國
立
交
通
大
學
應用化學研究所
博 士 論 文
多面體聚矽氧烷與高分子複合材之氫鍵作用力與高分子電解
質之研究
The Study on Hydrogen Bonding Interactions and Polyelectrolyte Properties of
Polyhedral Oligomeric Silsesquioxane/Polymer Composites
研 究 生:嚴英傑
多面體聚矽氧烷與高分子複合材之氫鍵作用力與高分子電解質之研究
The Study on Hydrogen Bonding Interactions and Polyelectrolyte Properties of
Polyhedral Oligomeric Silsesquioxane/Polymer Composites
研 究 生:嚴英傑 Student:Ying-Chieh Yen
指導教授:張豐志 Advisor:Feng-Chih Chang
國 立 交 通 大 學
應 用 化 學 系
博 士 論 文
A ThesisSubmitted to Department of Applied Chemistry College of Science
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Doctor of Philosophy
in
Applied Chemistry
October 2008
多面體聚矽氧烷與高分子複合材之氫鍵作用力
與高分子電解質之研究
學生:嚴英傑
指導教授:張豐志 教授
國立交通大學應用化學研究所 博士班
摘 要
多面體聚矽氧烷 (POSS) 衍生物於許多領域均有應用性。然而,探討其於高 分子電解質及質子傳導膜方面(能源儲存及來源方面)應用之論文卻十分稀少。本 論文中,改質後之多面體矽氧烷經由共價鍵結(化學鍵)及非共價鍵結(物理鍵) 之方法導入高分子中,形成新型高分子電解質及質子傳導膜。此外,我們也利用Painter-Coleman association model (PCAM) 理論計算改質後多面體聚矽氧烷分子
內作用力常數及其與高分子間作用力常數。
本研究首先探討具八酚官能基多面體聚矽氧烷分別與聚甲基丙烯酸甲酯
[poly(methyl methacrylate) (PMMA)]、聚乙烯必喀烷酮 [poly(vinyl pyrrolidone)
(PVP)],及兩者之共聚物 (PMMA-co-PVP) 形成摻合物之相溶性及作用力機制。
常數為 29,此常數小於聚乙烯苯酚 [poly(vinyl phenol) (PVPh)] 與聚甲基丙烯酸
甲酯間之作用力常數 (37) 及乙基苯酚 [ethyl phenol (EPh)] 與聚甲基丙烯酸甲
酯間之作用力常數 (101)。由上述可知,具八酚官能基多面體聚矽氧烷之酚官能 基與聚甲基丙烯酸甲酯之羰基作用之機率較其他二者低。此外,我們也發現,具 八酚官能基多面體聚矽氧烷之摻入可增加過氯酸鋰 (LiClO4) 與甲基丙烯酸甲酯 -乙烯必喀烷酮共聚物 (PMMA-co-PVP) 高分子電解質之離子導電度。 接著觀察具八酚官能基多面體聚矽氧烷摻入過氯酸鋰與聚甲基丙烯酸甲酯 高分子電解質後對於熱性質、形態,及作用力之影響。具八酚官能基多面體聚矽 氧烷與聚甲基丙烯酸甲酯二相摻合物中,具八酚官能基多面體聚矽氧烷傾向自身
聚集。因此,導致整體玻璃轉移溫度 (glass transition temperature) 下降。但於聚
甲基丙烯酸甲酯、過氯酸鋰,與具八酚官能基多面體聚矽氧烷三相摻合物中,具 八酚官能基多面體聚矽氧烷可形成約 20 奈米大小之聚集,導致整體之玻璃轉移 溫度上升。傅立葉轉換紅外線光譜 (FTIR) 量測結果指出,過氯酸鋰有助於增加 具八酚官能基多面體聚矽氧烷與聚甲基丙烯酸甲酯形成氫鍵作用力之機率。場發 射掃描式電子顯微鏡 (SEM) 、熱示差掃瞄卡量計 (DSC) ,及 X 光繞射儀 (XRD) 之結果均指出,過氯酸鋰之摻入是可改善具八酚官能基多面體聚矽氧烷於聚甲基 丙烯酸甲酯中之分散情形並使具八酚官能基多面體聚矽氧烷之物理效應由稀釋 之角色轉為物理交聯之角色。 在本研究之最後一部分中,我們將多面體聚矽氧烷導入磺化聚醚醚酮
[sulfonated poly(ether ether ketone) (SPEEK)] 中,形成一新型交聯型質子傳導
膜。在傳導膜中,奈米級交聯劑之分散性受其上有無磺酸根之影響而產生不同之
分散程度,導致親水區域之分散及聯結程度不同。其中,含 17.5 wt% 交聯劑之
新型質子傳導膜,具有高質子導電度 (0.0153 S/cm) 、低甲醇穿透率 (1.34 x 10-7
The Study on Hydrogen Bonding Interactions and
Polyelectrolyte Properties of Polyhedral
Oligomeric Silsesquioxane/Polymer Composites
Student:Ying-Chieh Yen
Advisors:Dr. Feng-Chih Chang
Department of Applied Chemistry
National Chiao Tung University
ABSTRACT
Polyhedral oligomeric silsequioxane (POSS) derivatives have the potential for the
application in several fields, however, they had rarely been used in the filed of polyelectrolyte
and proton exchange membrane which play a critical role in the people’s life because of the
need for energy storage and cleaning energy source. In this thesis, POSSs were modified to be
non-covalently and covalently incorporated into polymer matrix to form the new
polyelectrolyte and proton exchange membrane. In addition, the Painter–Coleman association
model (PCAM) was employed to theoretically study the intra-association and
Firstly, we investigated the miscibility behavior and mechanism of interaction of
poly(methyl methacrylate) (PMMA), poly(vinyl pyrrolidone) PVP, and PMMA-co-PVP
blends with octa(phenol)octasilsequioxane (OP-POSS). For the PMMA/OP-POSS binary
blend, the value of the association constant (KA = 29) was smaller than that in the poly(vinyl
phenol) (PVPh)/PMMA (KA = 37.4) and ethyl phenol (EPh)/PMMA (KA = 101) blend systems,
implying that the phenol groups of the OP-POSS units in the PMMA/OP-POSS blends
interacted to a lesser degree with the C=O groups of PMMA than they did in the other two
systems. In addition, the ionic conductivity of a LiClO4/PMMA-co-PVP polymer electrolyte
was increased after blending with OP-POSS.
Secondly, the thermal properties, morphologies, and interactions within the binary and
ternary blends of poly(methyl methacrylate) (PMMA), octa(phenol)octasilsesquioxane
(OP-POSS), and LiClO4 were described. In the binary PMMA/OP-POSS blends, the
OP-POSS molecules tend to aggregate and result in a decrease (19 °C) in the glass transition
temperature. In the ternary PMMA/LiClO4/OP-POSS blends, however, the OP-POSS
molecules form small sphere-like domains (20 nm) leading to the composite’s glass transition
temperature increasing by up to 30 °C. Based on these FTIR spectra, the addition of LiClO4
influenced the probability of hydrogen bonds formed between PMMA and OP-POSS and
these SEM micrographs, DSC, and XRD data indicated that the addition of LiClO4 is a
where the presence of LiClO4 changes the physical effect of OP-POSS from that of a diluent
role to a cross-linker role.
