中 國 醫 藥 大 學
專題研究計畫成果報告
計畫名稱:以材料工程暨天然物萃取技術開發血管塗
支架,子計畫 4:探討天然物塗藥支架改
善氣球擴張誘導血管再阻塞之分子機轉
計畫編號:CMU98-NCTU-06
執行期限: 99 年 4 月 1 日 至 100 年 3 月 31 日
單位名稱:藥學系
主持人:吳介信
中
華 民 國
100
年
6
月
25
日
Specific Aims:
About thirty years ago, percutaneous transluminal coronary angioplasty (PTCA) has been first introduced to restore the lives from patients suffered from myocardial infarction
(Landau et al., 1994). It has been estimated more than 300,000 cases of coronary angioplasty are performed in the United States each year (Topol et al., 1993). Approximately 30% to 50%
of the patients receiving a successful PTCA were affected by restenosis within six months (Gruentzig et al., 1987; Leimgruber et al., 1986; Bell et al., 1992; Sturek and Reddy, 2002).
Restenosis remains the major restraint on the long-term success of coronary angioplasty (PTCA). Stents are therefore developed to decrease restenosis; however, 20 to 30 % of the
patients are still affected by restenosis after coronary stenting (Sturek and Reddy, 2002). With the advent of drug-eluting stents such as sirolimus and paclitaxel eluting stent, the restenosis
rate has been reduced to single digit (Eisenberg, 2006).
Many gene encoding proteins responsible for vascular relaxation and free radical
scavenging have been studied to prevent restenosis, such as vascular endothelium growth factor (VEGF) (Isner et al., 1996; Chen et al., 1999; Moulton, 2001), superoxide dismutase
(SOD), catalase (Sozmen et al., 2003), cyclooxygenase (COX) (Speir et al., 1998), FAK (Zhao et al., 1998; Richard and Parson, 1996; Gilmore and Romer, 1996), basic FGF (Ghiselli
et al. 2003) and extracellular nitric oxide synthase (ecNOS) (Varenne et al., 1998). A remarkable 20-fold increase in superoxide anion production occurs immediately after balloon
injury and continues for up to 10 minutes when compared to uninjured arteries (Souza et al., 2000). Pretreatment of porcine with antioxidants was found to reduce coronary stenosis
(Nunes et al., 1995; Nunes et al., 1997; Hornig, 2002). Therefore, the etiology of restenosis is definitely a complicated process involving a significant number of proteins interacted to each other. A systemic analysis of the proteins responsible for restenosis is then a “must” to
explore its molecular mechanisms thus to develop an effective therapeutic remedy against this devastating disease.
Nitric oxide (NO) is a mediator involved in the regulation of vascular tone, neurotransmission, and host defense response (Furchgott and Zawadzki, 1980; Moncada et al.,
1991; Cooke and Dzau, 1997). NO is synthesized from L-arginine by the enzyme NO synthase, that can be competitively inhibited by analogs of L-arginine which have substitution
at the terminal guanidine nitrogen. Defects in the NO synthetic pathway have been implicated in several clinical disorders including atherosclerosis (Ross, 1993; Vanhoutte, 1997). Risk
factors for the development of atherosclerosis, such as hypertension, hypercholeserolemia, and smoking, have been shown to cause endothelial vasodilator dysfunction and, when
coexisting, have an additive effect (Vita et al., 1990; Quyyumi et al., 1995). Treatment of these factors restores endothelial function and decreases cardiovascular mortality (Kuscher et
al., 1996). These observations suggest a relationship between endothelial vasodilator dysfunction and risk factors for atherosclerosis, although the pathophysiological mechanisms
for endothelial dysfunction are not fully elucidated (Harrison, 1997).
The endothelial cells (ECs) play a pivotal role in control of vascular tone by releasing
several vasoactive substances including nitric oxide. In addition to its action as a vasodilator, NO also inhibits platelet aggregation, leukocyte adhesion, and smooth muscle cell
proliferation (Moncada and Higgs, 1993; Adams et al., 1997). The reduction in NO bioavailability may be due in part to the action of a circulating endogenous NO synthase
(NOS) inhibitor, NG,NG-dimethylarginine (asymmetric dimethylarginine or ADMA) (Kakimoto and Akazawa, 1970; Vallance et al., 1992). ADMA is an endogenous competitive
inhibitor of all three isoforms of NO synthase (Vallance et al., 1992). ADMA is thought to be derived from proteins that have been posttranslationally methylated and subsequently
hydrolyzed to release ADMA (Kakimoto and Akazawa, 1970). A number of cells elaborate ADMA, including human endothelial cells (Fickling et al., 1993). Intra-arterial administration
of ADMA causes vasoconstriction in forearm vessels via inhibition of endothelium-derived NO synthesis (Calver et al., 1993). Current studies have demonstrated that high levels of
ADMA in urine from hypertensive rats (Matsuoka et al., 1997), in plasma from hypercholesterolemic rabbits (Bode-Boger et al., 1996), in patients with peripheral arterial
occlusive disease (Boger et al., 1997), and in the regenerating endothelium of balloon-injured vessels (Azuma et al., 1995). Accordingly, ADMA may participate in this endothelial
dysfunction and lead to vascular diseases.
Methylarginines such as ADMA and NG-monomethyl-L-arginine (L-NMMA) are
degraded to citrulline and methylamines in the endothelial cells and blood vessels by a unique enzyme dimethylarginine dimethylaminohydrolase (DDAH) (Kimoto et al., 1995; Leiper et
al., 1999). DDAH specifically metabolises the asymmetric methylarginines and does not hydrolyse SDMA (symmetric dimethylarginine). Regulation of asymmetric methylarginine
concentration by DDAH may provide a novel mechanism for the regulation of NOS activity in vivo. MacAllister recently demonstrated that inhibition of DDAH in isolated vascular rings
induces a gradual vasoconstriction that is reversible by L-arginine. Therefore, decreased activity of DDAH may lead to local accumulation of ADMA and inhibition of NO synthase.
DDAH activity in the endothelial cells is decreased to almost 60% of baseline values by incubation with oxLDL or TNF-. In addition, DDAH activity in several tissues is
significantly decreased in rabbits fed with a high-cholesterol diet (Ito et al., 1999). These results provide a novel mechanism with the notion that the endothelial vasodilator dysfunction
in hypercholesterolemia may be due to reduced degradation of ADMA, which may contribute to the development of atherosclerosis.
