國立高雄大學應用化學系(研究所)
碩士論文
以HPLC-UV-MS分析ε-聚離胺酸之實驗參數探討
Investigation of experimental conditions for analyzing
ε-polylysine with HPLC-UV-MS
研究生:黃怡郡
指導教授:何永皓 博士
I
目錄
中文摘要……….…………..1 英文摘要………...2 第一章 前言……….…..4 第二章 文獻探討………..….…6 2.1 Polylysine 的介紹………...….6 2.1.1 Polylysine 的合成……….7 2.1.1.1 α-Polylysine 的合成方法………8 2.1.1.2 ε-Polylysine 的生物合成………9 2.1.2 ε-Polylysine 的特性……….12 2.1.3 ε-Polylysine 的應用……….…14 2.2 分析與鑑定方法………....17 2.2.1 蛋白質的分析方法……….17 2.2.1.1 結構分析………...….17 2.2.1.2 膠體電泳分離技術………..………18 2.2.1.3 質譜法………..20 2.2.2 ε-Polylysine 的分析方法………...23 2.3 蛋白質的構型………31 2.3.1 溶劑對構型的影響……….32 2.3.2 酸對構型的影響………...…..33 第三章 實驗方法……….……37 3.1 藥品………37II 3.2 儀器設備………….………...…39 3.3 分析方法開發……….…...41 3.3.1 測定樣品化學性質….……….…...41 3.3.2 進樣方法……….…....42 3.3.3 HPLC-UV-MS 分析條件……….…...43 3.3.4 流動相中 TFA 濃度的配製……….….….44 3.3.5 流動相中添加不同種類的酸……….……….…….…..…45 3.3.6 以 Zetasizer 量測ε-PL 的粒徑大小………...……46 3.4 樣品前處理………47 3.4.1 樣品的 pH 值….………..…...47 3.4.2 誘導ε-PL 構型的轉變………..47 3.4.2.1 溶劑………..47 3.4.2.2 溫度………..48 3.4.2.3 添加 EDTA………..…48 第四章 結果與討論……….…..…..50 4.1 分析方法開發………...….…50 4.1.1 以 UV/Vis 比對不同樣品中ε-PL 含量……….…..50 4.1.2 HPLC 流動相沖提強度的影響………...…..52 4.1.3 流動相中 TFA 濃度的影響………...62 4.1.4 離子對試劑的影響……….68 4.1.5 管柱差異對分析結果的影響……….76 4.1.6 以 Zetasizer 量測ε-PL 的粒徑大小………78
III 4.2 樣品前處理………..………..80 4.2.1 不同樣品來源的差異性………...……….….……80 4.2.2 樣品基質的影響……….…82 4.2.3 溫度對ε-PL 構型的影響………..86 4.2.4 添加 EDTA 對ε-PL 的影響……….89 4.2.5 稀釋液的影響………..…...94 第五章 結論……….…..…..95 第六章 參考文獻………...…..99
IV
表目錄
表 3-1 不同濃度 TFA 的 pH 值……….44 表 4-1 分析物 A 與分析物 B 水溶液的 pH 值……….51 表 4-2 在不同濃度 TFA 下得到的魔術數字………64
V
圖目錄
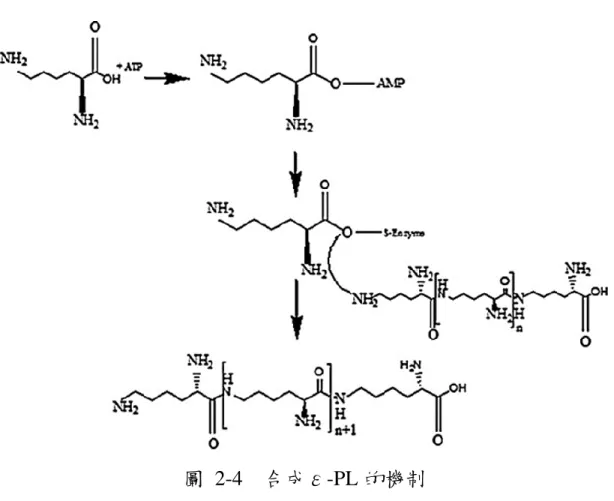
圖 2-1 L-、D-離胺酸及四種聚離胺酸的結構………...………7 圖 2-2 α-polylysine 的結構式………...……….……7 圖 2-3 ε-polylysine 的結構式………....………7 圖 2-4 合成ε-PL 的機制………...…………11 圖 2-5 ε-PL 是通過細菌發酵產生的離胺酸的小型天然生物聚合物… …….…..………...………...12 圖 2-6 2D-PAGE 的分離原理………...…..19圖 2-7 電噴灑游離法(Electrospray ionization, ESI)的原理…..…..….21
圖 2-8 介質輔助雷射脫附游離法(MALDI)的原理……….22 圖 2-9 HPLC 測定ε-PL 的層析圖……….…...24 圖 2-10 K.kifunense 菌株 MN-1、麥角菌 Epichloe sp.菌株 MN-9 產生的 聚合物與ε-PL 的 MALDI-TOF 質譜比較圖………...…25 圖 2-11 上方為ε-PL 水解產物,下方為待測反應產物的質譜圖….…26 圖 2-12 由 Streptomyces sp. DES20.中分離產物的 IR 光譜圖…….…...27 圖 2-13 由 Streptomyces sp. DES20.中分離產物的 1H–NMR 圖譜...28 圖 2-14 由 Streptomyces sp. DES20.中分離產物的 13C–NMR 圖譜….28 圖 2-15 由 Streptomyces sp. DES20.中分離產物的 HMBC 圖譜……….29
圖 2-16 由 Streptomyces sp. DES20.中分離產物的 MALDI-TOF 圖譜… ………...……..30
VI
圖 2-18 溶菌酶(A)和轉鐵蛋白(B)在不同 TFA 濃度下的層析圖
……….34
圖 2-19 溶菌酶在定組成沖提模式中的質譜圖………...………36
圖 3-1 HPLC-UV Detector 並聯 Q-TOF 裝置圖………..…….39
圖 3-2 VESI 自動進樣裝置示意圖………..….40 圖 4-1 樣品 L 與樣品 S 的 UV/Vis 吸收圖譜………...51 圖 4-2 樣品 L 與樣品 S 倍數稀釋後的 UV/Vis 吸收圖譜………….…..52 圖 4-3 以 MeOH 與 0.1% TFA 做為流動相,樣品 S 在不同梯度沖提條件 下,得到不尋常分離現象的層析圖………..…..53 圖 4-4 以以 MeOH 與 0.1% TFA 做為流動相,樣品 S 在不同梯度沖提條 件下,得到不尋常分離現象的層析圖……….………..54 圖 4-5 樣品 L 在流動相 MeOH/0.1%TFA 組成比例為(A)50/50(B) 30/70(C)25/75(D)20/80 的 HPLC-UV-MS 分析結果.……..56 圖 4-6 流動相 MeOH : 0.1% TFA=25:75 條件下,樣品 L 於不同滯留 時間的質譜圖……….………..59 圖 4-7 樣品 S 在流動相 MeOH/0.1%TFA 組成比例為(A)26/74(B) 25/75 的 HPLC-UV-MS 分析結果……….….60
圖 4-8 流動相為 MeOH 與 0.05% TFA 的 HPLC-UV-MS 分析結果…..63
圖 4-9 樣品 L 與 S 在不同濃度 TFA 流動相的 UV 圖譜及 TIC 圖...…64
圖 4-10 在流動相 MeOH 與 TFA 組成比例為 25:75 下,不同濃度 (A)0.1%(B)0.05%(C)0.02% TFA 對ε-PL 滯留時間的影 響………..……….65
VII
圖 4-11 樣品 L 在不同濃度(A)0.1%(B)0.05%(C)0.02% TFA 流 動相下的分離結果……….…..66 圖 4-12 樣品 S 在不同濃度(A)0.1%(B)0.05%(C)0.02% TFA 流
動相下的分離結果……….……….……..67 圖 4-13 流動相 MeOH : 0.5% AcOH 條件下,樣品的 TIC 及質譜圖
……….……….……….….68 圖 4-14 流動相 MeOH : 0.5% FA 條件下,ε-PL 的 TIC 及質譜圖 ……….69 圖 4-15 流動相 MeOH : 0.8% FA 條件下,ε-PL 的 TIC 及質譜圖 ………..………...70 圖 4-16 TFA 的結構式………...71 圖 4-17 TFPA 的結構式……….……..………..71 圖 4-18 流動相 MeOH : 0.15% TFPA 條件下,ε-PL 的 TIC 及質譜圖
………72 圖 4-19 流動相 MeOH : 0.15% TFPA = 18 : 82 條件下,(A)ε-PL 的
TIC 圖及(B)400.2、(C)413.0、(D)425.8、(E)438.6 m/z
的 EIC 圖………..…………..73 圖 4-20 流動相 MeOH : 0.15% TFPA=18:82 條件下,樣品 L 的質譜圖
……….74 圖 4-21 以(A)0.02% TFA 以及(B)0.15% TFPA 作為流動相,n=30
的ε-PL 質譜圖的差異………..…75 圖 4-22 不同管柱對有效分離ε-PL 聚合度的條件具差異性………....77
VIII
圖 4-23 以粒徑分析儀對溶於 MeOH : 0.1% TFA(50 : 50)的樣品 S 進行
分析的五重複實驗結果………..79
圖 4-24 以直接進樣方式,(A)樣品 L 與(B)樣品 S 的質譜圖...…80
