國
立
交
通
大
學
資訊科學與工程研究所
碩
士
論
文
利 用 圖 形 表 示 的 基 因 規 劃 法
找 尋 核 醣 核 酸 的 共 同 結 構 元
Prediction of RNA common structural motifs by
Genetic Programming with graphical expressions
研 究 生:許登貴
指導教授:胡毓志 博士
利用圖形表示的基因規劃法找尋核醣核酸的共同結構元
Prediction of RNA common structural motifs by Genetic Programming
with graphical expressions
研 究 生:許登貴 Student:Deng-Guei Shiu
指導教授:胡毓志 Advisor:Yuh-Jyh Hu
國 立 交 通 大 學
資 訊 科 學 與 工 程 研 究 所
碩 士 論 文
A ThesisSubmitted to Institute of Computer Science and Engineering College of Computer Science
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master
in
Computer Science June 2006
Hsinchu, Taiwan, Republic of China
利用圖形表示的基因規劃法
找尋核醣核酸的共同結構元
研究生:許登貴 指導教授:胡毓志博士
國 立 交 通 大 學 資 訊 科 學 與 工 程 研 究 所
摘要 隨著人們對 RNA 功能的知識增加,最近對 RNA 的研究較以往吸引了更多的 注意。如同生物體中的其它大分子一樣,RNA 的功能取決於它們的結構。由於 直接從三級立體結構出發的技術在效用與效率上仍受限制,因此有各式各樣的計 算機方法被提出。在這篇論文裡,我們將目標放在 RNA 的二級結構預測。基於 需要被預測的 RNA 結構的數量不同,計算的方法可被分類為單一序列結構預測 以及多重序列結構預測。一般而言,單一序列結構預測被用來尋找一條序列可能 的整個二級結構,而多重序列結構預測則是被用來尋找同一 RNA 家族序列的共 同區域性二級結構。目前大部分多重序列結構預測的方法皆侷限於找到相對較短 的共同結構元素,它們無法找出較長的共同結構,而這些共同結構很可能在生物 學上扮演重要的角色。我們提出了一個多重策略的方法,結合了單一序列結構預 測以及多重序列結構預測的方法。藉由使用單一序列結構預測系統的預測結果轉 換成我們定義的 RNA 二級結構的圖形化模型,我們可以提升多重序列結構預測 的能力。為了驗證我們系統的效用與效率,我們從 Rfam 下載數個真實世界裡的 RNA 家族來做測試,實驗顯示本方法能有不錯的表現。Prediction of RNA common structural motifs by Genetic Programming
with graphical expressions
Student:Deng-Guei Shiu Advisor:Dr. Yuh-Jyh Hu
Institute of Computer Science and Engineering College of Computer Science
National Chiao Tung University Hsinchu, Taiwan, Republic of China
Abstract
As the increase of knowledge of RNA functions, the research on RNA has recently attracted more attentions than ever. Like other biopolymers, the functions of RNA are dependent upon their structures. Since the effectiveness and efficiency of ab initio 3D structure determination Technologies are still limited, various computational approaches have been proposed. In this thesis, we are focused on RNA secondary structure prediction. Based on the number of RNA for which to predict the structures, computational methods can be classified as single-sequence prediction and multiple-sequence prediction. In general, single-sequence prediction is aimed to find the probable global secondary structures, and on the other hand, multiple-sequence prediction is aimed to identify the common local secondary structures in a given RNA family. Most of the current approaches to multiple-sequence prediction are limited to finding relatively short common structure elements. As a consequence, they fail to identify those longer common structures that may play important biological roles. We propose a multi-strategy method that combines the advantages of both single-sequence and multiple-sequence prediction. By using the prediction results of single-sequence predictors as the basis to form the graphical models of RNA secondary structures, we can improve the performance in multiple-sequence prediction. To demonstrate the efficiency and effectiveness, we tested our new approach on several real-world RNA families downloaded from Rfam. The experiments showed some promising results.
致謝 能夠順利完成這篇論文,首先必須感謝我的指導教授:胡毓志老師,在這兩 年來的指導與督促,讓我能在短時間內進入生物資訊這領域,並且學到做研究的 方法與態度,這對將來工作或是在為人處事上都有極大的幫助。 感謝生物資訊實驗室的所有成員,讓我在這兩年內度過難忘的時光。感謝世 彥、音璇、勁伍、昀君和秉蔚各位學長姊的提攜與解惑,貫中和豐茂的互相督促 與勉勵,繼養學弟的協助與幫忙,以及博班學長子緯、巽昌和鈞木的加油與打氣, 還有實驗室豐富的資源與熱鬧的氣氛,這些都是讓我能順利畢業的大功臣。 另外,還要感謝同時在為碩士論文拚命的姿伶、偉民、志鵬、立泓等人,可 以與我互吐苦水、互相激勵。還有感謝眾多親朋好友的鼓勵,讓我擁有繼續努力 下去的動力。 最感激的還是家人的支持與關心,幾句簡單的問候便是減輕心理壓力的最佳 良藥。 順利畢業了,謝謝身邊的各位,以及感謝交大土地公的保佑。
目錄 摘要...i Abstract...ii 致謝... iii 目錄...iv 第一章、前言...1 1.1 研究動機...1 1.2 研究假設...3 1.3 研究目的...4 1.4 論文架構...4 第二章、文獻探討...5 2.1 核醣核酸簡介...5 2.1.1 核醣核酸的重要性 ...5 2.1.2 核醣核酸結構基本單位元 ...6 2.1.3 核醣核酸二級結構 ...6 2.2 預測核醣核酸結構的相關方法...8 2.2.1 單一核醣核酸序列二級結構預測 ...8 2.2.2 根據多重序列排比結果進行摺疊來預測核醣核酸共同結構元 ....10 2.2.3 同時考慮序列排比與摺疊的資訊來預測核醣核酸的共同結構元 11 2.2.4 對核醣核酸摺疊結構進行排比來預測核醣核酸共同結構元 ...14 2.2.5 其他相關工具 ...15 2.3 核醣核酸資料庫...17 2.3.1 Rfam...17