Finally, polyhedral oligomeric silsequoixane (POSS) was incorporated into sulfonated
poly(ether ether ketone) (SPEEK), forming a new cross-linked proton exchange membrane
(PEM). The distribution of these nano-scale cross-linkers were affected by their sulfonic acid
groups and dictated the water behavior and the dispersion and connectivity of hydrophilic
domains within these PEMs. A PEM formed by incorporating 17.5 wt% of the cross-linkers
(containing POSS molecules and sulfonic acid groups) into SPEEK exhibited high proton conductivity (0.0153 S/cm), low methanol permeability (1.34 × 10–7
cm2/s), and high
Acknowledgment
經過 2004 年夏天的研究所考試,我考上了交大應化所,從沒離開過家的我,帶著 簡單的行李來到交大展開研究生涯。幸運地,張豐志老師願意收留我這個考試生,大學 成績十分不理想的考試生。碩一時,在老師及郭紹偉學長的指導下,進行高分子間氫鍵 作用力的研究。當時,老師經常和我說一個故事,一個博士和碩士有何不同的故事 (這 故事我到現在都還常和學弟提起) ,且經過和最支持我的家人,爸爸和媽媽,討論後, 我報名了直升博士班的甄試,於 2005 年順利直升博士班。從直升後到現在,已有三年 半的時間,期間老師對於我的影響非常大,無論是課業、人生觀,甚至我對未來的計畫。 在老師自由的學風下,我對研究的興趣及渴望與日俱增,這種感覺,我想是其他實驗室 的同學無法體會的。因此,我也決定繼續在老師的實驗室進行博士後研究,繼續做我喜 歡做的事。我更希望,這就是我未來的職業。此外,老師待人處事豁達的態度,讓我對 人生有了不同的體會,也學會了忍讓。謝謝老師這四年來的指導,未來的日子裡,也請 老師多多指教。 這四年中間,林振隆學長及邱俊毅學長是幫助我最大最大的人,沒有你們二位,就沒 有今天的我,雖相處時間只有短短二年,但無論實驗及生活上你們都給了我極大的幫 助。智嘉及昀昇學弟,如果沒有你們,我也不會有現在多元化的成果,我相信我們的堅 持是對的,希望可以一直合作下去,也期待未來會磨擦出更多火花。我的好朋友,振華, 謝謝你在課業上及生活中的幫助,希望你也能順利地走下去。同學們及其他實驗室學弟妹和學長姊們,非常感謝大家的幫忙,讓我在研究及課業上均可順利進行。
感謝女友芝嫻八年半來的陪伴,容忍我的怪脾氣,過去的日子有歡笑也有淚水,我
會努力,讓未來的日子只有歡笑。最後,謝謝我的家人,爸爸、媽媽、外婆、乾媽、弟
弟、妹妹,有你們的支持,我才能專心地進行研究工作。僅以此論文獻給所有曾經幫助
Outline of Contents
Pages
Abstract (in Chinese)
Abstract (in English)
Acknowledgment Outline of Contents List of Tables List of Schemes List of Figures Chapter 1 Introduction
1.1. Introduction to Polyhedral OligomericSilsesquioxane (POSS)
References
1.2. Introduction to Hydrogen Bonds in Polymer/POSS blends
1.2.1. Polymer/POSS blends
1.2.2. Introduction to Painter-Coleman Association Model
1.2.3 Ternary Polymer Blends
References 1.3. Introduction to Polyelectrolyte References I IV VII IX XIV XV XVI 1 1 3 7 7 7 8 11 12 15
1.4. Introduction to Proton Exchange Membrane (PEM) Applied in
Direct Methanol Fuel Cell (DMFC)
References
Chapter 2 Miscibility and Hydrogen Bonding Behavior in Organic/Inorganic
Polymer Hybrids Containing Octaphenol Polyhedral Oligomeric
Silsesquioxane
Abstract
2.1. Introduction
2.2. Experimental Part
2.2.1. Materials
2.2.2. Synthesis of Octa(phenol)octasilsequioxane-POSS (OP-POSS)
Oligomer
2.2.3. Syntheses of PMMA-co-PVP Random Copolymers
2.2.4. Blend Preparations
2.2.5. Characterizations.
2.3. Results and Discussion
2.3.1 PMMA-co-PVP Copolymer Characterization
2.3.2. Analyses of OP-POSS/Homopolymer Binary Blends
2.3.3. Analyses of Binary Blend OP-POSS/Copolymers.
19 21 25 25 25 26 26 27 27 28 28 29 29 30 36
2.3.4. Analyses of Ionic Conductivity
2.4. Conclusions
References
Chapter 3 Effect of LiClO4 on the Thermal and Morphological Properties of
Organic/Inorganic Polymer Hybrids
Abstract
3.1. Introduction
3.2. Experimental Part
3.2.1. Materials
3.2.2. Characterization
3.3. Results and Discussion
3.3.1. Morphologies
3.3.2. Thermal Properties
3.4. Conclusions
References
Chapter 4 Effect of Sulfonic Acid Groups on Properties of New
Organic/Inorganic Cross-linked Proton Exchange Membrane
Abstract 4.1. Introduction 37 38 39 59 59 59 60 60 61 61 61 63 64 66 76 76 76
4.2. Experimental Part
4.2.1. Materials
4.2.2. Octakis(dimethylsilyloxypropylglycidyl
ether)octasilsesquioxane (OG-POSS) Oligomer
4.2.3. Synthesis of 4,4´-Diaminodiphenyl Ether-2,2´-disulfonic Acid
(ODADS)
4.2.4. Sulfonation of PEEK
4.2.5. Membranes Preparations
4.2.6. Characterization
4.3. Results and Discussion
4.3.1. Morphologies of Cross-linkers
4.3.2. Thermal Analysis
4.3.3. Membrane Morphologies
4.3.4. Relationship between water sorption and membrane miscibility
4.3.5. Proton conductivity, methanol permeability, and selectivity
4.4. Conclusions References Chapter 5 Conclusions Publication List 77 77 77 78 78 78 79 81 81 82 82 83 85 87 88 102 103
List of Tables
Pages
Table 2-1 PMMA-co-PVP Copolymer Compositions and Molecular Weights
Table 2-2 Curve Fitting of the C=O stretching bands in the FTIR spectra of
PVP/OP-POSS and PMMA/OP-POSS blends at room temperature
Table 2-3. Self-association and Inter-Association Equilibrium Constants and
Thermodynamic Parameters for PMMA/OP-POSS blends at 25°C
Table 2-4. Curve fitting of the C=O stretching bands in the FTIR spectra of
MMA61/OP-POSS (120oC) blends
Table 3-1. Compositions, 2θ (degrees), and Tg of the binary and ternary blends.
Table 4-1. Compositions of semi-interpenetrating network proton exchange
membranes
Table 4-2. Properties of crossl-linked proton exchange membranes
44 45 46 47 70 94 95
List of Schemes
Pages
Scheme 2-1. Synthesis and chemical structure of OP-POSS
Scheme 2-2. Synthesis of PMMA-co-PVP random copolymer
Scheme 4-1. Structure of the organic/inorganic networks
42
43
List of Figures
Pages
Figure 1-1. Structures of POSS.
Figure 1-2. Schematic illustration of a lithium rocking chair battery with
graphite and spinel as intercalation electrodes and its electrode
reactions
Figure 1-3. Schematic of the segmental motion assisted diffusion of Li+ in
the PEO matrix. The circles represent the ether oxygen atoms of
PEO
Figure 1-4. Basic membrane electrode assembly of DMFC
Figure 2-1. Kelen–Tudos plot for the PMMA-co-PVP copolymers
Figure 2-2. DSC scans for (a) PMMA/OP-POSS and (b) PVP/OP-POSS
blends of various compositions
Figure 2-3. Plots of Tg versus (a) the PVP content of PVP/OP-POSS blends
and (b) the PMMA content of PMMA/OP-POSS blends
Figure 2-4. Partial IR spectra (OH stretching region) of OP-POSS and P
MMA/OP-POSS and PVP/OP-POSS blend systems
Figure 2-5. Partial IR spectra (C=O stretching region) of (a) PVP/OP-POSS
blends at 120 °C and (b) PMMA/OP-POSS blends at 25 °C
6 17 18 24 48 49 50 51 52
Figure 2-6. Fraction of the hydrogen-bonded C=O groups plotted with respect
to the blend composition: (■) FTIR spectroscopy data; (-)
theoretical values for PMMA/OP-POSS blends (KA = 2) at 25
°C
Figure 2-7. Fraction of the hydrogen-bonded C=O groups plotted with respect
to the blend composition: (■) FTIR spectroscopy data; (-)
theoretical values for PMMA/OP-POSS blends (KA = 29) at 25
°C.
Figure 2-8. Partial IR spectra (1800–1625 cm–1) of the MMA61/OP-POSS
blend containing various OP-POSS contents, recorded at 120 °C
Figure 2-9. DSC scans for MMA61/OP-POSS blends of various compositions
Figure 2-10. Plots of Tg versus (a) the MMA81 content of MMA81/OP-POSS
blends, (b) the MMA61 content of MMA61/OP-POSS
blends, and (c) the MMA53 content of MMA53/OP-POSS
blends
Figure 2-11. Plots of ionic conductivity with respect to the MMA content in
PMMA-co-PVP copolymers for (▓) LiClO4/polymer binary
blends and (□) LiClO4/OP-POSS/MMA61 ternary blends
53 54 55 56 57 58
Figure 3-1. FE-SEM micrographs of (a) PMMA/LiClO4/OP-POSS (100/0/5)
and (b) PMMA/LiClO4/OP-POSS (100/25/5). (c, d) Schematic
representations of the proposed mechanisms of formation of the
various OP-POSS domains
Figure 3-2. IR spectra of various PMMA/LiClO4/OP-POSS blends recorded
at 120 °C
Figure 3-3. XRD patterns of these binary and ternary blends
Figure 3-4. DSC curves of these binary and ternary blends
Figure 4-1. SEM micrographs (cross-sectional views) of the (a) OG15 and (b)
SOG15 membranes
Figure 4-2. TGA curves of the (a) SPEEK, OG03, and OG10 membranes and
the (b) OG03 and SOG03 membranes. (c) Corresponding loss factors (tan δ)
Figure 4-3. TEM micrographs of the (a) SPEEK, (b) OG10, and (c) SOG10
membranes
Figure 4-4. (a) IEC values (b) water uptakes, and (c) values of λ plotted
with respect to the cross-linker content for the SPEEK, OG, and
SOG membranes
Figure 4-5. (a) Proton conductivities, (b) methanol permeabilities, and (c)
72 73 74 75 96 97 98 99 98 100
selectivities plotted with respect to cross-linker content for
SPEEK, OG, and SOG membranes
Figure 4-6. Proposed mechanisms of proton transfer within the (a) SPEEK (b)
OG, and (c) SOG membranes
Chapter 1 Introduction
1.1. Introduction to Polyhedral OligomericSilsesquioxane (POSS)
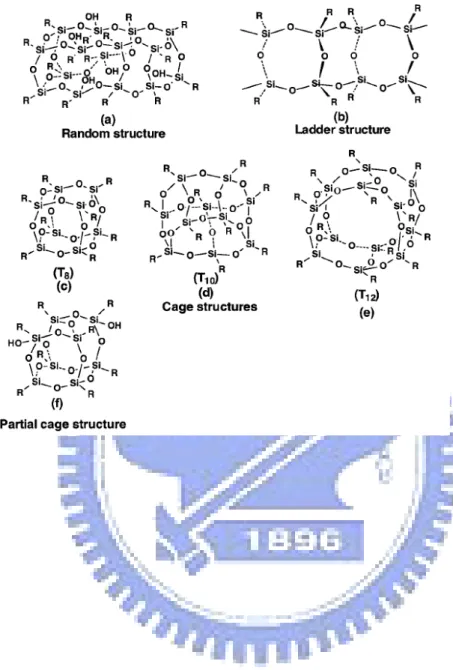
Polyhedral oligomeric silsesquoixanes (POSSs), which have the general formula (RSiO1.5)n, are prototypical organic/inorganic systems composed of inorganic cores with external organic substituents where R can be hydrogen or any alkyl, alkylene, aryl, and arylene groups, or their derivatives of which structure can be the random structure, ladder structure, cage structure, and partial cage structure as shown in Figure 1-1.1 In 1946, Scott et al. first isolated the oligomeric organosilsesquioxanes through the co-hydrolysis of methyltrichlorosilane and dimethylchlorosilane.2 However, the further development of POSS was started until the discovery of the polymerizable POSS and its related copolymer proposed by Lichtenhan and the American Air Force Research Laboratory in 1991.3,4 Later, Feher, Laine, Sellinger,5 and Mather et al.6 incorporated these POSS derivatives into polymer systems, resulting improvement in the physical and mechanical property, sequentially, the interest in lowering the cost and enlarging the quantity of the production was also growing. In 1998, the start-up of Hybrid plastics in Fountain Valley, CA, turned the scale of the production of the material into large, the industrial production.
