紫質在溶液中、生物與奈米環境下的光譜與緩解動力學研究
133
0
0
全文
(2) 紫質在溶液中、生物與奈米環境下的光譜與緩解動態學研究 Spectroscopy and Relaxation Dynamics of Porphyrins in Solution and under Biological and Nanostructural Environments. Student:Liayng Luo. 研究生:駱立揚. Advisor:Dr. Wei-Guang Diau. 指導教授:刁維光博士 國立交通大學. 應用化學研究所 博士論文 A Thesis Submitted to Department of Applied Chemistry College of Science National Chiao Tung University In Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy In Applied Chemistry October 2006 Hsinchu, Taiwan, Republic of China. 中華民國九十五年十月.
(3) 紫質在溶液中、生物與奈米環境下的光譜與緩解動態學研究. 學生:駱立揚. 指導教授:刁維光博士 國立交通大學應用化學所博士班. 中文摘要 本論文利用時間相關單光子技術與飛秒螢光上轉移技術來研究紫質分子在溶 液中與固相薄膜上的緩解動態學。在液相溶液中的紫質分子之研究,主要為 ZnBPP 和 H2BPP 及其溴化物衍生物。ZnBPP 在 S1 態的緩解過程可以分成兩種,第一種屬 於高能態緩解過程,另一種為低能態的緩解過程。高能態的緩解過程主要發生在 540-570 nm 的螢光光區,而低能態的緩解則發生在 580 nm 以後的光區。對於溴化 鋅離子的紫質與原紫質分子的研究,我們發現分子內的重原子效應會影響 S2 與 S1 能態上的緩解過程或 Qx 態上的分子內振動能重新分配與其他的振動弛緩過程。另 一方面,ZnPP 在緩衝溶液中易形成聚集體,其在 S1 能態的緩解過程的生命期小於 100 fs,但 ZnPP-Mb 錯合物生成,則因蛋白質使得 ZnPP 免於在緩衝溶液中產生聚 集體,而觀測到 ZnPP 單體在蛋白質中的 S1 能態的緩解過程。因 ZnPP-Mb 激發後 的振動能量易使蛋白質改變本性,由 α 螺旋結構變成鏈型結構,而使得內部的 ZnPP 裸露在緩衝溶液中,故我們亦觀測到部分聚集體的衰減過程。. 在固相中的研究則以 ZnCA(PE)1-4BPP 與 ZnCATPP 為主,此分子利用羧基化 學吸附在 TiO2 的奈米粒子上。因為紫質分子易在固相薄膜中產生聚集體,故我們 利用大量的 PMMA 聚合物來使紫質分子均勻地分散在聚合物中,其所測得的結果 與在溶液中的緩解過程相似。當紫質分子直接塗布在玻璃上時,其螢光瞬態光譜 顯示有兩組去激化過程,其主要以 J-聚集體猝滅為主。若紫質分子吸附在二氧化 鈦奈米晶相薄膜上時,其光譜訊號衰減速率變快,因為其過程中包含了介面電子 轉移過程。此外我們亦發現紫質分子與二氧化鈦之間的介面電子轉移過程包含經 由化學鍵傳遞或經由空間穿遂的方式來達成。經由化學鍵的介面電子轉移與紫質 分子上π電子是否為定域化有關,而空間穿遂的電子轉移則與激發電子的能態和 聚集體或分子與二氧化鈦粒子間的距離有關。. i.
(4) Spectroscopy and Relaxation Dynamics of Porphyrins in Solution and under Biological and Nanostructural Environments.. Student:Liyang Luo. Advisor:Dr. Eric Wei-Guang Diau. Institute of Applied Chemistry National Chiao Tung University. ABSTRACT The excited-state relaxation dynamics of zinc and free-base porphryins in solutions and on solid films were measured with time-correlated single photon counting and femtosecond fluorescence up-converted techniques. ZnBPP, H2BPP and its bromides were studied the dynamics of the excited states in solution. The relaxation processes of the S1 excited state of ZnBPP involve two types of relaxations. One is due to the hot-relaxations which were obtained at 540-570 nm and another is due to the cold-relaxations which were obtained above 580 nm. We also found intramoleuclar heavy-atom effect that affects relaxations of the S2 and S1 excited states in zinc bromo-porphyrins. In addition, the intramoleuclar heavy-atom effect also influences the intramoleuclar vibrational energy redistribution and other relaxations of the Qx excited state for free-base bromo-porphyrins. Besides ZnBPP and its derivates, we also measured fluorescence transients of ZnPP-Mb in buffer solution. The transients show two components of relaxations, one component is contributed to a unfolding protein which enables a ZnPP molecule to form H-aggregate and another component is contributed to the relaxations of a folding ZnPP-Mb complex. ZnCA(PE)1-4BPP and ZnCATPP were studied on solid films. To avoid aggregating, we used a lot of PMMA polymers to mix with zinc porphyrins. The transients of ZnCAPEBPP/PMMA show the dynamics which is like that in THF. However, the transients of porphyrin/glass are different from that in THF or PMMA. The relaxations of porphyrin/glass are assigned to H- and J-aggregation quenchings, but the major quenching is J-aggregates. According to the transients of porphyrin/TiO2, the interfacial electron transfers (IET) are complicated. The interfacial electron transfer contain two ways that are through chemical bond or space tunneling. IET by way of chemical bond is concerned with the delocalized π electrons on porphyrin, however, IET by way of space tunneling is concerned with the distance between porphyrin and TiO2 and the energy of an excited electron. ii.
(5) 誌 謝. 首先感謝共同研究者:暨南大學應化系林敬堯教授與其研究生提供紫 質化合物;交通大學生物科技系吳東昆教授與張晉豪同學提供紫質-蛋白 質錯告物。感謝指導教授刁維光教授在學識及研究上的指導使我受益良 多。感謝研究室內所有伙伴們在研究上與生活上的幫助。 感謝口試委員鄭博元教授、吳東昆教授、洪政雄教授、胡景瀚教授與 葉鎮宇教授的精闢見解讓論文的內容更為嚴謹與充實。 最後感謝我的家人在讀博士學位過程中的支持與鼓勵. iii.
(6) 目錄 中文摘要. …………………………………………………………. i. 英文摘要. …………………………………………………………. ii. 誌謝. …………………………………………………………. iii. 目錄. …………………………………………………………. iv. 表目錄. …………………………………………………………. vii. 圖目錄. …………………………………………………………. viii. Chapter 1. 緒論……………………………………………………. 1. 1-1. 染料敏化太陽能電池的發展…………………………. 1. 奈米晶相二氧化鈦的結構與釕錯合物在 DSSC 上的應. 2. 1-2. 用…………………………………………………….. 1-3. 紫紫質分子在染料敏化太陽能電池之相關研究回顧. 3. 1-3-1. 紫質分子的吸收光譜原理……………………………. 4. 1-3-2. 紫質分子的聚集原理與種類…………………………. 10. 1-4. 飛秒量級的超快動態…………………………………. 12. 1-5. 第一章的參考資料……………………………………. 13. Chapter 2. 實驗儀器與測量方法…………………………………. 18. 2-1. 樣品的製備……………………………………………. 18. 2-2. 紫外-可見光吸收光譜儀………………………………. 18. 2-3. 螢光光譜儀……………………………………………. 19. 2-4. 時間相關單光子計數系統……………………………. 19. 2-4-1. 時間相關單光子計數系統原理與裝置圖……………. 19. 2-4-2. 時間相關單光子計數系統單電子元件………………. 21. 2-4-2-1. 分數式時間鑑別器……………………………………. 21. 2-4-2-2. 時間-振幅轉換器………………………………………. 21. 2-4-2-3. 類比-數位轉換器………………………………………. 22. 2-4-2-4. 多頻道分析儀…………………………………………. 22. 2-4-2-5. 可調節式延遲器………………………………………. 23. 2-4-2-6. 雙光柵光譜儀…………………………………………. 23. 2-4-2-7. 二極體雷射光源………………………………………. 23. 2-5. 皮秒時間—解析螢光非等向性光譜技術……………. 23. 2-5-1. 非等向性原理…………………………………………. 23. 2-5-2. 時間解析非等向性螢光光譜…………………………. 26. iv.
(7) 2-6. 飛秒雷射系統…………………………………………. 27. 2-6-1. 摻鈦藍寶石雷射光譜…………………………………. 28. 2-6-2. 超快雷射脈衝振盪器…………………………………. 28. 2-6-3. 自動校正儀……………………………………………. 32. 2-7. 飛秒時間解析螢光上轉移技術………………………. 33. 2-8. 資料分析………………………………………………. 35. 2-9. 第二章的參考資料……………………………………. 36. Chapter3. 紫質分子在溶液與蛋白質中的光譜與動力學研究…. 38. 3-1. 原紫質分子與鋅離子紫質分子的光譜與動力學研究回. 38. 顧…………………………………………………… 3-2. ZnBPP、ZnBPPB 和 ZnBPPBr2 在溶液中的光譜與動力學. 42. 研究……………………………………………… 3-2-1. ZnBPP、ZnBPPBr 與 ZnBPPBr2 在苯液中的穩定吸收與. 42. 螢光光譜…………………………………………… 3-2-2. ZnBPP、ZnBPPBr 與 ZnBPPBr2 在第二激發態上的動力. 44. 學研究……………………………………………… 3-2-3. ZnBPP 在第一激發態的瞬態螢光光譜研究…………. 45. 3-2-4. ZnBPPBr 與 ZnBPPBr2 在第一激發態的瞬態螢光光譜研. 49. 究………………………………………………… 3-3. H2BPP、H2BPPBr 和 H2BPPBr2 在溶液中的光譜與動力學. 52. 研究……………………………………………… 3-3-1. H2BPP、H2BPPBr 和 H2BPPBr2 在苯液中的靜態吸收光譜. 53. 與螢光光譜……………………………………… 3-3-2. H2BPPBr 與 H2BPPBr2 在第二激發態與第一激發態上的. 54. 動力學研究………………………………………… 3-4. ZnPP 在四氫呋喃、緩衝溶液、與脫輔基肌紅蛋白中的光. 58. 譜與動力學研究………………………………… 3-4-1. ZnPP 在四氫呋喃、緩衝溶液、與脫輔基肌紅蛋白中的靜. 59. 態吸收光譜、螢光光譜與旋光光譜…………… 3-4-2. ZnPP 在四氫呋喃與脫輔基肌紅蛋白中的時間解析-螢光. 63. 非等向性光譜…………………………………… 3-4-3. ZnPP 在四氫呋喃、緩衝溶液、與脫輔基肌紅蛋白中的瞬. 64. 態飛秒時間解析螢光光譜……………………… 3-5. 第三章之參考文獻…………………………………… v. 69.