2.3.2 The RNase P Database ...17
2.3.3 tRNA Compilation 2000...18 2.3.4 RAG ...18 2.3.5 RNABase...19 2.3.6 SCOR ...20 2.3.7 其他常見資料庫 ...20 第三章、研究方法...21 3.1 系統設計目的與概念...21 3.2 核醣核酸結構描述語言...22 3.3 系統主架構...27 3.3.1 輸入序列及系統參數 ...28 3.3.2 產生背景序列 ...28 3.3.3 預測二級結構 ...29 3.3.4 分析二級結構及轉換為描述語言 ...31 3.4 基因規劃法...33
3.4.1 產生初代個體 ...34 3.4.2 適應函數 ...36 3.4.3 母代挑選機制 ...37 3.4.4 演化計算子 ...38 3.4.5 終止條件 ...39 3.4.6 後處理 ...39 第四章、實驗...40 4.1 實驗評估標準...40 4.2 實驗測試資料...41 4.3 實驗結果...43 4.3.1 與 RNAshapes 比較 ...44 4.3.2 實驗結果分析 ...45 第五章、結論與未來方向...47 5.1 結論...47 5.2 未來研究方向...47 5.2.1 處理共同結構元莖幹數較多之家族 ...47 5.2.2 對多個家族做分群 ...48 第六章、參考文獻...49
第一章、前言
1.1 研究動機
核醣核酸(RNA)在生命體中扮演很重要的角色,其中最為人知的信使核醣核 酸(mRNA)傳遞核醣核酸的資訊到核糖體,合成所需要的蛋白質。其他常見的還 有轉錄核醣核酸(tRNA)、核糖體核醣核酸(rRNA)、微核醣核酸(microRNA)等。這 些 RNA 會褶疊成特定的形狀來輔助生命機制,如催化化學反應及調控基因表現 等等。 從已知的生物知識可知,褶疊形狀相似的核醣核酸很有可能也會有相似的功 能。因此,若能由已知的核醣核酸序列來預測其褶疊而成的二級結構,進而猜測 其功能,將能更迅速的瞭解生命運作的機制。 然而,在生物實驗室裡進行實驗來決定一個核醣核酸的結構是很費時的,單 用人工的方式實驗非常沒有效率。因此,我們希望利用已知序列上的資訊,加入 能量預測二級結構的資訊,藉由電腦的輔助以提供一個快速的方法,希望能預測 出核醣核酸的結構,更進一步的從一個家族的核醣核酸序列中,預測出他們的共 同結構元(motif),因為這些共同的結構在生物演化上可能是有意義的,他們可能 控制著一種重要的生物機能,所以在經過長時間的演化之後,這些結構仍然保留 至今。研究核醣核酸二級結構預測(RNA secondary structure prediction)的方法有很 多,例如使用動態程式規劃(dynamic programming)的方法尋找化學上能量最為穩 定的結構;或是以排比(alignment)的方式,利用一條已知二級結構核醣核酸序列 上的資訊,去預測另外一條結構未知的相關核醣核酸序列;以及用基因演算法 (genetic algorithm)的方式尋找二級結構和摺疊路徑(folding pathway)等。以上的方法 都是只針對單一條核醣核酸序列提供唯一的最佳二級結構預測結果,或是包含多 個次佳解的結果。
近年來,對於核醣核酸二級結構的研究主題多在預測同一核醣核酸家族的共 同結構元,目前常見的方法有三大類[Paul PG et al., 2004]:
(1)先對所有核醣核酸序列做多重排比(multiple sequence alignment),再將 排比好的序列利用單一核醣核酸序列的二級結構預測系統進行摺疊 (folding),最後所得的摺疊結構即為該家族的預測共同結構元。 (2)以 Sankoff algorithm 為基礎,使用動態程式規劃同時考慮序列排比與 摺疊的資訊來預測同一家族序列的共同結構元。 (3)利用單一核醣核酸序列的二級結構預測系統,對此家族的每一條核醣 核酸序列各自進行單一序列的摺疊,再對所有產生的結構進行結構排 比(structure alignment)。 本研究與上述的第三類方法有點相似,在前半段使用單一核醣核酸序列的二級結 構預測系統作為前處理器,來預測單一核醣核酸序列的完整二級結構。然而在後 半段,也是最主要的核心部分,我們並非只是對其產生的結構進行排比,而是將 其預測的結構轉換成圖形表示語言,再利用基因規劃法(genetic programming)預測 出此家族序列的共同結構元。
1.2 研究假設
關於核醣核酸二級結構的預測,本研究設定了兩個合理的基本假設: 假設一:同一家族的核醣核酸序列有共同的二級結構。 一群核醣核酸序列之所以會被視為同一家族,就是因為他們有類似的功能。 由化學的角度來看,當結構有些許改變就很有可能影響分子結合的能力,因而影 響其功能,所以我們認為,一群功能相同的核醣核酸序列行使功能之區域,其二 級結構必定極為相似。 本研究假設一群被歸類為同一家族的相關核醣核酸序列中,從在某些共同的 結構,而這些共同的結構則是決定此家族核醣核酸所行使的功能。 假設二:行使功能的共同結構元不容易出現在隨機產生的序列中。 本研究要尋找的共同結構應該具有演化上的意義,在演化的過程中,核醣核 酸的序列及結構可能會經過多次的突變,但是其重要結構仍被保留下來,表示這 些結構在演化的過程中必定扮演很重要的假設。 因此我們假設這樣的結構應該不是偶然形成,也就是說在我們隨機產生的核 醣核酸結構中不應該會經常出現。1.3 研究目的
在過去的研究中,預測核醣核酸二級結構的共同結構元用到許多不同的方 法,包含動態程式規劃、隱藏式馬可夫模型(Hidden Markov Model)、序列排比、 圖論方法以及演化式計算等等。每一個研究所切入的角度都不太一樣,對於不同 的家族的共同結構元預測能力也不太相同,但目前的系統大多只能預測出長度較 短的共同結構元。 而在本研究中,我們同樣使用基因規劃法,試圖找出同一家族的共同結構 元,加入能量的資訊縮小搜尋空間以節省搜尋時間,而資料結構的表示法則以圖 形(graph)的概念表示,希望可以藉此找出較長或者是更複雜的共同結構元。
1.4 論文架構
本篇論文包含六個章節: 第一章為前言,介紹本研究的動機、背景、此研究所使用的方法及其基本假設, 以及主要的研究目的。 第二章為文獻探討,將介紹核醣核酸的背景知識,以及此研究過去的發展。 第三章為研究方法,是本篇論文的核心,詳細介紹本研究設計的方法流程與細節。 第四章為實驗結果,整理所有實驗的內容與實驗的結果。 第五章為結論與討論,分析本實驗的優缺點。 第六章參考文獻,則列出本研究參考的相關文獻。第二章、文獻探討
2.1 核醣核酸簡介
長期以來,人們對於核醣核酸(ribonucleic acid, RNA)的瞭解不多,僅知道 RNA 在 合 成 蛋 白 質 的 過 程 中 扮 演 著 “ 遺 傳 信 使 " 的 角 色 : 將 去 氧 核 醣 核 酸 (deoxyribonucleic acid, DNA)所攜帶的訊息帶到核醣體,作為轉譯(translation)蛋白質 使用。最近幾年隨著對 RNA 的研究發現愈來愈多,RNA 在生物學上的地位也愈 來愈為重要。
2.1.1 核醣核酸的重要性
除了早期所知的信使核醣核酸(messenger RNA, mRNA)外,有其他重要功能的 核醣核酸也陸續被發現,如許多未編碼的核醣核酸(non-coding RNAs, ncRNAs), 其中有些甚至可以促進生化反應,控制細胞內蛋白質(酶)的合成,這類的核醣核 酸包括轉錄核醣核酸(transfer RNA, tRNA)和核醣體核醣核酸(ribosomal RNA, rRNA) 等。 還有能夠調控基因表現的核醣核酸,如微核醣核酸(microRNAs)。微核醣核 酸是一群非常短,長度約二十多個鹼基的核醣核酸,最明顯的特徵就是所有微核 醣核酸的先質(precursor)都具有一個類似髮夾的構造,而這些構造在基因體裡是 非常穩定的。微核醣核酸在後轉錄時期(post-transcription)參與調控,其影響包含 控制細胞凋亡、組織生長、肥胖代謝,以及決定某些基因的表現時間。 在科學(Science)雜誌所刊載的 2002 年研究表明,一些長度較短的核醣核酸, 即所謂的小分子核醣核酸(Small RNA),能夠對細胞和基因的很多行為進行控制, 比如打開和關閉多種基因,刪除一些不需要的 DNA 片段等。它們在細胞分裂過 程中更是發揮了至關重要的控制作用,可指導染色體中的物質形成正確的結構, 防止 DNA 片段位移出錯。若 DNA 功能的產生錯亂,可能是引發癌症的一個重要 原因。