圖 4-25 以 Loop injection 方式,(A)樣品 L 與(B)樣品 S 的質譜圖 ………..81 圖 4-26 樣品 S 在(A)pH 值= 4.65 及(B)8.35 時的 TIC 圖………82 圖 4-27 樣品 S 在(A)pH 值= 4.65、(B)6.65 及(C)8.35 時的 TIC 圖………83 圖 4-28 樣品 L 在(A)pH 值= 4.65 及(B)8.35 時的 TIC 圖………83 圖 4-29 聚合度為 32 的樣品 S 於(A)pH 值= 4.65、(B)6.65 及(C) 8.35 環境下所得質譜圖………..………...84 圖 4-30 在(A)pH 值= 8.36 與(B)4.65 環境下,樣品 L 聚合度為 22 的質譜圖比較………...………..85 圖 4-31 溫度變化對樣品 L 影響的 TIC 圖……….…..86 圖 4-32 溫度變化對樣品 S 影響的 TIC 圖……….…..….87 圖 4-33 在 60℃下,加熱時間對樣品 L 影響的 TIC 圖………..….88 圖 4-34 在 60℃下,加熱時間對樣品 S 影響的 TIC 圖………..….88 圖 4-35 添加 EDTA 影響樣品 L 分離結果的 TIC 圖………...90 圖 4-36 添加 EDTA 影響樣品 S 分離結果的 TIC 圖……….…..90 圖 4-37 於樣品中添加 EDTA 的質譜圖……….………..….91 圖 4-38 樣品 L 經(A)加熱(B)未加熱後添加不同比例 EDTA 的質譜 圖……….92
IX
圖 4-39 樣品 S 經(A)加熱(B)未加熱後添加不同比例 EDTA 的質譜 圖……….………93 圖 4-40 以 H2O、MeOH、IPA 以及 ACN 作為溶劑的分析結果……...94
1
以HPLC-UV-MS分析ε-聚離胺酸之實驗參數探討
指導教授:何永皓 博士(教授) 國立高雄大學應用化學系研究所 學生:黃怡郡 國立高雄大學應用化學所 (碩士班) 摘要 本研究原意為發展一套能檢視不同市售ε-聚離氨酸(ε-polylysine;ε-PL)樣品的 HPLC 方法,然而於研究過程當中發現預計發展的 HPLC 方法耐變性不佳,檢視比對 不同ε-PL 樣品時有可能產生誤判,因此針對造成耐變性不佳的原因進行深入的探討。 首先發現要能有效分離不同聚合度的ε-PL,添加離子對試劑三氟醋酸(TFA)於流動相 中是必要的,以甲酸或醋酸調節流動相 pH 至與 TFA 水溶液相同的 pH,並無法分離 不同聚合度的ε-PL。流動相中 TFA 與 ε-PL 形成離子對的多寡會改變ε-PL 構型,以 及所帶的靜電荷,進而改變不同聚合度ε-PL 的滯留時間。流動相中 MeOH/0.1%TFA 水溶液比例由 50/50 改至 20/80,會使 ε-PL 的滯留時間由短於完全不滯留時間,變到 聚合度很小的ε-PL,經過了一小時都無法被沖提出。此外,意外發現兩種市售樣品(L 和 S) 中相同聚合度ε-PL,於相同層析條件下,卻有不同表現,推論 L 和 S 樣品中 ε-PL 的構型並不相同。基於構型不相同的論點,我們進行了以下樣品前處理方式: (1) 將原本 pH 不同的 L 和 S 溶液調整至相同的 pH;(2)加熱 L 和 S 溶液到不同溫度後冷 卻;(3)加熱添加 EDTA 的 L 和 S 溶液後冷卻;(4)以不同有機溶劑稀釋 L 和 S 溶液, 而後觀察這些處理後ε-PL 於層析圖中的相對表現。由實驗結果當中獲致下列結論: (1) 樣品的基質影響ε-PL 的構型,改變樣品的基質會誘導 ε-PL 構型進行變化,但是過程 緩慢,可能要耗費數小時以上;(2) 緩慢的構型轉換使得來自不同基質的 ε-PL,得以 在層析的過程中仍保有部分原始構型的特性; (3) ε-PL 構型於 L 中傾向蜷曲,S 中傾 向延展,因此 L 中的 ε-PL 對基質的變化較為敏感。最後我們發現在實驗條件一切相 同下,使用相同廠商、規格的管柱也會有顯著不同的差異,說明以 HPLC 進行不同聚 合度ε-PL 的分析,其耐變性遠低於一般 HPLC 在分析它類樣品的應用。 關鍵字: HPLC-UV-MS、ε-聚離氨酸、聚合度、構型、三氟乙酸、離子對2
Investigation of experimental conditions for analyzing
ε-polylysine with HPLC-UV-MS
Advisor: Dr. Yeung-Haw Ho Institute of Applied Chemistry National University of Kaohsiung
Student:Yi-Chun Huang Institute of Applied Chemistry National University of Kaohsiung
ABSTRACT
Preliminary goal of this research is to develop an effective HPLC method for analyzing ε-polylysine (ε-PL) from various suppliers. After the method was developed, we found that the robustness of the method is very poor and can easily misinterpret experimental results. Therefore, we further investigated the factor that cause the poor robustness of the HPLC method. Trifluoroacetic acid (TFA) is an essential mobile phase modifier for analyzing ε-PL with various degree of polymerization. Using formic acid and acetic acid as the modifier cannot resolve ε-PL with various degree of polymerization. TFA acted as an ion-pair reagent. Number and strength of multi-interactions between TFA and ε-PL will affect the conformation and the net charge of ε-PL---TFAs adduct resulted in different retention time. ε-PLs coeluted before the retention time of mobile phase while MeOH/0.1% TFA=50/50 was used as mobile phase. ε-PLs were retained in column while MeOH/0.1% TFA=20/80 was used as mobile phase. Accidentally, we noticed ε-PL from two different vendors (L and S) had different retention time under same experimental conditions. We speculated that matrix effect was the cause of disagreement. We studied the effect of several sample pretreatment on separation of ε-PLs. The pretreatments included (1) adjusted pH of L and S to same pH; (2) heated up L and S then cool down them to room temperatures; (3) heated up L and S then added EDTA into the solutions and cool down the solution to room temperatures; (4) diluted L and S with various organic solvents. The experimental results showed: (1) the matrix in sample did affect the retention time of ε-PL in column; (2) the mobile phase cannot overcame the matrix effect originated from sample L and S. In conclusion, results indicated ε-PL exhibited molten globule conformation properties in L and extended conformation properties in S.
3
Keywords: HPLC-UV-MS, ε-polylysine, the degree of polymerization, conformation,
4
第一章 前言