(8) Chapter 4. 鋅離子紫質衍生物在奈米晶相二氧化鈦上的光誘導電子. 72. 轉移與其動態學之研究…………………………. 4-1. 染料敏化奈米晶相光電化學太陽能電池之簡介與其動態. 72. 學相關文獻回顧………………………………… 4-2. 鋅離子紫質衍生物分子的結構與名稱………………. 75. 4-3. ZnCAPEBPP 在液相與固薄膜之動態學與. 77. ZnCAPEBPP/TiO2 之介面電子轉移機制………… 4-3-1. ZnCAPEBPP 在液相與固薄膜之靜態吸收與螢光光. 77. 譜……………………………………………………… 4-3-2. ZnCAPEBPP 在四氫呋喃溶液中的緩解動力學……. 78. 4-3-3. ZnCAPEBPP/PMMA 在固相薄膜中的緩解動力學…. 79. 4-3-4. ZnCAPEBPP 在固相薄膜中的緩解動力學…………. 81. 4-3-5. ZnCAPEBPP 在奈米晶相 TiO2 薄膜中的介面電子轉移過. 83. 程與其緩解動力學……………………………… 4-4-5. ZnCAPEBPP 在液相與固相中的動態學結論………. 88. 4-4. ZnCATPP 在液相與固薄膜之動態學與 ZnCATPP / TiO2. 89. 之介面電子轉移機制…………………………… 4-4-1. ZnCATPP 在液相中之靜態吸收與螢光光譜…………. 90. 4-4-2. ZnCATPP 在在固相薄膜中的緩解動力學……………. 91. 4-4-3. ZnCATPP 與 ZnCAPEBPP 在二氧化鈦粒子上的介面電子. 94. 轉移機制………………………………………… 4-5. ZnCA(PE)1-4BPP 在液相與固薄膜之動態學與. 100. ZnCA(PE)1-4BPP/TiO2 之介面電子轉移機制……… 4-6. 第四章的參考資料……………………………………. 104. Chapter 5. 結論……………………………………………………. 107. 5-1. ZnBPP 在苯溶液中的緩解動態過程…………………. 107. 5-2. 雙苯基鋅離子紫質分子的分子內重原子效應………. 107. 5-3. 雙苯基原紫質分子的分子內重原子效應……………. 107. 5-4. ZnPP 在脫輔基肌紅蛋白中的緩解動態過程. 108. 5-5. ZnCAPEBPP 在固相薄膜上的去激化過程…………. 108. 5-6. ZnCATPP 在固相薄膜上的去激化過程………………. 108. 5-7. ZnCA(PE)1-4BPP 在二氧化鈦奈米晶相薄膜上的介面電. 108. 子轉移過程………………………………………… 附錄. …………………………………………………………. vi. 110.
(9) 表目錄 表 3-1. ZnPP 在苯液中以 400 nm 激發後,利用螢光上轉移技術. 48. 所測量到的各緩解過程之生命期與其相對振幅強度。 表 3-2. 原紫質分子衍生物在苯液中的吸收及螢光光譜之頻帶位. 52. 置 表 3-1. H2BPPBr 與 H2BPPBr2 在苯液中,以 417 或 422 nm 激發. 57. 至 S2 能態,利用螢光光轉移技術所測量到各緩解過程之 生命期 表 4-1. ZnCAPEBPP 在三種不同環境下的螢光上轉移光譜擬合. 93. 結果之衰減時間係數與各過程之相對強度大小。激發波 長在 430 nm 與偵驗波長在 620 nm。 表 4-1. ZnCATPP 在三種不同環境下的螢光上轉移光譜擬合結 果之衰減時間係數與各過程之相對強度。激發波長在 430 nm 與偵驗波長在 600 nm。. vii. 93.
(10) 圖目錄 圖 1-1. 三種常見釕離子多吡啶錯合物結構. 3. 圖 1-2. 紫質分子的基本骨架圖。. 3. 圖 1-3. 環 18 碳共軛多烯的自由電子模型(free electron model)與. 5. 電子在 HOMO 與 LUMO 之間的躍遷形式圖。 圖 1-4. 四苯基紫質在的吸收光譜。左圖為原四苯基紫質. 5. (H2TPP),右圖為鋅離子四苯基紫質(ZnTPP)。 圖 1-5. 紫質分子的四軌域理論之分子軌域。藍色與紫色為 π 軌. 6. 域之不同的相位,其對稱的節面以虛線表之。 圖 1-6. 在金屬離子四苯基紫質中,以不同的金屬離子來取代所. 7. 得的振子強度(Oscillator strength,f)對 Q(0,0)頻帶的能量 之相關圖。 圖 1-7. 原紫質分子的結構圖。. 7. 圖 1-8. 金屬離子紫質與原紫質的電子躍遷概念圖,圖中藍色線. 8. 為 y 軸偏振方向的電子激發躍遷,而紅色線為 x 軸偏振 方向的電子激發躍遷。 圖 1-9. D4h 的特徵標表。. 10. 圖 1-10. 不同形態之聚集體與其躍遷偶極力矩的五種排列方式。. 11. 圖 1-11. 激子偶和模型所預測的雙體在不同排列方式下之能階. 12. 圖。 圖 2-1. 時間相關單光子計數的工作原理. 19. 圖 2-2. Fluo Time 200 儀器配置圖。. 20. 圖 2-3. 當輸入鑑別器的訊號低於一特定門檻的電壓高度時,則. 21. 被分數式時間鑑別器視為雜訊去除。 圖 2-4. TAC 偵測單一光子的計時機制。. 22. 圖 2-5. 單分子被 z 軸方向的偏振光激發後,在 y 軸的位置上所. 24. 觀測到的平行與垂直於激發光電場之螢光強度概念圖。 圖 2-6. 當激發光的偏振方向為 z 軸方向,溶液中被激發的分子. 25. 的分布圖(假設此分子的激發躍遷矩與螢光躍遷矩是同 方向的)。 圖 2-7. 測量螢光非等向性光譜之實驗示意圖. 27. 圖 2-8. 摻鈦藍寶石晶體的吸收與螢光光譜。. 28. 圖 2-9. 超快雷射脈衝振盪器(Mira900D)。. 29. viii.
(11) 圖 2-10. 克爾透鏡鎖模示意圖。. 30. 圖 2-11. 群速分散補償裝置示意圖。. 31. 圖 2-12. 自動校正儀裝置圖。. 32. 圖 2-13. 螢光上轉移的原理示意圖。. 34. 圖 2-14. CDP-FOG100 螢光上轉移偵測系統圖。. 34. 圖 3-1. H2TPP 與 ZnTPP 的結構圖. 38. 圖 3-2. H2TPP(A)與 ZnTPP(B)的吸收與螢光光譜。. 39. 圖 3-3. ZnTPP 的去激化機制圖。. 40. 圖 3-4. H2TPP(A)與 ZnTPP(B)在苯液中的去激化機制圖。. 41. 圖 3-5. ZnBPP、ZnBPPBr 和 ZnBPPBr2 的分子結構與名稱。. 42. 圖 3-6. ZnBPP、ZnBPPBr 與 ZnBPPBr2 在苯液中的吸收光譜與. 43. 螢光光譜。 圖 3-7. ZnBPP、ZnBPPBr 與 ZnBPPBr2 在苯溶液中的螢光瞬態. 45. 光譜。激發波長為 400 和 422 nm。偵測波長為 435 與 457 nm。圓圈表示原始數據,實線為用單指數函數擬合之結 果,虛線代表激發脈衝的干擾訊號。 圖 3-8. ZnBPP 苯溶液中的擬合短時間尺度(0-1000 fs)下之 3D-. 46. 螢光瞬態光譜。激發波長為 400 nm。偵測波長 540-680 nm。。 圖 3-9. ZnBPP 在苯溶液中的螢光瞬態光譜。激發波長為 400. 47. nm。偵測波長為 580 (A)與 640 nm (B)。圓圈表示原始數 據,實線為用單指數函數擬合之結果,點線、虛線和點 虛線則代去迴旋積分之後的各緩解過程的訊號曲線。 圖 3-10. ZnBPP 苯溶液中的擬合長時間尺度(0-120 ns)下之 3D-螢. 48. 光瞬態光譜。激發波長為 400 nm。偵測波長 540-680 nm。 圖 3-11. ZnBPP 在苯溶液中的 Jablonski diagram。. 49. 圖 3-12. ZnBPPBr 在苯溶液中的螢光瞬態光譜。激發波長為 422. 50. nm。偵測波長為 593 與 640 nm。圓圈表示原始數據,實 線為用 A→B→C→D 的連續反應動力學模型擬合之結 果,點線、虛線和點虛線則代去迴旋積分之後的各緩解 過程的訊號曲線。 圖 3-13. ZnBPPBr2 在苯溶液中的螢光瞬態光譜。激發波長為 422. 51. nm。偵測波長為 607 與 659 nm。圓圈表示原始數據,實 線為用 A→B→C 的連續反應動力學模型擬合之結果。 圖 3-14. H2BPP、H2BPPBr 和 H2BPPBr2 的分子結構、名稱與簡稱。 ix. 52.