Endothelial NO synthase (eNOS), the key enzyme for NO production in ECs, is tightly regulated not only at the transcriptional level but also by several post-translational
mechanisms. The enhanced phosphorylation of Ser1179 of bovine eNOS (Ser1177 in humans) leads to increased eNOS activity. A great number of evidence indicated that the
phosphorylation of Ser1177/1179 can be enhanced by shear stress (Dimmeler et al., 1999; Gallis et al., 1999; Boo et al., 2002). Use of the phosphatidylinositol 3-kinase (PI3K) inhibitor
wortmannin and LY 294002 has demonstrated that Akt phosphorylates eNOS Ser1177/1179 in response to shear stress (Dimmeler et al., 1999; Gallis et al., 1999). However,
dominant-negative mutants of Akt were unable to block the shear stress–stimulated Ser1179 phosphorylation (Boo et al., 2002). Further, H89, a protein kinase A (PKA) inhibitor, and an
adenovirus-expressing PKA inhibitor (PKI) blocked the eNOS Ser1179 phosphorylation, which indicates the involvement of PKA (Boo et al., 2002a; Boo et al., 2002b; Lungu et al.,
2004; Dixit et al., 2005).
Functioning as a metabolic master switch, AMP-activated protein kinase (AMPK) senses
and regulates the cellular energy status in various cell types. AMPK is activated by several physiological and pathological stresses such as exercise, hypoxia, and nutrient depletion that
result in increased AMP-to-ATP ratio. Once activated, AMPK switches on the catabolic pathways that produce ATP while alleviating the ATP-consuming processes. LKB1,
identified as a gene mutated in human Peutz-Jeghers syndrome, has been shown recently to be the upstream kinase phosphorylating AMPK (Hawley et al., 2003; Woods et al., 2003; Shaw
et al., 2004). In addition to phosphorylating multiple target proteins involved in energy regulation, such as acetyl coenzyme A carboxylase (ACC) and
3-hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase, AMPK also phosphorylates eNOS at Ser1177/1179 (Chen et al., 1999). Indeed, AMPK is required for
adiponectin-, thrombin-, and histamine-induced eNOS phosphorylation and subsequent NO production in ECs. (Chen et al., 2003; Ouchi et al., 2004; Thors et al., 2004). Activation of
AMPK by the pharmacological activators 5'-aminoimidazole-4-carboxamide ribonucleoside or carbonyl cyanide m-chlorophenylhydrazone also results in eNOS activation in ECs (Thors
From our preliminary studies, we have also identified the differential expression levels of several proteins from balloon injured arteries as compared to those from the uninjured
vessels by two dimensional gel electrophoresis followed by MALDI/TOF/TOF analysis. These proteins are PDIA3 (disulfide-isomerase A3), GLUD 1 (glutamate dehydrogenase 1),
PGK1 (phosphoglycerate kinase 1) and GSTp2 (glutathione S-transferase, pi2 ).
PDIA3 is a member of PDI (protein disulfide isomerase) family. It is also known as
ERp57, ER-60 or GRP58. This protein is responsible for disulfide bond formation, which is important for the proper protein folding. There are several reports indicating that PDIA3 was
able to cooperative with calnexin and calreticulin to fold the protein structure properly (Elliott et al.,1997). There are also several studies demonstrating the regulatory effects of calreticulin
on the activity of metal metalloproteinase (MMP) and angiogenesis (Michalak et al.,2004; Wu et al., 2007). The upregulation of calreticulin can also be modulated by p38MAPK to protect
cardiomyocytes against ischemia/reperfusion and hypoxia damage (Wu et al., 2007). In an earlier study, Garbi et al. found that PDIA3 was involved in MHC class 1 formation (Garbi et
al., 2006). We therefore expect that PDIA3 activates MMP or MAPK probably by itself or in conjunction with calreticulin chaperon to trigger cell migration which in turn results in
neointimal formation.
GLUD1 is primarily involved in metabolism of nitrogen and glutamate as well as the
homeostasis of energy. There were reports indicating that hyperinsulinemia was closely reated to GLUD1 mutation (Tanizawa et al., 2002). This mutation may produce excessive amount of α-ketoglutarate, an intermediate substrate in the TCA cycle, through oxidizing glutamate to
elicit insulin secretion. This increased level of insulin can then activate the signal
transduction pathway, like MAPK pathway, leading to cell proliferation through binding to IGFR (insulin growth factor receptor) (Jung et al., 2000).
PGK1 is a kinase for glycolysis to regulate the ATP production. PGK1 have been reported to overexpress in many cancer tissue as well as in the hypoxia condition (Kress et al.,
1998). It was therefore found to be associated with HIF-1 (hypoxia induced factor-1)(Kress et al., 1998). Since HIF-1 has been reported to regulate the expression levels of VEGF
(vascular endothelial growth factor), there is a possibility that HIF-1 can be induced by ischemia/reperfusion in the angioplasty process thus shed the important role of PGK1 in
restenosis.
GSTp2 is a pi class member of GST (glutathione S-transferase) family to detoxify the
free radical damage by its reduced form of glutathione (Hayes, J. D.,1995). The GSTp monomer can bind JNK and prevent its phosphorylation, resulting in the inactivation of c-Jun
(Adler et al., 1999). Because of this finding, many cancer research results have reported that activation of p53/c-Jun by JNK can affect the transcription activity of GSTp (Polyak et
al.,1997). Based on the findings that GSTp can not only scavenge free radicals but also inhibit JNK activation, GSTp may play an important role in regulating neointimal formation.
