to the rate of propagation is
essential for a successful controlled radical polymerization. GPC curves of macroinitiator (Br-PEG 600-Br) and triblock copolymers, Br-PMMA-b-PEG
600-b-PMMA-Br, were presented in Figure 8. The macroinitiator (Br-PEG 600-Br)
and triblock copolymers showed a unimodal type of GPC curve (A) and (B) in Figure 8, respectively.Thermal Property
The second DSC heating curves of amphiphilic triblock copolymers were shown in Figure 9. The crystalline phase-transition temperatures of the Br-PMMA-b-PEG
600-b-PMMA-Br, Br-PMMA-b-PEG 400-b-PMMA-Br and Br-PMMA-b-PTMEG 2000-b-PMMA-Br were 20.2, 18.2 and 23.2 ℃, respectively. The crystalline
phase-transition temperatures of amphiphilic triblock copolymers increase with increasing repeating unit of ethylene glycol.第二年度
Synthesis and Characterization of Macroinitiator Br-D2000-Br、Br-ED2003-Br and Br-PCL2000-Br
In Scheme 3 and 4, the synthesis of Br-D2000-Br、 Br-ED2003-Br and
Br-PCL2000-Br is outlined. These macroinitiators were synthesized by coupling
poly(propylene oxide) diamine, poly(propylene oxide- ethylene oxide) diamine and polycaprolactone diol to 2-bromo isobutyryl bromide. The detail of experiments was presented in Experimental Section. Also, the structure of the macroinitiators was confirmed by IR and NMR spectroscopies. The IR spectrum of Br-D2000-Br showed absorption bands at 1717 cm-1 (C=O) , 3318 cm-1 (N-H) and 1157 cm-1 (C-O-C), confirmed the presence of poly(propylene oxide) and amide groups in the structure. The1H NMR spectrum and assignment of the signals for the macroinitiator Br-D2000-Br,
Br-ED2003-Br and Br-PCL2000-Br is depicted in Figure 3 and Figure 4. In
1H NMR the amine group (-NH2) disappears, and the new signals at 6.99 and 1.95 ppm appear for the Br-D2000-Br macroinitiators in CDCl3 after the esterification. To enable the amine groups to be completely substituted, the feed molar ratio of 2-bromoisobutyryl bromide to D2000, ED2003 and PCL2000 was five to six times. So, these evidences can decide that the amine group is completely substituted and the macroinitiator is a difunctional initiator. The spectra agree satisfactorily with the proposed structure.Synthesis and Characterization of triblock Copolymers
ATRP allowed the preparation of well-defined triblock copolymers via PPO-base, PPO-PEO-base and PCL-base initiator. The CuBr/bipy catalyst system was used in the polymerization of MMA, TFMA and PFS. The 1H NMR spectrum of triblock copolymers was obtained after ATRP of macroinitiator (Br-D2000-Br) and MMA exhibited signals at 3.60 ppm due to proton of OCH3 group of PMMA segment and 1.02 ppm and 0.83 ppm due to proton of CH2C(CH3) group of PMMA segment. This observation confirmed the blocking of macroinitiator with MMA. Because the peak of poly(propylene oxide) moiety was overlapped the peak of OCH3 and CH2C(CH3) of MMA unit in the 1H NMR spectrum, the quantity of MMA incorporated can not calculate from 1H NMR spectrum.
Solubility
Solubility of various triblock copolymers was checked in various organic solvents. All the triblock copolymers exhibited excellent solubility in various solvents such as methylene chloride, chloroform, benzene, toluene and tetrahydrofuran (THF) at ambient temperature.
GPC Analysis
GPC curves of macroinitiator (Br-D2000-Br) and triblock copolymers,
PMMA-b-D2000-b-PMMA, were presented in Figure 10. The macroinitiator
(Br-D2000-Br, Mn=2,800, polydispersity = 1.06) and triblock copolymers (PMMA-b-D2000-b-PMMA, Mn=7,400, polydispersity = 1.18) showed a unimodal type of GPC curve (A) and (B) in Figure 10, respectively. GPC analysis of the purified triblock copolymers indicated no signal of residual Br-D2000-Br macroinitiator and the monomer MMA in the elution traces. (Figure 10). According to the Figure 10, the number of PMMA segment was 46 in the triblock copolymer (PMMA-b-D2000-b-PMMA).