聚合物(polymers)是指具有非常大分子量的化合物,分子間由結 構單位(structural unit)或單體由共價鍵連接在一起,包括了很多種適 用於不同目的及用途的人工合成及自然生成的材料,而蛋白質、核酸和 多肽也是相當重要的生物聚合物。就結構和功能而言,蛋白質是所有生 物分子中最複雜的,其結構範圍從一級到四級結構,許多蛋白質就算維 持了一級到三級結構,四級結構一旦遭到破壞即失去其原有的生物功 能。 聚離胺酸(polylysine, PL)有α-和ε-兩種不同的結構異構物,α- 聚離胺酸來自人工合成,可以有不同的聚合度,ε-聚離胺酸(ε-polylysine,ε-PL)則是一種特殊的鹼性生物聚合物,只能由天然的生物 合成方式產生,其特色是在ε-氨基和α-羧基之間具有酰胺鍵,並且一 般有穩定的聚合度。ε-PL 有高水溶性、熱穩定性、具多陽離子、可生 物降解、不具毒性,廣泛的被應用在醫藥、食品添加劑、農業及工業領 域等,目前有大量的研究投入ε-PL 和其衍生物的合成以及應用開發。 工業上所使用的ε-PL 因為來源不同而有溶液與固體粉末兩種包 裝,兩者都宣稱主成分為ε-PL。初步利用 loop injection 方式讓分析物 隨著流動相不經過分離直接進入質譜儀,針對兩種樣品所含的ε-PL 進 行質譜法分析,卻發現兩者所展現的平均分子量,以及不同聚合度ε-PL 的分布輪廓並無顯著差異。於是更進一步以 LC/MS 方式分離樣品中 的成分,意外發現不同聚合度ε-PL 的分離效果對 HPLC 流動相的組成 極為敏感,初步判定只有在特定的流動相組成才可以分離不同聚合度的5
ε-PL,否則不同聚合度ε-PL 不是同時沖提出,就是長期滯留於管柱內 無法被沖提出。
本研究為了找出能夠分離不同聚合度ε-PL 的最佳條件,參考已知 分析生化高分子的 HPLC 方法,將在 HPLC 流動相中加入離子對試劑 trifluoroacetic acid (TFA)以獲得較佳的分離效果,並規劃探討的 HPLC 流動相組成變因,包含 (1) 不同的流動相溶劑組成比例;(2) 流 動相中 TFA 的濃度;(3) 不同的離子對試劑等條件。並依據所蒐集的資 料推導不同聚合度ε-PL 在 HPLC 分離過程中造成異常分析結果的關鍵 變因。 另外,本研究採用的分析方式為 HPLC-UV-MS,在這套系統中 Q-TOF 質譜儀的資料,可以協助我們觀察各種不同聚合度ε-PL 的滯留時 間變化,包含特定聚合度ε-PL 的電荷分布等資料,HPLC-UV 層析圖 則提供我們在特定化合物量的變化上有較精確的判讀,了解經過管柱後 ε-PL 的回收效果,是否完全沖提出。 更有趣的是,對於不同來源的分析物,在相同的分析條件下我們卻 得到了不同滯留時間的分析結果,經由初步的量測,我們發現兩分析物 的 pH 值存在極大的差異,並接續利用不同的樣品前處理法,在相同分 析條件下進行分析,觀察兩種分析物中不同聚合度ε-PL 在 LC/MS 中層 析及質譜圖上的表現,找尋造成差異性的原因並加以證實。
6
第二章 文獻探討
隨著聚合物(polymers)在各個領域的快速發展及廣大的應用,人 們對具有各種結構、特徵的新型聚合物的發現與開發有了更大量的投入 與研究。而聚胺基酸(Polyamino acids)也是聚合物的一種,由相同類 型且多個胺基酸通過酰胺鍵(amide bonds)連接,它們表現出優異的生 物相容性和對生物材料具有相當重要的性質,特別是應用在生物醫藥及 製藥領域。其中,聚離胺酸(Polylysine)因其各種優點及特殊性是目前 研究最多的聚胺基酸之一。2.1 Polylysine 的介紹
聚離胺酸(Polylysine)是指幾種類型的離胺酸聚合物,它們在立體 化學和連接位置方面可以彼此不同(圖 2-1)。其前體胺基酸為(Lysine,Lys, K),與精胺酸(Arginine, Arg, R)和組胺酸(Histidine, His, H)一
樣,屬於鹼性胺基酸;離胺酸是一種α-胺基酸,含有兩個氨基,一個 在α-碳上,一個在ε-碳上,兩者都可以是聚合的位置,產生α-聚離胺 酸(α-polylysine,α-PL)或ε-多聚離胺酸(ε-polylysine,ε-PL)。 離胺酸能以 L-或 D-的立體異構型式存在,但只有 L-型存在於生物 的蛋白質中,D-型離胺酸不具有生物活性且能被轉化成 L-型離胺酸,因 此所有商業化生產的離胺酸均為 L-型離胺酸。
7 圖 2-1 L-、D-離胺酸及四種聚離胺酸的結構
2.1.1 Polylysine 的合成
Polylysine 含有兩個氨基,一個在α-碳上,一個在ε-碳上,這兩者 都可以是聚合的位置,分別產生α- polylysine(圖 2-2)和ε-polylysine (圖 2-3),兩者在合成方法上也不同,α-PL 可以利用人工化學合成方 法製備得到,而ε-PL 卻只能以天然的方式生產無法被化學合成。 圖 2-2 α-polylysine 的結構式 圖 2-3 ε-polylysine 的結構式8
2.1.1.1 α-polylysine 的合成方法
α-聚胺基酸(Poly-α-amino acids)是由通過胜肽鍵連接的α-氨基 酸單體組成的合成聚合物,並伴隨著應除去的不同量的副產物,其合成 通常是以典型的一步聚合方法完成的,並且能獲得比較高分子量的聚合 物1,2,合成的α-聚胺基酸作為蛋白質模型的巨大價值在於它們對天然 蛋白質的複雜性相對而言是簡單的3。α- polylysine 是透過鹼性縮聚反 應製備而成,利用羧酸和胺的縮合聚合反應逐步的增長聚合,並失去副 產物水,主要由 50 個單體連接而成。 許多研究致力於α-PL 在藥物輸送系統中的各種應用,例如 1978、1979 年的 Shen 和 Ryser 4,5以及 1982 年的 Gad 6等;然而,其實 Sela 和
Katchalski 於 1959 年就已經發表α-PL 對人體具有毒性3,因此,現在
的研究多投入於天然且不具毒性的ε-polylysine,希望能用來代替化學 合成的α-PL。
9
2.1.1.2 ε-polylysine 的生物合成
ε-polylysine 的生產方式只能透過微生物發酵,且能分泌ε-PL 的 微生物主要集中在鏈黴菌屬、北里孢菌屬和芽孢桿菌屬;最早被發現產 生ε-PL 的菌種為鏈黴菌屬微生物包括小白鏈黴菌(Streptomyces
albulus)、澱粉酶產色鏈黴菌(Streptomyces diastatochromogenes)、灰褐
鏈黴菌(Streptomyces griseofuscus)等;但是天然生成的ε-PL 產量極 低,因此越來越多研究投入期望達到較高的生產率並使其工業化以供應 廣大的需求。
最早於 1977 年被 Shima 和 Sakai 7發現,是由 Streptomyces albulus
346 一種鏈黴菌產生的細胞外陽性物質。另外,Shima 和 Sakai 也持續地 進行了分類學、發酵培養8與化學性質9的研究,更加確認了ε-PL 有別 於α-PL 的聚合度,且雖然離胺酸其天然存在的異肽鍵不罕見,但其均 聚合物攜帶了異肽鍵也就是異於其他蛋白質,其酰胺鍵不在肽鍵典型的 α-氨基和羧基之間,而是在ε-氨基和羧基之間,這在當時還是個未知 的物質。在基礎培養基中通常需要在 30℃下培養 48 小時才能產出 0.3 g/L 的ε-PL;然而透過進行了兩步培養的方法,Shima 等人在 1983 又 發表了在 8 或 9 天內可以產生大量(約 4-5 g/L)的ε-PL10。 在後續的研究中,Hiraki 等在 1998 年發現了菌株 346 的突變體能 產生比原方法四倍量的ε-PL11;為了增強ε-PL 的產量,Kahar 等人採 用了 pH 控制的策略,所使用的菌種為 Streptomyces albulus 410 (S410)12,該方法成功的將ε-PL 的產量從 5.7 g/L 增加至 48.3 g/L。
10
直到近期,約 2002 年才由 Nishikawa 和 Ogawa 提出由其他細菌菌
株或真核生物合成ε-PL 的方法13,原本是為了開發篩選ε-PL 生產者
的方法,卻意外地找到 Kitasatospora 與 Epichloe,尤其是 Epichloe sp. MN-9 是一種可以產生具有 24-29 個離胺酸單體的ε-PL,屬於麥角菌是 真核生物中第一個被發現的ε-PL 生產者。 在工業上,ε-PL 主要由微生物發酵生產,與需要重複保護/去保護 反應或許多化學化合物的化學方法相比,微生物對於ε-PL 生產更實 用、有效且是對環境友善的。近年來,隨著基因工程、生物信息學、先 進精密儀器和檢測設備的發展,研究人員試圖了解ε-PL 的合成機制並 投入研究中。 透過使用放射性同位素標記方法發現ε-PL 生物合成的前體是 L-離 胺酸10。2003 年,Kawai 等人提出了可能的合成機制,在無細胞系統中 進行ε-PL 的生物合成研究,結果顯示ε-PL 的生物合成是由膜中的非 核醣體肽合成酶(NRPS)催化而來;在體外,發現ε-PL 合成依賴於 ATP 並且不受核糖核酸酶、卡那黴素或氯黴素的影響14,在ε-PL 生物 合成的第一步 L-lysine 在其自身的羧基腺苷酸化具有 ATP-PPi 交換反 應,酶的硫醇基的活性位點形成活性氨酰基硫酯中間體,導致活化的離 胺酸單體縮合15。 從菌株中分離的基因被鑑定為具有腺苷酸化和硫醇化結構的 NRPS 膜蛋白,它不像傳統的縮合或硫酯酶結構,而是具有串聯結構能使游離 的 L-lysine 聚合物作為受體和ε-PL 合成酶結合的 L-lysine 作為供應體 持續地催化 L-lysine 聚合,從而產生不同長度的鏈(圖 2-4)16。另外,