From sub-group 1 and 2, we will screen a great amount of herbal medicines, their extracts, furthermore the pure compounds as the targets of those proteins related with
restenosis. These proteins contain AMPK, DDAH, GLUD1, GSTp, PDIA3 and PGK1 as the targets. From our previous studies, we have been applied Magnolia officinalis to study its
effects on the proliferation of smooth muscle cells. Magnolia officinalis is a commonly prescribed Chinese medicine for many treatments, including as an anti-biotic and for blood
circulation. It has been shown to possess antioxidant activity approximately 1,000 times greater than alpha-tocopherol (Lo et al., 1994). Earlier studies have shown that pretreatment
with antioxidants can significantly reduce balloon injury-induced neointima formation (Pollman et al., 1999; Szocs et al., 2002; Liu et al., 2002). Therefore, it is expected that
Magnolol, the active compound of Magnolia officinalis, might play an important role in preventing balloon injury-induced restenosis. Previous study has shown that Magnolol
exerted vascular relaxation effect by releasing endothelium-derived relaxing factor (EDRF, NO) (Teng et al., 1990). Our previous studies also demonstrated that Magnolol can be an
effective remedy in preventing outgrowth of smooth muscle cells (In the preliminary data of this proposal). We therefore propose in the first year of this study to evaluate whether the
vascular relaxation effect of the natural compounds from sub-project 1 and 2 are through enhancing the activity of AMPK and DDAH or suppressing ADMA content. Also determine
the activity of eNOS and the expression levels of proteins involved in cell proliferation by these natural compounds. These proteins include PDIA3, GLUD1, PGK1 and GSTp2. The
increase of NO production in endothelial cells may prevent platelet adhesion, thrombosis and down-regulate the expression of adhesion molecules such as ICAM-1 and VCAM, which in
turn regulating smooth muscle cell proliferation (Furchgott and Zawadzki, 1980; Moncada et al., 1991). From the research of the first year, we could finalize potential canditates for the
animal stdies.
In the second year of this proposal, we will continue investigate if AMPK, DDAH,
PDIA3, GSTp, GLUD1 and PGK1 will be affected by these potential canditate natural compounds from first year. And, explore their signal pathway. The experiment will be further
conducted on the effects of natural compounds on the balloon injury-induced smooth miscle proliferation. The natural compounds will be administered via osmotic pump, and verify their
effects on inhibition of neointima formation. Since different shear stress may regulate the AMPK or DDAH (Dimmeler et al., 1999; Gallis et al., 1999; Boo et al., 2002), we will
evaluate the protein and gene expression levels of these molecules in aorta arch (low shear) and thoracic aorta (high shear). The vascular tissue at the branching points of the abdominal
aorta will be collected by PALM Microlaser System (P.A.L.M. Microlaser Technologies AG, Germany) to further study the effects of shear stress on the gene expression of AMPK, DDAH,
PDIA3, GSTp, GLUD1 and PGK1. Histochemistry analysis of these proteins will be applied to localize these proteins in the branch area of aorta.
In the third year, the PTCA model of abdominal aortic of the New Zealand rabbit will be conducted to study how the natural compounds-eluting stent affect the neointina formation.
The pharmacokinetics of the natual compounds will also be investigated. These natural products will be applied on the stent through the regulation of sub-project 3. Sub-project 3
will develop a new generation of drug eluting stent through a combination of biomaterials technology, coating, and scaffolding technology. This new-generation drug-eluting stent will
be fully explored and developed via the development of a new drug-carrying biomaterial: An amphiphilically-modified chitosan (Taiwan and US patent, pending), which will be employed
as drug carrier.
AIM 1: To determine whether the natural compounds can regulate the transcriptional, translationalor posttranslational levels of AMPK, DDAH, PDIA3, GSTp, GLUD1 and PGK1 with subsequent increase in NO production in endothelial and smooth muscle cells.
ECs and smooth muscle cells will be treated with various doses of the natural
compounds and subjected to gene and protein analysis. RNA will be extracted from cells to study the gene expression of AMPK, DDAH, PDIA3, GSTp, GLUD1 and PGK1 using
quantitative Q-PCR analysis. Proteins extracted from treated cells will be analyzed about the translational and posttranslational levels of AMPK, DDAH, PDIA3, GSTp, GLUD1 and
PGK1, including phosphorylation of these proteins. In addition to these proteins, 2-D protein gel electrophoresis will also be performed to acquire a comprehensive effect of natural
compounds on other protein expression in ECs and smooth muscle cells. The end product of NOS, eg NO, under the effects of natural compounds will be examined as well.
AIM 2: To elucidate the biological effects of natural compounds on balloon injury-induced vascular smooth muscle proliferation
Rats will be subjected to various concentrations of natural compounds for 14 days via osmotic pump canulated to carotid arteries. The rat aortic arch and thoracic aorta with
differential shear stress will be collected to evaluate the transcriptional, translational and posttranslational levels of AMPK, DDAH, PDIA3, GSTp, GLUD1, PGK1 and eNOS. The
microdissection of the tissue around the renal arteris branching out from the abdominal aorta using Laser Capture Microscopy (LCM, PALM Microlaser System) will be subjected to
real-time PCR analysis to evaluate if the gene expression of the above molecules under different shear stress can be affected by natural compounds. A matrix-assisted laser
desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) image analysis can be applied to view the differential protein expression at the branch point as compared to
the laminar regions of the aorta sections.
AIM 3: To evaluate the therapy of natural compound-eluting stent on abdominal aortic balloon injury-induced neointimal formation
Rabbits receiving balloon injury at the abdominal aortid arteries will be grouped into those with or without natural compounds treatment via drug-eluting stent for 14 days. After
confirming the regulatory effects of balloon injury and natural compounds on these molecules in the second year of this proposal, a therapeutic remedy can then be evaluated by the natural
compounds-eluting stent.
Methods:
To determine whether natural compounds can regulate the transcriptional, translational or posttranslational levels of AMPK, DDAH, PDIA3, GSTp, GLUD1 and PGK1 with
subsequent increase in NO production in endothelial and smooth muscle cells.
A. Cell Culture
Human aortic endothelial cells (HAEC) are obtained as cryopreserved cultures from Cascade Biologics (OR, USA). HAEC cells are grown in culture flasks in Medium 200 supplemented with low serum growth supplement. The cells are cultured at 37 ℃ in a
smooth muscle cells (VSMCs) derived from rat thoracic aorta are obtained from Food Industry Research and Development Institute (Hsinchu, Taiwan). The cells will be cultured in
10 cm dishes containing DMEM supplemented with 10% FBS, 3.7 g/l NaHCO3, 1.028 g/l N-acetyl L-alanyl-L-glutamine, 1% Na-pyruvate, 4.5 g/l D-glucose, 100 units/ml penicillin G,
and 100 µg/ml streptomycin sulfates. The culture medium will be replaced every two days and the cells will be passaged every week. In the last passage, the same numbers of cells will
be subcultured to 6 well plates. The cells that became at least 80% confluent will be starved for 24 h in DMEM containing 0.5% FBS for subsequent treatment. Either HAEC or A10 cells
will be treated with the natural compounds from sub-project 1 and 2 for 24h, 48h or 72h.