2.1.2 核醣核酸結構基本單位元
我們已知核醣核酸的功能與其結構息息相關,結構的多樣性讓核醣核酸具備 多重的生物功能。因此,在核醣核酸的相關研究上,我們對於核醣核酸的結構所 產生的興趣,遠大於對於序列的分析。談結構之前,還是必須先對序列組成有基 本的了解。 核醣核酸由四種含氮鹼基組成,分別是腺嘌呤(Adenine) 、胞嘧啶(Cytosine)、 鳥糞嘌呤(Guanine)、尿嘧啶(Uracil),習慣上常分別以 A、C、G、U 來代表這四種 含氮鹼基。 核醣核酸常以單股存在於生物體中,透過分子間的作用力,會自己摺疊成特 定的結構,產生摺疊的作用力主要來自於 C≣G 三個氫鍵的鏈結以及 A=U 兩個 氫鍵的鏈結,此兩組鏈結的個別配對稱之為標準鹼基對(canonical base pair)。此外 還有一個搖擺鹼基對(wobble base pair)G-U,為一個氫鍵的鏈結,此結構較不穩 定,需要週圍的鹼基對輔助。由 A、G、C、U 各自配對所產生的摺疊形成了 RNA 的基本結構,稱之為 RNA 的二級結構。2.1.3 核醣核酸二級結構
a. 莖幹結構(stem) 核醣核酸序列中,連續鹼基配對所形成的一個長狀形狀,稱之為莖幹結構。 b. 髮夾環狀結構(hairpin loop) 當一個連續的非配對區域不是出現在序列的終端,而且僅與一個莖幹相鄰的 話,該區域就是一個髮夾環狀結構。而此環狀結構與相鄰的莖幹則合稱一個 髮夾結構。 c. 內部環狀結構(internal loop) 一個連續的非配對區域恰與兩個莖幹相連,而且兩側都有未配對鹼基,則該 區域即為內部環狀結構。內部環狀結構又可分為對稱性(symmetrical)與非對稱 性(asymmetrical),當兩側未配對鹼基個數相同時,則稱為對稱性內部環狀結 構,反之則稱為非對稱性內部環狀結構。 d. 突起結構(bulge) 在莖幹中僅一邊有未配對的鹼基,而另一邊都是連續的鹼基對,則稱這些未 配對的區域為突起結構。 e. 多分支環狀結構(multi-branched loop) 類似內部環狀結構,但當該環狀結構與三個以上的莖幹接觸時,則稱為多分 支環狀結構。 f. 擬結結構(pseudo-knot) 擬結結構是一種比較特別的結構,形成的主因是莖幹交錯配對。當莖幹間的 鹼基會與莖幹外的鹼基形成配對時,該結構就稱之為擬結結構。 圖 2. 擬節結構範例
2.2 預測核醣核酸結構的相關方法
研究核醣核酸的目的是希望能夠了解核醣核酸在生物體裡所擁有的功能,而 讓這些核醣核酸有其功能的原因不在於它的一級結構(序列),而是它所折疊而成 的二級結構。目前生物學家認為,分子的結構是影響核醣核酸功能的關鍵,例如 常見的轉移核醣核酸,其結構都是很穩定的苜蓿葉(cloverleaf)結構:包含四個莖 幹而形狀類似四瓣的苜蓿葉。另外,擁有相同生化功能的同一家族成員,也常會 擁有相似的二級結構片段。 因此,若能利用計算機的輔助,能夠迅速的發現同一家族成員中的共同結構 元,這對生物學是很有幫助的。這不僅能協助生物學家快速找出該家族行使其功 能的結構片段,亦能利用已知的共同結構,檢驗未知功能的序列,來推論出其功 能,進而找到所屬家族。 過去研究核醣核酸二級結構預測的方法很多,本節簡述過去幾個比較具代表 性的方法:2.2.1 單一核醣核酸序列二級結構預測
給定一條核醣核酸序列,我們希望預測出它摺疊而成的二級結構,最常見的 方法則是使用熱力學(thermodynamics)的知識來推論此序列可能折疊而成的形狀, 以下為幾個代表的系統: 2.2.1.1 Mfold [Zuker M, 2003]Mfold 是一套單一核醣核酸序列的二級結構預測系統,實作 Zuker 與 Stiegler 所提供的演算法,利用動態程式規劃法(Dynamic Programming)計算出核醣核酸序 列擁有最小自由能量(minimal free energy, MFE)的摺疊結構,以預測最為穩定的結 構。當序列的長度為 n 時,系統所需的時間複雜度為 O(n3
),所需要的空間複雜 度為 O(n2
)。此系統可以依照自由能量的大小,由小到大輸出數個可能的二級結 構供使用者參考。
然而核醣核酸在摺疊的過程中可能受到某些因素或是受到其他分子的影 響,使得理論上最穩定的結構無法形成,單純依靠最小能量來斷定結構形狀仍會 有很大的不足。另外,Mfold 無法摺疊成擬結結構,這也是其缺點之一。但在許 多相關的研究上,Mfold 仍被廣泛應用,其確實提供了相當程度的資訊。
2.2.1.2 RNAfold [Hofacker et al., 1994]
RNAfold 是維也納 RNA 研究團隊(Vienna RNA package)所實作的單一核醣核 酸序列的二級結構預測系統,其運作原理與 Mfold 一樣皆是建立在 Zuker 與 Stiegler 所提供的演算法上,以動態程式規劃法找出能量最小而最為穩定的二級 結構。 RNAfold 與 Mfold 的運作原理相同,差別只在於實作的方式不同,兩者所預 測出來的核醣核酸二級結構結果差異性很小,從兩者的比較研究顯示,這兩個不 同的單一核醣核酸序列二級結構預測系統,從準確性上看來沒有重大的差別存 在。
2.2.1.3 Sfold [Ding Y et al., 2003]
Sfold 實作了另一種以能量為基礎的單一序列折疊演算法,給定一條核醣核 酸序列,利用統計的方法取出其二級結構的樣本,接著依據給予的熱力學參數產 生核醣核酸二級結構的相稱分割函數(equilibrium partition functions),根據分割函 數使用條件機率對所有可能的結構進行遞迴取樣,而後產生二級結構的統計上典 型樣本,最後使用分群(clustering)的技術獲得可能的結構。可根據最小自由能取 出前幾名可能的結構以供使用者參考。
根據先前的研究分析,從精確度上看來,Sfold 的結果與 Mfold 和 RNAfold 產生的結果非常相似,但 Sfold 相較於其他兩者而言,它的結果的變異性(variance) 有稍微高出了一些。
2.2.2 根據多重序列排比結果進行摺疊來預測核醣核酸共同結構元
預測核醣核酸共同結構元的一類逼近方法,先同時對所有核醣核酸序列進行 多重排比,再將排比結果的序列摺疊成二級結構。而進行多重排比的方法,最常 見的為 ClustalW,其不僅擁有長久的歷史,且其結果也優於許多其他類似的工 具。而摺疊的方法則是各有明顯的差異。
2.2.2.1 RNAalifold [Hofacker et al., 2002]
RNAfold 是維也納 RNA 研究團隊(Vienna RNA package)所開發的系統之一, 可預測出多條已排比好序列的一致結構,其原理為 Zuker-Stiegler 演算法的延伸, 摺疊結構時同時考慮最小自由能(MFE)和共變(covariation)關係。當資料有 N 條序 列,而最長序列長度為 n 時,本系統的時間複雜度為 O(N.n2 +n3 ),空間複雜度 為 O(n2 )。
2.2.2.2 Pfold [Knudsen B et al., 2004]
Pfold 使用隨機前後無關文法(stochastic context free grammar, SCFG),產生核醣 核酸結構的先前機率分配(prior probability distribution),針對輸入的已排比核醣核 酸序列和系統發生的樹狀結構(phylogenetic tree), 計算出此結構的後端機率 (posterior probabilities),而後進行行(column)的排比或行的配對。最後在 SCFG model 中找到最大可能發生樹(maximum-likelihood tree),產生最有可能的核醣核酸二級 結構。
2.2.2.3 ILM [Ruan J et al., 2004]
ILM(iterated loop matching)使用熱力學和相互資訊(mutual information)的結合 產生一個二級結構,接著產生所有可能的莖幹,根據熱力學和相互資訊的結合分 數對莖幹進行排序。選擇分數最高的莖幹,更新分數,然後將與被選上的莖幹有 衝突的莖幹移除,之後再選擇分數第二高的莖幹,接著一直重複此動作直到沒有 其他莖幹剩下,最後的所有莖幹則決定了結構。
2.2.3 同時考慮序列排比與摺疊的資訊來預測核醣核酸的共同結構元
Sankoff algorithm 是一種合併了做序列排比與做結構摺疊的動態程式規劃方 法,它可以被用來獲得排比結果和一致性的共同結構。而最原始的 Sankoff algorithm 實作雖然可以同時做結構摺疊與序列排比,但其負擔卻是相當的大,當 資料有 N 條序列而最長序列長度為 n 時,其運作所需的時間複雜度為 O(n3N ),空 間複雜度為 O(n2N )。因此,為了減少系統運作的負擔,則有了一些新的實作方法, 針對原始的 Sankoff algorithm 加了一些限制,而能在預測核醣核酸的共同結構元 時仍有不錯的表現。2.2.3.1 Foldalign [Gorodkin J et al., 1997]