11
Yamanaka 等人在 2010 年提出,ε-PL 合成酶功能受細胞內 ATP 調節, 並且需要在酸性 pH 條件下使細胞內 ATP 積累,而不是抑制ε-PL 降解
酶所致17。
12
2.1.2 ε-Polylysine 的特性
由於α- polylysine 被報導出具有毒性,現在研究多趨向於天然的ε -polylysine。ε-PL 是一種不常見的陽離子生物聚合物,屬於天然存在的 鹼性聚酰胺,其特色是在ε-氨基和α-羧基之間具有酰胺鍵,主要由 25-35 個單體所組成(圖 2-5),ε-PL 的分子量一般受菌株自身調控,不同 菌株合成ε-PL 聚合度的分佈範圍存在差異;外觀呈現淡黃色、略帶有 苦味。 該生物聚合物具有優於其他現有離胺酸形式的各種優點,ε-PL 具 有高水溶性及穩定性,即使當ε-PL 溶液在 100℃下煮沸 30 分鐘或在 120℃下高壓滅菌 20 分鐘時,也未觀察到降解現象並且保持其聚合物長 度18。 圖 2-5 ε-PL 是通過細菌發酵產生的離胺酸的小型天然生物聚合物13 天然存在的ε-PL 可生物降解也可食用,在大鼠的急性口服毒性研 究中顯示ε-PL 是無毒的19;而大鼠的慢性餵養研究中,在飲食中即使 給予高劑量的ε-PL 也沒有顯著的不良反應20;另外,Neda 等於 1999 年的研究顯示ε-PL 在繁殖、神經及免疫功能,還有對胚胎和胎兒發育 中均未引起毒性19,2003 年 Hiraki 等在藥物動力學中 ADME 的研究中 表示給藥後在 168 小時內ε-PL 可通過排泄離開人體,並通過全身自動 放射攝影技術並未觀察到任何組織或器官中有ε-PL 的累積20,這些研 究皆表示ε-PL 是不具毒性的。 ε-PL 是水溶性、熱穩定、可生物降解和具陽離子的生物聚合物, 對人體健康與對環境無毒。基於這些優勢,在近幾年來,ɛ-PL 及其衍生 物在食品、醫藥和電子工業上皆有相當廣泛的應用。
14
2.1.3 ε-Polylysine 的應用
ε-PL 分子是陽離子表面活性劑,因為其具有在水中帶正電荷的氨 基,在內部有疏水性亞甲基,在極性溶液中有分子外部的親水性羧基和 氨基。陽離子表面活性化合物通常會抑制微生物的增殖,通過 Kito 等在 酵母、真菌和革蘭氏陽性或革蘭氏陰性菌種的生長抑制研究已證明它是 一種有效的抗菌劑21。 在食品的應用上,ε-PL 常被用作食品添加劑,添加大量的ε-PL 會產生苦味,但由於其高抗微生物活性,食品保存所需的ε-PL 濃度 低。19 世紀 80 年代,ε-PL 在日本首次被批准用作食品抗菌防腐劑, 然後逐漸被引入美國、韓國和其他國家;在日本食品中通常含有 10-500 ppm 的ε-PL,包括米飯、麵條、蔬菜、壽喜燒、蛋糕等;而在美國ε-PL 被用作米飯和壽司飯中的防腐劑,建議的添加量為 5-50 ppmε-PL22。 另外,透過將ε-PL 與其他食品添加劑相結合,防腐活性有大幅提 高的效果。例如將ε-PL 和從柑橘類水果種子中提取的殺菌物質結合, 可以安全地用於表面或物品的消毒,或是清潔碗盤,廚房用具等。 儘管ε-PL 可以用作天然抗菌劑,但是將其應用於加工食品中的一 個實際問題是它傾向於與蛋白質和酸性多醣相互作用,導致可能喪失抗 微生物活性;此外,ε-PL 的乳化性能差,所以其用途主要限於澱粉類 食品23,24。在 2000 年 Ho 等人將ε-PL 通過梅納反應(Maillard reaction)與葡聚醣結合改善了乳化活性,除了優於市售乳化劑(Sunsoft15 SE-11 和 Q-18S),還保留了ε-PL 原有的抗菌活性,可用作食品加工中 的雙功能食品添加劑25。 在食品中,除了用作防腐、抗菌劑及乳化劑之外,ε-PL 還有其他 功用。在預防肥胖疾病中,主要的治療方法是低脂飲食,另一種是攝入 限制腸道對膳食脂肪吸收的天然物。胰脂肪酶在腸道脂質的吸收中扮演 重要角色(Duan, 2000),所以抑制胰脂肪酶活性的天然物可以抑制膳食 脂肪從小腸吸收26。在 Kido 等人的研究中,含有膽鹽和磷脂酰膽鹼且 濃度範圍為 10-1000 ppm 的ε-PL 下,能夠抑制並破壞人和豬的胰脂肪 酶活性;另外,在確定分子中是由α-或ε-氨基位置在抑制脂肪酶活性 的實驗中,ε-PL 與α-PL 的不同之處在於與胰蛋白酶、α-胰凝乳蛋白 酶和胃蛋白酶等消化酶一起培養後,其對胰脂肪酶的抑制活性得以維 持,而α-PL 則無法27。這些結果表明ε-PL 會抑制消化道中的脂肪 酶,可被作為膳食劑使用。 除此之外,ε-PL 還能被應用在生物電子學的領域中,它是一種新 興技術,主要利用生物分子代替無機材料,目前能使用聚陽離子化合物 建構生物晶片28,29,又或作為塗層材料30,並用做奈米電子/光子傳導元 件、電路等,具有小型化、低功率、高效率等優點。 ε-PL 的氨基能通過交聯作用與多醣合成具有高水結合能力的水凝 膠31,也因為ε-PL 對人體的安全性,所以高分子凝膠廣泛用於農業、 食品及醫藥等領域。在醫學上,ε-PL 還可做為干擾素誘導劑、脂肪酶 抑制劑、藥物遞送載體、基因傳遞載體、脂質體、奈米粒子、塗料等。
16
由於ε-PL 眾多的特點及優勢,包括高水溶性、具多陽離子、無毒 性、可食用即可生物降解,使其在各大領域皆有廣泛的應用,對現今工 業來說是非常感興趣的生物聚合物,其發展在經濟和環境方面都具有相
17
2.2 分析與鑑定方法
2.2.1 蛋白質的分析方法
蛋白質的分離和鑑定在蛋白質體學(proteomics)中佔有相當大的 重要性,有別於傳統使用溶劑沉澱或鹽析方法分離特定的蛋白質,現今 以高通量的分離技術、高靈敏度的檢測能力和龐大的數據庫分析,更有 助於我們更快地辨識蛋白質。2.2.1.1 結構分析
在所有生物分子中,就結構和功能而言蛋白質是最複雜的。蛋白質 的結構範圍從初級到四級結構,接近 90%的蛋白質結構是透過 X 射線 晶體學的方法測定的,它可以通過測定蛋白質分子在晶體中電子密度的 空間分布,在一定解析度下解析蛋白質中所有原子的三維坐標;另外大 約 9%是由核磁共振(Nuclear Magnetic Resonance, NMR)技術來進行解 析,也可以用於測定蛋白質的二級結構。 對於蛋白質的二級結構,最常用的方法為圓偏光二色性光譜 (Circular dichroism, CD),利用紫外線波長範圍 170-250 nm,在獲得的 光譜吸收曲線上,α螺旋結構會在 208 nm 和及 222 nm 兩處同時出現極 小值,若在 204 nm 和 207 nm 處出現單個極小值則分別為無規捲曲和β 褶板;另一個常用的方法是傅立葉轉換紅外光譜(Fourier-transforminfrared spectroscopy, FT-IR),它可以偵測因氫鍵所造成胺基的震盪;而
光譜法中,測定二級結構最準確的方法是利用 NMR 所紀錄的化學位 移。
18
2.2.1.2 膠體電泳分離技術
十二烷基硫酸鈉聚丙烯酰胺凝膠電泳33,34(Sodium dodecyl sulfate
polyacrylamide gel electrophoresis, SDS - PAGE)是一種廣泛應用於生物 化學、分子生物學、鑑識科學和遺傳學的膠體電泳技術,通常用於分離 分子量在 5-250KDa 之間的蛋白質。 在 SDS-PAGE 的實驗中,需將蛋白質溶液與作為界面活性劑的十二 烷基硫酸鈉(SDS)混合,SDS 是陰離子洗滌劑會破壞蛋白質的二級結 構使其變性,並為蛋白質分子提供均勻的負電荷。接著將變性的線性蛋 白質分子加載到聚丙烯酰胺凝膠(PAGE)上,並置於具有合適電解質 的電泳緩衝液中,施加電壓導致帶負電荷的分子往帶正電的陽極方向移 動;凝膠就像一個篩子,小蛋白質相對容易通過凝膠,而較大的蛋白質 則可能被滯留,因而造成不同分子量的分離;最後再將 PAGE 凝膠以染 料染色以便後續的辨識與鑑定。 對於蛋白質分級分離 SDS-PAGE 可用在純化過程中的鑑定和監測蛋 白質,並評估純化級分的同質性;但由於它同時能分離的蛋白質數目不 夠多,於是發展了 2D-PAGE 的技術35。 二維凝膠電泳是一種高通量蛋白質分離技術,能夠在單個凝膠中 分離多達 10,000 種蛋白質。主要是使用蛋白質的兩種性質來分離複雜的 樣品混合物,在第一維中,蛋白質由等電點 pI 值分離;第二維則由相 對分子量分離(圖 2-6)。
19 圖 2-6 2D-PAGE 的分離原理。A)第一維:等電點的等電聚焦;B) 第二維:分子量的 SDS-PAGE 分離35。 2D-PAGE 可以對細胞中的蛋白質總數進行編目,該目錄還可用在 相關樣品間找到差異的蛋白質,例如: 健康的和患有疾病的、接受治療 的與對照組。來自 2D-PAGE 圖像可用於檢測某些轉譯後修飾,如糖基 化、磷酸化等,也可用於發現僅在特定條件下存在的蛋白質;另外,在 2D-PAGE 上分離的蛋白質具有相當高的純度,因此它們可以被切下來 用於蛋白質測序以及進行質譜分析。
20
2.2.1.3 質譜法
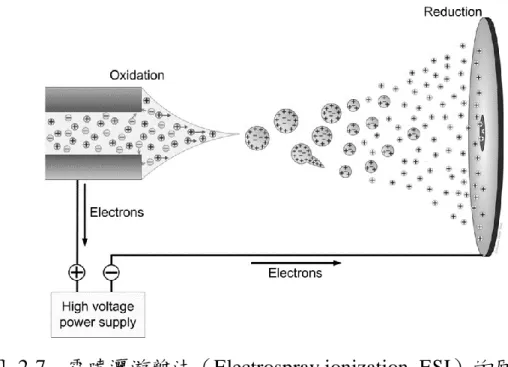