B. Cytotoxicity assay
MTT assay was performed to measure the cytotoxicity of natural compounds on A10 and HAEC cells. A10 cells were seeded in 96-well plates with 1×104 cells/well in F12K
supplemented with 15% FBS. After 24 h, cells were washed with phosphate-buffered saline (PBS) and then exposed to either 15% FBS alone or serial dilutions of natural compounds.
After 12, 24, 48 and 72 h, the number of viable cells was determined. Briefly, MTT (3 mg/ml in PBS)wasadded to each well(25 μlper200 mlmedium),and theplatewasincubated at
37℃ for 2 h. Cells were then spun at 300×g for 5min, and the medium was carefully aspirated. A 50 μlaliquotofDMSO wasadded,and theabsorbance at570 nm wasmeasured foreach
well on an ELISA reader (Anthos 2001, Anthos Labtec, Salzburg, Austria).
C. Flow cytometric analysis
Cellular total DNA contents of the treated cells will be assessed using flow cytometry following propidium iodide (PI) staining. Cells will be harvested with trypsin-EDTA, washed
twice with 10 ml ice-cold PBS, fixed in 70% ethanol, and kept at 4°C prior to FACS analysis. For DNA content analysis, cells will be centrifuged and resuspended in 0.3 ml of DNA
staining solution [100 Ag/ml PI, 0.2% Nonidet P-40, and 1 mg/ml RNase A (DNase-free) in PBS lacking Ca2+and Mg2+; at a 1:1:1 ratio by volume]. The cell suspension will be stored on
ice in a dark room for a minimum of 30 min and analyzed within 2 h. Cells will be analyzed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). PI fluorescence
will be linearly amplified and both the area and width of the fluorescence pulse will be measured. Ten thousand events will be acquired, and the percentages of hypodiploid
(apoptotic, sub-G1) events and percentages of cells in G0/G1, S, and G2-M phases will be determined using the DNA analysis software ModFitLT, version 2.0 (Verity Software,
Topsham, ME, USA).
D. Western blot analysis
A10 cultured in 6-well plates were incubated with various concentrations of natural compounds in F12K containing 15% FBS for 24 h. The cells were then lysed in a buffer
containing 2% SES, 50 mM DTT, 62.5 mM Tris-HCl, pH 6.8, followed by incubation at 95°C for 5 min. Samples were separated using SES-PAGE, transferred to PVDF membranes,
blocked with 5% nonfat dry milk in PBS-Tween for 1 h, and then probed with the desired antibodies (anti-AMPK, anti-DDAH, anti-GLUD1, anti-GSTp, anti-PDIA3 and anti-PGK1;
Novus Biologicals, Littleton, CO, USA) overnight at 4°C. The blots were then incubated with horseradish peroxidase-linked secondary antibody for 1 h followed by development with the
ECL reagent and exposure to Hyperfilm (Amersham, Arlington Height, IL, USA).
HAECs will be seeded in six-well plates and grew to 90% confluency. On the next day,
the cells will be treated with different doses of natural compounds for 24 h. Each well of cells will be lysed into 200 μloflysisbuffercontaining 20mM Tris, pH 7.4, 150 mM NaCl, 1 mM
EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerol phosphate, 1 mM Na3VO4, 10 μg/mlofeach proteaseinhibitorsaprotinin, leupeptin and pepstatin, and
1 mM phenylmethylsulfonyl fluoride. Twenty-five micrograms of protein was loaded to each lane and separated by 8% SDS–polyacrylamide gel and transferred to nitrocellulose
membrane. The membrane will be blocked with 5% non-fat powdered milk in TBST (50 mM Tris, pH 7.5, 150 mM NaCl, and 0.05% Tween 20), and then incubated with the polyclonal
anti-phospho-eNOS (Ser1177) and anti-eNOS antibody (Cell Signaling Technology, Beverly, MA. 1:1000 dilution) overnight at 4 °C. The membrane was washed with PBST and incubated
with anti-rabbit antibody horseradish peroxidase conjugate (Amersham Biosciences, Piscataway, NJ). Actin will be detected using anti-human β-actin antibody in the same
membrane after the primary detecting antibody will be stripped as described previously (Shen et a., 2004). Bands were visualized with ECL (Amersham Biosciences).
E. Two dimension-gel electrophoresis (2-DE)
Aliquotes of protein lysates from the previous preparation will be subjected to 2-DE
analysis to explore a comprehensive interaction of proteins affected by natural compounds. The protein sample will be loaded to the Readystrip IPG strips (Bio Rad
Laboratory, Hercules, CA, USA) in the pH range of 3-10NL (nonlinear) and layered with 0.8 ml cover oil to prevent gel from drying or urea crystalization. Gel was run on
the PROTEAN IEF cell (Bio Rad Laboratory, Hercules, CA, USA) with 30 V to rehydrate the gel strip for 12 h followed by the runnig programas: 30 V for 12 h with 360 Vh, 500 V for 1
h with 500 Vh, 1000 V for 1 h with 1000 Vh, 8000 V for 2 h with 1600 Vh. The voltage can ramp automatically based on the increasing resistance from the strip as excess ions move out
of the strip. After the first diemsion isoelectric focusing (IEF), the strip was washed to remove the cover oil and then equilibrated in 5 ml of equilabration buffer containing 50 mM Tris-HCl,
pH 8.8, 6 M urea, 2% SDS, 30% glycerol, 1% DTT for 12~15 min. The strip will be then subjected to second equilabration with 5 ml equilabration buffer with 1.5% iodoacetamide
substituted for the DTT for additional 12~15 min followed by SDS electrophoresis at 200 V for 4 h. At the end of electrophoresis, gel will be then stained with 0.25% (w/v) coomassie brilltant blue (Amersham Biosciences, UK), 50% (v/v) methanol, 10% (v/v) acetic acid for
30 min followed by destaining with 10% (v/v) methanol, 0.07% (v/v) acetic acid to remove
excessive stain. The spots with differential expression level between control and experimental groups will be picked manually and digested by trypsin for the subsequent MALDI-TOF-TOF
analysis.