Foldalign 可被視為一個區域性排比(local alignment)與鹼基配對數最大化 (maximum number of base-pairs) 演 算 法 的 混 合 體 , 它 使 用 了 與 CLUSTAL 和 CONSENSUS 相似的啟發式方法(heuristics),由兩條序列的排比與鹼基配對的關係 建立了分數矩陣(scoring matrix),使用由 Sankoff algorithm 延伸的動態程式規劃法 求出兩條最佳配對排比結果(pairwise alignment)。而系統將所有序列兩兩成對個別 求出其排比結果,從中取出分數最高的排比結果,再個別與其他序列進行排比, 從中再取出最好的配對排比結果,此時的配對排比結果即為三條序列的最佳配對 排比結果。之後再依此方法持續循環下去,最後所得即為所有序列的最佳配對排 比結果。
Foldalign 將 Sankoff algorithm 延伸實作,但限制了尋找的共同結構元最大長 度,而且禁止了多分支環狀結構(multi-loops)的產生,因此可以降低系統運作的負 擔。當資料有 N 條序列而最長序列長度為 n 時,其運作所需的時間複雜度為 O(n4
N)。
Foldalign 被專門設計來預測短區域的調控共同結構元,例如 IREs(iron response element)中的髮夾結構(hairpin structures),因此在找尋全域性(global)的結 構與多分支環狀結構上的表現不佳。
2.2.3.2 Dynalign [Mathews D et al., 2002]
Dynalign 結 合 了 自 由 能 最 小 化 (free energy minimization) 與 比 較 序 列 分 析 (comparative sequence analysis),依此找出兩條序列低自由能的共同結構。系統先 對兩條序列進行排比,再分別對兩條序列進行摺疊,而摺疊的結構其鹼基可以產 生配對的條件為:必須兩條序列在排比結果的同個位置上皆能產生標準鹼基對, 亦即使兩條序列可以摺疊成相同的結構。 Dynalign 的目的將整個系統的總自由能做最小化,總自由能的求法為:
G
total o ∆ 表示整個系統的總自由能,G
sequence o 1 ∆ 與G
sequence o 2 ∆ 分別為序列 1 與序列 2 的構造自由能(conformational free energy),∆G
ogap為兩條序列排比產生 的缺口(gap)造成的處罰值(penalty),此值根據經驗設置。Dynalign 使用全能量模型(full energy model),進行 Sankoff algorithm 的動態 程式規劃法對系統的總自由能做最小化,但在進行演算時則限制了兩條序列在進 行排比時的最大距離,即當序列 1 的第 i 個鹼基要與序列 2 第 j 個鹼基排比在一 起,則 i 與 j 的差值必須小於由使用者設定的 M 值。使用這樣的限制可以使系統 的時間複雜度降為 O(n3 M3),而空間複雜度則為 O(n2M2),其中 n 為較短序列的 序列長度。 Dynalign 只能同時找兩條序列的共同結構元,儘管可以擴展至多條序列,但 會造成系統的嚴重負擔,例如當序列數為三條時,系統的時間複雜度會增至 O(n3M6),而空間複雜度則增為 O(n2M4)。 由實驗的測試結果顯示,Dynalign 在較短的且較多樣性的 tRNA 預測上有比 較好的表現。
圖 3. Carnac 演算法三步驟範例圖
2.2.3.3 Carnac [Perriquet O et al., 2003]
Carnac 同時考慮區域相似性(local similarity)、莖幹能量(stem energy)和共變關 係(covariation),產生序列的共同摺疊二級結構。此系統採用啟發式的演算法,概 略圖如圖 3.,演算法步驟如下(設有 N 條序列): Step1: 對所有的序列分別找出每條序列所有可能的莖幹,再使用熱力學的知 識,利用動態程式規劃法計算出每條莖幹的自由能,留下能量低於預設門檻值的 所有莖幹。 Step2: 將所有的序列兩兩成對,分別建立所有可能的 N*(N-1)/2 個序列對成 對摺疊(pairwise foldings)。方法為先找出兩條序列鹼基高度相似的區域,考慮區 域相似性與共變關係找出成對的莖幹(pairwise stems),然後根據所選到的所有莖 幹,考慮能量最小化使用類似 Sankoff algorithm 的動態程式規劃法找出最佳的共 同摺疊。而此動態程式規劃法與 Sankoff algorithm 的差異點在於 Carnac 將序列的 莖幹視為基本單位元去運作,而不是像一般皆以含氮鹼基視為基本單位元。因此 找兩條序列的共同結構元的時間複雜度只需要 O(n2
),所需要的空間複雜度亦為 O(n2
Step3: 此步驟將 Carnac 擴展至可以同時找多條序列的共同結構元。在經過 step 2 之後,每條序列皆得到 N-1 個預測結構,為了得到最有可靠度(reliable)的莖 幹,於是建立了一套新的資料結構,稱之為莖幹圖(stem graph)。在莖幹圖中的所 有點(vertices)的集合表示所有序列預測出來的所有莖幹的集合,觀察序列 1 中的 任一莖幹 A 與序列 2 中的任一莖幹 C,若配對(A,C)出現在 step 2 產生的成對摺疊 中,則在莖幹圖中的點 A 與點 C 建立一致邊(identity edge)。觀察完每條莖幹,建 立完整的莖幹圖,再對莖幹圖中的每個連通單元(connected component)進行排序, 排序的依據則考慮各個莖幹圖的幾個拓撲特徵(topological features):(i)莖幹圖中節 點的數目,(ii)每條序列的莖幹數目,(iii)所有邊的總數,以及(iv)各個一致邊的數 目。最理想的情形是連通單元可以構成一個頂點數為 N 的完全圖(complete graph),而每個頂點皆來自於不同的序列。最後則根據排序好的最佳連通單元完 成其二級結構。 Carnac 在鹼基對的預測測試中發現其結果有很高的選擇性(selectivity),然而 它的敏感性(sensitivity)一般而言卻偏低,儘管可以藉由限制最小自由能的摺疊來 提高其敏感性,但如此一來則相對的會因此降低其選擇性,而相關性(correlation) 則有非常些微的提高。
2.2.4 對核醣核酸摺疊結構進行排比來預測核醣核酸共同結構元
當已知序列有可信賴的二級結構時,我們可以考慮藉由結構提供的資訊進行 多重結構排比,由此預測核醣核酸的共同結構元。而每一條序列的二級結構,可 以使用 2.2.1 節裡介紹的單一核醣核酸序列二級結構預測工具來取得,其中的 Mfold 與 RNAfold 皆常被各相關研究引進使用。2.2.4.1 RNAforester [Höchsmann M et al., 2003]
RNAforester 建立樹狀排比模型(tree alignment model),依此推論核醣核酸二級 結構的多重排比,只考慮核醣核酸分子的二級結構而不需要知道其序列的相似 性。系統使用其他單一核醣核酸序列二級結構預測工具將序列轉為二級結構,再 將預測的二級結構轉換成樹狀結構(tree)或森林結構(forest)的輪廓圖(profile),之後
將 ClustalW 多重序列排比的演算法延伸為多重結構排比,以此演算法對所有序列 轉換成的輪廓圖進行多重結構排比,由排比結果可得預測的核醣核酸共同結構 元。 當實驗的資料序列有 N 條,其平均的長度為 n,設 d 值為輪廓圖中的樹狀結 構節點的最大分支度(degree),則此系統運作的時間複雜度為 O(n2 d2 N2 ),空間複雜 度為 O(Nn+N2 +n2 d)。
2.2.4.2 MARNA [Siebert S et al., 2003]
MARNA 同時考慮核醣核酸一級序列與二級結構產生 RNA 的多重排比,它 建立了權重排比邊(weighted alignment edges)的集合,而這些邊的權重則反映了序 列的和結構的共通性(conservation),其計算方法須考慮到序列與結構兩部分,而 結構部份則參考由單一核醣核酸序列二級結構預測工具產生的預測結構。之後將 這些邊的集合輸入 T-coffee 系統,產生多重排比的結果,最後可從此結果擷取出 一致性的序列與一致性的結構。 當實驗的資料序列有 N 條,假設每條序列長度皆接近為 n,設 E 值為每條序 列所產生的預測結構個數(此值通常極小),則此系統運作的時間複雜度為 O(E2 N2 n4 )+O(N3 n2 )。
2.2.5 其他相關工具
