國
立
交
通
大
學
材料科學與工程研究所
碩
士
論
文
合成予體–受體型高分子含噻吩并[3,4-c]吡咯-4 ,6 -二
酮之性質分析及其太陽能電池之研究
Synthesis and Characterization of Donor–Acceptor
Conjugated Polymers containing
Thieno[3,4-c]pyrrole-4,6-dione unit for Bulk
Heterojunction Solar Cell Applications
研究生:鄭宇欣 Yu-Hsin Cheng
指導教授:韋光華 教授 Dr. Kung-Hwa Wei
合成予體–受體型高分子含噻吩并[3,4-c]吡咯-4 ,6 -二酮之性質分析及 其太陽能電池之研究
Synthesis and Characterization of Donor–Acceptor Conjugated Polymers containing Thieno[3,4-c]pyrrole-4,6-dione unit for Bulk Heterojunction
Solar Cell Applications
研 究 生:鄭宇欣 Student:Yu-Hsin Cheng 指導教授:韋光華 Advisor:Dr. Kung-Hwa Wei
國 立 交 通 大 學 材料科學與工程研究所
碩 士 論 文
A Thesis
Submitted to Department of Material Science and Engineering College of Engineering
National Chiao-Tung University in Partial Fulfillment of the Requirements
for the Degree of Master
in
Material Science and Engineering June 2011
Hsinchu, Taiwan, Republic of China
摘要 本論文以拉電子基團 thieno[3,4-c]pyrrole-4,6-dione(TPD)為受體, 與平面性良好之推電子基團 3,6-dioctylthieno[3,2-b]thiophene(TT)為 予 體 , 合 成 出 具 有 結 晶 性 之 共 軛 高 分 子 PTTTPD , 同 時 也 與 4-(heptadecan-9-yl)-4H-dithieno[3,2-b:2',3'-d]pyrrole(DTP),合成具有低 光學能隙之共軛高分子 PDTPTPD。 此外,為降低立體障礙,在 TT 與 TPD 單體間和 DTP 與 TPD 單 體間插入噻吩做為 spacer,合成出 PDTTTPD、PDTPDTTPD。 PTTTPD、PDTTTPD 具有高結晶性,高熱穩定度,PTTTPD 高分 子與 PCBM 混摻製成塊狀異質介面太陽能元件,其光電轉換效率為 0.25 %。將 PDTTTPD 高分子與 PC71BM 混摻後,同時再加入添加劑 1 % DIO,製成塊狀異質介面太陽能電池元件,具良好之元件特性,效率 可達 5.1 %,Voc可達 0.86 V。 PDTPTPD、PDTPDTTPD 則具低光學能隙及高熱穩定性質,將此 兩高分子分別與 PCBM 混摻製成塊狀異質介面太陽能電池元件,其能 量轉換效率為 1.86 %與 0.76 %,PDTPDTTPD 加入 1% DIO 添加劑後, 有些許的提升至 1.16 %。
Synthesis and Characterization of Donor–Acceptor
Conjugated Polymers containing
Thieno[3,4-c]pyrrole-4,6-dione unit for Bulk Heterojunction
Solar Cell Applications
Abstract
In this thesis, we have synthesized a conjugated polymer containing
electron-withdrawing thieno[3,4-c]pyrrole-4,6-dione (TPD) and
electron-donating thieno[3,4-c]pyrrole-4,6-dione units (TT), PTTTPD. We also synthesized TPD based low band-gap polymers conatining electron-donating 4-(heptadecan-9-yl)-4H-dithieno[3,2-b:2',3'-d] pyrrole (DTP), PDTPTPD. Besides, we also intrduced the thiophene as the spacer between donor and acceptor, and then we synthesized PDTTTPD and PDTPDTTPD.
The PTTTPD and PDTTTPD exhibited high crystalline characteristics and excellent thermal properties. Bulk heterojunction (BHJ) photovoltaic devices of PDTTTPD incorporating [6,6]-phenyl-C71-butyric acid methyl
ester (PC71BM) and 1% DIO, than it displayed high power conversion
efficiency (PCE) of 5.1 %. But the devices of PTTTPD didn’t, it just only 0.25 %. Moreover, both of PDTPTPD and PDTPDTTPD exhibited low band-gap properties and excellent thermal properties. BHJ photovoltaic devices of PDTPTPD and PDTPDTTPD incorporating PCBM and they displayed PCEs of 1.86 % and 0.76 %, respectively. Furthermore, when the devices of PDTPDTTPD incorporating with small amount of additives into these blends, the PCE of PDTPDTTPD were enhanced to 1.16 %.
誌謝
讓我能順利於二年的碩士生涯畢業,最要感謝的是我的指導教授 韋光華 老師,老師不辭辛勞的爭取實驗計劃,讓我們擁有豐富的實驗 資可以享用,並且給與我們指導,讓我在二年中不只學習到如何做研 究,更讓我們學習如何待人處世,實在受益良多。亦要感謝許千樹 教 授、林宏洲 教授、黃華宗 教授於百忙之中抽空指導,讓本論文更加 完善。 在這二年的研究生涯中,在實驗上與生活上要感謝實驗室裡的學 長姐們的指導及學弟妹們相互鼓勵,才使這篇論文能順利的完成,首 先要感謝陳冠宇學長,二年中在實驗上給與許多指導及幫助,並且讓 實驗室充滿著歡樂的氣氛;蘇明鑫學長,他對實驗的熱情,讓人感到 佩服;感謝陳家閔學長幫忙我量測 XRD,使我的實驗數據更加完整; 感謝江建銘學長,時常會與我討論在實驗上所碰到的困難,並幫助我 解決,論文能夠順利完成,都要感謝他教導我如何將內容寫的更完善 及順暢;感謝許昌隆學長,幫助我量測 TGA,才能使數據更加完整; 感謝劉志明學長,他時常讓我們搭便車,參加實驗室的聚會;感謝陳 修成學長,多謝他教我如何操作 AFM,讓我在畢業前又能多學到一項 技能;謝宗軒學長,我們時常一同討論實驗的操作,教學相長;吳任 宥學長,扮演著實驗室開心果的角色;我的同學,周亦任,要不是有你幫忙元件的製作,我想我的論文可能就開天窗了;小天兵學妹 王淑 民,時常出其不意的做一些很天的事,讓實驗室笑聲不斷,未來實驗 室的歡樂氣氛也要靠妳啦!運動健將的學弟楊博安,認真做實驗之 餘,不忘在戶外揮灑熱情的汗水。很高興認識你們,從你們的身上學 習到很多的事情,不論是研究上、做人處事上,大大的說聲,謝謝你 們! 最後,最辛苦的就是我的父母親,從小的時候就希望我們將來長 大能有所成就,現在我碩士畢業了,也希望我能成為你們的驕傲,爸、 媽,你們辛苦了。也要感謝一路從大學陪伴著我的紹華,要不是有你 的一路相伴、與鼓勵,我想我也無法順利的走到這一步。 感謝所有在我身旁支持我、幫助我、鼓勵我的人,謝謝你們!
目錄
中文摘要... ii 英文摘要... iii 誌謝 ... v 目錄 ... vii 圖目錄 ... xii 表目錄 ...xvi 第一章 緒論 ···1 1-1 太陽能電池的崛起··· 1 1-2 高分子太陽能電池元件介紹 ··· 2 1-2-1 太陽能電池元件結構與操作原理···2 1-2-2 太陽能電池元件材料的選擇 ··· 3 1-2-3 高分子太陽能電池元件設計 ···4 1-2-4 元件效率符號定義 ··· 6 1-3 共軛高分子的發展··· 9 1-3-1 分子結構的特性 ··· 9 1-3-2 分子結構與其能隙之關聯 ··· 10 1-4 低能隙高分子···11 1-4-1 低能隙高分子的設計概念 ··· 11 第二章 文獻回顧···13 2-1 高效率高分子材料的演進 ··· 132-2 高電洞遷移率之共軛高分子 ··· 14 2-2-1 TT 衍生物 ···14 2-2-2 DTP 衍生物 ···17 2-3 主鏈予體-受體系列之共軛高分子 ···20 2-3-1 TPD 衍生物 ···20 2-4 研究動機 ···22 第三章 實驗 ···25 3-1 試藥 ··· 25 3-2 使用儀器與性質測量 ··· 27
3-2-1 核磁共振光譜儀(Nuclear Magnetic Resonance,NMR) ··· 27
3-2-2 凝膠滲透層析儀(Gel Permeation Chromatography,GPC) ··· 27
3-2-3 紫外-可見光吸收光譜儀(UV-Vis Absorption Spectrometry) ···27