對於蛋白質體學來說,質譜(Mass spectrometry)是一種不可或缺 的技術,被用於蛋白質鑑定、定量、測序、定位及鑑定轉譯後修飾36。 質譜儀的結構可分為五大部分,包括樣品導入系統、離子源、質量 分析器、偵測器、及數據處理系統,並且需要在高真空的條件下運行。 其中游離源的功能是使原本是中性的分子變成帶電荷的離子,而質譜儀 是利用偵測物質的質量與電荷比值大小來分析離子。傳統上的游離方法 有電子游離法(EI)、化學游離法(CI)、熱灑法(TS)、場游離法 (FI)、場脫附法(FD)、快速原子撞擊法(FAB)、電漿脫附法(PD) 等。但這些傳統的游離法並不能有效地游離巨大的生化分子,直到電噴 灑游離法(ESI)與介質輔助雷射脫附法(MALDI)的出現。 於 1984 年首度由 Fenn 提出37,電噴灑游離法(Electrospray ionization, ESI)為大氣壓游離法的一種,在出口端毛細管上加一正的高 電壓,當溶液流經會因電場產生電泳現象,使帶正電的離子會遠離毛細 管,而帶負電的離子則向毛細管移動,形成電荷分離,帶正電荷的液面 往低電場方向拉長形成圓錐狀並且在毛細管尖端匯聚形成泰勒錐 (Taylor cone)。當尖端的庫倫排斥力大於表面張力時,溶液會從尖端噴 灑而出,被分散成許多細小帶電的液滴,並受電場吸引朝入口飛去,溶 劑會在飛行過程中揮發,只剩下帶電荷的分析物進入質譜儀進行分析 (圖 2-7)。21
圖 2-7 電噴灑游離法(Electrospray ionization, ESI)的原理 38
電噴灑游離法(ESI)屬於軟性游離方法,很少促發分析物的自發 碎裂,並且能使分析物帶多電荷(Multiple charges),它能使數萬分子量 的 m/z 值落在幾千的範圍內,擴大了一般質譜儀所能偵測的範圍,這是 其他的游離方式所無法做到的;也因為接上了許多不同數目的質子,因 此會得到一系列的離子訊號,產生了更多的圖譜峰資訊,這對蛋白質的 分析與鑑定相當的重要。 另外,因為 ESI 對大部分的化合物皆有很好的離子化效率,在離子 化的過程中溶劑會大量的揮發,也能使離子在經由溶液相到氣相的過程 中依然維持離子的完整性,因此非常適合與高效能液相層析儀 (HPLC) 、毛細管電泳儀(CE)等連接使用。藉由此種的組合方式, ESI 已成為一種非常普遍且可做為分析多種巨大生化分子的方法。 ESI 游離源對無機鹽類十分敏感,會導致信號抑制和加成物的生 成,降低靈敏度和訊雜比,例如磷酸鹽可能會在 ESI 加熱的毛細管中沉
22
澱導致其永久性損壞。在蛋白質的分析中,主要使用正電模式,常需要 添加甲酸或三氟乙酸(TFA)做為離子對試劑以促進離子形成。
介質輔助雷射脫附游離法(Matrix-assisted laser desorption ionization,
MALDI)39,40,使用雷射作為離子源,將分析物配製於適當的有機小分 子介質中,介質在吸收雷射光後將能量轉移到分析物上,造成分析物脫 附或是氣化,並且經由介質離子進行質子交換而達到離子化的目的(圖 2-8);由於介質會吸收大部分雷射光的能量,所以能將雷射光對分析物 的傷害降到最低。 圖 2-8 介質輔助雷射脫附游離法(MALDI)的原理38 相較於 ESI,MALDI 對鹽類及汙染物的忍受度較高,較適合混合物 的分析,但是當鹽類濃度過高時還是會產生抑制的現象。由於其分析速 度、靈敏度、準確性、耐受性和易於自動化的特點,目前被廣泛應用於 蛋白質大分子的分析與鑑定。
23
2.2.2 ε-Polylysine 的分析方法
許多學者透過 NMR、IR 和 CD 光譜進行ε-PL 的分子結構和構型 的研究。Lee41等人測定了結晶ε-PL 的熔沸點,觀察到玻璃轉化溫度 (Tg)和熔點(Tm)分別為 88℃和 172.8℃。在 IR、CD 光譜以及 1H 和 13C-NMR 的化學位移結果中,顯示ε-PL 在鹼性水溶液中呈現β褶板的 構型42;在酸性 pH 下,由於質子化的α-氨基的排斥,ε-PL 呈現靜電 擴張的構型,而在高於α-氨基的 pKa的 pH 下,則轉變為與反平行β褶 板相似的構型43,44。Maeda45等人於 2003 年,利用 IR 和 Raman 光譜顯 示固態的ε-PL 呈現β褶板的構型;固態13C-NMR 則表示ε-PL 以兩種 結晶形式的混合物存在。 而最早用來測定培養液中 PL 濃度的方法為 Itzhaki 於 1972 年提出 的比色法46,利用甲基橙(Methyl orange)作為染料,陽離子 PL 與過 量的陰離子甲基橙之間相互作用形成水不溶性複合物,再以吸收波長 465 nm 進行偵測,PL 的濃度可以從殘留在上清液中的甲基橙的吸光度 換算而來。2001 年 Kahar12等人發表了利用 HPLC 檢測培養液中 ɛ-PL 的 方法,分析管柱為 Tsk gel ODS-120 T(4.6 mm × 250 mm),其流動相所 使用的是 0.1% H3PO4,流速為 0.4 mL/min,偵測波長是 215 nm。 Itzhaki 的比色法和 Kahar 的 HPLC 檢測方法,在後續由微生物生產ε-PL 的研究中大量被使用到。 另外,同樣是利用比色法進行濃度測定的是 Shen47,他在 1984 年 提出利用台盼藍(trypan blue)代替甲基橙,取上清液以分光光度計波 長 580 nm 進行偵測,進而換算出ε-PL 的濃度;可測量範圍為 1 至 1024 µg/ml,比甲基橙沉澱法低約 10 倍。 為了從生產ε-聚離胺酸鏈黴菌菌株中純化與鑑定ε-聚離胺酸降解 酶,Kito48利用 HPLC 來測定ε-PL 的聚合度,使用的管柱為 Spherisorb S5ODS 管柱,4.6 mm × 150 mm,流速為 1 mL/min,管柱溫度保持在 50 ℃,UV 偵測器的偵測波長設定為 215 nm。流動相 A 為 10 mM NaH2PO4(pH 2.6) 加上 100 mM NaClO4和 10 mM 辛烷磺酸鈉;流動 相 B 為 50%的乙腈水溶液中添加 20 mM NaH2PO4 (pH 2.6)、200 mM NaClO4及 20 mM 辛烷磺酸鈉,並且以梯度沖提方式將流動相 B 由 0% 在 45 分鐘內增加到 75%。該方法的層析圖如下(圖 2-9),將純化後的 酶與ε-PL 一同培養,分析ε-PL 形成的時間和聚合物的長度。另外又 以 Itzhaki 的比色法進行定量;以及 Laemmli 於 1970 發表的 SDS-PAGE 方法估計其分子量。 圖 2-9 HPLC 測定ε-PL 的層析圖 48 (a) 反應前 0 小時;(b) 反應後 1 小時,數字表示ε-PL 聚合度 Nishikawa 和 Ogawa49在測定土壤中微生物產生抗菌ε-PL 聚合物 的分佈實驗中,在分析的部分透過薄層層析(TLC)來確認單體的連接 模式,以及利用 SDS-PAGE 電泳方法測定分子量,並且以介質輔助雷射
25 脫附游離法連接飛行式時間質譜儀(MALDI-TOF)進行更詳細的聚合 物解析,可以用作比較不同菌種分別是 K.kifunense 菌株 MN-1 和麥角 菌 Epichloe sp.菌株 MN-9 產生的聚合物,與市售ε-PL 標準品的差異 (圖 2-10)。 圖 2-10 K.kifunense 菌株 MN-1、麥角菌 Epichloe sp.菌株 MN-9 產生的聚合物與ε-PL 的 MALDI-TOF 質譜比較圖。 n 值表示離胺酸單體的數量49 於 2008 年 Yamanaka 等人50在ε-PL 的分散度由非常不尋常的非核 醣體肽合成酶控制的研究中,利用質譜的優勢鑑定反應的聚合產物是否 為ε-PL,通過使用 1N HCl 製備ε-PL 水解產物(0.1mg/ml)作為標準
26 品,並且進入 HPLC/ESI-MS-MS 進行分析,經由質譜資訊的比對確認 產物為聚合度約 14 個單體的ε-PL(圖 2-11)。 圖 2-11 上方為ε-PL 水解產物,下方為待測反應產物的質譜圖50 Gao 和 Luo51在 2016 年,於鏈黴菌屬產生ε-PL 的生物合成、分離 和結構鑑定研究中,使用了 Itzhaki 的比色法和 Kahar 的 HPLC 檢測方 法,MALDI-TOF 用以確定ε-PL 的聚合度,而在結構鑑定的部分則是 以 IR 光譜法及1H 和13C-NMR 進行量測。 IR 光譜圖中(圖 2-12),1670 cm-1、1561 cm-1和 1280 cm-1分別代 表在醯胺基團中 C=O 雙鍵的伸縮振動、N-H 鍵的彎曲振動和 C-N 鍵的 伸縮振動,而 3402 cm-1和 3247 cm-1是指 N-H 鍵的不對稱伸縮振動和對
27 稱伸縮振動;在 2936 cm-1和 1398 cm-1表示了-CH 和-CH 2的存在,另 外,在 1161 和 657 則表示了-NH2和-NH 的存在。 圖 2-12 由 Streptomyces sp. DES20.中分離產物的 IR 光譜圖 51 在 NMR 氫譜測定結果中有五種不同的氫原子化學位移被觀察到 (圖 2-13),訊號面積比為 1:2:2:2:2 分別代表 CHα 、CHε 、CH β 、CHδ 和 CHγ ;在碳譜的解析中,發現了六種不同的碳原子化學位移 (圖 2-14),分別為 170 ppm、53 ppm、39 ppm、31 ppm、28 ppm 和 22 ppm,各代表 C=O、Cα、Cε、Cβ、Cδ和 Cγ;另外,利用 HMBC
(Heteronuclear multiple bond coherence)(圖 2-15)進行更進一步的鑑
定,C=O 和 Hα
間有強烈的鍵結,Hε
28
脫水縮合反應,由1H、13C-NMR 和 HMBC 可以確定產物非α-PL 而是
ε-PL。