以下介紹與本論文設計之系統較為相關的工具: 2.2.5.1 RNAshapes [Steffen P et al., 2006]RNAshapes 是一套使用抽象形狀(abstract shapes)表示法的軟體套件,其中包含 了三項核醣核酸的分析工具:形狀代表物分析(analysis of shape representatives)、形 狀機率計算(calculation of shape probabilities)、以及找尋一致性形狀(consensus shapes)。另外,RNAshape 亦包含了一些實用的特色:如使用正確的懸蕩能量 (dangling energies)找出摺疊次佳候選解、輸出二級結構圖形、找尋形狀的配對、 以及提供了圖形化的使用者介面。
RNAshapes 的主要核心是它的抽象形狀表示法,即使用 RNAcast[Reeder J and Giegerich R, 2005]工具將二級結構轉為抽象形狀(shapes),轉換方法則是將目前常 用來表示二級結構的點-括號(dot-bracket)表示法根據 RNAcast 定義的語言轉換 成形狀的抽象表示法。如以下為點-括號表示法: 根據其語言定義,將連續對稱的圓括號(“("and“)")以方括號(“["and “]") 表示,而連續的未配對區域則以底線(“_")表示,以下則為其轉換結果: 此為最低層級的 type 1 抽象形狀表示法。為了提高其抽象的層級,於是將未配對 區域拿掉,而連續巢狀(nested)的方括號則合併,以上兩例皆可轉換成 type 5 的抽 象形狀表示法,如下: RNAshapes 則利用此種抽象形狀的表示法應用到 RNA 的相關研究中。以下 列舉與本研究相關的兩項: 形狀摺疊(shape folding) 對單一核醣核酸序列,考慮其自由能摺疊出能量最小的結構,並且可以輸出 次佳的數個候選,以能量低的結構為排序優先。而 RNAshapes 的特點則是在輸出 候選時限制每種形狀(shape)的組成只能出現一次,而其輸出則是以該形狀能量最 小的結構為代表。
一致性形狀(Consensus shapes)
RNAshape 所提供的找尋一致性形狀的功能則為 RNAcast 原本有的功能。對 於同一家族的核醣核酸序列,RNAcast 對其中每條序列分別進行摺疊,列出接近 最小能量的各種抽象形狀,存於一個抽象形狀空間(abstract shape space),再從所 有序列產生的抽象形狀空間中找出交集,取出其共同的抽象形狀,則為此家族的 共同一致性形狀。
2.3 核醣核酸資料庫
由於核醣核酸的相關研究蓬勃發展,已知的核醣核酸序列及結構資料量快速 地成長,於是有許多相關的生物資料庫收集了分散在各個文獻的資料,以各自設 計的方法系統化地將核醣核酸的資料分門別類整理,公開提供給所有相關的研究 人員使用。目前核醣核酸相關的資料庫有許多,以下則簡單介紹幾個常用的資料 庫。2.3.1 Rfam [Griffiths-Jones et al., 2003]
(http://www.sanger.ac.uk/Software/Rfam/)
Rfam(RNA families database of alignments and CMs)資料庫儲存了多數家族的核 醣核酸資料,包含家族成員各自的鹼基資料、家族的多重序列排比結果、以及家 族二級結構的共同結構元等,是一個廣泛被使用的資料庫。其中每個家族的排比 結果分為兩組資料,"SEED"集合裡的排比結果是以手動的方式完成,參與排 比的序列有高度的相似性;而另一組"FULL"集合裡的排比結果則是以共變模 型(covariance model, CM)的方法[Eddy SR and Dubrin R, 1994]所產生。
2.3.2 The RNase P Database [Brown, 1999]
(http://www.mbio.ncsu.edu/RNAseP/home.html)
序列摺疊的二級結構、以及部分序列的 3D 摺疊立體結構。其中的序列與二級結 構則以生物有機體做為分類,每條序列皆有連結可連至 NCBI(National Center for Biotechnology Information)網頁查詢較完整的資訊,而二級結構則有 4 種檔案格 式可供使用。
2.3.3 tRNA Compilation 2000 [Sprinzl et al., 1996]
(http://www.staff.uni-bayreuth.de/~btc914/search/)
資料庫收集了大量的轉錄核醣核酸序列,亦包含了明確的結構資訊。資料庫 中提供了查詢的功能,可在 11 個界(kingdom)中選擇適當的分類,再從界之下的 有機體(organism)分立中做選擇,最後再查詢是攜帶哪一種胺基酸的轉錄核醣核 酸,這比較適合有生物背景的使用者使用。
2.3.4 RAG [Gan HH et al., 2004; Fera D et al., 2004]
(http://monod.biomath.nyu.edu/rna/rna.php)
RAG(RNA-As-Graphs web resource)是一個存放 RNA 二級結構的資料庫,利用 圖學理論(Graph Theory)的結果,提供了一個量化的方法可以對 RNA 二級結構的 拓撲(topology)進行分類,相較於其他 RNA 的資料庫,RAG 容易用於比較相異二 級結構的相似與相異處。
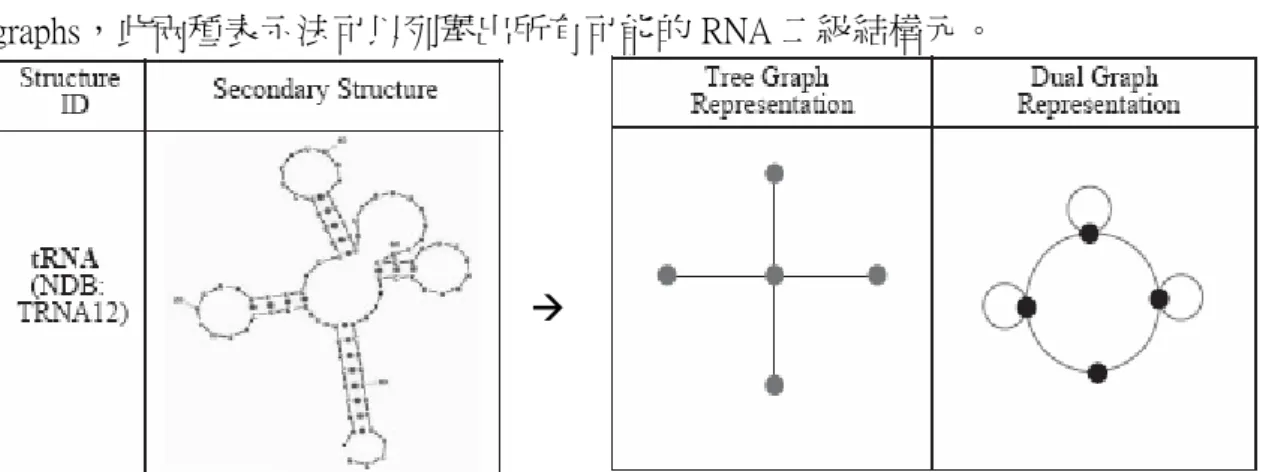
RAG 提供了兩種二級結構拓撲的表示法:RNA tree graphs 及 RNA dual graphs,此兩種表示法可以列舉出所有可能的 RNA 二級結構元。
Æ
RNA tree graphs:
將突起結構與所有環狀結構都視為一個點(vertex),而莖幹結構則視為一個邊 (edge),如此便能將一個 RNA 二級結構表示成一個 RNA tree graph。但此表示法 無法表示擬結結構。
RNA dual graphs:
將莖幹結構視為一個點,而突起結構或環狀結構的單股(single strand)則視為 一個邊,如此便能將一個 RNA 二級結構表示成一個 RNA dual graph。此表示法可 以表示所有可能的 RNA 二級結構,包含了擬結結構。而 RAG 的接續下來的研究 [Kim N et al., 2004]也都著墨在 RNA dual graphs 的特性。
RAG 提供了圖學中圖形拓撲的表示法,然而擁有同一種拓撲的相異 RNA, 其二級結構還是很有可能會有很大的不同,因為缺少了每個點跟邊的長度資訊, 即使在每個點跟邊都只有些微差異的情況下,累積起來的差異依舊不小,這對尋 找 RNA 二級結構的共同結構元影響頗大。這是目前 RAG 的圖形拓撲表示法較為 不足的地方。
2.3.5 RNABase [Murty et al., 2003]
(http://www.rnabase.org/)
RNABase(The RNA Structure Database)資料庫整合了 Protein Data Bank(PDB)與 Nucleic Acid Data Base(NDB)兩者的核醣核酸資料,再依功能與結構的不同來做分 類。此資料庫的主要特色是能提供核醣核酸的 3D(three-dimensional)結構圖,另外 還能執行結構的分析與檢測。
2.3.6 SCOR [Klosterman et al., 2002; Tamura et al., 2004]
(http://scor.lbl.gov/)
SCOR(Structural Classification or RNA)提供了核醣核酸共同結構元的階級分 類,分別以生物功能、二級結構元和三級立體結構為依據,提供了三種不同的 分類方法。而生物功能類別則分別以分子功能、結構元功能與結構模型向下細 分;二級結構元類別則分類成髮夾結構和內部環狀結構,而各類別底下再依據結 構的形狀做更小的細分;三級立體結構類別則以各種形狀不同的相互作用作為細 分的依據。
2.3.7 其他常見資料庫
PseudoBase [Batenburg et al., 2000]
(http://wwwbio.leidenuniv.nl/~Batenburg/PKB.html)收集了擬節結構的核醣核酸相關 資料,包含了序列、結構與生物功能三類資訊。
5S ribosomal RNA database [Szymanski, 2002] (http://rose.man.poznan.pl/5SData/) 專門為 5S 的核醣體核醣核酸所建置,提供了這些序列的排比資訊與二級結構。 另外也提供了與這些核醣核酸結合蛋白質資訊。
miRBase(http://microrna.sanger.ac.uk/)收集了微核醣核酸序列,可依物種分類瀏 覽,此資料庫亦包含了各微核醣核酸的先質(precursor),也有提供搜尋介面,使 用者可根據序列片段、編號或名稱進行搜尋。