(12) 圖 3-15. H2BPP、H2BPPBr 與 H2BPPBr2 在苯液中的吸收光譜與螢. 53. 光光譜。 圖 3-16. H2BPPBr(A)與 H2BPPBr2(B)在苯溶液中的螢光瞬態光. 55. 譜。激發波長各為 417 與 422 nm。偵測頻帶在 Qx(0,0) 頻帶上,插圖為 B 頻帶與 Qx(0,1)頻帶的螢光瞬態光譜。 圓圈表示原始數據,實線為用連續反應動力學模型擬合 之結果。 圖 3-17. H2BPPBr (A 和 B)與 H2BPPBr2 (C 和 D)在苯溶液中的螢. 56. 光瞬態光譜。激發波長各為 417 與 422 nm。其偵測波長 為(A) 649 nm;(B) 680 nm;(C) 663 nm 和(D)710 nm。圓 圈表示原始數據,黑實線為用連續反應動力學模型擬合 之結果,藍、綠、橘和洋紅色實線為去迴旋積分後的擬 合曲線。 圖 3-18. H2BPPBr 在苯溶液中的 Jablonski diagram。. 57. 圖 3-19. ZnPP 的分子結構與重組新成的 ZnPP-Mb 人工蛋白質結. 58. 構示意圖。 圖 3-20. ZnPP 在三種不同環境下的吸收光譜,這些環境分別為:. 59. ZnPP-Mb/buffer 溶液(實線)、ZnPP/buffer 溶液(虛線)、 ZnPP/THF 溶液(點線)。 圖 3-21. ZnPP 在三種不同環境下的螢光光譜,這些環境分別為:. 60. ZnPP-Mb/buffer 溶液(實線)、ZnPP/buffer 溶液(虛線)、 ZnPP/THF 溶液(點線)。激發波長為 435 nm。 圖 3-22. ZnPP-Mb 在緩衝溶液中被 430 nm 雷射光照射不同時間. 61. 下的吸收光譜圖。 圖 3-23. ZnPP-Mb 在緩衝溶液中被 430 nm 雷射光照射不同時間. 62. 下的旋光光譜。a-e 光譜為 ZnPP-Mb/buffer 在激發時間 各為: (a) 0 min,(b) 5 mins,(c) 30 mins 與(d) 60 mins。f 光譜為 ZnPP/buffer。 圖 3-24. ZnPP/THF(圓圈)與 ZnPP-Mb/buffer(三角形)的時間解析-. 63. 螢光非等向性光譜。激發波長為 435 nm,而偵測波長為 590nm,兩者的溫度均在 25oC。 圖 3-25. ZnPP 與 ZnPP-Mb 在緩衝溶液中的螢光去偏極化機制圖。. x. 64.
(13) 圖 3-26. ZnPP 在四氫呋喃溶液之飛秒時間解析螢光瞬態光譜,紅. 65. 圓圈為原始數據,黑線為擬合的結果,藍色與墨綠色線 為去迴旋積分之後的結果。其激發波長為 430 nm,偵測 波長為(A)588 nm 和(B)638 nm。 圖 3-27. ZnPP 在緩衝溶液之飛秒時間解析螢光瞬態光譜,紅圓圈. 66. 為原始數據,黑線為擬合的結果,藍色與墨綠色線為去 迴旋積分之後的結果,綠色線為原始資料與擬合結果之 差。。其激發波長為 430 nm,偵測波長為(A)598 nm 和 (B)655 nm。 圖 3-28. ZnPP-Mb 在緩衝溶液中的飛秒時間解析螢光瞬態光譜,. 68. 紅圓圈為原始數據,黑線為擬合的結果,藍色與墨綠色 線為去迴旋積分的結果,綠色線為原始資料與擬合結果 之差。。其激發波長均為 430 nm,偵測波長為(A)600 nm 和(B)680 nm。 圖 4-1. 染料敏化太陽能電池的簡單示意圖,圖中的染料分子為. 72. N3 染料。 圖 4-2. 染料敏化太陽能電池的循環機制。. 73. 圖 4-3. 鋅離子紫質分子的結構、名稱與簡稱。. 76. 圖 4-4. ZnCAPEBPP 在四氫呋喃中收光與螢光光譜,其濃度為. 78. 2.6×10-6 M。實線表示吸收光譜,虛線表示螢光光譜。插 圖為 ZnCAPEBPP 直接鍍在玻璃上(點線)、鍵結在二氧 化鈦薄膜上(虛線)與壓克力混合(1:5000 = ZnCAPEBPP:PMMA)後旋轉塗布在玻璃上(實線)的相對 吸收光譜,其相對吸收光譜以 Soret 頻帶之最大吸收值 做歸一化。 圖 4-5. ZnCAPEBPP 在四氫呋喃中螢光瞬態光譜,其濃度為 1×10-5 M。其偵測波長各是在 470 nm(圓圈)、620 nm(方 形)與 675 nm(三角形),黑色實線為擬合結果,墨綠色實 線與桃紅色實線各表示分析後各組成的擬合結果,綠色 線為原始資料與擬合結果之差。. xi. 79.
(14) 圖 4-6. ZnCAPEBPP 與 PMMA 混合後塗布在玻璃上的螢光瞬態. 80. 光譜,其混合重量比為 1:5000 = ZnCAPEBPP : PMMA。 其偵測波長各是在 460 nm(圓圈)、620 nm(方形)與 680 nm(三角形),黑色實線為擬合結果,墨綠色實線與桃紅 色實線各表示分析後各組成的擬合結果,綠色線為原始 資料與擬合結果之差。 圖 4-7. ZnCAPEBPP/glass 的螢光瞬態光譜。其偵測波長是在 470. 81. nm,激發波長為 420 nm,圓圈為原始數據,為黑色實線 為擬合結果,虛線代表示 ZnCAPEBPP / PMMA 在 S2 能 態上的螢光瞬態光譜的擬合曲線。 圖 4-8. ZnCAPEBPP/glass 的螢光瞬態光譜。其偵測波長是在. 82. 620(A)、640(B)、660(C)與 680(D) nm,激發波長為 430 nm,紅色圓圈為原始數據,為黑色實線為擬合曲線,藍 色與墨綠色曲線為去迴旋積分後個別去激化過程。綠色 線為原始資料與擬合結果之差。 圖 4-9. ZnCAPEBPP 敏化在奈米二氧鈦薄膜上的吸收光譜。將. 83. 奈米二氧鈦薄膜浸泡在 3.7×10-5 M (黑色曲線)、3.7×10-5 M (黑色曲線)與 3.7×10-5 M (黑色曲線)。其浸泡溶液為共 溶液(THF : CH2Cl2 = 1:14 (v:v))。 圖 4-10. ZnCAPEBPP 敏化奈米二氧化鈦半電極的螢光瞬態光. 84. 譜。其偵測波長是在 470 nm,激發波長為 420 nm,圓圈 為原始數據,為黑色實線為擬合結果,點線表示為空白 的奈米二氧化鈦薄膜。 圖 4-11. ZnCAPEBPP 敏化太陽能電池在不同的激發光下之光能. 85. 轉換效率圖。 圖 4-12. 三種不同濃度之 ZnCAPEBPP 敏化奈米二氧化鈦半電極. 86. 的螢光瞬態光譜。其偵測波長是均在 620 nm,激發波長 為 430 nm,紅色圓圈為原始數據,為黑色實線為擬合結 果,藍色、墨綠色與粉紅曲線為去迴旋積分後各別的緩 解過程曲線。 圖 4-13. 三種不同濃度之 ZnCAPEBPP 敏化奈米二氧化鈦半電極 的螢光瞬態光譜。其偵測波長是均在 620 nm,激發波長 為 430 nm,紅色圓圈為原始數據,為黑色實線為擬合結 果,藍色、墨綠色與粉紅曲線為去迴旋積分後各別的緩 解過程曲線。 xii. 87.
(15) 圖 4-14. ZnCAPEBPP 敏化奈米二氧化鈦半電極在 Q(0,0)與. 88. Q(0,1)態上的衰減過程與介面電子轉移過程之機制圖。 圖 4-15. ZnCAPEBPP 敏化奈米二氧化鈦半電極被激發至 S2 能態. 89. 後,其緩解途徑與介面電子轉移過程之機制示意圖。 圖 4-16. ZnCAPEBPP 與 ZnCATPP 的結構圖。. 90. 圖 4-17. ZnCAPEBPP(A)與 ZnCATPP(B)在 THF 中的靜態吸收(黑. 91. 實線)與螢光(紅色虛線)光譜。插圖為兩紫質分子在二氧 化鈦奈米晶相薄膜中的吸收光譜。 圖 4-18. ZnCAPEBPP (A)與 ZnCATPP (B)的螢光瞬態光譜。激發. 92. 波長為 430 nm,偵測波長各別為 620 與 600 nm。綠色代 表鋅離子紫質分子在四氫呋喃溶液中;紅色代表鋅離子 紫質分子塗布在玻璃上;藍色代表鋅離子紫質分子吸附 在二氧化鈦奈米晶相薄膜上。圓圈為原始數據,實線為 擬合的結果。 圖 4-19. ZnCAPEBPP 與 ZnCATPP 的理論計算之分子結構。利用. 93. B3LYP/LANL2MB 來計算兩鋅離子紫質分子在基態的最 低能量結構。上圖為分子結構俯視圖,下圖為由羧基方 向來看的側面圖。藍色球代表氮原子,紅色球代表氧原 子,黑色球代表碳原子和白色球代表氫原子,紫質環的 中央為鋅離子。 圖 4-20. ZnCAPEBPP (A)與 ZnCATPP (B)在 Q(0,1)頻帶的螢光瞬. 95. 態光譜。激發波長為 430 nm,偵測波長各別為 680 與 650 nm。紅圓圈為原始數據,黑色實線為擬合曲線,藍色、 墨綠色與洋紅色曲線為去迴旋積分後個別去激化過程。 圖 4-21. ZnCAPEBPP 與 ZnCATPP 的理論計算之分子軌域圖。利. 96. 用 B3LYP/LANL2MB 來計算兩鋅離子紫質分子在基態 中的最低能量結構,以此結構來顯示紫質分子在基態與 激發態的分子軌域。左圖為 HOMO 的分子軌域圖,右圖 為 LUMO 的分子軌域圖。 圖 4-22. ZnCAPEBPP (A)與 ZnCATPP (B)在 Q(0,0)頻帶的螢光瞬. 97. 態光譜。激發波長為 430 nm,偵測波長各別為 620 與 600 nm。紅圓圈為原始數據,黑色實線為擬合曲線,藍色、 墨綠色與洋紅色曲線為去迴旋積分後個別去激化過程。 圖 4-23. 紫質分子與二氧化鈦之間經由空間穿遂傳遞電子的相對 位能示意圖。 xiii. 98.
(16) 圖 4-24. ZnCAPEBPP 與 ZnCATPP 敏化太陽能電池的 IPCE 與. 99. APCE 圖。 圖 4-25. ZnCA(PE)1-4BPP/TiO2 的吸收光譜圖。. 100. 圖 4-26. ZnCA(PE)1-4BPPP 在四氫呋喃溶液中的螢光瞬態光譜。. 101. 左圖的偵測波長是均在 460 nm,激發波長為 420 nm;右 圖的偵測波長為 680 nm,激發波長為 430 nm。紅色圓圈 為原始數據,黑色實線為擬合結果,藍色、墨綠色曲線 為去迴旋積分後個別去激化過程。 圖 4-27. ZnCA(PE)1-4BPPP 敏化奈米二氧化鈦半電極的螢光瞬態. 102. 光譜。其偵測波長是均在 470 nm,激發波長為 420 nm, 紅色圓圈為原始數據,為黑色實線為擬合結果,藍色、 墨綠色曲線為去迴旋積分後個別去激化過程。 圖 4-28. ZnCA(PE)1-4BPPP 敏化奈米二氧化鈦半電極的螢光瞬態 光譜。其偵測波長是均在 680 nm,激發波長為 430 nm, 紅色圓圈為原始數據,為黑色實線為擬合結果,藍色、 綠色與粉紅曲線為去迴旋積分後個別去激化過程。. xiv. 103.