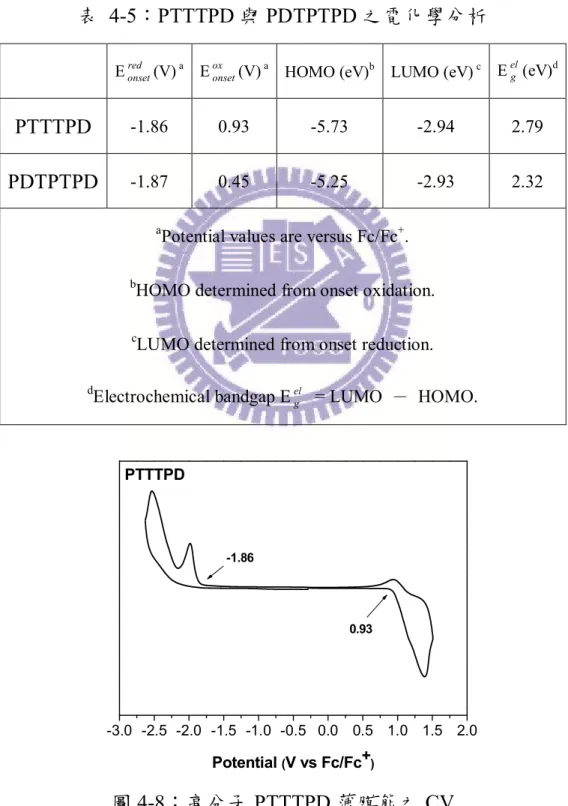
3-2-4 循環伏特安培計(Cyclic Voltammetry,CV) ··· 28
3-2-5 微差掃瞄式卡計( Differential Scanning Calorimeter, DSC ) ··· 28
3-2-7 旋轉塗佈機(Spin Coater) ··· 29 3-2-8 模擬光源···29 3-2-9 光強度偵測計··· 29 3-2-10 電性分析儀 ··· 30 3-2-11 原子力顯微鏡 ··· 30 3-2-12 探針式表面輪廓儀··· 30 3-3 合成 ··· 30 3-3-1 單體 M1 ···30 3-3-2 單體 M2 ···35 3-3-3 單體 M3···36 3-3-4 單體 M4 ···38 3-3-5 單體 M5 ···44 3-3-6 單體 M6 ···47 3-3-7 高分子 PTTTPD 之聚合 ··· 50 3-3-8 高分子 PDTTTPD 之聚合 ··· 51 3-3-9 高分子 PDTPTPD 之聚合··· 52 3-3-10 高分子 PDTPDTTPD 之聚合 ···52 第四章 結果與討論 ···54 4-1 含 TT 單體與 DTP 單體共聚 thieno[3,4-c]pyrrole-4,6-dione(TPD)
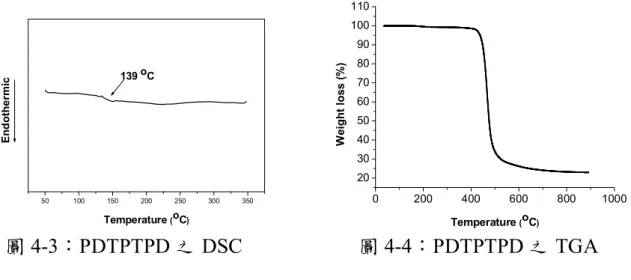
單體之共軛高分子 PTTTPD、PDTPTPD 分析與研究 ···54 4-1-1 GPC 量測 ···54 4-1-2 DSC 和 TGA 量測 ··· 54 4-1-3 溶解度測試···56 4-1-4 低掠角入射 X-ray 繞射(GIXD)分析 ···56 4-1-5 光學性質(UV-vis 分析測量)···57 4-1-6 電化學性質-氧化與還原電位量測 ···59 4-1-7 電洞遷移率量測 ··· 62 4-1-8 太陽能元件光伏性質 ··· 63 4-2 TT 單體與 DTTPD 單體及 DTP 單體與 DTTPD 單體共聚之共軛 高分子 PDTTTPD、PDTPDTTPD 分析與研究 ···70 4-2-1 GPC 量測 ···70 4-2-2 DSC 和 TGA 量測 ··· 70 4-2-3 溶解度測試···72 4-2-4 低掠角入射 X-ray 繞射(GIXD)分析 ···72 4-2-5 光學性質(UV-vis 分析測量)···73 4-2-6 電化學性質-氧化與還原電位量測 ···75 4-2-7 電洞遷移率量測 ··· 78 4-2-8 太陽能元件光伏性質 ··· 79
第五章 結論 ···87 參考文獻 ···88 附圖目錄 ···93
圖目錄
圖 1-1:高分子太陽能電池元件結構 ... 2
圖 1-2:光電轉換詳細作用機制 ... 3
圖 1-3:高分子太陽能電池雙層結構示意圖... 5
圖 1-4:高分子太陽能電池塊狀異質介面結構示意圖 ... 6
圖 1-5:aromatic form 與 quinoid form... 10
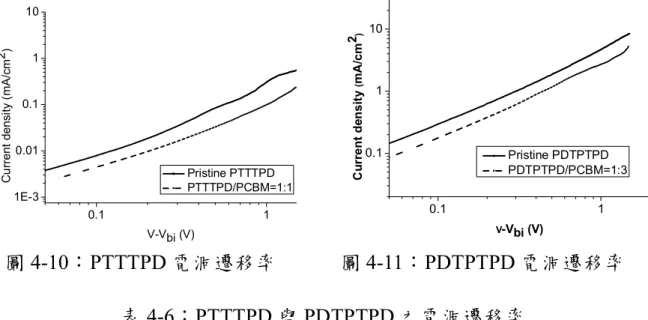
圖 1-6:聚合常用的推電子材料 ... 12 圖 1-7:各種常見的聯環分子 ... 12 圖 2-1:P3HT 衍生物的分子結構... 13 圖 2-2:TT 衍生物之分子結構 ... 14 圖 2-3:PQTT、PQPDTT、PQTPDTT 高分子結構 ... 14 圖 2-4:DPP 與 TT 共聚之高分子結構 ... 16 圖 2-5:DTP 衍生物之分子結構... 17 圖 2-6:PDTPDTBT 之分子結構 ... 18 圖 2-7:PDDTPDPP 之分子結構 ... 19 圖 2-8:TPD 衍生物之分子結構... 20 圖 2-9:BDT 與 TPD 共聚之高分子結構圖 ... 21 圖 2-10:PDTPTPD 與 PTTTPD 之分子結構... 23 圖 2-11:PDTPDTTPD 與 PDTTTPD 之分子結構 ... 24
圖 4-1:高分子 PTTTPD 之 DSC ... 55 圖 4-2:高分子 PTTTPD 之 TGA ... 55 圖 4-3:PDTPTPD 之 DSC ... 55 圖 4-4:PDTPTPD 之 TGA ... 55 圖 4-5:高分子 PTTTPD 的 X-ray 繞射圖... 56 圖 4-6:PTTTPD 在溶液態與薄膜態之吸收光譜 ... 59 圖 4-7:PDTPTPD 在溶液態與薄膜態之吸收光譜... 59 圖 4-8:高分子 PTTTPD 薄膜態之 CV ... 61 圖 4-9:高分子 PDTPTPD 薄膜態之 CV... 62 圖 4-10:PTTTPD 電洞遷移率... 63 圖 4-11:PDTPTPD 電洞遷移率 ... 63 圖 4-12:PTTTPD 之 J-V 曲線圖 ... 64 圖 4-13:(a) 以 o-DCB 為溶劑,在 PTTTPD:PCBM = 1:1 的比例下製備 薄膜的 AFM 圖,上邊圖為 height image,下邊圖為對應的 phase image。 ... 65
圖 4-14:PTTTPD 之 EQE... 66
圖 4-15:高分子 PTTTPD 與 PDTPTPD 之 J-V 曲線圖... 67 圖 4-16:以 o-DCB 為溶劑,在 PDTPTPD:PCBM = 1:1 的比例下製備薄 膜的 AFM 圖,上邊圖為 height image,下邊圖為對應的 phase
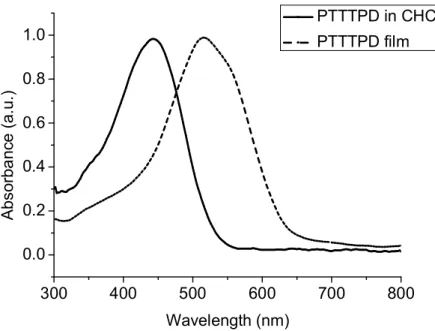
image。 ... 68 圖 4-17:PDTPTPD 之 EQE ... 69 圖 4-18:PDTTTPD 之 DSC ... 71 圖 4-19:PDTTTPD 之 TGA ... 71 圖 4-20:PDTPDTTPD 之 DSC... 72 圖 4-21:PDTPDTTPD 之 TGA ... 72 圖 4-22:高分子 PDTTTPD 的 X-ray 繞射圖 ... 73 圖 4-23:PDTTTPD 之溶液態與薄膜態吸收光譜圖... 75 圖 4-24:PDTPDTTPD 之溶液態與薄膜態吸收光譜圖... 75 圖 4-25:PDTTTPD 薄膜態之 CV ... 77 圖 4-26:PDTPDTTPD 薄膜態之 CV ... 78 圖 4-27:PDTTTPD 電洞遷移率... 79 圖 4-28:PDTPDTTPD 電洞遷移率... 79 圖 4-29:PDTTTPD 之 J-V 曲線圖 ... 81 圖 4-30:以 o-DCB 為溶劑,在 PDTTTPD:PCBM = 1:1 的比例下製備薄 膜的 AFM 圖,上邊圖為 height image,下邊圖為對應的 phase image。 ... 82
圖 4-31:以 o-DCB 為溶劑,在 PDTTTPD:PC71BM = 1:1 的比例下並加
image,下邊圖為對應的 phase image。... 82 圖 4-32:PDTTTPD 之 EQE... 83 圖 4-33:PDTPDTTPD 之 J-V 曲線圖 ... 84 圖 4-34:以 o-DCB 為溶劑,PDTPDTTPD:PCBM = 1:4 的比例下製備薄 膜的 AFM 圖,上邊圖為 height image,下邊圖為對應的 phase image。 ... 85 圖 4-35:以 o-DCB 為溶劑,PDTPDTTPD:PCBM = 1:4 的比例下並加入 1 % DIO 添加劑,製備薄膜的 AFM 圖,上邊圖為 height image,下邊圖為對應的 phase image。... 86 圖 4-36:PDTPDTTPD 之 EQE... 86
表目錄 表 2-1:PQTT、PQPDTT、PQTPDTT 高分子效率示意表... 15 表 2-2:DTP 衍生物應用在 FET 之電洞遷移率示意圖... 18 表 2-3:TPD 衍生物的元件特性表現 ... 20 表 2-4:高分子元件特性表現 ... 21 表 4-1:高分子 PTTTPD、PDTTTPD 之分子量... 54 表 4-2:高分子 PTTTPD、PDTPTPD 之熱性質數據表 ... 55 表 4-3:高分子溶解度測試 ... 56 表 4-4:PTTTPD、PDTPTPD 之吸光性質整理 ... 58 表 4-5:PTTTPD 與 PDTPTPD 之電化學分析 ... 61 表 4-6:PTTTPD 與 PDTPTPD 之電洞遷移率 ... 63 表 4-7:PTTTPD 之元件特性表現... 65 表 4-8:PTTTPD 與 PDTPTPD 之元件特性表現 ... 68 表 4-9:高分子 PDTPTPD、PDTPDTTPD 之分子量 ... 70 表 4-10:高分子 PDTTTPD、PDTPDTTPD 之熱性質數據表 ... 71 表 4-11:高分子溶解度測試 ... 72 表 4-12:PDTTTPD、PDTPDTTPD 之吸光性質整理 ... 74 表 4-13:PDTTTPD 與 PDTPDTTPD 之電化學分析 ... 77 表 4-14:PDTTTPD 與 PDTPDTTPD 之電洞遷移率 ... 79
表 4-15:PDTTTPD 之元件特性表現 ... 81 表 4-16:PDTPDTTPD 之元件特性表現 ... 85
第一章 緒論
1-1 太陽能電池的崛起 近年來太陽能電池逐漸受到人們的重視,主因來自於全球陷入缺乏能 源的危機中,人類所使用的能源來自於煤、石油、天然氣…等石化燃料, 其中又以石油為最大宗,但目前石油在全球的含量已大量減少,各國都已 有危機意識,紛紛開始尋找替代能源。除了能源危機外,因石化燃料經燃 燒後會產生二氧化碳、氮氧化合物…等廢氣,進而造成了溫室效應…等, 使得全球災害頻傳。 除了能源缺乏的危機再加上環保意識抬頭,各國的學者逐漸投入心力 在發展太陽能電池上,人們希望利用太陽能做為另一種能源來源,並且減 少對地球的傷害。 最早太陽能電池之材料為矽,矽晶太陽能電池又分為單晶矽[1]、多晶矽 [1] 、非晶矽[1]、微晶矽四種;再來是化合物半導體,以 GaAs、CdTe、GaInP… 等。此種無機太陽能電池已有相當長的研究歷史,不但效率高且性能穩定, 目前已有量產並被廣泛應用,但其製作複雜、製作成本高…等缺點,使得 科學家又朝向另一個方向發展。進而發展出有機染料敏化太陽能電池(Dye-Sensitized Solar Cell)、有機