第三章、研究方法
3.1 系統設計目的與概念
當核醣核酸折疊成二級結構時,其莖幹的兩股在序列上的位置可能相距很 近,也可能相距很遠。若共同結構元的莖幹其兩股在序列上相距太長,在先前的 相關研究則很不容易找到。如下圖 5 所示,這是 RNaseP 家族的結構示意圖假設 它的共同結構元是方框所為起來的莖幹(P5, P7),由於該莖幹後面還有很長的一段 序列,並且折疊成數個莖幹結構(P8, P9, P10, P12),以先前定義的結構描述語言無 法單獨表示 P5, P7 所形成的莖幹。因此我們設計了一套新的結構描述語言,以圖 論中的圖形(Graph)來表示,則將使莖幹兩股在序列上的距離所造成的影響大大減 少。 圖 5. RNaseP 家族的結構示意圖本系統的目標在預測核醣核酸家族的共同結構元,當核醣核酸轉換成圖形, 找出共同結構元的目標則轉換為類似圖論中找出共同子圖的問題,此問題的難度 為 NP-Complete,會有相當大量的搜尋空間,將會需要大量的搜尋時間,因此我 們採用演化式計算(evolutionary computing)中的基因規劃法(genetic programming)來 減少系統的搜尋時間。 在演化式計算中,有個良好的演化起點可以加快搜尋時間,並且可以使得搜 尋結果容易趨向最佳解,因此我們採用現有的單一核醣核酸序列二級結構預測工 具做為前處理器,這些預測工具考慮了最小自由能,個別產生每條序列可能的結 構,我們再從這些結構中擷取子圖,以做為演化中初代的個體(individual)。
3.2 核醣核酸結構描述語言
本研究針對核醣核酸的二級結構設計一套圖形表示的描述語言,語言的定義 如下:1. 以 RNA dual graph 的概念表示核醣核酸的二級結構,以二維陣列儲存 RNA dual graph 的資訊,其中包含了結構拓樸及結構長度的資訊。 2. 結構拓樸中的莖幹辨別是根據前置器的設定而得到,不一定只是 C≣G、 A=U、G-U 三種鹼基配對。 3. 一個莖幹中可包含特定長度以下的內部環狀結構或突起結構,長度大小 可由使用者自由設定,系統預設值為 1。 4. 莖幹結構與每個環狀結構分別給一個長度範圍,表示該結構元所涵蓋的 結構大小。 5. 內部環狀結構或突起結構的長度不包含在莖幹長度中。 6. 環狀長度的結構可能為零,供區別相鄰的兩個莖幹。 透過上述定義,任何的二級結構能輕易的轉換為描述語言,以下舉例說明, 下頁圖 6 為 DF4600(tRNA)執行 Mfold 後所輸出的第一個候選結構:
圖 6. DF4600(tRNA)由 Mfold 產生之第一候選結構
我們將此結構圖轉為 RNA dual graph 的概念圖,所得的概念圖如下頁之圖 7。 轉換的方法則先分別將每個莖幹結構皆視為一個圖形(graph)的點(vertex),而上圖 中有四個莖幹,則可以轉換成四個點。上圖中的‧為 RNA 的 5'端,我們從 5' 端出發往 3'端走,根據先後遇到的莖幹分別命名為莖幹 1、莖幹 2、莖幹 3 與 莖幹 4,而其建立的對應點則為點 1、點 2、點 3 以及點 4。由於莖幹 1 到莖幹 2 之間有環狀結構,故建立一條由點 1 往點 2 的邊(edge);莖幹 2 到莖幹 3 之間有 環狀結構,故建立一條由點 2 往點 3 的邊;而從莖幹 3 走髮夾環狀結構回到莖幹 3 本身,故在點 3 建立一條自身邊(self-edge)回到點 3 自己。莖幹 3 走環狀結構到 莖幹 2,然後走環狀結構到莖幹 4,再走髮夾環狀結構回到莖幹 4 本身,於是我 們建立從點 3 到點 2 的邊、點 2 到點 4 的邊以及從點 4 走回到點 4 的自身邊。而 從莖幹 4 到莖幹 1,雖然其中沒有環狀結構,但為了區別莖幹 4 與莖幹 1,我們 建立了一條從點 4 到點 1 邊,雖然其長度為零,但仍視為存在此邊。
圖 7. 由圖 6 轉換之 RNA dual graph 根據此概念圖,我們欲建立一個資料結構來表示此圖,在圖學上,習慣以鄰 接串列(adjacency-list)表示有向圖(directed graph),如下圖 8 所示,最左邊的陣列依 照索引(index)分別表示點 1、點 2、點 3 與點 4,而陣列中每個點右邊的鏈結,其 內容表示該點可連結到的點,而內容為 0 則表示此點亦為 3'端終點。在此圖表 示點 1 可以連結到點 2,而點 1 亦為終點;點 2 根據由 5'端到 3'端的走法,第 一次走到點 2 時接下來則連結到點 3,而第 2 次走到點 2 時則連結到點 4;點 3 的第一個連結則是回到點 3 自己,第二個連結則是到點 2;點 4 的第一個連結則 是回到點 4 自己,第二個連結則是到點 1。如此便可由此鄰接串列清楚的表達其 RNA dual graph 的概念圖。
然而 RNA dual graph 的缺點便是缺少了點跟邊的長度資訊,我們將此鄰接串 列擴充,根據前面的二級結構圖,把點跟邊的長度資訊納進資料結構中,所得新 的鄰接串列如下圖 9。 圖 9. 將圖 8 的表示法擴充之鄰接串列表示法 最左邊的陣列內容表示該點(即莖幹)的長度,而其中會有兩個值則指的是長 度範圍,[X,Y]表示其長度最小為 X,長度最大為 Y,在此的鄰接串列表示的是一 個確切的結構,因此 X 與 Y 值會相同。由於我們的資料結構是為了找出共同結 構元所設計,而共同結構元意指會出現在序列上的相似結構,所以我們將長度擴 展為長度區間來表示長度範圍,如下頁圖 10 中表示,例如點 1 的長度範圍變為 [6, 8],以較有彈性的資料結構來進行基因規劃法。 而陣列的第一個鏈結(link)內容表示到下一個點的邊(即環狀結構)的長度範 圍,陣列的第二個鏈結內容則表示第一個可連結的點。第三個鏈結內容表示到達 第二個連結點的邊的長度範圍,第四個鏈結內容則表示第二個連接點。在上圖 中,取陣列中索引為 2 的點舉例說明:陣列中的值為 5,表示點 2(即莖幹 2)的長 度為 5;第一個鏈結內容為 8,第二個鏈結內容為 3,表示點 2 可以經過長度為 8 的邊到達點 3;第三個鏈結內容為 1,第四個鏈結內容為 4,表示點 2 可以經過 長度為 1 的邊到達點 4。
圖 10. 將圖 9 表示法中各點與邊隨機擴展長度後之表示法 由於在實作時需要不斷的取出資料結構中的值來使用,在演化過程中亦會不 斷的改變資料結構中的內容,而鄰接串列中的鏈結串列在資料存取時較為不便, 需花費很多時間,也需要額外的空間來做鏈結。因此我們想到利用陣列有隨機存 取(random access)的優點,以陣列(array)模擬鄰接串列實作。 以下圖 11 的陣列表示法即模擬實作了鄰接串列,相對的位置則代表著相同 的意義。陣列中的每一列儲存著與每一個點相關的資訊,例如從第二列,我們可 以知道:點(莖幹)2 的長度範圍為[3, 7],在經過長度範圍為[6, 10]的邊(環狀結構) 可以到達點 3,而點 2 亦可由經過長度範圍為[1, 4]的邊到達點 4。 圖 11. 以矩陣表示法表示圖 10 中之鄰接串列
3.3 系統主架構
下圖為本系統的主要流程圖,系統先讀入使用者輸入的序列及系統參數,根 據輸入序列產生背景序列,再以預測工具對輸入序列與背景序列產生個別的二級 結構,之後分析結構,將這些結構轉為本系統所使用的描述語言。接著進行系統 核心的基因規劃法,由演化的方法預測出核醣核酸的共同結構元,再由後處理對 預測的結果做修正,最後輸出共同結構元。 在以下的各節將做更詳細的說明: 圖 12. 系統流程圖3.3.1 輸入序列及系統參數
一開始讀入使用者輸入的核醣核酸序列以及系統參數,使用者可以使用系統 預設的參數,若使用者對於輸入的核醣核酸序列的生物背景有些了解,則可以藉 由自行設定參數來使實驗達到更好的效果。 使用者可以自行定義的參數包含系統能預測的莖幹數目最大上限、莖幹內允 許環狀結構的大小、負面背景資料的產生倍數等,以及在執行基因規劃法時所需 要的各種參數,如族群的大小(population size)、一個個體(individual)產生突變 (mutation)的機率、兩個個體進行互交(crossover)的機率、個體產生重製(reproduction) 的機率,還有系統進行演化的代數(generation)等。 而輸入系統的核醣核酸序列檔案以 FASTA 格式表示,檔案內容第一行以大 於( > )符號開始,後面則接第一條序列名稱與註解,第二行則為此序列的內容, 以各鹼基所組成的序列表示。第三、四行以同樣表示法表示第二條序列,第五、 六行以同樣表示法表示第三條序列,後面各序列皆類似。以下為 ctRNA_pND324 家族序列的一部份: 圖 13. FASTA 格式範例3.3.2 產生背景序列