These ladder-like POSSs [Figure 1-1 (b)] proposed by Baney et al. and others possess outstanding thermal stability and oxidative resistance at high temperature (more than 500 oC),6-12 thus it can be used in the filed, including photoresist for electronics and optical devices,13,14 dielectrics and protective films for semiconductor,15,16 liquid crystal display materials,17 recording media,18 gas separation film,19 and ceramics binder.20 Recently, the specific cage structures [Figure 1-1 (c)] have attracted great interest because it is a nanoparticle with diameter varied from 1 to 3 nm and organic substituents on its outer surface leading to the formation of miscible blend with polymers, biological systems, and surfaces.
Through appropriate designing of the functionality of these organic substituents, it is possible to create both mono- and octa-functional macromonomers for desired applications. These functionalized POSS derivatives can be blended21-23 or attached covalently to linear thermoplastics24-31 or thermosetting networks25,26,32-37 to form high-performance hybrid materials.38-43 Based on the above, these POSS derivatives have the potential for the application in several fields, however, they had rarely been used in the filed of polyelectrolyte and proton exchange membrane which play a critical role in the people’s life because of the need for energy storage and cleaning energy source. In this thesis, POSS were modified to be non-covalently and covalently incorporated into polymer matrix to form the new polyelectrolyte and proton exchange membrane. In addition, the Painter–Coleman association model (PCAM) was employed to theoretically study the intra-association and inter-association between the modified POSS and polymer.
References
1. Pittman, Jr. C. U.; Li, G. Z.; Ni, H. Macromol. Symp. 2003, 196, 301. 2. Scott, D. W. J. Am. Chem. Soc. 1946, 68, 356.
3. Lichtenhan, J. D.; Feher, F. J.; Gilman, J. W. Macromolecules 1993, 26, 2141. 4. Lichtenhan, J. D.; Otonary, Y.; Carr, M. J. Macromolecules 1995, 28, 8435. 5. Laine, R. M.; Sellinger, A. Macromolecules 1996, 29, 2327.
6. Baney, R. H.; Itoh, M.; Sakakibara, A.; Suzuki, T. Chem. Rev. 1995, 95, 1409. 7. Zhang, X.; Shi, L. Chin. J. Polym. Sci. 1987, 5, 197.
8. Xie, Z.; He, Z.; Dai, D.; Zhang, R. Chin. J. Polym. Sci. 1989, 7, 183.
9. Maciel, G. E.; Sullivan, M. J.; Sindorf, D. W. Macromolecules 1981, 14, 1607.
10. Engelhavdt, G.; Jancke, H.; Lippmaa, E.; Samoson A. J. Organomet. Chem. 1981, 210, 295.
11. Frye, C. L.; Collins, W. T. J. Am. Chem. Soc. 1970, 92, 5586.
12. Belot, V.; Corriu, R.; Leclerq, D.; Mutin, P. H.; Vioux, A. Chem. Mater. 1991, 3, 127.
13. Adachi, H.; Hayashi, O.; Okahashi, K. Japanese Patent Kokoku-H-2-15863. 14. Adachi, H.; Hayashi, O.; Okahashi, K. Japanese Patent Kokai-S-60-108841. 15. Adachi, E.; Aiba, Y.; Adachi, H. Japanese Patent Kokai-H-2-277255.
16. Aiba, Y.; Adachi, E.; Adachi, H. Japanese Patent Kokai-H-3-6845. 17. Shoji, F.; Sudo, R.; Watanabe, T. Japanese Patent Kokai-S-56-146120. 18. Imai, E.; Takeno, H. Japanese Patent Kokai-S-59-129939.
19. Saito, Y.; Tsuchiya, M.; Itoh, Y. Japanese Patent Kokai-S-58-14928. 20. Mine, T.; Komasaki, S. Japanese Patent Kokai-S-60-210570.
21. Kuo, S. W.; Lin, H. C.; Huang, W. J.; Huang, C. F.; Chang, F. C. J. Polym. Sci., Polym.
22. Lee, Y. J.; Huang, J. M.; Kuo, S. W.; Chang, F. C. Polymer 2005, 46, 10056. 23. Zhang, H.; Kulkarni, S.; Wunder, S. L. J. Phys. Chem. B 2007, 111, 3583.
24. Kopesky, E. T.; Haddad, T. S.; Cohen, R. E.; McKinley, G. H. Macromolecules 2004, 37, 8992.
25. Li, G. Z.; Wang, L.; Toghiani, H.; Daulton, T. L.; Koyama, K.; Pittman, C. Y.
Macromolecules 2001, 34, 8686.
26. Matejka, L.; Strachota, A.; Plestil, J.; Whelan, P.; Steinhart, M.; Slouf, M.
Macromolecules 2004, 37, 9449
27. Lin, H. C.; Wang, C. F.; Kuo, S. W.; Tung, P. H.; Huang, C. F.; Lin, C. H.; Chang, F. C. J.
Phys. Chem. B 2007, 111, 3404.
28. Leu, C. M.; Chang, Y. T.; Wei, K. H. Chem. Mater. 2003, 15, 2261. 29. Leu, C. M.; Chang, Y. T.; Wei, K. H. Chem. Mater. 2003, 15, 3721.
30. Phillips, S. H.; Haddad, T. S.; Tomcrzak, S. J. Curr. Opin. Solid State Mater. Sci. 2004, 8, 21.
31. Li, G. Z.; Wang, L. C.; Ni, H. L. J. Inorg. Organomet. Polym. 2001, 11, 123.
32. Chen, W. Y.; Wang, Y. Z.; Kuo, S. W.; Huang, C. F.; Tung, P. H.; Chang, F. C. Polymer
2004, 45, 6897.
33. Tamaki, R.; Choi, J.; Laine, R. M. Chem. Mater. 2003, 15, 793.
34. Lee, Y. J.; Huang, J. M.; Kuo, S. W.; Lu, J. S.; Chang, F. C. Polymer 2005, 46, 173. 35. Ye, Y. S.; Chen, W. Y.; Wang, Y. Z. J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 5391. 36. Liu, Y. L.; Chang, G. P.; Hsu, K. Y.; Chang, F. C. J. Polym. Sci., Part A: Polym. Chem.
2006, 44, 3825.
37. Do Carmo, D. R.; Paim, L. L.; Dias, N. L. Appl. Surf. Sci. 2007, 253, 3683. 38. Voronkov, M. G.; Lavrent’ev, V. I. Top. Curr. Chem. 1982, 102, 199.
40. Provatas, J. G. Trends Polym. Sci. 1997, 5, 327. 41. Loy, D. A.; Shea, K. J. Chem. Rev. 1995, 95, 1431.
42. Lichtenhan, J. D.; In Polymeric Materials Encyclopedia, Salamone, J. C. Ed.; CRC Press: Boca Raton, FL, 1996, 7768.
1.2. Introduction to Hydrogen Bonds in Polymer/POSS blends 1.2.1. Polymer/POSS blends
The production of polymers was growing rapidly in the past 50 years and their versatility guarantees that they are suitable for not only the traditional use but also the use in high technology. Because covalent copolymerization can be a complicated and time-consuming process, polymer blending is usually considered to be a simpler and more convenient means of preparing new polymer materials with improved properties and has been developed for a long time. Recently, the incorporation of nanoparticle into polymer matrix has attracted great interest because the resulted composite exhibited the advantage over both inorganic and organic compounds with the unique physical properties of POSS nanoparticle. The properties of these polymer/POSS composites are strongly depend on the miscibility between the incorporated polymer and POSS derivatives which can be affected by the correlation between the polymer matrix and the externally covered functionality of POSS. The correlation in the polymer/POSS composite is similar to that in polymer blend which has been widely concerned with the following intermolecular or inter-segment forces:
(a) Strong dipoles (b) Hydrogen bonds
(c) Charge transfer complexes (d) Ionic interactions in ionomers
The most common and important correlation are the strong hydrogen bond and/or strong dipole interactions. Coleman and Painter proposed the association models and the related theories in 1995,1 resulting in significant progress in the field of hydrogen bonded in polymer blends. In this thesis, the model proposed by them was employed to obtain the equilibrium association constant.