小分子太陽能電池(Organic Solar Cell)、高分子太陽能電池(Polymer Solar
能製造出更輕薄短小且價格低廉的太陽能電池。但目前學者在高分子太陽 能電池上的效率表現仍無法超越第一代矽晶太陽能電池 25 %,而Ⅲ-Ⅴ族 單晶電池更可達 28.8 %[2],所以高分子太陽能電池仍需更多研究與改進。 1-2 高分子太陽能電池元件介紹 1-2-1 太陽能電池元件結構與操作原理[3] 圖 1-1:高分子太陽能電池元件結構 高分子太陽能電池元件結構如上圖 1-1 右邊所示,最上層為一塗 佈 ITO 的玻璃基材,再塗佈電洞傳輸層(PEDOT:PSS)跟主動層(Active layer),最後蒸鍍上金屬電極,其主動層大部份都為 P 型的共軛高分子 與 N 型 C60衍生物所組成。 光主要由共軛高分子吸收,由於共軛高分子材料具高的吸收係 數,其元件的厚度約為 100 nm,為最輕薄的太陽能電池。
其光電轉換詳細作用機制如圖 1-2 所示,利用 P 型與 N 型的材料 混滲,藉由太陽光的照射,產生電子與電洞對(Electron/Hole Pair), 擴散至 P-N 介面後,並在 P-N 介面產生電子與電洞分離,並分別經由 電子與電洞的傳導材料,傳輸至陰陽電極而形成電壓降,以產生電能。 圖 1-2:光電轉換詳細作用機制 由於有機半導體材料與無機材料相比有較高的束縛能(Binding Energy),其束縛能約大上一到兩個 order,故於室溫條件下,有機材料 無法形成自由的電子或電洞,必須藉由 N 型與 P 型材料界面的勢能 差,才能達到電子與電洞分離的效果。 1-2-2 太陽能電池元件材料的選擇[3] 太陽能電池中的每一層,其材料的選擇上有其限制條件與其不同 的功能,其中 ITO 為照光面的透明電極材料,必需具備高導電度(< 20 Ω)及高穿透度功能(於可見區域穿透度 T %> 85 %)。而 PEDOT:
PSS 主要功能,為修飾 ITO 的 Work Function(減少 Hole Injection
擋(Electron Blocking)的角色。ITO/PEDOT:PSS 及 Al 的電極的選擇, 亦是在能階考量下所搭配出,如圖 1-2 所示,ITO/PEDOT:PSS 的 Work Function 必須配合 P3HT 的 HOMO(High Occupied Molecular Orbital) 能階,才能有效將電洞引導出來,而 Al 的選擇也是符合由 PCBM 的 LUMO(Lowest Unoccupied Molecular Orbital)引導出電子需求的緣 故。在蒸鍍 Al 前鍍上 Ca 之原因為藉由這層材料導入,可以增進 electron injection,幫助效率的提昇。 1-2-3 高分子太陽能電池元件設計 電池元件的設計依其結構的不同分成,雙層結構(bilayer devices) 如圖 1-3 所示、塊狀異質介面圖如圖 1-4 所示(bulk heterojunction devices)。 雙層結構(bilayer devices): 其電池內的主動層分為二層,一層以 P 型材料,一層以 N 型材料, 將二層堆疊後常用於矽薄膜太陽能電池,但運用在有機高分子材料 上,效率表現較塊狀異質介面差[4],其原因在於高分子材料之激子擴散
長度(exciton diffusion length)不夠所致,高分子在吸光後所產生之激
子在擴散 10-20 nm[5]後釋放出能量回到基態,但主動層之厚度通常需
100 nm 才能有效率地吸收光子,所以當高分子吸光後,其所產生的激 子只有在 P-N 介面 10~20 nm 間的介面處進行電荷分離而產生自由載
子,其他區域所產生之激子皆以輻射或非輻射方式回到基態,故元件 效率較差。但此種設計的優點在於載子的傳導和收集,較另一種塊狀 異質介面結構佳以及製程較簡單和易於控制。
圖 1-3:高分子太陽能電池雙層結構示意圖 塊狀異質介面(bulk heterojunction devices):
其電池內的主動層只有一層,是將 P 型與 N 型材料做混合後,塗 佈在 ITO 玻璃基材上,與雙層結構最大的不同在於主動層中不僅僅只 有一個 P-N 介面,將二種材料混摻後形成無數個 P-N 介面,所以高分 子吸光後產生之激子可立即進行電荷分離而形成自由載子,分別在陰 極和陽極有效地收集,但缺點在於材料混合後,使主動層缺乏一個完 整的內建電場梯度,自由載子只能依區間的電場變化移動,增加了電 子與電洞再結合的機率,故載子收集率較低,但綜觀其表現仍優於雙 層結構。 由於載子只能隨區間電場變化移動,所以主動層之形貌成為影響 元件表現的主要因素,適當的相分離可以減低電子與電洞再結合[6]的機 Al Fullerene Polymer PEDOT:PSS ITO Glass 主動層
率,部份元件經回火後會形成相分離,而提升效率,但不適用於所有 的元件,因為不同的高分子會有不同的情況產生。 圖 1-4:高分子太陽能電池塊狀異質介面結構示意圖 1-2-4 元件效率符號參數定義 測量元件時,有幾個指標性的數值可判別元件的優劣,分別是「開 路電壓 Voc」、「短路電流 Jsc」、「填充因子 FF」、「轉換效率 η」。
開路電壓(open circuit voltage, Voc):[7]
所謂開路電壓即是太陽能電池在負載無限大的狀況下,也就是外 部電流斷路時所量到的電壓。太陽能電池的電流-電壓特性為: I=I0(e(qV/Kt)-1)-IL 當輸出的電流=0 時,根據上式可得開路電壓為: Voc= 1 ln 0 L I I q kT 經計算當電流為 0 時,此時的開路電壓為最大值。而開路電壓的 大小可由 P 型材料與 N 型材料的準費米能階差來調整。有機高分子 中,P 型係指推電子材料的 HOMO 與 N 型的拉電子材料的 LUMO, PC61BM Polymer Al PEDOT:PSS ITO 主動層 Glass
二者間的能階差來決定 Voc。
短路電流(short circuit current, Jsc):[7]
所謂短路電流即是太陽能電池在無負荷狀態下,也就是外部電路 短路時的輸出電流,此時電壓為 0。在理想的狀態下,太陽能電池的短 路電流即等於照光時所產生的電流,但實際情形中,電流會因薄膜的 形貌或缺陷而產生損失或傳遞不易,所以電流值會比理論值來的低。 在理想狀態下,短路電流可由下列公式定義: Jsc=ne μE
n 表示電荷載子的濃度,e 表示為單位電荷(elementary charge),
μ 表示移動率(mobility),E 表示電場強度,假設元件的內部量子效率
(Internal Quantum Efficiency)能達到 100%,n 即為每單位體積吸收的光 子數。電流主要受到電荷載子濃度與載子移動速率的影響,而載子移 動速率受本身的材料所限制,如果高分子的平面性好,移動速率會較 高,相反的若扭曲的太厲害,也會降低其移動速率。除了材料本身, 塗佈在元件上薄膜其形貌[8]也是要素之一,而形貌又受溶劑的種類、溶 劑的揮發速度及蒸鍍的方法影響,必須經由不斷嘗試不同的實驗參 數,才能得到最好的電流值。 分流電阻 (shunt resistance): [7] 用來定義太陽電池的漏電流大小,也就是 Rsh≡ V / Ileak。分流電阻
越大,就表示漏電流越小。如果考慮串聯電阻 Rs 和分流電阻 Rsh,太 陽電池的電流-電壓關係則可寫成下列公式: ( )
[
V IRs VT1]
s s L shV
IR
I
I e
I
R
填充因子(fill factor, FF):[7] 在照光下的電流-電壓特性曲線中,任何一個工作點的輸出功率等 於該點所對應的電流、電壓的乘積,在量測時會量到一點 Pm(Vm、Jm 的乘積),此時的輸出功率為此元件能達到之最大值,而定義 Vm、Jm 乘積結果與 Voc、Jsc的乘積之比,叫做填充因子。 填充因子會受到元件上之薄膜的表面形貌、電子電洞遷移率的平 衡…等等影響,若薄膜粗糙度太大,可能就會使電池無法發揮其最大 功率,亦或電子、電洞其遷移率相差太大,會使分離的電子電洞對在 元件中產生再結合的情形,而無法將電子順利的傳遞出來,意即本身 材料的性質不錯,若因受到其他因素無法使元件完美(例:回火的溫 度、塗布的均勻度…等),也無法發揮出材料的最大功率。 轉換效率 η(efficiency):[7] 最能表現出太陽能電池好壞的指數,將能量轉換效率定義為輸出 能量與入射能量之比,用方程式表示為: sc oc m m J V J FF V * * η= in sc oc in m P FF J V P V *Im * * Pin:入射光之總功率 1-3 共軛高分子的發展 共軛高分子具有單、雙鍵交替,可藉由 π 軌域的重疊,使 π 電子在分 子間或分子內呈現非定域化(delocalization),使得 π 電子在分子鏈上可做 自由移動,而產生導電的可能性。 從過去的文獻中得知,具有導電性質的高分子在 1960 年代相繼被發 現。1910 年時首次出現無機聚合物,poly(sulphur nitride)[9],直至 1962 年發 現一個半導體高分子[10],此一材料隨後即被加以證實在溫度為 0.4 K時具 有導電的現象[11]。 1-3-1 分子結構的特性[12] 聚乙炔等線性共軛高分子,有二個缺點,是:熱穩定性及抗氧化性 都差,使分子鏈在激態下易裂解,但以芳香環為主鏈之共軛高分子較無 此缺點,如poly(p-phenylene) (X=-(CH=CH)-) [13]、poly(pyrrole)(X= ─(NH)─)[14]、poly(thiophene)(X=-(SH)-)[15],此類高分子分為兩 種不同能量大小之共振結構,第一種較穩定之結構稱為aromatic form, 其 π 電子之移動受限於芳香環內,因而無法利用共振移動於環與環之 間,因此具較高的分子能隙,若主鏈上之芳香環具較大共振穩定能 ( resonance energy,Er) 時,會偏向於aromatic form。
另一則是相對不穩定之結構稱為quinoid form,其主鏈上芳香環內 的 π 電子不會被受限在芳香環中,π電子可以沿著主鏈呈未定域化
(delocalization),使主鏈上之雙鍵變單鍵,同時,單鍵也會變為雙鍵,