(17) 第一章 緒論 1-1 染料敏化太陽能電池的發展 自 18 世紀工業革命以來,人類對能源的需求日益增加。目前,能源的消耗主要來 自化石燃料,由此引發的能源危機和環境汙染成為極待解決的嚴重問題。20 世紀 70 年 代的石油危機使這一矛盾更加突出。因此,各種可再生能源包括生質能源、生物能、風 能、水能、核能以及太陽能成為解決全球性的能源危機和環境問題的幾個重要途徑。這 其中,太陽能作為一種可再生能源,具有其他能源所不可比擬的優點:與化石燃料相比, 太陽能取之不盡,用之不竭;與核能相比,太陽能更為安全,其應用不會對環境造成任 何汙染;與水能、風能相比,太陽能利用的成本較低,而且不受地理條件的限制。 太陽每年向地球輻射的能量大約為 5.4×1024 J1。全世界每年需要的最終能源相當於 8×109 噸的煤,也就是 1.09×1020 J 的能量(1 噸的煤大約可產生 2.93×1010 J 的能量)2。如 果地球上一小部分的太陽能被利用的話,許多能源問題都可以迎刃而解。因此利用太陽 能的研究和應用受到世界各國政府的重視。目前,太陽能的轉換主要有四種形式:太陽 能轉換成熱能、太陽能轉換成熱電能、光電太陽能轉換與化學太陽能轉換。其中光電太 陽能轉換是將太陽能直接轉成電能,其為世界各國最重視的研究課題之一。 光伏效應可追溯到 166 年前。1839 年,Becquerel 等人將兩個電極放在電解液裏, 光照其中的一個電極,能夠檢測到光電壓的產生 3。在此後相當長的時間裏,光伏效應 僅僅是一種現象而已,沒有實用化器件的產生。五十年代以來,以矽為代表的半導體材 料得到迅速的發展。1954 年,轉換效率為 6%的 p-n 型太陽能電池在貝爾實驗室誕生 4, 這是一個實用化的光電轉換器件。從此,半導體矽太陽能電池得到蓬勃的發展,並且廣 泛應用於衞星、航空、軍事以及偏遠地區的通訊和電力供應。由於這種光電轉換系統非 常穩定,開始人們很少關心它的生產成本。70 年代,工業國家發生石油危機,人們更加 重視光伏電池作為陸地電力供應的可能性。目前,提高效率、降低成本以及提高穩定性 是太陽能電池三個主要核心課題。 與此同時,人們也在積極尋找更為廉價的光電轉換系統。各種光電太陽能電池應運 而生,如 CuInSe2、CuInGaSe5、CdTe6 等以及染料敏化太陽能電池 7。對於前三種太陽 能電池是屬於三五族的光伏電池,其面臨的主要問題是它們的劇毒以及對環境的汙染。 染料敏化太陽能電池(dye-sensitized solar cell, DSSC)雖然目前光電轉換效率相對較低,穩 定性需要進一步提高,但是由於它的綠色環保、價格低廉而受到各國研究者的高度重 視。第一個染料增強的光伏效應可以追溯到 19 世紀,也就是 Becquerel 發現光伏效應半 個世紀之後,Vienna 大學的 Moster 報導了第一個染料敏化的光電效應,這些結果很快 被應用到成像領域,並最終促進了彩色成像的形成。然而經過了一個世紀之後,染料敏 -1-.
(18) 化的光電效應才在光伏領域展開了研究。1976 年 Tshubomura 使用多晶 ZnO 粉末代替單 晶半導體增加電極的表面積,並將電極浸泡在染料溶液中,其光電轉換效率可達 1.5%, 並且他們發現 I-./I3-氧化還原電對是實現電極電效率的有效電解體系 8。 染料敏化太陽能電池早期的研究工作發現:由一個低功函金屬、一個有機層和一個 高功函金屬(或導電玻璃)組成夾心式電池,便會觀察到光伏效應。電池器件通常的結構 為:玻璃/ITO(三氧化銦)/染料/金屬電極。用於染料敏化太陽能電池的有機染料通常具有 以下特性: (1) 製備成本低,容易提純,比較穩定。 (2) 在可見光區域內有較高的吸收係數。 (3) 在適當環境中,其基態和激發態可以發生氧化還原反應。 太陽光首先透過透明電極(導電玻璃)照射到有機薄膜上;有機薄膜吸收光之後,產生許 多電子/電洞對(即正負電荷對),稱之為激子;這種電子/電洞在薄膜中遷移到界面或某個 表面;激子發生分離(即正負電荷分離),正電荷向負極移動,負電荷向正極移動,這種 已經分離的電荷統稱電荷載流子,電荷載流子到達電極提供外電路時,表現為電流。. 1-2 奈米晶相二氧化鈦的結構與釕錯合物在 DSSC 上的應用 二氧化鈦有三種晶體結構,這些結構的共同點是其組成結構的基本單元是[TiO6]8八面體。這些結構的區別在於,是由[TiO6]8-八面體通過共同頂點還是共邊組成骨架。金 紅石的結構是建立在氧的最密堆積,儘管它的結構不是一種最密堆積;板鈦礦的結構也 是由氧最密堆積而成的,鈦原子處在八面體中心位置,不同於金紅石結構。而銳鈦礦結 構是由[TiO6]8-八面體共邊組成,而金紅石和板鈦礦結構則是由[TiO6]8-八面體共頂點且共 邊組成。銳鈦礦的結構實際上可以看做一種四面體結構,而金紅石和板鈦礦的結構則是 晶格稍有畸變的八面體結構。由於銳鈦礦的結構不如金紅石穩定,因此銳鈦礦具有良好 的光催化活性。二氧化鈦晶體的導電帶與價帶之間的能隙為 3.0 eV(金紅石相)和 3.2 eV(銳鈦礦相)。半導體的光吸收值(λg)與能隙寬度(Eg)的關係為公式 1-1 所示: λg (nm) = 1240 / Eg (eV). (1-1). 我們研究染料敏化太陽能電池中所使用的二氧化鈦是銳鈦礦相,而且是奈米級大小。因 此奈米級銳鈦礦相二氧化鈦的最大吸收峰的位置小於 387 nm 是屬於紫外光區,但是在 地球上的太陽光譜在紫外光區的光線因被臭氧層所吸收,所以紫外光強度很小無法直接 利用太陽光來產生光電流,所以利用染料分子來當光敏化劑來吸收可見光與紅外光範圍 的波段,有機染料由於分子小,消光係數(extinction coefficient)大、成本低,則有良好的 實用性。現今最常使用的染料分子為釕的多吡啶之錯合物 9 如圖 1-1 所示。尤其以 N3 染料為最熱門的光敏化染料,其相關的研究與報導非常的多 10-16。雖然其化合物的光電 轉換效率高,但是因為釕元素在地球上的含量非常稀少,使得合成釕錯合物的成本較 -2-.
(19) 高,因此有許多科學家還在尋找或試驗其他的有機染料分子與無機量子點 17, 18 來代替 N3 染料。 HOOC. HOOC. COOH N. HOOC. N. HOOC N. N. NCS. N. Ru. Ru. N. N. N N. HOOC. NCS N. HOOC. COOH. COOH. COOH. cis-Ru(dcbby)2(NCS)2. Ru(dcbby)3. N3 dye HOOC. COOH N. HOOC. N. N. Ru NCS. SCN NCS. Ru(tctpy)(NCS)3 black dye. 圖 1-1 三種常見釕離子多吡啶錯合物結構. 1-3 紫質分子在染料敏化太陽能電池之相關研究回顧 Y. 1. 2. I. δ. α. N 3. 8. IV. II. N. N. 7. X 4. N. β. r III 6. 5. 圖 1-2 紫質分子的基本骨架圖。. 在自然界中紫質(porphyrin)的衍生物扮演著重要的角色,如光合作用中的葉綠素與 血液中的原血紅素等。紫質分子的基本結構如圖1-2所示,是由四個吡咯(pyrrole)以四個 未飽合碳橋相連接而成。若在紫質環中II與IV(或I與III)的氮原子上有接氫原子,我們稱 -3-.
(20) 這種紫質分子為原紫質(free base porphyrin);假如紫質環的中央利用氮原上的孤對電子 嵌合住金屬離子,我們稱此種紫質為金屬離子紫質(metal ion porphyrin),金屬離子紫質 的種類有很多,像葉綠素的金屬離子為鎂離子,而原血紅素中的金屬離子為鐵離子。大 部分的金屬離子紫質中的金屬為過渡元素,有些金屬離子紫質在基態上的氧化還原過程 就非常的複雜,如鐵離子紫質,可瞭解到其激發態的去激化過程中會包含許多不同氧化 態之間的緩解過程或電子轉移,不利於瞭解紫質在固相中的非輻射緩解與電子轉移過 程。因為鋅離子的氧化態只存在+2價,沒有其他的氧化態干擾,所以我們選擇鋅離子紫 質衍生物來研究光激介面電子轉移與其在激發態的去激化機制與衰減時間係數。 利用紫質分子來當DSSC中的光敏化物質之相關研究在近幾年才開始展開19-39,雖然 有一些紫質分子的光電轉換效率不錯,但為什麼效率高的原因並不清楚,而且紫質分子 與TiO2之間的介面電子轉移與其相關動態學的研究卻非常稀少40-42。G. Ramakrishna等人 利用TPP與TiO2奈米粒子結合來研究兩者之間的介面電子移轉。他們發現了正向的電子 轉移過程與逆向電子轉移過程,但是其過程無法擬合在染料敏化太陽能電池的條件中, 因為TPP-TiO2的系統是在水溶液中並非是在固相的薄膜中40。另外,Naoto Tamai教授等 人利用TPPS在水溶液中與TiO2結合,並利用酸鹼值來控制紫質分子的聚集狀態,主要是 在探討逆向的電子轉移過程41。Yasuhiro Tachibana等人測量了釕錯合物與紫質分子在二 氧化鈦薄膜上的電子轉移過程,發現這兩種化合物的電子轉移速度是相似的,均為小於 100 fs的時間尺度上42,其相關的動態學與機制於第四章節討論之。 為了系統化的研究紫質分子在不同的環境下的動態學與介面電子轉移過程,我們以 雙苯基鋅離子紫質(ZnBPP)的衍生物來討論不同的取代基與取代基長度之間的關係。在 紫質環上的取代基位置有 1-8 與 α-δ 的位置,α-δ 的位置亦稱之為 meso 位置,本研究所 有的紫質衍生物分子除了 ZnPP 以外,均在 meso 位置上有苯環或在苯環上再接共軛的 直鏈取代基與羧基,其結構圖在第三章與第四章中。. 1-3-1 紫質分子的吸收光譜原理 紫質分子的光譜原理首先來自於共軛環多烯類(cyclic polyene),對於紫質環上的共 振結構可分為兩類,這兩類的共軛結構為類似 16 碳與 18 碳的環共軛多烯化合物而且紫 質的吸收躍遷與共軛環多烯類相同為 ππ*躍遷,由圖 1-3 左圖可知,因為有 18 個 π 電子, 因此照能階順序填之後,發現最高填滿電子的軌域(HOMO)的磁量子數為±4,而最低未 填電子的軌域(LUMO)的磁量子數為±5。因此電子的躍遷只會發生兩個簡併的 HOMOs 與兩個簡併的 LUMOs,其電子躍遷方式如圖 1-3 右圖所示。但是如果紫質分子的軌域 能量如共軛環多烯類的話,其吸收光譜在紫外光區的 Soret 頻帶亦稱之為 B 頻帶的吸收 強度與可見光區的 Q 頻帶的吸收度強度應該相同,不會像在 meso 位置上有四個苯基之 紫質分子(H2TPP 與 ZnTPP)如圖 1-4 所示 43,其莫耳消光係數在 Soret 頻帶峰比較大,而 且對於 H2TPP 而言,Q 頻帶分裂成 Qx 與 Qy 兩頻帶。此現象是無法用共軛多烯的自由電 -4-.