Thermal Property
The temperatures (Td10) of 10 % weight loss of these triblock copolymers were in the range of 255-355 ℃ and 254-313 ℃ in nitrogen and air, respectively. The theramogravimetric analysis diagram of triblock copolymer was shown in Figure 11.
Among the various triblock polymers, PPFS-D2000-PPFS, containing PFS and D2000
group had the highest thermal stability in nitrogen and air atmospheres. Obviously, the incorporation of the PFS and D2000 groups into the polymer backbone enhanced the thermal stability. Most of triblock copolymers were shown higher Td10 in nitrogen than in air atmosphere.
第三年度
Synthesis and characterization of a new macroinitiator, poly(NBMECl)
Ring-opening metathesis polymerization(ROMP) of NBMECl could be carried out under catalyst (I) and catalyst (II) system. Influences of [M]/[I] ratio on polymerization time, molecular weight and polydispersity(PDI) of poly(NBMECl) were investigated. The results of this investigation were summarized in Table 3. However, gelation was observed after ROMP of NBMECl with same monomer concentration by using Ru(II) catalyst. By decreasing monomer concentration to 2×10-2 mole.l-1, an organo-soluble polyNBMECl with high molecular weight 8.46×104 could be obtained after 15 mins(entry 4, Table 3). However, the resulting polymer with broad molecular weight distribution was observed. Hence, Ru catalyst (II) is not suitable to prepare macroinitiators because of broad polydispersity. For catalyst I, the higher ratio of [M]/[I], the higher and other, the molecular weight and polydispersity would depends on ratio of [M]/[I]. The macroinitiator with narrow molecular weight distribution was obtained when the polymerization was carried out for 3 hours by using Ru catalyst(I) with low [M]/[I] ratio of 40.62) 1H and 13C NMR spectra analyses were carried out to confirm the chemical structure of poly(NBMECl). In the 1H NMR spectrum of poly(NBMECl), the olefinic signals around δ5.90-6.20 ppm present in the monomer were disappeared and new signals due to olefinic hydrogen appeared around δ 5.19-5.33 ppm, corresponding to the hydrogen of double bond of the ring-opened polymer(cis/trans=86/14)[Figure 12(A)]. This confirmed the formation of macroinitiator [poly(NBMECl)].
Synthesis and Characterization of a new highly stable macroinitiator, poly(HNBMECl)
Hydrogenation agent, p-toluenesulfonylhydrazide (TSH) was used as a
convenient precursor to generate diimide in situ 63). The new highly stable macroinitiator poly(HNBMECl) could be prepared by hydrogenation of poly(NBMECl) (Scheme 7). The GPC data for poly(NBMECl) and poly(HNBMECl) were summarized in Table 4. It was found that molecular weight of polymer increased after hydrogenation. The 1H and 13C NMR spectra of poly(HNBMECl) were employed to confirm the formation of a highly stable macroinitiator[Figure 12(B)]. It can be seen that there was no olifinic resonance visible after hydrogenation around δ 5.19-5.33 ppm and δ 129.23–134.90 ppm in 1H NMR spectrum and 13C NMR spectrum, respectively. This confirmed the complete saturation of double bond in the polynorbornene backbone and formation of highly stable macroinitiator.
The thermal properties, glass transition temperature (Tg) and 10 % weight loss temperature (Td10), were evaluated, respectively, by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The results are tabulated in Table 4.
As expected, Tg slightly decreased and Td10 remarkably increased after hydrogenation of polymers 64,65). The decomposition of macroinitiators [poly(NBMECl) and poly(HNBMECl)] occurred in two steps. The macroinitiator [poly(NBMECl)] with C=C bond in the polynorbornene backbone showed high char yield (20%) than the saturated macroinitiator [poly(HNBMECl)](9%) which might be due to the crosslinking reaction between C=C bonds of polynorbornene main chains at high temperature.