所以能讓π電子沿著主鏈傳導;若要使 π 電子能在主鏈上移動,則必須 破壞其芳香性,故quinoid form較不穩定。
圖1-5:aromatic form 與 quinoid form 1-3-2 分子結構與其能隙之關聯
分子的結構對其能隙(Eg)之影響可用下列式子表示:
能隙主要被五個因素所影響:分別是Eδγ
、Eθ
、ESub、ERes、EInt。
Eδγ代表當分子在變化時,其單鍵與雙鍵的鍵長差,而此為決定能 隙最主要的因素。 Eθ代表主鏈上芳香環間之共平面程度,共平面性越好,π電子能夠 共振的機會越大,亦即π電子越易未定域化,而π電子就能夠沿主鏈而 移動,Eθ越大則Eg將越大。 ERes代表每個芳香環的共振穩定能,π電子要能在芳香環內移動必
ESub表 示 側 鏈 取 代 基 對 Eg的 影 響 , 為 調 整 高 分 子 的 HOMO 及 LUMO,會設計在側鏈接上取代基,通常推電子基會使HOMO能階提 升,拉電子基會使LUMO能階下降。 以上四種影響的為單一條高分子鏈的情形,當高分子在 ITO 玻璃 上成膜時,是許多條高分子所堆疊而成,最後一個因素EInt,是指當高 分子堆疊時[16],其分子鏈間的作用力,因高分子相互堆疊,使π電子可 在分子鏈間移動,增加共軛長度降低分子能隙。 1-4 低能隙高分子 現今許多研究團隊紛紛研究如何將高分子設計成低能隙,原因為太陽 光最大光流量密度約在700 nm附近(約1.77 eV),根據公式E (eV)=1240/吸 收波長 (nm),若想吸收長波長約700 nm以上的光子,則能隙需在1.77 eV以 下,所以許多科學家都想辦法設計出低能隙之高分子。 1-4-1 低能隙高分子的設計概念 高分子設計分為二個單體片段,一為予體一為受體,藉由共聚一個 推電子基團與一拉電子基團,產生 intramolecular charge transfer 效應, 使得電子易於在二者間共振,能隙降低。
常 見 到 的 推 電 子 基 團 有 thiophene 、 fluorene 、 carbazole 、 cyclepentadithiophene…等如圖 1-6 所示,拉電子基團則為下述介紹的聯 環系統。
S S R R R R N R S Flourene
Cyclopenta[2,1-b;3,4-b′]-dithiophene Carbazole Thiophene
圖 1-6:聚合常用的推電子材料
亦或設計讓分子結構形成 Quinoid form,擁有較高共振穩定能,也 能得較低之能隙。具有低能隙之特性的高分子,大部份以聯環系統作為 單體如:Isothianaphthene[17]、Thieno[3,4-b] pyrazine[18]、Thieno [3,4-b]
thiophene[19],如圖 1-7 所示。 S S N N S S
Isothianaphthene Thieno [3,4-b] pyrazine Thieno [3,4-b]thiophene
第二章 文獻回顧
2-1 高效率高分子材料的演進 P3AT [Poly(3-alkylthiophene)]及其衍生物 P3AT 因絕佳的光電性質、熱穩定性、化學穩定性,所以常被運用在發 光二極體、場效電晶體和高分子太陽能電池上。McCullough 研究團隊將 P3HT 分子鏈位向規則性提高到 95 %以上[20],使其吸收波長產生紅位移、 吸收係數、載子移動率提高[21] ,但位向規則性過高的 P3HT 往往在熱退火下 與 PCBM 產生過度的相分離,即最適化形貌的熱穩定性較差,因此藉由位 向規則性適度的降低,可提高元件之熱穩定性如圖 2-1 所示[22] 。 S C6H13 S C6H13 S C6H13 C6H13 m n n regioreaularity = 96% m/n = 96/4 annealing at 150 °C, 30 min annealing at 150 °C, 300 min η=4.3% η=3.5% η=2.6% η=4.4% 圖 2-1:P3HT 衍生物的分子結構 上述之 P3HT 與 MDMO-PPV 因受限於相對大之分子能隙,故再經過 許多元件上的最佳化後,效率已無法顯著提高,因此予體-受體組合之低 能隙高分子已成目前高分子結構設計的主流。2-2 高電洞遷移率之共軛高分子 2-2-1 TT 衍生物 圖 2-2:TT 衍生物之分子結構 具有推電子特性的 thieno[2,3-b]thiophene(TT)[23]擁有良好的平面 特性且其為二個噻吩並聯之結構,剛硬不易扭曲。將其導入高分子主鏈 後,使得高分子在電洞遷移率上有 0.2–0.6 cm2 V–1 s–1表現。 PQTT、PQPDTT、PQTPDTT:[24] 圖 2-3:PQTT、PQPDTT、PQTPDTT 高分子結構
陸續有相關 TT 的文獻應用,其中 Doo-Kyung Moon 研究團隊將其 設計為予體-受體的形式,並將其應用在塊狀異質介面太陽能電池,設 計出 PQTT、PQPDTT、PQTPDTT 三種高分子,三者間的差異為在 TT 單體加上長碳鏈以增加溶解度,以及在予體與受體間插入一個噻吩,發 現插入噻吩後的 PQTPDTT 其分子堆疊性比 PQTT 良好,傳導電子與電 洞也比 PQTT 有效率。此外,PQTPDTT 在 UV 吸收的面積也比 PQTT 大,表示所吸收到的光子較多,致使電流提升 2 倍,最好效率可達到 2.27%。
表 2-1:
PQTT、PQPDTT、PQTPDTT 高分子效率示意表Polymer D:A Ratio(w/w) Voc (V) Jsc
(mA/cm2) FF (%) PCE (%) PQTT/PCBM 1:3 0.61 4.12 0.33 0.84 PQPDTT/PCBM 1:3 0.36 1.37 0.32 0.16 PQTPDTT/PCBM 1:3 0.7 4.63 0.39 1.28 PQTT/PC71BM 1:3 0.69 5.29 0.38 1.39 PQPDTT/PC71BM 1:3 0.37 2.60 0.31 0.30 PQTPDTT/PC71BM 1:3 0.71 8.8 0.36 2.27
DPP 與 TT 共聚之高分子:[25] 圖 2-4:DPP 與 TT 共聚之高分子結構 Iain McCulloch 研究團隊將 TT 單體導入高分子鏈中,同時應用在 場效電晶體和塊狀異質介面太陽能電池,此作者的設計概念為希望利用 DPP 擁有低能隙之特性,期望吸收光譜位置可接近紅外光範圍,作者同 時參考了 Prashant Sonar 團隊[26],此團隊以 DPP 做為受體,TT 做為予 體,兩者中央再插入了噻吩作為 spacer 製成 FET,其電洞遷移率可達 0.88 cm2 V–1 s–1,因此作者便將插入的噻吩替換成 TT,期望因為 TT 的 共平面性,使得電洞的傳遞比單一噻吩更順利,其中做到最好的電洞遷 移率可達 1.88 cm2 V–1 s–1,同時此結構做成塊狀異質介面太陽能電池, 在效率上可達 5.4 %,電流可達 15 mA/cm2。
2-2-2 DTP 之衍生物[27] 圖 2-5:DTP 衍生物之分子結構 N-alkyldithieno[3,2-b:2′,3′-d]pyrroles 其擁有堅硬且平面的結構,加 強了π-π共軛,避免了鏈-鏈折疊。此外 DTP 為高度共平面推電子基 團,同時 DTP 也有降低分子 reorganization energy 的特性,使分子內的 電子轉移更加容易。Richard D. McCullough 團隊將其應用在場效電晶體 (FET),期望藉由堅硬且平面的結構,使電洞遷移率提升,並且研究 接上不同碳鏈之噻吩其各項性質的不同,作者發現不同碳鏈的長短並不 會影響其 UV 波長的吸收位置,但 P5 因 3、4 號位置同時接上了碳鏈, 立體障礙的緣故而導致扭轉,使得有效共軛長度降低,所以 UV 吸收有 藍位移的現象。同時提出非規則的表面形態也可表現出好的電洞遷移 率,反而是經由退火後的規則表面形態,電洞遷移率有下降的趨勢,其
中最好的電洞遷移率可達 0.13 cm2 V–1 s–1。 表 2-2:DTP 衍生物應用在 FET 之電洞遷移率 DTP 首次使用在太陽能元件高分子: 圖 2-6:PDTPDTBT 之分子結構 Kazuhito Hashimoto 的研究團隊[28]試著將 DTP 與 DTBT 做成塊狀異 質介面太陽能電池,2008 年時並無任何團隊將其應用在塊狀異質介面 太陽能電池,此高分子擁有寬廣的吸收波長範圍從 350 nm-800 nm,以
Polymer Codition μ(cm2 V–1 s–1)average on/off ratio
P4 as-cast 0.026 4 Annealed 0.0058 20 P6 as-cast 0.13 120 Annealed 0.058 1500 P7 as-cast 0.0035 140 Annealed 0.00019 150
致能夠吸收更多的光子而提高電流。在效率表現上,開路電壓並不高, 只有 0.52 V,但電流因 UV 吸收範圍有相當寬廣的吸收,且吸收的位置 近似於太陽光譜,使得電流可達 9.47 mA/cm2,效率為 2.18 %。 PDDTP-DPP 表現出高電洞遷移率 圖 2-7:PDDTPDPP 之分子結構 Richard D. McCullough 研究團隊[29]再次利使用 DTP 單體與 DPP 單 體去聚合,想同時結合具有強拉電子特性、且同時擁有 fuse ring 系統的 DPP,以增加高分子的平面性,再加上 DTP 強推電子之特性可以使得 二者間的π-π共軛延長且產生強的 ICT 效應,共平面特性使排列更加 規則,而擁有高電洞遷移率,此高分子在 FET 表現上,電洞遷移率最 高可達 0.41 cm2 V–1 s–1,平均約為 0.29 cm2 V–1 s–1。
2-3 主鏈予體-受體系列之共軛高分子 2-3-1 TPD 衍生物[30] 圖 2-8:TPD 衍生物之分子結構 拉電子基團 thieno[3,4-c]pyrrole-4,6-dione(TPD)具高度共平面與 強拉電子等特性,故導入高分子主鏈後,能得到較低 HOMO 能階,使 開路電壓提高,此外許多文獻顯示導入 bithiophene 單體後,具有結晶 性且高電洞遷移率,當二者共聚後,此高分子具有高度結晶性,此文獻 由於導入 TPD 後,擁有良好之開路電壓,可達 0.95 V,在效率上也可 達 4.7 %。 表 2-3:TPD 衍生物的元件特性表現 Ratio (w/w) Jsc (mA/cm 2 ) Voc (V) FF(%) PCE(%) 1:1 -7.23 0.95 58 4.0 1:1.5 -8.02 0.95 62 4.7 1:2 -7.16 0.95 63 4.3