根據本研究的假設,具有生物意義的二級結構元不會任意出現在隨機產生的 序列中,因此我們採用監督學習法(supervised learning),藉由使用負面背景資料來 抑制太過普遍的結構。負面背景資料的產生方法則依據輸入序列的資料產生,負面背景序列的長度 模擬輸入序列的長度,數量則以使用者輸入的倍數參數做決定,設為一至三倍。 當背景序列倍數設為三倍時,第一到三條背景序列的長度會取輸入序列第一條的 長度,第四到六條背景序列的長度會取輸入序列第二條的長度,依此類推。 由於已知自然界中的核甘酸序列,相鄰的鹼基對之間是有相關性的,因此使 用一級(first order)序列產生法產生背景序列的鹼基,亦即每一條序列的第一個鹼 基是由四個鹼基個別出現的機率來決定,之後的每個鹼基則必須考慮前一個鹼基 的種類,由條件機率決定出現的鹼基。
3.3.3 預測二級結構
目前現有許多可以將序列折疊成二級結構的預測工具可供使用,我們可將輸 入的序列及系統產生的背景序列分別輸入這些工具來預測二級結構,再取得每條 序列產生的數個候選結構。 當使用 Mfold 做為我們的前處理器時,Mfold 輸出的檔案有數種檔案格式, 而我們取其中的 ct 檔來使用,下頁圖 14 中為 ctRNA_pND324 家族中的 AL592102.1 序列片段,經由 Mfold 預測出來的第二個預測結構的 ct 檔。 在 ct 檔中第一列分別表示序列長度、結構能量、以及序列名稱,之後的第 一行表示索引(index)、第二行為序列的鹼基、第五行則為鹼基配對的所在位置, 我們可由下頁圖中看出有(7, 19)(8, 18)(9, 17)(10, 16)四個連續的鹼基對所形成的一 個莖幹。 而在本研究的實驗中,為了方便與其他相關研究做比較,因此不限於使用 Mfold 做為前處理器,亦可能使用其他的序列折疊預測工具。3.3.4 分析二級結構及轉換為描述語言
在此步驟則將前處理器產生之二級結構轉換成本系統的描述語言,讀入的檔 案預設為 ct 檔,若前處理器的輸出不是 ct 檔則必須先進行轉換。系統根據 ct 檔 中的鹼基配對資訊,研判產生莖幹結構長度、環狀結構長度,以及莖幹結構與環 狀結構之間的相對關係。 在 ct 檔中,我們可以看出鹼基之間的配對關係,當遇見連續有配對的鹼基, 若其配對的鹼基也連續,則將這些連續的鹼基與其配對的鹼基視為一個莖幹結 構;若配對的鹼基不連續,則會產生兩個以上的莖幹。當遇見不連續的鹼基,則 視為環狀結構。 例如系統可以根據上頁圖 14 中序列 AL592102.1 的 ct 檔,得到如下的資料結 構表示法: 圖 15. 由圖 14 所得之資料結構表示法 其對應的概念圖為: 圖 16. 對應圖 15 之概念圖而此結構的摺疊形狀如下: 圖 17. 序列 AL592102.1(ctRNA_pND324)由 Mfold 產生之第二候選 觀察此結構,其中存在有兩莖幹間的內部環狀結構長度為 1 的情形,另外還 有莖幹長度為 2 的情形發生。我們在系統的設計上,可以忽略以上的情形,也就 是說,將內部環狀結構長度為 1 的兩相鄰莖幹視為一個莖幹,以及將長度小於 3 的莖幹拆開成環狀結構。如此一來,使得整個結構的莖幹數減少而降低了複雜 性,亦使本系統增加了些彈性,可以找出差異性較大的共同結構元。 當略過上述的情形後,上頁圖中的概念圖可以簡化為如下: 圖 18. 簡化後之概念圖
上頁圖 18 的概念圖對應的資料結構表示法則為:
圖 19. 對應圖 18 之資料結構表示法
亦即將頂點數為 11 個的圖形資料結構簡化成 8 個點。
3.4 基因規劃法
基因規劃法源自於基因演算法(genetic algorithms, GA),為 John Koza 在 1992 年首先提出,基本架構遵循演化式計算的精神,模擬生物演化過程中「物競天擇, 適者生存」的概念,自隨機產生的初代個體中,透過突變、互交、複製等演化過 程,並且經過與其他個體的互相競爭,逐漸演化留下個體適應度高的個體。經由 一代一代的演化,最後留下來適應度最高的個體,即代表了此問題領域的最佳可 能解。 基因規劃法分為五個主要部份: 1. 產生初代個體 (initial population)、 2. 適應函數 (fitness function)、 3. 母代挑選機制 (selection)、 4. 演化運算子 (genetic operator)、 5. 終止條件 (termination criterion)。 系統從得到的二級結構中擷取初代個體,計算所有個體的適應分數,透過挑 選機制產生母代,並經由演化運算子得到子代個體,直到個體數目達到族群數 量。再如此反覆的一代一代得進行演化,直到滿足終止條件為止。最後則輸出最 佳的幾個個體。
本系統的基因規劃法流程如下: 圖 20. 基因規劃法流程圖 以下各小節將詳述故部分的實作方法:
3.4.1 產生初代個體
在演化的過程中,有個良好的演化起點可以加快搜尋的效率。由於共同結構 元表示會出現在各序列上的相似結構,因此我們從輸入序列的候選結構中擷取子 結構來做為第一代。而取子結構的方法,則相當於在候選結構的 RNA dual graph 概念圖中取出子圖一樣,先在圖中隨機取一個頂點,然後隨機沿著邊走向其他頂 點,然後將走過的頂點與和這些點相關的邊取出來構成子圖,並將子圖調整成一 個合理 RNA dual graph。最後再將子結構中的每個長度隨機往外拓展,形成一長 度區間。圖 21. 產生初代個體:左圖為候選結構,中圖為隨機取得之子結構,右圖則為將中圖補上適當邊 之後所得之合理的 RNA dual graph。
我們取前面候選結構的概念圖(如上圖 21 左圖)來做例子,假設我們隨機取到 的點為頂點 3,而從頂點 3 出發,沿著邊隨機走的路徑為:3Æ7Æ8Æ8Æ2Æ3, 依此路徑取得了子圖(如上圖 21 中圖),接著補上應該有的邊,並計算其長度,再 重新調整頂點編號(如上圖 21 右圖),得到一個合理的 RNA dual graph。則此子圖 的資料結構表示法如下:
圖 22. 對應圖 21 右圖之資料結構表示法
其表示的子結構如下圖方框部份。
最後我們將此子結構中的每個長度隨機向外拓展,所得即為演化的初代個 體,如下圖 24 所示。 圖 24. 將圖 22 表示之結構隨機拓展長度之資料結構表示法
3.4.2 適應函數
適應函數是演化式計算中最核心的部份,不同的適應函數會影響整個族群演 化的方向,因而影響到最後演化的結果。好的適應函數不僅能將族群個體發展導 向正確的演化方向,亦能減少不必要的搜尋,縮小搜尋空間,提升搜尋的效率及 品質。 本研究所定義的適應函數包含了兩部份:一部分是能兼顧正確率(precession) 與擷取率(recall)的 F-score,另一方面則是考慮配對的鹼基對數的影響。 在相關的研究顯示,若為了增加該實驗的正確率則會使得擷取率下降,若為 了提高擷取率則會犧牲部分正確率,因此要同時考慮兩者來設計適應函數才能確 保演化過程是走向正確的方向。而 F-score 則是取正確率與擷取率的調和平均數, 預期當正確率與擷取率分數都高時才會拉高整體的分數,這不會因為只有某一個 值的增加就過度拉高整體的分數。 F-score 定義如下:C
N
M
M
C
M
N
M
M
I
F
+
+
=
⎥
⎥
⎥
⎦
⎤
⎢
⎢
⎢
⎣
⎡
+
+
=
2
1
)
(
1
2
1
1
)
(
其中M 代表輸入序列中,包含結構元I的個數;N 代表背景序列包含結構元I 的個數;C 為輸入序列的總個數。 N M M + 則表示正確率, C M 表示擷取率。 然而背景資料對於較大的共同結構元影響不大,當 F-score 相近時,我們會 傾向選取結構較大且較完整的個體,亦即會傾向找鹼基配對數較多的結構。因此 定義P(I)表示鹼基配對數所產生的影響,也就是將結構元 I 符合序列的鹼基數除 以鹼基配對最多的結構的鹼基數,如此可以使P(I)正規劃在 0 到 1 之間。 本系統的 fitness function 則定義為: 其中α為一個介於 0 到 1 之間的值,當α愈大時,系統會偏好被更多輸入序列包 含的結構元,但可能受到雜訊的影響,只能找到共同結構元的子結構。而當α愈 小時,則系統會偏好找到鹼基配對數較多的結構,但很有可能受到少數幾條序列 的影響,找到的結構只符合少數幾條序列,而非是全體的共同結構元,當輸入序 列中每條序列的長度差異性較大時,此類的影響更為明顯。 而在本系統的測試中,當α設定為 0.7 時,可以滿足絕大部份的資料。
3.4.3 母代挑選機制
產生新一代個體時,我們先保留一定比例適應分數高的個體到下一代,剩餘 的個體則透過挑選機制選出母代,執行演化運算後產生新一代的個體。本系統採 用的挑選機制為競賽法(tournament),競賽法可以保留隨機的機制,也符合適者生 存的原理,是目前最普遍採用的選取方法。其挑選的方法為:自母代群體中隨機 挑選出一定數量的個體,個體之間彼此比較適應分數,其中分數最高者被挑選出 來作為母代個體,進而產生子代。).