F. MALDI-TOF-TOF analysis
MALDI-TOF-TOF allowed high-throughput MS analysis. The isotope-coded affinity tags (ICAT) labeling reagent for MS analysis was composed of three functional elements: a
reactive group that binds to Cys residues of proteins, a biotin-containing affinity tag that binds irreversibly to avidin, and a linker region that contains eight stable isotopes (8 hydrogen (H1) atomsin the“light”version vs.8 deuterium (D2)atomsin the“heavy”version).Based on our
ICAT-labeling strategy, identical peptides derived from each of the paired sample were
modified with either the light version or the heavy version of ICAT, respectively. Thus, they differ in mass (by 8 Daltons) and appear as doublets in the MS spectra. By measuring the
ratios of the peak intensities, we can quantify relative protein expression levels due to natural compounds treatment. As to defining the identity of the peptide of interest, we subjected it to
the second MS analysis (i.e. MS/MS, peptide MS fingerprinting) to get the sequence information of the peptide. Then the database (MASCOT) will be searched to get the most
probable protein which is consistent with the given peptide sequence.
G. Detection of NO and cGMP
Nitrite (NO2-), a stable breakdown product of NO, will be measured as an index of NO
production. The aliquot of culture media from ECs with various treatments is mixed with an
equal volume of Griess reagent and then incubating it at room temperature for 15 minutes. The azo dye production was analyzed by use of a spectrophotometer with absorbance set at
540 nm. Sodium nitrite was used as a standard. Intracellular levels of cGMP in ECs were assessed over 4 hours. After removal of culture media, ECs were lysed, and the extracts were
collected and centrifuged for 5 minutes at 5000g. cGMP level was determined by use of a cGMP enzyme immunoassay kit (R&D System, Minneapolis, Minnesota, USA), then
Results:
This proposal was originally designed as a three-year project to complete the whole
assay to identify the major active components from the natural herbs. Yet it was only funded for one year. We have then initiated the first part of the whole proposal to focus on one herb.
We have identified three major compounds from the natural compound Curcuminoid which is naturally acquired from the rhizome of the plant Curcuma. These three compounds to be
studied in this proposal were Curcumin, Demothoxycurcumin and Bisdemethoxycurcumin. We have carried both in vitro and in vivo assay to elucidate their efficacies in preventing
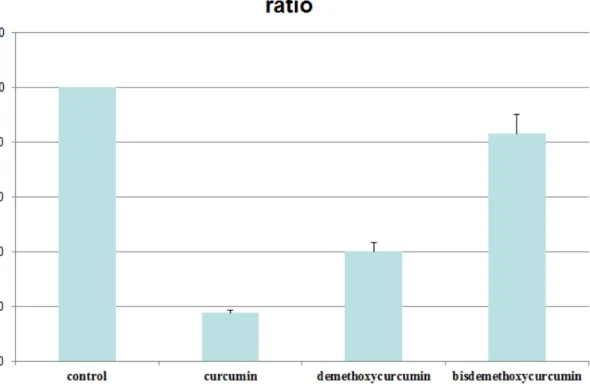
restenosis. We have first carried out the experiment on vascular smooth muscle cell (VSMCs, A7r5) viability by MTT assay (Fig 1). Curcumin, demethoxycurcumin and
bisdemethoxycurcumin were then tested on A7r5 for their effects on cell cycle by flow cytometeric assay (Fig 2). Effects of curcumin, demethoxycurcumin and
bisdemethoxycurcumin on migration of VSMCs were demonstrated in Fig 3. Wound for determining cell migration was introduced by scraping confluent cell layers with a pipet tip.
Effects of curcumin, demethoxycurcumin and bisdemethoxycurcumin on transwell was another approach for migration assay on vascular smooth muscle cells (Fig 4). We further
evaluate the molecular mechanisms of these three compounds on cell proliferation and migration. We found that there was a dose-dependent effect of curcumin, demethoxycurcumin
and bisdemethoxycurcumin on protein expression of FAK, the phosphorylated FAK, ERK1/2, the phosphorylated ERK1/2, PI3K,Akt , MMP 2/9 and the phosphorylated Akt (Fig 5 and Fig
6). We finally conducted an in vivo assay using these three compounds suspended in the pluronic gel topically applied on the surface of the wounded arteries in rat models. Figure 7
demosntrated that the neointimal formation was most significantly suppressed by curcumin rather than demethoxycurcumin and bisdemethoxycurcumin.
Fig1.
Vascular smooth muscle cell (A7r5) viability was examined using MTT assay. VSMCs was
seeded in a 96-well plate at density of 8×10
3
cells/well. The cells was allowed to proliferate for 24 h. 100 μlof the MTT reagent solution was added, and incubated for 4 h at 37 °C. Absorbance of the samples was measured with an ELISA reader at a wavelength of 590 nm.
Fig2A
Flow cytometric results of VSMCs treated with vehicle only (control) or with curcumin, demethoxycurcumin and bisdemethoxycurcumin for 24 hr.
Fig2B
Cell cycle distribution (percentage of total) in the treatment groups. Data represent mean ± SD, n = 3, * p < 0.05 compared with 15% FBS DMEM. * * p < 0.01 compared with 15% FBS DMEM.
Fig3
Effects of curcumin, demethoxycurcumin and bisdemethoxycurcumin on wound healing migration of VSMCs.Wound was introduced by scraping confluent cell layers with a pipet tip. VSMCs were incubated with different concentrations of Cur, DMC and BDMC 18h, and the wound area were calculated. Migration cell were measured by Image J software. The mean number of migration cell is quantified by three independent experiments. Values (means ± SD, n = 3).
* ( p < 0.05 compared with 15% FBS DMEM medium.) * * ( p < 0.01 compared with 15% FBS DMEM medium.) * * * ( p < 0.001 compared with 15% FBS DMEM medium.)
Fig4
Effects of curcumin, demethoxycurcumin and bisdemethoxycurcumin on transwell migration assay vascular smooth muscle cells. VSMCs were incubated with different concentrations of Cur, DMC and BDMC for 24 h. Photos of the migration VSMCs cells were taken under a microscope (100-fold magnification).The transwell migration cells were calculated. The mean number of migration cells is quantified by three independent experiments. Values (means ± SD, n = 3).
* ( p < 0.05 compared with 15% FBS DMEM medium.) * * ( p < 0.01 compared with 15% FBS DMEM medium.) * * * ( p < 0.001 compared with 15% FBS DMEM medium.)
Fig5
Dose-dependent effects of curcumin, demethoxycurcumin and bisdemethoxycurcumin on the protein expression level of FAK, the phosphorylated FAK, ERK1/2, the phosphorylated ERK1/2, PI3K,Akt and the phosphorylated Akt. In the dose-dependent assay, VSMCs cells were treated different concentrations of Cur, DMC and BDMC for 24 h. The expression of FAK, the phosphorylated FAK, ERK1/2, the phosphorylated ERK1/2, PI3K,Akt and the phosphorylated Aktwere analyzed by Western blotting.β-actin was used as a loading control.