1:2.5 -5.12 0.93 61 2.9 BDT 與 TPD 共聚之高分子:[31] 圖 2-9:BDT 與 TPD 共聚之高分子結構圖 此篇文獻作者在 TPD 單體上接上不同的長碳鏈,其中包含立體障礙較 大的分支碳鏈及立體障礙較小的直鏈,發現當導入八個碳的直鏈時,其效 率及電流有較好的表現,最好效率可達 6.6 %。 表 2-4:高分子元件特性表現
Polymer Jsc (mA/cm2) Voc (V) FF (%) PCE (%)
P1 8.1 0.87 56 3.9
P2 9.7 0.81 67 5.4
2-4 研究動機 鑑於文獻中顯示,將高分子設計為予體-受體,已密集的使用在塊狀異 質結構太陽能電池,因其易於調整其高分子之電化學性質、傳遞電子電洞 的能力、並有效地降低光學能隙。此外,同時希望設計出具有合適的 LUMO 能階,而能夠克服激子的束縛能,並且設計具有平面結構,使之具有結晶 性並且提高其電洞遷移率。 受體的部份,我們挑選了 TPD 單體,因為其可使 HOMO 降低,提升 開路電壓,從文獻中得知 TPD 擁有平面且剛硬的結構、強拉電子的特性, 使得 π 電子在分子間或分子內呈現非定域化(delocalization),使得 π 電子 在 主 鏈 上 可 做 自 由 移 動 , 此 外 也 提 高 予 體 與 受 體 間 的 電 荷 轉 移 (intramolecular charge transfer)效應。
予體的部份,挑選了具有推電子特性的 thieno[2,3-b]thiophene(TT)其 有良好的平面性及其剛硬不易扭曲的特質,期望擁有良好的排列,提升電 洞遷移率,進而提升電流。許多文獻表示導入 TT 單體之高分子應用在場效 電晶體上,都擁有高結晶性及高電洞遷移率,應用在太陽能電池上,其光 電轉換效率也有 2.3 %。此外,也挑選同樣具有良好平面特性之 DTP,從 上述文獻得知導入 DTP 的 PDTPDTBT 其 UV 擁有寬廣的吸收波長範圍從 350 nm-800 nm,進而電流較高,但其能隙較小,開路電壓僅有 0.52 V,致 使光電轉換效率只有 2.18 %,但電流有 9.47 mA/cm2之良好表現。
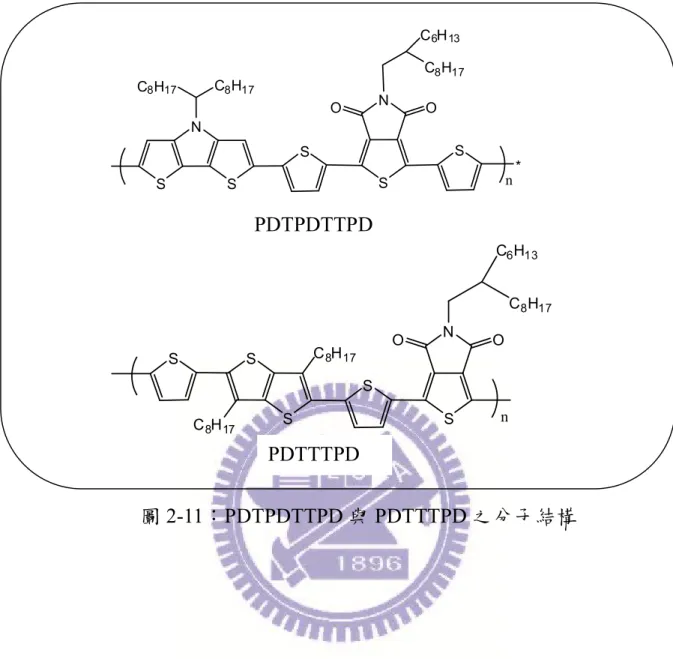
綜觀以上各單體之優點(高平面性及剛硬結構、強拉電子特性),我們 以 TT 單體及 DTP 單體為予體及 TPD 單體為受體,在 TPD 單體上之碳鏈, 參考文獻後我選用了八個碳的直鏈,再使用 Stille coupling 聚合法合成出新 穎之予體-受體型共聚高分子 PTTTPD 及 PDTPTPD(圖 2-10),期望此高 分子有良好的排列性、低 HOMO 能階及高開路電壓之優點。 圖 2-10:PDTPTPD 與 PTTTPD 之分子結構 同時在二個單體之間插入噻吩做為 spacer,此一動機及想法來自於 Doo-Kyung Moon 研究團隊,他們發現插入噻吩後,其排列性較為規則,且 在傳導電洞與電子的表現上也較好,結果也顯示,隔開後的高分子有較好 的表現,所以同樣地,我將 PTTTPD 及 PDTPTPD,都照樣插入噻吩合成出 PDTTTPD 及 PDTPDTTPD(圖 2-11)以期效率及電流值會因此而改善並提 升,同時在 TPD 的碳鏈上我也做了改變,因為導入噻吩後,使溶解度大幅 度的下降,因此我將直鏈的碳鏈改為分支的碳鏈,藉此提高溶解度以利進 行聚合反應及元件的製作。 N S S C8H17 C8H17 S N O O C8H17 n S S C8H17 S N O O C8H17 C8H17
圖 2-11:PDTPDTTPD 與 PDTTTPD 之分子結構 S S C8H17 S N O O C8H17 S S n C8H17 C6H13 N S S C8H17 C8H17 S N O O S S * n C8H17 C6H13 PDTTTPD PDTPDTTPD
第三章 實驗
3-1 試藥
實 驗 中 所 用 的 溶 劑 皆 購 自 Merck 、 Fischer Scientific 、 Aldrich 、 Mallickrodt、聯工等公司。無水四氫呋喃(tetrahydrofuran THF)及無水乙 醚(ether)皆以鈉金屬除水,並加入二苯甲酮(benzophenone)為指示劑, 在 氮 氣 條 件 下 迴 流 一 日 後 蒸 餾 出 使 用 , 無 水 甲 苯 ( toluene )、 氯 苯 (chlorobenzene)、鄰二氯苯(o-dichlorobenzene)皆以氫化鈣除水,在氮氣 條件下迴流一日後蒸餾出使用。 所使用之藥品來源如下: Thiophene Aldrich
Trimethyltin chloride Aldrich
Bromine Across
Zinc powder Fluka
Copper powder Showa
Ethylene glycol TEDIA
Oxalyl dichloride Alfa Aesar
1-bromooctane Acros
Copper(I) cyanide Sigma-Aldrich
LDA Acros
N-bromosuccinimide Acros
tri(o-tolyl)phosphine TCI
Octylamine Fluka
Ferric chloride Aldrich
Potassium hydroxide Sigma-Aldrich
2-(tributystannyl)thiophene Aldrich
nonanoyl chloride fluka
Potassium carbonate Sigma-Aldrich
18-crown-6 fluka
ethyl mercaptoacetate sigma
Sodium hydroxide Sigma-Aldrich
Tetrabutylammonium iodide fluka
Quinoline Aldrich
triethylamine Sigma-Aldrich
3-bromothiophene Aldrich
Copper(II) chloride Aldrich
sodium-butoxide Aldrich
Tris(dibenzylideneacetone)dipalladium(0) Aldrich
3-2 使用儀器與性質測量
3-2-1 核磁共振光譜儀(Nuclear Magnetic Resonance,NMR)
使用 Varian Unity-300、500MHz 核磁共振儀。以 d-chloroform 為溶 劑時,氫譜的溶劑訊號 δ=7.26 ppm 為內標準,碳譜則以 δ=77 ppm 為 內標準。而 d-DMSO 為溶劑時,氫譜的溶劑訊號 δ=2.49 ppm 為內標準。 光譜資料中:s 符號代表單峰(singlet),d 代表二重峰(doublet),t 代
表三重峰(triplet),q 代表四重峰(quartet),m 代表多重峰(multiplet)。
3-2-2 凝膠滲透層析儀(Gel Permeation Chromatography,GPC) 使用 Water 410 Didderential Refractometer 及 Water 600 Controller (Waters Styragel Column),以 polystyrene(PS)標準品製作分子量校
正曲線。樣品溶液之配製為每 2.0 mg 溶於 1.0 mL chloroform(CHCl3)
中,並以 0.2μm 的[poly(tetrafluoroethylene), PTFE] filter 過濾。測試時
以 CHCl3為沖提液,流速 1 mL/min 並保持於 40 ℃的恆溫槽中。
3-2-3 紫外-可見光吸收光譜儀(UV-Vis Absorption Spectrometry) 製造商:HP,型號:Agilent-8453。
Solution: 利用逐步稀釋法配置樣品濃度在各別溶液中,使其 UV-vis 的 最大吸收值介於 0.05 左右。
Film: 配置樣品濃度在各別溶液中的濃度為 1.0 wt%,以 2.5×2.5×0.15
石英玻璃。
3-2-4 循環伏特安培計(Cyclic Voltammetry,CV)
使用 Autolab 的 AVD 164 型電位儀來紀錄氧化-環原電位。以 Acertonitrile 為溶劑,配置 0.1 M tetrabutylammonium hexafluorosphate
(TBAPF6) 的電解液 10 mL,通入氮氣 5 分鐘,將待測的樣品配製為 1mg /200μL (溶於 CHCl3中),塗佈於工作電極上,Ag/Ag + 為參考電極,並 以 ferrocene/ferrocenium(Fc/Fc+)為內參考電極,以碳電極為工作電極, 白金絲為輔助電極,掃瞄速率為 100 mV/s,範圍-200~2000 mV。 3-2-5 微差掃瞄式卡計( Differential Scanning Calorimeter,DSC )
使用 Perkins Elmer Pyris DSC1 及水浴系統。溫度以 indium 及 tin 做校正,取樣品 2—5 mg 的樣品裝入鉑製 cell 中,在通入氮氣流速 20 mL/min 的條下,做九階段式升溫 DSC 測試: 1. 恆溫於 40 ℃下,固定 5 min 2. 升溫速率 20 ℃/ min,範圍為 40℃~ 450℃ 3. 恆溫於 450 ℃下,固定 5 min 4. 升溫速率-40 ℃/ min,範圍為 450℃~ 40℃ 5. 恆溫於 40 ℃下,固定 5 min 6. 升溫速率 20 ℃/min,範圍為 40℃~ 450℃ 7. 恆溫於 450 ℃下,固定 5 min
8. 升溫速率-40 ℃/min,範圍為 450℃~ 40℃ 9. 恆溫於 40 ℃下,固定 5 min
3-2-6 熱重分析儀(Thermal Gravimetric Analyzer,TGA)