In the Painter-Coleman association model proposed by Painter and Coleman, a contribution of hydrogen bonding interaction was added to the Flory-Huggins equation for the free energy of mixing: RT G n N N RT G H A B A A B B m = Φ + Φ + Φ + Δ Δ χ ln ln (1-1)
where NB and NA are the number of polymer molecules and nB is the total number of B
segments. The first four terms correspond to the classic Flory-Huggins equation. The contribution of hydrogen bonds (the last term of the equation) can be derived from a simple model: 1 1←⎯→⎯ + + K h h B B B B A B A B h K h A ⎯ ⎯→ ← +
where KB and KA correspond to the “chain like” self-association and inter-association
equilibrium constant, respectively, thus
RT GH
Δ
can be expressed as:
+ + + Φ + + Φ + Φ ∑ = Δ
∑
h AB h BB A A A h A B B B H n n r n r h B n h n RT G h h h ln( ) ln( ) ln( ) 1 1 (terms in z and σ) A (1-2) h AB B h BB A A B B n n K n K n ln ln ] ln ln [ Φ + Φ − − −where the square bracket corresponded the excess entropy term. The association model describes the mixing of small molecules, or in effect, describes the mixing of disconnected polymer segments. With the “physical” force derived from these solubility parameters, ΔGH
can be obtained from the equilibrium constants and enthalpies of hydrogen bond formation. As the combinatorial entropy is very small, the free energy of mixing (the miscibility) is dominated by the “physical” force and enthalpy of hydrogen bond formation.
1.2.3. Ternary Polymer Blends
The reasonable development of the association model is the extension of the model to describe the phase behavior of ternary blend. However, the accuracy and the unambiguous
interpretation of experiment become problems when changing the system from binary to ternary blends.
Considering a ternary blend system composed of three polymers, including PolyA, PolyB, and PolyC, PolyB is self-associated and contains proton donor groups and PolyA and PolyC are both not self-associated and both exhibit acceptor groups [there are no strong intermolecular interactions (hydrogen bond) PolyA and PolyC]. These hydrogen bonds within this ternary blend can be described through the equilibrium conastnts, KA, KB, and KC.
1 1←⎯→⎯ + + K h h B B B B A B A Bh + ←⎯→⎯KA h C B C B h K h C ⎯ ⎯→ ← +
The equation describing the mixing free energy of the ternary blend is very similar to that of the binary blend, except the extra terms corresponded to the additional component of the blend: RT G N r N r N r RT G H BC C B AC C A AB B A C C C C B B B B A A A A m Δ + Φ Φ + Φ Φ + Φ Φ + Φ Φ + Φ Φ + Φ Φ = Δ χ χ χ ln ln ln (1-3)
where , , and correspond to the volume fractions of PolyB, PolyA, and PolyC,
respectively, and Ni is the degree of polymerization. These parameters,
A Φ ΦB ΦC B A A V V r = and B C C V V
r = are the ratios of the segment molar volumes of PolyA to PolyB and PolyC to PolyB,
respectively. Because the interaction parameter, χij, obtained from the group molar attraction and molar volume constant designed to specifically exclude contributions from hydrogen
bonding is positive or equal to zero,2 the term,
RT GH
Δ
the mixing free energy from the hydrogen bonding interaction.3-12 The further study on the calculation of these equilibrium constants within these binary and ternary blends is described in chapter 2.
References
1. Coleman, M. M.; Painter, P. C. Prog. Polym. Sci. 1995, 20, 1.
2. Coleman, M. M.; Graf, J. F.; Painter, P. C. Specific Interactions and the Miscibility of Polymer
Blends; Technomic Publishing, Inc.; Lancaster, PA, 1991.
3. Painter, P. C.; Park, Y.; Coleman, M. M. Macromolecules 1988, 21, 66. 4. Painter, P. C.; Park, Y.; Coleman, M. M. Macromolecules 1989, 22, 570. 5. Painter, P. C.; Park, Y.; Coleman, M. M. Macromolecules 1989, 22, 580. 6. Coleman, M. M.; Xu, Y.; Painter, P. C. Macromolecules 1994, 27, 127. 7. Coleman, M. M.; Pehlert, G. J.; Painter, P. C. Macromolecules 1996, 29, 6820.
8. Pehlert, G. J.; Painter, P. C.; Veytsman, B.; Coleman, M. M. Macromolecules 1997, 30, 3671. 9. Painter, P. C.; Veytsman, B.; Kumar, S.; Shenoy, S.; Graf, J. F.; Xu, Y.; Coleman, M. M.
Macromolecules 1997, 30, 932.
10. Coleman, M. M.; Painter, P. C. Macromol. Chem. Phys. 1998, 199, 1307. 11. Zeman, L.; Patterson, D. Macromolecules 1972, 5, 513.
1.3. Introduction to Polyelectrolytes
Rechargeable Li-ion cells are key components of the portable, entertainment, computing and telecommunication equipment required by today’s information-rich, mobile life. Polymer electrolytes formed through dissolving salts into polar and high-molecular-weight polymers are the subject of intensive research because of their potential for use as the solid polymer electrolyte (SPE) in rechargeable lithium batteries.1-6 Both the salt and the neat polymer are solids and the complex-forming reaction can be expressed as follows:7
n m
n MX RY
RY
mMX +(− −) →( ) •(− −) (1-4)
where (–RY–) corresponds to the repeat unit of polymer. The kinetics of eq (1-4) are unfavorable, even when the complex is stable. The most commonly used method to dissolve or suspend both the MX salt and the host polymer is dissolving them in a common solvent and then removing the solvent, producing the solvent-free polymer electrolyte.8 Obviously, the dissolving reaction will be thermodynamically favorable (negativeΔG°) only if the Gibbs energy of dissolving the salt into polymer is large enough to overcome the lattice energy of the salt, thus three parameters are important for the controlling these salt/neutral molecule interactions: (a) electron pair donicity, (b) acceptor number, and (c) an entropy term. The electro pair donicity is corresponded to the ability of the solvent donating electrons to solvate the cation as Lewis acid, thus the incorporated polymer should exhibit donor site such as oxygen, sulfur, or nitrogen either in the backbone or in the side chain. The acceptor number represents the possibility for anion (base) solvation. For instance, PEO, a polyether, exhibits strong donor and its donicity is close to 20, additionally, ethers are very poor acceptors because they lack hydrogen bonding for anion salvation.9 Thus, PEO can effectively solvate cation possessing bulky delocalized counter anions such as I-, ClO4-, BF4- or CF3SO3-which require little or no salvation.
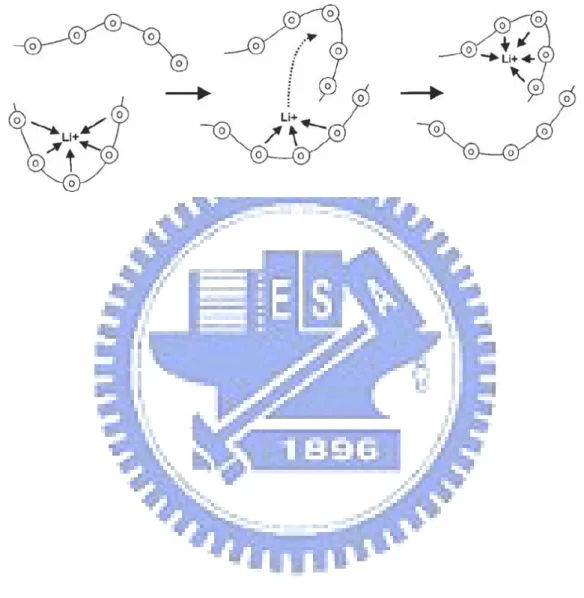
electrochemical devices spurred considerable interest in the ion-transport properties of these materials.10-15 Besides the advantage of flexibility, polyelectrolyte can also be cast into thin films and since thin films while minimizing the resistance of the electrolyte also reduces the volume and the weight, use of polymer electrolytes can increase the energy stored per unit weight and volume. As shown in Figure 1-2, the polyelectrolyte serves as a medium to transport the ions in the cell. In addition, a separator isolating the anode from the cathode electronically can be ceramic or polymeric separator as using liquid electrolytes. Both functions, ion conduction and separation, can be realized in a single thin membrane when polymer electrolytes are used.