Fig6
Dose-dependent effects of curcumin, demethoxycurcumin and bisdemethoxycurcumin on the protein expression level of MMP-2 and MMP-9. In the dose-dependent assay, VSMCs cells were treated different concentrations of Cur, DMC and BDMC for 24 h. The expression of MMP-2 and MMP-9 wereanalyzed by Western blotting.β-actin was used as a loading control.
Figure 7. The inhibitory effects of curcumin, demethoxycurcumin and bisdemethoxycurcumin on balloon-injured common carotid arteries in rats. Red arrows indicate the thickness of
neointimal formation.
Conclusion:
Our in vitro studies suggested that all three compounds can suppress cell proliferation, migration and protein expression levels of p-FAK, PI3K, p-AKT and MMP2/9. However, the
in vivo study suggested that only curcumin but not demethoxycurcumin and bisdemethoxycurcumin had the most significantly preventive effects on balloon
injury-induced neointimal formation. A further study on larger animal, like porcine model, may be needed to identify its efficacy for being a potential intervention for restenosis.
Refernces:
Adams MR, McCredie R, Jessup W, Robinson J, Sullivan D, Celemajer DS. Oral L-arginine improves endothelium-dependent dilation and reduces monocyte adhesion to endothelial cells
in young men with coronary artery disease. Atherosclerosis. 1997;129:261-269.
Azuma H, Sato J, Hamasaki H, Sugimoto A, Isotani E, Obayashi S. Accumulation of
endogenous inhibitors for nitric oxide synthesis and decreased content of L-arginine in regenerated endothelial cells. Br J Pharmacol. 1995;115:1001-1004.
Baas, A.S. & Berk, B.C. (1995). Differential activation of mitogen-activated protein kinases by H2O2 and O2- in vascular smooth muscle cells. Circ. Res. 77(1), 29-36.
Bell, M.R.; Berger, P.B.; Bresnahan, J.F.; Reeder, G.S.; Bailey, K.R. & Holmes, D.R. Jr. (1992). Initial and long-term outcome of 354 patients after coronary balloon angioplasty of
total coronary artery occlusions. Circulation 85, 1003-1011.
Bellas, R.E.; Lee, J.S. & Sonenshein, G.E. (1995). Expression of a constitutive NF-kB-like
activity is essential for proliferation of cultured bovine vascular smooth muscle cells. J. Clin. Invest. 96, 2521-2527.
Bleeke, T.; Zhang, H.; Madamanchi, N.; Patterson, C. & Faber, J.E. (2004). Catecholamine-induced vascular wall growth is dependent on generation of reactive oxygen
species. Circ. Res. 94(1), 37-45.
Bode-Böger S.M., Böger R.H., Kienke S, Junker W, Frölich JC. Elevated
L-Arginine/dimethylarginine ratio contributes to enhanced systemic NO production by dietary L-Arginine in hypercholesterolemic rabbits. Biochem Biophys Res Commun.
1996;219:598-603.
Böger RH, Bode-Böger SM, Thiele W, Junker W, Alexander K, Frölich JC. Biochemical
evidence for
Circulation. 1997;95:2068-2074.
Boo YC, Hwang J, Sykes M, Michell BJ, Kemp BE, Lum H, Jo H. (2002b). Shear stress
stimulates
phosphorylation of eNOS at Ser(635) by a protein kinase A-dependent mechanism. Am J
Physiol Heart Circ Physiol 283, H1819–1828.
Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J, Jo H. (2002a). Shear stress
stimulates
phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent
mechanisms: role of protein kinase A. J Biol Chem. 277, 3388–3396.
Bourcier, T.; Sukhova, G. & Libby, P. (1997). The nuclear factor kB signaling pathway
participates in
dysregulation of vascular smooth muscle cells in vitro and in human atherosclerosis. J. Biol.
Chem. 272, 15817-15824.
Calver A, Collier J, Leone A, Moncada S, Vallance P. Effect of local intra-arterial asymmetric
dimethylarginine (ADMA) on the forearm arteriolar bed of healthy volunteers. J Human Hypertension. 1993;7:193-194.
Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of
nitric oxide in vascular endothelial cells. J Biol Chem. 2003;278:45021–45026.
Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power
DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289.
Chen, Y.X.; Nakashima, Y.; Shiraishi, S.; Nakagawa, K.; Sueishi, K. & Tanaka, K. (1999). Immunohistochemical expression of vascular endothelial growth factor/vascular permeability
factor in atherosclerotic intimas of human coronary arteries. Arterioscler. Thromb. Vasc. Biol. 19, 131-9.
Cohen, G.M. (1997). Caspases: the executioners of apoptosis. Biochem. J. 326 ( Pt 1), 1-16. Cooke JP, Dzau VJ. Derangements of the nitric oxide synthase pathway, L-arginine, and
cardiovascular diseases. Circulation. 1997;96:379-382.
Degertekin, M.; Regar, E.; Tanabe, K.; Lemos, P.; Lee, C.H.; Smits, P.; de Feyter, P.; Bruining,
N.;
Sousa,E.; Abizaid, A.; Ligthart, J. & Serruys, P.W. (2003). Evaluation of coronary remodeling
after sirolimus-eluting stent implantation by serial three-dimensional intravascular ultrasound. Am. J. Cardiol. 91(9), 1046-50.
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. (1999). Activation of nitric
oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605. Dixit M, Loot AE, Mohamed A, Fisslthaler B, Boulanger CM, Ceacareanu B, Hassid A, Busse
R, Fleming I. (2005). Gab1, SHP2, and protein kinase A are crucial for the activation of the endothelial NO synthase by fluid shear stress. Circ Res 97, 1236–1244.
Fairman, M.P. (1990). DNA polymerase delta/PCNA: actions and interactions. J. Cell Sci. , 1-4. Fickling S, Leone AM, Nussey SS, Vallance P, Whitley GSJ. Synthesis of NG,N’G-dimethy1-arginine
by human endothelial cells. Endothelium. 1993;1:137-140.
Freyschuss, A.; Stiko-Rahm, A. & Swedenborg, J. (1993). Antioxidant treatment inhibits the development of intimal thickening after balloon injury of the aorta in hypercholesterolemic
rabbits. J. Clin. Invest. 91, 1282-1288.