(21) 子模型來解釋的。. 圖 1-3 環 18 碳共軛多烯的自由電子模型(free electron model)與電子在 HOMO 與 LUMO 之間的躍遷形式圖。. 圖 1-4 四苯基紫質在的吸收光譜。左圖為原四苯基紫質(H2TPP),右圖為鋅離子四苯基紫質(ZnTPP)43。. 因此 Gouterman 在 1961 年發展了四軌域理論(four-orbital-theroy)來說明紫質分子的 吸收光譜特徵 44。四軌域指的是紫質的 HOMOs 與 LUMOs 這四個分子軌域,其分子軌 域的對稱性與其 π 電子的相位圖如圖 1-5 所示。HOMO 的兩個分子軌域的對稱性分別為 a2u 與 a1u。我們將 a2u 對稱的分子軌域的代號指定為 b1,a1u 對稱的分子軌域的代號指定 為 b2。另外,LUMO 的分子軌域的對稱性為 eg,將具有 x 軸的對稱面的分子軌域指定 為 c1,另一個具有 y 軸對稱面的分子軌域指定為 c2。b1 與 b2 的分子軌域有四個節面,c1 與 c2 的分子軌域有五個節面,因此 c1 與 c2 的分子軌域的能量比較高。從理論計算的結 果中,b1 軌域的能量比 b2 軌域高,所以 Soret 或 B 頻帶是電子由 b2 軌域激發到 c1 或 c2 軌域的躍遷,而 Q 頻帶的吸收是由電子由 b1 軌域激發到 c1 或 c2 軌域的躍遷。對於金屬 離子紫質而言,金屬離子以自身的 p 軌域電子與紫質環上的 π 電子發生共軛現象 45,由 圖 1-5 可知,金屬離子的 p 軌域的電荷密度會影響 a2u 對稱軌域的能量,若金屬離子的 -5-.
(22) 正電荷性質愈強,則會使得 a2u 對稱軌域(b1)的能量被提高,因此會造成 Q 頻帶紅位移如 圖 1-6 所示 45。. 圖 1-5 紫質分子的四軌域理論之分子軌域。藍色與紫色為π軌域之不同的相位,其對稱的節面以虛線表之。. -6-.
(23) 圖 1-6 在金屬離子四苯基紫質中,以不同的金屬離子來取代所得的振子強度(Oscillator strength, f)對 Q(0,0) 頻帶的能量之相關圖 45。. y. I N. IV. NH. NH. II. X. N. III. 圖 1-7 原紫質分子的結構圖。. 在靜態光譜的研究中,Weigl 利用螢光去偏振(fluorescence depolarization)的實驗中 發現 Qx 的螢光偏振方向與 Qy 的螢光偏振方向互相垂直 46。Rimington 等人以非常純的 原紫質在低溫下,發現 Soret 頻帶分裂成 Bx 與 By 兩頻帶 47。以上現象均可以四軌域理 論來解釋。對於原紫質分子而言,其中兩個氮上接有氫原子,在理論計算中,不管是 I 與 II 的吡咯上為相鄰的氫或是在 II 與 IV 吡咯上的相對位置的氫之間的能量是相同的, 但是若原紫質中氮上的氫原子在第 II 與 IV 的吡咯上,也就是在 x 軸上(如圖 1-7),會造 成 Qx 頻帶的能量比 Qy 頻帶的能量比較低,其原因可由四軌域理論來解釋。因為氫的位 置在 II 與 IV 吡咯的氮上,所以將 c2 軌域能量降低下來(因為 c2 軌域的 π 電子定域化在 x -7-.
(24) 軸方位的氮,但是 c1 軌域的 π 電子則是定域在 y 軸方位的氮上),造成原紫質分子之四 軌域的能階與金屬離子紫質的不相同,其相對的分子軌域能階如圖 1-8 所示。. 圖 1-8 金屬離子紫質與原紫質的電子躍遷概念圖,圖中藍色線為 y 軸偏振方向的電子激發躍遷,而紅色線 為 x 軸偏振方向的電子激發躍遷。. 原紫質分子的吸收躍遷以線性組合的方式來產生 B0x,y 與 Q0x,y 態的能量,其公式如 1-2 與 1-3 所示: Bx0 ⎫ ⎬ = [(b1c2 ) ± (b2 c1 )] / 2 Qx0 ⎭. (1-2). By0 ⎫⎪ ⎬ = [(b1c1 ) ± (b2 c2 )] / 2 Qy0 ⎪⎭. (1-3). (b1c2)代表 b1 軌域到 c2 軌域的電子單重態躍遷,同理,(b2c1)、(b1c1)與(b2c2)各代表 b2→ c1、b1→c1 與 b2→c2 的躍遷。如果 b1 與 b2 的能量相同,而且 c1 與 c2 的能量也相同的話, 則 B0(Soret)頻帶的躍遷矩不為零,為允許躍遷(allowed transition),其振子強度(oscillator strength)幾乎等於 1,但是 Q0 頻帶的躍遷矩為零,為禁制躍遷(forbidden transition),其 理論上振子強度為零。對於金屬離子紫質而言(圖 1-8),因為 c1 與 c2 的軌域能量是相等 的,所以 b1c2+b2c1=b1c1+b2c2,因此 Bx0 與 By0 是相同能量的,造成吸收光譜上只能看見 一個 B 頻帶而沒有分裂成 Bx 與 By 兩頻帶,又因電子組態的交互作用(configuration interaction, C.I.)下 B 頻帶為 Bx 與 By 的線性混合,所以對於金屬離子的 B 頻帶的躍遷偶 矩的極化方向具有 x 與 y 軸兩個方向。此外,對於 Q 頻帶的吸收躍遷也因為 b1c2-b2c1 = b1c1-b2c2,所以亦沒有分裂成 Qx 與 Qy 兩頻帶,如同 B 頻帶的原理,經 CI 後所產生的 Q 頻帶的躍遷偶矩的極化方向也具有 x 與 y 軸兩個方向。若未經微擾因素的話,吸收光譜 上 B 頻帶因為允許躍遷吸收,故其莫耳消光係數大,但是 Q 頻帶為禁制躍遷故應無法 看見,但是真實的吸收光譜中我們可以觀察到 Q 頻帶的吸收,這是因為微擾理論 (perturbation theory)或電子振動偶合 (vibronic coupling)的效應,使得 Q 頻帶並非完全禁 -8-.
(25) 制躍遷。其微擾的公式如下所示: Qx = Qx0+λxBx0. (1-4). Qy = Qy0+λyBy0. (1-5). λx = {[ε(c2)- ε(b1)]- [ε(c1)- ε(b2)]}/2Δ. (1-6). λx = {[ε(c1)- ε(b1)]- [ε(c2)- ε(b2)]}/2Δ. (1-7). 其中 Δ 為允許躍遷頻帶(Bx 或 By)與禁止躍遷頻帶(Qx 或 Qy)的能量差,ε(x)代表 x 軌域的 能量,Qx 與 Qy 頻帶的吸收強度與 λx2 和 λy2 成正比。 對於原紫質而言,因為兩個氫原子放在 x 軸方位上的氮上,造成了 c2 軌域的能量小 於 c1 軌域的能量,因無法判別 b1 與 b2 軌域的能量相對大小,所以其電子激發的躍遷圖 與其軌域能量的相對關係圖如 1-8 所示,(A)圖為當 b1 軌域能量小於 b2 軌域的能量,四 軌域之間的相對關係;(B)圖與(A)圖相反,為 b2 軌域的能量小於 b1 軌域的能量。不管是 圖(A)或圖(B),由於 b1c2+b2c1=b1c1+b2c2 所以對於 B 頻帶依然沒有分裂,但是 b1c2-b2c1 ≠b1c1-b2c2,所以 Q 頻帶分裂成 Qx 與 Qy 兩頻帶。如果原紫質的軌域相對能量大小如圖 1-8(A)所示的話,因為 x 極化方向的電子躍遷 b1→c2 與 b2→c1 的能量相似,又 Qx= b1c2-b2c1,所以 Qx 頻帶的吸收度比 Qy 頻帶的吸收度弱。若其軌域能量的相對大小如圖 1-8(B)的話,就會造成 Qx 頻帶的吸收度比 Qy 頻帶的吸收度強。 由圖 1-4 可知,Qx 頻帶的吸收度比 Qy 頻帶的吸收度弱,所以 H2TPP 的四軌域能階 的分布應如圖 1-8(A)所示,b2 軌域的能量比 b1 軌域的能量小。利用四軌域理論亦可解釋 當紫質環上有取代基時,會影響到光譜吸收的變化,例如在紫質環上 1、2、5、6 的位 置上放置烷基或在 3、4、7、8 的位置上放置烷基,前者會造成 c2 軌域的能量提升,後 者會造成 c1 軌域的能量提升。 在此也利用群論的概念來說明紫質分子吸收光譜,允許躍遷必須要瞬態偶極力矩 (transient dipole moment)不為零,而瞬態偶極力矩的計算公式如 1-8 所示: μ fi,Z = f μ Z i = − e ∫ ψf * zψi dτ. (1-8). 其中 μz 為電子偶極矩,Ψf 與 Ψi 各為 f 態的波函數與 i 態的波函數,因為波函數相常的 複雜,故以群論的觀點,當瞬態偶極矩不為零時,Γ(Ψf) ⊗ Γ(μz) ⊗ Γ(Ψi) 以其群軌域的對稱操作,應可約簡成全對稱元素(A1)。對於金屬離子紫質的對稱為 D4h, 其 HOMO 的軌域對稱性為 a2u 與 a1u,LUMO 的軌域對稱性為 eg,所以其基態(ground state) -9-.