The goal for the SPE research is the development of highly ionic conductive (ca. 10–4 S/cm), dimensionally stable, and flexible SPE materials under ambient condition. Since Wright reported that poly(ethylene oxide) (PEO) can be the candidate for use in the SPEs at 1975, it has became one of the most studied materials16 and polymer electrolytes were proposed for batteries in 1978 because they exhibited the advantages of solid state electrochemistry with the ease of processing inherent to plastic materials.17 Since that time, the number of contributions to the field of SPEs related to PEO has grown enormously, these PEO based polyelectyrolytes were prepared through numerous physical and chemical procedures including non-covalent blending,10-15 covalent copolymerization,18-21 and grafting22 for studying their interaction mechanism and for improving their flexibility and chemical and physical properties without detrimentally affecting the ionic conductivity. The ionic conductivity σ can be roughly expressed by the following equation:
∑
= i i i iz n μ σ (1-5)where ni, zi, and μi are the effective number of mobile ions, the elementary electric charge,
and the ion mobility, respectively, indicating that the fraction of “free” ions is an important parameter, a high degree of dissociation of the salt in the polymer is needed for obtaining
highly ionic conductive polyelectrolyte. High Li+transference number is also needed, i.e., a high ratio of the charge transfer which has been an important subject of research in recent years.23-24 The molecular dynamic simulation shown in Figure 1-3 indicated that the Li+ ions are complexed to PEO through approximately five ether oxygens of the PEO chain, and thus mobility of the cations is decreased considerably,25 implying that the mobility of the Li+ cation is related to the motion of the complexed PEO chain. In summary, the polymer required for the SPE application should possess that (1) high concentration of polar (basic) groups and (2) low cohesive energy and high flexibility to solvate the salt effectively, resulting high ionic conductivity.26-30
References
1. Croce, F.; Appetecchi, G. B.; Persi, L.; Scrosati, B. Nature 1998, 394, 456.
2. Gray, F. M. Polymer Electrolytes (RSC Materials Monographs, The Royal Society of Chemistry, Cambridge, 1997).
3. Scrosati, B. (ed.) Applications of Electroactive Polymers (Chapman & Hall, London, 1993).
4. Bruce, P. G. (ed.) Solid State Electrochemistry (Cambridge University Press, Cambridge, 1995).
5. Fenton, D. E.; Parker, J. M.; Wright, P. V. Polymer 1973, 14, 589.
6. Ratner, M. A. in Polymer Electrolytes Reviews-1 (eds MacCallum, J. R.; Vincent, C. A.) 173–236 (Elsevier Applied Science, London, New York, 1987).
7. Ratner, M. A.; Shriver, D. F. Chem. Rev. 1988, 88, 109.
8. LaNest, J. F.; Cheradame, H.; Dalard, F.; Deroo, D. J. Appl. Electrochem. 1986, 16, 75. 9. Gutmann, V. The Donor Acceptor Approach to Molecular Interactions; Plenum Press: New
York, 1978.
10. Druger, S. D.; Ratner, M. A.; Nitzan, A. Solid State Ionics 1983, 9/10, 1115.
11. Armand, M. B.; Chabango, J. M.; Duclot, M. J. in Fast Ion Transport in Solids (eds Vashishta, P., Mundy, J. N.; Shenoy, G. K.) 131–136 (North-Holland, Amsterdam, 1979).
12. Chiu, C. Y.; Yen, Y. J.; Kuo, S. W.; Chen, H. W.; Chang, F. C. Polymer 2007, 48, 1329. 13. Gorecki, W. Solid State Ionics 1998, 28, 1018.
14. Chiu, C. Y.; Hsu, W. H.; Yen, Y. J.; Kuo, S. W.; Chang, F. C. Macromolecules 2005, 38, 6640.
15. Chiu, C. Y., Chen, H. W., Kuo, S. W. & Chang, F. C. Macromolecules 2004, 37, 8424. 16. Wright, P. V. Br. Polym. J. 1975, 7, 319.
17. Armand, M.; Duclot, M. French Patent 1978, 7832976.
18. Cho, B. K.; Jain, A.; Gruner, S. M.; Wiesner, U. Science 2004, 305, 1598.
19. Epps, T. H.; Bailey, T. S.; Waletzko, R.; Bates, F. S. Macromolecules 2003, 36, 2873. 20. Singh, M.; Odusanya, O.; Wilmes, G. M.; Eitouni, H. B.; Gomez, E. D.; Patel, A. J.; Chen,
V. L.; Park, M. J.; Fragouli, P.; Iatrou, H.; Hadjichristidis, N.; Cookson, D.; Balsara, N. P. Macromolecules 2007, 40, 4578.
21. Zhang, H.; Kulkarni, S.; Wunder, S. L. J. Phys. Chem. B 2007, 111, 3583.
22. Gavelin, P.; Jannasch, P.; Wessle´n, B. J. Polym. Sci. Part A: Polym. Chem. 2001, 39, 2223.
23. Armand, M. Solid State Ionics 1994, 69, 309.
24. Bruce, P. G.; Vincent, C. A. Faraday Discuss. Chem. Soc. 1989, 88, 43. 25. Müller-Plathe, F.; Van Gunsteren, W. F. J. Chem. Phys. 1995, 103, 4745.
26. Vincent, C. A.; MacCallum, J. R. In Polymer Electrolyte Reviews; Mac Callum, J. R., Vincent, C. A., Eds., Elsevier: London, 1987.
27. Armand, M. B.; Chabagno, J. M.; Duclot, M. J. In Fast Ion Transport in Solids; Duclot, M. J., Vashishta, P., Mundy, J. N., Shenoy, G. K., Eds.; North-Holland:
New York, 1979.
28. Shriver, D. F.; Papke, B. L.; Ratner, M. A.; Dupon, R.; Wong, T.; Brodwin, M. Solid State
Ionics 1981, 5, 83.
29. Paper, B. L.; Ratner, M. A.; Shrever, D. F. J. Phys. Chem. Solids 1981, 42, 493. 30. Paper, B. L.; Ratner, M. A.; Shrever, D. F. J. Electrochem. Soc. 1982, 129, 1694.
Figure 1-2. Schematic illustration of a lithium rocking chair battery with graphite and spinel
Figure 1-3. Schematic of the segmental motion assisted diffusion of Li+ in the PEO matrix.
1.4. Introduction to Proton Exchange Membrane (PEM) Applied in Direct Methanol Fuel Cell (DMFC)
Recently, advanced energy technology reducing fossil fuel consumption and keeping the environment clean for human activities is becoming intensively important because of the worldwide concern about the environmental consequences of fossil fuel use in the day-to-day production of electricity and for the propulsion of vehicles. More importantly, the societal awareness concerning issues of environmental pollution increased over the last few decades. Therefore, the renewable energy sources such as wind, sun, and water were proposed and developed, but one has to remember that the complete generation process should be taken into account that these sources are not suited to satisfy the base demand. Fuel cell exhibits the potential to become an important energy conversion technology because it is a clean energy producing only water (hydrogen fuel cell) or water and carbon dioxide (direct methanol fuel cell) to overcome drawbacks of the previous energy generation. Fuel cells can usually classified by temperature and electrolyte employed in the cell, thus there are low temperature fuel cells such as Alkaline Fuel Cell (AFC), Phosphoric Acid Fuel Cell (PAFC), and DMFC, etc and high temperature fuel cells, including Molten Carbonate Fuel Cell (MCFC) and Solid Oxide Fuel Cell (SOFC), etc.1-10
In this thesis, we tried to focus on the polymer employed for the application in DMFC, a promising power source in today’s society because of its convenient power generation procedure which is suited to today’s mobile life. The basic membrane electrode assembly of DMFC was shown in Figure 1-4,10 displaying that the proton exchange membrane plays a critical role to separate the fuel from the oxidant. These reactions involved in the power generation within DMFC were expressed as follows:
Anode: − + + + → +H O CO H e OH CH3 2 2 6 6
Cathod: O H e H O2 6 6 3 2 2 3 + ++ − → Overall: O H CO O OH CH3 2 2 2 2 2 3 + → +
where the overall reaction only produced water and carbon dioxide representing that DMFC was a clean energy. The proton exchange membrane suitable for the application in DMFC should possess (1) high protonic conductivity, (2) low electronic conductivity, (3) low permeability to fuel and oxidant, (4) low water transport through diffusion and electro-osmosis, (5) oxidative and hydrolytic stability, (6) good mechanical properties in both the dry and hydrated states, (7) cost, and (8) capability for fabrication into MEA.10 Nafion, one of the most studied material for application as PEM, exhibits both chemical and physical stability at moderate temperature and high proton conductivity through its highly interconnected hydrophilic channels.10 Sulfonated polymers, such as sulfonated poly(ether sulfone) (SPES), poly(benzimidazole) (SPBI), poly(ether ether ketone) (SPEEK), and polyimide (SPI), are also potential candidates for use in PEMs,11-22 but their high cost and high methanol permeability limit their applications.10-22 To overcome these drawbacks, numerous new materials possessing reinforced sulfnoated polymers are often employed.23-30 Cross-linked PEMs have shown significant advantages in controlling the water behavior, improving the dimension stability, and thermal stability.31-40 The incorporation of inorganic materials into sulfonated polymers by cross-linking reaction has also been reported.41-48 Although numerous works to develop proton exchange membrane had been done, the development of more-efficient methods for improving the chemical and mechanical stabilities of sulfonated polymer membranes—without detrimentally affecting the proton conductivity and methanol crossover—remains an important challenge.