(
)
1
(
)
(
)
(
I
F
I
P
I
fitness
=
α
×
+
−
α
×
3.4.4 演化計算子
以下各小節介紹本研究所使用的各演化運算子: 3.4.4.1 突變(mutation) 突變包含了長度突變與結構突變:長度突變是在挑選的結構中,隨機挑選其 概念圖中的一個點或一個邊,然後改變其長度範圍。 而結構突變則是隨機改變挑選結構的結構形狀,或是在原本結構多加一個點 其對應邊。 Æ 改變結構 Æ 加點及對應邊 3.4.4.2 互交(crossover) 隨機選擇兩個結構,各選擇其中一個點,依此為依據,交換部分子結構,其 概念圖如下。 Æ Æ 互交不僅可以改變結構拓璞,莖幹數也隨之改變,而當挑選到的莖幹恰好是 共同結構元上的不同子結構時,透過互交將兩者結合起來,可以使跳脫區域最佳 解的機會大增。3.4.4.3 重製 除了保留適應分數高的個體外,我們也保留一些機會讓母代能完整保留下 來,以增加族群的多樣性。 3.4.4.4 清除重複個體 當演化到一定程度後,可能會往某幾個區域最佳解逼近,而使整個族群充斥 著特定的個體。而族群變異度太小,缺乏多樣性,容易使演化結果侷限在區域最 佳解,因此我們每五代之後,會將過多的重複個體清除,而再以產生初代個體的 方法,隨機產生新的個體,補足刪除掉的個體。
3.4.5 終止條件
通常系統在 20 代內即能收斂到不錯的結果,並且考量時間因素,我們將中 止條件設定在演化 20 代。3.4.6 後處理
我們透過基因規劃法找出找到輸入序列的共同結構元後,找出共同結構元在 每條序列上的位置,然後考慮配對的莖幹能否往外或往內延伸幾個鹼基對。另外 再針對沒被找出結構的序列,搜尋判斷是否可能有共同結構元的存在。最後再將 完整的結果輸出給使用者。第四章、實驗
4.1 實驗評估標準
與目前多數預測二級結構的研究一樣,我們採用 Matthews 的相關係數 (Matthews correlation coefficient)來做為評估的標準。其原始定義如下:
其中 Pt為正確正預測的總鹼基對數(true positive),即系統預測到的鹼基對也 出現在正確答案中的鹼基對數;Pf為錯誤正預測的總鹼基對數(false positive),即 系統預測到的鹼基對沒有出現在正確答案中的鹼基對數;Nt為正確負預測的總鹼 基對數(false negative),即沒有出現在預測結果上也沒有出現在正確答案上的鹼基 對數;Nf為錯誤負預測的總鹼基對數(false negative),即沒有出現在預測結果上卻 有在正確答案上出現的鹼基對數。 化簡後的式子如下: 其中 表示系統的精確率(precession),又稱為選擇性(selectivity), 而 表示系統的擷取率(recall),又稱為敏感性(sensitivity)。 f t t f t t
P
P
P
N
P
P
C
+
+
≈
)
)(
)(
)(
(
t f t f t f t f f f t tP
P
N
P
P
N
N
N
N
P
N
P
C
+
+
+
+
−
=
f t tN
P
P
+
f t tP
P
P
+
4.2 實驗測試資料
本研究使用多種核醣核酸家族作為實驗測試的資料,包含過去文獻常用來評 估的資料,以及其他各種多樣化的資料。簡介如下: S. cerevisiae tRNA-PHE tRNA-PHE 為攜帶胺基酸 PHE 的轉移核醣核酸,其共同結構元包含四個莖幹 結構,形狀像是四瓣的苜蓿葉,這對於使用熱力學來進行結構摺疊的方法是一種 比較困難的資料。實驗測試的資料來自於先前的文獻[Griffiths-Jones et al.,(2003), Sundaralingham & Rao,(1975)],其中包含兩組資料:一組為高相似的序列,其序列相似度為 84.4%, 此組資料為 11 條序列,序列長度皆為 73;另一組為中相似度的序列,其序列相 似度為 60.0%,此組資料為 11 條序列,序列長度皆為 75。 E. coli RNase P RNase P 是一種普遍存在內切核醣核酸(endoribonuclease),家族成員在序列與 結構上皆有相對比較高的多變性,在測試的資料中含有 11 條莖幹,其中還包含 了一個擬結結構。
實驗測試資料來自於 The RNase P Database[Brown, 1999],其中包含兩組資 料,一組為高相似性的資料,其序列的相似度為 81.5%,此組資料內有 9 條序列, 其平均長度為 335,其中五條序列則將原始資料在 5'與 3'兩端切去一部份以增 加測試資料的困難性,另一組資料為中相似度資料,其序列的相似度為 67.1%, 內有 11 條序列,平均長度為 359,。
Hepatitis C virus 3'X element
HCV_X3 家族為 C 型肝炎病毒,其共同結構元包含三條相鄰的莖幹,其中一 條莖幹有長度為 1 的突起(bulge),第二條莖幹有長度為 3 的突起,第三條莖幹則
分別在莖幹兩股不對稱的位置各有一個長度為 1 的突起。實驗的測試資料從 Rfam 的 HCV_X3 家族中取出 seed 裡的 16 條序列,其平均長度為 100。
ctRNA
ctRNA(counter-transcribed RNA)可嵌入 RepB 蛋白質的信息核醣核酸(mRNA) 中,而會產生轉譯抑制的功能。本實驗測試的資料為 Rfam 資料庫裡的 48 條 ctRNA_pND324 序列,平均長度為 85,其共同結構元為兩個相鄰的莖幹,其中的 一個莖幹比較有變化性,在部分序列中含有突起或是內部環狀結構,而此莖幹在 每條序列中的長度為 5~10 不等。 本實驗還準備了另一組測試資料,將原本的 48 條序列根據真實資料分別向 兩端延長,左右延長的長度隨機但兩端延長總和為 100,讓此組資料更有變化性, 而更難以預測。 Purine riboswitch Purine 的共同結構元為三分支的環狀結構,而整個家族中有數條序列的莖幹 存在長度為 1~2 的未配對鹼基,而莖幹長度在每條序列中也有些變化。我們測試 資料來自於 Rfam 資料庫,取自 seed 裡的 35 條序列,並且根據真實資料向序列 兩端延長,延長長度為隨機,但限制總和為 100,而其中兩條序列未延長,可測 試系統是否會略過此兩條長度較短的序列。
Enterovirus cis-acting replication element
Entero_CRE 的共同結構元為 1 個莖幹的髮夾結構,其中包含了一個不對稱的 內部環狀結構,兩端的環狀結構長度分別為 1 與 2。而此實驗的測試資料取自於 Rfam 的 Entero_CRE 家族中的 56 條 seed 的序列,並且根據真實資料向其兩端延 長,延長長度總和為 100~300 不等,全部序列中長度最短為 111,最長為 361, 平均長度為 262。
mir-160 microRNA precursor family
的內部環狀結構,而其髮夾環狀結構也相對的長了許多,可用來測試本系統對於 兩股相距較長之莖幹的預測能力。此實驗的測試資料取自於 Rfam 的 mir-160 家 族中全部(full)19 條序列,全部序列中長度最短為 85,最長為 136,平均長度為 97。 所有測試資料整理如下表: 資料名稱 序列數量 平均長度 結構元莖幹數目 tRNA(high) 11 73 4 tRNA(med) 11 75 4 RNaseP(high) 9 335 11 RNaseP(med) 11 359 11 HCV_X3 16 100 3 ctRNA 48 85 2 ctRNA_ext. 48 185 2 Purine 35 220 3 Entero_CRE 56 262 1 mir-160 19 97 1 表 1. 測試資料整理表
4.3 實驗結果
本系統以 C 語言實作,測試環境的作業系統為 Mandrake Linux 10.1,電腦配 備為 Pentium IV 3.2GHz 的中央處理器與 2Giga-bytes 的記憶體。 所有實驗基因規劃法的參數:族群數量為輸入序列之 100 倍,結構突變率為 45%,長度突變率 45%,互交率 5%,重置率 5%。結構部份不多加限制,但有濾 掉長度小於 3 的莖幹以及忽略莖幹內長度為 1 的環狀結構。 實驗所使用的前置器為 RNAshapes 的 folding 功能,實驗結果的數據為實驗 數次所取得的平均值,每次的實驗皆以 linux 系統時間做為隨機種子(random seed),可確保每次實驗不會重複。4.3.1 與 RNAshapes 比較
由於 RNAshapes 與我們的系統皆是以各序列個別摺疊的結果作為找尋核醣 核酸共同結構元的出發點,而且 RNAshapes 在部分的實驗測試上表現不錯,因此 以 RNAshapes 來與我們的系統做比較。 本系統(FGGP)與 RNAshapes[本文 2.2.5.1 小節]的實驗結果比較如下表: dataset RNAshapes FGGPSelectivity(%) Sensitivity(%) MCC(%) Selectivity(%) Sensitivity(%) MCC(%) tRNA(high) 33.61 34.63 34.12 88.53 83.55 86.00 tRNA(med) 78.81 80.52 79.66 83.40 84.85 84.12 RNaseP(high) NA NA NA 81.34 16.59 37.64 RNaseP(med) NA NA NA 49.76 13.73 26.13 HCV_X3 52.29 20.35 51.31 97.40 79.33 87.40 ctRNA 70.23 90.53 79.74 79.96 87.61 83.70 ctRNA_ext. 61.07 73.31 66.91 71.57 66.67 69.07 Purine 46.12 56.19 50.91 82.58 91.02 86.70 Entero_CRE NA NA NA 64.64 70.59 67.55 mir-160 63.21 98.67 78.97 85.27 97.56 91.21 表 2. RNAshapes 與 FGGP 的實驗結果比較表 在表 2 中,selectivity 表示在預測的結果中預測正確的比率,sensitivity 表示 在正確答案中有預測出來且正確的比率,MCC 則表示 Matthews 相關係數[本文 4.1 小節],而表中的 NA 表示 RNAshapes 無法產生結果。 在表 2 中可以看出 RNAshapes 對部分測試資料表現極差,這可以歸咎於 RNAshapes 的前處理器對這些資料的表現不佳,RNAshapes 對於其前處理性的依 賴性相當大,因此結果受前處理器的影響相當大。而我們的系統 FGGP 雖然也會 受到前處理器的影響,但於由於有演化的過程,因此對前處理器的依賴性不會太 大。在以上的各組測試資料中,我們的系統(FGGP)所得到的結果皆比 RNAshapes 好,而在 tRNA(high)、tRNA(med)、HCV_X3 與 Purine 的四組測試資料比較中,我 們系統所得結果的 selectivity、sensitivity 與 MCC 值皆明顯比 RNAshapes 的結果好 上許多。