(26) 的對稱性元素(Γ(Ψi))為 A1g(因基態的電子組態為 a2u2a1u2,由圖 1-9 中的特徵標表可算出)。. 圖 1-9 D4h 的特徵標表。. 其激發態的電子組態有兩種,一為 a2u1eg1 ,另一為 a1u1eg1。因此 a2u1eg1 與 a1u1eg1 的對稱 性元素(Γ(Ψf))均為 Eu 對稱。由 1-9 可知電子偶極矩的對稱元素(Γ(μz))為 A2u 與 Eu。將 Γ(Ψf) ⊗ Γ(μz) ⊗ Γ(Ψi)三者的對稱元素相乘之後可得: Γ(Ψ f ) ⊗ Γ(μ) ⊗ Γ(Ψ i ) = Eu. A2u Eu. A1g =. Eg A1g + A 2g + B1g + B2g. 因此,金屬離子紫質在 x 與 y 軸的瞬態偶極矩是不為零,也就是允許之躍遷。. 1-3-2 紫質分子的聚集原理與種類 (Theory and type for porphyrin-aggregates) π 共軛分子在高濃度、薄膜和晶體中可通過分子間的 π-π 互相作用形成聚集體。和 稀溶液的單體光譜相比,這種聚集體的光譜均會發生位移。根據 Kasha 公式 48,如果發 色團平面排列使其躍遷矩平行排列時,連接發色團中心的連線和躍遷矩之間的夾角 α > 54.7o 時,其吸收頻帶會藍位移(blue-shifted),但是若 α < 54.7o 時則其吸收頻帶會紅位移 (red-shifted)。當夾角 α > 54.7o 時發色團分子是屬於面對面(face-to-face)的排列,形成所 謂的 H-聚集體;夾角 α < 54.7o 時,發色團分子是頭對尾(head-to-tail)的排列,形成所謂 的 J-聚集體。與單體相比 H-聚集體的藍位移幅度愈大,表示面對面的 π-π 相互作用力愈 強;J-聚集體的紅位移幅度愈大,表示頭對尾的 π-π 相互作用力愈強。 聚集體之分子間作用力可以電子和聚集分子的原子核間庫倫型 (coulomb type)位能 表示,但其中牽扯到許多複雜的數學計算。為了簡化計算,一般利用激子偶合模型 (exciton coupling model)理論來解釋分子聚集在吸收光譜上所造成的變化 49。激子偶合模 型為描述一聚集的系統被激發後,其激發態彼此間之共振作用力(resonance interaction), 隨著不同系統而有不同的形式。例如,考慮系統為原子、離子或分子單元,並考慮外在 作用力的干擾、分子間的作用力、聚集分子的結構、及激發態間的作用力等等。此理論 之主要目的是希望藉簡單的古典力學模型來描述聚集分子的激發態行為。. - 10 -.
(27) 根據激子偶和的模型,當一堆聚集的分子一起被激發後,這些聚集分子的激發態稱 為激子 (exciton)49。激子偶合主要分為以下三種:強型偶合(strong coupling)、弱型偶合 (weak coupling)及中間型偶合(intermediate coupling)50。其中強型偶合使得聚集分子之光 譜有極明顯變化,而造成如此大的變化,可用分子間的偶極—偶極作用力(dipole-dipole interaction)來解釋。如圖 1-10(A)、(B)和(C)所示,一組分子對中其躍遷偶極力矩 (transition dipole moment) 以相同方向的方式排列 (包括 card-pack pair 和 head-to-tail pair );另外, 圖 1-10(D)與(E)中表示分子群的躍遷偶極力矩以相異方向的方式來排列。其中聚集體的 躍遷選擇率(selection rule)則是由分子對的躍遷偶極力矩的總和來決定,總和若不為零即 為允許躍遷,反之若總和為零,則為禁制躍遷 51。因此圖 1-10(A)、(B)和(C)的分子偶極 力矩的排列方式可以產生躍遷,但是圖-10(D)與(E)兩個分子偶極力矩加起來為零則為禁 制躍遷。圖 1-11 為激子偶和模型所預測的雙體在不同排列方式下之能階圖 49。由此圖可 以知當分子的偶極力矩排列為平行 (parallel) 時,因為分子間偶極力矩較穩定的排列為 禁制(forbidden)的躍遷,但是分子間偶極力矩為同方向(分子間為偶極排斥力,其能量較 高)為允許之躍遷,所以造成吸收光譜藍位移;而當分子的排列方式為頭對尾(head-to-tail) 時則剛好相反,使得吸收光譜紅位移;最後當分子的排列介在兩者之間時,則為 oblique 型,由於兩種排列方式均為允許(allowed)的躍遷,使得其吸收光譜之頻帶會產生兩個吸 收峰的情況。目前已有許多的研究利用此模型理論來解釋分子的聚集 52-56。由激子偶和 模型可知分子群的偶和力矩平行時,分子的聚集體堆疊方式屬於 H-聚集體(α < 54.7o), 造成吸收頻帶藍位移;若分子的偶和力矩為頭對尾時,此時分子群的排列為 J-聚集體(α > 54.7o),會造成吸收光譜紅位移。. 圖 1-10 不同形態之聚集體與其躍遷偶極力矩的五種排列方式。. - 11 -.
(28) 圖 1-11 激子偶和模型所預測的雙體在不同排列方式下之能階圖。. 1-4 飛秒量級的超快動態學 化學反應中的過渡態ゝ光化學動力學ゝ生物光合作用和染料敏化電池中的電子轉 移過程等都是屬於分子體系處於運動變化之中的過程,有些是皮秒和飛秒量級的超快過 程,需要用超快瞬態光譜方法才能研究。1987 年,隨著飛秒脈衝雷射的出現,Zewail 教授等科學家開創了分子化學反應動力學的一個新的研究領域-飛秒化學(femtosecond chemistry)。人們開始真正的觀測到分子內原子的運動,甚至試圖控制分子化學反應和生 物化學反應中的斷鍵、成鍵、質子轉換和電子轉移過程等基本過程,飛秒化學為分子化 學反應動力學打開了新的大門 57-60。 當有機分子被光激發到電子激發態後,所發生的光物理(photophysics)和光化學 (photochemistry)過程可分為兩大類。一是分子內(intramoleuclar)的過程,即斷鍵、生成 鍵、電荷遷移(charge transfer, CT)、電子態間的內轉移(internal conversion, IC)、系間交叉 (intersystem crossing, ISC)、不同振動能階間的能量再分配(intramoleuclar vibrational relaxation, IVR)和振動弛緩(vibrational relaxation, VR)等。其二為分子間(intermoleuclar) 的過程,即分子間的能量轉移(energy transfer)與電子轉移(electron transfer)、氫鍵作用、 偶極-偶極相互作用及激發態的溶劑效應等。這兩類的過程往往會同時發生造成激發態 衰減行為的複雜化。 飛秒化學研究是利用超快雷射光脈衝獲得飛秒時間解析的瞬態光譜。隨著飛秒雷射 光技術的迅速發展,獲得飛秒時間解析光譜的方法愈來愈多。例如螢光上轉移 - 12 -.
(29) (fluorescence up-conversion)61, 62 技術、飛秒激發-偵測(Femtosecond pump-probe)技術 63、 瞬態拉曼光譜(transient raman spectroscopy64 等。這些方法各有其特點,但對雷射系統與 探測設備的要求都很高。 我們主要以飛秒時間解析螢光上轉移與皮秒時間解析-時間相關單光子技術來測量 激發態紫質分子在凝態相中的緩解過程與紫質分子和二氧化鈦的奈米粒子間的介面電 子轉移過程的速率常數,藉此研究來發展出高光電轉換效能的紫質敏化太陽能電池。. 1-5 第一章的參考資料 (Reference for chapter one) 1. Sorensen, B., Renewable Energy. London, 1979. 2. Winter, C.-J.; Sizmann, R. L.; Vant-Hull, L. L., Solar Power Plants. Springer-Verlag.: Berlin, Heidelberg, 1991. 3. Becqurel, E.; Acad, C. R., Sci. Paris 1839, 9, 561. 4. Chapin, D. M.; Fuller, C. S.; Pearson, G. L., J. Appl. Phys. 1954, 51, 676. 5. Schock, H. W.; Noufi, R., Prog. Photovolt. Res. Appl. 2000, 8, 151. 6. Meyers, P.; V Albright, S. P., Prog. Photovolt. Res. Appl. 2000, 8, 161. 7. Regan, B. O.; Gratzel, M., Nature 1991, 353, 737. 8. Tsubomura, H.; Matsumura, M.; Nomura, Y.; Amamiya, T., Nature (London) 1976, 261, 402. 9. Nazeeruddin, M. K.; Kay, A.; Rodicio, I.; Humphrybaker, R.; Muller, E.; Liska, P.; Vlachopoulos, N.; Gratzel, M., J. Am. Chem. Soc. 1993, 115, (14), 6382. 10. Jang, S. R.; Vittal, R.; Lee, J. W.; Jeong, N.; Kim, K. J., Chemical Communications 2006, (1), 103. 11. Katoh, R.; Furube, A.; Yoshihara, T.; Hara, K.; Fujihashi, G.; Takano, S.; Murata, S.; Arakawa, H.; Tachiya, M., Journal of Physical Chemistry B 2004, 108, (15), 4818. 12. Lu, Y. F.; Choi, D. J.; Nelson, J.; Yang, O. B.; Parkinson, B. A., Journal of the Electrochemical Society 2006, 153, (8), E131. 13. Murai, M.; Furube, A.; Yanagida, M.; Hara, K.; Katoh, R., Chemical Physics Letters 2006, - 13 -.