References
1. Carrette, L.; Friedrich, K. A.; Stimming, U. CHEMPHYSCHEM 2000, 1, 162. 2. Barnett, B. M.; Teagan, W. P. J. Power Sources 1992, 37, 15.
3. Melle, F. D. J. Power Sources 1998, 71, 7. 4. Hart, D. J. Power Sources 2000, 86, 23.
5. Gallow, G. D. J. Power Sources 1999, 80, xvii. 6. Bauen, A.; Hart, D. J. Power Sources 2000, 86, 482.
7. “Clean fuel cell energy for today”: Platinum Met. Rev. 1999, 43, 14. 8. Chalk, S. G.; Miler, J. F.; Wanger, F. W. J. Power Sources 2000, 86, 40. 9. Dufour, A. U. J. Power Sources 1998, 71, 19.
10. Hickner, M. A.; Ghassemi, H.; Kim, Y. S.; Einsla, B. R.; McGrath, J. E. Chem. Rev. 2004,
104, 4587.
11. Wang, F.; Hickner, M.; Kim, Y. S.; Zawodzinski, T. A.; McGrath, J. E. J. Membrane. Sci.
2002, 197, 231.
12. Rozière, J.; Jones, D. J.; Annu.Rev. Mater. Res. 2003, 33, 503.
13. Xing, P. X.; Robertson, G. P.; Guiver, M. D.; Mikhailenko, S. D. Kaliaguine, S.
Macromolecules 2004, 37, 7960.
14. Wang, Z.; Ni, H. Z.; Zhao, C. J.; Li, X. F.; Zhang, G.; Shao, K.; Na, H. J. Membrane. Sci.
2006, 285, 239.
15. Schönberger, F.; Jochen Kerres, J. J. Polym. Sci Pol Chem. 2007, 45, 5237. 16. Patric Jannasch, P. Curr. Opin. Colloid. Interface Sci. 2003, 8, 96.
17. Fu, Y. Z.; Manthiram, A. Guiver, M. D. Eelctrochem. Commun. 2007, 9, 905.
18. Xing, P. X.; Robertson, G. P.; Guiver, M. D.; Mikhailenko, S. D.; Wang, K. P.; Kaliaguine, S. J. Membrane. Sci. 2004, 229, 95.
Macromolecules 2002, 35, 9022.
20. Zhou, Z. L.; Dominey, R. N.; Rolland, J. P.; Maynor, B. W.; Pandya, A. A.; M. DeSimone, J. M. J. Am. Chem. Soc. 2006, 128, 12963.
21. Miyatake, K.; Chikashige, Y.; Eiji Higuchi, E.; Masahiro Watanabe, M. J. Am. Chem. Soc.
2007, 129, 3879.
22. Asano, N.; Aoki, M.; Suzuki, S.; Miyatake, K.; Uchida, H.; Watanabe, M. J. Am. Chem.
Soc. 2006, 128, 1762.
23. Xu, K.; Li, K.; Khanchaitit, P.; Wang, Q. Chem. Mater. 2007, 19, 5937.
24. M. Tsui, E. M.; Cortalezzi, M. M.; Wiesner, M. R. J. Membrane. Sci. 2007, 306, 8. 25. Subbaraman, R.; Ghassemi, H.; Zawodzinski, T. A. J. Am. Chem. Soc. 2007, 128, 2238. 26. Yamaguchi, T.; Zhou, H.; Nakazawa, S.; Hara, N.; Adv. Mater. 2007, 19, 592.
27. Grunzinger, S. J.; Watanabe, M.; Fukagawa, K.; Kikuchi, R.; Tominaga, Y.; Hayakawa, T.; Kakimoto, M. J. Power. Sources. 2008, 175, 120.
28. Yang, Y. S.; Shi, Z. Q.; Holdcroft, S. Macromolecules 2004, 37, 1678. 29. Shi, Z. Q.; Holdcroft, S. Macromolecules 2005, 38, 4193.
30. Farhat, T. R.; Hammond, P. T. Adv. Funct. Mater. 2005, 15, 945.
31. Ding, F. C.; Wang, S. J.; Xiao, M.; Meng Y. Z. J. Power. Sources. 2007, 164, 488.
32. Cho, K. Y.; Jung, H. Y.; Shin, S. S.; Choi, N. S.; Sung, S. J.; Park, J. K.; Choi, J. H.; Park, K. W.; Sung, Y. E. Electrochim. Acta. 2004, 50, 589.
33. Lee, C. H.; Park, H. B.; Chung, Y. S.; Lee, Y. M.; Freeman, B. D. Macromolecules 2006,
39, 755.
34. Fu, T. Z.; Zhao, C. J.; Zhong, S. L.; Zhang, G.; Shao, K.; Zhang, H. Q.; Wang, J.; Na, H. J.
Power. Sources. 2007, 165, 708.
35. Qiao, J. L.; Hamaya, T.; Okada, T. Chem. Mater. 2005, 17, 2413.
37. Gasa, J. V.; Boob, S.; Weiss, R. A.; Shaw, M. T. J. Membrane. Sci. 2006, 269, 177. 38. Yamaki, T.; Kobayashi, K.; Asano, M.; Kubota, H.; Yoshida, M. Polymer 2004, 45, 6569. 39. Park, H. B.; Lee, C. H.; Sohn J. Y.; Lee, Y. M.; Freeman, B. D.; Kim, H. J. J Membrane.
Sci. 2006, 285, 432.
40. Kim, D. S.; Guiver, M. D.; Nam, S. Y.; Yun, T. I.; Seo, M. Y.; Kim, S. J.; Hwang, H. S.; Rhim, J. W. J. Membrane. Sci. 2006, 281, 156.
41. Kim, D. S.; Liu, B. J.; Guiver, M. D. Polymer 2006, 47, 7871. 42. Chen, W. F.; Kuo, P. L. Macromolecules 2007, 40, 1987. 43. Shahi, V. K. Solid. State. Ionics. 2007, 177, 3395.
44. Vona, M. L. D.; Marani, D. D’Ottavi, C.; Trombetta, Marcella.; Traversa, E.; Beurroies, I.; Knauth, P.; Licoccia, S. Chem. Mater. 2006, 18, 69.
45. Kim, D. S.; Park, H. B.; Rhim, J. W.; Lee, Y. M. J. Membrane. Sci. 2004, 240, 37.
46. Su, Y. H.; Liu, Y. L.; Sun, Y. M.; Lai, J. Y.; Wang, D. M.; Gao, Y. Liu, B.; Guiver, M. D. J.
Membrane. Sci. 2007, 296, 21.
47. Chen, S. W.; Holmberg, B.; Li, W. Z.; Wang, X.; Deng, W. Q.; Munoz, R.; Yan Y. S. Chem.
Mater. 2006, 18, 5669.
48. Chang, Y. W.; Wang, E.; Shin, G.; Han, J. E.; Mather, P. T. Polym. Adv. Technol. 2007, 18, 535.
Chapter 2
Miscibility and Hydrogen Bonding Behavior in Organic/Inorganic Polymer Hybrids Containing Octaphenol Polyhedral Oligomeric Silsesquioxane
Abstract In this study, we investigated the miscibility behavior and mechanism of interaction
of poly(methyl mechacrylate) (PMMA), poly(vinyl pyrrolidone) PVP, and PMMA-co-PVP blends with octa(phenol)octasilsequioxane (OP-POSS). For the PMMA/OP-POSS binary blend, the value of the association constant (KA = 29) was smaller than that in the poly(vinyl
phenol) (PVPh)/PMMA (KA = 37.4) and ethyl phenol (EPh)/PMMA (KA = 101) blend systems,
implying that the phenol groups of the OP-POSS units in the PMMA/OP-POSS blends interacted to a lesser degree with the C=O groups of PMMA than they did in the other two systems. In addition, the ionic conductivity of a LiClO4/PMMA-co-PVP polymer electrolyte was increased after blending with OP-POSS.
2.1. Introduction
Composite materials comprising organic polymers and inorganic materials have attracted great interests in recent years for both their fundamental scientific behavior and industrial applications. Polyhedral oligomeric silsesquoixanes (POSSs), which have the general formula (RSiO1.5)n, are prototypical organic/inorganic systems composed of inorganic cores with external organic substituents. Through appropriate designing of the functionality of these organic substituents, it is possible to create both mono- and octa-functional macromonomers for desired applications. These functionalized POSS derivatives can be blended1-3 or attached covalently to linear thermoplastics4-11 or thermosetting networks5,6,12-17 to form high-performance hybrid materials.18-23 The physical properties of POSS/polymer hybrid materials are strongly influenced by the miscibility of the host polymer and the POSS derivative. Hydrogen bonds are often exploited as a favorable interaction to improve the miscibility of blend systems24–26. In a previous study,27 we observed a dramatic increase in the
glass transition temperature when strong hydrogen bonds existed between the POSS moieties and polymer containing proton acceptors.