Furchgott RF & Zawadzki JV (1980). The obligatory role of endothelial cells in the relaxation
of arterial smooth muscle by acetylcholine. Nature 288, 373-376.
Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, Figeys D, Harrison DG,
Berk BC,
synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. J Biol Chem.
274, 30101–30108.
Gomez, R.E. & Vazquez-Ramos, J.M. (2003). Maize DNA polymerase alpha is
phosphorylated by a
PCNA-associated cyclin/Cdk complex: effect of benzyladenine J. Plant Physiol. 160(9),
983-90.
Gong, K.W.; Zhu, G.Y.; Wang, L.H. & Tang, C.S. (1996). Effect of active oxygen species on
intimal
proliferation in rat aorta after arterial injury. J. Vasc. Res. 33, 42-46.
Gruentzig, A.R.; King, S.B. 3rd.; Schlumpf, M. & Siegenthaler, W. (1987). Long-term follow-up after
percutaneous transluminal coronary angioplasty. The early Zurich experience. New Eng. J. Med. 316(18), 1127-32.
Harrison DG. Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest.1997;100:2153-2157.
Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. (2003).
Complexes between the LKB1 tumor suppressor, STRAD [alpha]/[beta] and MO25 [alpha]/[beta] are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2, 28.
Hornig, B. (2002). Vitamins, antioxidants and endothelial function in coronary artery disease. Cardiovasc. Drugs Ther. 16(5), 401-9.
Isner, J.M.; Walsh, K.; Rosenfield, K.; Schainfeld, R.; Asahara, T.; Hogan, K. & Pieczek, A.(1996).
Clinical Protocol: Arterial gene therapy for restenosis. Hum. Gene Ther. 7, 989-1011.
endothelial
dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. Circulation.
1999;99:3092-3095.
Jonsson, Z.O.; Hindges, R. & Hubscher, U. (1998). Regulation of DNA replication and repair
proteins through interaction with the front side of proliferating cell nuclear antigen. EMBO. J. 17(8), 2412-25.
Josson, Z.O. & Hubscher, U. (1997). Proliferating cell nuclear antigen: more than a clamp for DNA
polymerases. BioEssays 19, 967-975.
Kakimoto Y, Akazawa S. Isolation and identification of NG,NG- and NG,N’G-dimethy1-arginine,
N-epsilon-mono-, di-,and trimethyllysine, and glucosylgalactosyl- and
galactosyl-delta-hydroxylysine from human urine. J Biol Chem. 1970;245:5751-5758.
Karim, M.A.; Miller, D.D.; Farrar, M.A.; Eleftheriades, E.; Reddy, B.H.; Breland, C.M. &
Samarel,A.M. (1995). Histomorphometric and biochemical correlates of arterial procollagen gene expression during vascular repair after experimental angioplasty. Circulation 91,
2049-2057.
Kimoto M, Whitley GS, Tsuji H, Ogawa T. Detection of NG,NG-dimethylarginine
dimethylaminohydrolase in human tissues using a monoclonal antibody. J Biochem. 1995;117:237-238.
Küscher TF, Tanner FC, Noll G. Lipids and endothelial function: effects of lipid-lowering and other
therapeutic interventions. Curr opin Lipidol. 1996;7:234-240.
Labinaz, M.; Pels, K.; Hoffert, C.; Aggarwal, S. & O'Brien, E.R. (1999). Time course and
importance of neoadventitial formation in arterial remodeling following balloon angioplasty of porcine coronary arteries. Cardiovasc. Res. 41, 255-266.
Landau, C.; Lange, R.A. & Hillis, L.D. (1994). Percutaneous transluminal coronary angioplasty. New
Eng. J. Med. 330(14), 981-93.
Landry, D.B.; Couper, L.L.; Bryant, S.R. & Lindner, V. (1997). Activation of the NF-kB and
IkB system in smooth muscle cells after rat arterial injury: induction of vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1. Am. J. Pathol. 151, 1085-1095.
Leimgruber, P.P.; Roubin, G.S.; Hollman, J.; Cotsonis, G.A.; Meier, B.; Douglas, J.S.; King, S.B. Jr. &
Gruentzig, A.R. (1986). Restenosis after successful coronary angioplasty in patients with single-vessel disease. Circulation 73, 710-717.
Leiper JM, Santa Maria J, Chubb A, MacAllister RJ. Charles IG, Whitley GS,
Vallance P. Identification of two human dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochemical J.
1999;343:209-14.
Levine GN, Keanney JFJ, Vita JA. Cholesterol reduction in cardiovascular disease. Clinical
benefits and possible mechanisms. N Engl J Med. 1995;332:512-521.1995.
Liu, B.; Fisher, M. & Groves, P. (2002). Down-regulation of the ERK1 and ERK2
mitogen-activated
protein kinases using antisense oligonucleotides inhibits intimal hyperplasia in a porcine
model of
coronary balloon angioplasty. Cardiovasc. Res. 54(3), 640-8.
Liu, G.T.; Zhang, T.M.; Wang, B.E. & Wang, Y.W. (1992). Protective action of seven natural phenolic compounds against peroxidative damage to biomembranes. Biochem. Pharmcol. 43,
147-152.
and Honokiol isolated from Magnolia Officinalis protect rat heart mitochondria against lipid peroxidation. Biochem. Pharmacol. 47(3), 549-553.
Lungu AO, Jin ZG, Yamawaki H, Tanimoto T, Wong C, Berk BC. (2004). Cyclosporin A inhibits
flow-mediated activation of endothelial nitric-oxide synthase by altering cholesterol content in caveolae. J Biol Chem. 279,48794–48800.
Matsuoka H, Itoh S, Kimoto M, Kohno K, Tamai O, Wada Y, Yasukawa H, Iwami G, Okuda S, Imaizumi T. Asymmetrical Dimethylarginine, an endogenous nitric oxide synthase inhibitor,
in experimental hypertension. Hypertension. 1997;29:242-247.
Meneveau, N.F.; Klugherz, B.D.; Chaquor, B.; Golden, M.A.; Jouille, M.M.; Macarek, E.;
Weisz, P.B. & Wilensky, R.L. (2003). Separate and combined effects of local and continuous intravenous administration of beta-cyclodextrin tetradecasulfate on intimal hyperplasia after
angioplasty in porcine coronary arteries. J. Cardiovasc. Pharmacol. Ther. 8(1), 53-60.
Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med.
1993;329;2002-2012.
Moncada S, Palmer RMJ, higgs EA. Nitric oxide: physiology,
pathophysiology, and pharmacology. Pharmacol Rev 1991;43:109-42.
Moulton, K.S. (2001). Plaque angiogenesis and atherosclerosis. Curr. Atheroscler. Rep. 3,
225-33.
Nunes, G..L.; Sgoutas, .DS.; Redden, R.A.; Sigman, S.R.; Gravanis, M.B.; King, S.B. 3rd &
Berk, B.C. (1995). Combination of vitamins C and E alters the response to coronary balloon injury in the pig. Arter, Throm. Vasc. Biol. 15(1), 156-65.
Nunes, G.L.; Robinson, K.; Kalynych, A.; King, S.B. 3rd.; Sgoutas, D.S. & Berk, B.C. (1997). Vitamins C and E inhibit O2-production in the pig coronary artery. Circulation 96, 3593-3601.
Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, Funahashi T, Walsh K. Adiponectin
stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem. 2004;279:1304–1309.
Pollman, M.J.; Hall, J.L. & Gibbons, G.H. (1999). Determinants of vascular smooth muscle cell
apoptosis after balloon angioplasty injury: influence of redox state and cell phenotype. Circ. Res. 84, 113–121.
Quyyumi AA, Dakak N, Andrews NP, Husain S, Arora S, Gilligan DM, Panza JA, Cannon RO. Nitric
oxide activity in the human coronary circulation: impact of risk factors for coronary atherosclerosis. J Clin Invest. 1995;95:1747-1755.
Ross R. Rous-Whipple Award Lecture: atherosclerosis: a defense mechanism gone awry. Am J Pathol. 1993;143:987-1002.
Selzman, C.H.; Shames, B.D.; Reznikov, L.L.; Miller, S.A.; Meng, X.; Barton, H.A.; Werman, A.;
Harken, A.H.; Dinarello, C.A. & Banerjee, A, (1999), Liposomal delivery of purified IkBa inhibits tumor necrosis factor-a-induced human vascular smooth muscle proliferation. Circ.
Res. 84, 867–875.
Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The
tumor
suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in
response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335.
Y.H. Shen et al., Human cytomegalovirus causes endothelial injury through the ataxia
telangiectasia mutant and p53 DNA damage signaling pathways, Circ. Res. 94 (2004), pp. 1310–1317
Souza, H.P.; Souza, L.C.; Anastacio, V.M.; Pereira, A.C.; Junqueira, M.L.; Krieger, J.E.; da Luz, P.L.;
Augusto, O. & Laurindo, F.R. (2000). Vascular oxidant stress early after balloon injury: evidence for increased NAD(P)H oxidor eductase activity. Free. Radic. Biol. Med. 28,
1232–1242.
Sozmen, E.Y.; Kerry, Z.; Uysal, F.; Yetik, G.; Yasa, M.; Ustunes, L. & Onat, T, (2003).
Antioxidant
enzyme activities and total nitrite/nitrate levels in the collar model. Effect of nicardipine. Clin.
Chem. Lab. Med. 38(1), 21-5.
Speir, E.; Yu, Z.X..; Ferrans, V.J.; Huang, E.S. & Epstein, S.E. (1998). Aspirin attenuates
cytomegalovirus infectivity and gene expression mediated by cyclooxygenase-2 in coronary artery smooth muscle cells. Circ. Res. 83(2), 210-6.
Stanger, B.Z.; Leder, P.; Lee, T.H.; Kim, E. & Seed, B. (1995). RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell
death. Cell 81(4), 513-23.
Sturek, M. & Reddy, H.K. (2002). New tools for prevention of restenosis could decrease the “oculo-stent”reflex.Cardiovasc.Res.53,292–293.
Szöcs, K.; Lassegue, B.; Sorescu, D.; Hilenski, L.L.; Valppu, L.; Couse, T.L.; Wilcox, J.N.;
Quinn, M.T.; Lambeth, J.D. & Griendling, K.K. (2002). Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler. Thromb. Vasc.
Biol. 22, 21–27.
Teng CM, Yu SM, Chen CC, Huang YL, Huang TF. EDRF-release and Ca+(+)-channel
blockade by
magnolol, an antiplatelet agent isolated from Chinese herb Magnolia officinalis, in rat
thoracic aorta. Life Sci. 1990; 47(13):1153-61.
Thors B, Halldorsson H, Thorgeirsson G. Thrombin and histamine stimulate endothelial
nitric-oxidesynthase phosphorylation at Ser1177 via an AMPK mediated pathway independent of PI3K-Akt. FEBS Lett. 2004;573:175–180.
Topol, E.J.; Ellis, S.G.; Cosgrove, D.M.; Bates, E.R.; Muller, D.W.; Schork, N.J.; Schork, M.A. & Loop,F.D. (1993). Analysis of coronary angioplasty practice in the United States with
an insurance-claims data base. Circulation 87(5), 1489-97.
Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an Endogenous
Inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572-75.
Vanhoutte PM. Endothelial dysfunction and atherosclerosis. Eur Heart J. 1997;18(suppl
E):E19-E29.
Varenne, O.; Pislaru, S.; Gillijns, H.; Van Pelt, N.; Gerard, R.D.; Zoldhelyi, P.; Van de Werf, F.;
Collen,D. & Janssens, S.P. (1998). Local Adenovirus-Mediated Transfer of Human Endothelial Nitric Oxide Synthase Reduces Luminal Narrowing After Coronary Angioplasty
in Pigs. Circulation 98(9), 919-926.
Vita JA, Treasure CB, Nabel EG, McLenachan JM, Fish RD, Yeung AC,
Vekshtein VI, Selwyn AP, Ganz P. Coronary vasomotor response to acetylcholine relates to risk factors for coronary artery disease. Circulation. 1990;81;491-497.
Wei, G.L.; Krasinki, K.; Kearney, M.; Isner, J.M.; Walsh, K. & Andres, V. (1997). Temporally and
spatially coordinated expression of cell cycle regulatory factors after angioplasty. Circ Res 80, 418–426.
Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T,
Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008.
Wu CH, Chang WC, Chang GY et al., (2004) The inhibitory mechanism of YC-1 on smooth muscle cell proliferation: an in vitro and in vivo study. J Pharmacol Sci 94, 252–260.