29 圖 2-14 由 Streptomyces sp. DES20.中分離產物的 13C–NMR 圖譜 51 圖 2-15 由 Streptomyces sp. DES20.中分離產物的 HMBC 圖譜 51 由 MALDI-TOF 的資訊提供了ε-PL 的聚合度約為 26-33(圖 2-16),n 則代表單體的數目,ε-PL 的聚合度以及相對分子量由下列公式 表示,146.19 是離胺酸的相對分子量,18.02 為水的相對分子量。 MW = 146.19 n - 18.02 (n - 1)
30
圖 2-16 由 Streptomyces sp. DES20.中分離產物的 MALDI-TOF 圖譜51
對於ε-PL 的分析方法研究,常以 CD、IR 及 NMR 應用於構型的 鑑定,在定量的部分大多使用的是比色法,而膠體電泳法包括 SDS-PAGE 和 2D-SDS-PAGE 則用來測定其分子量,隨著質譜法的技術發展對蛋 白質鑑定也具有相當大的功能與貢獻,除了定性也可被用做觀察ε-PL 的聚合程度,比對實驗中不同聚合產物及ε-PL 衍生物之間的差異等。 另外,ε-PL 的構型變化也相當的有趣,隨著液體或固體型態、環境 pH 值的影響,都可能造成不同的表現。
31
2.3 蛋白質的構型
蛋白質主要由碳、氫、氧、氮、硫等化學元素組成。所有蛋白質都 是由 20 種不同的 L 型α胺基酸連接形成的多聚體,在形成蛋白質後, 這些胺基酸又被稱為單體。蛋白質透過大量的非共價相互作用正確摺疊 成一個特定構型,如氫鍵,離子鍵,凡得瓦力、雙硫鍵和疏水作用。蛋 白質的分子結構可劃分為四級,組成蛋白質多肽鏈的線性胺基酸序列稱 為一級結構;二級結構則是依靠不同胺基酸之間的氨基和醯基可以形成 氫鍵,使得主鏈具有一定的構型,包括α-螺旋、β-摺板和無規捲曲 等; 在二級結構的基礎上進一步盤曲摺疊,形成一個完整的空間構 型,稱為三級結構;而不同多肽鏈(亞基)間通過非共價鍵聚集而成的 空間結構,則稱為四級結構。 變性作用(Denaturation)可能會導致蛋白質失去生物活性,使其分 子內部結構和性質改變,變性的原因可分為物理及化學兩種,物理因素 像是加熱(高溫)、紫外線及 X 射線照射、超音波、劇烈振盪或攪拌 等;而化學方法有加酸、鹼、重金屬鹽、尿素、丙酮等。蛋白質構型可 能隨有機溶劑、離子對添加劑、溶劑 pH 值等因素而變化,當反應條件 劇烈持久蛋白質的變性為不可逆的,若非則可能會有可逆現象發生,並 恢復其天然構型和生物活性,這種現象稱之為復性(Renaturation)。32
2.3.1 溶劑對構型的影響
有機溶劑被廣泛用作結構誘導的共溶劑,已知醇類能將多肽或蛋白 質的骨架誘導為α-螺旋構型52,乙腈則能穩定並誘導使其呈現β-摺板 構型53。在 Arunkumar54等人的研究中,發現乙腈會誘導聚離胺酸構型 產生轉變,對於 pH 7.2 和 11.5 的聚離胺酸影響是完全不同的,並指出 乙腈不能被視為一般的β-摺板誘導溶劑,在多肽或蛋白質的構型研究 中會具有風險。 已知在中性 pH 值下的聚離胺酸為無規捲曲,當 pH 為 11.5 時則是 以α-螺旋構型存在55。經一系列實驗結果發現乙腈在不同濃度和 pH 條 件下會引發聚離胺酸不同構型的轉變。在 pH 7.2,濃度大於 80%(v / v)時,乙腈將聚離胺酸的主鏈從無規捲曲轉變為螺旋。在 pH 11.5 時, 可以看到完全相反的效果,乙腈使聚離胺酸中的α-螺旋構型不穩定。 當用乙腈滴定經熱誘導呈現β-摺板構型的聚離胺酸時,濃度低於 50% (v / v),乙腈誘導/穩定β-摺板構型;超過 60%(v / v)和高達 80% (v / v)的濃度,乙腈將β-摺板轉化成α-螺旋構型;最後,將乙腈濃 度增加至 90%(v / v)時,聚離胺酸的構型被轉化為無規捲曲。乙腈在 聚離胺酸中誘導的結構轉變可以總結如下圖所示(圖 2-17)。 圖 2-17 用乙腈滴定時,在聚離胺酸中發生構型轉變的示意圖5433
2.3.2 酸對構型的影響
對於逆相層析法應用於蛋白質和多肽類的分析,流動相中的離子對 試劑的類型與濃度具有相當的重要性,並且生物大分子通常選擇在酸性 條件下分離。在蛋白質或肽的等電點(Isoelectric Point, pI)之下,鹼性 胺基酸單體,像是離胺酸、精胺酸、組胺酸以及 N-端將變為質子化, 並且與流動相中存在的陰離子形成離子對,離子配對增加了蛋白質的疏 水性,改變與逆相的相互作用力。三氟乙酸(TFA)是一種廣泛使用的 離子對添加劑,但其對蛋白質結構和保留機制的影響尚不清楚。 2014 年 Bobály56等人對於流動相中添加的酸濃度對兩種蛋白質(轉 鐵蛋白和溶菌酶)分離的影響進行研究,並且觀察到了不尋常的層析結 果。HPLC 的流動相 A 為水、B 為乙腈,並且分別添加濃度範圍為 0.001% - 0.3%(v/v)的 TFA,將 B 相從初始的 30%以梯度沖提方式每 分鐘提高 5%到最終的 60%,流速為 0.3 mL/min。結果如下圖(圖 2-18),在低 TFA 濃度下滯留時間有顯著的變化,兩種蛋白質皆在 t0前被 沖提出;當 TFA 濃度略微增加時,層析圖中出現了兩個訊號,一個在 t0 前,另一個在 t0之後,經質譜確認確定為該蛋白質而非雜質;當濃度增 加(> 0.01%),蛋白質滯留時間皆出現在 t0之後。
34
圖 2-18 溶菌酶(A)和轉鐵蛋白(B)在不同 TFA 濃度下的層析圖
(1)t0前的峰;(2)溶劑干擾;(3)t0後的峰56
即使是微小的 TFA 濃度增加也會引起分離結果的變化,在較低濃
35 強度隨之降低;當 TFA 濃度逐漸增加產生 t0後的訊號,且其相對強度 會隨著濃度增加而增強,滯留時間延後。在兩種蛋白質的表現上皆有相 同的趨勢,但產生轉變的 TFA 濃度範圍不同,因此又以甲酸配製到與 TFA 相同的 pH 值進行試驗,結果顯示相同的 pH 值下兩種酸之間僅有 微小的差異。 透過質譜進行解析,該研究觀察到電荷的分佈也會受 TFA 濃度而 影響(圖 2-19),因而能證實蛋白質的構型確實受到影響。在 0.001% TFA 濃度下,溶菌酶在 t0前沖提出,相對高的電荷數且具有廣泛的分 佈,主要由酸性誘導而非天然的未摺疊的構型;當 TFA 濃度增加到 0.05% 時出現兩個訊號,由質譜判別兩訊號皆為溶菌酶,表示溶液中可 能存在兩種構型,並受到與固定相的相互作用影響,質譜呈現低電荷且 分佈較窄的狀態;再增加 TFA 濃度到 0.08% 時,t0前的訊號消失,質 譜呈現低電荷的狀態此時構型受到摺疊轉變為球體。 利用甲酸代替 TFA 進行更進一步試驗,結果顯示 TFA 與甲酸皆會 影響構型,並由電荷狀態分佈的變化監測的構型變化可能受所用酸的 pH 值、類型和質量的影響,並且可能涉及離子對形成,所需的濃度則 取決於預分析的蛋白質。
36
37
第三章 實驗方法
3.1 藥品
溶劑
藥品名稱 英文名稱 等級 廠牌 CAS Number
甲醇 Methanol LC/UV 級 Macron 67-56-1
異丙醇 Isopropanol LC/UV 級 Macron 67-63-0
乙腈 Acetonitrile LC/UV 級 Macron 75-05-8
酸類 藥品名稱 英文名稱 純度 廠牌 CAS Number 三氟乙酸 2,2,2-Trifluoroacetic acid,TFA 99% Alfa 76-05-1 三氟丙酸 3,3,3-Trifluoropropionic acid,TFPA 98% Alfa 2516-99-6
甲酸 Formic acid,FA 98-100% Sigma 64-18-6
醋酸
Acetic acid, AcOH
38 添加劑 藥品 名稱 英文名稱 分子量 廠牌 CAS Number 乙二胺 四乙 酸•二 胺鹽 Ethylenediaminetetraacetic acid diammonium salt,
EDTA•2NH3 326.3 ALDRICH 20824-56-0 本次實驗主要的ε-PL 為ε-Polylysine (ε-PL),分別有兩個來源並 在後續的實驗中將液體ε-PL 標示為樣品 L;而粉末ε-PL 則標示為樣 品 S,皆分裝保存於-20℃的環境中,待實驗時取出退冰至室溫進行配 製,而配製後的儲備溶液則保存於室溫下即可。 樣品 藥品名稱 英文名稱 分子量 性狀 來源編號 聚離胺酸 ε-Polylysine 3000-4500 Da 液體 L 粉末 S 實驗中所使用的水為 18.2 MΩ/cm 超純水(DI-Water)。
39
3.2 儀器設備
高效能液相層析儀(High Performance Liquid Chromatography,
HPLC)
HPLC 系統由 HITACHI 公司的 L-7000 Interface、L-7100 Pump 與 L-7420 UV-VIS Detector 組合而成。
質譜儀(Mass Spectrometry, MS)
為 Micromass Q-TOF,游離源為電噴灑游離裝置(Electrospray
ionization mass spectrometry, ESI),ESI 所使用的霧化氣體(Nebulizer
Gas)流速為 20 liters/hour,而去溶劑氣體(Desolvation Gas)的流速為 300 liters/hour;質量分析器為四極桿質譜分析器(Quadrupole mass analyzer)與時間飛行器(Time of flight, TOF)串聯使用。
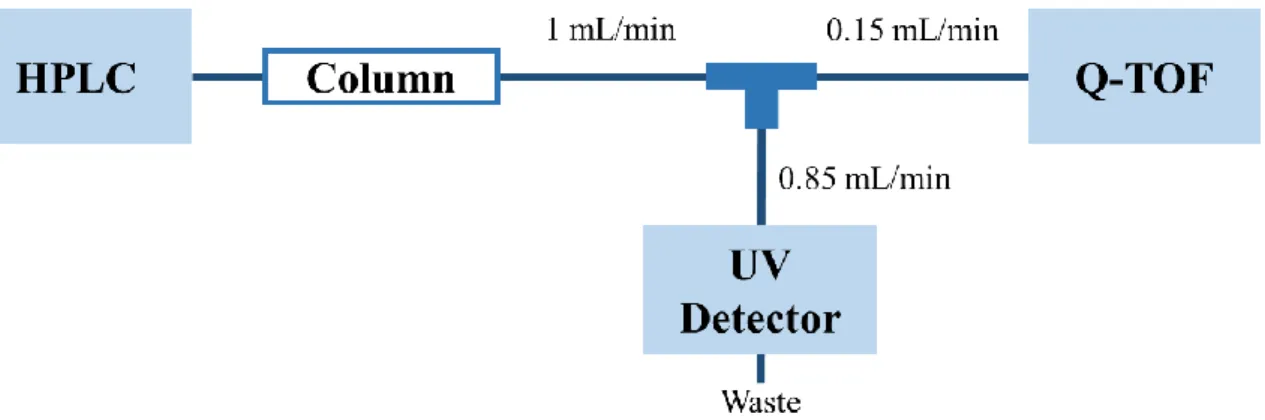