In this study, the POSS was functionalized as a strong proton donor [i.e., octa(phenol)octasilsesquioxane (OP-POSS)] to improve its miscibility with polymers containing proton acceptors. In previous studies,27,28 the thermal properties of poly(methyl methacrylate) (PMMA) and poly(vinylpyrrolidone) (PVP) polymers were enhanced through copolymerization with POSS derivatives. Because covalent copolymerization can be a complicated and time-consuming process, polymer blending is usually considered to be a simpler and more convenient means of preparing POSS/polymer hybrid materials. Our aim in this present study was to compare the miscibility and hydrogen bonding behavior of the PMMA/OP-POSS, PVP/OP-POSS, and PMMA-co-PVP/OP-POSS blends. Furthermore, POSS-based electrolytes for rechargeable lithium batteries have been reported:29 the incorporation of POSS derivatives improved their potential applicability of these systems as solid state electrolytes. Thus, the ionic conductivity of the LiClO4/OP-POSS/ PMMA-co-PVP ternary blends was also investigated. We employed differential scanning calorimetry (DSC), Fourier transform infrared (FTIR) spectroscopy, and ac impedance measurement as a battery of techniques to investigate the hydrogen bonding, miscibility, and ionic conductivity behavior of these systems.
2.2. Experimental Part
2.2.1. Materials. Ethyl ether, benzene, N,N-dimethylformamide (DMF), tetrahydrofuran
(THF), azobisisobutyronitrile (AIBN), N-vinyl-2-pyrrolidone (VP), platinum divinyltetramethyldisiloxane complex [Pt(dvs)], 4-acetoxystyrene (AS), lithium perchlorate (LiClO4), and methyl methacrylate (MMA) were purchased from Aldrich Chemical Co.
Q8M8H was obtained from Hybrid Plastics Co. AIBN was purified through recrystallization from ethanol. Benzene and DMF were fractionally distilled from calcium hydride. The MMA
and VP monomers were purified through vacuum distillation from calcium hydride. Ethyl ether, THF, Q8M8H, Pt(dvs), and AS were used as received.
2.2.2. Synthesis of Octa(phenol)octasilsequioxane-POSS (OP-POSS) Oligomer. The Q8M8H oligomer (1.96 mmol) was placed in a dry 50-mL round-bottom flask equipped with a stirrer bar. Toluene (30 mL), AS (16.66 mmol), and Pt(dvs) (one drop) were added sequentially over 10 min. The reaction mixture was then heated to 80 °C under a nitrogen atmosphere for 4h. After cooling to room temperature, the solution was filtered and then evaporated, and finally the residue was dried under vacuum until reaching a constant weight. The product, octa(acetoxystyryl)octasilsequioxane (AS-POSS, Scheme 2-1, yield was calculated to be 80%), was obtained as a colorless, viscous liquid.30 AS-POSS was dissolved in THF under a nitrogen atmosphere and then NaOH (10 %) was added dropwise. The mixture was stirred for 48 h at room temperature. After the reaction, ethyl ether and deionized water (1:1) were added; then aqueous hydrochloric acid (10 %) was added dropwise to the mixture with stirring until the pH reached 8. Residual ethyl ether and water were evaporated under vacuum to provide octa(phenol)octasilsequioxane (OP-POSS, yield was calculated to be 82%). The final product, OP-POSS (Scheme 2-1), which is a lightly brown and viscous liquid was filtered and dried in a vacuum oven for 96 h at 80 °C.30
2.2.3. Syntheses of PMMA-co-PVP Random Copolymers. The PMMA-co-PVP random
copolymers were prepared through the free radical polymerization using AIBN as the initiator (Scheme 2-2). The reactions were performed in benzene at 80 °C under a nitrogen atmosphere in a dry 100-mL round-bottom flask equipped with a stirrer bar. To determine the reactivity ratio, samples of the copolymers were removed from the reaction mixture during the early stages of the copolymerization, when the degrees of conversion remained relatively low (between 4–9%).31 After 24 h, the mixtures were cooled to room temperature and the product copolymers were purified through precipitation into ethyl ether. The filtered product
copolymers were dried until they reached a constant weight. The molecular weights and molecular weight distributions of the PMMA-co-PVP copolymers were characterized through GPC at 50 °C using DMF as the eluent and polystyrene standards for calibration. The compositions of the copolymers were characterized using 1H NMR spectroscopy and elementary analysis (EA).
The 1H NMR spectrum of the copolymer was recorded from a CDCl3 solution at 25 °C using a Varian UNITY INOVA-400 NMR spectrometer. EA was performed in an oxidative atmosphere at 1021 °C using a Heraeus CHN-O Rapid Elementary Analyzer. The MMA and VP units in the PMMA-co-PVP copolymers correspond to repeating units of C5H8O2 and C6H9NO, respectively, thus, the MMA content (mol%) was determined using the Eq. (2-1), based on the contents of C and N atoms.32
(
)
100 6 7 30 1 % × − − = N C N mol MMA (2-1)where N and C refer to the contents of N and C atoms, respectively, in the copolymer.
2.2.4. Blend Preparations. Several binary PMMA/OP-POSS, PVP/OP-POSS, and
PMMA-co-PVP/OP-POSS blends were prepared. Desired amounts of PMMA, PVP, PMMA-co-PVP, and OP-POSS were dissolved in DMF and stirred continuously for 24 h at 60 °C. The solutions were cast into Teflon dishes and maintained at 80 °C for 24 h to remove most of the solvent and then the blends were dried under vacuum maintained at 120 °C for 96 h.
2.2.5. Characterizations. Thermal analyses were performed using a DuPont TA2010 DSC
instrument calibrated with indium standards. The analyses were conducted under a nitrogen atmosphere at a scan rate of 20 °C/min over a temperature range from –60 to 200 °C. The glass transition temperature (Tg) was obtained as the inflection point of the heat capacity jump.
FTIR spectra of KBr disks were recorded over the range 4000–400 cm–1 using a Nicolet Avatar 320 FT-IR spectrometer, 32 scans were collected at room temperature and a resolution
of 1 cm–1. Each DMF solution was cast onto a KBr disk and then most of the solvent was evaporated at 80 °C for 24 h; a vacuum (0.2 torr) was applied and the blend was then heated at 120 °C for an additional 96 h to completely remove the solvent. The frequency-dependent impedance properties (from 10 MHz to 10 Hz) of the polymer complexes were measured using an Autolab instrument designed by Eco Chemie. The samples were pressed into disks and loaded into a sealed conductivity cell between stainless-steel blocking electrodes; the films had thicknesses varying from 0.50 to 0.15 mm for these conductivity measurements. The impedance response was measured at 30 °C and the conductivity was calculated from the bulk resistance according to the Eq. (2-2):33,34
b AR
L
=
σ (2-2)
where σ is the conductivity, L is the thickness of the electrolyte film, A is the section area of the stainless-steel electrode, and Rb is the bulk resistance.
2.3. Results and Discussion
2.3.1 PMMA-co-PVP Copolymer Characterization. A series of copolymers was prepared
using various VP and MMA monomer concentrations. Table 2-1 lists the MMA contents (mol%) of the copolymers, determined through 1H NMR spectroscopy and EA. Because of the compositions of the copolymers determined through 1H NMR spectroscopy were affected by the interaction between water and PMMA-co-PVP, their VP contents (mol%) are slightly overestimated. EA provided better accuracy, although the H atom contents determined this way are inaccurate because of compositions of the copolymers were affected by the presence of water. Accordingly, we applied only the EA-determined N and C contents to calculate the VP content using Eq. (2-1). In the following discussion, the sample codes for these copolymers are based on the MMA contents obtained through EA.
We calculated reactivity ratios (r1 for MMA; r2 for VP) using the methodology of Kelen
of the copolymers. To minimize errors resulted from changes in the feed ratios, the polymerization was terminated at monomer conversions of less than 10%. The values of r1
and r2 are the ratios of the homo-propagation and cross-propagation rate constants for each
monomer (i.e., k11/k12 and k22/k21, respectively). Figure 2-1 displays the Kelen–Tudos plot for
the PMMA-co-PVP copolymers. The values of r1 and r2 were 0.94 and 0.97, respectively. In a
previous study,38 we defined a copolymerization to be “ideal” when the product r1×r2 was
unity. When r1 and r2 both equal 1, the two monomers possess equal reactivity toward both
propagating species; the behavior of the resulting copolymer is referred to as random or Bernoullian. Thus, the copolymers synthesized through free radical polymerization in this study were essentially random. (i.e., close to an ideal copolymer: r1 × r2 = 0.91).
2.3.2. Analyses of OP-POSS/Homopolymer Binary Blends. Figure 2-2 presents DSC
thermograms of the PMMA/OP-POSS and PVP/OP-POSS blends. The star-shaped OP-POSS was similar to a linear oligomer of poly(vinyl phenol), i.e., it has a degree of polymerization equal to 8. The glass transition temperature of OP-POSS (25 °C) was lower than that of a typical high-molecular-weight (Mn = 10,000 g/mol) PVPh (150 °C) because of molecular weights and structural differences. Single values of Tg existed in both blends, implying that all of these binary and ternary blends are miscible. Several equations have been suggested to predict the variation of the glass transition temperature of a random copolymer or miscible blend as a function of its composition. In this study, we employed the Kwei equation to predict the variation of the glass transition temperature:
2 1 2 1 2 2 1 1 W qW kW W T kW T W Tg g g + + + = (2-3)
where W1 and W2 are the weight fractions of the compositions, Tg1 and Tg2 represent the glass
transition temperature of the corresponding blend components, and k and q are fitting constants. Furthermore, the value of q, a parameter corresponding to the strength of hydrogen
bonds in the blend, correlated to the balance between the breaking of the self-association and the forming of the inter-association hydrogen bonds. Figure 2-3 displays the dependence of Tg
on compositions of the PVP/OP-POSS and PMMA/OP-POSS blends. We obtained the values of k and q based on non-linear least-squares best fits. In the PVP/OP-POSS blends, q had a value of +100, revealing the presence of a strong intermolecular interaction between PVP and OP-POSS. On the other hand, a negative value of q (–40) was obtained for the PMMA/OP-POSS blends, indicating that the intermolecular hydrogen bonding was weaker than the intramolecular hydrogen bonding.