(30) 423, (4-6), 417. 14. Persson, P.; Lundqvist, M. J., Journal of Physical Chemistry B 2005, 109, (24), 11918. 15. Stergiopoulos, T.; Karakostas, S.; Falaras, P., Journal of Photochemistry and Photobiology a-Chemistry 2004, 163, (3), 331. 16. Walter, B. J.; Elliott, C. M., Inorganic Chemistry 2001, 40, (23), 5924. 17. Landi, B. J.; Castro, S. L.; Ruf, H. J.; Evans, C. M.; Bailey, S. G.; Raffaelle, R. P., Solar Energy Materials and Solar Cells 2005, 87, (1-4), 733. 18. Robel, I.; Subramanian, V.; Kuno, M.; Kamat, P. V., Journal of the American Chemical Society 2006, 128, (7), 2385. 19. Akiyama, T.; Matsushita, M.; Kakutani, K.; Yamada, S.; Takechi, K.; Shiga, T.; Motohiro, T.; Nakayama, H.; Kohama, K., Japanese Journal of Applied Physics Part 1-Regular Papers Short Notes & Review Papers 2005, 44, (4B), 2799. 20. Fritsch, C.; Homey, B.; Stahl, W.; Lehmann, P.; Ruzicka, T.; Sies, H., Photochemistry and Photobiology 1998, 68, (2), 218. 21. Fungo, F.; Otero, L. A.; Sereno, L.; Silber, J. J.; Durantini, E. N., Journal of Materials Chemistry 2000, 10, (3), 645. 22.Fungo, F.; Otero, L. A.; Sereno, L.; Silber, J. J.; Durantini, E. N., Dyes and Pigments 2001, 50, (3), 163. 23. Hasobe, T.; Imahori, H.; Kamat, P. V.; Fukuzumi, S., Journal of the American Chemical Society 2003, 125, (49), 14962. 24. Jasieniak, J.; Johnston, M.; Waclawik, E. R., Journal of Physical Chemistry B 2004, 108, (34), 12962. 25. Kim, H. S.; Kim, C. H.; Ha, C. S.; Lee, J. K., Synthetic Metals 2001, 117, (1-3), 289. 26. Milanesio, M. E.; Gervaldo, M.; Otero, L. A.; Sereno, L.; Silber, J. J.; Durantini, E. N., Journal of Physical Organic Chemistry 2002, 15, (12), 844. 27. Saha, S.; Flood, A.; Griffiths, K.; Tseng, H. R.; Stoddart, J. F.; Bonet, F.; Dunn, B., - 14 -.
(31) Abstracts of Papers of the American Chemical Society 2004, 227, U141. 28. Schaafsma, T. J., Solar Energy Materials and Solar Cells 1995, 38, (1-4), 349. 29. Takahashi, K.; Hashimoto, K.; Komura, T.; Murata, K., Chemistry Letters 1994, (2), 269. 30. Takahashi, K.; Higashi, M.; Tsuda, Y.; Yamaguchi, T.; Komura, T.; Ito, S.; Murata, K., Thin Solid Films 1998, 333, (1-2), 256. 31. Takahashi, K.; Nakajima, I.; Imoto, K.; Yamaguchi, T.; Komura, T.; Murata, K., Solar Energy Materials and Solar Cells 2003, 76, (1), 115. 32. Takahashi, K.; Nakamura, J.; Yamaguchi, T.; Komura, T.; Ito, S.; Murata, K., Journal of Physical Chemistry B 1997, 101, (6), 991. 33. Takahashi, K.; Nakatani, S.; Yamaguchi, T.; Komura, T.; Ito, S.; Murata, K., Solar Energy Materials and Solar Cells 1997, 45, (2), 127. 34. Takahashi, K.; Nakatani, S. I.; Matsuda, T.; Nanbu, H.; Komura, T.; Murata, K., Chemistry Letters 1994, (11), 2001. 35. Takahashi, K.; Nanbu, H.; Komura, T.; Murata, K., Chemistry Letters 1993, (4), 613. 36. Takahashi, K.; Takano, Y.; Yamaguchi, T.; Nakamura, J.; Yokoe, C.; Murata, K., Synthetic Metals 2005, 155, (1), 51. 37. Takechi, K.; Shiga, T.; Motohiro, T.; Akiyama, T.; Yamada, S.; Nakayama, H.; Kohama, K., Solar Energy Materials and Solar Cells 2006, 90, (9), 1322. 38. Wamser, C. C.; Kim, H. S.; Lee, J. K., Optical Materials 2003, 21, (1-3), 221. 39. Wamser, C. C.; Kim, H. S.; Lee, J. K., Optical Materials 2004, 25, (4), 445. 40.Ramakrishna, G.; Verma, S.; Jose, D. A.; Kumar, D. K.; Das, A.; Palit, D. K.; Ghosh, H. N., Journal of Physical Chemistry B 2006, 110, (18), 9012. 41. Yang, X. J.; Dai, Z. F.; Miura, A.; Tamai, N., Chemical Physics Letters 2001, 334, (4-6), 257. 42. Trachibana, Y.; Haque, S. A.; Mercer, I. P.; Durrant, J. R.; Klug, D. R., Journal of Physical Chemistry B 2000, 104, (6), 1198. - 15 -.
(32) 43. Dorough, G. D.; Miller, J. R.; Huennekens, F. M., J. Am. Chem. Soc. 1951, 73, 4315. 44. Gouterman, M., J. Mol. Spectroscopy 1961, 6, 138. 45. Gouterman, M., J. Chem. Phys. 1959, 30, 1139. 46. Meigl, J. W., J. Mol. Spectroscopy 1957, 1, 133. 47. Rimington, C.; Mason, S. F.; Kennard, O., Spectrochim. Acta 1958, 12, 65. 48. Mdrae, E. G.; Kasha, M., Physical processes in radiation biology New York : Academic Press, 1964. 49. Kasha, M., Radiation Research 1963, 20, 55. 50. Zhang, Z.; Imae, T., Nano Letters 2001, 1, 241. 51. Kasha, M., In Physical and Chemical Mechanisms in Molecular Radiation Biology. Plenum Press:New York, 1991. 52. Zimmermann, J.; Siggel, U.; Fuhrhop, J. H.; Roder, B., J. Phys. Chem. B 2003, 107, 6019. 53. Gusev, A.; Rodgers, M. A., J. Phys. Chem. A 2002, 106, 1985. 54. Kim, Y. H.; Jeong, D. H.; Kim, D.; Jeoung, S. C.; Cho, H. S.; Kim, S. K.; Aratani, N.; Osuka, A., J. Am. Chem. Soc. 2001, 123, 76. 55. Hwang, I. W.; Kamada, T.; Ahn, T. K.; Ko, D. M.; Nakamura, T.; Tsuda, A.; Osuka, A.; Kim, D., J. Am. Chem. Soc. 2004, 126, 16187. 56. Cho, H. S.; Song, N. W.; Kim, Y. H.; Jeoung, S. C.; Hahn, S.; Kim, D., J. Phys. Chem. A 2000, 104, 3287. 57. Dantus, M.; Kim, S. B.; Williamson, J. C.; Zewail, A. H., J. Phys. Chem. 1994, 98, (11), 2782. 58. Herek, J. L.; Materny, A.; Zewail, A. H., Chem. Phys. Lett. 1994, 228, (1-3), 15. 59. Williamson, J. C.; Zewail, A. H., J. Phys. Chem. 1994, 98, (11), 2766. 60. Zewail, A. H., Abstracts of Papers of the American Chemical Society 1994, 207, 40. 61. Liu, Q. L.; Wang, J. K.; Zewail, A. H., Nature 1993, 364, (6436), 427. 62. Zewail, A. H., Journal of Physical Chemistry 1993, 97, (48), 12427. - 16 -.
(33) 63. Wang, Q.; Schoenlein, R. W.; Peteanu, L. A.; Mathies, R. A.; Shank, C. V., Science 1994, 266, (5184), 422. 64. Lenz, K.; Pfeiffer, M.; Lau, A.; Elsaesser, T., Chem. Phys. Lett. 1994, 229, (4-5), 340.. - 17 -.
(34) 第二章 實驗儀器與測量方法. 2-1 樣品的製備 本研究的紫質衍生物是由國立暨南大學應用化學系林敬堯教授實驗室所提 供,將紫質衍生物溶解在光譜級的溶劑中。為避免自我吸收的效應發生,吸收光 譜與螢光光譜的測量均使用 2 mm 的石英比色管,並將其濃度配製在小於 1×10-5 M 的濃度內。 敏化奈米二氧化鈦薄膜的製備是採用溶膠-凝膠(sol-gel)法 1, 2,溶膠-凝膠法在 形成薄膜和大塊固體方面有顯著的優點,在 1997 年 Gratzel 發表染料敏化的奈米 晶體太陽能電池的優異光電轉換效率,其二氧化鈦奈米晶體薄膜的製備也是利用 溶膠-凝膠法,其奈米粒子的平均大小為 10-20 奈米 3。本實驗採用的溶膠-凝膠法 的步驟如下所示:(1)先取異丙基鈦酸鹽(titanium isopropoxide, 97%, Aldrich) 25 mL 慢慢滴入 149 mL 去離子水與 1 mL 的硝酸(70%)中,此時會在稀硝酸溶液中形成 雪花般的白色二氧化鈦聚集體;(2)將此溶液加熱至 60~70 ℃,並維持此溫度 8 小 時後,將其乳白色溶液裝至迴旋濃縮儀中,將其溶液體積濃縮至原體積的 1/3;(3) 將濃縮後的二氧化鈦膠態溶液加入等量體積的聚乙烯基甘油(polyethylenglycol 600, Merck),並將其混合均勻;(4)將其混合膠態溶液取適當的量滴至 ITO 玻璃上,放 至在旋轉鍍膜儀上,使其轉速為 4000 rpm,旋轉 30 秒後放置入 450 ℃烤箱中加熱 40 分鐘;(5)重複(4)的步驟 9 次,在 ITO 玻璃上總鍍上 10 層二氧化鈦粒子薄膜; (6)將鍍好的二氧化鈦薄膜片放入 0.1 M 的氯化鈦(TiCl4)的水溶液中半小時後,用純 氮氣將其表面吹乾,再放入 450 ℃烤箱中加熱 30 分鐘;(7)將製備完的奈米二氧化 鈦薄膜片靜置在已配製好的紫質溶液中 4 小時,然後再用溶劑將其表面洗淨,放 入乾燥箱中避免水氣與光線的照射。. 2-2 紫外-可見光吸收光譜儀 本實驗之吸收光譜量測方法主要有兩部分,一部份為液態樣品的量測;另一 部份則為固相染料敏化奈米二氧化鈦薄膜樣品的量測,以下分別論述。 樣品的吸收光譜之測量是使用 Varian 公司,型號為 Cary50 的紫外-可見光光 譜儀。使用光源為氙燈,其光區範圍為 190 ~ 1100 nm,實驗所使用的掃描速率為 每秒 1200 nm,光譜解析度為 0.25 nm。對於液相樣品的參考樣品,使用與溶液樣 品相同的溶劑當做參考樣品;而對於染料敏化奈米二氧化鈦薄膜的測量,則以空 白的奈米二氧化鈦薄膜來當作參考樣品,因為敏化奈米二氧化鈦薄膜的穿透性良 - 18 -.