Macroinitiator [poly(NBMECl) and poly(HNBMECl)] commenced decomposition respectively at 240 ℃ and 310 ℃, respectively.
Synthesis and Characterization of comb-shaped grafted copolymer
The influences of the ratio of concentration of monomer to concentration of macroinitiator on the grafting copolymerization of macroinitiator [poly(HNBMECl)]
with MMA and polymerization time on the molecular weight, polydispersity index of the polymer were discussed. The atom transfer radical polymerization was carried out by using CuCl/bipyridine complex as a reducing agent. The molecular weight of the polymers was analyzed by GPC using THF as an eluent. Polystyrene was used as a standard.
Grafting copolymerizations of macroinitiators [poly(NBMECl) and poly(HNBMECl)] with MMA were carried out in solution and bulk condition. Grafting copolymerization of macroinitiator [poly(HNBMECl)] with MMA in bulk condition
afforded gel- like insoluble products. GPC curves of macroinitiator [poly(HNBMECl)]
and grafted copolymers of poly(HNBMECl) with MMA were presented in Figure 7.
The macroinitiator [poly(HNBMECl)] showed a unimodal type of GPC curve [Figure
7(A)] whereas grafted copolymers exhibited bimodal type of GPC curve. Similar
bimodal nature in GPC curve of ethylene/vinyl acetate(EVA) copolymers grafted with acids has been observed earlier 66).The 1H NMR spectra of macroinitiators and grafted copolymers are presented in
Figure 12. The
1H NMR spectrum of comb-shaped copolymer obtained from grafting copolymerization of macroinitiator [poly(NBMECl)] and MMA exhibited signals at δ 3.58 ppm due to proton of OCH3 group , δ0.83-1.00 ppm due to proton of PMMA segment, and the 13C NMR spectra also exhibited signals at δ 51.76 ppm due to carbon of OCH3 group and δ 16.45-18.69 ppm due to carbon of PMMA segments.This observation confirmed the grafting copolymerization of macroinitiator with MMA.
The 1H NMR spectrum of comb-shaped copolymer obtained from ATRP reaction of highly stable macroinitiator [poly(HNBMECl)] and MMA exhibited signals at δ 3.57 ppm due to proton of OCH3 group , δ 0.81–0.99 ppm due to proton of PMMA segment, and the 13C NMR spectrum also exhibited signals at δ 51.72 ppm due to carbon of OCH3 and δ 16.43–18.64 ppm due to carbon of PMMA segments. This observation confirmed the grafting copolymerization of highly stable macroinitiator [poly(HNBMECl)] with MMA and formation of new comb-shaped copolymer[poly(HNBMECl-g-MMA)]. Comb-shaped copolymer [poly(HNBMECl-g-MMA)] started decomposition at 190 ℃ due to PMMA segment 67) and subsequently poly(HNBMECl) segment underwent decomposition at high temperature. The comb-shaped copolymer showed low char yield. There is no observation of Tg in DSC curve of comb-shaped copolymer [poly(HNBMECl-g-MMA)].
Solubility of macroinitiators [poly(NBMECl) and poly(HNBMECl)] and comb-shaped coplymers [poly(NBMECl-g-MMA) and poly(HNBMECl-g-MMA)] was listed in Table 5. All the macroinitiators and comb-shaped copolymers showed excellent solubility in organic solvents such as THF, pyridine, 1,2-dichlorobenzene, methylene chloride, chloroform, toluene, xylene, benzene, DMAc, NMP, and DMF.
Particularly comb-shaped copolymers showed better solubility than the corresponding macroinitiators.