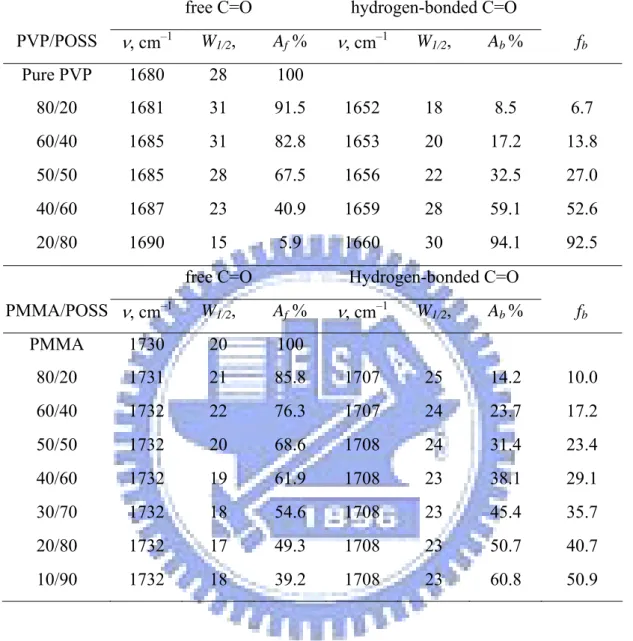
Figure 2-4 displays partial IR spectra (2700–3700 cm–1) of the PMMA/OP-POSS and PVP/OP-POSS blends. The pure OP-POSS exhibits two bands in the OH stretching region in the IR spectrum; one corresponding to the hydrogen-bonded OH groups (a broad band centered at 3350 cm–1) and the other to “free” OH groups (a shoulder centered at 3525 cm–1). When the PMMA (PVP) was mixed with OP-POSS and the C=O oxygen atoms of PMMA (PVP) interacted with the OH groups of OP-POSS, thus the broad band shifted to higher (lower) frequency at 3450 (3190) cm–1. This behavior reflected the competition between the hydroxyl–hydroxyl and hydroxyl–carbonyl interactions. Additionally, the hydroxyl–carbonyl interactions predominated over the hydroxyl–hydroxyl interactions in the PMMA (PVP)-rich blends; thus, we assigned the band at 3450 (3190) cm–1 to be the OH groups interacting with the C=O units. Moskala et al. used the frequency difference (Δ ν ) between the hydrogen-bonded and free OH absorptions to estimate the average strength of the intermolecular interaction.39 Accordingly, we used the free OH stretching at 3525 cm–1 as a reference, the hydroxyl–carbonyl inter-association was weaker than the hydroxyl–hydroxyl self-association interaction in the PMMA/OP-POSS blends, but stronger in the PVP/OP-POSS blends. This finding is consistent with the negative and positive values of q for the PMMA/OP-POSS and PVP/OP-POSS blends, based on Kwei equation.
Figure 2-5 displays the C=O stretching regions in the IR spectra of PVP/OP-POSS and PMMA/OP-POSS blends. In Figure 2-5(a), pure PVP exhibits a broad band centered at 1680 cm–1, corresponding to the “free” C=O groups. Painter et al.40 reported that the pyrrolidone group strongly self-associates through transitional dipole coupling. Therefore, the signal for “free” C=O groups at 1680 cm–1 is not that of “truly free” C=O groups, which would be centered at 1708 cm–1. The signal for C=O stretching was split into two bands at 1680 and 1650 cm–1 corresponding to “free” and the hydrogen-bonded C=O groups, respectively; these signals fitted the Gaussian function well. As the concentration of OP-POSS increased, the probability of PVP/OP-POSS interactions increased, resulting in an increased intensity of the hydrogen-bonded C=O band at the expense of the “free” C=O band. We calculated the fraction of the hydrogen-bonded C=O groups (fb) using Eq. (2-4):41
f A b A b A O C b f + = = 3 . 1 / 3 . 1 / (2-4)
where Ab and Af denote the peak areas corresponding to the hydrogen-bonded and “free” C=O
groups, respectively. In this case, we employed a ratio for the two absorptivities (a2/a1) of 1.3 based on a previous calculation.40 Table 2-2 summarizes the results of curve fitting for the PVP/OP-POSS blends. As expected, the fraction of hydrogen-bonded C=O groups increased upon increasing the OP-POSS content.
Figure 2-5(b) reveals that the PMMA blend system has a sharp (compared with that of the pure PVP) IR band at 1730 cm–1 and a shoulder at 1710 cm–1, representing the free and the hydrogen-bonded C=O groups, that also fitted the Gaussian function well. We applied the method described above to analyze the PMMA/OP-POSS blends, but in this case, we used a value of a2/a1 of 1.5.42 Table 2-2 lists the results of curve fitting of the PMMA/OP-POSS system. Again, the fractions of hydrogen-bonded C=O groups increased upon the increase of the OP-POSS content.
In previous studies,27,28 we confirmed that certain interactions occur between the POSS moieties and OH group. In this study, the situation was more complicated than those in previous studies because the OH groups were attached to the POSS cage. To further understand the interaction phenomena, we employed the Painter–Coleman association model (PCAM) to analyze these systems:41
2 1 1 B B B + ←⎯→K2 ) 2 ( B B B + 1←⎯→B h+1 h≥ K h C B C B c 1 h K h + ←⎯→ A B A B A 1 h K h + ←⎯→⎯
where A, B, and C are descriptors representing the siloxane groups, the phenol groups of the POSS cages, and PMMA, respectively; KA, KB, and KC are their respective association
equilibrium constants; K2 is the equilibrium constant of forming dimers between phenol
groups. These equilibrium constants can be expressed in terms of volume fractions: (2-5) (2-6) ⎥ ⎦ ⎤ ⎢ ⎣ ⎡ + + Γ = C 1 C C A 1 A A 2 1 B B 1 r K r K Φ Φ Φ Φ
[
A B1 1]
1 A A =Φ 1+ Φ Γ Φ K ΦC =ΦC1[
1+KCΦB1Γ1]
(2-7) where ⎟⎟ (2-8) ⎠ ⎞ ⎜⎜ ⎝ ⎛ Φ − + ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − = Γ B1 B B 2 B 2 1 1 1 1 K K K K K(
)
⎟⎟⎠⎞ ⎜⎜ ⎝ ⎛ Φ − + ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − = Γ 2 B1 B B 2 B 2 2 1 1 1 K K K K K (2-9) Where ΦA, ΦB, and ΦC are the volume fractions of the repeat units in the blend, ΦA1, Φ B1, and ΦC1 are the volume fractions of the isolated units in the blend, rA (VA/VB) and rC (VC/VB) are the ratios of the segmental molar volumes.28 Furthermore, we adopted the self-association equilibrium constants of PVPh45 (K2 = 21 and KB = 66.8), for phenol groups ininter-association equilibrium constant (KC) of the PVPh/PMMA blend has been reported
previously to be 37.4.44 Using the value of KC together with the phenol group self-association
equilibrium constants (K2 and KB), we obtained a theoretical curve for the fraction of
hydrogen-bonded C=O groups at 25 °C as a function of the weight fraction of OP-POSS content (Figure 2-6). When the value of KA was equal to 2, the experimental data agreed fairly
well with the predictions of the PCAM. A slight deviation was observed at weight fractions less than 0.4 because the most accurate range for determining the fraction of hydrogen-bonded C=O groups was from 0.4 to 0.7, where the bands for both the free and hydrogen bonded C=O bands were well separated and had significant absorbances.45 The ratio of the inter-association equilibrium constant (KA) corresponding to the interaction between
OH and siloxane groups of POSS cages to the inter-association equilibrium constant (KC),
was 0.05, implying that inter-association between the phenol and siloxane groups of the POSS cages was insignificant and, thus, could be ignored. The structure of the OP-POSS and the hydroxyl–hydroxyl interaction formed through the phenol groups are the reasons for this behavior because both the arms of OP-POSS which were steric barriers and the presence of hydroxyl–hydroxyl interaction blocked the OH groups from interacting with the siloxane groups.
Using the value of the KB and K2 above and ignoring inter-association between the phenol
and siloxane groups of OP-POSS, we employed the PCAM again to determine the “real” value of KA for the PMMA/OP-POSS blend. The approximate equations were simplified as
follows:46,47 ⎥ ⎦ ⎤ ⎢ ⎣ ⎡ + Γ = A A A B B r K 1 2 1 1 Φ Φ Φ (2-10)







![HPSH [ 分子間作用力 - 氫鍵 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)