使用 Du Pont TGA-2950 熱重分析儀。實驗時秤取樣品 2—10 mg 於 鉑製 cell 中,在通入氮氣流速為 100 mL/min 的條件下,以 10 ℃/min 的 升溫速度,從 40℃升溫至 900℃來觀察裂解情形。 3-2-7 旋轉塗佈機(Spin Coater) 製造商:LAURELL,型號:MODEL WS-400B-6NPP/LITE。 用以將配製好的高分子溶液,以旋轉塗佈的方式在石英片、ITO 玻 璃等基材上製成薄膜。 3-2-8 模擬光源
製造商:New port,型號:6902 Oriel 150W Xenon lamp solar simulator。 太陽光會受到各種因素干擾,使用模擬太陽光源可將實驗參數固定 於相同條件下,且不受天氣因素影響。利用氙氣搭配透鏡可以模擬出光 源頻譜強度為 AM 1.5 的均勻穩定光源。 3-2-9 光強度偵測計 製造商:OPHIR thermalpie。利用熱感應的方式來測量光的強度, 用來搭配模擬太陽光源,可以校正光源的強度來達到所要求的強度與頻
譜。 3-2-10 電性分析儀 製造商:Keithley,型號:Model 236。用以量測元件的 I-V 曲線, 並計算光電效率。靈敏度 10 fA,10μV。 3-2-11 原子力顯微鏡 製造商:Nanoscope IIIa。 以 tapping mode 的方式掃描樣品的表面形貌( 5×5 m2 ) 3-2-12 探針式表面輪廓儀 製造商:Veeco,型號:Dektak 150。用以量測樣品之膜厚。 3-3 合成 3-3-1 單體 M1 2,3,4,5-tetrabromothiophene(1) [32] 在氮氣下,將 thiophene(20.00 g, 237.70 mmole)、CHCl3 30.0 ml 加入雙頸瓶中。於 0℃下,再將 Bromine(159.54 g, 998.33 mmole)緩 慢加入,接著於室溫下攪拌一小時,接著恆溫在 60 ℃~70 ℃間,反應 12 小時。冷卻至室溫,將反應液倒入 Na2S2O3水溶液中清洗,將白色固 體產物過濾,濾液再以 EtOAc 萃取多次,收集有機層,再將白色固體 產物加入有機層中以溶解之,再以 Na2S2O3水溶液清洗有機層,收集有 機層,以無水硫酸鎂除水,濃縮,再以乙醇作再結晶,得白色針狀固體
S Br Br 71.26 g,產率 75.0%。 13 C NMR (300MHz, CDCl3):δ 116.9, 110.3(附圖一) 1 3,4-dibromothiophene(2) [32]
將 1(50.00 g, 125.08 mmole)、Zn(31.57 g, 482.80 mmole)、glacial
acetic acid 80.0 ml、水 160.0 ml 加入雙頸瓶中,於室溫下攪拌一小時, 接著恆溫於 120℃下,反應 3 小時。冷卻至室溫,過濾除去結塊的鋅粉, 將 200.0 ml EtOAc 加入濾液,在以 EtOAc 萃取多次,有機層再以食鹽 水清洗,收集有機層,以無水硫酸鎂除水,濃縮,以 n-hexane 進行管柱 層析,得到無色液體 22.64 g,產率 74.8%。 1 H NMR (300 MHz, CDCl3):δ 7.30 (s, 2H). (附圖二) 13 C NMR (300MHz, CDCl3):δ 123.8, 113.9.(附圖三) 2 3,4-dicyanothiophene(3) [33] 在氮氣下,將 2 (18.80 g, 77.71 mmole)、CuCN(25.00 g, 279.14 S Br Br Br Br
mmole)及 anhydus DMF 30.0 ml 置於雙頸瓶中,並恆溫在 150 ℃下, 反應 12 小時。冷卻至室溫。在此同時將 FeCl3(90.00 g, 554.87 mmole)、 HCl(aq)(2.0 M, 120.0 mL)加入錐形瓶中,並恆溫在 60 ℃~70 ℃間攪 拌 30 分鐘。接著將反應液倒入 FeCl3 /2M HCl(aq)中並恆溫於 60 ℃~70 ℃ 間 3 小時。冷卻至室溫,過濾除去多餘 FeCl3黑色固體,濾液再以 CH2Cl2 進行多次萃取,有機層以飽合 NaHCO3水溶液清洗,接著食鹽水清洗, 收集有機層,以無水硫酸鎂除水,濃縮,再以 EtOAc:n-hexane = 1: 5 進行管柱層析,得到白色固體 4.69 g,產率 45.0 %。 1 H NMR (300 MHz, CDCl3):δ 8.06 (s, 2H).(附圖四) 13 C NMR (300MHz, CDCl3):δ 137.0, 113.0, 111.8(附圖五) 3 thiophene-3,4-dicarboxylic acid(4) [33]
在氮氣下,將 3(4.60 g, 34.29 mmole)、KOH(18.30 g, 326.15 mmole)
及 ethylene glycol 70.0 ml 加入雙頸瓶中,於 197 ℃下攪拌 12 小時。冷
卻至室溫,之後將反應液倒入 HCl(aq)(5.0 M, 80.0 ml)中攪拌一小時,
接著將粉紅色固體產物過慮,濾液再以 EtOAc 萃取多次,收集有機層,
S
縮,再以熱水作再結晶,得到粉色針狀固體 3.60 g,產率 61.0%。 1 H NMR (300 MHz, d-DMSO):δ 13.64 (br, 2H), 8.14(s, 1H).(附圖六) 13 C NMR (300MHz, d-DMSO):δ 164.1, 152.3, 133.2 4 2,5-dibromothiophene-3,4-dicarboxylic acid(5) [34]
在氮氣下,將 4(3.00 g, 17.43 mmole)、glacial acetic acid 40.0 ml 加入雙頸瓶中。於室溫下攪拌,再將 Bromine(20.06 g, 125.50 mmole) 緩慢加入,反應 12 小時。將反應液倒入 NaHSO4水溶液中清洗,再以 EtOAc 萃取多次,收集有機層,有機層再以食鹽水清洗,收集有機層, 接著以無水硫酸鎂除水,濃縮,再以熱水作再結晶,得到白色固體 3.74 g,產率 65.0%。 1 H NMR (300 MHz, d-DMSO):δ 13.64 (br, 2H). (附圖七) 13 C NMR (300MHz, d-DMSO):δ 162.1, 134.7, 114.0(附圖八) 5 S HOOC COOH S HOOC COOH Br Br
2,5-dibromothiophene-3,4-dicarboxylic acid chloride(6) [34]
在氮氣下,將 5 (3.70 g, 11.21 mmole)、甲苯 40.0 ml 及一滴 anhydrous DMF 加入雙頸瓶中攪拌,接著緩慢加入 oxalyl chloride(11.38
g, 89.66 mmole),然後恆溫於 90℃下,反應一小時。冷卻至室溫,加熱
抽真空除去剩餘的 oxalyl chloride、甲苯,再加入 150.0 ml EtOAc,以無 水硫酸鎂除水,濃縮,65℃下抽真空,得到咖啡色固體 3.08 g,產率 75.0%。
MS (m/z): [M]+ calcd for C6Br2Cl2O2S, 365.7; found, 365. (附圖九)
6
1,3-dibromo-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione(M1)[35]
在氮氣下,將 6(0.40 g, 1.09 mmole)、octan-1-amine(0.26 g, 1.09 mmole)
加入雙頸瓶中攪拌,並恆溫於 140℃下,反應 6 小時。冷卻至室溫,以
飽和 NaHCO3(aq)清洗,再以 EtOAc 萃取多次,收集有機層,萃取的有
機層再以食鹽水清洗,收集有機層,接著以無水硫酸鎂除水,濃縮,再 以 EtOAc:n-hexane = 1:10 進行管柱層析,得到白色固體 0.41 g, 產率 70.0%。 S Br Br O O Cl Cl
1.24-1.29 (m, 10H), 0.83-0.87 (t, J=6.4 Hz, 3H)(附圖十) 13 C NMR (75 MHz, CDCl3): 160.4, 134.8, 112.9, 38.8, 31.7, 29.7, 29.1, 28.2, 26.8, 22.6, 14.1(附圖十一) Anal. Calcd: C,39.74; H,4.05; N,3.31. Found: C,40.00; H,4.56; N,3.16 M1 3-3-2 單體 M2[35] 1,3-dibromo-5-(2-hexyldecyl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (M2) 在氮氣下,將 6(0.40 g, 1.09 mmole)、2-hexyldecan-1-amine(0.26 g, 1.09 mmole)加入雙頸瓶中攪拌,並恆溫於 140 ℃下,反應 6 小時。
冷卻至室溫,以飽和 NaHCO3(aq)清洗,再以 EtOAc 萃取多次,收集有
機層,萃取的有機層再以食鹽水清洗,收集有機層,接著以無水硫酸鎂 除水,濃縮,再以 EtOAc:n-hexane = 1:10 進行管柱層析,得到白 色固體 0.41 g,產率 70.0 %。 1 H NMR (300 MHz, CDCl3): 0.81–0.94 (m, 6H), 1.10–1.40 (m, 24H), S N O O C8H17 Br Br
1.75–1.85 (m, 1H), 3.48 (d, J = 7.2 Hz, 2H).(附圖十二)
13
C NMR (75 MHz, CDCl3): 14.1, 22.6, 26.2, 29.3, 29.5, 29.9, 31.4, 31.8,
36.8, 43.1, 112.9, 134.7, 160.6.(附圖十三)
MS (m/z): [M]+ calcd for C22H33Br2NO2S, 535.1; found, 535. (附圖十 四)
Anal. Calcd: C, 49.36; H, 6.21; N, 2.62. Found: C, 49.64; H, 6.16; N, 2.62.