結論:
第一年度
X-PEG 600-X, X-PEG 400-X and X-PTMEG 2000-X (X = Br or Cl) were
successfully prepared with high purity and used in the synthesis of the amphiphilic triblock copolymers (X-PMMA-b-PEG 600-b-PMMA-X, X-PMMA-b-PEG400-b-PMMA-X and X-PMMA-b-PTMEG 2000-b-PMMA-X, X = Cl and Br) by
ATRP. These amphiphilic triblock copolymers exhibited excellent solubility in various organic solvents. From GPC data the system in Br- macroinitiator-Br was more easily controlled polymerization than that in Cl- macroinitiator-C l. Also, all of triblock copolymers showed a unimodal type of GPC curve. The crystalline phase-transition temperatures of amphiphilic triblock copolymers increase with increasing repeating unit of ethylene glycol.第二年度
Br-D2000-Br, Br-ED2003-Br and Br-PCL2000-Br were successfully prepared
with high purity and used in the synthesis of the triblock copolymers by ATRP. These triblock copolymers exhibited excellent solubility in various organic solvents. From GPC curves the triblock copolymers showed a unimodal type of GPC curve and it indicated no signal of residual macroinitiator and the monomer MMA in the elution traces. The triblock polymers, PPFS -D2000-PPFS, containing PFS and D2000 groups had the highest thermal stability in nitrogen and air atmospheres.第三年度
Novel macroinitiators for ATRP, poly(NBMECl) and poly(HNBMECl), were derived from a difunctional monomer NBMECl , which was synthesized from the reaction of 5-(hydroxymethyl)bicyclo[2.2.1]hept-2-ene (NBCH2OH) and 2-chloropropionyl chloride. The highly stable macroinitiator, poly(HNBMECl), indicated better thermalstability than poly(NBMECl). The Td10 value of poly(HNBMECl) (Td10=331oC) is higher than poly(NBMECl) (Td10=286oC). Novel comb-shaped copolymers such as poly(NBMECl-g-MMA) and poly(HNBMECl-g-MMA) were successfully obtained by combination of ROMP and
ATRP. Novel macroinitiators and their comb-shaped copolymers showed excellent solubility in organic solvents.
計畫成果自評:
本計畫完全依照計畫內容執行,執行之相關內容已發表或投稿在下列著名期 刊:
(1) Polymer, Vol. 48, pp. 3694-3702 (2007)
(2) J. Polym. Sci., Part A: Polym. Chem., Vol. 45, pp. 3022-3031 (2007) (3) Macromol. Symp., Vol. 245-246, pp. 68-76 (2006)
(4) J. Polym. Sci., Part A: Polym. Chem., Vol. 44, pp. 6287-6298 (2006) (5) J. Polym. Sci., Part A: Polym. Chem., Vol. 44, pp. 4428-4434 (2006) (6) Polymer, Vol. 47, pp. 4613-4621 (2006)
(7) Polymer, Vol. 47, pp. 3057-3064 (2006)
(8) J. Polym. Sci., Part A: Polym. Chem., Vol. 44, pp. 3382-3392 (2006) (9) J. Polym. Sci., Part A: Polym. Chem. Vol. 44, pp. 2901-2911 (2006) (10) Polym. Prepr. Jpn., Vol. 54(2), pp. 2515 (2005)
(11) J. Polym. Sci., Part A: Polym. Chem., Vol 43, pp. 4233-4247 (2005) (12) Macromolecules. Vol. 38, pp. 3533-3538 (2005)
本計畫之新型含原冰片烯單體、巨起始劑、聚合物及共聚合物皆使用儀器鑑 定其結構,鑑定結果均符合結構,另外其聚合物皆具有優良物性(如溶解性、熱
性質及機械性質),可供學術界及工業界參考,為一具有發展潛力之高分子材料。
致謝:
感謝行政院國家科學委員會補助專題計劃 NSC 95-2221-E-011-067 之經費補 助,使本三年期計劃之期末報告得以順利執行。
參考文獻:
1. M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 78, 2656 (1956) 2. Kennedy, J. P. and S. Jacob, Acc. Chem. Res., 31, 835 (1998)
3. Matyjaszewski, K., Ed. Cationic polymerizations: mechanisms,synthesis and applications, New York: Marcel Dekker (1996)
4. Szwarc, M., Carbanions, living polymers and electron transfer processes, New York:Interscience (1968)