在本實驗中將上述 HPLC-UV 與 Q-TOF 連接使用,連接的裝置如簡 圖(圖 3-1)所示,ε-PL 經由管柱分離後以並聯方式分別導入 UV 偵測 器及 Q-TOF 同時進行偵測與圖譜蒐集。
40
自動進樣系統採用本實驗室所開發的文式管電噴灑游離法57
(Venturi electrospray ionization mass spectrometry,VESI),VESI 不需要 改造現有的電噴灑游離源(ESI)界面,利用 ESI 中高速流動的
nebulizer gas 在 ESI tip 前端所形成 Venturi effect,造成管線兩端具有壓 力差來推動樣品液,而不需要以外加幫浦等動力設備方式進樣。VESI 具有線上即時偵測和用量少等優點。 VESI 裝置的組成為取一口徑為 130µm 長度 10 公分的 PEEK 毛細 管作為自動進樣的連接管線,穿過經打洞裝有待測物溶液的樣品瓶瓶 蓋,並將毛細管末端沒入液面下,另一端以轉接頭連接到質譜儀的電噴 灑游離源前端,示意圖如圖 3-2。隨著實驗需要也可在樣品瓶中放入攪 拌子,並在樣品瓶下方架設升降台與磁力攪拌器,又或是將樣品瓶更換 成微量離心管(eppendorf)。利用此系統作為進樣方式不但方便又快 速,所需的樣品量也非常的少。 圖 3-2 VESI 自動進樣裝置示意圖
41
3.3 分析方法開發
3.3.1 測定樣品化學性質
ε-PL 的最大吸收波長
以 HITACHI 公司的 U-3010 紫外光/可見光光譜儀(Ultraviolet/Visible
Spectrophotometer, UV/Vis),測定ε-PL 的最大吸收波長,作為實驗中 HPLC-UV 的偵測波長。 ε-PL 濃度 市售樣品 L 和 S 中的ε-PL 含量,並未準確提供,研究中樣品 L 和 S 中的ε-PL 含量是以其 UV/Vis 吸光度做為參考。實驗時液體樣品 L 是 以 Water 稀釋,粉末狀樣品 S 則時以天平秤取適當重量後加入 DI-Water 溶解配製成水溶液,稀釋的樣品 L 水溶液和樣品 S 水溶液的 UV/Vis 吸光度,控制在具有約略相同的強度。 ε-PL 的 pH 值 將以 DI-Water 稀釋的樣品 L 與以 DI-Water 溶解的樣品 S,以 pH-meter 測定其 pH 值。
42
3.3.2 進樣方法
直接進樣 利用 VESI 將稀釋的樣品 L 水溶液和樣品 S 水溶液直接導入 ESI 游 離源,即可以記錄其所相對應的樣品全質譜圖(VESI-MS)。兩種不同 樣品的 VESI-MS,可以快速比較兩種樣品中ε-PL 聚合度的差異性。 Loop injection HPLC 與質譜儀連接使用(LC/MS),但是沒有接上 HPLC 分離管 柱,樣品於 HPLC 的六向閥處注入,此方式就是 loop injection。使用時 以 MeOH 與 0.1%TFA 作為流動相,將ε-PL 帶入質譜儀進行分析。相 較於直接進樣,loop injection 所得到的質譜圖,可以扣除或釐清質譜圖 中一些基質的訊號。 HPLC-UV-MSHPLC-UV-MS 與 loop injection 進樣方式大致相同,差異在於前者的 HPLC 系統有接上分離管柱,因此 UV 或 MS 可以記錄樣品中經過分離 的成分。相反的,直接進樣和 loop injection 進樣所記錄的資料是樣品成 分未經分離的總表現。
43
3.3.3 HPLC-UV-MS 分析條件
使用的 HPLC 分離管柱為 Agilent, ZORBAX SB-C18(4.6 mm × 150 mm,膜厚 5 μm),流動相為 MeOH 與 0.1% TFA(aq)的混合液,流速為 1 mL/min。以 HPLC 分析蛋白質的研究中,TFA 是常用的流動相調質 劑,其功能為作為離子對試劑,用於調整不同蛋白質於管柱中的滯留時 間。UV 的偵測波長為 215 nm。流動相的操作方式主要以定組成方式進 行,研究中也曾以梯度方式進行,但無法對不同聚合度的ε-PL 進行有 效分離,因此沒有進一步探討。 質譜使用的為正電模式,分析時質譜儀的最佳參數為 Source Block Temp: 100℃、Desolvation Temp: 200℃、TOF: 7200 V、MCP: 2700 V、 Capillary Voltage: 2600 V、Cone: 30 V。流速為 0.15 mL/min,質量掃描 範圍是 100-1100 m/z。 樣品的配製為將液體的樣品 L 直接以 DI-Water 稀釋 10 倍作為儲備 溶液,而粉末的樣品 S 則以天平秤取 0.0040 g 後加入 4 mL 的 DI-Water 進行溶解,配製成 0.1 %(w/w)的儲備溶液。 進樣方式為手動注射,使用 HPLC 微量注射針取樣品溶液 20 µL 進 行注射,樣品經管柱分離後以並聯方式於 UV 偵測器及質譜儀進行圖譜 資料蒐集。44
3.3.4 流動相中 TFA 濃度的配製
在實驗中進行了流動相 TFA 水溶液濃度的調控,分別以 0.1%、 0.05%及 0.02%進行實驗,觀察在不同濃度的 TFA 流動相條件下,對分 析結果是否會產生影響。在每次實驗前皆需重新配製流動相水溶液,量 測 pH 值控制在相同範圍內,並且取新鮮的 DI-Water 以避免長菌的情況 發生。 0.1%的 TFA 水溶液配製方法為取 1 L 新鮮的 DI-Water,以 1000 µL 的微量移液管加入 1 mL 的 TFA,至超音波震盪器(Sonicator, Brandsonic 5510R-DTH)震盪混合均勻及除氣,再以 pH-meter 量測其 pH 值並記錄。 0.05%的 TFA 水溶液配製方法為取 1 L 新鮮的 DI-Water,以 100 µL 的微量移液管加入 0.5 mL 的 TFA;0.02%的 TFA 水溶液配製方法為取 1 L 新鮮的 DI-Water,以 100 µL 的微量移液管加入 0.2 mL 的 TFA,其餘 步驟皆與上述相同,實驗時視當日所需的流動相使用量以同比例放大或 縮小倍數進行配製。 表 3-1 不同濃度 TFA 的 pH 值 TFA 濃度 0.1% 0.05% 0.02% pH 值 1.90 2.25 2.5545
3.3.5 流動相中添加不同種類的酸
本研究中共取了幾種酸配製成流動相,包括了三氟乙酸(TFA)、
醋酸(Acetic acid, AcOH)、甲酸(Formic acid, FA)及三氟丙酸
(Trifluoropropionic acid, TFPA),觀察不同的酸是否皆能作為有效的離 子對試劑,且對分析的結果又會造成如何的影響。 0.5%醋酸水溶液的配製: 以 1000 µL 的微量移液管取 5 mL 的醋酸 加入 1 L 新鮮的 DI-Water 中,放至超音波震盪混合均勻及除氣,溶液 pH 值為 2.88。此 pH 值雖然高於 TFA 水溶液的 pH 值,但仍可以用於觀 察將流動相中的 TFA 置換成醋酸後,對ε-PL 分離效果有何影響。 0.5%甲酸水溶液的配製: 以 1000 µL 的微量移液管取 5 mL 的甲酸 加入 1 L 新鮮的 DI-Water 中,放至超音波震盪混合均勻及除氣,溶液 pH 值為 2.55。此 pH 值與 0.02% TFA 水溶液的 pH 值相同,將應用於觀 察相同 pH 值下,將流動相中的 TFA 置換成甲酸,對ε-PL 分離效果有 何影響。 0.15%三氟丙酸水溶液的配製: 以 1000 µL 的微量移液管取 1.5 mL 的三氟丙酸加入 1 L 新鮮的 DI-Water 中,放至超音波震盪混合均勻及除 氣,配製成 0.15%的三氟丙酸水溶液。溶液 pH 值為 2.55。此 pH 值與 0.02% TFA 水溶液的 pH 值相同,將應用於觀察相同 pH 值下,將流動 相中的 TFA 置換成三氟丙酸,對ε-PL 分離效果有何影響。
46
3.3.6 以 Zetasizer 量測ε-PL 的粒徑大小
本研究所使用的奈米粒徑及電位分析儀(Malvern Zetasizer Nano
ZS),是利用 DLS 動態光散射法量測布朗運動由此得出顆粒大小,且因
分子的運動會受溫度以及所存在溶劑的黏度影響,必須注意溫度與黏度 兩項參數的設定值。以多分散係數(Polydispersity Index, PDI)作為實 驗結果可信度的依據,數值介於 0~0.05 只有標準樣品或真正單分散樣品 才有這麼小的分散度;0.05~0.08 表示幾乎為單分散;0.08~0.7 為中度分 散,絕大部分樣品所測得值皆在該範圍,並利用非負最小二乘法(NNLS) 計算法可得到分布曲線;若數值大於 0.7 則表示粒徑分布極寬,通常 DLS 不適用於這類樣品,因此對於測得圖譜資訊的解釋必須非常小心。 先將樣品 L 與 S 配製成水溶液,並以超音波震盪 30 分鐘避免分析 物有團聚現象,量測樣品中ε-PL 初始狀態的粒徑尺寸。接著再分別將 樣品 L 與 S 稀釋或溶解於三種流動相組成中,包括 MeOH 與 0.1% TFA 組成比例為(1)50 : 50、(2)25 : 75、(3)10 : 90,並以超音波震盪 30 分鐘,觀察在不同流動相組成中是否會跟層析圖譜結果相符,因ε-PL 構形的變化因而量測到不同的粒徑尺寸。
47
3.4 樣品前處理
3.4.1 樣品的 pH 值