M2 3-3-3 單體 M3 5-(2-hexyldecyl)-1,3-di(thiophen-2-yl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione(8)[36] 將 M2(0.3 g, 0.56 mmol)、tributyl(thiophen-2-yl)stannanl(0.54 ml, 1.68 mmol),用 THF 10 ml 溶解,並在 50 ℃,purged 10 分鐘後,再加 入 PdCl2(PPh3),升溫至 80 ℃,迴流反應一天後,冷卻至室溫,倒入水 中,用 CH2Cl2萃取,收集有機層,並用硫酸鎂除水,減壓濃縮抽乾。 再以 CH2Cl2:n-hexane=1:1 進行管柱層析,得一黃色固體,產率 75 %。 S Br Br N O O C8H17 C6H13
1 H NMR (300 MHz, CDCl3): 8.03 (dd, J=3.6 Hz, 2H), 7.44 (dd, J=5.1 Hz, 2H), 7.13 (t, J = 4.2 Hz, 2H), 3.56 (d, J = 7.2 Hz, 2H), 1.89 (m, 1H), 1.25–1.48 (m, 20H) , 0.84–0.88 (m, 6H)(附圖十五) 13 C NMR (75 MHz, CDCl3): 162.6, 135.1, 133.7, 131.1, 129.7 128.6, 116.7, 37.0, 31.9, 31.8, 31.6, 31.5, 29.9, 29.7, 29.5, 29.3, 22.7, 22.6, 14.1, 14.0.(附圖十六) 8 1,3-bis(5-bromothiophen-2-yl)-5-(2-hexyldecyl)-4H-thieno[3,4-c]pyrrole -4,6(5H)-dione(M3)[36] 將 8(0.1 g, 0.18 mmol)置入雙頸瓶中,並加入 CHCl3:Acetic acic=1:1(7ml)溶劑,並將反應瓶浸在冰浴中,分批在冰浴下加入 NBS(0.07 g, 0.41 mmol),加完 NBS 後,移除冰浴,在室溫下反應 24 小時,再倒入水中,用 Chloroform 萃取,收集有機層,加入硫酸鎂除 水,減壓濃縮後,用 CH2Cl2:n-hexane = 4:1 進行管柱層析,得一黃 色固體,產率 78 %。 1 H NMR(300 MHz, CDCl3 , δ ): 7.66 (dd, 2H, J = 3.9 Hz), 7.08 (dd, 2H, J = S N O O C8H17 C6H13 S S
5.1 Hz), 3.53 (d, 2H, J = 7.2 Hz), 1.86 (m, 1H), 1.25 (m, 20H), 0.84-0.88 (m, 6H).(附圖十七) 13 C NMR (75 MHz, CDCl3): 162.6, 135.1, 133.7, 131.1, 129.7 128.6, 37.0, 31.9, 31.8, 31.6, 31.5, 29.9, 29.7, 29.5, 29.3, 22.7, 22.6, 14.1, 14.0.(附圖十 八) Anal. Calcd: C,51.50; H,5.33; N,2.00. Found: C,50.54; H,5.41; N,1.78 M3 3-3-4 單體 M4 2,3,5-tribromo-4-octylthiophene(9)[24]
在氮氣下將 1(10.00 g, 50.93 mmol)、glacial acetic acid 20ml 置入 雙頸瓶中,混合均勻後,再將 bromine (10.97 ml, 213.28 mmol)緩慢 滴入,接著於室溫下反應 4 小時,再加溫至 70 ℃,並保持此溫度反應 overnight。冷卻至室溫,將反應液倒入 Na2S2O3水溶液中清洗,再以 NaOH 水溶液調至中性,並用 EtOAc 萃取多次,萃取的有機層再用食鹽水清 水,收集有機層,以無水硫酸鎂除水,並減壓濃縮,以 n-hexane 進行管 S N O O C8H17 C6H13 S S Br Br
柱層析,得到無色液體 31.6g,產率 77 %。 13 C NMR (75 MHz, CDCl3) : δ141.5, 115.7, 109.4, 108.0, 31.8, 29.7, 29.3, 29.2, 29.1, 28.5, 22.7, 14.1(附圖十九) 9 3-bromo-4-octylthiophene(10)[24] 將 2(15.6 g, 36.02 mmol)置入雙頸瓶中,並抽真空灌氮氣三次, 加入無水 THF 90 ml,降溫至-78 ℃,慢慢滴入 n-BuLi(28.82 mL, 72.05 mmol),並保持此溫度 10 分鐘。將反應液倒入水中,並以 EtOAc 萃取, 收集有機層,以無水硫酸鎂除水,並減壓濃縮,以 n-hexane 進行管柱層 柱,得到無色液體 4.46 g,產率 45 %。 1 H NMR (300 MHz, CDCl3): δ 7.20 (s, 1H), 6.93 (s, 1H), 2.55 (t, J=8.1 Hz, 2H), 1.57-1.62 (m, 2H), 1.24-1.31 (m, 10H), 0.88 (t, J=6.9 Hz, 3H)(附圖二 十) 13 C NMR (75 MHz, CDCl3) : δ 141.7, 122.6, 120.5, 112.8, 31.9, 29.9, 29.7, 29.4, 29.3, 29.2, 22.7, 14.1(附圖二十一) S C8H17 Br Br Br
10
1-(3-bromo-4-octylthiophen-2-yl)nonan-1-one(11)[24]
於氮氣下,加入 3(10.00 g, 36.36 mmol)、AlCl(9.6 g, 72.72 mmol)3 、
CH2Cl2 50 ml,混合均勻後,再慢慢滴入 nonanoyl chloride(6.55 ml, 36.36 mmol),並於室溫下反應 2 小時後,倒入鹽酸水溶液 6M 200 ml,並用 CH2Cl2萃取,並用食鹽水清洗有機層,收集有機層,以無水硫酸鎂除水, 並減壓濃縮。此步得到為混合物,但無純化即進行下一步。 11 ethyl 3,6-dioctylthieno[3,2-b]thiophene-2-carboxylate(12)[24] 於氮氣下,加入 4(13.18 g, 31.72 mmol)、K2CO3(9.36 g, 65.03 mmol)、催化劑 18-crown-6、DMF(65 ml),混合均勻後,加溫至 65 ℃
後 , 再 加 入 ethyl mercaptoacetate ( 3.56 ml, 31.72 mmol), 並 反 應 overnight。冷卻至室溫,並倒入水中,並以 EtOAc 萃取多次,收集有機 S C8H17 Br S C8H17 Br O C8H17
層,以無水硫酸鎂除水,並減壓濃縮,此步得到為混合物,但無純化即 進行下一步。
12
3,6-dioctylthieno[3,2-b]thiophene-2-carboxylic acid(13)[24]
在氮氣下,將 5(12 g, 27.47 mmol)、NaOH 19.7 ml (10%)、THF
(45 ml)、MeOH(7.7ml)、催化劑 tetrabutylammonium iodide 加入並混
合均勻,加溫至 80℃,並反應 3 小時。冷卻至室溫,倒入水中,加入 HCl 10ml,以 EtOAc 萃取多次,收集有機層,以無水硫酸鎂除水,並 減壓濃縮,以 n-hexane:EtOAc=9:1 進行管柱層柱,得到棕色液體 2.8 g,產率 25 %。 1 H NMR (300 MHz, CDCl3): δ 7.31 (s, 1H), 3.20 (t, J=7.8 Hz 2H), 2.76 (t, J=7.8 Hz 2H), 1.61-1.79 (m, 4H), 1.22-1.38 (m, 20H), 0.92 (t, J=6.9 Hz 6H) (附圖二十二) 13 C NMR (75 M Hz, CDCl3) : δ 180.1, 168.7, 157.9, 145.7, 142..6, 141.0, 135.6, 125.9, 34.1, (附圖二十三) S S C8H17 C8H17 EtOOC
13 3,6-dioctylthieno[3,2-b]thiophene(14)[24] 在氮氣下,將 6(2.8 g, 6.85 mmol)、copper powder(0.28 g, 4.52 mmol)、quinoline 20 ml,混合均勻後加溫至 250 ℃,並反應 1 小時。冷 卻至室溫,倒入水中,加入 HCl 水溶液 2M 100 ml,以 hexane 萃取多 次,收集有機層,以無水硫酸鎂除水,並減壓濃縮,以 n-hexane 進行管 柱層柱,得到無色液體 1.5 g,產率 60 %。 1 H NMR (300 MHz, CDCl3): δ 6.93 (s, 2H), 2.69 (t, J=7.8 Hz 4H), 1.67-1.75 (m, 4H), 1.24-1.32 (m, 20H), 0.86 (t, J=6.3 Hz 6H)(附圖二十四) 13 C NMR (75 M Hz, CDCl3) : δ 139.2, 135.4 120.8, 31.9, 29.8, 29.7, 29.4, 29.2, 28.7, 22.7, 14.1(附圖二十五) 14 S S C8H17 C8H17 HOOC S S C8H17 C8H17
2,5-dibromo-3,6-dioctylthieno[3,2-b]thiophene(15)[24]
在氮氣下,將 7(1.4 g, 3.83 mmol)、CH2Cl2 60 ml、glacial acetic cid
20 ml,混合均勻後,再將 N-Bromosuccinimide(1.37 g, 7.68 mmol)分 四次加入,每次約間隔 10 分鐘,反應 3 小時後。將反應液倒入水中, 並以 EtOAc 萃取,收集有機層,以無水硫酸鎂除水,並減壓濃縮,以 n-hexane 進行管柱層柱,得到無色液體 1.4 g,產率 70 %。 1 H NMR (300 MHz, CDCl3): δ 2.67 (t, J=8.1 Hz 4H), 1.66 (m, 4H), 1.27-1.31 (m, 20H), 0.88 (t, J=6.6 Hz 6H)(附圖二十六) 13 C NMR (75 MHz, CDCl3) : δ 136.0, 134.4, 109.4, 31.9, 29.7, 29.3, 29.1, 28.9, 28.1, 22.6, 14.1(附圖二十七) 15 (3,6-dioctylthieno[3,2-b]thiophene-2,5-diyl)bis(trimethylstannane)(M4) [24] 將 10(0.5 g, 0.728 mmol)置入雙頸瓶中,抽真空灌氮氣三次,打 入無水 THF 10ml,降溫至-78 ℃,並慢慢滴入 n-BuLi(0.64 ml,1.6
mmol),保持此溫度二小時,再打入 trimethyltin chloride(1.82 ml, 1.82
S S C8H17 C8H17 Br Br
mmol),再保持在-78 ℃二小時,並自然回溫,反應 overnight。將反應 液減壓濃縮除去 THF,再加入 n-hexane,將鹽類固體過濾除去,再濃縮, 以 triethylamine 清洗 2 小時;重複上述步驟多次直到沒有鹽類固體產 生,濃縮除去 triethylamine,加入 n-hexane,再以食鹽水清洗兩次,收 集有機層,接著以無水硫酸鎂除水,濃縮,得黃色固體 0.25g,產率 40 %。 1 H NMR (300 MHz, CDCl3): δ 2.65 (t, J=7.8 Hz 4H), 1.66-1.71 (m, 4H), 1.24-1.31 (m, 20H), 0.88-0.84 (m, 6H), 0.28-0.46( m, 18H) (附圖二十八) Anal. Calcd: C, 48.72; H, 7.59; Found: C, 49.54; H, 8.14 M4 3-3-5 單體 M5 3,6-dioctyl-2,5-di(thiophen-2-yl)thieno[3,2-b]thiophene(16) 將 8 ( 1.57g,3.00mmol )、 2-(tributystannyl)thiophene ( 2.4mL,
7.5mmol)、tris(dibenzylideneacetone)dipalladium(0)(56mg, 0.06mmol)、
tri(o-tolyl)phosphine(146mg, 0.48mmol),置入後,purged 十分鐘,再
S S C8H17 C8H17 Sn Sn
反應 1 小時。減壓濃縮除去 toluene,以 n-hexane 進行管柱層析,並再 以 hexane 再結晶,得黃色固體 1.4g,產率 88%。 1 H NMR (300 MHz, CDCl3): δ 7.34 (d, J=5.1 Hz 2H), 7.17 (d, J=3.6 Hz 2H), 7.09 (td, J=1.5, 3.6 Hz 2H), 2.87 (t, J=8.4 Hz 4H), 1.73-1.78 (m, 4H), 1.25-1.41 (m, 20H), 0.86 (t, J=7.2 Hz 6H)(附圖二十九) 13 C NMR (75 MHz, CDCl3) : δ 138.5, 136.5, 131.9, 131.3, 127.5, 126.1, 125.7, 31.9, 29.7, 29.3, 29.2, 29.1, 28.7, 22.6, 14.1(附圖三十) 16 2,5-bis(5-bromothiophen-2-yl)-3,6-dioctylthieno[3,2-b]thiophene(17)