5. Hsieh, H. L. and R. P. Quirk, Anionic polymerization: principles and practical applications, New York: Marcel Dekker (1996)
6. Matyjaszewski, K., Chem. Eur. J., 5, 3095 (1999)
7. Patten, T. E. and K. Matyjaszewski, Acc. Chem. Res., 32, 895 (1999)
8. Matyjaszewski, K., Ed. Controlled radical polymerization, Washington, D. C.:
American Chemical Society (1998)
9. Matyjaszewski, K., Ed. Controlled/ living radical polymerization, Washington, D. C.: American Chemical Society (2000)
10. Otsu, T. and M. Yoshida, Makromol. Chem. Rapid Commun., 3, 127 (1982) 11. Nair, C. P. R. and G. Clouet, J. Macromol. Sci., Rev. Macromol. Chem. Phys.,
C31(2-3), 311 (1991)
12. Solomon, D. H., E. Rizzardo and P. P. Cacioli, Eur. Pat. Appl. EP (1985) 13. Rizzardo, E., Chem. Aust., 54, 32 (1987)
14. Georges, M. K., R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Trends Polym. Sci., 2, 66 (1994)
15. Hawker, C. J., J. Am. Chem. Soc., 116, 185 (1994)
16. Greszta, D. and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 35, 1857 (1997)
17. Puts, R. D. and D. Y. Sogah, Macromolecules, 29, 3323 (1996) 18. Goto, A. and T. Fukuda, Macromolecules, 32, 618 (1999)
19. Farcet, C., M. Lansalot, B. Charleux, R. Pirri and J. P. Vairon, Macromolecules, 33, 8559 (2000)
20. Burguiere, C., M. A. Dourges, B. Charleux and J. P. Vairon, Macromolecules,
32, 3883 (1999)
21. Benoit, D., V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc.,
121, 3904 (1999)
22. Benoit, D., S. Grimaldi, S. Robin, J. P. Finet, P. Tordo and Y. Gnanou, J. Am.
Chem. Soc., 122, 5929 (2000)
23. Wailand, B. B., G. Poszmik, S. L. Mukerjee and M. Fryd, J. Am. Chem. Soc.,
116, 7943 (1994)
24. Arvanitopoulos, L. D., M. P. Greuel, B. M. King, A. K. Shim and H. J.
Harwood, Am. Chem. Soc. Symp. Ser., 685, 316 (1998)
25. Gaynor, S. G., J. S. Wang and K. Matyjaszewski, Macromolecules, 28, 8051 (1995)
26. Matyjaszewski, K., S. Gaynor and J. S. Wang, Macromolecules, 28, 2093 (1995)
27. Goto, A., K. Ohno and T. Fukuda, Macromolecules, 31, 2809 (1998)
28. Lansalot, M., C. Farcet, B. Charleux, J. P. Vairon and R. Pirri, Macromolecules, 32,7354 (1999)
29. Chiefari, J., Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T.
A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H.
Thang, Macromolecules, 31, 5559 (1998)
30. Kato, M., M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules,
28, 1721 (1995)
31. Granel, C., P. Dubois, R. Jerome and P. Teyssie, Macromolecules, 29, 8576 (1996)
32. Wang, J. S. and K. Matyjaszewski, J. Am. Chem. Soc., 117, 5614 (1995) 33. Patten, T. E., J. Xia, T. Abernathy and K. Matyjaszewski, Science, 272, 866
(1996)
34. Matyjaszewski, K., T. E. Patten and J. Xia, J. Am. Chem. Soc., 119, 674 (1997)
35. Qiu, J. and K. Matyjaszewski, Macromolecules, 30, 5643 (1997) 36. Percec, V. and B. Barboiu, Macromolecules, 28, 7970 (1995)
37. Coca, S., K. Davis, P. J. Miller and K. Matyjaszewski, Polym. Prepr. Am.
Chem. Soc.,Div. Polym. Chem., 38 (1), 689 (1997)
38. Davis, K. A., H. J. Paik and K. Matyjaszewski, Macromolecules, 32, 1767 (1999)
39. Wnng, J. L., T. Grimaud and K. Matyjaszewski, Macromolecules, 30, 6507 (1997)
40. Haddleton, D. M., C. B. Jasieczek, M. J. Hannon and A. J. Shooter,