市售樣品 L 與 S 因為有不同的基質,導致兩者具有不同的 pH 值, 研究中觀察到兩者在相同層析分析條件下,有不同的分離效果,為了釐 清分離效果的差異性與樣品 pH 值間的關係,因此將樣品 L 和 S 調整至 相同的 pH 值,觀察在相同的分析條件下,樣品 L 和 S 是否會有相同的 分析結果。 樣品 L 為溶液,以 DI-水稀釋十倍後的 pH 值為 8.36,添加甲酸於 此溶液中調節 pH 至 4.65 ,則與 0.1% S 水溶液的 pH 相同。相反的, 0.1% S 水溶液以氨水調整 pH 至 8.36,則與稀釋十倍的 L 溶液 pH 相 同。3.4.2 誘導
ε-PL構型的轉變
3.4.2.1 溶劑
有機溶劑具有誘導蛋白質構型轉變的效果,不同的溶劑會使蛋白質 轉變為不同的構型,本研究選用 DI-水、甲醇、異丙醇與乙腈作為稀釋 或溶解樣品的溶劑。 首先,取樣品 L 與 S 分別以 DI-水、甲醇、異丙醇及乙腈進行稀釋 或溶解,在溶解過程中可發現 DI-水與甲醇可以完全將樣品溶解為澄清 溶液。異丙醇則會使樣品溶液呈略為混濁的狀態,需經過離心後取得澄 清液,至於沉澱物是ε-PL 或是樣品中的基質,則沒有確認。樣品 S 無 法溶於乙腈中,需要先以少量 DI-水溶解後再加入乙腈稀釋,我們分別48 配製了含有 90%與 50%乙腈的樣品溶液。
3.4.2.2 溫度
加熱使蛋白質有足夠的能量將構型延展開,研究中將樣品溶液裝入 微量離心管(Eppendorf)中,以乾式恆溫加熱器(Thermolyne 17600)加 熱至 40℃或 60℃。以室溫約 25℃作為對照組,比較溫度是否會對樣品 中ε-PL 的構型造成不可逆的變化,進而改變 ε-PL 於 HPLC 管柱中的分 離效果。 另外也探討加熱時間的長短對 ε-PL 的構型是否有影響,採用 60℃ 下將樣品恆溫加熱半小時、1 小時、2 小時及 3.5 小時,再分別取樣分析 比較結果。3.4.2.3 添加 EDTA
乙二胺四乙酸(ethylenediaminetetraacetic acid, EDTA)為常用的有 機螯合劑,具有兩個胺基和四個羧基,共有六個配位基能與金屬離子螯 合形成穩定的錯化合物。EDTA 於水溶液中有許多不同的形態,如:
H6Y2+、H5Y+、H4Y、H3Y-、H2Y2-、HY3-、Y4-(其中 Y: EDTA),在鹼
性環境下,大多以 Y4-形態存在;中性環境下以 H
2Y2-、HY3-型態存在;
酸性環境下,氫離子競爭力較強則以 H6Y2+、H5Y+、H4Y、H3Y-形態存
在。研究中嘗試於樣品溶液中添加 EDTA,若基質含有金屬離子, EDTA 則可以與這些離子螯合,或許會間接影響 ε-PL 構型的改變。
49 2NH4)配製成濃度為 10-3 M 的水溶液,pH 值為 5.8,另將樣品 L 與 S 水溶液裝入 Eppendorf 中,並利用乾式恆溫加熱器加熱至 60℃後,加入 與樣品溶液等體積的 10-3 M EDTA 水溶液,等待溫度降回室溫後分別取 樣進行分析。樣品 L 添加 EDTA 後溶液 pH 值為 7.6,而樣品 S 添加 EDTA 後溶液 pH 值為 4.8。
50
第四章 結果與討論
目前有關ε-PL 的研究,大多著重在生物合成的條件和 ε-PL 及其衍 生物的應用,至於不同聚合度ε-PL 的分析方法,目前則無較深入的報 導。當我們以 HPLC 開發分離不同 ε-PL 聚合度的方法,過程中發現層 析圖顯示ε-PL 的分離效果,對所選用的實驗參數異常的敏感,因此接 續以 HPLC-UV/MS 進行深入探討,不同實驗參數對 ε-PL 聚合度的分離 效果,嘗試歸納出造成異常分離現象的主因。4.1 分析方法開發
4.1.1 以 UV/Vis 比對不同樣品中ε-PL 含量
本研究所取得的工業用 ε-PL 來自不同供應商,樣品有溶液(L)和 固體粉末(S)兩種不同物態,溶液樣品中 ε-PL 濃度小於 1000 ppm,固 體粉末中的ε-PL 含量未知。為了確保進行分析時,不同樣品的ε-PL 濃 度相近,首先我們比對 L 和 S 的 UV/Vis 圖,發現稀釋 10 倍的 L 水溶 液和 0.1 %(w/w)S 水溶液的吸收度相近(圖 4-1),後續將以 215 nm 為 HPLC-UV 的偵測波長。51 圖 4-1 樣品 L 與樣品 S 的 UV/Vis 吸收圖譜 UV/Vis 吸收圖譜中,L 和 S 的 λmax有些許的差距,檢視 L 和 S 的 pH,發現 10%(v/v) L 溶液呈現鹼性,但是 0.1 %(w/w)S 水溶液卻 為酸性(表 4-1)。因此 L 和 S 雖然主成分都是ε-PL,但是 pH 卻有極 大的差異,這或許是兩者的λmax不相同的原因。 表 4-1 樣品 L 與樣品 S 水溶液的 pH 值 樣品 L S pH 8.36 4.65 實驗中也觀察到不論是 L 或 S,λmax都隨著樣品稀釋倍數的增加而 持續有藍位移(圖 4-2)。根據文獻資料58,推測可能原因為溶劑的黏度 對ε-PL 所造成的影響,因此當樣品中水含量增加造成黏度降低,使的 ε-PL 的 λmax產生藍位移的現象,但確切的原因仍需近一步的證明。
52 圖 4-2 樣品 L 與樣品 S 倍數稀釋後的 UV/Vis 吸收圖譜
4.1.2 HPLC 流動相沖提強度的影響
一般以 HPLC 分離蛋白質常以 MeOH 和 TFA 做為流動相組成,本 研究選擇以 MeOH(A)與 0.1% TFA(B)做為流動相,初步以 HPLC-UV 系統進行分析方法開發,試圖找出分離樣品 L 與 S 中不同聚合度 ε-PL 的最佳參數。 以梯度沖提方式將流動相由 10% MeOH 在 30 分鐘內提升到 90%, 得到的層析圖如下(圖 4-3,A 圖),分析物會在 11.12 分鐘被沖提出; 調整初始沖提比例,當流動相由 20% MeOH 在 30 分鐘內逐漸增加至 90% MeOH,意外的在層析圖上竟出現兩析出峰(圖 4-3,B 圖)第一 個析出峰滯留時間為 1.59 分鐘,第二個析出峰延遲至 8.26 分鐘;當梯 度沖提條件由 30% MeOH 在 30 分鐘內增加到 90% MeOH,分析物則在53 1.38 分鐘也就是 t0(~1.5 分鐘)之前就被完全沖提出來(圖 4-3,C 圖),且訊號強度比 B 圖中的第一析出峰要強。 圖 4-3 以 MeOH 與 0.1% TFA 做為流動相,樣品 S 在不同梯度沖提條件 下,得到不尋常分離現象的層析圖 此外,利用 ACN 替代 MeOH 作為流動相,我們也得到了相似的分 離結果(圖 4-4),當沖提條件由 10% ACN 在 30 分鐘內提升到 90%, 分析物在 6.63 分鐘被沖提出來;若初始沖提比例調整為 15% ACN,層 析圖上卻出現多個析出峰,分別為 1.33、2.11、4.30 及 6.11 分鐘。結果 顯示分析物滯留時間會隨著 0.1% TFA 的比例增加而逐漸提前。
54 圖 4-4 以以 MeOH 與 0.1% TFA 做為流動相,樣品 S 在不同梯度沖提條 件下,得到不尋常分離現象的層析圖 進一步將圖 4-3 的 B 圖中兩析出峰蒐集並以質譜儀量測,我們竟得 到了相同的質譜資訊,表示兩析出峰皆為ε-PL,而非樣品中的不純物。 接續上述實驗結果,我們將 HPLC 系統與 Q-TOF 並聯,並改用相 對梯度更為單純的定組成操作方式,以 MeOH 與 0.1% TFA 作為流動 相,對於該特異性的分離結果進行更深入且詳細的探討。
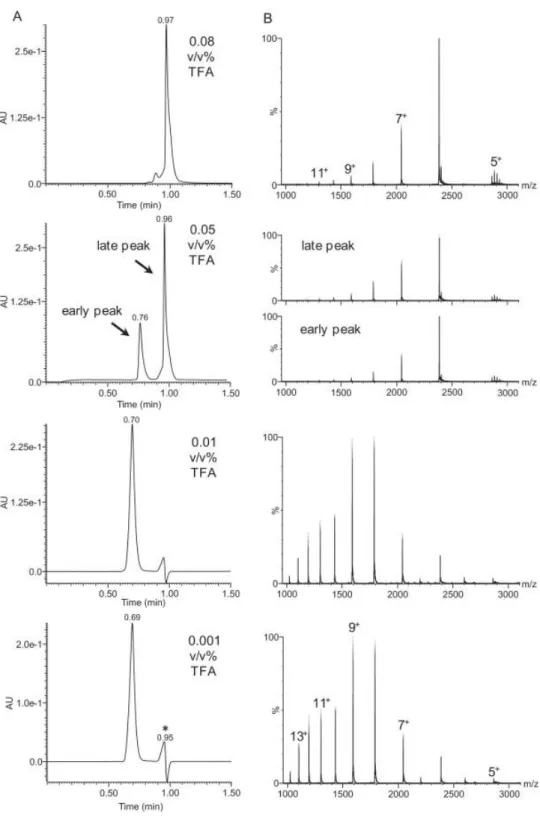
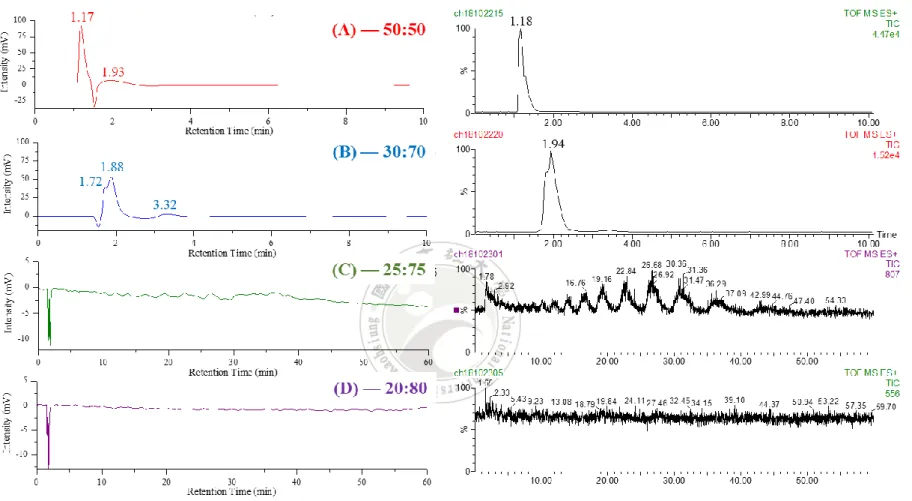
55 當流動相 MeOH/0.1% TFA = 50 /50 時,樣品 L 的層析圖顯示只有 一支析出峰,出現的滯留時間為 1.17 分鐘,這個時間較 t0(~1.5 分鐘) 來的短(圖 4-5)。將 MeOH/0.1% TFA 比例調整為 30 : 70 時,第一支析 出峰滯留時間移至 t0後面,約為 1.88 分鐘左右並且波型分叉,顯示多於 一種化合物被析出。後續於 3.32 分鐘又出現一支新的析出峰。將流動相 中的 MeOH 含量繼續減少,MeOH/0.1% TFA 比例為 25/75 時,
HPLC/UV 層析圖,以及與 UV 偵測器並聯的 Q-TOF 所記錄的總離子強 度層析圖(total ion chromatography, TIC),都顯現連續多個波峰起伏 (圖 4-5)。 檢視 TIC 圖中各個滯留時間的質譜圖,發現聚合度(n)為 15~34 ε-PL 單體的聚合物可以被有效分離,結果顯示滯留時間 2 分鐘左 右的析出液,含有 n 為 5~10 ε-PL 的聚合物混合液,2.4 分鐘附近則為 n 為 10~14 ε-PL 的聚合物混合液。但是 n 為 15~34 ε-PL 的聚合物,則可 以在 2.8~46 分鐘這個範圍內被有效的分離(圖 4-6)。後續再將
MeOH/0.1% TFA 比例微調至 20/80 時,原有在 HPLC/UV 層析圖和 TIC 圖顯現的連續波形消失了,出乎意料之外,原本滯留時間分布在 2~46 分鐘的不同聚合度ε-PL 化合物,竟然過了 60 分鐘後仍然沒有被沖提 出,也就是 5%MeOH 含量的變化,就讓不同聚合度的ε-PL 化合物滯 留時間大增。
56
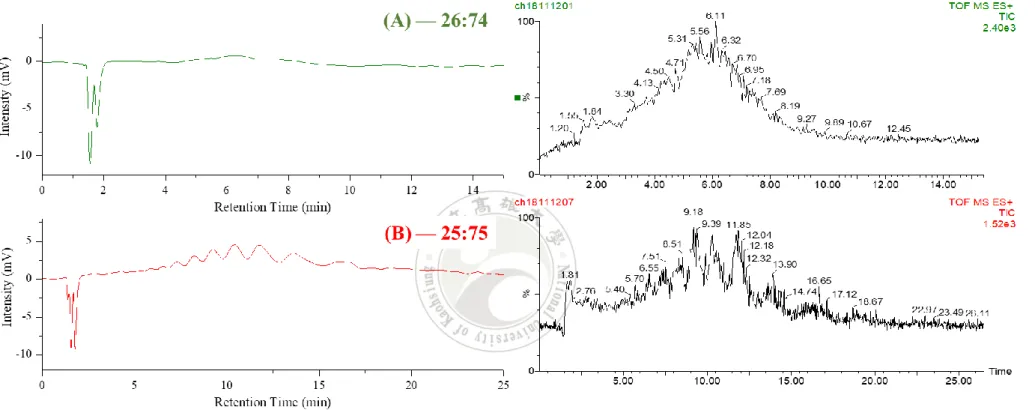
圖 4-5 樣品 L 在流動相 MeOH/0.1%TFA 組成比例為(A)50/50(B)30/70(C)25/75(D)20/80 的 HPLC-UV-MS 分析結果