在氮氣下,將 9(1.46g, 2.76mmol)、CH2Cl2 110mL、glacial acetic
acid 45mL,混合均勻後,再將 N-Bromosuccinimide(1.01g, 5.66mmol) 分四次加入,每次約間隔 10 分鐘,反應 3 小時後。加入 methanol 後, 產生絲狀黃色固體,抽氣過濾,收集濾餅,並以 hexane 再結晶,得黃 色絲狀固體 1.1g,產率 58%。 1 H NMR (300 MHz, CDCl3): δ 7.04 (d, J=3.9 Hz 2H), 6.9 (d, J=3.9 Hz 2H), 2.82 (t, J=8.4 Hz 4H), 1.70-1.75 (m, 4H), 1.28-1.39 (m, 20H), 0.88 (t, J=6.6 S S C8H17 C8H17 S S
Hz 6H)(附圖三十一) 13 C NMR (75 MHz, CDCl3) : δ 138.6, 137.9, 132.4, 130.6, 130.3, 126.5, 112.4, 31.9, 29.6, 29.3, 29.2, 29.0, 28.7, 22.7, 14.1(附圖三十二) 17 (5,5'-(3,6-dioctylthieno[3,2-b]thiophene-2,5-diyl)bis(thiophene-5,2-diyl)) bis(trimethylstannane)(M5) 將 10(0.5g, 0.728mmol)置入雙頸瓶中,抽真空灌氮氣三次,打入 無水 THF 10ml,降溫至-78℃,並慢慢滴入 n-BuLi(0.64mL,1.6mmol), 保持此溫度二小時,再打入 trimethyltin chloride(1.82mL, 1.82mmol), 再保持在-78℃二小時,並自然回溫,反應 overnight。將反應液減壓濃 縮除去 THF,再加入 n-hexane,將鹽類固體過濾除去,再濃縮,以 triethylamine 清洗 2 小時;將鹽類固體過濾除去,再濃縮,再加入 n-hexane,將鹽類固體過濾除去,再濃縮,以 triethylamine 清洗 2 小時, 重複上述步驟多次直到沒有鹽類固體產生,濃縮除去 triethylamine,加 入 n-hexane,再以食鹽水清洗兩次,收集有機層,接著以無水硫酸鎂除 S S C8H17 C8H17 S S Br Br
水,濃縮,得黃色固體 0.25g,產率 40%。 1 H NMR (300 MHz, CDCl3): δ 7.27 (d, J=3.3 Hz 2H), 7.16 (d, J=3.3 Hz 2H), 2.88 (t, J=8.1 Hz 4H), 1.74-1.76 (m, 4H), 1.25-1.42 (m, 20H), 0.87 (t, 6.4 Hz 6H)(附圖三十三) 13 C NMR (75 MHz, CDCl3) : δ 142.3, 138.5, 138.4, 135.5, 131.5, 131.4, 127.0, 31.9, 29.7, 29.3, 29.2, 29.0, 28.8, 22.7, 14.1, -8.2(附圖三十四) Anal. Calcd: C, 50.60; H, 6.61; Found: C, 51.98; H, 7.21 M5 3-3-6 單體 M6 heptadecan-9-amine(16) 由本實驗室陳冠宇學長提供。 1 H NMR (300 MHz, CDCl3): δ 2.64 (s, 1H), 1.25-1.36 (m, 28H), 0.85 (t, 6.3 Hz 6H)(附圖三十五) S S C8H17 C8H17 S S Sn Sn
13 C NMR (75 MHz, CDCl3):51.2, 38.1, 31.7, 29.8, 29.6, 29.3, 26.2, 22.6, 14.1(附圖三十六) 16 3,3'-dibromo-2,2'-bithiophene(17)[26] 將 雙 頸 瓶 抽 真 空 , 並 灌 氮 氣 , 重 覆 此 動 作 3 次 後 , 打 入 3-bromothiophene(2 g,12 mmol),再加入 THF(20 ml),降溫至-78 ℃, 慢慢滴入 LDA(6.1 ml, 12 mmol),滴完後並保持在低溫 1 小時,此時 再加入氯化銅(1.6 g, 12 mmol),再保持在低溫 2 小時後,回溫至室溫 反應 overnight。將水溶液倒入 2M HCl 200 ml 水溶液中,以 EtoAC 萃 取並收集有機層,用硫酸鎂除水,並抽乾,以 n-hexane 進行管柱層析, 再以 hexane 再結晶。 1 H NMR (300 MHz, CDCl3): δ 7.39 (d, J=5.1 Hz 2H), 7.06 (d, J=5.4 Hz 2H), (附圖三十七) 13 C NMR (75 MHz, CDCl3):130.8, 128.9, 127.5, 112.6 (附圖三十八) S S Br Br C8H17 C8H17 NH2
4-(heptadecan-9-yl)-4H-dithieno[3,2-b:2',3'-d]pyrrole (18)[26]
將 19(0.5 g, 1.55 mmol),sodium-butoxide(0.36 g, 3.72 mmol),
Pd2dba3(35 mg, 0.038 mmol),BINAP(96 mg, 0.15 mmol),放入雙頸
瓶中,再將 heptadecan-9-amine(compound 16)以 4 ml 的 dry toluene 溶解後,加入雙頸瓶中,升溫 110 ℃,反應 overnight。反應不需降溫, 直接倒入水中,以 ether 萃取,收集有機層,並以硫酸鎂除水,抽乾。 以 n-hexane 過 cloumn,得白色固體。 1 H NMR (300 MHz, CDCl3): δ 7.08 (d, J=5.4 Hz 2H), 6.99 (d, J=5.1 Hz 2H), 4.15-4.25 (m, 1H), 1.99-2.02 (m, 4H), 1.76-1.85 (m, 4H), 1.15-1.46 (m, 20H), 0.84 (t, J=6.6 Hz 6H)(附圖三十九) 13 C NMR ( 75 MHz, CDCl3):144.1, 122.3, 114.7, 111.8, 59.7, 35.1, 31.7, 29.3, 29.2, 29.1, 26.5, 22.6, 14.1(附圖四十) 18 4-(heptadecan-9-yl)-2,6-bis(trimethylstannyl)-4H-dithieno[3,2-b:2',3'-d] pyrrole(M4)[26] 將 19 (0.3 g, 0.72 mmol),置入雙頸瓶中,並打入 THF(10 ml), N S S C8H17 C8H17
降溫至-78 ℃後,打入 n-Buli(0.63 ml, 1.58 mmol),並保持在低溫下 1 小時,再移除低溫,保持在室溫攪拌 3 小時,再降至-78 ℃,打入 1M Trimethyltin chloride ( 1.8 ml, 1.8 mmol ) 後 , 使 其 回 至 室 溫 反 應 overnight,將反應液減壓濃縮除去 THF,再加入 n-hexane,將鹽類固體 過濾除去,再濃縮,以 triethylamine 清洗 2 小時;將鹽類固體過濾除去, 再濃縮,再加入 n-hexane,將鹽類固體過濾除去,再濃縮,以 triethylamine 清洗 2 小時,重複上述步驟多次直到沒有鹽類固體產生,濃縮除去 triethylamine,加入 n-hexane,再以食鹽水清洗兩次,收集有機層,接 著以無水硫酸鎂除水,濃縮,得一深綠色液體,產率 72 %。 1 H NMR (300 MHz, CDCl3): δ 6.97 (s, 2H), 4.15-4.25 (m, 1H), 1.99-2.02 (m, 4H), 1.76-1.85 (m, 4H), 1.15-1.46 (m, 20H), 0.84 (t, J=6.6 Hz 6H): 0.28-0.48 (m, 12H)(附圖四十一) 13 C NMR (75 MHz, CDCl3):147.0, 134.9, 122.7, 122.0, 118.8, 111.9, 59.6, 35.1, 31.8, 29.7, 29.3, 29.2, 26.6, 22.6, 14.1, -8.1(附圖四十二) Anal. Calcd: C,50.09; H,7.46; N,1.88. Found: C,52.98; H,7.75; N,1.78 M6 N S S C8H17 C8H17 Sn Sn
3-3-7 高分子 PTTTPD 之聚合
在氮氣下,將單體 M1(100 mg, 0.24 mmol)、M4(166 mg, 0.24
mmol)、triphenylphosphate(5.8 mg, 0.019mmol)、無水 chlorobenzene
加入雙頸瓶中,待其攪拌至完全溶解,並進行除氧充氮氣 10 分鐘在 60 ℃下。再加入 Tris(dibenzylideneacetone)dipalladium(4.4 mg , 0.005mmol) 後,加熱至 130 ℃反應 48 小時後,再加入 2-tributylstannylthiophene(0.16 ml, 0.492 mmol),使其再反應 3 小時後,再加入 2-bromothiophene(0.05 ml, 0.492 mmol),再反應 3 小時進 end-capping 反應。冷卻至室溫,將 反應液滴入甲醇做再沉澱,抽氣過濾收集沉澱的固體,再用三氯甲烷將 其溶解,並以 0.5 μ PTFE filter 過濾,並再次滴入甲醇中進行再沉澱, 抽氣過濾收集沉澱的固體,罝於 60 ℃真空烘箱烘乾後,將固體置入 thimble filter 作 Soxhlet Extraction(甲醇清洗二天→正己烷清洗二天→ 丙酮清洗二天),收集到的高分子罝於 60℃真空烘箱烘乾,收集到 PTTTPD 194 mg,產率 73 %。 1 H NMR (300 MHz,CDCl3):δ3.66 (br, 2H), 2.99 (br, 4H), 2.74 (br, 2H), 1.68-1.81 (br, 12H), 1.27 (br, 24H), 0.86-0.88 (br, 9H)(附圖四十三) Anal. Calcd: C,69.57; H,8.76; N,2.13 Found: C,67.92; H,8.10; N,2.37
3-3-8 高分子 PDTTTPD 之聚合
合成與純化方式同高分子 PTTTPD。使用的藥品為單體 M2(100 mg, 0.18
mmol)、單體 M5(160 mg, 0.18 mmol)、triphenylphosphate(4.3 mg, 0.014
mmol)、Tris(dibenzylideneacetone)dipalladium(3.3 mg, 0.0036 mmol)、
2-tributylstannylthiophene(0.12 ml, 0.37 mmol)、2-bromothiophene(0.04
ml, 0.37 mmol),最後收到 PDTTTPD 177 mg,產率 68 %。 1 H NMR (300 MHz, CDCl3):δ7.89 (br,2H), 7.01 (br,2H), 3.56 (br,2H), 2.62 (br,4H), 1.23-1.56 (m,48H), 0.87 (s,12H). (附圖四十四) Anal. Calcd: C,69.21; H,7.93; N,1.55. Found: C,68.17; H,8.09; N,0.99 3-3-9 高分子 PDTPTPD 之聚合 合成與純化方式同高分子 PDTTPD。使用的藥品為單體 M1(100 mg,
0.24 mmol)、單體 M6( 176 mg, 0.24 mmol)、triphenylphosphate(5.8 mg,
0.019mmol ) 、 Tris(dibenzylideneacetone)dipalladium ( 4.4 mg , 0.005mmol )、 2-tributylstannylthiophene ( 0.16 ml, 0.492 mmol )、
2-bromothiophene(0.05 ml, 0.492 mmol),最後收到 PDTPTPD 193 mg,
產率 70 %。
1
H NMR (300 MHz, CDCl3):δ8.97 (br,1H), 7.05 (br,1H), 4.38 (br,2H),
四十五)
Anal. Calcd: C,69.44; H,8.53; N,3.95. Found: C,69.59; H,8.00; N,3.82
3-3-10 高分子 PDTPDTTPD 之聚合
合成與純化方式同高分子 PTTTPD。使用的藥品為單體 M3( 100
mg, 0.14 mmol)、M6(106 mg, 0.14 mmol) 、triphenylphosphate(3.4 mg,
0.01mmol ) 、 Tris(dibenzylideneacetone)dipalladium ( 2.6 mg ,
0.003mmol )、 2-tributylstannylthiophene ( 0.1 ml, 0.28 mmol )、
2-bromothiophene(0.03 ml, 0.28 mmol),最後收到 PDTPDTTPD 146 mg,產率 71 %。 1 H NMR (300 MHz, CDCl3):δ6.91-7.13 (br, 4H), 5.34 (br, 1H), 4.22 (br, 1H), 3.58 (br, 2H), 1.9-2.3 (br, 8H), 1.25 (br, 44H), 0.86(s, 12H)(附圖四十 六) Anal. Calcd: C,69.30; H,8.00; N,2.89. Found: C,65.56; H,7.98; N,2.44