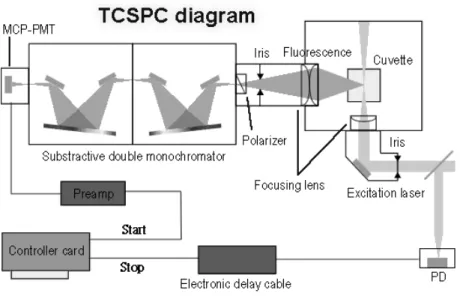
(35) 好,故以穿透式的方式來測量其吸收光譜。. 2-3 螢光光譜儀 本實驗螢光光譜的測量利用脈衝式二極體雷射為激發光源,經由光纖打入樣 品,在 90°方向上以光纖收集樣品的放光並傳送到 CCD 光譜儀(USB2000-FLG, Ocean Optics)進行光譜的偵測。偵測波長範圍 378-1050 nm,入射孔徑為 200 μm, 解析度為 10 nm,偵測器為 2048-圖素線性 CCD 列陣。. 2-4 時間相關單光子計數系統 2-4-1 時間相關單光子計數系統原理與裝置圖 本實驗主要利用 PicoQuant 公司所生產的時間相關單光子計數系統來進行時 間-解析螢光光譜(time-resolved fluorescence spectra)的測量。其工作原理如圖 2-1 所示 4, 茲簡述如下。利用脈衝光源週期性激發樣品以進行單一光子的偵測,精 確的記錄所偵測到的光子訊號與雷射同步觸發訊號(synchronization trigger,SYNC trigger)的相對時間,並對單一光子所測得的相對時間進行累計,以重建螢光訊號 波形。由於”pile up”的效應 5, TCSPC 技術所要求的實驗條件為”每個週期所偵測 到的光子數目遠小於 1”,在此偵測條件下才可正確地重現實際放光訊號的時間波 形 (temporal profile)。. 圖 2-1. 時間相關單光子計數的工作原理. - 19 -.
(36) TCSPC 之儀器配置如圖 2-2 所示。所使用之激發光源為脈衝式的二極體雷射 (pulsed diode laser) ,其最小半高寬(full width half maximum,FWHM)約為 80 ps 的脈衝。當雷射輸出時,由光源本身的控制器(driver;PDL 800-B,PicoQuant)送 出同步觸發訊號,經由分數式時間鑑別器(constant fractional discriminator,CFD) 判別訊號有效與否並設定訊號到達時間,再由可調節式延遲器(variable delay line; 425A,ORTEC)調整進入 TCSPC 模組(SPC630,PicoQuant)的延遲時間。激發光 源經由一面反射鏡導向樣品室(sample chamber),可經由調整鏡面上的兩個旋鈕控 制導向樣品的雷射光方向,反射鏡前具有一光圈(iris)可調節進入樣品室的雷射光 通量,接著通過一個聚焦透鏡(focusing lens)聚焦到樣品上。樣品的放光利用兩面 透鏡進行收集,再經由一個光圈調節收集的光通量及一可選擇垂直(perpendicular) 、 水平(horizontal)或魔術角度(magic angle)偏振方向的偏振器(polarizer),接著 再 通 過 相 減 式 的 雙 光 柵 光 譜 儀 (subtractive double monochromator ; 9030DS , Sciencetech),而進入偵測器。. 圖 2-2. Fluo Time 200 儀器配置圖。. 偵 測 器 為 微 頻 道 光 電 倍 增 管 ( micro-channel plate photon-multiplier tube , MCP-PMT ; R3809U-57 , Hamamatsu ), 其 輸 出 的 電 流 值 經 由 一 個 前 置 放 大 器 (Pre-Amplifier)將訊號轉換成正電壓值並放大以達到 TCSPC 模組可以接受的範 圍,接著進入 TCSPC 模組進行訊號計時的工作。 利用兩個分數式鑑別器(constant fractional discriminator,CFD)分別判別螢光 及觸發訊號有效與否並決定其所到達的時間,再以標準訊號(NIM)送入時間-振 幅轉換器(time-to-amplitude converter,TAC)中。當 TAC 收到螢光訊號後,內部 的電容便開始充電,直到收到下一發觸發訊號時停止充電,並產生電壓輸出,所輸 出的電壓振幅正比於兩訊號輸入 TAC 的相對時間差。最後以類比-數位轉換器 - 20 -.
(37) (analog-to-digital converter,ADC)將電壓振幅轉換為個別的時間頻道(channel), 再送入多頻道分析儀(multi-channel analyzer,MCA)進行個別時間頻道的累計, 完成一次單一光子計時的偵測。針對單一光子持續進行計時並累積,可將螢光隨時 間的分佈完整重現。. 2-4-2 時間相關單光子計數系統單電子元件 2-4-2-1. 分數式時間鑑別器 時間鑑別器的作用在於辨別訊號是否為有效,並且判定其到達時間。在一般的 時間鑑別器中,其判別方式如下所述:設定一特定門檻(threshold)的電壓高度, 當外來訊號超過此特定之電壓高度時,便可被觸發而認知此訊號。如果輸入鑑別器 的訊號低於此特定電壓,則視為電路中的雜訊,此訊號將被完全忽略,如圖 2-3 所 示。. 5. 4. 2. 3. Noise Single photon events. 1 Disc / LLD ULD. Multi- photon events. 圖 2-3 當輸入鑑別器的訊號低於一特定門檻的電壓高度時,則被分數式時間鑑別器視為雜訊去除。. 分數式時間鑑別器判讀訊號到達時間的方式則如下所述:將輸入的脈衝訊號 分成兩部分,其中之一的電壓振幅以一固定比例縮小;另一個電壓則以訊號反轉 (invert)的方式並且延遲 Td 時間,此時前者的振幅出現在後者前緣相同振幅的位 置上,接著再將處理後的訊號加總,將電壓值為零的時間作為原始脈衝的到達時 間。. 2-4-2-2. 時間-振幅轉換器 TAC是一電容裝置,功能類似精確的碼錶,利用TAC可精確得知雷射脈衝激發 樣品後,產生之單一光子的時間。其作用機制為:接收到「開始的訊號」時開始 充電,直到接收到「停止的訊號」時停止,並產生一類比輸出電壓,此電壓振幅 正比於兩訊號的輸出時間差,如圖2-4所示。. - 21 -.
(38) Photon detected. ready for next photon. next SYNC. TAC setting time TAC starts. TAC stops. start-stop time. ADC starts. reset. TAC ready. TAC dead time. 圖 2-4 TAC偵測單一光子的計時機制。. 在單一個脈衝週期中,若 TAC 同時接收到兩個螢光光子,此時 TAC 在針對第 一顆光子進行充電時,對進來的第二顆光子形同「blind」,因此時間較慢的光子將 被忽略使得光譜的形狀變形,造成所得到的螢光訊號比真實的螢光訊號更快的衰 減,為了避免此現象,必須確定每個激發脈衝所產生的螢光光子被偵測器偵測到的 機率遠小於一,如此可避免 TAC 在同一個週期中接收到兩個光子。因此,TCSPC 之實驗條件則需要求螢光光子數必須小於脈衝重複頻率的 1/100,可藉由控制雷射 光的強度及光圈(iris)大小來調整。 TAC有兩種充電模式:一為正常的開始-結束模式(normal start-stop mode), 以同步觸發訊號為開始的訊號,螢光訊號為停止的訊號;另一種則為反轉的開始結束模式(reverse start-stop mode) ,此時以螢光訊號為開始的訊號,同步觸發訊號 作為結束。為了取得真實的訊號必須降低每週期收到螢光的機率,而TAC若以正常 的開始-結束模式運作,則在大部分週期內無法接收到光子,使得整個系統不斷地 空轉。本系統採用相反的TAC充電模式,如此可保證TAC每次充電皆可收到結束的 訊號,減少dead time(等待系統回到可進行下一回偵測所需的時間)及提高讀取訊 號的速率。. 2-4-2-3. 類比-數位轉換器 由 TAC 所產生的電壓後,進入類比-數位轉換器,此類比-數位轉換器之功能為 將電壓振幅轉換成相對應的時間頻道。. 2-4-2-4. 多頻道分析儀 MCA 將每次螢光時間偵測結果紀錄在個別的時間頻道中,藉著重複螢光時間 - 22 -.
數據




+7





Outline
相關文件
E-B3 具備藝術創作與欣賞的基 本素養,促進多元感官的發 展,培養生活環境中的美感體 驗。. 音樂與
E-B3 具備藝術創作與欣賞的基 本素養,促進多元感官的發 展,培養生活環境中的美感體 驗。. 音樂與
合流(confluence)或混淆 :自我與環境的 分化,無法有清楚的知覺,導致內在經驗與外在
一、寵物美容基本常識 二、寵物相關法規認識 三、寵物保健衛生 四、寵物行為認知. 五、寵物美容工作環境使用與維護
A-1-1參照課程綱要 與學生特質明定教 學目標,並研擬課 程與教學計畫或個
」競賽,是結合生物科技與工程概念,以應用與設計為導向 的最新生物科學,為解決人類周遭生活問題。iGEM
式中 、 、 為隨物質而定的常數﹐表面張力隨液體性質不同可有很大差別。例 如 20 C 時有機液體苯的表面張力是 28.88
在上 一節中給出了有單位元的交換環 R 上的模的定義以及它的一些性質。 當環 R 為 體時, 模就是向量空間, 至於向量空間中的部分基本概念與定理, 有些可以移植到模上來。 例如 子
